Embed Size (px)
Citation preview

SELF-ASSEMBLY OF RECOMBINANT HUMAN ELASTIN POLYPEPTIDES
WiTJiI POTENTIAL FOR USE IN BIOMATERIALS APPLICATIONS
Catherine M. Bellingham
A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosopy
Graduate Department of Chexnical Engineering aad Apptied Chemistry
University of Toronto
O Copyright by Catherine M. Bebngham 2001

Acquisitions and Acquisitions et Sibliogmphic SeMces services bibliographiques
The author has granted a non- exciusive licence allowing the National Lrkary of Canada to reproduce, loan, &'bute or seU copies ofthis thesis in microform, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts fiom it may be prineâ or othemise reproduced withouî the author's permissim.
L'auteur a accordé une licence non exclusive pexmettant a la Biôliotheque nationale du Canada de reproduire, prêter, disüibuer ou vendre des copies de cette thése sous La f m e de microfiche/nim, de reproduction sur papier on sur format électronique.
L'auteur conserve la propriété du droit d'auteur qiii protège cette thèse. Ni la thèse ni des errtraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.

SELF-ASSEMBLY OF RECOMBINANT HUMAN ELASTIN POLYPEPTIDES
WITB POTENTLAL FOR USE IN BIOMATERLALS APPLICATIONS
PhD. Degree, 2001
Catherine M. Bellingham
Department of Chemical Engineering and AppIied Chemistry
University of Toronto
Abstract
Eiastin is an extracellular matrix protein found in a nurnber of tissues, including the
large arteries such as the aorta, imparting the characteristics of extensibility andelastic recoil.
Once laid down in tissues, polyrneric elastin is not subject to turnover but is able to sustain its
mechanical tesilience through billions of cycles of extension and recoil. The process of
ordered assembly of elastin into its extracellular, polymeric form remains one of the Ieast
weH-understood steps in the biosynthesis of elastin. During this step, side chains of lysine
residues in elastin monomers must be oxidatively dearninated and brought into juxtaposition
in preparation for crosslinking. In vivo, several factors have been proposed to contribute to
the alignment of elastin monomers in the formation of polymeric elastin, including a
mimfibrillar scaffold and a cell-surface elastin binding protein.
We have used a series of srnall, recombinant polypeptides based on sequences of
human elastin to investigate the roles of various hydrophobic domains in promoting self-
aggregation, and to determine whether this self-aggregation facilitates specific alignment of
elastin polypeptides alIowing crosslink formation at lysine residues. Our resuIts demonstrate
that polypeptides with as few as three hydrophobic and two crosslinking domains are able to

self-aggregate into fibrillar stnrcnires essentialfy identical in apparance to those fomed by
the full-length elastin monomer, mpoelastin. Moreover, oxidation of lysine residues
following aggregation, using a simple oxidizing agent (pyrroloquinoline quinone), results in
spontaneous formation of lysine-derived covalent crosslinks between polypeptides, including
desmosine and isodesmosine. Fabrication of these covalently crossiinked elastin potypeptides
into membrane structures has aIso allowed assessment of their physical properties. Such
membranes possess an elastic modulus, and extensibility and recoil properties sirnilar to
those of native insoluble elastin.
These results strongly support the view that, independent of the influences of other
factors, monomers of elastin possess an intrinsic ability to organize themselves into
polymenc structures, aligning lysine residues for covalent crosslinking and forming matrices
with elastomeric properties. Understanding the basis of the self-organizational ability of
etastin-based polypeptides may provide important ches for the general design of self-
assembling biomaterials.

Acknowledgements
In completion of this work, 1 have been very fortirnate to have had the opportunity to
work with some wonderfui people, Fust, my supervisors Dr. Fred Keeley and Dr. Kim
Woodhouse. With different backgrounds and unique interests and strengths, 1 have had the
opportunity to l e m much about science, engineering and life from each of them. In common
was their unfailing enthusiasm for my work. Thank you both. 1 wodd also like to thank my
committee memkrs, past and present- First, Dr. AA Chakrabartty for his vduable input
throughout my thesis. Next, Dr. Michael Lee for encomging me to work with Dr. Keeley and
Kim when his own lab was moving to W f a x and for his contributions to my thesis in its eariy
stages. FinalIy, 1 would like to thank Dr. Elizabeth Edwards for agreeing to join my committee
toward the completion of my thesis and for her contributions to it.
On opposite coasts, Dr. Glenda Wright and Dr. Margo Lillie, both inspiring and
enthusiastic scientists (and wonderful women), helped me put my work into perspective and gave
me a fresh look at science. Thank you both. At the Hospital for Sick Children, 1 would like to
thank Dr. Shashi Joshi, Dr. Kostas Stathakis and Rey Interior for sharing their science wisdom
and techniques. 1 wouid iike to thank Dr. Maurice Ringuene, my teaching mentor. Maurice's
enthusiasm for teaching and his wonderfui sense of humour have inspired me to achieve
excellence in teaching. 1 would like to thank my friends and colieagues, past and present, at the
hospital and at UofT for their support and friendship. A special note of thanks to Eva and
Richard for their heip with my research. 1 would also like to thank the support staff at Sick Kids
and in Chemical Engineering for dl of their administrative help.
1 would Lke to thank my family for their support. My mom, Sylvia Bellingham, for
passing down her love of science to me. Had she had the opportunity, 1 know she would have
been a rernarkable scientist, Also, my sister Susan and her husband Stuart for sharing their home
and their two beautiful boys, Andrew and Jonathan, Donna and Ji, for their support when 1
needed them. And Carola, for treating me as if 1 was her own daughter. 1 am honoured.
1 have had the pleasure of spending time with many others on this road, some of whom
remain in my life and others whose lives have taken a separate path. For those who have gone, 1
wiU remember you. To my €iiends, thank you 1 consider myseif fortunate indeed to be part of
your lives.
Finally, to Brian, for sharing his life with me, enriching mine in the process. And to
Jackie, who believed in me and helped me believe in myseff. Thank you.

Chapter 2:
Chapter 3:
Chapter 4:
Chapter 5:
The EP20-24, EP20-24-24, and m l - 2 3 cDNA constructs were made by
Paul Robson and Lynne Cameron in the laboratory of Dr. Stephen Rothstein at
the University of Guelph, Guelph, Ontario.
The EP20-24' cDNA consûuct was made, expressed, purifie4 and
characterized by Richard Stahl at the Hospital for Sick Children.
1 did the transmission electron microscopy with the assistance of Dr. Glenda
Wright and Dorota Wadowska at the University of Pnnce Edward Island,
Charlottetown, PEI.
Transmission electron microscopy of the EP20-24-24 membrane was done by
Christine Campbell and Dorota Wadowska, in the laboratory of Dr. Glenda
Wright at the University of Prince Edward Island, Charlottetown, PEI.
Identification of desrnosine and isodesmosine by radioimmunoassay was done
by Dr. Barry Starcher at the University of Texas, Texas, USA. Crosslink
identification by ion exchange chromatography was done in the laboratory of
Dr. Allen Bailey at the University of Bristol, Bristol, UK. 1 did the mechanical
testing of the polypeptide membranes in Dr. John Gosline's laboratory at the
University of British Columbia, Vancouver, BC, with the assistance of Dr.
Margo LiUie. Polypeptide expression and purification were done by Eva Sitan
and Richard Stahi at the Hospital for Sick Children.

Chapter 1 ......................................................................................................... Introduction 1
General Introduction to Elastin and Elastic Fibres ............................... 2 UItnrsfnrcfure of ElastUI ..................................................................... 3 Mechanicd Properfies of Eiastin ...................................................... 4
Tropoelastin: Nature of the Gene .......................................................... 6
Tropoelastin: Protein Characteristics ...................................................... 7 In Vivo Assembly and Crosslinking ........................................................ 7 SecondarJi Structure ............................................................................ l i
........................................................................ Self-Aggregation of Elastin 12 ......................................................................................... Coacervation 12
M e c h k m of Coacervation ............................................................. 12 ............................. Factors Influencing the Coacervation Temperatrrre 13
........................................................... ulh.aslrucirrre of the Coacervate 14
Aggregation as an Ordering Process ...................................................... 14
........................................................................................................ Rationale 16
..................................................................................................... Hypothesis 17
...................................................................................................... Objectives 17
Chapter 2 Production. Purlftcation and Characterization of Recombinant Human Elastin Polypeptides ................................. .... ............................................ 18
Introduction ................................................................................................... 19
Materials and Methods ................................................................................ 21 ..................................... PCR Amplification of Human Elaslin cDNA's 21
................................................................................. Rimer Sequenees 21 .............................................................. Elastin Polppeptide Constnrcts 22
Elastin PoIypeptide (EP) 20-24 ........................................................ 22 Eiastin PoIrpeptide (EP) 20-24-24 ................................................... 27 Eiascin Polypeptide (Em 20-24-26 ................................................... 27

Elastin PoIypeptide (EP) 20-24-30 ................................................... 27 Elastin Poiypeptide (EP) 21-23 .................................................. 28
............................ Expression and Funfiation of Elastin Polypeptides 28 ............................. Production of Antibodies to EP2&24 and EP21-23 -30
................................................................................... Western Blo#ing 31
.......................................................................................................... Results 32
..................................................................................................... Discussion 39
Chapter 3 Self-Assernbly Characteristics of Recombinant Human Elastin
............................................................................................................ Polypeptf des 4 2
Introduction ............................................................................................... 43
................................................................................ Materials and Methods 46 .................................................................... Coacewation Experiments 46
Polypeptide Concentration .................................................................... 47 .............................................................................. SfafrSticuf Analysk 4 7
............................................................................................................ Results 48
..................................................................................................... Discussion 57
Chapter 4 Ultrastructure end
Introduction ............................. Self-Assembly .. .................................... 62
................................................................................................... 63
................................................................................ Materials and Methods 65 Coacmotion Experiments ................................................................... 65 Transmission Elecîron Microscopy (TEM) ........................................... 65
...................................................................... Circrrlar Dichmisnt (CD) 66
Discussion ................................................................................................. 76 h t r u c t u r e ........................................................................................ 76 Revenibk tuui Irreversible S&ges of Cwcewokokon .............................. 78 Mode1 of EGastin Assembly .................................................................... 79

Chapter 5 Alignmsnt of Mechanics
Elastin Polypeptides: Crosslinking and Polymer ......................... ,. ................................................................................ 81
Introduction ................................................................................................... 82
Materials and Methods ............................................... 83 Oxidative Deaminaibn of Lysine Redues Using HorserasLrh Perondnre ........................................................................ 83 h ù M v e haminuîion of Lysine ReMues Using Pymloquinoline Quihone (PQe) ..................................................... 83
................................................ Fabrication of Polypeptide Membranes 84 Transmission Elecaon Mimscopy of an EPSib2424 Membrane ........................................................................................... 85 lnsolubilitp of Membranes Prepiwedfiom Polypeptides ....................... 85 &termination of Mechankul Roperîies .............................................. 85
.......................................................................... Sample Mounting 86 .......................................................... ..................... Tensile Tests ..,,. 86
Analysis ........................................................................................... 89 Saristicai Analysis .......................................................................... 90
Resuits ............................................................................................................ 91 ..................................................................... Evidence for Alignmenf 9 1
Natute of Crosslinks Fonned ............................................................. 94 Charrrcterization of Membmnes .......................................................... 98 Mechnicd Roperîiés ..................................................................... 98
Discussion .................................................................................................. 109 Aügnment ............................................................................................ 109 Membrane Fobricaîion and ~ c î u r e ......................................... 110 Mechankm of l?h&c@ .................................................................. 110
Chapter 6 Conclusions and RecommendatIons ............................................................ 113
Conctusions and Recommendations ................................................... 114 General Summmy ............................. ,., ..,,., ........................................ 114 Role of the Hydrophobie Lbmui'ns in Assembiy .................................. 115 Repirentents forAssembly into un ûrgonk44 Elasth-LiRo ManLr ..................... ,. ............................................... 116

Importance of Sequence of the Hydrophobie Domains in ............................................................................................. Assembiy 117
............................................. Ultrastructure and a Mode1 of Assembly 118 Potential Uses of the Elartin Poiypeptides m Bwmaterhh
........................................................................................ Applications 119
............................................................................................................... References 122

List of Figures and Tables
Figure 1.1,
Figure 13.
Figure 2.1.
Figure 2.2.
Figure 23.
Figure 2.4.
Figure 2.5.
Figure 2.6.
Figure 3.1.
Figure 33.
Figure 33.
Figure 3.4.
Figure 3.5.
Figure 3.6.
Figure 4.1.
Sequence similarities ainong a number of self-assembling matrix ............................................................................................ proteins 8
Formation of covalent crossLnks in elastin. ......................................... 10
Recombinant DNA technology used to produce the polypeptide constructs, and rnethodology for expression and purification of the polypeptides ..................................................................................... 23
Rep~sentations of the elastin polypeptide expression constructs .......... 24
Polypeptide sequences for each of the exons with repeating peptide motifs bracketed ................................................................................... 25
SDS polyacrylamide gel electrophoresis of recombinant elastin pdypeptide EP20-24 at various stages of purification .......................... 34
............................................................. Mass spectrometry of EP20-24 37
...................................... Sequencing of EP2 1-23 by mass spectrometry 38
Coacervation is followed by monitoring turbidity by Lght scattering at 440nm. ................................................................................................. 45
Comparison of coacervation characteristics of purified recombinant elastin polypeptides EP21-23, EP20-24 and EP'O-24-24 (representative curves) ......................................................................... 50
Effect of NaCl concentration on coacervation temperatures of recombinant elastin poLypeptides EP20-24 and EP20-24-24. ................ 52
Effect of polypeptide concentration on coacervation temperahires of recombinant elastin polypeptides EP20-24 and EP20-24-24 ................. 53
Effect of polypeptide concentration and NaCI concentration on the rate of coacervation .................................................................................. SS
Effect of polypeptide concentration and NaCl concentration on the maximum absorbace achieved during coacervation ............................ 56
Representative coacervation curves for EP20-24-24 indicating that reversibiity of coace~ation is he-dependent ...................................... 69

Figure 4.2.
Figure 43.
Figure 4.4.
Figure 4.5.
Figure 4.6.
Figure 4.7.
Figure 4.8.
Figure 4.9.
Figure 5.1.
Figure 5.2.
Figure 5.3.
Figure 5.4.
Figure 5.5.
Figure 5.6.
Figure 5.7.
Prior to coacervation the elastin polypeptides have a globular structure, visible with transmission electron microscopy. ..................................... 70
Above the coacervation temperature, open filamentous networks are visible with transmission elecmn microscopy ...................................... 71
Filaments of EP20-24-24 formed folIowing coacervation show some directionality .................................................................................... 72
Overnight incubation above the coacervation temperature results in fibrillar coacervates . ...................... .. ......... ...... .............................. 73
Cornparison of ERO-24, =O-24-24 and tropoelastin structures at three stages of coacervation .................................................................. 74
Changes in secondary structure of EP20-24 are coincident with coacervation ..... ................................................................................... 74
Fibre structures of coacervated mû-24 and EP20-24-24 as a reflection of opportunities for overlap of hydrophobic domains during self-aggregrition .............................................................................. 77
A mode1 of elastin seIf-assernbly showing reversible and irreversible stages ............................................................................................. 80
Mechanical testing apparatus ..................... .............. .... ......... ................ 87 Schematic of the membrane mounting procedure ............................. 88
Formation of covalent crosslinks in EP20-24-24 following coacervation and oxidative deamination of lysine residues with horseradis h peroxidase . ... .... .... .. ............... ............... . . . ............. 92
Crossiink formation in the coacewated polypeptides using PQQ to oxidatively deaminate the lysine residues ................... - ......................... 93
Coacewation is required to align Iysine residues for crosslink formation ................................................ + .......................................... 95
Crossiink formation does not occur in EP21-23, the polypeptide Iacking hydrophobic domains . .. ........................... ........................... 96
Chromatographie cornparison of crosslink profdes for EP20-24-24 and insoluble elastin ......................................................................... 97

Figure 5.8.
Figure 59.
Figure 5.10.
Figure 5.11.
Figure 5.12
Figure 5.13.
Figure 5.14.
Figure 5.15.
EP20-24-24 and EP20-24' membranes fabricated using the cuvette ............................................................................................... protocol .99
............................................................ EP20-24 membrane fragments 100
EP2û-24-24 membranes visualized using tight microscopy before and ........................................................... after CNBr or NaOH treatment IO1
An EP20-24-24 membrane appears amorphous using transmission ........................................................................... electron rnicroscopy 102
Stages of tensile testing of an m0-24-24 membrane ......................... 103
Representative stress-strain curve of an EP20-24-24 membrane ................................................................................... loaded to break 104
Representative stress-strain c w e for cyclic loading of an ....................................................................... EP20-24-24 membrane 105
Representative stress-strain curve showing the third cycle of cyclic ioading of an EP20-24-24 membrane .................................................. 106
Table 2.1. Summary of polypeptide characteristics including the number of residues per mole, number of hydrophobic domains, molecular mass and hydropathy values .......................................................................... 26
Table 2.2. Representative amino acid compositions of purified recombinant humart elastin polypeptides (actual) compared to expected compositions based on known peptide sequences (amino acid residueshoIe) ................... ... 35
Table 23. Cornparison of molecular mass data calculated from the amino acid composition (expected) versus the value determined using mass spectrometry (actual) ............................................................................ 36
Table 3.1. Coacervation temperatures, rates of coacervation and maximum absorption values for the five elastin polypeptides together with the number of hydrophobic domains, molecular m a s and hydropathy values for each polypeptide. ................................................................ 5 1
Table 5.1. Summary of the mechanical properties of the elastin pdypeptides. ..... 108

List of Abbreviations
A
AEBSF
CD
cDNA
CNBr
Des
DMSO
m DTr
EBP
EP
e-PTFE
G
GST
HRP
HSC
1
IDes
rnG
K
L
LB
LNL
MAGP
MeroDes
MW
P
PAGE
PBS
alanine
4-(2-aminoethy1)-benzenesulfonyl fluoride hydrochloride
circular dichroism
complementary DNA
cyanogen bromide
desmosine
dimethy lsuifoxide
deox yribonucleotide triphosphate
dithiothreitol
elastin binding protein
elastin polypeptide
extended polytetraflouroethylene
glycine
glutathione S-transferase
horseradish peroxidase
Hospital For Sick Children
isoleucine
isodesmosine
isopropyl fbgalactopyranoside
lysine
leucine
Luria-Bertani (culture medium)
lysinonorleucine
mimfibril-associated glycoprotein
merodesrnosine
molecular weight
pdine
polyacrylamide gel electrophoresis
phosphate buffered saline

PCR
PQQ SDS
STE
TBS-T
TEM
v Y
YT
polymerase chah reaction
pyrroloquinoline quinone
sodium dodecyl sulfate
sodium 15s EDTA buffer
tris buffered saline with tween
transmission electron mimscopy
valine
tyrosine
yeast tryptone

List of Sy m bols
density
engineering strain
engineering stress
eliipticity
absorbance
original cross sectional area
concentration
optical pathway
eIastic madulus
molar extinction coefficient
force
original length
change in Length (extension)
molecular mass of chains between crosslinks
Newton
Pascal
universal gas constant
temperature (Kelvin)
coacervation temperature (C)

Location of Suppliers Amencan National Cm., Chicago, nlinois, USA
Amersham Pharmacia Biotech, Inc., UppsaIa, Sweden
BioRad Labontories, Hercules, California, USA
Canemco Supplies, Quebec, Canada
Elastin Products Company Inc., Owensville, Missouri, USA
Fisher Scientific Ltd, Nepean, Ontario, Canada
Gibco BRL, Rockville, Maryland, USA
Pierce, Rockforci, IlIinois, USA
Qiagen Inc., Valencia, California, USA
Sigma-Aldrich Co., St. Louis, Missouri, USA
Stratagene, La Jolla, California, USA

Chapter 1
lntroduct ion
Well begun is haff &ne. Mary Poppins

Elastin is an extracellular matrix molecule found in tissues requiring extensibility and
elastic recoif. Tissues containing elastin include the lungs, the skin, the arteries, and the
elastic ligaments, aii tissues which mechanicdly must be able to extend under an applied
load and subsequently recoil when the load is removed. The amount by dry weight of eIastin
in tissues varies, making up approxirnately 40% of the aorta and approximately 80% of the
ligarnentm nuchae (Ayer, 1964; Gosline, 1976; Harkness, 1968). Mechanically, elastin is
extremely durable and, in the large arteries for example, can undergo billions of loading and
untoading cycles without fdure. This is ail the more remarkable given that. under normai
ci~umstances, eIastin is laid down only during devetopment and subsequently has an almost
complete lack of turnover (Davis, 1993).
Whik the importance of elastin for the mechanical properties of tissues is clear,
ment evidence suggests that it may pIay other roles during development. In the homozygous
elastin knockout mouse, mice died shoaly after birth from artenal obstruction caused by
proliferation of srnwth muscle celis (Li et al., 1998). These results suggest that elastin also
plays a role in arterial development, perhaps through regdation of ceU proliferation.
Similarly, in Iung tissue, elastin appears to be involved in regulating terminai ainvay
bmching during pulmonary deveIopment (Wendel et al., 2000).
Elastic fibres are made up of two major components: an eiastin component and a
microfibrillar component. The elastin component is synthesized as tropoelastin, the soluble
ptecursor to insoluble, crosslinked elastin. Tropoelastin has a molecular weight of
approximateIy 70 Da. The microfibriliar component, consisting of 10-12 nm filaments, is
made up of at lem five distinct proteins but thought to be predominantiy the 350 kDa protein
fibriiiin, of which there are fwo foms, fibillin-1 and fibriliin-2 (Mecham et al., 1994; Sakai
et al., 1986; Shang et ai., 1994). The rnicrofiôrils also include two mi~rofibril-associated
glycoproteins (MAGP-1 and MAGP-2) (Gibson et ai., 19%; Gibson et al., 1991). Elastic
fibres may also include a number of other components including a 64 kDa chaperone protein
known as the elastin binding protein (EBP), (Hinek and Rabinovitch, 1994; Hinek et ai.,
1988), and lysyl oxidase, the enzyme that catalyzes oxidative deamination of lysine residues
in preparation for crosdink formation (Kagan and Trackman, 1991).

Ulfrartnrcture of Elastut
Early studies on the uioastmcture of elastic fibres from bovine ligamentum nuchae
and rat aortic tissue showed two components, a peripheral fibrillar component and a central
amorphous component (AIbert, 1972; Greenlee Jr. et al., 1966; Ross and Bomstein, 1969).
The fibrillar component was believed to be glycoprotein in nature, whereas the centrai
amorphous component had the amiuo acid composition of elastin (Ross and Bomstein,
1969). These snidies used standard epoxy embedâed sections to examine elastic tissue by
electron microscopy and led investigators to believe that elastin had an arnorphous structure.
In addition, the lack of birefiingence in single elastin fibres examined with polarized light
microscopy also suggested that insoluble elastin was amorphous in structure (Aaron and
Gosline, 1980).
Other investigators desaibed insoiuble elastin as having a fibrillar structure, with
single fibre diameter ranging h m 4 to 7 pm (Gotte et ai., 1972). Scanning electron
microscopy on both native bovine ligamenhm nuchae and purified insoluble elastin from
ligamennim nuchae showed fibrillar structures paraltel to the main axis of the ligament,
suggesting that the structure of elastin was ordered (Gotte et ai., 1972). In addition, fine
suspensions of insoluble elastin from Ligamenfum nuchae stained with various negative stains
showed filaments, 3.5 to 10 nm in diameter, aligned approximately in parallel (Gotte et aI.,
1972; Gotte et ai., 1974, Mecham and Heuser, 199 1). Similar observations were made by
Serafini-Fracassini and coworkers who ais0 showed ordered structure with low-angle X-ray
diffraction of stretched specimens (Serafini-Fracassini et ai., 1976). More defined reflections
seen in the stretched specimens were speculated to be due to the alignment of fibnls on
stretching. Investigations using fieeze-Fracture techniques and scanning force microscopy to
study elastin structure also reveaied fibriIIar structures that were more apparent after
stretching (Pasquali Ronchetti et al., 1998; PasquaIi Ronchetti et al., 1979). Based on these
resuits, it was suggested that the filaments making up the larger fibres were themsetves
composed of globular structures iinked in one dimension.
To resolve the question of the u l ~ û u c t u r e of elastin as amorphous or fibrillar,
Quintarelli and coworkers cornpanxi eIectron mimscopic images of epoxy embedded
fragments of elastin with suspensions of elastin placed onto carbon coated grids. The
observations that the embedded samples appeared amorphous whereas the dispersed samples

were filamentous prompted the authors to suggest that these diffetences in structurai
appearance of insoluble elastin might be explained by the different preparation methods used
in the studies (Quintarelü et al., 1973).
Mechanicd h p e r t i e s of Elasfin
The mechanical properties of a material are determined by the mechanicai properties
of each component in the material, the relative mount of each component, the orientation of
the components and the connections within and between cornponents (Ward, 197 1). In most
tissues (e.g. the aorta) elastin is found in association with collagen. Arteries exhibit
non-linear stress-strain behaviour, initially showing low stiffness at low extension followed
by higher stiffness at increased extension. The initial Iow stiffness component represents
extension of elastin and uncrimping of collagen fibres in the tissue. The increased stiffness
component represents m i t m e n t of collagen fibres to bear the load (Roach and Burton,
1957). The collagen will recrirnp on unioading and the elastin recoils, restoring the original
tissue configuration. Thus elastin provides elasticity and elastic recoil to these tissues
whereas collagen provides tende strength. Under a continuous load, elastin does not
experience creep and, under cyclic loading, eIastin efficientiy stores energy, showing Iittle
hysteresis in the loading/unloading curves (Gosline, 1976; Harkness, 1968).
The organization of the components within a composite materiai is aIso important for
its mechanicd properties. The controversy over the ultrastructue of elastin, amorphous
versus ordered or fibrillar, has consequences for the explanation of the mechanism
underlying the mechanical properties of insolubb elastin. Several models have been
proposed to explain the elastic behavior of insoluble elastin. A common feature of aü the
modeis is that the restoring force is enmpy driven.
The earliest model, proposed in 1958, suggested that elastin is a random network of
kinetically free molecules with mechanicd properties consistent with a Iightiy cmsslinked
rubber (Hoeve and Rory, 1974; Hoeve and Hory, 1958). As with rubber, the elastic
mechanism would be driven by changes in conformationd entropy (Rosen, 197 1). Due to the
decrease in avaiIable chah configurations upon an appiied force, the random network goes
from a state of high entropy to one of lower entropy. Upon release of the force. the network
returns to a higher state of entropy, providing energy for elastic recoil. However, the

meçhanism of elasticity for elastin has been proposed to differfiom that of rubber, with
respect to contributions h m the hydrophobic interactions in elastin or solvent entropy
(Gosline, 1978). Under an applied load, the hydrophobic residues becorne exposed to water,
furthet ordering water around non-polar residues exposed on stretching. This ordering then
contributes to the stored elastic energy and hence the restoring force (Gosline, 1978;
Vrbvski and Weiss, 1998).
Other models of elasticity, including the liquid drop model, the oiled coil model, and
the librationai model, assume order. The liquid drop model is an extension of the tw+phase
structural model of Partridge. Partridge assumed that elastin is composed of crosslinked
globular molecules with the hydrophilic groups on the surface of the globules and the
hydrophobic; groups buricd inside, protected from contact with water (Pwûidge, 1966). The
liquid drop rnodel also assumes that tropoelastin molecules are globular and are held together
by intermolecuIar crosslinks. However, both hydrophobic and hydrophilic residues are
suggested to occur at the water-globule interface since the number of hydrophilic residues in
elastin are not sufficient to cover the entire exposed surface (Weis-Fogh and Andersen.
1970). The mechanism of elasticity is still entropie, but includes contributions h m both
conformational and solvent entropy.
A second model, the oiled coil model, also assumes an ordered structure. In this
model, tropoelastin molecuIes are assumed to be fibriiiar, made up of rigid a-helical
crosslinking regions altemating with flexible hydrophobic regions termed 'oiled coils'. The
oiled coil is described as a broad coil consisting of a series of 0-tums, with glycine residues
on the outside of the coil in contact with water, and the hydrophobic residues, including
proline and vdine, buried Again, sotvent entropy contributes to the mechanism of elasticity,
with the hydrophobic residues on the interior of the coi1 coming into contact with water upon
stretching (Gray et al., 1973).
The most recent model of elasticity is the iibrational entropy mode1 (Urry, 1984;
Urry et ai., 1983). This model is based on extensive work on synthetic polypeptides modetied
on the hydrophobic sequences of elastin. Urry proposes that the hydrophobic regions of
eIastin form a bspiraI structure, defined as a series of b s forrning a hetical structure.
During coacervation, the hydrophobic surfaces of the B-spirals interact to form fibrillar
structures. The bspirais are interspersed with crosslinking regions coniaining the

intemolecular crosslinks. Urry suggests that in the relaxed state fhpirals undergo interna1
chah dynamics, rocking motions, which he calls librations. When stretched, these librational
motions become damped, decreasing entropy, and thereby providing the restoring elastic
force (Uny, 1984).
While it appears that the mechanism of elastic recoil is entropy driven, and that
models of elastin structure assuming an ordered, perhaps fibrillar structure are Iikely to better
represent the structure and properties of insoluble elastin, it is clear that M e r work will be
required to fully understand the relationship between structure and mechanics of insoluble
elastin.
Tropoelastin: Nature of the Gene
The human elastin gene is approximately 45 kb in size, with an exon to intron ratio of
1:20. There are 34 exons in the human gene. With the exception of exon 1, containing the
signal peptide, and exon 34, containing the C-terminal domain and 3'-untranslated region, al1
of the exons code for either a hydrophobic or a crosslinking domain (Bashir et ai., 1989).
While there is a single gene for elastin, a number of mRNA splice variants have been
reported which result in several isoforms of tropoelastin. Exons subject to splicing are
hydrophobic exons 22,24,32 and 33 and msslinking exon 23 (Boyd et ai., 1991; Indik et
ai., 1989; indik et al., 1987; Yeh et al., 1987). In human cDNA, exon 22 has not been found,
indicating it is dways spliced out. The role of these various isoforms is not clear. Exon 26A,
a hydrophilic exon with a sequence atypical of elastin, is found only infrequentIy in human
elastin, and Weiss and coworkers found little difference in physical properties between
tropoelastin containing 26A and the isoform lacking it (Jensen et al., 2000). Others have
suggested that the isoform containing exon 26A is a less efficient substrate for lysyl oxidase
(Bedell-Hogan et al., 1993), which may afiect crosslinking patterns within the moIecule.
Gene structures of elastin between species are similar in nature, particularly in their
arrangement of hydrophobic and crosslinking exons. Exons 34 and 35 are rnissing from the
genes of humans and some primates, and exon 26A has only been found in the hurnan gene.

