Embed Size (px)
Citation preview

SEMIEXPERIMENTAL EQUILIBRIUM STRUCTURES FOR THE EQUATORIAL CONFORMERS OF N-
METHYLPIPERIDONE AND TROPINONE BY THE MIXED ESTIMATION METHOD
JEAN DEMAISON, Laboratoire de Physique des Lasers, Atomes et Molécules, Université de Lille I, 59655 Villeneuve d'Ascq Cedex, France
NORMAN C. CRAIG, Department of Chemistry and Biochemistry, Oberlin College, Oberlin, Ohio 44074
EMILIO J. COCINERO, Departamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco, Ap. 644, E-48080 Bilbao, Spain
JENS-UWE GRABOW, Institut für Physikalische Chemie and Elektrochemie, Lehrgebiet A, Gottfried-Wilhelm-Leibniz Universität, Callinstrasse 3A, D-30167 Hannover, Germany
ALBERTO LESARRI, Departamento de Química Física y Química Inorgínica, Facultad de Ciencias, Universidad de Valladolid, E-47011 Valladolid, Spain
H. D. RUDOLPH, Department of Chemistry, University of Ulm, D-89069 Ulm, Germany

PreliminaryMixed estimate structure for cis,trans-1,4-difluorobutadienea
17 parameters; rot. consts. for each 13C and 2H species from MW
aDemaison, J. F.; Craig, N. C. J. Phys. Chem. A 2011, 115, 8049-8054.

N-methylpiperidone
tropinone

N-Methylpiperidone (19 atoms)plane of symmetry28 parameters: 11 bond lengths, 17 bond angles
Tropinone (23 atoms)plane of symmetry34 parameters: 14 bond lengths, 20 bond angles

Ground state rotational constants from microwave spectroscopy with natural abundance for all the different heavy atom substitutions.1,2
Converted to equilibrium rotational constants by vibration-rotation constants (alphas) computed with the quadratic and cubic force constants from a B3LYP/cc-pVTZ model. At most 1% adjustments. Peter Groner did these calculations with his VIBROT program.
1 N-Methylpiperidone. Evangelisti, L.; Lesarri, A.; Jahn, M. K.; Cocinero, E. J.; Caminati, W.; Grabow, J.-U. J. Phys. Chem. A 2011, 115, 9545-9551.
2 Tropinone. Cocinero, E.J.; Lesarri, A.; Écija, P.; Grabow, J.-U.; Fernández, J. A.; Castaño Phys. Chem. Chem. Phys. 2010, 12 , 6076-6083.

Heavy atom Cartesian coordinates for N-methylpiperidone
Red final values; black from Kraitchman equations
a/Å b/Å c/Å O 2.6194(6) 0.0000 -0.3959(11) 2.619 0.021 -0.395 C3 carbonyl 1.4991(16) 0.0000 0.0632(26) 1.498 0.019i 0.068 C2 0.7366(14) 1.2671(5) 0.3697(12) 0.736 1.267 0.369 C1 -0.6779(19) 1.1923(6) -0.2005(22) -0.678 1.193 -0.196 N -1.3632(18) 0.0000 0.2783(16) -1.363 0.011 -0.278 C7 -2.7457(11) 0.0000 -0.1672(22) -2.745 0.013i -0.168
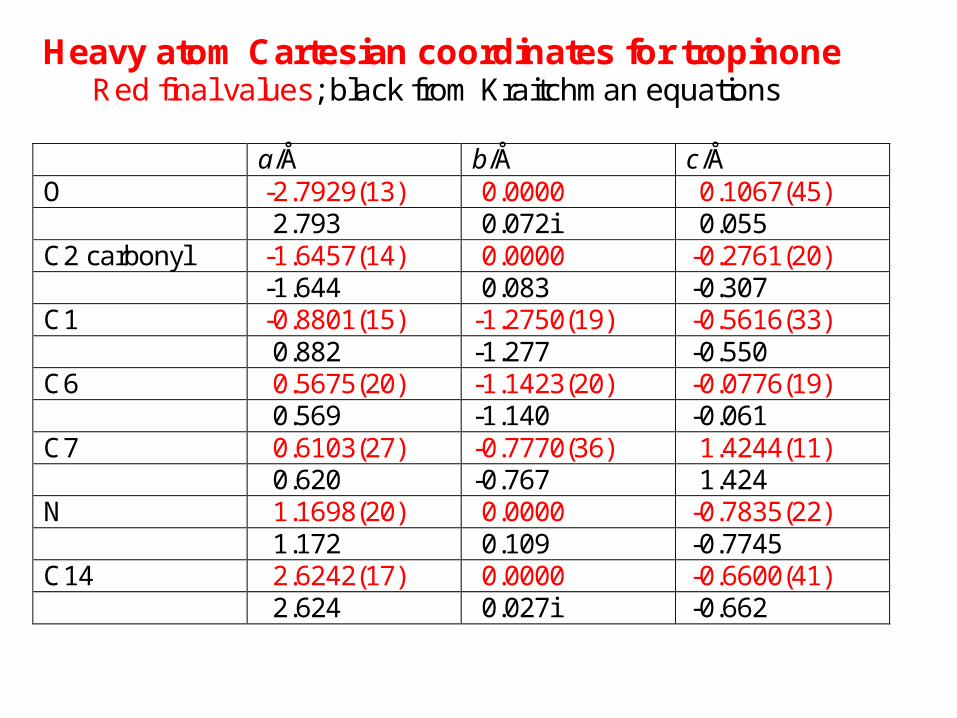
Heavy atom Cartesian coordinates for tropinone Red final values; black from Kraitchman equations
a/Å b/Å c/Å O -2.7929(13) 0.0000 0.1067(45) 2.793 0.072i 0.055 C2 carbonyl -1.6457(14) 0.0000 -0.2761(20) -1.644 0.083 -0.307 C1 -0.8801(15) -1.2750(19) -0.5616(33) 0.882 -1.277 -0.550 C6 0.5675(20) -1.1423(20) -0.0776(19) 0.569 -1.140 -0.061 C7 0.6103(27) -0.7770(36) 1.4244(11) 0.620 -0.767 1.424 N 1.1698(20) 0.0000 -0.7835(22) 1.172 0.109 -0.7745 C14 2.6242(17) 0.0000 -0.6600(41) 2.624 0.027i -0.662