Tropoelastin: Protein Characteristics Tropoelastin has a repeating domain structure, alternating between hydrophobic and
crosslinking regions. In general, tropoelastin is extremely hydrophobic with approximateiy
80% of the total residues made up of proline (P), valine (V), giycine (G), leucine (L),
isoleucine O and alanine (A). One third of the amino acid residues are glycine, These non-
polar amino acid residues are found predominantIy in the hydrophobic regions of
tropoelastin. TypicaI sequences found in these domains, often present in tandem repeats,
include PGGV, PGVGV, PGVGVA, and GGLGV. These types of repetitive motifs are found
in elastin of a11 species aithough the specific sequences may Vary. For example, the sequence
encoded by exon 24 contains the hydrophobic peptide motif PGVGVA repeated seven times
in tandem in human and bûboon elastin (Szabo et ai., 1999). In cornparison, exon 24 of
bovine eiastin contains the sequence PGVGV repeating four times in tandem. In chicken
elastin this sequence is repeated ten times in this exon (Figure 1.1).
The crosslinking regions are rich in alanine and also contain the lysine (K) residues
fiom which the crosdinks of elastin are formed. Alanines and lysines are typically found in
sequences such as AAAKAAKAA or AAAKAAAKAA where the lysine residues occur in
alanine-rich sequences and are usudly separated by two or three alanine residues (Gerber and
Anwar, 1974). In human elastin, lysine makes up 4% of the total amino acid residues with
typicd crosslinking regions containing either two or three lysine residues. Aromatic residues
(tyrosine or phenylalanine) are found C-terminai to some of these lysine residues, and have
been suggested to play a role in formation of some types of crossIinks (Baig et ai., 1980;
Gerber and Anwar, 1975).
In Viio Assentbly and Crosslinking
Tmpoelastin is beiieved to be escorted through intracellular compartments and
pmented on the surface of the ce11 for orderly incorporafion into the elastic fibres by the
elastin binding protein (Hinek and Rabinovitch, 1994; Hinek et al., 1988). It has been
speculated that microfibrils act as scaffolding for elastin deposition in the extraceIIu1ar matrix
(Ross and Bomstein, 1%9), and that the C-termind of mplastin, containing a conserved
tetrabasic sequence may bind to the N-terminai region of MAGP-I, helping to ahgn the
tropoelastin molecdes (Brown-Augsburger et ai., 1996; Rosenblwm et al., 1993)- Once in

Chicken PGVGV
Human PGVGVA
ELASTIN (exon 30)
Rat
Chicken
LAMPRIN
03 (sea lamprey)
CHORION CLASS B PROTEINS (domestic silkmoth)
OOTHECIN (American cockroach)
SPIDROIN 1 (spider)
PRE-COL-D (mussel byssus)
PGVGV PGVGV PGVGV PGVGV PGVGV PGVGV PGVGV PGVGV PGVGV
PGVGVA PGVGVA PGVGLA PGVGVA PGVGVA PGVGVA
GLGGA GGLGA GGLGA GGLGA GGLGA GGLGA GGLGA GGVI. .PGAVGLG
GVPGA GVPGV GGIP. GGLGV GGLGV GGLGA GGLGA . . . . . . . . . . GVG GLVGA AGL.. GGLGV GGLGV . . . . . . . . . . . . . . . . . . . . .PGVGGLG
LGHPV GGLGY GGLGY GGLGY GGLGA AGLGY GGLGY
IGCGRGC GGRGY GGLGY GGLGY GGLGY GGLGG GCGRG
YGGY GGLGY GGLGY GGLGY GGLGY GGLGY GGLGY GGLGY GGLGY
GQGGY GGLGS QGAGR GGLGG QGA
PGVGP GGLGG LGGLG AGGLG GGLGG GLGGL GGAGG LGGGL GGLGG
Figure 1.1: Sequence similarities among a number of self-rissembling mlitrix proteins. Exon 24 of chicken tropoelastin contains the reperitive sequence PGVGV ten times in tandem, A similar sequence, PGVGVA, repais seven times in tandem in human tropoelastin ai the same site. Elastin also contains the GGLGVJA repeat sequence, ii sequence similar to the GGLGY sequence that is present in other self-assembling proteins including lamprin, the major mairix protein of Iümprey cariilage.

the extraceildar matrix, lysyI oxidase oxidatively deaminates the lysine residues found in the
mssiinking regions, allowing intermolecular crosslinking into a stable elastin matrix (Eyre
et al., 1984; Kagan and Trackman, 1991). Once crosslinked, tropoelastin cannot be
solubilized from the matrix even under harsh extraction conditions such as boiling in 0.1M
NaOH for 45 minutes (Lansing et al., 1952). Treatment with oxalic acid or potassium
hydroxide does not release soluble tropoelastin h m insoluble elastin, but nther only
disrupts peptide bonds giving soluble fragments of elastin (a-elastin and fl-elastin, or K-
elastin respctively), leaving the crosslinks intact (Jacob and Robert, 1989; Pariridge et al.,
1955). The rnechanisms of elastin crosslinking have not ken fully elucidated. However, it
has been weii-established that crosslinks occur through the lysine residues, ultimately
forming desmosine and isodesmosine, the predominant crossiinks of elastin.
Four Lysine residues are involved in each desmosine or isodesmosine crosslink. The
initiai step in crosslink formation is the enzyme-cataiyzed oxidative deamination of the
lysine residues. in vivo, the enzyme that catalyzes the deamination of the lysine residues is
lysyl oxidase, a copper4ependent amine oxidase (Ksigan and Trackman, 199 1). Animais
with a dietary deficiency of copper show incomplete crosslinking of tropoelastia to insoluble
elastin, and increased proportions of tropoelastin (Sandberg et al., 1969).
In formation of desrnosine and isodesmosine crosslinks, lysyl oxidase oxidatively
deaminates three of the four lysine residues involved to allysine, an
u-aminoadipic-6semialdehyde. It has been speculated that the presence of a tyrosine or
phenylalanine adjacent to a lysine residue in eiastin may prevent deamination of that lysine
(Baig et al., 1980; Foster et al., 1974; Reiser et al., 1992), providing the nitrogen in the
pyidinium ring of desmosine. Deamination is followed by spontaneous condensation with
other modified and unmodified lysines to form the crosslinks of elastin. A schematic of the
cmslink pathways is shown in Figure 1.2.
There are aiso a srnall nurnber of reduced crosslinks present in insoluble elastin,
incIuding lysinonorleucine, formed by the condensation of allysine and lysine side chains,
allysine aldol, formed by the condensation of two allysine side chains, and merodesrnosine,
formed by the addition of a lysine to allysine aldol. The two possible pathways leading to
desmosine formation have been suggested to be by the spontaneous condensation of
dehydrolysinonorleu~ne and allysine aldol or by the addition of an ailysine to


merodesrnosine. Whether desrnosine or isodesmosine forms may depend on the site of the
unmodified lysine residue in the sequence of the crosslinking region. Desmosine and
isodesmosine were initially thought to be unique to elastin, but have subsequently been
identified in smaller quantities in eggshell membrane proteins of the iguana and the chicken
(Cox et al., 1982; Starcher and King, 1980).
While it is believed that crosslinks usually form between two tropoelastin molecules
(Foster et al., 19741, the possibility of crosslinks involving three tropoelastin moIecules has
been suggested. At Ieast two hydrophobic domains contain three lysine residues, theoretically
pennitting formation of a desmosine crosslink between two tropoelastin molecules, and
leaving the third lysine in each molecule to form a crosslink with another tropoelastin
moiecule (Brown-Augsburger et ai., 1995). Formation of crosslinks involving three
tropoelastin molecutes would have important implications for the assembly of elastin.
Secondary Structure
Based on the prirnary sequence of mpoelastin, the secondq structure of both human
and bovine tropoelastin has been predicted to be predominantl y structure for the
hydrophobic domains, alternating with a-helicd segments correspondhg to the alanine-rich
crosslinking regions (Debelle et al., 1992). The overall conformation has been predicted to be
approximately 76% ~stmcture, 6% random coi1 and 18% a-helix (Debelle et ai., 1992).
The alanine-rich sequences of the crosslinking domains are predicted to be a-helical
in structure. Such a-helicai structure is supprted by the fact thrit typicd sequences in the
crosslinking domains contain two lysine residues separated by either two or three alanine
residues. If these domains are a-helical in nature, the spacing of the lysine residues wouId
bring the lysines to the same side of the helix, a position that would be favourable for
crosslink formation (Debelle et ai., 1992; Gray et al., 1973).
ExperimntaUy, the circdar dichroism spectra of recombinant tropoelastin show a
large proportion of structure, but Iess a-helix than predicted (Vrhovski et al., 1997).
Similar results were found using soluble tropoelastin isoIated from bovine tissue (DebeIIe
and Alix, 1995), bovine and human ~elastin (Debelle et ai., 1995; Debelle et al., 1998), and
a-elastin (Foster et al., 1976; Mamrni et ai., 1968; Starcher et al., 1973; Tamburro et al.,
1977), where K-elastin and a-elastin are heterogeneous mixtures of hydrolyzed elastin.

Peptides based on the hydrophobic domains of elastin, for example poly(VPGVG), also
showed significant amounts of B-stnicture (Abdel Rahman et ai., 1987; Uny et al., 1985a;
Urry et ai., 198%). The circular dichroism spectra of these polypeptides were interpreted as
an ail-fl protein, contaking distorted or short fbsheetlfbturn regions (Debelle and Aüx, 1995;
Manavalan and Johnson Jr., 1983). Urry has speculated that these sequences form a series of
pturns, a helicai arrangement termed a B-spiral (Urry, 1983).
Self-Aggregat ion of Elastin Coacervation
One of the striking features of elastin is its ability to self-aggregate. This property of
elastin has generalIy been investigated in virro using measurements of the ability to
coacervate. Coacervation is defined as a reversible phase separation in which a protein in
solution forms molecular aggregates upon an increase in temperature, and separates h m the
solvent as a separate phase (Bungenberg de Jong, 1949; Uny, 1982). This process has been
described for both tropoelastin and a-elastin (Bressan et ai., 1983; Cox et ai., 1974; Cox et
al., 1973; Vrhovski et al., 1997). Tropoelastin or polypeptides derived from insoluble elastin
are sohble at low temperatures but aggregate upon an increase in temperature, spontaneously
forming fibrillar-type structures. Direct evidence that the hydrophobic domains are involved
in the aggregation pmess cornes from a number of investigators who have synthesized
polypeptides based on sequences found in the hydrophobic domains of elastin (Castiglione
Morelli et ai., 1993; Reiersen et al., 1998; Urry et al., 1974). One such polypeptide,
poIy(PGVGV), was reported to coacervate and form similar fibrillar structures to those seen
with tropoelastin and a-elastin (Tamburro et aI., 1995; Urry et ai., 1974).
Mechanisni of Coacervation
Coacervation has been suggested to take place by the following mechanism (Urry,
1 995). In solution, elastin molecules are shielded by water moIecuIes. These water
molecules, unable to interact with the non-polar residues of elastin, fom a clathrate smcture
of hydrogen bonds surrounding the elastin molecules and separate h m the bulk water. With
increasing temperature, the hydrogen bonds become disntpted and the dathrate structures

become disordered. This disordering decreases the shielding of the elastin molecules,
allowing the hydrophobie domains of elastin to interact and fom a coacervate (Urry, 1995).
UnIike most proteins that undergo denahiration with increased temperature, becoming
less ordececi, elastin appears to become more ordered through the process of coacervation.
For this reason, Urry has termed the coacervation process for elastin an inverse temperature
transition (Urry, 1995). This increase in order appears inconsistent with the second law of
themodynamics, which States that systems will spontaneously tend toward more disorder,
resulting in increased entropy. However, while the elastin molecules becomes more ordered,
decreasing the entropy of this component of the system, there is a net increase in entropy due
to the disordering of the water molecules that initiaity surround the elastin molecules (Urry,
1995).
Factors Influencing the Coacervation Temperature
The temperature at which coacervation takes place is affected by a number of factors:
pH, ionic strength, protein concentration and relative hydrophobicity. Experirnents with (II-
elastin have shown the lowest temperature for appearance of turbidity and the highest rate of
turbidity formation around the isoelectric point (Kaibara et al., 1992; Pamidge, 1955;
Podrazky and Jackson, 1976). At the isoelectnc point there is no charge on the protein,
elirninating any potentiai hindtances to interaction between molecules due to charge
repulsion.
The ionic strength, usuaily represented as salt concentration, also affects the
coacervation temperature. As the ionic strength of the coacervation buffer increases, the
coacervation temperature decreases. The explanation of this effect Likely invoIves hydration.
With an inmase in the ionic strength of the solution, water becomes preferentially bound in
hydration sheils around the salt ions, dehydrating the elastin. With less water ordered around
the elastin molecules the energy required to disrupt the remaining clathrate water is reduced,
allowing coacervation to occur at a lower temperature (Urry, 1995). This effect has ken
demonstrated using recombinant trapoeIastin as well as tropoelastin isolated from tissues of
copper-deficient animais and aeIastin (podrazky and Jackson, 1976; Vrhovski et ai., 1997).
Similar effects are seen with an increase in protein concentration. An increase in protein
concentrahm d t e d in a decrease in the coacervation temperature for both recombinant

tropoelastin and the polypentapeptide poly(VPGVG) (Kondo et al., 1987; Vrhovski et al.,
1997).
Finally, the relative hydrophobicity of the protein &O affects its coacervation
characteristics. Urry and coworkers have show that increasing the relative proportions of
hydrophobic residues in a given peptide resulted in a decrerise in the coacervation
temperature (Urry et al., 1991; Urry et al., 1993).
Ulfrrrrtnrcture of the Coacervate
Coacervation of tropoelastin, a-eIastin, and hydrophobic poiypeptides based on the
sequences found in the hydrophobic domains of elastin resulted in coacervates with fibrillar
structures resernbling those of native, insoIuble elastin when viewed using electron
microscopy (Cleary and Cliff? 1978; Cox et ai., 1974; Cox et al., 1973; Urry et al., 1974).
Optical diffraction of coacervates of tropoelastin and a-elastin indicated a value of 5 nm for
the diameters of the parailei fiIaments making up the coacervate fibre (VoIpin et al., 1976).
Bressan and coworkers showed fibrillar structures formed frorn coacervates of tropoelastin
(Bressan et ai., 1983). Their studies also suggested that, prior to coacervation, tropoelastin
exists in solution as globular monomers and small oligomers. Imrnediately following
coacervation, a network of filaments is seen, transfoming into a more ordered fibriIIar
structure after overnight incubation at the coacervation temperature (Bressan et al., 1986).
Aggregation as an Ordering Process Bressan and coworkers specdated that the process of aggregation of elastin was an
ordering mechanism through which the hydrophobic domains interact aligning the lysine
residues to ailow crosslink formation (Bressan et aI., 1986). In support of this suggestion,
Narayanan has shown that formation of desmosine crosslinks between tropoelastin molecdes
in vitro, after oxidation with lysyl oxidase, takes pIace only at temperatures above the
coacervation temperature (Narayanan et ai., 1978).
Recombinant DNA technology has dowed the e x p s i o n of tropoeIastin, making
larger quantities avaiIabIe for the study of assembly in virro (BedeU-Hogan et al., 1993; hdik
et al., 1990; Martin et al., 1995). In one study, recombinant tropoeIastin was coacervated and
subsequently incubated with lysyl oxidase. After 24 hours, alysine aldol,

&hydrolysinonorIeucine and desmosine were detected in the crossünked material (Bedell-
Hogan et al., 1993). These results also support the hypothesis that the coacervation process
aligns lysine residues, allowing the formation of a crosslinked, polymeric elastin matrix.
Other hydrophobic matrix proteins have also shown the ability to seIf-aggregate.
These proteins including lampnn (the major matrix protein of lamprey cartilage), silkmoth
chorion proteins, musse1 byssus threads and spidroin 1 (a spider dragline siik protein)
(Hamodrakas et al., 1985; Qin et al., 1997; Robson et al., 1993; Xu and Lewis, 1990). These
matrix proteins share repetitive sequence motifs with elastin, including repeats of GGLGY
(Figure 1.1). These sequences have been suggested to adopt short fJ-sheet(&turn or &spiral
structures, and it has been suggested that these sequences may promote self-organization by
interdigitation or stacking of the hydrophobic amino acid side chahs, perhaps in a 'lego'-
like manner (Robson et al., 1993). In this way, these types of hydrophobic motifs may have
general implications for the ability of proteins to self-assemble into organized, polyrneric
matrices, which may be extended into other naturai and 'biornimetic' biomaterials.

Rationale The process of ordered assembly into its extracellular, polymeric form remains one of
the least well-understood steps in the biosynthesis of elastin. During assembly, the side
chains of lysine residues in tropoelastin mu t be oxidatively deaminated and brought into
juxtaposition in preparation for crosslinking. Moreover, d l this mu t be accomplished with
an overall architectural organization consistent with the physical properties of an elastomer.
Several factors have k e n proposed to contribute to the alignment of elastin monomers in the
formation of polymeric elastin, including the proteins of the microfibrillar scaffold and a ceIl-
surface elastin binding protein (Hinek and Rabinovitch, 1994; Hinek et al., 1988; Ross and
Bomstein, 1969). However, in vitro, recombinant tropoelastin, oxidized by lysyl oxidase,
assembled into a crosslinked maûix in the absence of these factors (BedeIl-Hogan et al.,
1993).
The ability of elastin to self-aggregate through a process called coacervation, a
property based on the hydrophobicity of the protein, has been known for many years.
Suggestions have been made that this self-association may not be a random process, but
rather plays an organizational role in the aiignment of elastin molecules for assembly into a
crossiinked matrix. However, the details of the process of self-assembly, and the role of
specific hydrophobic and crosslinking sequences, have been diitïcult to determine due to the
complex nature of tropoelastin. Therefore, we have used a series of small, recombinant
human elastin polypeptides to investigate the roles of various hydrophobic domains in
promoting self-assembly and to determine whether this self-assembly facilitates aiignment of
the elastin polypeptides, dlowing crosslink formation at the lysine residues. Understanding
the basis of the self-organizational ability of elastin-based polypeptides may provide
important clues for the general design of self-assembling biomateriais.

Hypothesis PoIypeptide regions of human elastin, expressed using recombinant DNA technology,
will self-align to allow crosslinking and will have modifiable mechanical properties.
Objectives
1. To express and puri@ representative regions of human elastin using recombinant
DNA technology.
2. To determine the aggregation characteristics of the polypeptides and the role of
specific hydrophobic and crosslinking regions.
3. To investigate the ultrastructural properties of the elastin polypeptides.
4. To investigate alignment of elastin using the expressed polypeptides.
5. To characterize the mechanical properties of materials made from the crosslinked
elastin polypeptides.

Chapter 2
Production, Purification and Characterization of
Recombinant Human Elastin Polypeptides
Two roads diwirged in a wood. and I - I bak the one iesa travelled by, and that bas made al1 Vie differenœ. Fmm The Road No1 Taken" by Robert F m

The in vivo assembly of elastin is suggested to involve a nurnber of factors, including
the elastin binding protein, a microfibrillar network, and lysyl oxidase. The elastin binding
protein is believed to escort tropoelastin to the extracellular matrix, depositing it ont0 the
microfibriilar network which acts as a scaffold for the developing matrix. Lysyl oxidase is
then required to modify the Lysine residues to ailow the formation of a crossiinked network.
However, Little is known about the mechanisms of this assembIy and how tropoelastin
molecules associate to fom a stable, insoluble matrix.
Attempts to investigate the mechanisms of assembly had k e n hampered by two
factors: the lack of sufficient quantities of monorneric tropoelastin and the complexity of the
molecule. It is onIy recently that sufficient quantities of tropoelastin have becorne available,
synthesized using recombinant DNA technology. Tropoelastin is especially well-suited for
bacterial expression as it does not require post-translational modifications such as
glycosylation. WhiIe in vivo, 14% of prolines in elastin are hydroxylated to hydroxyproline,
this modification does not appear to have any functionai consequences (Rosenbloom et al.,
1993). Seved investigators have expressed human tropoelastin in E. coli (Bedell-Hogan et
al., 1993; Grosso et al., 1990; Indik et ai., 1990; Martin et al., 1995; Vrhovski et al., 1997).
Weiss and coIleagues have pmduced nlatively large yields of tropoelastin by modifying the
third position of some human codons to reflect common codon usage in bacteria (Martin et
ai., 1995).
Experimental evidence suggests that recombinantly produced tropoelastin has similar
properties to native mpoeIastin, including similar coacervation characteristics (Vrhovski et
al., 1997) and chernotactic propeaies (Indik et al., 1990; Senior et al., 1982). In addition,
antibodies to native tropoelastin recognize recombinant tropoelastin (Indik et al., 1990).
Rosenbloom and coworkers (Bedell-Hogan et ai., 1993) expressed recombinant tropoelastin
and showed self-aggregation, msslinking, and fibre formation in the presence of lysyl
oxidase. Their results indicate that whiIe the mimfibrillar component and possibly other
factors are important for abgnment in vivo, assembly, crosslinking and polymerization can
occur in their absence.
Though availabiIity of tropoelastin is now less of an issue because of the possibility
of recombinant synthesis, the complexity of the tropoelastin molecule remains an

impediment to determining the mechanisms of assembly and the roles of specific
hydrophobic and crosslinking domains in this process. It has k e n proposed that the
hydrophobic domains can fom short &sheet/&turn structures (Abdel Rahman et al., 1987;
Castiglione-Moreili et al., 1990; Debelle et ai., 1992) or pspirals (Reiersen et ai., 1998; Urry,
1983), allowing interaction of the hydrophobic domains (Robson et al., 1993). Bressan has
suggested that these interactions align the crosslinking domains to facilitate crosslinking
through lysine residues (Bressm et al., 1986).
The hydrophobic domains of eIastin contain repetitive peptide sequences similar to
sequences found in a number of other self-assembling proteins. The repetitive GGLGY motif
is found in lamprin, the major matrix protein of larnprey cartilage. Similar sequences are
found in silkmoth chorion pruteins, musse1 bysuss threads and spidroin 1, a spider dragline
silk protein (Figure 1.1) (Harnodrakas et ai., 1985; Qin et ai., 1997; Robson et ai., 1993; Xu
and Lewis, 1990). Hydrophobie domains of elastin contain many penta-, hexa- and higher-
order tandem repeats, including PGVGVA repeating seven times in exon 24 of human
tropoelastin (Indik et al., 1 987). These sequences rnay be found over a broad phyiogenetic
range because they play an important role in the assembly of these hydrophobic rnatrix
proteins (Robson et al., 1993).
To investigate the rnechanisrns of assembly and the contributions of the hydrophobic
and msslinking domains we have used a series of recombinant human elastin polypeptides.
This chapter describes the choice of polypeptides, the methodology for their production and
purification, and their general characterization.

Materials and Methods PCR AmplijkEation of Human Elaslin cDNAs
Human elastin cDNAs for use as PCR templates were obtained from Dr. Charles
Boyd, Pacific Biomedical Research Center, University of Hawaii, Honolulu, USA.
Amplifications were carried out in a 50 pi reaction volume containing 0.5 pM of each
primer, 8-10 ng of template, 5% DMSO, 0.2 rnM dNTPs, 1.5 mM MgC12, 2.5 units of Taq
polymerase (Qiagen Inc.), and lx PCR buffer supplied with the enzyme. PCR reaction
mixtures were initially denatured at 94OC for 4 minutes followed by amplification using 30
cycles of denaturation (45 seconds at 94"C), annealing (45 seconds at 50-65°C depending on
the specific primes used), and extension (1 minute at 72OC), with the final extension lasting
8 minutes. The PCR products were separated on a 1.5% agarose gel. Bands correspondhg to
the desired PCR product were purified from the gel using a QIAEX II Gel Extraction Kit
(Qiagen Inc). PCR reactions were carried out on a GeneAmp PCR System 2400 (Perkin
Elmer Corp., USA).
Primer Sequences
A: 5' CrGcTAGGGGGATCCATGmCcCGGCTITGGTG 3'
B: 5' CTGCCTAGGGAAmCCTAAGGGCCAATCGCGGG 3'
C: 5' GCTTCGGGCCCAATCGCGGGAGCCAC 3'
D: 5' CGATTGGGCCCGAAGCTCAGGCAGCAGCTC 3'
E: 5' AAGCCCAGCTGCAGCTCGçCCMACTGGGCGGmG 3'
F: 5' AAAGCCGCCCAG'IITGGGCGACGTGCA~GGGmG 3'
G: 5' CCGGMTTCITATGCCCCGAAGCCAGG 3'
H: 5' CGGGGATCCATGGAAGCTCAGGCAGCAGC 3'
1: 5' CGGGAATTrnACCCAAACTGGGCGGClTTG 3'
J: 5' AGCGGCTCCCACTAGGCCCCCAAACTGGGCGGCZTTG 3'
K: 5' AAAGCCGCCCAGTTTGGGGGCCTAGTGGGAGCCGCTG 3'
L: 5' CCGGAATr'mATCCAAGGCCCCCAC 3'

Elash'n Polypeptide &pression Constructs
Al1 elastin polypeptide constmcts were designed as glutathione S-transferase (GST)
fusion pmteins. Recombinant proteins are Erequently expressed as fusion proteins in which
the protein of interest is coupled to a second pmtein sequence that can improve the solubility
of the desired protein andor provide a specific binding site for purification by
chromatography (Figure 2.1). The structures of these constructs are given in Figure 2.2 and
the polypeptide sequences are given in Figure 2.3. in general, elastin polypeptides were
designated according to the hydrophobic exons that they contained, with the exception of
EP21-23, which contained only crosslinking exons. Table 2.1 provides a surnmary of
polypeptide characteristics including the number of residues and hydrophobic domains,
moIecular weight, and hydropathy value of each polypeptide. Mean hydropathies were
calculated by summing the Kyte-Doolittle hydropathies of al1 of the amino acids in the
polypeptide and dividing by the total number of amino acid residues (Kyte and Doolittie,
1982). Hydropathy values reflect the overall hydrophobicity of a protein or polypeptide.
EIastin polypeptide (EP) 2 b î 4
Polypeptide EP2&24 corresponded to exons 20-21-2324 of human elastin. This
sequence was produced by PCR using forward primer A, containing a BamHI restriction site
and a methionine codon, and reverse primer B, which contains a stop codon and EcoRi
restriction site. Human fetaI aortic elastin clone H-6 was used as the PCR template for these
reactions. The H-6 cIone contained exons 20 to 36, excluding exons 22,26A, 32,34, and 35.
The BamHI and EcoRI sites allowed for directional cloning into the pGEX-2T vector
(Amersham Phmacia Biotech, Inc.). After ligation, DHSa cells (Gibco BRL) were
transformed with the pGEX-2T vector. Transformed cells w m plated on LB plates, and
colonies were picked and inoculated into 5 ml cultures. Plasmid prepmtions were digested
with BamHI and EcoRI and then electrophoresed on a 1.5% agarose gel to identiQ colonies
containing the insert, reflected by an increase in plasmid size. Inserts fiom positive colonies
were sequenced using an AB1 Prism automated sequencing system (Centre for Applied
Genomics, Hospital for Sick Children WC), Toronto, ON). After confiation of the
scqucncc, BUl(DE3) p ~ y s ~ b cells (Süatagene) were transformed with the construct for
expression of the polypeptides.

1. ligate GST 2. transform
3. select B + - (=) a fusion protein
human elastin + other proteins pGEX-2T cDNA
h) W
CNBr + other proteins --) h
Figure 2.1: Recombinsnt DNA achnology uscd to produce the polypeptide co~isuucts, and methodology for expression and purification of the polyppiides, Human elustin cDNA corresponding to the &sired cxons wns incorponted into the pGEX-2T vector containing GST and an ampicillin resisiance gene. After ligation, E. coli were iransformcd with ihe recombinunt vector and bacteria contnining the vecior werc selected using resistance to ampicillin. Human elastin was expressed by the bacterin as a GST-fusion protein along wiih other bacterial proteins. To separate the bacterial proteins from the fusion protein, gluuithione-Agarose kvds were idded to the pmtein mixture, selectively binding [O the fusion pmtein. GST was cleaved from the clnstin polypepide ut a methionine residuc between the GST and the polypeptide by CNBr treatmenl. CNBr also cleaved the GST into nine srnaller fragments and tlicse were sepürnted from the elastin polypeptide using si= exclusion cliromatagraphy.

S-transferaseT CNBr cleavage thrombln oleavage PC'
I alutathlone 49 as
Molecular Mass (Ba)
10,010
Figure 2.2: Representations of the elastin polypeptide expression constructs. AH peptides were generaied as glutathione S- transferase (GST) fusion proteins. Thromhin and cyanogen bromide (CNBr) cleavüge sites are indicaied only for EP20-24 but rire similar for other constmcts. Exon nurnbers and number of umino acids corresponding to each exon are also indicated, together with the molecular mass of ench of the elastin peptides after purification. Crosslinking exons are shuded,


Table 2.1 : Summary of polypeptide characteristics including the riurnber of residues per mole, number of hydrophobie domains, molecular mass and hydroparhy values.
Number of Hydrophobic Domains
O
Molecultu Mass Mean Hydropathy (Da) (Kyk-Do01 i tt le) 2,973 0.22
10,010 0.94
16,992 0.92
16,385 O. 86

Elastin polypeptide (EP) 2&2&24
EP2&24-24 corresponded to exons 20-21-23-24-21-23-24 of human elastin. Using
the cDNA template described above, exons 20-21-23-24 were produced by PCR using
forward primer A, and reverse primer C which contained an ApaI restriction site. Exons
21-23-24 were produced h m the H-6 cDNA template by PCR using reverse primer B
together with forward primer D that contained an Apd restriction site. The pGEX-2T vector
was digested with BamHI and EcoRI and the PCR products were digested with ApaI and
either BamHI or EcoRI. The two inserts were ligated together into the vector. Transformation
and al1 subsequent steps were the same as for EP2%24.
Eiastii polypeptide (EP) 2M4-26
EP20-24-26 corresponded to exons 20-21-23-24-2 1-23-26 of human elastin.
Exons 2G21-23-24-21-23 were produced by PCR with forward primer A and reverse
primer E. Reverse primer E annealed to exon 23 and included 18 bases of exon 26.
EP2û-24-24 was used as the template. A second PCR reaction used forward primer F, which
annealed to the 5' end of exon 26 and included 18 bases of exon 23, and reverse primer G
containing a stop codon and an EcoRI site, with the H-6 clone as the template. After
purification of hese products, a third PCR step used fonvard primer A, reverse primer G,
with the PCR products of the previous two PCR reactions as templates. This final PCR
reaction resutted in a product corresponding to exons 20-21-23-24-21-23-26.
Transformation and ail subsequent steps were the same as for EP20-24 except DHSa celIs
were used for polypeptide expression.
Elastin polypeptide (EP) 2û-%3û
EP20-24-30 corresponded to exons 2&21-23-2&21-23-30 of human e1ast.n.
Exons 2&21-23-24-2143 were produced by PCR with fonvard primer A and reverse
primer J. Reverse primer J annealed to exon 23 and inchded 18 bases of exon 30.
EP20-24-24 was used as the template. A second PCR reaction used forward primer K, which
anneded to the 5' end of exon 26 and contained 18 bases of exon 23, and reverse primer L
containing a stop codon and an EcoRX site, with H-6 human eIastin cDNA as the template.
After purification of these products, a third PCR step used forward primer A and reverse

primer L, with the PCR products of the previous two PCR reactions as templates. This final
PCR reaction resulted in a product comsponding to exons 2&21-23-24-21-2M6.
Transformation and al1 subsequent steps were the same as for EP20-2426.
Elastin polypeptide (EP) 21-23
EP21-23 corresponded to exons 21-23 of human elastin. This constnict was
produced by PCR using forward primer H containing a BamHI resûiction site and a
methionine codon, and reverse primer 1 containing a stop codon and an EcoRi restriction site.
The H-6 human cDNA was used as the template. Transformation and al1 subsequent steps
were the same as for EP20-24.
Expression and Purification of Elastin Polypepruies
E. cdi transformed with the recombinant pGEX-2T vector were plated on LB
medium with ampicillin (50 pgiml) and incubated overnight at 37°C. For expression of
polypeptides, 25 ml of 2 x YT broth containing ampicillin (50 pgM) was innoculated with a
single colony of E. coli and incubated for 18 hours at 37°C with shaking. This culture was
then used to inocuiate 2 x YT culture medium containing 20% glucose, 100 Wml ampicillin,
34 pg/ml chloramphenico1 (for BL21 cells only) , and allowed to grow at 37°C with shaking
until the optical density at 600 nm ( O D d was 0.8-1.0.
Polypeptide expression was induced by addition of isopropy1-a-D-
thiogalactopyranoside (IPTG) to a final concentration of O.lmM. After an incubation period
of 3 hows, the flasks were chiiled on ice and the culture medium centnfhged for IO minutes
at 7500g. The supernatirnt was removed and the pellet resuspended in STE (50mM Tris
buffer, pH 8.0, containing l00mM NaCi, 1mM EDTA and lmg/ml Lysozyme) (Martin et al.,
1995) and held on ice for 30 minutes. A protease inhibitor, 4-(2-aminoethy1)-bellzenesulfonyl
fluoride hydrochloride (AEBSF), was then added to a final concentration of 0.1 mM and the
resuspended pellet was h z e n oveniight, The cells were thawed, DNase I (10 unitdm1 of
STEJ and MgC12 were added to a f i a l concentration of lOmM, and the lysate was vortexed
and heId on ice for 45 minutes. For the DIEU cells, the Iysates were frozen and thawed three
times prior to the addition of the DNase 1 and MgQ. The Iysate was then centrifuged at
13800g for 20 minutes at 4OC and the supernatant was removed

The GST-fusion proteins were purified by an affinity method using glutathione-
Agarose beads (Sigma-Aldrich Co.) or giutathione-Sepharose beads (Amershm Pharmacia
Biotech, Inc.) A 50% sIuny of beads (v/v) was added to the supernataut (LAO,
slurrysupernatant) and the suspension was mixed by rotation at room temperature for 45
minutes. This suspension was then centrifuged and the supernatant removed. The pelleted
beads were washed six times with phosphate-buffered saline (PBS) and stored at 4°C
overnight, A second 50% glutathione-Agarose slurry was added to the supernatant (120
sIwry to supernatant) and mixed by rotation overnight at 4°C. This mixture was centrifuged
and the supernatant removed. The pelleted beads were washed six times with PBS, combined
with the beads h m the first extraction and lyophilized.
Cyanogen bromide (CNBr) was used to cleave the GST-fusion protein and release the
elastin polypeptide (Figure 2.1). Cyanogen bromide cleaves specifically at methionine
residues. A methionine residue was placed just upstream of the N-terminal of the elastin
polypeptide in the constructs, and in the absence of intemal methionine residues in the elastin
polypeptide, cyanogen bromide cleaved the elastin polypeptide from the fusion protein. The
GST portion of the fusion protein contains 8 methionine residues that, on complete cleavage
with CNBr, results in polypeptide fragments mging in size h m 0.34 kDa to 8.2 kDa (2.1).
For al1 of the polypeptides with the exception of EP21-23, CNBr (50 m g h i in 70%
formic acid, under NJ was added to the lyophilized beads containing the bound fusion
protein and incubated at m m temperature in sealed tubes in a fume hood for 6 hours. The
cleavage products were then diluted -10 fold with distilled water and the tubes were left
uncovered ovemight in the fume hood to alIow residual HCN to disperse. The samples were
dialyzed against distilled water using 3500 MW cutoff dialysis tubing (Pierce or Fisher
Scientific Ld) for EPXM4, and 7000 MW cutoff dialysis tubing (Pierce) for EPSû-24-24,
EP2û-24-26, and EP2û-24-30. The retained material was IyophiIized, then redissolved in
O.OSM acetic acid and centrifuged for 10 minutes at 18000g. The supernatant was removed
and loaded ont0 a gravity-fed column (65 cm x 3cm) of Bio-Gel P30 (BioRad Laboratories)
which had been equilibrated with 0.05M acetic acid, Fractions were eluted with 0.05M acetic
acid and monitored for absocbance at 230 and 280 nm. The elution fractions containing the
elastin polypeptides were separated by SDS-PAGE, transferred electrophoreticaiIy to
nitroceHuIose, and identified by Western blottkg with an anti-tropoeiastin antibody (Hastin

Products Company Inc.). The identity and purity of the etastin polypeptides was confinned
by amino acid anaiysis and mass spectrometry (Biotechnology Service Centre, HSC,
Toronto, ON).
Because of the small size of EP21-23, an alternate purification method was utilized,
In this case the elastin polypeptide was cleaved from the GST fusion protein using the
thrombin cleavage site which is present in the pGEX-2T vector three amino acid residues
upstream h m the start of the elastin polypeptide, The beads containing bound fusion
proteins were hydrated with PBS to give a 50% slurry, and thrombin was added to a final
concentration of 50 units/ml. After mixing overnight at room temperature, the suspension
was centrifuged, the supernatant removed, and the beads washed three times with PBS. The
supernatant from each wash was added to the original supernatant and lyophilized. The
product was redissolved in O.OSM acetic acid and loaded ont0 a gravity-fed column of Bi*
Gel Pl0 (BioRad Laboratones) equilibrated with O.05M acetic acid. Fractions eluted with
0.05M acetic acid were rnonitored for absorbante at 230 and 280 nm. Fractions comprising
the first eluted peak, containing EP21-23, were combined and lyophilized. This lyophilized
product was then treated with CNBr, as described above, to cleave at the methionine residue
just N-terminai to the ml-23 polypeptide. M e r CNBr cleavage, the samples were
dialyzed against distilled water using 1000 MW cutoff diaiysis tubing, and the retained
material was analyzed by amino acid analysis and mass spectrometry as described above.
Production of Antibodies to EP2&24 and EP2I-23
Lyophilized EP20-24 was dissolved in sterile PBS to a final concentration of
1 mg/ml, and emulsified with an quai volume of Freund's cornpletc adjuvant (Gibco BRL).
New Zealand White rabbits were injected subcutaneousIy with 400 pl of the antigedadjuvant
emulsion. A booster injection containing 400 pi of polypeptide emuisified with an equal
amount of Freund's incomplete adjuvant (Gibco BRL) was given two weeks Iater, followed
by a second booster injection given after another two weeks. Four weeks d e r the finai
booster injection the rabbits were exsanguinated and the semm coliected Antibodies to
EP21-23 were made using the same protocol as for antibodies against EP20-24. Monocionai
antibodies to PGVGVA were a gift of Dr. Robert Mecham, Washington University Medical
Center, St. Louis, Missouri, USA.

Western Blottihg
Samples were boiied for 10 minutes in sampk 5 i i t r gris, 2% SDS, Dm, glycerol,
and bromophenol blue) and then loaded ont0 SDS-polyacrylamide gels (5.7% stacking,
12.5% separating) and electrophoresed (SDS-PAGE) using a BioRad Mini-Gel apparatus
(BioRad Laboratories). Foilowing electrophoresis, gels were transferred onto nitrocellulose
membranes using a BioRad Mini-Transfer apparatus (BioRad Laboratories). After transfer,
membranes were incubated for one hour at room temperature in blocking buffer (TBS buffer
with 0.5% Tween (TBS-T) and 5% skim milk), washed in TBS-T and then incubated
overnight at 4°C with the primary antibody in TBS-T and 5% skim milk. Membranes were
washed and then incubated for one hour at room temperature with the secondary antibody
(horseradish peroxidase conjugated goat anti-rabbi t an tibodies in TBS-T and 5% skim milk).
Membranes were washed, treated with enhanced chemiluminescence solutions (Arnersharn
Phannacia Biotech), and exposed to film.

Results Polyacrylamide gel electrophoresis of EP30-24 at different stages of purification is
shown in Figure 2.4. Gels are stained with Coomassie blue or transferred for Western
blotting using antibodies to EP20-24 or to the PGVGVA sequence. Antibodies either to
wpoelastin (not shown) or to EP20-24 identified a 36 kDa protein ( m w , Lane 1) that bound
to glutathione beads and corresponded to the expected size of the GST fusion protein. A
second, unidentified band at -30 kDa in this fraction dso reacted with the antibody. The 30
D a band was present in a11 of the elastin polypeptide preparations at this stage, but was
removed during subsequent purification steps. After CNBr treatment and chromatography,
ody a single band with a molecular weight of - 10 kDa could be detected either with an
antibody to EP20-24 (lane 2) or with the monoclonal antibody to the PGVGVA sequence
(lane 4). Coomassie blue staining of the protein that was bound to the glutathione beads (lane
5) also showed the 36 kDa fusion pmtein and the 30 kDa bands seen in lane 1. However, as
expected because of its hydrophobie character, phfied EP20-24 (lane 6) did not stain with
Coomassie blue, nor were other bands visible in this lane, indicating the absence of
detectable arnounts of contaminating proteins in this final product. Sirnilar results were seen
for the other purified polypeptides, except that EEl-23 could not be detected on Western
blotting either with the antibody to EP2&24 or with the monoclonal antibody to PGVGVA,
and detection was only possible with the antibody to EP21-23.
The purity of the isolated polypeptides was assessed by amino acid analysis,
comparing expected to actual compositions (Table 2.2) and by mass spectrometric analysis to
determine the molecular mass of the poIypeptides (Tabte 2.3). in al1 cases the composition
and size of the elastin polypeptide products corresponded well with expected values,
confinning the identity of these products. The mass spectrometry spectra also c o n h e d the
absence of significant arnounts of contaminating proteins. A representative spectrum of
EP2&24 (Figure 2.5) shows one dominant peak at 10009.2 Da, the moIecular mass of the
polypeptide, with traiIing peaks corresponding to the polypeptide with adducts, the
noncovalent addition of ions to the polypeptide. Other peaks were present either as
backgruund or minor peaks. Dut to the small size of EP21-23 mass spectrometry was able to
&termine the sequence of the p d e d polypeptide. This sequence was identical to the

expected sequence, confirming the absence of codon mutations leading to amino acid
substitutions (Figwe 2.6).
Storage of EPXL24 in 0.05M acetic acid at 4°C for four weeks resdted in the
formation of oiigomeric products that had not been present upon initiai purification of the
polypeptide (Figure 2.4, Iane 3). This was the Eirst indication that even at low temperature
(4'C) and ionic strength some self-aggregation of these polypeptides couId take place.
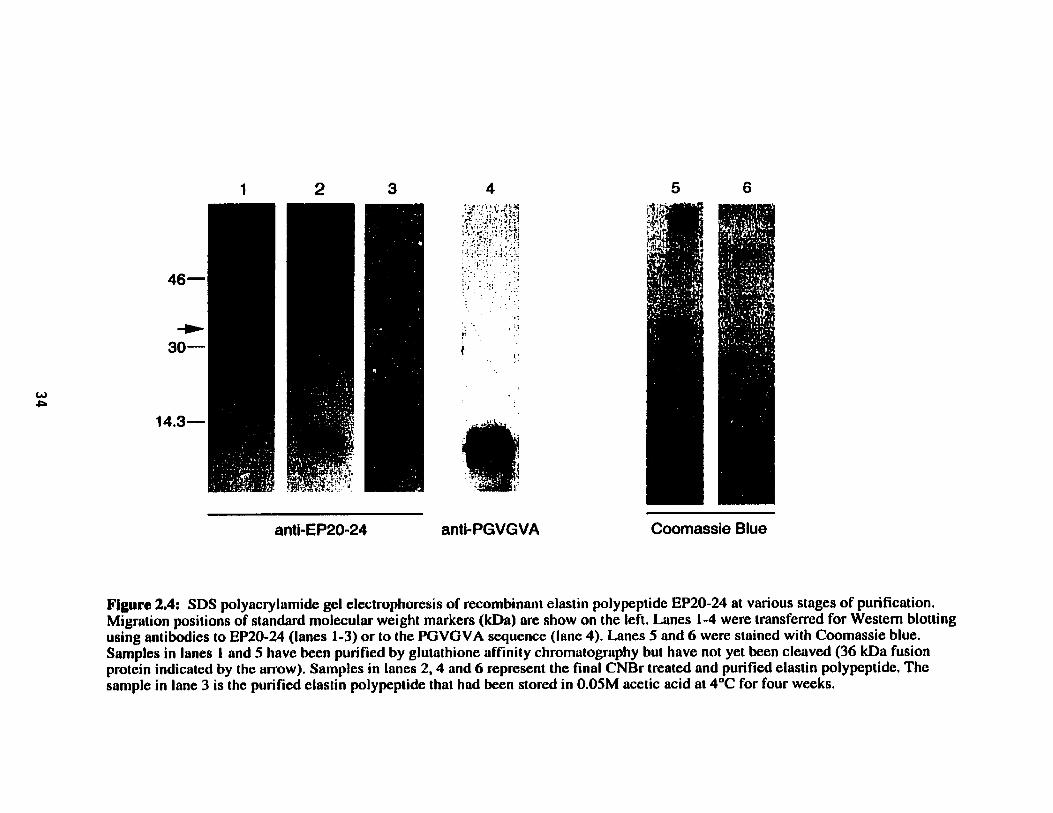
Coomassie Blue
Figure 2.4: SDS polyacrylamide gel electrophoresis of recombinant elastin polypeptide EP20-24 at various stages of purification. Migration positions of standard molecular weight markers ( k h ) are show on the left. Lanes 1-4 were transferred for Western blotting using antibodies to EP20-24 (lanes 1-3) or to the PGVGVA squence (lune 4). Lanes 5 and 6 were stained with Coomassie blue. Sarnples in lanes 1 and 5 have been purified by glutathione affiniiy chromatography but have not yet k e n cleaved (36 kDa fusion protein indicated by the arrow). Samples in lanes 2,4 and 6 represent the final CNBr treated and purified elastin polypeptide. The sarnple in lane 3 is the purified elastin polypeptide that had been stored in O.OSM acetic acid at 4°C for four weeks,

Table 2.2: Representative amino acid c~mpositions of purified recombinant human elasiin polypepiides (actual) compared to expected compositions based on known peptide sequences (amino acid residuedmole).
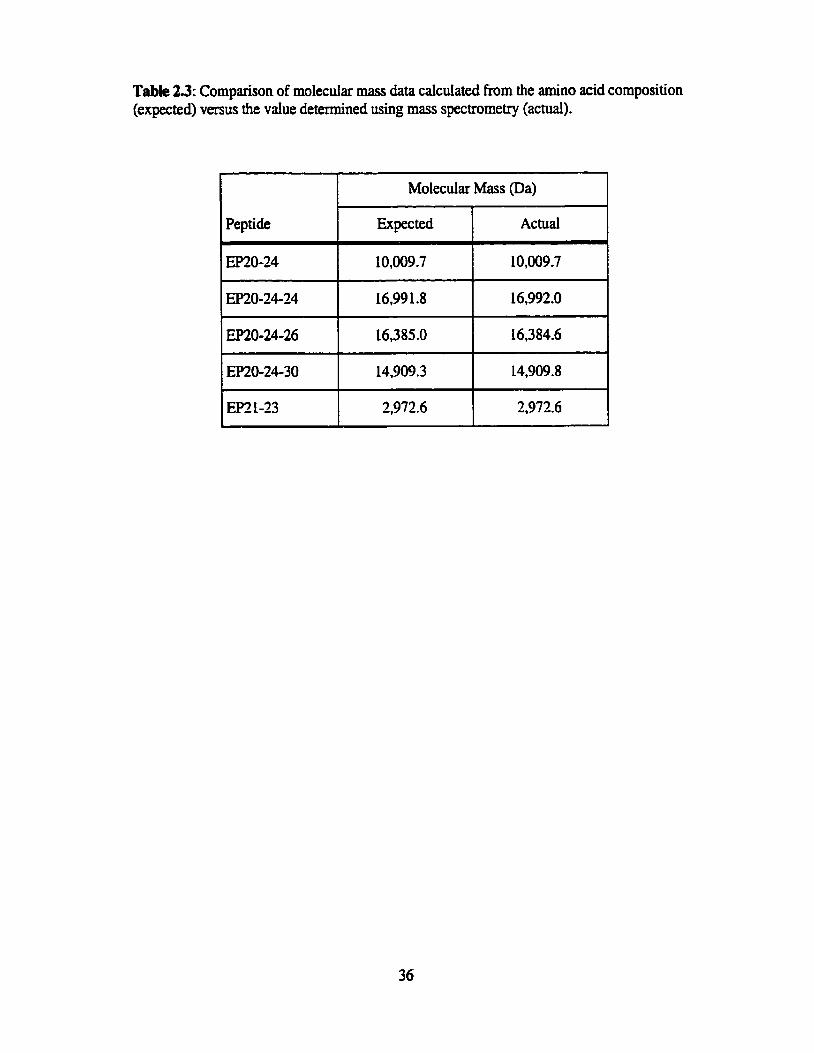
Table 23: Cornparison of molecular mass data calculated h m the amino acid composition (expected) versus the value determined using mass spectrometry (actual).