Mixed estimation method for semiexperimental structuresa
Predicate values for bond lengths and bond angles derived from QC calculations (MP2/cc-pVTZ model) after applying a correction for the C=O bond. The correction came from comparing predictions with equilibrium values for various small molecules.
Concurrent fit of predicate values with appropriate uncertainties and of equilibrium rotational constants with appropriate (tighter) uncertainties. A stepwise process.
a A similar method was first applied in electron diffraction. L. S. Bartell, D. J. Romanesko, T. C. Wong, in: Chemical Society Specialist Periodical Report No. 20: Molecular Structure by Diffraction Methods, Sims, G. A.; Sutton, L. E., Eds., The Chemical Society London, 1975, Vol. 3, pp. 72-79.

Uncertainties
Predicates
CH bond lengths: ±0.002 Å
Heavy atom bond lengths: ±0.003 Å
Bond angles: ±0.3°
Torsion angles: ±0.5°
Equilibrium rotational consts.: ±0.02 MHz
too small but gives relatively greater weight

Structure of N-methylpiperidone (distances in Å, angles in degree) Method MP2 B3LYP Least-squares fit Basis set cc-pVTZ 6-311+G Predicate 0 17 28 r(C7–N) 1.453 1.454 1.4521(4) 1.4521(7) 1.4525(22) r(N–C1) 1.455 1.458 1.4553(3) 1.4556(6) 1.4562(15) r(C1–C2) 1.527 1.533 1.5255(5) 1.5270(10) 1.5268(26) r(C2–C3) 1.509 1.515 1.5083(3) 1.5097(5) 1.5102(14) r(C3=O) a 1.218 1.209 1.2126(4) 1.2113(8) 1.2108(20) r(C1–H18) 1.090 1.090 1.0904(9) 1.0905(33) r(C1–H19) 1.104 1.105 1.1046(9) 1.1033(32) r(C2–H16) 1.088 1.089 1.0883(9) 1.0884(33) r(C2–H17) 1.092 1.094 1.0925(9) 1.0910(32) r(C7– H8) 1.088 1.090 1.0887(9) 1.0884(33) r(C7– H10) 1.101 1.103 1.1009(9) 1.1007(33) (NC7H10) 112.33 112.87 112.29(13) 112.41(48) (NC7H8) 109.63 109.78 109.62(13) 109.60(48) a Corrected value of 1.211 Å from a correlation between calculated and equilibrium values for small molecules.

Structure of N-methylpiperidone (angles in degree) Problematic angles. Method MP2 B3LYP Least-squares fit Basis set cc-pVTZ 6-311+G Predicate 0 17 28 (H8NC7H10) -120.54 -120.64 -120.5(2) -119.79(75) (OC3C2C1) 128.81 135.30 131.771(57) 132.03(11) 131.90(23) (C3C2C1H19) -66.99 -70.9 -68.17(14) -68.02(50) (OC3C2H17) -112.95 -105.67 -109.96(17) -109.99(46) (OC3C2H16) 5.99 11.593 8.51(20) 8.39(59)

Structure of tropinone (distances in Å, angles in deg) Method MP2 B3LYP Least-squares fit Basis set cc-pVTZ 6-311+G(3df,2pd) Predicate 0 34 r(C2=O) a 1.2188 1.209 1.207(10) 1.2110(15) r(N5–C6) 1.470 1.473 1.467(6) 1.4712(20) r(N5–C14) 1.459 1.458 1.456(6) 1.4595(21) r(C1–C2) 1.511 1.521 1.515(6) 1.5140(19) r(C1–C6) 1.530 1.536 1.529(10) 1.5300(20) r(C6–C7) 1.547 1.556 1.556(12) 1.5454(22) r(C7–C8) 1.546 1.551 1.533(10) 1.5561(74) r(C1–H22) 1.0889 1.090 1.0889(16) r(C1–H23) 1.0920 1.094 1.0919(16) r(C6–H13) 1.0897 1.089 1.0897(16) r(C7–H11) 1.0892 1.089 1.0891(16) r(C7–H12) 1.0884 1.089 1.0882(16) r(C14–H15) 1.0887 1.090 1.0886(16) r(C14–H17) 1.0978 1.100 1.0977(16) (NC14H17) 114.00 114.49 114.50(25) (NC14H15) 109.08 109.26 109.07(24) a Predicate value of 1.211 Å.

N-methylpiperidone
tropinone

ConclusionsRotational constants are available from MW spectroscopy for
heavy atom substitutions in natural abundance.An equilibrium structure for the heavy atom backbone obtained
by the Kraitchman substitution method is usually flawed. The mixed estimation method, in which calculated internal
coordinates () and “observed” equilibrium rotational constants () are fit concurrently, yields good semiexperimental equilibrium structures.
This method has been illustrated with N-methylpiperidone and tropinone.
Good agreement with the structure predicted by the MP2/cc-pVTZ model with a few exceptions.

Acknowledgements
Peter Groner, University of Missouri-Kansas City
Oberlin College, Department of Chemistry and Biochemistry
National Science Foundation for support of the Beowulf computer cluster at Oberlin College





