The fmt polypeptide used in this study, EP20-24, was chosen :O be representative of
the overall tropoelastin stnictwe of aiternating hydrophobic and crosslinking domains, yet
small enough to determine the d e s of these domains in self-assembly. Ems24 contains
cwo hydrophobic exons (exons 20 and 24) flanking a crosslinking sequence incorporating
exons 21 and 13. This region of the tropoelastin molecule, and more particuIarIy exon 24,
was chosen because it contains the most striking tandem repeat sequence in human elastin - a
PGVGVA sequence repeated seven times.
Similar tandem repeat sequences are found at the sarne location in elastins of many
other species. For example, baboon elastin also contains the PGVGVA sequence repeated
seven times in tandem in exon 24 (Szabo et ai., 1999) and chicken élastin contains a ten-fold
repeat of a PGVGV sequence at this site. The hexapeptide PGVGVAIV repeats four times in
bovine tropoelastin at this location, and the pentapeptide PGVGV is repeated eleven times at
an additional site. The general presence of these penta-, hexa- or higher-order peptide repeats
suggests that such sequences may have particular importance for the properties of elastin,
including the ability of this protein to self-assemble.
Support for the participation of these sequences in self-assembly cornes h m the
proposed rote of the elastin-binding protein (EBP). The PGVGVA repeat of tropoeIastin is
known to be one of the binding sequences recognized by the EBP (Hinek and Rabinovitch,
1994, Hinek et al., 1988). The EBP has ken suggested to act as a chaperone, binding
intraceilularly to tropoelastin, escorting it to the ce11 surface where the EBP interacts with
galactosugars on glycoproteins in the microfibrillar scaffold, and subsequentiy releasing
tropoeIastin to coaiesce onto the developing extracellular elastin network. By binding to
tropoelastin through the PGVGVA repeat, the EBP is believed to prevent premature
intracellular self-aggregation of tropoelastin (Hinek and Rabinovitch, 1994).
Arguments for the importance of these sequences in self-assembly cm ais0 be made
on the basis that several other structurai proteins contain similar tandem repeat sequences and
share a cornmon characteristic of self-aggregation. Hydrophobie sequences have k e n
identifieci in Imprin, the major matrix protein of lamprey cartilage (Robson et d., 2000;
Robson et ai., 1993). This protein contains the repetitive motif GGLGY. with similar
sequences also found in sillanoth chonon proteins (Hamodrakas et ai., 1989, musse1 byssus

threads (Qin et al., 1997) and spider dragline siik proteins (Xu and Lewis, 1990). While
lamprin does not contain the PGVGVA sequence of elastin, nevertheless monoclonal
antibodies to this sequence were found to crossreact with lamprin, suggesting similarities in
the secondary structure of these proteins (Robson et al., 1993). It has been suggested that
these repetitive motifs adopt short fi-s heet/$-turn structures that interact hydrophobically
resulting in self-aggregation (Robson et al., 1993).
Having selected exon 24 as a potentially important hydrophobic domain, in order to
preserve the altemating domain structure of tropoelastin it was necessary to use either exon
23 or exon 25 as the adjacent crosslinking domain. Exon 25 was not chosen as the
crosslinking domain because it contained a senne residue that is less common in msslinking
regions. However exon 23 is a typicai crosslinking domain of elastin, alanine-rich with two
lysine residues separated by three aianine residues. The adjacent hydrophobic domain, exon
22 is a short exon present in the elastin gene but not found in any human elastin cDNAs
reported to date. Therefore, exon 20, which also contains a repetitive motif, was used as the
second hydrophobic domain. A second crosslinking domain, exon 21 was incorporated into
mû-24, preserving the natural sequence of tropoelastin and its aiternating domain
structure.
A second polypeptide, m0-24-24, was designed to investigate the effect of size and
number of hydrophobic domains on the ability of the polypeptide to self-aggregate. This
polypeptide, approximately twice the molecular mass of EP2&24, included one additional
hydrophobic domain. The arrangement of three hydrophobic domains interspersed with two
crosslinking domains might provide more opportunity for overlap during aggregation,
increasing the propensity of this polypeptide to aggregate compared to EPW24. Two
additional polypeptides based on EP20-2624 were produced to investigate the importance
of exon 24 and the repetitive PGVGVA sequence in self-aggregation. First, EF2û-2626 was
designed by replacing the final exon 24 of E R S 2 6 2 4 with exon 26. Because this
polypeptide is approximately the same size as EP2&24-24, it controiied for the effect of
molecular mass on seIf-aggregation propensity. Exon 26 aiso contains a repetitive sequence,
a nonapeptide which repeats three times in tandem with one consenative substitution,
ailowing direct comparison of the effect of sequence on the ability of the polypeptides to
seif-aggregate.

The second polypeptide baed on EP2624-24 was EP20-24-30. EP20-24-30 has a
slightly smalIer molecular mass than EP2&24-24 but contains the sequence GGLGV twice
in tandem, a sequence similar to those found in other self-aggregating proteins (Hamodrakas
et al., 1985; Qin et al., 1997; Robson et ai., 1993; Xu and Lewis, 1990). As with
EP2&24-26, mû-24-30 was designed to investigate the importance of the PGVGVA
repeat in exon 24, and to determine whether the GGLGV sequence changed the propensity
for aggregation of the polypeptide.
Findy, EP21-23, containing oniy crossLinking domains, was designed to investigate
the general requirement of hydrophobie domains in self-aggregation and alignrnent.
Al1 of the polypeptides were successfully produced using E. coli and purified using
the biochemical techniques described in the Methods section. Yields ranged h m 2-10 mg of
polypeptide per litre of culture, sirnilar to the yields obtained by investigators expressing
recombinant tropoelastin (Grosso et al., 1990; Indik et ai., 1990). Polypeptide purity was
consistently estimated at at least 90% by amino acid anaiysis. Mass spectrometry also
confmed the absence of any major contaminanis in the purified samples. The precision of
mass spectrornetry in determining the mass of the polypeptide was sufficient to indicate the
absence of codon mutations Ieading to amino acid substitutions. Absolute confirmation of the
EP2 1-23 sequence was given by sequencing using mass spectrometry.
The original polypeptide, EP20-24, showed a propensity for seIf-aggregation even
under Iow h i c strength and low temperature conditions (Figure 2.4). This was the first
indication that these polypeptides, in spite of their smail size relative to tropoelastin, had
suficient structurai information to aggregate, at least into smail oligomers. The following
chapter describes the detailed characterization of the coacervation properties of these
polypeptides.

Chapter 3
Self-Assembly Characteristics of
Recombinant Human Elastin Polypeptides
Evolutton & smarter than you are. Attributed to Francis Crick

Introduction The process of elastin assembly in the extracellular matrix has been suggested to
invoIve the aggregation of tropoelasun monomers, although details of the mechanisms
involved in this association are not known. For some time it has been known that
polypeptides related to elastin self-aggregate through a process calied coacervation (Partridge
et ai., 1955). Coacervation has subsequently been used as a simple in vitro measure of the
aggregation process.
Coacervation is defined as a reversible phase separation (Bungenberg de Jong, 1949;
Uny, 1982). A protein in solution at low temperatures will aggregate upon an increase in
temperature, forming a second phase (coacervate), and sepmte from the solvent.
Coacenation is usually monitored by spectrophotornetric mesurement of turbidity at a light
scattering wavelength (Figure 3.1). Below the coacervation temperature the protein solution
is clear. At the coacenation temperature (Tc) the solution becomes cloudy or turbid, with a
corresponding increase in absorbance. Typical coricervation cuves show coacervation taking
place over a narmw temperature range, suggestive of cooperative kinetics (Jensen et al.,
2000). The temperature at which coacervation OCCLUS is influenced by a number of factors,
including ionic strength of the solvent, protein concentration, molecular mas, and the overail
hydrophobicity of the protein (Jensen et al., 2000; Podrazky and Jackson, 1976; Urry et al.,
1991; Uny et al., 1992; Vrhovski et al., 1997).
While the earliest observation of efastin coacervation used a-elastin (Partridge et al.,
1955), both tissuederived tropoelastin and recombinant tropoelastin have also show the
ability to coacervate (Cox et al., 1974; Vrhovski et aI., 1997). Of particular interest is the
observation that the coacervates of both a-elastin and tropoelastin form fibriilar structures
(Bressan et al., 1986; Cox et al., 1974; Cox et ai., 1973; Vrhovski et ai., 1997). Urry has
term~d the coacervation process for elastin an inverse temperature transition because, unlike
most proteins which denature upon an increase in temperature, becoming less ordered,
tropoeIastin becomes more ordered at higher temperanues, fomüng fibrilla. structures (Urry,
1995).
In addition, some polypeptides based on the hydrophobie sequences of elastin aiso
showed the abiiity to coacervate, forming fibrillar stnictures similar to those of aelastin and
tropoelastin coacervates (vny et al., 1974). The ability of these polypeptides to coacervate

suggests that the hydrophobic domains are of particular importance in this aggregation
process. Bressan speculated that the association of the hydrophobic domains may be
important for aiigning the crosslinking regions, facilitating the formation of the crosslinked
polyrneric matrix (Bressan et al., 1986). However, details of the roles of specific sequences in
the mechanism of elastin assembly remain unclear. Here, we have used a series of
recombinant human elastin polypeptides ta characterize the process of coacervation of elastin
and ro investigate the contribution of specific hydrophobic domains to the propensity of
elastin polypeptides to self-assemble.

Absorbance (440 nm)
Absorbance
Temperature 1 "Chin
1
Temperature (OC)
Figure 3.1: Caacewation is followed by monitoring turbidity by light scatîering ai 440nm. Before co;icervation the protein solution is clear, At coacervation the solution becomes cloudy, resulîing in an increase in absorbance, The temperature was raised nt 1 'C per minute and the coacervation temperature (Tc) was defined as the onset of turbidity, detennined as the temperature corresponding to the intersection of the tangent to the curve prior to coacervation and the tangent to the initial increase in absorbance (dashed lines),

Co~u:erVafion Experùrtents
Coacervation experiments were perfomed using a Cary 3 spectrophotometer
equipped with a temperature controuer (Varian hc., Victoria, Australia). Polypeptides were
dissoived in coacervation buffer (50 mM Tris, 1.5 M NaCI, 1 mM CaC1: pH 7.5) to a
concentration of 25 pM and stored at 4°C ovemight. Solutions were placed into a quartz
cuvette, inserted into the sample ce11 of the spectmphotometer and allowed to equilibnte at
15°C for 5 minutes. The temperature was then increased at a rate of 1°C per minute with
gentie stimng. Absorbance was monitored at 440 nm, and coacervation was detected as the
onset of turbidity appearing as an increase in absorption. The coacervation temperature (Tc)
was taken as the temperature correspondhg to the intersection of the tangent to îhe curve
before coacervation and the tangent to the initial inmase in absorbance (the highest rate of
change) on a temperature versus absorbance plot (Figure 3.1). The rate of coacervation was
measured as the slope of the steepest part of the coacervation curve. Each coacervation
expriment used elastin polypeptides pmduced h m independent batches of expressed
protein, in order to take into account variability in characteristics between batches.
Coacervation characteristics were also detennined for an additional polypeptide,
EP2û-24-24-2424 (EPLO-24'), with a rnoIecular mus of -3 1 kDa This polypeptide is
sirnilar to EF2&24-24 with three additional repeats of exons 21-23-24, giving it five
hydrophobic domains flanking four crosslinking domains.
To study the effect of pdypeptide concentration on coacervation temperature,
EP2û-24 and EP2û-24-24 were dissolved in coacervation buffer (50 rnM Tris, 1.5 M NaCl,
1 mM CaC12, pH 7.5) at concentrations ranging from 2 - 100 pM, then were subjected to
coacervation. Expiments studying the effects of ionic strength utilized varied
concentrations of NaCl in the coacervation buffer. NaCl was added to Tris buffer (50 mM
Tris, 1 mM CaC12) to a fina1 concentration of 1.5 M, 1.0 M, 0.5 M, and 0.2 M, and the pH
adjusted to 7.5. Coacervation experiments were then camed out as described above.

Polypeptide Conceniraïion
Initially, Beers law (A=ecd) was used to detefinine polypeptide concentrations (c).
Absohance (A) was measured at a wavelengt!! of 275 nm, corresponding to the maximal
absorption of tyrosine residues. EP2&24 and EP21-23 each contain a single tyrosine residue
and EP20-24-24, EP2û-2426 and EP2û-24-30 each contain two tyrosine residues.
polypeptide concentration was calculated assuming a molar extinction coefficient (e) of
1390 M"cm-', and an optical pathway (d) of 1 cm. This calculation also assumes no
interactions between tyrosine residues. This proved not to be the case when concentrations
were compared to those detennined by amino acid analysis, using norleucine as an internai
standard to correct for polypeptide loss, with the exception of concentrations of EP21-23.
This was likely because the absence of hydrophobic domains prevented aggregation of
EP21-23, whereas aggregation of polypeptides containing hydrophobic domains would have
affected the molar extinction coefficient (Freifelder, 1982). For this reason, to ensure
accurate concentration values, arnino acid anaiysis was consistently used to determine
polypeptide concentration.
StdsiiCd Andysis
Standard errors for the coacervation data were calculated using Statview software
(version 4.01) (Abacus Concepts, Inc., Berkeley, California). Differences between groups
were assessed by ANOVA.

The propensities of these various elastin polypeptides to undergo self-aggegation, as
indicated by the coacewation temperature, were compared using a polypeptide concentration
of 25 pM and the standard coacewation buffer (50 mM Tris, 1.5 M NaCI, 1 m M CaCIL pH
7.5) as describecl Typical coacewation cuves for EP2û-24, EP2û-24-24 and EP21-23 are
shown in Figure 3.2. Results for al1 polypeptides are summarized in Table 3.1. EP 21-23,
containing no hydrophobic domains, did not coacervate under these conditions.
The average coacervation temperature for EP2û-24-24 was approximately 12°C
lower than that of EP20-24, and the coacervation temperature of ERû-24' was
approximately 8°C lower than that of EP20-24-24. While it has been shown that the overall
hydrophobicity of polypeptides can affect their coacervation temperature (Urry et al., 1991),
in this case the hydropathies of these three polypeptides were essentially identical, indicating
that hydrophobicity alone was not the factor determining the difference in coacervation
temperatures (Table 3.1). In order to assess the contributions of molecular rnass and specific
polypeptide sequences on the ability of these polypeptides to coacervate, similar coacervation
studies were done using EP2û-24-26 and EP20-24-30, with the results summarized in
Table 3.1.
EP7&24-26 has approximately the sarne moIecular mass and generai hydropathy as
EP2&24-24, but does not contain a duplication of exon 24. In spite of this, the average
coacervation temperature of EP2û-24-26 was approximately 4'C Iower than that of
EP2û-24-24 (Table 3.1). Similarly, the coacervation temperature of EP20-24-30, in which
the second exon 24 was replaced by hydrophobic exon 30, was essentiaily identicd to that of
EP2û-24-24 (TabIe 3.1) in spite of the lower molecuIar weight of this pdypeptide.
As expected, coacewation temperatures of these polypeptides were dependent on
ionic strength of the solution (Figure 3.3). For example, decreasing the concentration of NaCI
from 1.5 M to 1.0 M increased the coacewation temperature of EP2&24-24 by
approxirnately 13'C. A fuaher decrease in NaCi concentration to 0.5 M caused an
approximately 16°C inmase in coacervation temperature. At 0.2 M NaCI EPSb24-24 did
not coacewate. Similar effects were seen on the coacervation temperature of EP2&24
(Figure 3.3).

The temperature of coacervation of these polypeptides was also dependent on
polypeptide concentration (Figure 3.4A/B), with coacervation temperature decreasing with
increased pdypeptide concentration in an apparently non-linear relationship (Figure 3.4CtD).
As a result of this rdationship, the impact of changes in polypeptide concentration on
coacervation temperature is smaller at higher polypeptide concentrations as compared to
lower potypeptide concentrations. Sirnilar results were reported by Vrhovski et ai. in
coacervation studies of recombinant tropoeIastin (Vrhovski et al,, 1997). A cornparison of
slopes of semi-logarithmic plots of coacervation temperature against polypeptide
concentration (Figure 3.4E) indicated clearly that the effect of increased concentration on
coacervation temperature was greater for EP20-24 than for EP2û-24-24.
A significant difference was found between the rate of coacervation of EP20-24 and
EP20-24-24, with EPîO-24-24 showinp a significantly higher rate of coacewation than
EP2û-24 (p4.05). Although the sample size was limited, EP20-24' appeared to have a
higher rate of coacervation than EP20-24-24 (data not shown). In addition, the rate of
coacervation appeared to be higher for EP20-24-24 than for both EP20-24-26 (p4.05) and
EP2û-24-30 (Table 3.1). Both polypeptide concentration (Figure 3.5A) and NaCl
concentration (Figure 3.93) affected the rate of coacervation. As the concentration of
polypeptide or NaCl increased, the rate of coacervation increased for both EP20-24 and
EP20-24-24.
The maximum absotbance reached by EP2û-24 appeared to be Iower than that of
EP20-24-24 and mû-24" although these differences were no! sigxificant (Table 3.1). The
maximum absohance aiso appeared to be dependent on the concentration of the polypeptide
(Figure 3.6A) and on the concentration of NaCI in the coacervation buffer mgure 3.6B) for
both EPîû-24 and EP20-24-24, with the maximum absorbance increasing with sodium
chloride concentration.
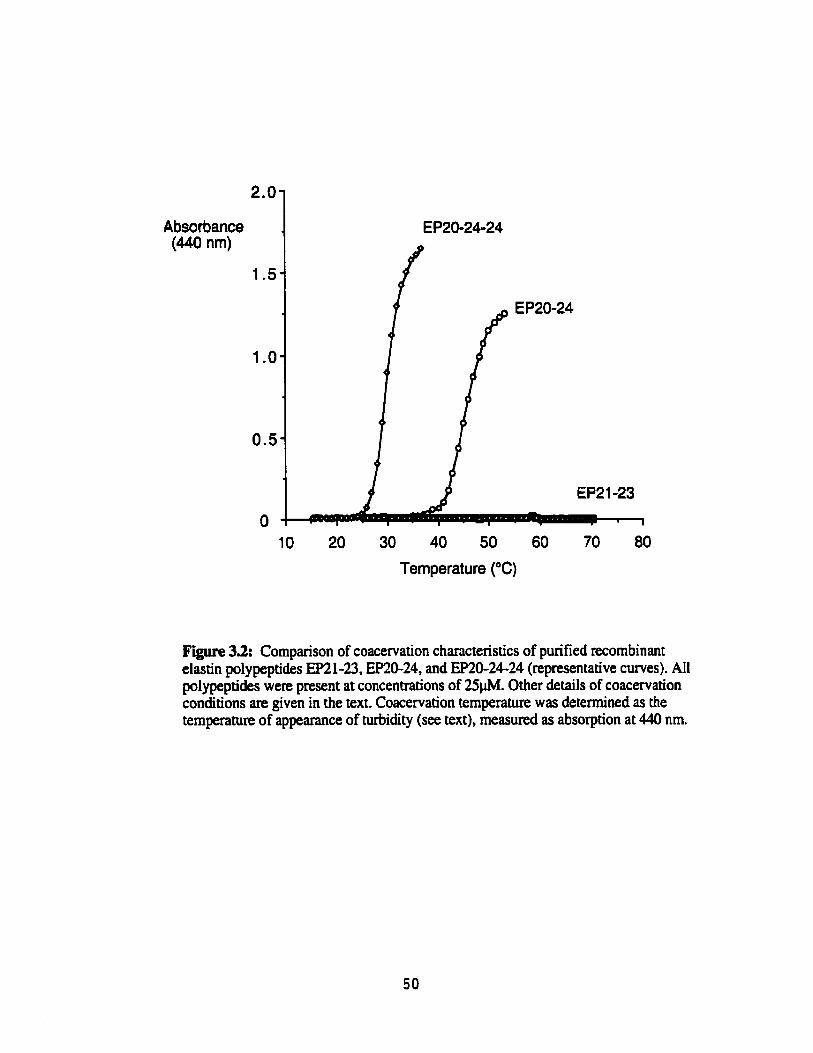
Absorbante (440 nrn)
10 20 30 40 50 60 70 80
Temperature ("C)
Figure 3.2: Cornparison of coacervation characteristics of purified recombinant elastin polypeptides EP21-23, EP20-24, and EP20-24-24 (representative curves). All polypeptides were present at concentrations of 25pM. Other details of coacervation conditions are given in the text. Coacervation temperahue was detennined as the temperature of appearance of turbidity (see text), measured as absorption at 440 nm.
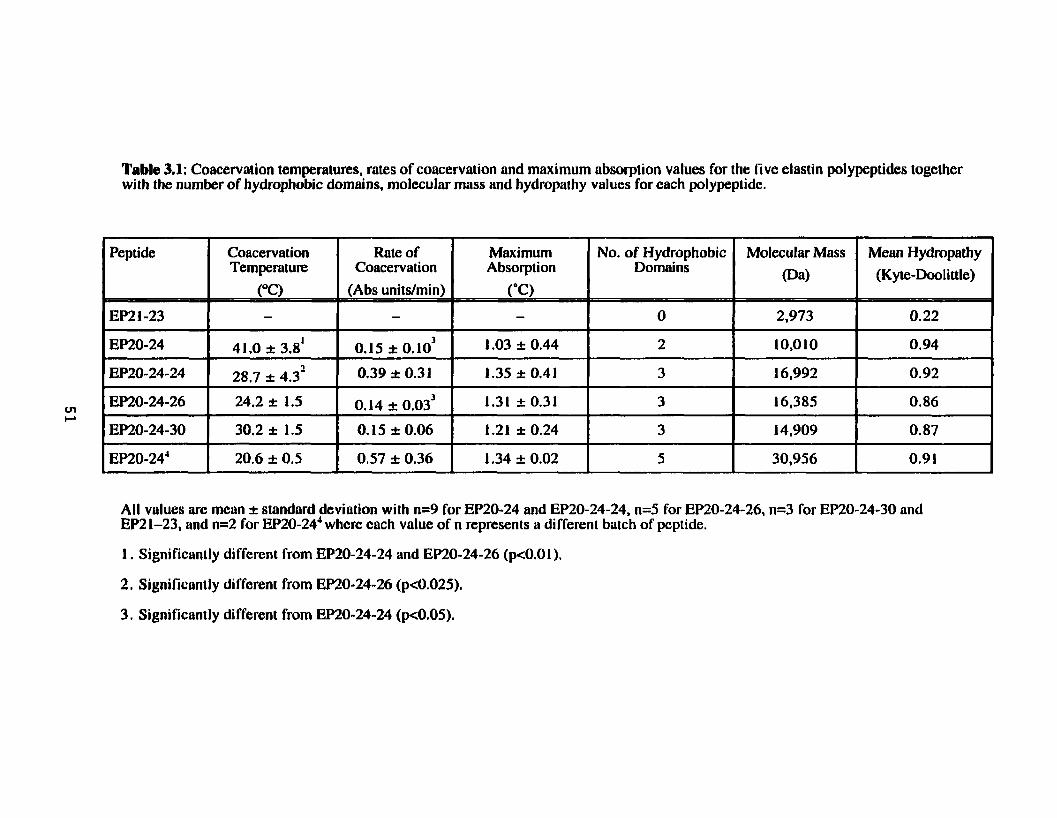
Table 3.1: Coacervatian temperatures, rates of coacervation and maximum absorption values for the rive elastin polypeptides together with the number of hydrophobie domains, molecular mass and hydroparhy values for each polypeptide.
Rote of Maximum No. of Hydrophobie Coacervation Absorption Domains
1 Peptide l ~ !
'EP2 1-23
EP20-24 I
EP20-24-24
EP20-24-26
EP20-24-30
(Abs unitdmin) ('0 - - O
0.15 *0.103 1 .O3 1: 0.44 2
0.39 I 0.31 1.35 I 0.4 1 3
0.14 i 0.03) 1.31 st 0.31 3
O. 15 t 0.06 1.21 t 0.24 3
Coacervation Temperature
(OC) -
41.0 t 3.8'
28.7 I 4.3'
24.2 st 1.5
30.2 st 1.5
Molecular Mass Mean Hydropathy
(Da) (Kyte-Dooli ttle)
All values are meun k standard deviation with n=9 for EP20-24 and EP20-24-24, n=5 for EP20-24-26, n=3 for EP20-24-30 and EP21-23, and n=2 for EP20-24'where each value of n represents a different bakh of peptide.
1. Significanily different from EP20-24-24 and EP2O-24-26 (p<0.01),
2, Significantly di fferent from EP20-24-26 (~~0.025) .
3. Significantly different from EP20-24-24 (pcO.05).
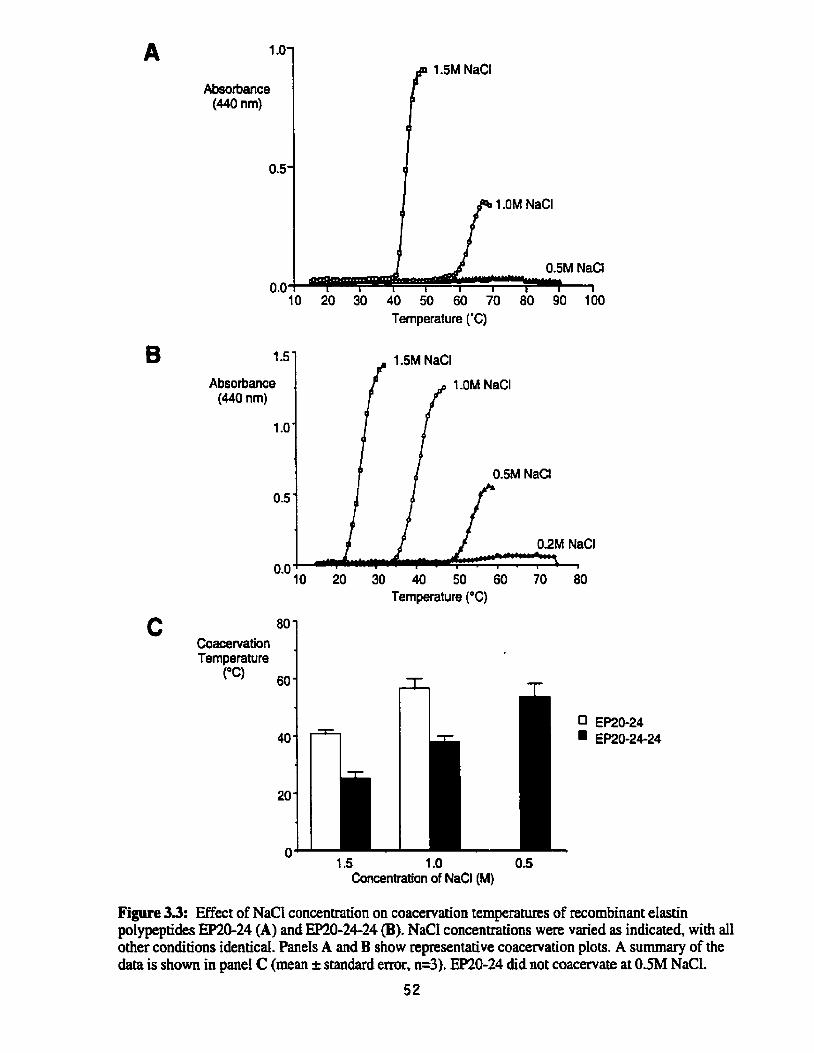
(440 nm) 1
Temperature ('Cl
Absorbante (440 nm) I
OPM NaCl 01 n n 5
Temperature ("C)
V 1.5 1 .O 0 5
Concentiatr-on of NaCl (M)
Figure 3.3: Effect of NaCI concentration on coacervaîion temperatures of recombinant elastin polypeptides EP2û-24 (A) and EP20-24-24 (B). NaCI concentrations were varied as indicated, with aU other conditions identical. PaneIs A and 3 show representative coacervation plots. A summary of the data is shown in panel C (mean +-standard error, n=3). EP20-24 did not coacervate at OSM NaCI.
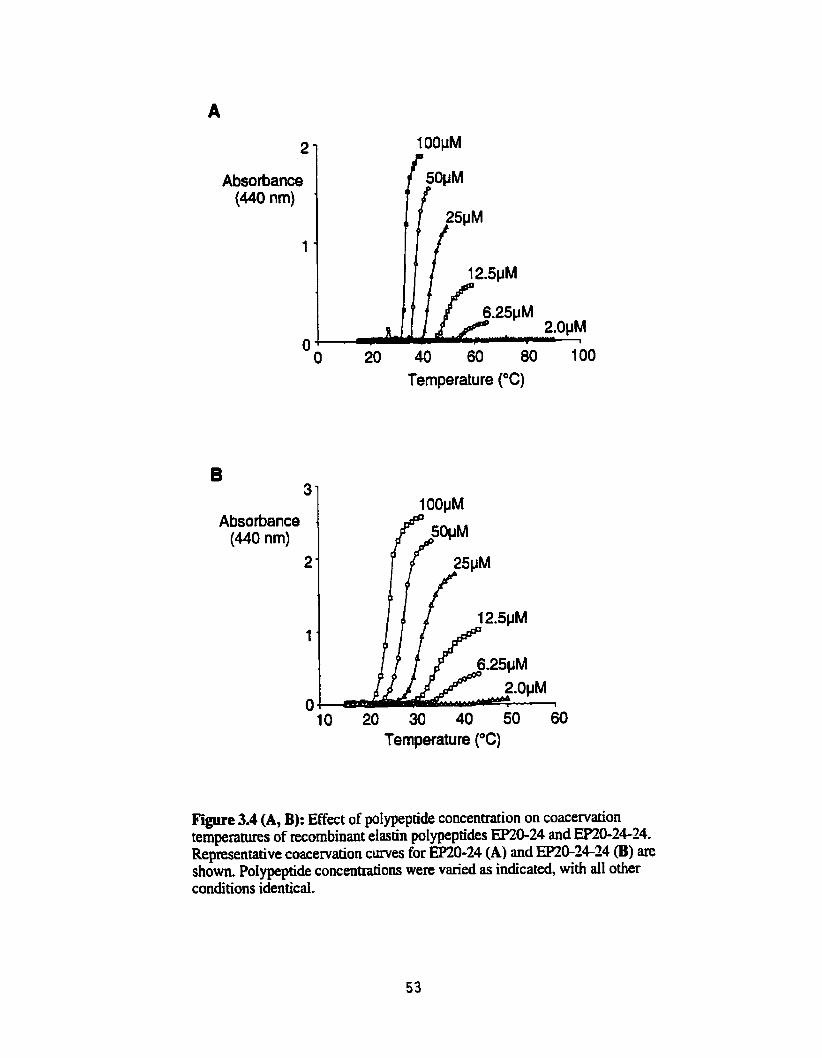
2
Absorbance (440 nm)
t
O O 20 40 60 80 100
Temperature ("C)
B 3
Absorbance (440 nm)
2
t
O 10 20 30 40 50 60
Temperature ("C)
Figure 3.4 (A, B): Effect of poIrpeptide concentration on coacervation temperams of recombinant elasth polypeptides EFZO-24 and EP20-24-24. Representative coacervation c w e s for EP20-24 (A) and EP20-24-24 (B) are show. PoIypeptide concentrations were varied as indicated, with ail other conditions identicd.

D Peptide Concentration (PM)
Peptide Concentration (PM)
2$4 -13 -12 -11 -10 -9
In Peptide Concentration
Figure 3.4 (C-E): Effect of pdypeptide concentration on coacervation temperatures of recombinant elastin polypeptides EP20-24 and EP20-24-24. PIots of coacervation temperature as a îünction of pdypeptide concentration are shown for EP2û-24 (C) and EP20-24-24 @). The bottom panel O compares the relationship of coacervation temperature to polypeptide concentration for WmG24 and =0-24-24 using a semi- logarithmic pIot.
5 4
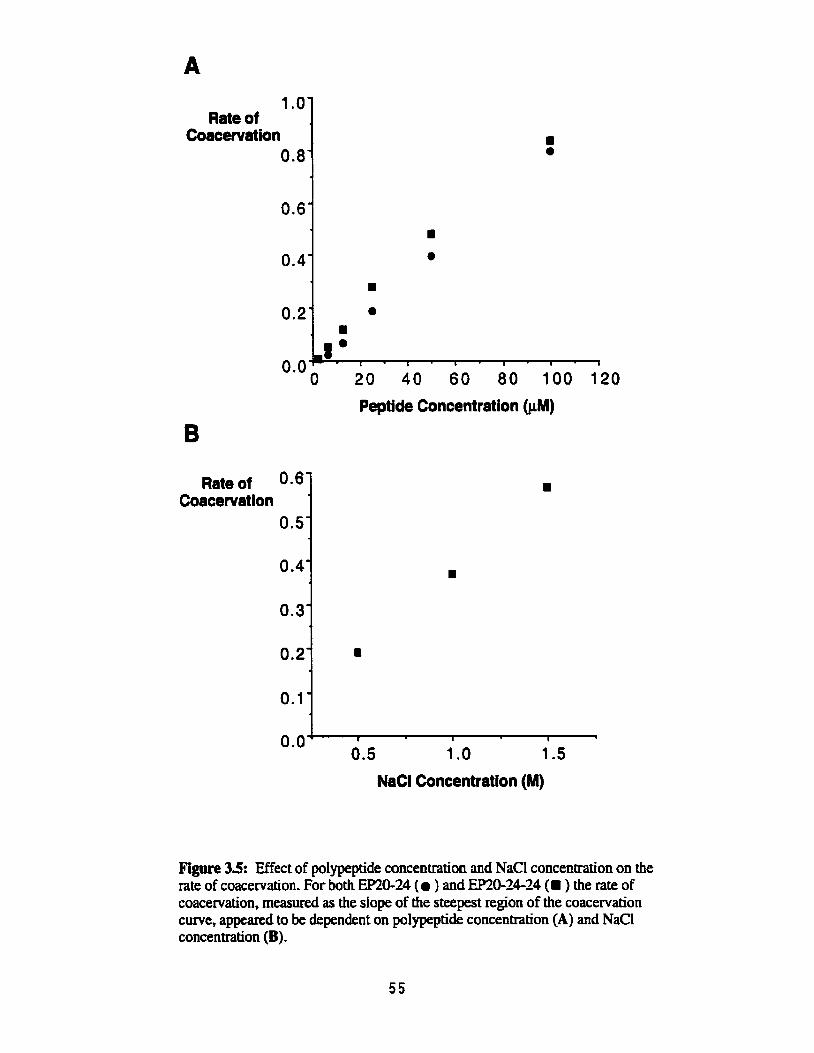
Rate of Coacewation
0.8
Peptide Concentration (PM)
Rate of 0.6' Coacewation
0.5'
O -4'
0.3'
NaC1 Concentration (M)
Figure 35: Effect of polypeptide concentration and NaCl concentration on the rate of coacervation. For both EP20-24 (O ) and EP2û-24-24 (. ) the rate of coacervation, measured as the dope of the steepest region of the coacervation curve, appeared to be dependent on polypeptide concentration (A) and NaCI concentration (B).

Maximum 1 Absorbance 1
Peptide Concentration (PM)
Maximum Absorbance
NaC1 Concentration (M)
Figure 3.6: Effect of polypeptik concentration and NaCl concentration on the maximum absorbance achieved during coacervation. Extent of coacervation, measured as the maximum absorbance achieved during coacervation, appeared to be dependent on the concentration of polypeptide (A) and NaCl concentration (B) for both EP20-24 (a ) and EP20-24-24 (. ).

In order to clarify the effects of polypeptide sequence, molecuk mass and number of
hydrophobic domains on elastin seif-assembly, we have constructed a series of mode1 human
elastin pdypeptides. EP2û-24, the initial polypeptide in the series, had previously showed
the ability CO form low rnolecular weight aggregates even under non-coacervating conditions
(Chapter 2, Figure 2.4). Here, using the polypeptides desMibed in Chapter 2. we have
characterized the ability of these potypeptides to aggregate, using coacervation as a measure
of propensity for self-aggregation.
T h e important principles have emerged. First. hydrophobic regions were required
for temperaturedependent aggregation of these polypeptides. Second, like mpoelastin,
mode1 polypeptides containing both hydrophobic and crosslinking domains showed the
ability to coacervate. Finally, while the number of hydrophobic exons, the general
hydropathy of the polypeptides, and their rnolecular mass infhence propensity for self-
aggregation, the amino acid sequence of the hydrophobic regions w u aIso an important
factor in determining assembly charactenstics.
A number of factors have previously been shown to influence the coacervation
temperature of solubilized elastin (Podrazky and Jackson, 1976) and tropodastin (Vrhovski
et ai., 1997). These include ionic strength of the coacervation buffer and polypeptide
concentration. For both EP20-24 and EP2&24-24, increased saIt concentration lowered the
coacervation temperature. This effect of ionic strength has ken suggested by Urry to be due
to disruption of hydration shelIs surrounding the poIypeptides, caused by the addition of
sodium and chloride ions, promoting polypeptide aggregation (Urry, 1995). Sirnilx to
vopoelastin (Vthovski et ai., 1997), the propensity of these polypeptides to self-aggregate
was directly related to their concentration in solution. As had previousty k e n reported for
tropoelastin (Vrhovski et al., 1997), the inverse reiationship between pdypeptide
concentration and cmcervation temperature was non-linear. That is, at tower polypeptide
concentraiions srna11 changes in polypeptide concentration had a relatively Iarge effect on
coacervation temperature as compared co the effect of sirnilar changes at high polypeptide
concenhations. This non-linear relationship benveen polypeptide concentration and
coacervation temperature was enhanceci in EP2tk24 as compared to E2&24-34.

The presence of hydrophobic exons appeared to be a fundamenta1 requirement for the
ability of elastin polypeptides to seif-assemble, since a polypeptide consisting of oniy the
crosslinking exons 21 and 23 at the same molar concentrations did not show the ability to
self-aggregate under the conditions investigated. Although this was an expected result, we
cannot completely rule out the possibility that self-aggregation might have taken place had
this polypeptide been of equivdent mass to the others used in this study.
Evidence in the literature suggests that increasing the molecular mass demases the
temperature of onset of coacervation (Jensen et al., 2000). Differences found in rhe
coacervation temperatures of EP20-24, EP20-24-24, and m&244 couId have been due to
the increase in the number of hydrophobic domains, which is also reflected in an increase in
the mokcular mass. However, number of hydrophobic domains or molecular mass did not
appear to be a complete explanation for these differences since mû-24-30, which was
smaller than EP20-24-24 by approximately 2000 Da, had essentiaily the same coacervation
temperature. Moreover, EP2&24-26 was approxirnately identical in size to mû-24-24 but
coacervated at a significantly iower temperature.
Fwther evidence for this is given by the cooperative nature of coacervation, as
reflected in the steepness of the slope of the coacervation cwve. The process of aggregation
is more cooperative in the larger polypeptides EP2&24-24 and EP2û-24* than in the smailer
EP2û-24. Weiss and coworkers aiso reported differences in rates of coacervation between
two of their recombinant polypeptides: the full tropoelastin molecule lacking exon 26A, and
a uuncated consmct containing exons 26 through 36 and aIso lacking exon 26A (Jensen et
al., 2000). The rate of coacervation of the truncated tropoelastin was only 34% of that for the
full tropoelastin molecule. This was suggested to be due to the increased number of
hydrophobic domains available for interaction in the larger molecule. The slower rate of
coacervation found for EPîû-24 may, therefore, be due to the lack of a third hydrophobic
domain found in the larger polypeptides. However, the rates of coacervation of E%%-24-30
and EP2û-24-26 appeared to be lower than that of EP2û-24-24, even though ai l of these
polypeptides have three hydrophobic domains. This indicates that the number of hydrophobic
domains is not the only determinant of the cwperativity of self-aggregation. We would
specdate that the sequence of those domains is dso important.

Hydropathy has also been shown to be important in the propensity of polypeptides to
coacervate. in particular, hydrophobic residues lowcr and hydrophilic residues raise the
coacervation temperature (Urry et al., 1991; Urry et al., 1992). However, in our experiments,
differences in propensity for coacervation were not related to polypeptide hydropathy.
Indeed, al1 poIypeptides, with the exception of EP21-23 that lacked hydrophobic exons, had
approximately the same general hydropathy. In fact, EP20-24, which coacervated at
approximately 41°C had a higher generd hydropathy than the other polypeptides, and
EP2&24-26 had a significantly lower coacervation temperature than EP20-2+24, in spite
of its somewhat lower general hydropathy.
On the basis of these results it appears that while both number of hydrophobic
domains and general hydropathy have an effect on determining propensity for self-assembly
of elastin pdypeptides, this property must also be influenced by the specific nature of the
sequences in the hydrophobic domains.
In designing our polypeptides, exon 24 was chosen because it contains the most
striking tandem repeat of the PGVGVA sequence, repeating seven times in tandem in this
exon. Similar sequences are found in other species of elastin, and we hypothesized that the
general conservation of these repeating motifs suggested they might be imponant in self-
assembly. if exon 24 was of particular importance for the self-assembly properties of elastin,
mû-24-24 would be expected to show an increased propensity for self-aggregation,
reflected in a lower coacervation temperature. This was indeed the case when comparing
EP2û-24-24 to EP2û-24. However, substitution of exon 26 for the second occurrence of
exon 24 (EP2Ck24-26) resulted in a polypeptide with an even lower coacervation
temperature. Similarly, substitution of exon 30 for the second iteration of exon 24
(EP20-24-30) resulted in a polypeptide with a coacervation temperature essentidly identical
to that of EP;!û-N-24. These results clearly indicated that the simple repetition of exon 24
was not a critical determinant of the ability of ttzese elastin polypeptides to coacervate.
The ability of synthetic hydrophobic polypeptides based on exons 24,26 and 30 to
coacervate, and the potential roles of these domains in promoting self-assembly of
tropoelastin have been the subject of some controversy. According to data from Urry (Urry,
19821, a synthetic poIy(PGVGVA) peptide was not able to coacervate, ahhough at higher
temperatures it would precipitate to fonn fibrilla. structures. Weiss and coworkers (Jensen et

al,, 2000) have reported that presence of exon 26 is required for coacervation of tropoelastin,
at least under physiologicai conditions, and have suggested that this domain forms an
exposed sequence in the tropoelastin molecule which is required for intermolecular
interactions.
Mecham and coworkers have investigated the role of exon 30 in self-assembly using
pigmented epitheliurn celis, transfected with tropoelastin consmcts containing various exon
deletions (Mecham, 1999). These cells produce both microfibrils and the elastin-binding
protein but not tropoelastin. They found that constnicts lacking exon 30 couid neither
associate with the microfibrils nor crosslink, indicating a role for exon 30 in elastin assembly
(Mecham, 1999). In contrast, Tamburro and coworkers reported that synthetic peptides based
on exon 30 of human elastin were unable to coacervate, aithough fibrillar structures could be
detected by transmission electron microscopy (Castiglione Morelli et al., 1993).
While the importance of these exons in self-assembly in vivo remains controversid,
the data presented here show clearly that polypeptides containing al1 of these hydrophobic
sequences in juxtaposition to crosslinking domains, the context in which they are present in
natural uopoelastin. were able to undergo self-assembly througb a classical coacervation
pmcess. However, there were differences in the propensity of these polypeptides to
coacervate, which we suggest to be due to the nature of the specific sequences found in these
hydrophobic exons. It is, therefore, possible that exon 26, with its nonapeptide repeat, may be
more important for coacervation of tropoelastin than exon 24.
The rate of coacervation (a mesure of the cwperativity of the aggregation process)
and the maximum absorùance (a measure of the size of the aggregates formed) both appeared
to be higher for EP2û-24-24 than EP2û-24. Weiss and coworkers aiso found a higher rate of
coacervation for recombinant tropoelastin lacking exon 26A compared to a tropoelastin
isoform containing ody exons 26 through 36, but also lacking exon 26A (Jensen et al.,
2000). They attnbuted the enhanced cwperativity of the full-length tropoelastin to the
additional hydrophobic domains present. In the case of our polypeptides, we suggest that the
third hydrophobic domain in EP2û-24-24 likely provided an additiond nucleation site for
aggregation, enhancing the rate of coacervation. More independent sites of coacervation
wodd dso result in a Iarger nurnber of smaU particles, possibly explainhg the higher
maximum absorbance (turbidity) as compared to EP2û-24. In contrast, with fewer nucleation

sites, EP2&24 would have a slower rate of coacewation and fewer larger particles, resulting
in a lower maximum absorbance.
Both the rate of coacervation and the maximum absorbance were dependent on the
ionic strength of the buffer and on the polypeptide concentration. As the concentration of
polypeptide or NaCl increased, the maximum absorbance and rate of coacervation aiso
increased for both EP2û-24 and EP2&24-24. Urry and coworkers found similar results for
a+Iastin and the polypentapeptide (UV and Long, 1977). The increased availability of
polypeptide for self-aggregation, resulting either from increased polypeptide concentration or
effective dehydration of the polypeptides by increasing ionic strength, wodd provide
additional nucleation sites for coacervation. Again, an increased number of independent
nucleation sites would enhance the rate of coacewation and result in a larger number of
smaller coacervate particles generated, reflected in an increase in the maximum absorbance.

Chapter 4
Ultrastructure and Self-Assem bly
The art d progress is to preserve order amid change, and to pieserve change amid order. -Alfred Norai Whitehead. rnathmatldan and philosopher (1861-1947)

Introduction The ultrastructure of elastin has long been a subject of controversy . Some
investigators argue that insokuble elastin is amorphous (Greedee Jr. et al., 1966; Ross and
Bomstein, 1969), consisting of a random network of protein chains. Others present evidence
for a more orientated structure made up of a fibrillar matrix (Bressan et al., 1986; Gotte et al.,
1974; Serafini-Fracassini et al., 1976). Models of elasticity of elastin have k e n developed
for both structurai representations (Gray et al., 1973; Hoeve and Flory, 1974; Urry et al.,
1984, Weis-Fogh and Andersen, 1970).
Early evidence for an amorphous structure came €rom elecbon microscopy data
(GreenIee Jr. et ai., 1966; Ross and Bomstein, 1969), visualizing insoluble elastin in elastic
fibres as an amorphous core surrounded by microfibrils. However more recent studies using
scanning and ~ s m i s s i o n electron microscopy described elastin matrices as fibrillar
structures with a substnicture of smaller filaments. Such observations have been made on
purified insoluble eIastin (Gotte et al., 1974; Gotte et al., 1972; Serafini-Fracassini et al.,
1976), coacervated tropoelastin (Bressan et al., 1983; Bressan et aI., 1986; Cox et al., 1974),
coacervated aelastin ('ïamburro et al., 1995; Volpin et al., 1976) and coacervated synthetic
polypeptides correspondhg to hydrophobie sequences of elas tin (Urry et al., 1974).
Overwhelming evidence now supports a fibrillar elastin structure and suggests that early
observations of eIastin as an amorphous matrix were probably an artifact of the visualization
process.
The mechanisms involved in assembling these fibrillar sîructures have also been
investigated using the in vitro process of coacervation as a mode1 of assembly. Classically,
coacervation has been defined as a reversible process. However, Jamieson has presented
evidence for both reversible and irreversible phases, suggesting elastin assembIy is a muiti-
stage process (Jamieson et al., 1972). A üansmission elecmn microscopy study of
üopoeiastin coacervation, capturing the ultrastructure of elastin at various times during the
aggregation process, provides additional support for multi-stage assembly (Bressan et ai.,
1986). This work gave rise to the speculation that fiIaments fonned during the earIy stages of
the coacervation process subsequently aIign into compact fibri1hr structures.

While the coacervation process has been useful as a general mode1 of eIastin
assembly, details of the mechanisms of the assembly process and fibre formation are still
lacking. Here we have used two of the recombinantly generated elastin polypeptides,
EP2û-24 and EP2&24-24, to further investigate details of the assembly pmcess during
coacervation.

Materials and Methods Coacervafion Erperinients
Coacervation experiments used the protocol deshbed in the Materials and Methods
section of Chapter 3. EP2624 and EP2&24-24 were dissolved in coacervation buffer (50
rnM Tris, 1.5 M NaCI, 1 m M CaC12, pH 7) to a final polypeptide concentration of 25 ph4
and the solutions stored ovemight at 4'C. In order to investigate reversibility, once the initial
coacervation was complete (Le. the maximum absorbance was attained), the temperature was
decteased immediately to 1YC, held for 5 minutes, then increased again at 1'C per minute.
Alternatively, after coacervation was complete the temperature was held above the
coacervation temperature for 18 hours, followed by cooling tu 15°C for 5 minutes, before re-
coacervation was attempted. Separate batches of polypeptide were used for each experiment
(n=3).
Transmission Electmn Microscopy (TEM)
EP2û-24 and ERû-24-24 were prepared for electron microscopy at three stages
during the coacewation process: precoacervation (Le. before the temperature was raîsed),
pst-coacervation (i.e. irnmediateIy after coacewation was complete), and following
overnight incubation of the coacervate at elevated temperature. Copper grids (400 mesh)
(Canemco Supplies) coated with formvar, dried and carbon coated in a Hitachi Hus-SGB
vacuum evaporator were used for al1 experiments.
For pre-coacervation samples, EP2624 and EPSS24-24 were dissolved in cold
coacewation buffer (50 m M Tris, 1.5 M NaCI, 1 mM CaC12, pH 7) to a final polypeptide
concentration of 250 pM, and were stored at 4°C overnight. The @ds for pre-coacervation
samples were pre-cooled to 4°C. A drop of sample was placed on parafilm (American
NationaI Cm), also pre-cooled to 4"C, and a grid was placed over the sample drop and
incubated for one minute. The grid was removed fiom the drop and blotted dry using filter
paper and lefi to dry fully. The samples were then negatively stained with uranyl acetate. A
drop of 2% acpeous m y i acetate was placed on parafilm and the grid placed sample side
down on the drop, mcubated for 30 seconds to 1 rriinute and blotted dry. The grid was
ailowed to dry completely before viewing in a Hitachi H6ûû transmission electron
microscope.

For pst-coacervation samples, EP20-24 and EP20-24-24 were dissolved in cold
coacervation buffer (50 mM Tris, 1.5 M NaCl, 1 mM CaC12, pH 7) to a final polypeptide
concentration of 250 pM, and were stored at 4°C overnight. The polypeptide samples were
placed in a 22°C water bath and the temperature was increased to either 39°C for
EP20-24-24 or 50°C for EP20-24. The coacervated polypeptide samples were incubated at
the elevated temperatm for 10 minutes. The grids for the post-coacervation samples were
preheated to either 39°C or 50°C. The samples were applied to the grids in the same manner
as the precoacervation samples. The g d s were dried and negatively stained as described
above.
For overnight samples, EP20-24 and EP20-24-24 were prepared in the manner
described for the post-coacervation samples, and following coacervation were incubated at
50°C or 39°C overnight. The coacervate becarne dense and settled to the bottom of the tube
after ovemight incubation. SarnpIes were then applied to preheated grids, as described for the
pre-coacervation sarnples. In some cases the samples were sonicated before application on
the grids in order to decrease the sample thickness prior to negative staining. The grids were
dried and negatively stained as described above.
Circulizr Dichroikm (CD)
Elastin polypeptide samples were dissolved in coacervation buffer (50 mM Tris, 1.5
M NaCl, 1 mM CaCl,, pH 7) to a final polypeptide concentration of 25 ph4, and were stored
at 4°C overnight. Secondary structure was monitored through the coacervation process using
an AVIV 62DS spectrometer with a temperature controller. Structure was monitored at
22011x11 Erom 15°C to 55°C for EP20-24, h m 15" to 45°C for EP20-24-24 and from 15" to
60°C for EP21-23. Typical CD spectra of soluble elastin show a minimum at 220 nm
(Debelle and ALix, 1995; Tarnburro et aI., 1977; Vrhovski et al., 1997) generally indicative of
structure in proteins (Manavalan and Johnson Sr., 1983). Temperature was increased at a
rate of 1°C per minute, consisting of a 6 second rise in temperature and an equilibration time
of 54 seconds at each temperature. Controis using buffer aione were subtracted h m the
sample curves to elirninate the effect of the buffer.

Results For both EP2û-24 and EP20-24-24, the process of coacervation could be reversed if
the solution temperature was decreased immediatdy after coacervation was compIete.
Polypeptide solutions that became M i d upon coacervation became clear again when the
temperature was decreased. A subsequent increase in solution temperature resulted in a
coacervation curve essentiaily identical to the initiai coacervation curve, with the same
coacervation temperature (Figure 4.1). In confrast, if the temperature of the coacervate was
held above the coacervation temperature for 18 hours after coacervation, the solution
rernained cloudy even after coohg, and contained substantidIy larger, flaky aggregates.
With a second increase in temperature there was no further coacervation, indicated by no
increase in absorbance (Figure 4.1). Together these results suggest that polypeptide
aggregation, as a resuit of coacervation, is a multi-stage process with only the first stage
king reversible on cooling.
Transmission electron microscopy was used to examine the ultrastnicture of
the polypeptides at the three stages of coacervation. Prior to coacervation, both EP2M4
(Figure 4.2A) and EP20-24-24 (Figure 4.m) appeared to form clustered globular structures
ranging h m 6nm to 25nm in diameter. A similar size range was reported by Bressan et al. in
electron micrographs of soluble tropoelastin (Bressan et al., 1986). Shortly after
coacervation, an open network of relatively unaiigned filamentous structures formed (Figure
4.3A for EP2û-24 and Figure 4.3B for EP2û-24-24). These filaments had diameters ranging
in size h m 3nm to 6nm. Even at this stage the network of filaments formed from
EP2û-24-24 appeared to be developing some directionaiity (Figure 4.4). Although simiiar
filaments formed h m ERû-24, such directionaiity was not evident.
For both EP2û-24 and EP20-24-24, following oveniight incubation at elevated
temperature, the open network of filaments appeared to compact into ordered fibrillar
structures (Figure 4.5). However, the finai fibrillar structures seen for EP2û-24 folIowing
ovemight incubation (Figure 4.5A) were clearly not as well compacted as those of
EP2û-24-24 (Figure 4.5B). The stages of coacervation for both elastin polypeptides are
show dong with micrographs for mpoeIastin published by Bressan (Bressan et ai., 1986)
and prepared in a similar manner to the polypeptides (Figure 4.6). Ultrastructures for both
polypeptides are very similar to those fomed h m mpoelastin at the pre-coacervation and

10 minutes pst-coacervation stages. However, the compacted, weii-orde~d fibres seen for
mû-2624 closely resemble those seen for tropoelastin, in contrast to the l a s weN-
organized fibres fonned from EP2û-24.
Circula. dichroism measurements using EP2&24 were done in an attempt to detect
structural changes taking place coincident with or immediately preceding coacervation. A
gradua1 increase in eilipticity at 220nrn with increasing temperature suggested that EPXb24
initially undergoes non-cooperative behaviour, unraveling prior to coacervation. Coincident
with coacervation, there was a sharp increase in ellipticity suggesting a more cooperative
structural change. (Figure 4.7A). In contrast. EP21-23 showed only non-cooperative
behaviour throughout the temperature range. Interpretation of these circuIar dichroism resuIts
was, however, difficult because of the rapid loss of signal as the solution became tubid on
coacervation figure 4.7B). Similar resuits were seen for EP3û-24-24 (data not shown).
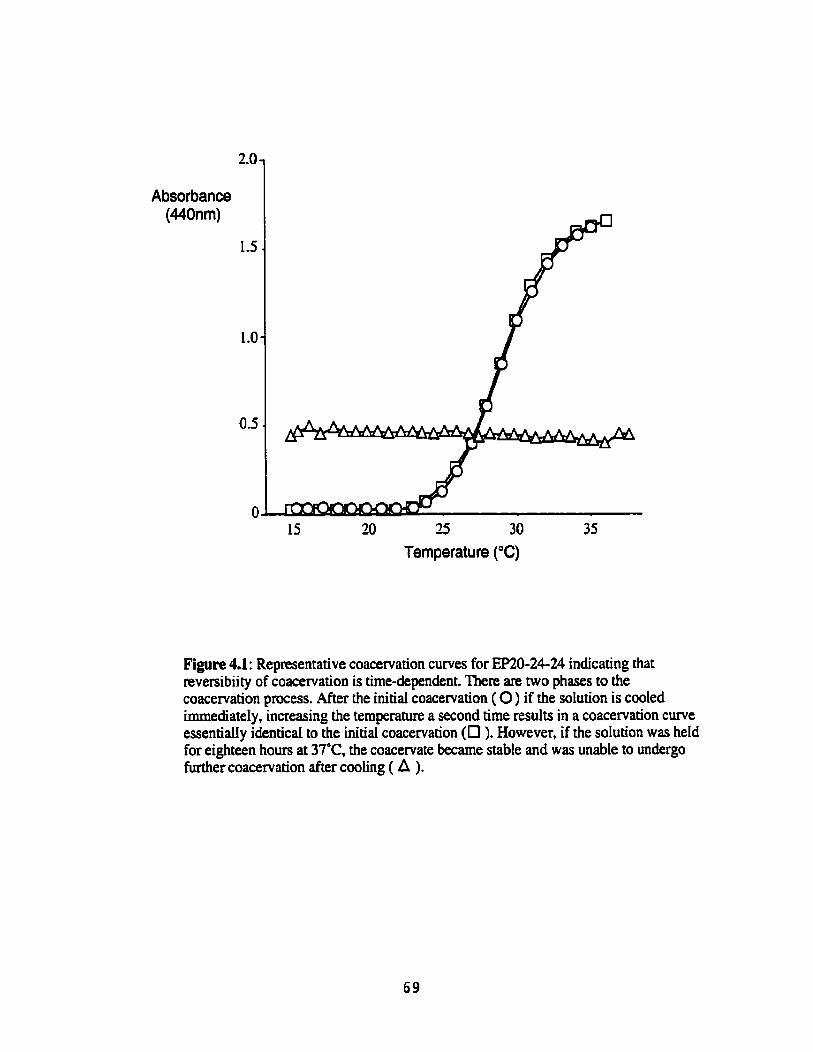
Absorbance (440nm)
15 20 25 30 35
Temperature ("Cl
Figure 4.1: Representative coacervation curves for EP20-24-24 indicating that reversibiity of coacervation is tirne-dependent. There are two phases to the coacervation p e s s . AFter the initial coacervation ( O ) if the solution is cooled immediately, increasing the temperature a second time results in a coacervation curve essentially identicd to the initial coacervation (O ). However, if the solution was held for eighteen hours at 37°C. the coacervate became stabIe and was unable to undergo further coacervation after cooting ( A ).

Figure 43: Prior to coacervation the eIastin polypeptides have a giobular structure, visibIe using transmission electron microscopy. Before coacervation both EP20-24 (A) and EP2û-24-24-24 (B) appear as giobdar structures (monomers and small oiigomers). The polypeptides are negatively stained with uranyl acetate, appeaing white in these transmission eIectron micrographs.
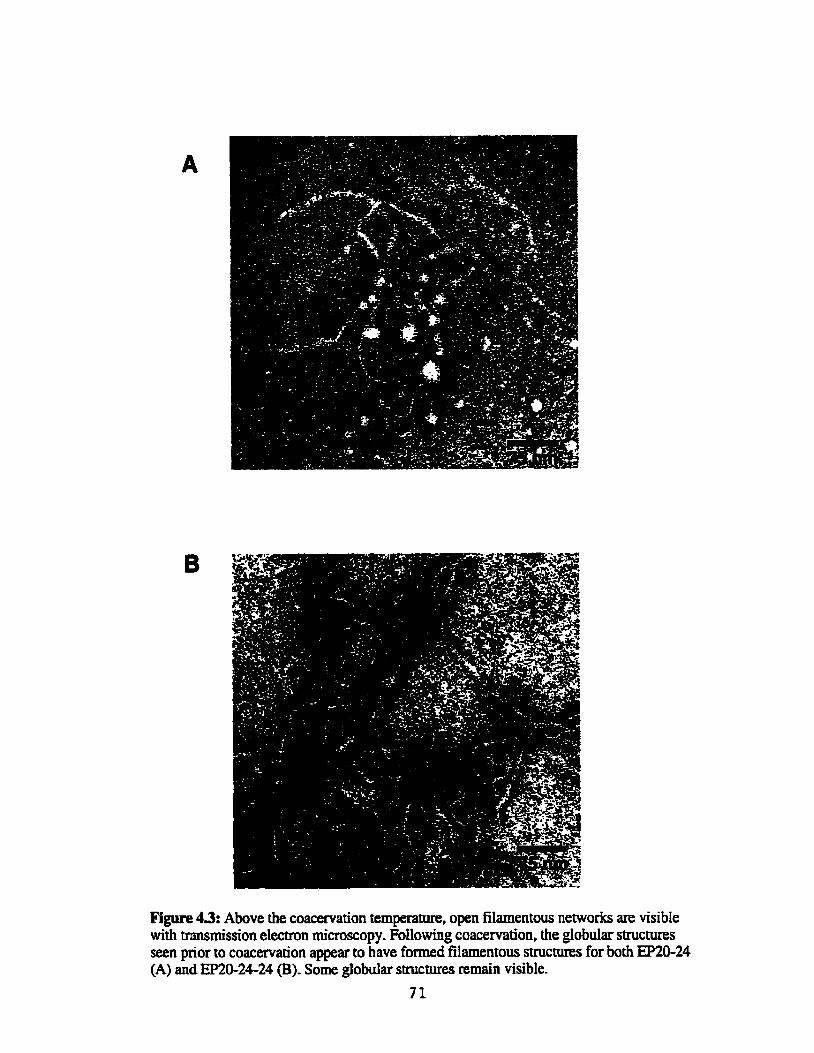
Figure 43: Above the coacervation temperame, open filamentous networks are visibIe with tmsmission electron mic~oscopy. FoUowing coacervation, the giobular stnicnires seen pior to coacervation appear to have fomed filamentous structures for both EP20-24 (A) and EPSO-24-24 (B). Some globular structures remain visible.
71
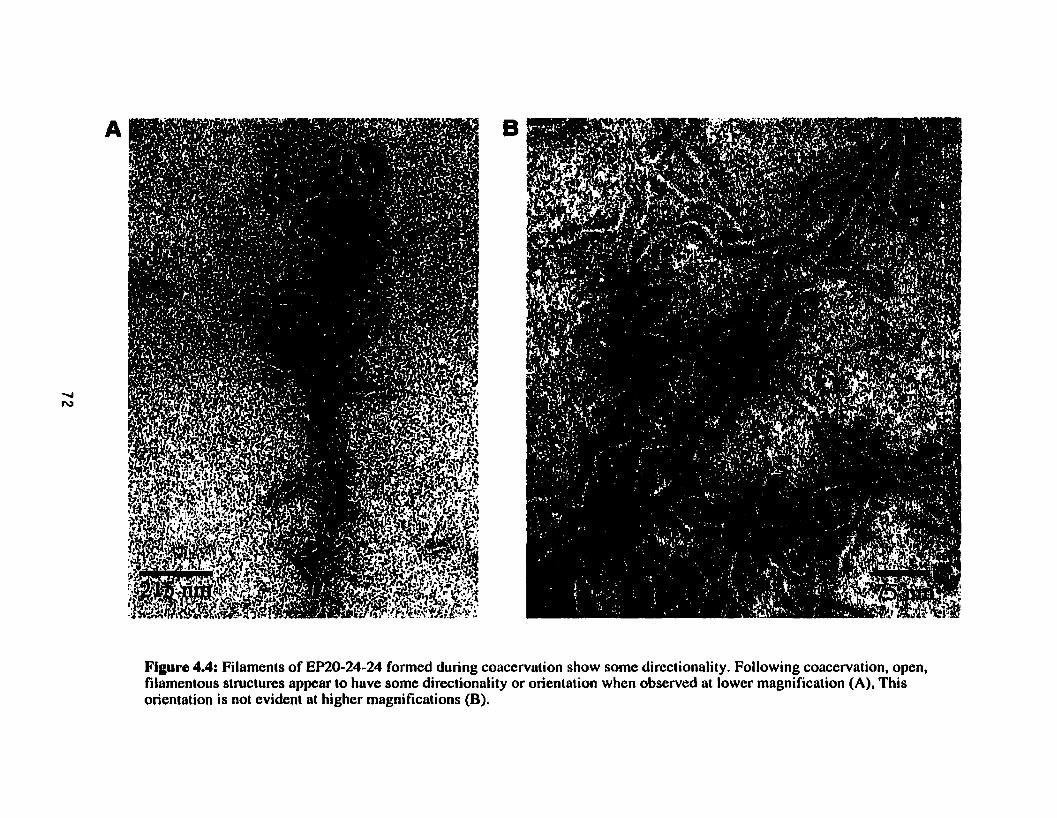
Figure 4.4: Filaments of EP20-24-24 fonned during coacervation show some direciionality. Following coacervation, open, filamentous structures appear to have some direciionality or orientation when observed at lower magnification (A), This orientation is not evident at higher magnifications (B).

Figure 45: Overnight mcubation above the coacervation temperature resuits in fibriuar coacervates. After incubation for 18 hours at 50'C for EP20-24 (A) or 39'C for EP20-24-24 (B) the open filamentous structures seen imrnediateiy foilowing coacervation appear to have aIigned in paraiiel to form fibdIar structures. EP20-24-24 fibrils appear more ordered than those of =û-24.

Figure 4.6: Cornparison of EP20-24, EP20-24-24 and iropoelastin structures at lhree stages of coacervation. Pre-coacervation and immediately following coacervation (10 minutes) the polypeptide structures are similnr to those of tropoelastin. However, iifter overnight incubation (overnight) the structure of EP20-24-24 closely resembles that of iropoelastin wherens EP20-24 is not as well organized, The scale bar is 100nm. The tropoelastin micrographs are from Bressan et al., 1986.

10 20 30 40 50 60 Temperature ("C)
0 30 Absorbance
Absorbance / '
--
10 20 30 40 50 60 Temperature (OC)
Figure 4.7: Changes in secondary structure of EP20-24 are coincident with coacewation. Circuiar dichroism monitored at 220nm with increasing temperature show a non4ooperative change in structure of E'1-23 throughout the temperature range. However, EP20-24 appears to undergo a cooperative change at the temperature corresponding to coacervation (A). The structural change in EP20-24 is coincident with an increase in absorbante, at the coacervation temperature (B).

Details of the process of aggregation and fibre formation of elastin and elastin
polypeptides during coacervation are largely unknown. Using mode1 polypeptides we have
investigated two aspects of the coacervation process, the ultrastructure during the aggregation
process and the stability of the coacervate, and have developed a mode1 of elastin assembly.
Ultrastructure
Insight into the mechanisms of elastin assembly was gained through the visualization
of the elastin polypeptides at various stages in the coacervation process. Transmission
electron microscopy prior to coacervation revealed that the polypeptides were in a globular
form with variable diameter size, indicating the presence of monomers and small oligomers.
Ten minutes after coacervation, an open, relatively unorganized network of filaments was
seen, suggesting codescence and restnicturing of the globular molecules. Finally, after
incubating the coacervate ovemight at elevated temperatUres, these structures spontaneously
reorganized into compact fibriilar arrays of parallel filaments. Remarkabty, the structures at
the various stages of coacervation were sirnilar to those seen for coacervation of tropoelastin
(Bressan et al,, 1986).
In spite of these similarities in ultrastructure, it was clear that m&24 did not
organize as comprictiy as EP2û-24-24 or tropoelastin. On the other hmd, fibres fonned from
EP3û-24-24 were indistinguishable from those fonned h m tropoelastin. Earlier data from
us (Chapter 3) and others (Bressan et al., 1986; Urry et al., 1974) suggested that aggregation
of elastin polypeptides required the presence of hydrophobic dornains. In this respect, it may
be significant that EP2&24 contains two hydrophobic domains as cornpared to the presence
of three hydrophobic domains in EP20-24-24. We have already shown (Chapter 3) that the
presence of this additional hydrophobic domain substantialIy inmases the propensity of the
elastin polypeptides for self-aggregation, as reflected by a lower coacervation temperature.
This additional hydrophobic domain now also appears to be important for the ability of the
polypeptide to self-assemble into compact, well-organized fibriUar stntctures itfter
coacervation. The decreased abiiity of EP2S24 to form compact fibres may be a reflection
of more Iimited opportu~ties for overlap in fibre formation resulting h m the presence of
only two hydrophobic domains (Figure 4.8).

Figure 4.8: Fibre struclures of coucervated EP2û-24 and EP20-24-24 as a reflection of opporiunities for overlap of hydrophobic domains during self-aggmgation, EP2Q-24-24 has an udditional hydrophobic domain thut müy allow il to organize into structures more closely resembling those of troplastin, The cartoans below the iransmission electron micrographs show the possibilities for aggregation with two or three hyhphûbic domains. The hydrophobic domains are represented by reclangular plates, interconnecied by crosslinking domuins.

Reversible a d Iweversible Stages of Coacervaîiun
Although coacervation has been classically considered a reversible process (Urry,
1982) results presented here indicate that the reversibility of coacervation of elastin
polypeptides is tirne-dependent. Using EP2&24 and EP2û-2624 as models of tropoelastin,
coacervation can be Mly reversed only if the solution temperature is lowered irnrnediately
after coacervation. However, after overnight incubation at temperatures above the
coacervation temperature, the coacervate has transformed into flaky particles and Iowering
the solution temperature no longer results in redissolution of the polypeptide. These resdts
are in contrast to those reported by Urry for the polypentapeptide, poly(PGVGV), the
coacervation of which could be reversed even after prolonged incubation above the
coacervation temperature (Urry, 1982).
Early work by Jamieson and coworkers investigated the reversibility of ~-ehStin
coacewation using light scattering methods (Jamieson et ai., 1972). Oligomers of these
peptides were detected at rwm temperature, which becarne larger aggregates upon further
increases in temperature. At the coacervation temperature, the aggregates were described as
decreased in size and the solution became turbid. This size reduction was interpreted as a
conformationai change involving compaction. if cwled imrnediately, the solution became
clear, indicating a reversible process. However, after holding the coacervate at temperatures
above the coacewation temperature for 10 to 12 hours, they reported that coacervation
became irreversible (Jamieson et ai., 1972).
We have also obsewed time-dependent reversible and irreversible stages of assembly.
However, our interpretation of the process of coacervation differs sornewhat from that of
Jarnieson. Jarnieson suggested that pnor to visibly detectable coacervation (Le. onset of
turbidity) large aggregates were aiready forming in solution. Subsequent coacewation was
due to a conformationai change in these aggregates to a more compact structure (Jamieson et
al., 1972). However, our transmission electron microscopy data, dong with that of Bressan
for tropoelastin (Bressan et al., 1986). suggest that coacervation involves a transition of
globular structures which may be associated in solution, into larger, more open filamentous
aggregates. This transition may be reflected in the abrupt change in the CD spectnun at the
point of coacervaîion seen for the elastin pdypeptides. Moreover, our data suggest that

compaction and parailel alignment of the filaments occurs only following prolonged
incubation at elevated temperatures.
Mo&l of Eiàstin Assembly
Based on these data we have proposed a model, adapted h m Jamieson (Jamieson et
al., 1972), of the reversible and irreversible stages of elastin assembly (Figure 4.9). Initially,
below the coacervation temperature, the polypeptides exist as monomers or smail otigomers.
Srna11 oligomers of this kind have k e n observed for the eIastin polypeptides on SDS
polyacrylamide gel elecmphoresis (Figure 2.5, lane 3). On coacervation, the polypeptides
undergo a ûansformation into a loosely organized, open network of filaments. This stage of
the coacervation process likely involves a conformational change of the polypeptides and is
reversible. However, during protonged incubation at an etevated temperature reorganization
of the filaments takes place invokving improved laterai alignment into stable, compact
fibrillar stnictures which can no longer be solubilized simpIy by lowering solution
temperature. Optimal reorganization into these fibrillar stnictures appears to require at least
three hydrophobic domains in the polypeptides.

oligomer formation
coacervation //
Figure 4.9: A model of elastin self-assembly showing reversible and irreversible stages. At room temperature, the elastin polypeptides exist as manoiners or smull oligomers. Pollowing coacervation, open filamentous networks form from the globular oligomers. This stage of coiicervation can be reversed, However, following overnight incubation fibrillar structures form, made up of aligned filaments. Ai this stage, coacervation can no longer be reversed. Adapted from Jarnieson et al., 1972.

Chapter 5
Alignment of Elastin Polypeptides:
Crosslinking and Polymer Mechanics
In order to make a petied and beauüiul machine, it is not requisiîe to know how to make it. -hKenzie 1868 in 'Damin's Dangernus Ideam by OC Oennen

There has been speculation that molecular aggregation of tropoelastin in the
extracellular mamx aIigns lysine residues, allowing the formation of lysine-derived,
intermolecular crossiinks (Bressan et al., 1986). In vitro. using coacervation to induce
aggregation of elastin, investigators have reported crosslinks forming in coacervated
tropoelastin modified by purified lysyl oxidase, supporting the hypothesis that alignment
occurs during self-aggregation (Bedell-Hogan et ai., 1993; Narayanan et al,, 1978).
The major crosslinks in insoluble elastin are desmosine and isodesmosine, involving
the participation of four lysine residues, with two conüibuted from each polypeptide chah.
In order for these crosslinks to form, the lysines must be presented in a specific spatial
orientation. In addition, only three of the four lysines are conveaed to semi-aldehydes or
ailysines (Figure 1.1). Once modified, spontaneous condensation teactions occur to form
dehydrolysinonodeucine and allysine aldol, which can subsequently condense ta fom
desmosine and isodesmosine crosslinks.
Crosslink formation convens tropoelastin into a stable, highly insoluble polymerized
matrix, and contributes to the mechanical properties of this matrix. Once crossIinked,
insoluble eIastin is resistmt to solubilization under most extraction conditions, except those
which hydrolyze the polypeptide backbone.
Mechanically, elastin provides tissues with the property of extensibility and elastic
recoil. There is generai agreement that the mechanism of elasticity in elastin is entropic in
nature. However, the controversy over whether elastin is amorphous or ordmd extends to
proposed models of elasticity. A number of models have been presented. The mode1 of
Hoeve and Flory (Hoeve and Flory, 1974; Hoeve and Flory, 1958) assumes a random
configuration of poIypeptide chahs. In contrast, several other models assume ordered or
Eibrillar stmctures (Gray et ai., 1973; Urry et al., 1984; Weis-Fogh and Andersen, 1970).
Using recombinant elastin polypeptides as toaIs, we have investigated the process of
ordered alignrnent of elastin monomers in elastin assembly. Specifically we have studied the
mle of coacervation in the alignment of lysine residues, the minimai requirements for
assembly into continuous, crossiinked polymers and the tensile mechanical properties of
polymeric membranes fabricated h m the pdypeptides.

OxiMve DeamirrUtion of Lysine Redues Ushg Horseradish Peroxidare
Horseradish peroxidase (HRP) (Sigma-Aidrich Co.) and catechol were dissolved in
0.01 M Tris buffer, pH 7.5. Polypeptides were dissolved in coacervation b a e r (50 mM Tris,
1.5 M NaCl, 1 mM CaCI,, pH 7) to a concentration of 10 mglml (lm. for ER(F-34 and
0.6mM for EP2û-24-24) and were incubated overnight at 4°C.
Poiypepu'de soIutions were placed in a waterbaih at ruom temperature and the bath
temperature was subsequently raised to approximately 10°C above the coacervation
temperature of the potypeptide ( 4 O C for EP2M4-24 and -5 1°C for EP2û-24). After
coacervation, the polypeptides were incubated at the eievated temperature for 10 minutes
prior to addition of crosslinking reagents. Catechol, hydrogen peroxide and horseradish
peroxidase were added ta final concentrations of 0.1 M, 0.15 M and 0.42. mglm1 respectiveIy.
As a conuol, only the horseradish peroxidase was added. Aliquots of the solution were
removed at three time points: immediately after the addition of the crossiinking reagents, and
after incubation for 5 and 15 minutes. These aliquots were centrifugeci, the supematant was
removed and a solution of 2% SDSI4 M urea in Tris buffer was added to the pellet. The
suspension was boited for 10 minutes, centrifuged and the materia1 so Iubiiiied by SDSfurea
was removed. The supematant and SDS/urea extract were analyzed by SDS-PAGE
eIecuophoresis foilowed by Western blotting with an antibody to EP20-24. Insolub[e
materiai remaining after extraction with SDS/urea was sent for amino acid andysis (HSC
Biotechnoiogy Service Centre).
Oxidatnte Deaminrilion of Lysine Residues Using Fyrroloqrrinoline Qirinone (Pm PQQ (Sigma-Aldrich Co.) was dissolved in sodium borate buffer (0.1 M sodium
borate, 0-15M NaCI, pH 8) to a concentration of 20 mM. Copper sulfate was dissolved in
water to a concentration of 100 mM. PoIypeptides W h 2 4 and EP20-24-24) were
dissdved in sodium borate buffer (0.1 M sodium borate, 1.5M NaCl, pH 8) to a
concentration of 50 pM and incubated overnight at 4OC.
As âescribed above, poIypepeide solutions were placed in a water bath at room
temperature and the bath temperature was raised to approximately 10°C above the

coacemation temperature of the polypeptide (-40°C for EP20-24-24 and -51°C for
EP2&24). The coacervates were incubated at the eIevated temperature for 10 minutes and
then crosslinking reagents were added- Copper sulfate and PQQ were added in a ratio of 1:2
to final PQQ concentrations of 0,0.2,0.4,0.9 and 2.9 mM. The solutions were incubated for
four hours at the elevated temperatures. After incubation, the solutions were centrifuge& the
supernatant removed and 2% SDS/4 M urea in Tris buffer was added to the pellet. After
boiling for 10 minutes the solutions were centrifugecl, the matenal solubilized by SDSIurea
was removed and the pellet was sent for amino acid analysis (HSC Biotechnology Service
Centre). Following separation by SDS-PAGE, soluble polypeptides were detected by
Western blotting using an antibody to mû-24.
To determine whether coacervation was necessary for crosslinking, polypeptides were
dissolved in buffer lacking sodium chloride, preventing coacervation, and the polypeptides
were oxidized as described above. Similar experiments were done with EP20-24,
EP20-24-24 and EP21-23. E E 1-23 contained only crosslinking domains and does not
coacervate un&r any conditions tested.
Crossiinked EP2&24 and EP20-24-24 samples were analyzed for desmosine and
isodesmosine using a specific radioimmunoassay (courtesy of Dr. Barry Starcher (Starcher,
1977)). Crosslinks in EP2û-24-24 were also analyzed by column chromatography in the
laboratory of Dr. Allen Bailey (Bailey et al., L970).
Fabricrdion of Polypeptide Membranes
In the initiai experiments, PQQ and copper suIfate were dissolved in sodium borate
buffer (0.1 M sodium borate, 0.15 M NaCl, pH 8) to final concentrations of 20m.M and
lOOmM respectivety. Lyophilized EP20-24-24 was dissolved in sodium borate buffer (0.1M
sodium borate, 1.5M NaCi, pH 8) to a final concentration of 1.0 m g h l (60pM). The
solutions were incubated in eppendorf tubes overnight at 4OC. The polypeptide solutions
were placed in a water bath at m m temperature and the temperature raised to 37°C. Once
coacervated, polypeptides were incubated for an additional 10 minutes at the elevated
temperature. Copper sulfate and PQQ were added to the coacervated polypeptide solution to
final concentrations of 1.5 mM and 3.0 mM respectively. Solutions were transferred to a
iked angle cenmfuge and centrifuged for one minute at 5000g. Foiiowing centtifugation, the

polypeptides had adhered to the waU of the eppendorf tube, forming a thin membrane. The
samples were further incubated overnight at 37'C and then removed from the waIi of the tube
using a gentle Stream of water. Initiai membranes produced by this method were very thin,
fragile and diff~cult to manufacture consistently.
Subsequently, improvements wete made to this process which increased the
membrane quality and consistency of production.
These improvements used the protocol described above except for the following
changes. Polypeptides were dissolved in sodium borate buffer to final concentrations of 2.3
mgmi for EP2&24 and EP2&24-24 and to 3.1 mghl for EP2û-244. Following overnight
incubation at 37'C the solutions were transferred to 1.5 ml glass cuvettes pre-warmed to
37°C and the cuvettes were seaied. The cuvettes were centrifuged at 3200g for one minute in
buckets of a swinging bucket centrifuge, also preheated to 37'C. Cuvettes containing the
polypeptides were then incubated overnight at 37°C. The membranes were removed from the
cuvettes using a gentle Stream of water and were stored in water at 4°C until use.
Transmissiun Electron Microscopy of an EP2&2&24 Membrane
An EP2CL2424 membrane was sent to Dr. Glenda Wright (University of Prince
Edward Island, PEI, Canada) for transmission electron rnimscopy (Morrison et al., 2000).
Insolubility of Membranes Prepamdfrom Polypepîides
CIassically insoluble elastin is purified fiom tissues by treatment with CNBr or
boiiing in NaOH. These treatments solubilize other matrix components, but leave insoluble
elastin intact. Membranes fabricated h m EP2&24-24 were tceated with either CNBr or
NaOH. For CNBr treatment, membranes were incubated in a soIution of 50 mghl CNBr in
70% forrnic acid for 18 hours (Johnson et al., 1993). AItemately, membranes made h m
EP2û-24-24 membranes were incubated in 0.1M NaOH at 100°C for 45 minutes.
Determination of Mechantcal Prrrpertîes
Tests were performed on an Instron Mode1 55R11î2 tensile test apparatus, with a
custom-built strain gage transducer (- 1 kg). The Instron was controlled by a Gateway PC

computer using Instron software (Merlin) (Figure 5.1). Membranes fabricated h m
EP2û-24-24 and EP20-24'using the cuvette protocol described above were tested.
Sampte Mounting
A piece of cardboard was cut to 25 x 28 mm with a 15 x 5 mm or 15 x 10 mm hole
cut in the middIe (Figure 5.2). The cardboard was placed in a glass dish, submerged in water
on top of a piece of Teflon to prevent the membrane from adhenng to the dish. The
membrane was placed into the water over the cardboard and maneuvered so that it was
centered over the gap in the cardboard. The cardboard was taken out of the dish with the
Teflon and the Teflon was peeIed away from the cardboard. The sample remained attached to
the carciboard above and below the hole in the cardboard. The top grip of the Instron was
placed over the cardboard and membrane and the grip was tightened. The top grip was
mounted on the instron and then lowered to ailow the bottom clamp to be tightened over the
cardboard and membrane. The sides of the cardboard were then cut and a chamber containing
water at room temperature was raised to submerge the sample during testing. The initiai
length was set to either 10 mm or 5 mm, depending on the sample length, and the load was
reset to zero.
T e d e Tests
To determine the ability of the membranes to recoil after extension, the membranes
were extended between 20% and 100% at a rate of 6.22 mm per minute and returned to zero
Ioad at the same rate. Each membrane was subjected to three loading and unloading cycles at
the highest extension. The membranes were then loaded to failure at a rate of 6.22 mm per
minute. M e r failure, the portion of the membrane between the grips was removed, dried and
sent for arnino acid analysis to determine the dry weight of the tested membrane.
As a control, cardboard alone was mounted in the instron grips and tested in the same
manner as the membranes.

Figure 5.1: Mechanicd testing apparam. The ïnstron tende testing apparatus is shown on the ieft. The arrow indicates the test chamber containhg a sample mounted and submerged in water, The hstron is controiied through a Gateway compter system, shown on the cight.


Anaiysis
After testing, the data was adjusted to account for any initial slack in the membrane.
L, was set as the length of the membrane in metres when the membrane became engaged,
taking up a load. The effects of the h g of the cardboard were accounted for in the analysis
by subtracting the load observed by the carciboard alone from the total load.
Load-extension data was converted to stress-strain data using formula for engineering
stress (a) and engineering strain (E):
where F is the force (Ioad) 0, A, is the original cross-sectional area (m3 and AL, is the
extension (m).
Cross sectional area, A,, could not be measured directly because of the small size of
the membranes. This parameter was, therefore, cdculated using the water content and density
of insoluble elastin, 0.6 g/g and 1.3 glml respectively, assuming the values for the
polypeptide membranes wouId be similar to those of native, insoluble elastin. Quantitation of
insoluble elastin in the polypeptide membrane between the test grips was obtained by amino
acid analysis (dry weight) using a norleucine standard. Quantitation by arnino acid analysis
twk into account the loss of the lysine residues in the formation of crosslinks.
[(dry weight / density elastin) + (dry weight * water content / density water)] % =
=O
Enmgy loss (%), or hysteresis, was caiculated h m the third cycIe of the cyclic stress-
strain data as follows:
(area under loading curve - area under unioading curve) Energy Loss (96) = * 100
(area under loading c m )

The rernaining calcdations were made using the load-to-break data. The elastic
moduius (Pa) of each membrane was caicdated as the dope of the stress-strain curve, using
linear regression between strains of 0.1 to 0.5 for EP2û-24-24 and between 0.4 and 0.8 for
EP2û-24'. Maximum stress (Pa) and strain were taken as the stress at break (breaking
strength) and the corresponding strain value.
Finaiiy, an estimate of the crosslink density was made by &terminhg the average
molecuiar weight of chains between crosslinks (Mc).
where E is the elastic modulus, p is the density of insoluble elastin (p =1.3 g/l), R is the
universai gas constant and T is the temperature (T = 295K or 22°C) (Aaron and Gosline,
198 1). This calculation assumes the elastin polypeptide matrices are rubber-like polymers
(Le. a lightly crosslinked, random network of chains).
Statistical Analysis
Standard emrs for the crosslinking and mechanicd data were caiculated using
Statview software (version 4.0 1) (Abacus Concepts, Inc., Berkeley, California). Differences
between groups were assesseci by ANOVA.

Evidence for Alignment
Formation of covalent crosslinks in elastin required ordering of the polypeptide
monomers during assembly in order to bring lysine residues into juxtaposition for
crosslinking. If the process of coacervation aisa serves to aiign eiastin monomen, oxidation
of lysine residues should result in the formation of crosslinks. h these experiments,
formation of crosslinks was detected by the appearance of oiigomers or an insoluble pellet
that could not be solubilized to the monomenc fonn by extraction with 2%SDS/4M wea.
Crosslinking with the horseradish peroxidase system indicated that dignment of the
lysines occuned during coacewation. After crosslinking EE&24-24 with the homradish
peroxidase, no soluble polypeptide remained in the supematant a& any time point investigated
(Figure 5.3A). Furthemore, extraction of this insolubIe peIlet with 2% SDS/4M urea was
unabte to re-sohbilize elastin polypeptides. in contrast, eIastin polypeptides remained soluble
in controls lacking horseradish peroxidase and catechol. Amino acid andysis of the peiIet
showed that the nurnber of lysines was reduced by appmxirnately hdf after crosslinking,
while the proportion of other amino acid residues was unaffected (Figure 5.3B). Figure 5.3
shows representative resuIts of crosslinking mû-2624. SimiIar results were found for
EP2û-24 (data not show).
Attempts to determine if coacervation was necessary for alignment were unsuccessful
using the horseradish peroxidase crosslinking method because addition of the catechoi
caused the polypeptide soiutions to become cloudy, either coacervating or precipitating the
polypeptide. For this reason, a second method for oxidative deamination was developed
which subsquentiy proved to be supenor in several respects. This alternative methoci
involved the use of PQQ and copper as an oxidation reagent.
Oxidative deamination of W-0-24 using the PQQ system dso demonstrated that
aIignment of lysine residues occurred with coacervation (Figure 5.4A), aliowing the
formation of mssiinks as detected by oiigomer formation and insolubiIization of the eIastin
polypeptides. Foiiowing coacervation, essentiaily no eIastin polypeptide remained in the
supematant. In the absence of PQQ treatment, subsequent extraction of the pellet with
SDS/urea produceci monomeric polypeptides.
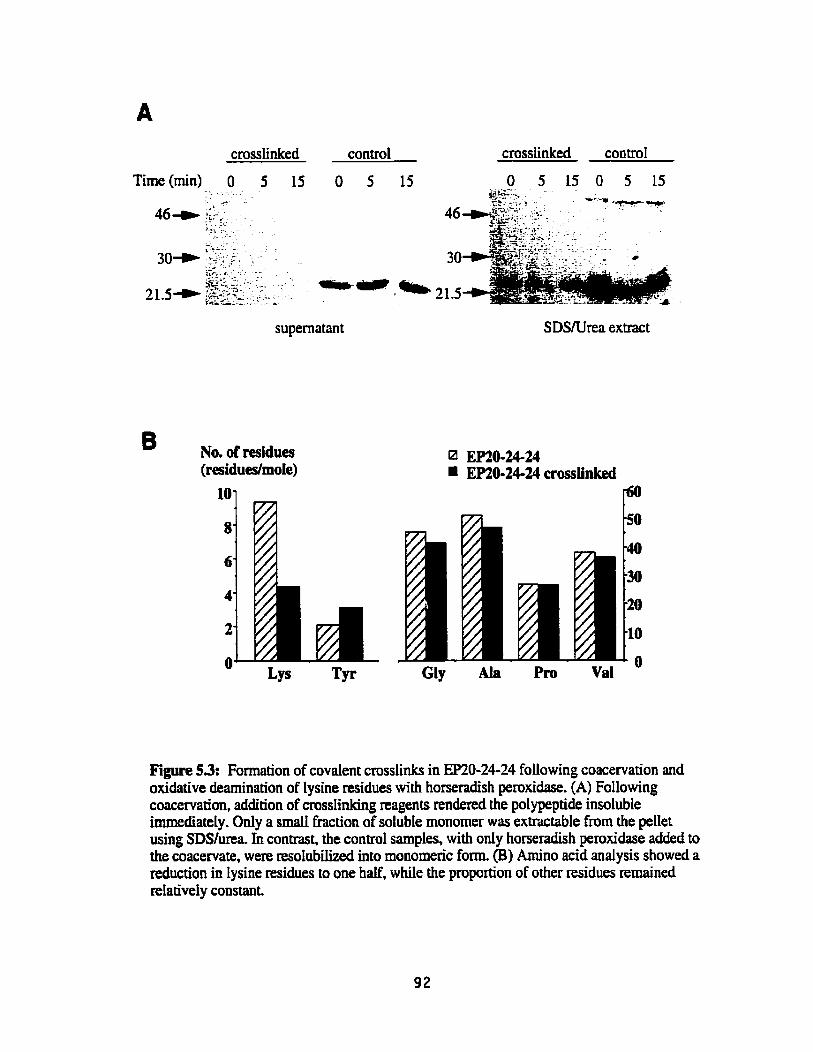
crosslinked control crossiinked control
Time(min) O 5 15 O 5 15 - - . . . - . . .
O 5 15 O 5 15 *ifi'-I,, ; . . . Ur-.. 3;-- , . ' c...; I .+ ? .
46- GY.: . . 9 . .CS . 3-7 . . 'F. . - . - . . . .. ~ ~
... -... &>. -. . . .
30+ i;:~- F-. , . . . -- . .- . . &P.,, . . , - - *.- * .:, , :.
%-y:- .. .: -- . 5- a 21.5 21.5- <AA.8~<??-.. - , , - ;z<-:.+ :, . 1 .~ ,,
supernatant SDSNrea extract
B No. of residues EP20-24-24 (residues/mole) EP20-24-24 crosslinked
10
8
6
4
2
O Gly Ala Pm Val LYS TF
Figure 53: Formation of covalent crosslinks in EP20-24-24 foliowing coacervation and oxidative deamination of lysine residues with horseradish peroxidase. (A) Following coacervation, addition of crossIinking reagents rendered the polypeptide insoluble immediately. Only a small k t i o n of solubIe monomer was extractable from the pellet using SDSfurea in contrast, the control samples, with only horseradish peroxidase added to the coacervate, were resolubiiized into monomaic form. (B) Amino acid andysis showed a reduction in lysine residues to one half, while the proportion of other residues rernained retatively constant.

Supernatant coacervated - 0.2 0.4 0.9 2.9
SDS/Urea Extract coacervated
Supernatant SDSNrea Extract coacervated
- coacervated 0.2 0.4 0.9 2.9 0 2 0.4 0.9 2.9
Figure 5.4: Crossiink formation in the coacervated polypeptides using PQQ to oxidatively deaminate the lysine residues. FoIlowing coacewation, essentially al1 of the polypeptide was absent from the supernatant. After SDSfurea extraction on the pellet, without PQQ, the polypeptide remained primarïly in monometic fonn. However, with the addition of PQQ, dimers, trimers, and higher ordered oligomers formcd, increasing in size with increasing concentrations of PQQ. Finally, at 2.9 mM, the insoluble polypeptide was unable to enter the gel. Representative Western blots are shown for EP20-24 (A) and EP20-24-24 (B).

Treatment with PQQ resulted in the formation of oligomers that could not be restored
to a monomeric species by treatment with SDSIurea Furthemore, increasing concentrations
of PQQ resulted in loss of the monomer and the presence of higher ordeï oligomers in the
SDSIurea extract. At higher concentrations of PQQ the pellet was completely insoluble in
SDSIurea. Similar results were seen for EP2&24-24 (Figure 5.4B)
In the absence of coacervation (no NaCl), no crosslinking occurred for either
EP2&24 or EP20-24-24, and the majority of the polypeptide remained in the supernatant
(Figure 5.5). However, this polypeptide had been modified by the PQQ, since only one lysine
of the original four lysines in E R 6 2 4 or only two of the original eight lysine residues of
EP20-24-24 remained following oxidation. These resuIts showing h e of four lysine
residues m-ed for each polypeptide were consistent over several experiments (n=3). The
fact that these lysine residues were modified by PQQ but crossünks did not fom suggests
that proper juxtaposition of lysine residues did not take place in the absence of coacervation.
Similarly, lysine residues in EPX-23 were oxidatively deaminated by PQQ treatment, but no
crosslinks were formed (Figure 5.6).
Nature of Crusstulks Fomted
Desrnosine and isodesmosine, crosslinks typical of native insoluble elastin, were
detected in crosslinked EP2û-24 and EPZO-24-24 using a radioimmunoassay technique.
Three repeats of the radioimmunoassay were performed for each of the two polypeptides.
However, the amounts of each of the crosslinks were variable between samples. Desrnosine
contents for each 2 mg sample were between 0.85 nmoIes to 1.7 moles (1.25 f 0.4 nmoles,
mean f standard deviation) for EP20-24-24 and between 0.25 nmoles to 2.1 nmoles (1.03 f
0.97 moles) for EP2G24. Isodesrnosine contents were also variable ranging from 1.4
moles to 4.1 nmoles (3.08 f 1.46 moles) for EP2&24-24 and 0.2 nmoles to 4.6 nmoles
(1.97 f 2.33 nmoIes) for EP20-24. Using ion exchange chromatography, the ratio of
desmosine to isodesmosine in EP2O-2624 was inverse to that found in native insoluble
elastin (Figure 5.7).
A membrane fabricated from EP20-2424, also exarnined by
desmosine/isodesniosine radioimmunoassay, contained 1-0 nmoles of desrnosine and
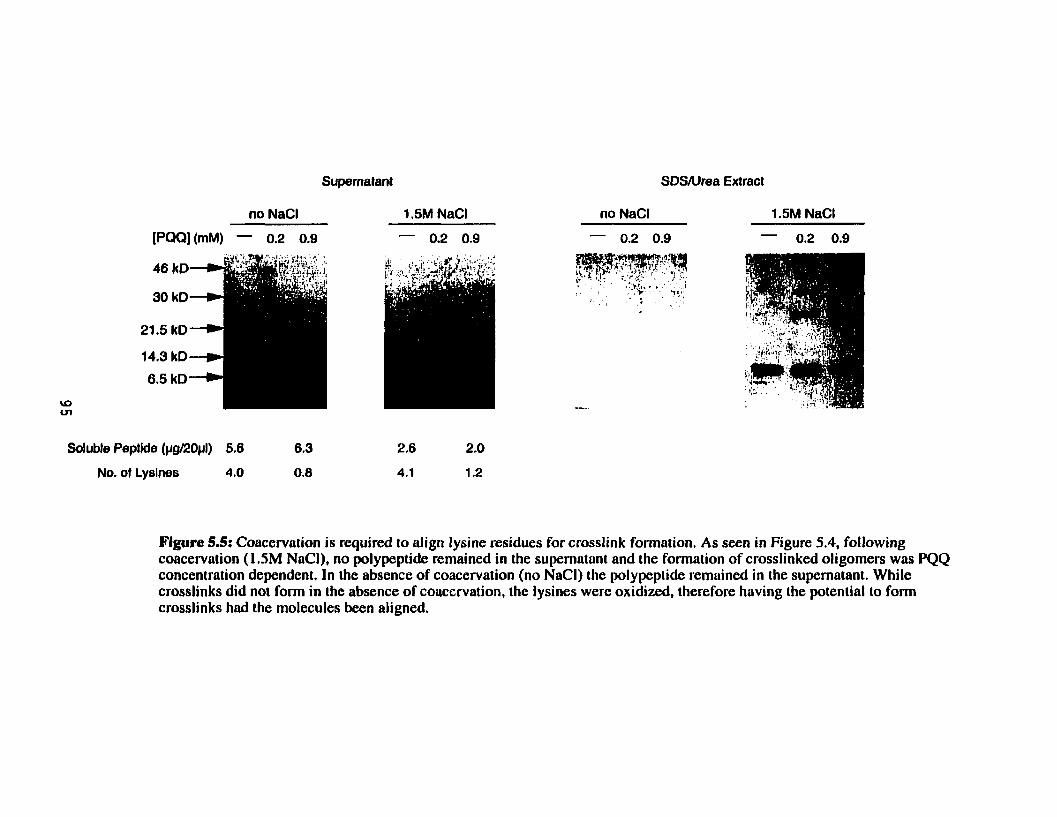
Supernatant
Soluble Peptlde (pg/20pI) 5.6 6.3 2.6 2.0
No. of Lysines 4.0 0.8 4.1 1.2
Figure 5.5: Coacervation is required to align lysine residues for crosslink formation. As seen in Figure 5.4, following coacervation (1 SM NaCI), no polypeptide remained in the supernatant and the formation of crosslinked oligomers was PQQ concentration dependent. In the absence of coacervation (no NaCI) the polypeptide remuined in the supernatant. While crosslinks did not form in the absence of coocervntion, the lysines werc oxidized, therefore having the potential to form crosslinks had the molecules been aligned.

supematant SDS/urea extract
[PQQ] (mM) - 0.2 0.9 2.9 - 0.2 0.9 2.9
Soluble Peptide (pgJ20pl) 2.9
No. of Lysines 2.5
Figure 5.6: Crosslink formation does not occur in EP21-23, the polypeptide lacking hydrophobic domains. EP2 1-23 was unable to coacervate because it lacked hydrophobic domains. The addition of PQQ modified the lysines but no crosslinks formed in the absence of aiignment. The addition of PQQ appeared to change the affinity of the antibody to EP21-23. However, amino acid anaiysis confirmed the polypeptide remained in the supematant.

l Des MeroDes?
Elastin
O 5 10 15 20 25 30 35 40 45 Time (rnins)
Figure 5.7: Chromatogruphic comparison of crosslink profiles for EP20-24-24 and insoluble elastin. Following ion exchange chromatography, isodesmosine and desmosine crosslinks were identified in crosslinked EP20-24-24. The ratio of isodesmosine to desmosine in EP20-24-24 is the inverse of thiit of insoluble elastin. Smaller peaks likely correspond to the bifunctional crosslinks lysinonorleucine (LNL) and merodesrnosine (MeroDes),

1.3 nmoles of isodesmosine per 2 mg of original elastin polypeptide, similar to the values
found for the non-membrane EP2û-24-24 samples.
Other peaks present in ion exchange analyses of crosslinks may correspond to
lysinonorleucine and merodesrnosine, crosslinks normally found in smaI1 amounts in native
insoluble elastin after reduction with sodium borohydride (Figure 5.7). No standards for
allysine aldol were available.
Characterization of Membranes
Membranes were successfulIy fabricated using both EP20-24-24 and EP20-24'
(Figure 5.8). Attempts to fom a membrane with EP2&24 were unsuccessful, due to the lack
of structurai integrity of the polypeptide membrane on the surface of the cuvette (Figure 5.9).
Treatment of EP20-24-24 membranes with either CNBr or hot NaOH left the
membranes essentially intact Wgure 5.10). Although in general the membranes remained
dense and without obvious structure, an open network could be seen at the edges of the
membrane following CNBr treatment, which was likely the resuIt of mechanical
manipulation. Sections of an EP2û-2424 membrane, observed in horizontai and transverse
directions under transmission electron microscopy, did not show any orientation of the
polypeptides in the membrane (Figure 5.11). Open, network-like smchires were seen at the
edges of the membrane under transmission election microscopy (Figure 5.1 1C and D).
Mechanical hopdes
Membranes fabricated h m both EP2û-24-24 and m0-24J were tested
mechanicaily. An EP2CL24-24 membrane is show loaded, extended and broken in Figure
5.12. Both EP2&24-24 and EP20-24' were able to extend and recoil. Representative test
curves are shown in Figures 5-13 to 5.15 for EP20-24-24. These curves show averaged data
to reduce noise in the collected data Similar curves were typical of the results for EP2û-24'
(data not shown).
Unlike native insoluble elastin, cyclic Ioading and udoading of the elastin
polypeptide membranes showed no pconditioning, indicating that no preliminary alignment
of the crosslinked elastin potypeptides was necessary (Fîgure 5.14).

Fignre 5.8: EPM-24-24 and mû-244 membranes fabncated using the cuvette protocoi. EPU)-24-24 (A) and EP2û-244(B) wcre hth able to fom continuons membranes following oxidation of lysine &dues with PQQ. The membranes appear dùcker at one end as a result of the fabrication protocoi.


40x rnagnification 40x rnagnification
Flgare 5.10: EP20-24-24 membranes visualized using iight microscopy befon and after CNBr or NaOH matment. Panel A shows an EP20-24-24 membrane prior to CNBr or NaOH treatment. The arrow indicates where the membrane has roUed up on itseif, on the right side. Treatment with CNBr (B) or Ming in 0.1M NaOH (C) left the membraues essentially intact. An open filamentous network can been seen toward the right side of the membrane in panel B (arrow).

horizontal section transverse section
transverse sections
Figure 5.11: An EP20-24-24 membrane appears amorphous using transmission electron microscopy. No orientation is seen in either the horizontal or transverse sections of the EP20-24-24 membranes (upper panels). As seen in the light micrograph taken following CNBr treatment (Figure 5. JOB), a less dense network of crosslinked polypeptide is seen at the edges af the membrane (lower panels),

A. Mounted, unloaded B. Extended C. Broken
er grip
Figure 5.12: Stages of tensile testing of an EP20-24-24 membrane. An EPZO-24-24 membrane (arrow) is shown mounted in the Jnstron grips prior to toading (A), after extension (B) and after breaking (C), The upper and lower grips are labelled in panel A, gripping both the membrane and the uncut cardboard. Following mounting, the cardboard was cut (B, C).

Stress (MPa)
1
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8
Strain ( d m ) Figure 5.13: Representative stress-suain curve of an EP20-24-24 membrane loaded to break. The elastic modulus was calculated aver the linear portion of the stress-strain curve. The stress increased relaiively linenrly, brenking ai a stress of approximately 0.18 MPa corresponding to a strain of approximately 65%.
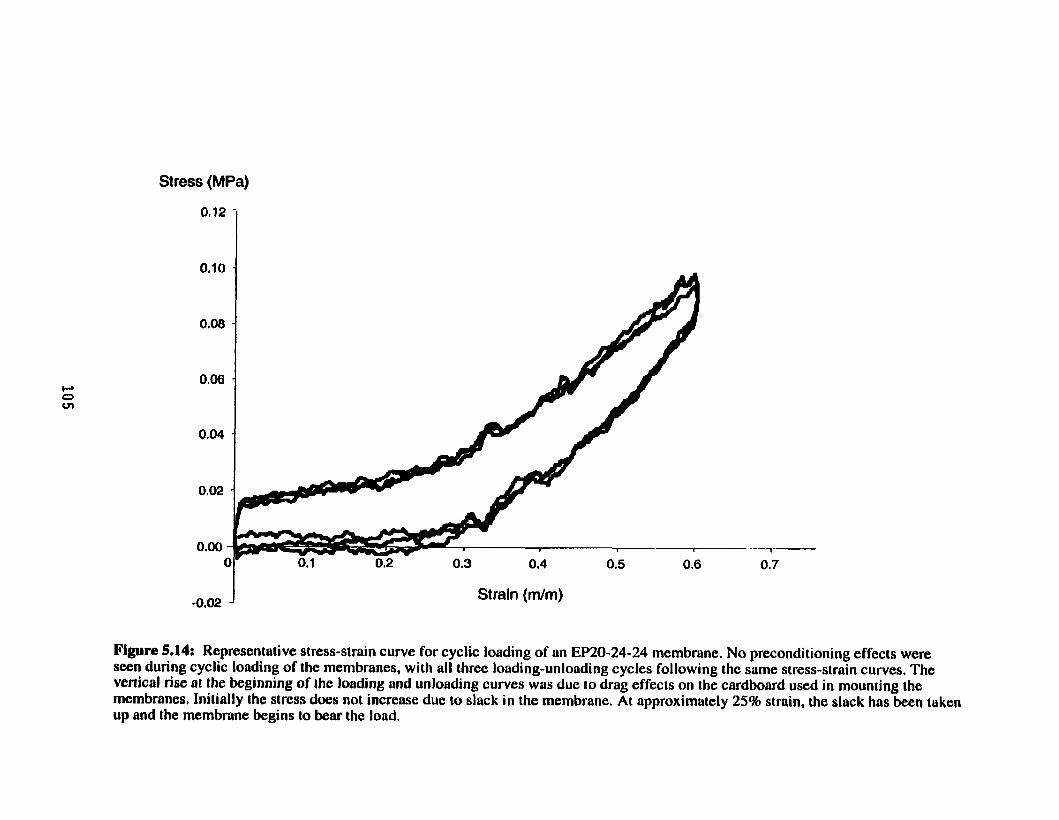
Stress (MPa)
Straln (dm)
Figure 5.14: Representative stress-strain curve for cyclic loading of an EP20-24-24 membrane. No preconditioning effecis were seen during cyclic loading of the membranes, wi th al l three loading-unloading cycles following the same stress-main curves. The vertical rise al the beginning of the loading and unloading curves was due to drag effecis on the cardboard used in mounting the membranes, Iniiiiilly the stress does not incrense due to slack in the membrane. At approximately 25% strain, the sluck has been taken up and the membrane begins to bear the laad.

Stress (MPa)
0.09 -
0.08 -
0.07
0.08 .
0,05 .
0.04
0.03 -
0,02 .
Strain (mhn)
Figure 5.15: Representative stress-strain curve showing the third cycle of cyclic loading of an EP20-24-24 membrane. The data has been adjusted to account for slack in the EP20-24-24 mernbrime and the drag effects of the cardbaard. The energy loss, or hysteresis, was calculoted as the ares under the unloading curve (lower curve) as u percentüge of the area under the loading curve (upper curve).

The effect of the carciboard on the bad is seen at the start of the loading and the unloading
c w e as a vertical change in load To determine the energy loss, this h g effect was removed
fiom the third cycle (Figure 5.15).
A summary of mechanical characteristics for the two polypeptide membranes and for
aortic elastin is given in Tabte 5.1. Remarkably, for similar ranges of strain, the elastic
modulus for the membranes was of the same order of magnitude as that of aortic elastin. In
fact, aU of the values for the mechanicd propemes of the elastin polypeptide membranes
were quite sirnilar to those of aortic elastin. No significant differences were found between
EP20-24-24 and EP20-24" in any of the mechanicd values determined. The average
molecular weight between crosslinks was cdcuiated from the elastic modulus to be
approximately 38 kDa for both EP20-24 and EP2û-244. In contrast, single elastin fibres
extracted from bovine ligarnenturn nuchae are calcuiated to have an average molecular
weight between crosslinks of between 6 to 7. L kDa (Aaron and Gosline, 198 1). indicating the
crosslink density of the membranes is 5 to 6 times lower than that of native insoluble elastin.
The standard deviations of the mechanical properties were large. This was iikely due
to variability in the manufacturing pmess, First, based on the crosslink data on non-
rnembranous EP2G24-24, the variability in msslink density was likely dso high in the
membranes and could contribute to the variability in mechanicd data. Second, the placement
of the cuvettes in the centrifuge was difficult and could have given rise to membranes that
were thicker atone end, or one side, than the other. Finally, estimations of cross-sectional
area using arnino acid analysis on membrane reaieved after mechanical testing may be
subject to larger errors.

Table 5.1: Summary of the mechanicd properties of the eIastin polypeptides. The values for aortic elastin are for strain rates between 0.2 and 0.6 using autoclaved aortic tissue without extracting the microfibds.
Values for the elastin polypeptides are mean * standard deviation with n=9 for EP20-24-24 and n=4 for EP20-24". No significant differences were found between the pdypeptides for any of the properties measured. Values for aortic elastin are fiom experiments performed by Dr. Margo Lillie, in the laboratory of Dr. John Gosline at the University of British Columbia, Canada. The source of aortic elastin was porcine tissue purified by autoclaving and was tested in rings (circumferentially).
Sample
EP20-24-24
Elastic Modulus (MW
0.25 + 0.10
Strain at Break (%)
86 + 42%
Energy Loss (%)
20 + 7%
Stress at Break W a )
O. 19 + 0.08

Discussion Using the elastin polypeptides a s models of elastin assembly, we have shown that
coacervation is required to align lysine residues to faditate crosslinking in elastin
polypeptides, that the crosslinks formed are similar to those seen in native insolubIe elastin,
and that polymenc membrane maûices formed h m these crosslinked eIastin polypeptides
had soIubility and mechanical characteristics similar to native insoluble elastin.
Aügnmenf
Results indicated that coacervacion was a requirement to align the lysine residues in
the crosslinking domains for crosslink formation to occur. In the absence of coacervation,
although lysine residues were modified, the etastin poIypeptides remained soluble. In the
presence of coacervation but without polypeptide oxidation, coacervated polypeptides could
k recovered as monomers by extraction with solutions of SDS/urea. Thwe results support
the hypothesis that the ptocess of coacervation not ody aggregated the elastin polypeptides,
but also jwtaposed lysine residues for zero-length crosslinking. Furthemore, crosslinks
present after coacervation and oxidative deamination included desmosine and isodesmosine,
the major crosslinks of native insoluble elastin. Others have aiso reported the itbility of
coacerviited tropoe1astin to crosstink after in vim treatment with Iysyl oxidase or PQQ (Shah
et al., 1992). Furthmore, in an earlier study, Naranaryan also suggested that coacervation
was required for such in vitro crosslinking of tropoelastin (Narayanan et al., 1978)
The presence of desmosine and isodesmosine crossIinks in the insoIubIe polypeptides
was surprising given the organizationai requirements for these types of crosdinks. Four
Lysines, only three of which have been modified to ddehydes, must be correctIy positioned to
form these covalent crosslinks. Both EP2G24 and EP2W4-24 showed the ability to aiign in
this manner. In tropoelastin it has been suggested that the unmodified lysine residue is
adjacent to an aromatic amino acid (tyrosine or phenyIaianine) (Baig et ai., 1980; Gerber and
Anwar, 1975). The crossIinking sequences present in these polypeptides (corresponding to
exons 21 and 23) contain ody a single lysine which is adjacent to an aromatic residue,
promoting the speculation that it is this lysine which may remain unmodified after PQQ
treatment.

The ratio of desmosine to isodesmosine was inverse in the crossiinked pdypeptides
as compared to native insoluble elastin, suggesting that alignment of the lysine residues in
the polypeptides occurs in a spatial orientation favouring isodesmosine over desmosine
formation. As well as desmosine and isodesmosine, lysinonorleucine and merodesrnosine
also appear to be present in crosslinked EP2û-24-24. The presence of allysine aldol
crosslinks rernains to be determined
Membrane Fabrication and üürastructure
Crusslinked membranes codd be formed from EP2û-24-24 and EP2û-24' but not
fiom EP2û-24. Previously we showed (Chapter 4) that the formation of well-organized,
compact fibrillar structures from these elastin polypeptides required the presence of at Ieast
three hydrophobic domains. These observations are supported by the fact that intact
membranes could not be foned from polypeptides with less than three hydrophobic
domains. Similar to insoluble elastin, membranes made from EP2û-24-24 could not be
solubiIized by CNBr treatment or by M i n g in O.1M NaOH for 45 minutes, both standard
methods for purifying insoluble elastin h m other extracellular matxix proteins in tissues.
Transmission electron micropphs of an embedded EP20-24-24 membrane showed
similar dense arrangements of polypeptide in d l directions: longitudinal (not show),
transverse, and horizontal. The lack of an oriented fibrillar network was not unexpected
given that the polypeptides were not allowed time to spontaneously fonn fibrillar structures,
but rather were oxidized by PQQ and forced into place by centrifugation at the earlier, open
filamentous stage of coacervation (Chapter 4).
Mechanhm of Elosticity
The mechanîcal properties of the crosslinked polypeptide membrane made with only
three hydrophobic domains were remarkabty similar to those of insoluble aortic elastin. The
mechanics of a material are affected by a number of factors, including the orientation of the
fibres wihin the material, the density of crosslinking and the properties of individuaf fibres
within the materiai. First, if the fibres within a materiai are oriented, the elastic moddus and
breaking strength will be higher in the direction of the fibre orientation (Ward, 1971). Based
on our electron rnicroscopic images, the membranes formed h m elastin polypeptides appear

to be isotropie, in contrast to most models of elastin ultrastructure. This suggests that the
matrix fabricated h m the elastin polypeptides rnay be Merent in structure from that of
native insoluble elastin, perhaps contributing to a lower modulus and breaking strength. The
lack of preconditioning in cyclic loading cuwes of the elastin polypeptides compared to
native insoluble elastin may also be an indication of structurai ciifferences.
Crosslink density also affects the mechanics of materials, with elastic modulus and
breaking strength directly related to crosslink density. The totai desmosine and isodesmosine
content of native insoluble elastin is equivaient to 2100 gram of crosslink per mole of
tropoelastin (Starcher and Galione, 1976). In cornparison, the EP2û-24-24 membrane had an
approximately 200 fold Lowa density of desrnosine and isodesmosine crosslinks. Unlike
insoluble elastin, ion exchange data suggests that the predominant crosslinks in the
polypeptides, and likely in the polypeptide membranes, are not desmosine and isodesmosine,
but lysinonorleucine and merodesmosine. Estimates of msslink density calculated h m the
elastic modulus of the membranes indicate that the membranes are not as highly crosslinked
as single elastin fibres. Calculation of crosslink density from elastic modulus accounts for ail
types of intermolecular crosslinks, not just desmosine and isodesmosine. Therefore, taking
into account al1 crosslinks, the membranes do not appear to be as highiy crosslinked as aortic
elastin. This may be a contributing factor to their lower elastic modulus and breaking
strength.
While the ultrastructure and cmsslink density certainly contribute to the mechanicai
propexties of elastin, the intrinsic structure of elastin at the molecular level may also play a
d e . The mechanism by which elastin extends and recoiIs is the subject of some controversy,
but there is general agreement that this mechanism is entropic in nature. That is, the entropy
of the elastin matrix is decreased upon stretch, and is returned spontaneously to maximum
entropy upon recoil. The entropy of the system is Mieved to decrease either by a
combination of configurational restrictions and ordering of water molecules around
hydrophobic regions that becorne exposed upon stretctiing or by decreasing the 'librationai'
motion of the hydrophobic regions by limiting their mobility on stretching. In addition, the
role of specific hydrophobic sequences in the mechanism of elasticity of elastin has been
demonstrated by the mechanical properties of membranes made h m different repetitive
sequences found in the hydrophobic domains of elastin. Urry has made materials h m elastin

polypentapeptide (PGVGV) and polynonapeptide (VPGFGVGAG), crosslinked with gamma
radiation. (Urry et ai., 1990; Uny et al., 1988; Urry et al., 1984). Subjected to the same
treatments and at the same crossiink density, the polynonapeptide membranes were stiffer
than the polypentapeptide membranes, indicating that specific hydrophobic sequences
contribute to the mechanicai properties of ehstin.
The mechanicai properties of EP2&24-24 membranes did not appear to differ h m
those of ~P20-24'. even though m0-244 had two additionai hydrophobic domains and
almost twice the molecular mas of EP2&24-24. Although no structural investigations or
crosslinking assays were conducted on EP20-244the ultrastructure and crosslinks of the two
types of membranes were likely similar. If specific hydrophobic dornains contribute to the
elastic properties of elastin, as suggested by Uny, then the similarities in mechanical
properties of the two types of membranes may be explained by the presence of the same
hydrophobic sequences in each polypeptide. Additionally, in spite of differences in
ultrastmcture and crosslink density, the mechanical properties of the polypeptide membranes
were quite sirnilar to those of aortic elastin, We woutd speculate that sequence of
hydrophobic dornains rnay play a role in determining the mechanicai properties of insoluble
elastin.

Chapter 6
Conclusions and Recommendations
We Shan flot œase from exploration. And the end of all wr axplohv~ will be 10 anive where we started and know the place for the first time. T.S. Eliot

Conclusions and Recommendations General Summmy
In the extracellular matrix, elastin provides tissues with the properties of extensibility
and elastic recoil. Zn vivo, the assembly of mpoelastin into this extracellular, polymeric
matrix is suggested to involve a nwnber of factors, including an elastin binding protein, a
microfibrillar scafioId, and lysyl oxidase (Hinek and Rabinovitch, 1994; Hinek et al., 1988;
Kagan and Trackman, 1991; Ross and Bornstein, 1969). However, in vitro investigations
using recombinant tropoelastin have shown elastin assembly in the absence of either the
elastin binding protein or the microfibrillar scaffold (BedeH-Hogan et al., 1993). A nurnber of
in vitro strategies have been used to investigate the requirements for organized, self-assembly
of elastin, yet the details of the mechanism for elastin assembly and their relationship to the
underlying mechanisms of elasticity remain elusive.
We have used a series of recombinant human elastin polypeptides to investigate the
role of various hydrophobic and crosslinking domains in self-assembly and to determine
whether self-asscmbly is an ordering pmcess, aligning the lysine residues for crosslink
formation. The choice of specific hydrophobic and crosslinking regions incorporated into the
polypeptides is outlined in Chapter 2, dong with the methods ofconstmcting, expressing and
purifying the polypeptides. The propensity of the polypeptides CO coacervate, which we used
as the in vitro measure of self-aggregation, was characterized pmviding information on the
requirements for aggregation and the relative contibutions and mles of specific hydrophobic
regions (Chapter 3). To further investigate the assembly pmess, the stability of the
coacervate was investigated and the dtrasûucturai organization of the polypeptides was
examined at various stages during the coacervation process, suggesting a multi-stage mode1
of assembly (Chapter 4). In addition, the ability of tfie poIypeptides to spontaneously form
crosslinks after oxidative deamination of lysine residues was used as a measure of specific
aügnment of the polypeptide chahs, as a result of the process of self-aggregation or
coacervation. Fxndy, the ability of at Ieast some of these polypeptides to form stable,
insoluble polymers ailowed evaluation of the mechanicai properties of these crosslinked
polypeptide matrices (Chapter 5).
Ou. original hypothesis that recombinant pdypeptide regions of human eiastin would
self-align to allow crosslinking into maîrices with elastin-like mechanicai properties has been

substantiated by the data presented in this thesis. All of the polypeptides manufactured had
the ability to coacervate. The two polypeptides that were studied in most depth, EP20-24 and
EP20-24-24, formed fibrillar aggregates as a resdt of coacervation and were also aligned
during the coacervation process such that Lysine residues were juxtaposed, allowing the
formation of covalent crosslinks. While crosslinking took place in aggregates of EP20-24,
this polypeptide could not be fabricated into a mechanicaily stable matrix. However,
crosslinked matrices of both EP2&24-24 and m&244 formed stable mechanical structures
with solubility characteristics and physicd properties remarkably sirnilar to insoluble aortic
elastin.
It was not determined whether the mechanical properties of these matrices were
modifiable. Somewhat surprisingly, membranes produced from mû-24' had physical
properties indistinguisible from EP2&24-24, in spite of the increased length and
substantially lower coacervation temperature of EP2@24". In the future, the effect of
variation of the sequence of the find domain on mechanicd properties could, however, be
tested using mû-24-26, EEZO-24-30 and other variations of the polypeptides sequences.
Role of the Hydrophobie Domains in Assembly
The presence of hydrophobic domains in the eIastin polypeptides was found to be a
requirement for self-aggregation, as measured by coacervation. Al1 of the recombinant elastin
polypeptides were able to coacervate with the exception of EP21-23, which lacked
hydrophobic dornains. These resuIts provide further evidence that elastin assembles through
the hydrophobic dornains (Castiglione Morelli et al., 1993; Reiersen et al., 1998; Urry et al.,
1974).
Furthermore, the coacervation process is not characterized by random aggregation of
tropoehstin molecules, but rather our results suggest that specific aiignrnent of polypeptides
occurs during coacervation. In the absence of coacervation, although the majority of Iysine
residues are modified with the addition of PQQ, no crossiinking takes place. However, if the
polypeptides were coacewated prior to the addition of PQQ, covalent crosslinks fom,
including desrnosine and isadesmosine, the predominant crosslinks found in native, insoluble
elastin.

The presence of desmosine and isodesmosine cmsslinks in the crosslinked elastin
polypeptides was remarkabie given the requirements for theu formation. Four lysine
residues, oniy ihre..% of which have been modified to allysine aldol, must be juxtaposed for
zero-length cmsslinking. Three of four lysine tesidues in the polypeptides were consistently
modified using PQQ. It has been suggested that the presence of an aromatic residue adjacent
to a lysine residue in human elastin may prevent the oxidative deaminaciun of that lysine
(Baig et ai., 1980; Foster et al., 1974; Reiser et al., 1992). In the polypeptides used in these
studies, the most C-terminal lysine in exon 21 is adjacent to a tyrosine residue, leading us to
speculate that it is this Iysine that remains umodified. To investigate this further, the single
tyrosine residue in exon 21 could be replaced with an alanine residue, a residue fiequently
found C-temiinal to lysine residues in crosslinking sequences. The oxidation of dl lysines in
this mutated polypeptide, and the inabihty of the polypeptide to fonn desrnosine and
isodesmosine crosslinks, would support this hypothesis.
Although, our data did not aIlow the calculation of thermodynrimic parameters, the
process of coacervation of both recombinant tropoelastin and hydrophobie polypeptides has
generally been shown to be endothennic and entropic (Luan et al., 1990; Vrhovski et al.,
1997). Consistent with this. our data showed that while some formation of small rnolecular
weight aggregates took place at 4'C after a prolonged period of time, addition of heat was
required to initiate the coacervation process, indicating that the process was endothermic.
Reqirirements for Assentbly into an Organùe4 ElasiUi-Làke Matrh
ûriginaily, we designed EP2û-24 to be tepresentative of huma. tropoelastin,
maintainhg its aiternating domain stnicttm, and hypothesized it wodd be capable of
assembly into a polyrnenc rnaûix. In fact, this polypeptide was able to aggregate into low
molecular weight ohgomers even under non-coacervating conditions (4'C in 50mM acetic
acid), suggesting that our hypothesis was correct. In addition, EP20-24 was able to self-
aggregate and, s i d a r to tropoelastin, its coacervation temperature was dependent on both
ionic strength and poIypeptide concentration. Further support for our hvpothesis came from
the visualization of EP2û-24 at various stages during the coacervation process using
transmission eiectron microscopy. Prior to coacervation and immediately following
coace~ation, the ulûastnicture of EP20-24 tesembled that of tropoelastin under similar

conditions (Bressan et al., 1986). However, foiiowing overnight incubation above the
coacervation temperature, while tropoelastin fomed aligned fibrillar structures, EP2û-24
was unable to form structures that were as weii organized (compact) or as extensive. In spite
of the relatively inferior nature of the fibriiiar coacervates formed from EP2C24, oxidation
by PQQ did result in crosslinks typical of mature, insoluble elastin. However, these
assembled structures fiom EP20-24 were not capable of forming a stable, extensive,
polymerized membrane.
In contrast, aggregation of EP2C24-34 produced fibrillar structures essentially
indistinguishable from those of tropoelastin following overnight incubation (Bressan et al.,
1986). in addition, continuous, crosslinked membranes fabricated using this polypeptide,
showed tensile mechanical properties similar to those of aor&ic elastin. The presence of a
third hydrophobic domain in EP20-24-24 had previously k e n shown to increase the
propensity of EP20-24-24 to coacervate, doing so at a lower temperature than EP2û-24
under the sarne conditions. It appears that the ability to form organized, crosslinked
membranes may also be attributed to the presence of this third hydrophobic domain,
providing the polypeptide with an enhanced ability to overlap during assembly. We wouid
now suggest that at least three hydrophobic domains are required for optimd organization
into fibriiiar structures and crosslinked polypeptide matrices.
Importance of Sequence of the Hydrophobie Domains in Assembly
in choosing the polypeptides, exon 24 was selected because it contains the most
striicing tandem repeat in human elastin -the PGVGVA motif, repeating seven times in
tandem in this exon. The presence of this and similar repetitive motifs in other species of
elastin suggests that these sequences may be important for the properties of elastin, including
its ability to self-assemble.
Replacing the final exon 24 in EP2CL24-24 with exon 30, the domain containing the
GGLGV sequence repeating twice in tandem, gave a polypeptide with a coacervation
temperature similar to that of EP2&24-24, in spite of its Iower molecular mass. Replacing
the finai exon 24 in EP2û-24-24 with exon 26, the domain containing a nonapeptide
sequence repeating t h e times in tandem, resulted in a polypeptide that coacervated at a
lower temperature than EPîû-24-24, although the molecular mass and the hydrophobicity of

these two polypeptides were comparable. Therefore, we suggest that the sequence of the third
hydrophobic domain in the polypeptide plays an important role in its propensity to aggregate.
While we had hypothesized that the PGVGVA sequence would be of particular importance
to self-assembly of elastin, it now appears that other sequences, such as the nonapeptide
sequence in exon 26, may be more important for tropoelastin assembly.
Crosslinked matends fabricated from polypeptides derived h m hydrophobic domain
sequences of elastin have been shown to have different mechanicai properties depending on
the sequence of the poIypeptide, Materials formed h m the polypentapeptide,
poly(PGVGV), were less stiff than those formed from the polynonapeptide,
poly(VPGFGFGAG), dthough the crosslink densities were simiIar (UT et al., 1990; Urry et
al., 1984). In addition, membranes fabricated from EP2û-24 and EP20-24', polypeptides
containing the same hydrophobic domains, showed comparable mechanical properties.
Investigation of the propeaies of membranes fabricated from EP2û-24-26 and EP2&24-30
couId shed additional light on the role of sequence in detennining mechanicd properties.
Ulinrtructure and a Mode1 of Assembly
Visuaiizing the coacervation process using transmission electron mimscopy has
allowed us to suggest that coacervation of the elastin polypeptides is a muiti-stage pmcess.
BeIow the coacervation temperature, the elastin poIypeptides fonn globular stnicttms,
becoming an open network of filaments following coacervation. With incubation above the
coacervation temperature for a prolonged period of time, the filaments appear to aiign in
paraltel, forming fibrillar structures, These results are in agreement with those of Bressan and
coworkers, who found similar types of structures fonned during the process of coacervation
of tropoelastin (Bressan et ai., 1986).
In addition, the stability of the coacervate, as measured by its abiiity to resolubilize
upon cooling, has been shown to be time-dependent for the elastin polypeptides. After
coacervation, if the solution is cooled immediately, the coacervate wiII resolubilize. In
contras& once the fibriliar structures form, possibïy by alignrnent of the initiai filamentous
network, the coacervate is stable, and cannot be soiubiIized
Taken together, these data support a multi-stage mode1 of assembly during
coacervation, consisting of revemile and irreversible stages (Chapter 4, Eigure 4.9), similar

to that first proposed by Jarnieson (Jamieson et al., 1972). Our model proposes that giobular
molecuks of polypeptide (monomers and smaiI oligomers) present below the coacervation
temperature, form an open network of filaments above the coacervation temperame. This
stage of coacervation can be reversed by cooling immediateIy following coacervate
formation. However, if the coacervate is incubated above the coacervation temperature for
prolonged periods of tirne, it becomes stable. This stage of the coacervation process is
irreversible under the conditions tested.
In agreement with previous work by Jamieson, ow model of assembly proposes that
the fmt stage of coacervation is reversible (Jamieson et al., 1972). However, Jamieson
suggests that large molecular aggregates fom prior to coacervation and that coacervation is
the compaction of these pre-formed, large aggregates (Jarnieson et al., 1972). In contrast, we
have evidence that this stage of the coacervation process is the aggregation of small
oligomers into large filamentous aggregates. This is the primary difference between the two
models. This could be investigated further by studying the ultrastructure of the polypeptides
present in coacervates that had been immediately resolubilized by lowenng of the soIution
temperrintre. The return of the less-ordered filamentous networks to globutar structures after
cooling would confum the reversibility of this first stage of coacervation in our model of
assembl y.
In contrast to the fibrillar structures formed during coacervation, EP2û-24-24
membranes appeared amorphous by ultrastructural examination. This was likety due to the
method of fabrication, since the polypeptide was forced by centrifugation against the side of
the cuvette at the F i t stage of coacervation when only the reIatively unordered filamentous
networks had formed, To further investigate the membrane strucnire, the membranes could
be tested mechanically in two orientations, offset by ninety &grees in the plane of the
membrane. Higher elastic modulus and breaking strength in one orientation in comparison to
the other would suggest the presence of ordered structure.
PotenM Uses of the Elartin Polypeptifes in Bionuûerials Applications
A number of investigators have begun using elastin in the development of new
biomaterials. Uny and coworkers have made crosslinked materiais from poIypeptides based
on the hydrophobic sequences of elastin. The polyteuapeptide, poly(VPGG), the

polypentapeptide, poly(VPGVG), and the polynonapeptide, poly(VPGFVGVAG), were
coacervated and crosshked with y-irradiation resulting in eIastomeric materials (Urry et al.,
1982; Urry et al., 1990; Urry et al., 1984). The addition of the polyhexapeptide sequence,
poly(PGVGVA), making a sequential, multi-component polypeptide, made a higher tensile
strength materiai and introduced a known binding site for the elastin-binding protein, found
on the membrane of smooth muscle cells and fibroblasts, into the material (Urry et al., 1988).
Soluble elastin peptides (a-elastin or K-elastin) have also been used in conjunction
with other proteins, including colIagens and fibrin, to fabricate composite matrix materials.
An elastin-fibrin material was used as a scaffold to repair a slit in the abdominal aorta of the
rabbit (Lefebvre et al,, 1989). After three months, the material was no longer present.
However, the slit had been closed and the arterial tissue restored. Materials formed with the
addition of collagen to the elastin-fibrin material have also been developed and show promise
in repairing tympanic membranes (Bonzon et al., 1995). Recently, reconstruction of elastic
cartilage was attempted using K-elastin incorporated into molded hydrogels consisting of
alginate and collagen (de Chalain et al., 1999). Chondrocytes, ernbedded in the matrix,
synthesized new cartilage essentially identical to native cartilage.
One potential drawback of using elastin as a biomateriai is that elastin hm ken
reported to calcify, at Ieast under sorne circumstances (Hollinger et al., 1988; Urry, 1976;
Urry et al., 1976a). However, the mechanism of calcification is unclear. Urry and coworkers
have used this property to fabricate calcified polypentapeptide matrices that they suggest may
have biomedical applications (Urry et al., 1976b). However, caicification of elastin wouid not
be a desirable property in other biomaterids applications. Retreatment with aluminum
chloride has been reported to inhibit elastin calcification (Vyavahare et al., 1999).
Elastin may also be useful as a coating for other biomaterids. Many of the cunently
available synthetic vascutar p f t materials, including Dacron, expanded
poIytetrafluoniethyIene (e-PITE), and polyurethanes, are highIy successful for grafts larger
than 6 mm in diameter. However, the ctinical use of synthetic vascular prostheses smaller
than 6 mm is currently Limited by luminal thromùosis and intima1 hyperplasia (Didisheim and
Watson, 1996). Intima1 hyperplasia is characterized by proliferation and migration of
vascula. smooth muscle cells and subsequent synthesis of extraceliular matrix proteins. The
majority of these small diameter grafts faiI within five years (Greenwald and Berry, 2000).

Elastin shows reduced platelet adsorption and aggregation in comparison to other
components of the extracelluIar ma& (Barnes and MacIntyre, 1979; Legrand and Fauvel,
1992; Ordinas et ai,, 1975). In addition, coacervated elastin polypeptides, a-elastin and K-
elastin, either alone or in conjunction with currently available biomatenals, have been shown
to prevent smooth muscle ce11 migration but allow proiiferation of endotheliai cells (Dutoya
et ai., 2000; Ito et al., 1998). Preiirninary investigations into the use of the elastin
polypeptides in bloodcontacting applications are in agreement with these results. Coating
EP20-24-24 ont0 the surface of materials cunently used in vascular applications reduced
platelet adhesion and activity in comparison with non-coated surfaces (Chen, 1999). These
results suggest that coating of currentIy used biomateriais with elastin poIypeptides may not
only present a less thrombogenic surface to the flowing blood, but also may suppress intimai
hyperplasia while promoting endotheiialization of the graft materid (Ito et ai., 1998; Ooyama
et al., 1987).
The ability to fabricate mCL24 and E~20-244 into stable structures with elastin-Iike
properties is also promising from a biomatefials point of view. However, like elastin, these
polypeptide membranes on their own have rather low tensile smngth and, therefore, would
not be useful in situations requiring load bearing. In this respect, it would be interesting to
attempt the fabrication of composite materids involving these polypeptides. For example,
polypeptides coacervated and crosslinked in the presence of collagen fibres might yield
materials with physicd properties more similar to those of colIagen4astin composites
present in tissues such as the aorta
The resuIts presented in this thesis represent only early stages in the understanding of
how recombinant human elastin polypeptides seIf-assemble into organized polymeric
matrices. NevertheIess, the remarkable abiiity of these relatively smalI polypeptides to self-
organize into biomateriais with propercies sirnilar to insoluble elastin suggest that further
investigations of the d e s of polypeptide size, structure and sequence holds the promise of
production of human elastin-like polymers with usefuI and modifiable mechanicd properties.

References Aaron, B. & Gosline, J. (1980) Optical properties of single elastin fibres indicate random
protein conformation. Nature 287,865-867.
Aaron, B. B. & Gosline, J. M. (1981) Elastin as a random-network elastomer: a mechanical
and optical analysis of single elastin fibers. Biopolymers 20,1247-1260.
Abdel Rahman, S., EIsafty, M, & Hattaba, A. (1987) A circular dichroism and infrared study
of Ftm formation in repeat peptides of elastin. Oriental Joumal of Chemistry 3-40-
45.
Albert, E. N. (1972) Developing elastic tissue: An electron microscopie study. Amencan
Journal of Pathology 69,89-102.
Ayer, J. P. (1964) Elastic Tissue. International Review of Connecrive Tisme Research 2,33-
100.
Baig, K. M., Vlaovic, M. & Anwar, R. A. (1980) Amino acid sequences C-terminal to the
cross-links in bovine elastin. Biochemical Journal 185.611-6.
Bailey, A., Peach, C. & Fowler, L. (1970) Chemistry of collagen cross-links: isolation and
characterization of two intermediate intermolecular crosslinks in collagen.
Biuchemicd Journal 117,s 19-83 1.
Bames, M. & MacIntyre, D. (1979) Platelet-rcactivity of isolated constituents of the blood
vesse1 walI. [Review]. Huernostasis 8,158-170.
Bashir, M., Indik, Z., Ornstein-Goldstein, N., Rosenbhom, J., Abrams, W., Fazio, M., Uitto,
J. & Rosenbloom, 1. (1989) Characterization of the complete human elastin gene.
Journal of Biological Chemistry 264,8887-889 1.
Bedeli-Hogan, D., Trac-, P., Abrams, W., Rosenbloom, J. & Kagan, H. (1993)
Oxidation, cross-linking, and insolubilization of recombinant tropoelastin by purified
Iysyl oxidase. Joumai of Biological Chemistry 268,10345-50.

Bonzon, N., Carrat, X., Deminiere, C., Daculsi, G., Lefebvre, F. & Rabaud, M. (1995) New
artificiai connective rnatrix ma& of fibrin monomers, elastin peptides and type 1 + JI collagens: structural study, biocompatibility and use as tympanic membranes in
rabbit. Biumtenals 16,88 1-885.
Boyd, C. D., Christiano, A. M., Pierce, R. A., Stolle, C. A. & Deak, S. B. (1991) Mammalian
tropoelastin: mdtiple domains of the protein define an evolutionariIy divergent amino
acid sequence. Munir 1 1,235-24 1.
Bressan, G., Castellani, L, Giro, M., Volpin, D., Fornieri, C. & Ronchetti, 1. (1983) Banded
fibers in tropoelastin coacervates at physiological temperatures. Journal of
Ultrastructure Research 82,335-340.
Bressan, G., Pasquali-Ronchetti, I., Fornieri, C., Mattioli, F., Castellani, 1. & Volpin, D.
(1986) Relevance of aggregation properties of tropoelastin to the assembly and
structure of elastic fibers. Journal of Ultrastructure Research 94,209-216.
Brown-Augsburger, P., Broekelmann, T., Rosenbloom, J. & Mecham, R. P. (1996)
Functionai domains on elastin and microfibril-associated glycoprotein involved in
elastic fibre assembl y. Biochemical Journal 3 18,149-55.
Brown-Augsburger, P., Tisdale, C., Broekelmann, T., Sloan, C. & Mecham, R. (1995)
Identification of an elastin cross-linking domain that joins three peptide chains.
Journal of Biological Chemistry 270,17778-17783.
Bungenberg de h g , H. G. (1949). In Colloid Science, VoI. 2 (ed. H. R. Kruyt). Amsterdam:
EIsevier.
Castiglione MorelIi, M., DeBiasi, M., DeSüadis, A. & Tamburro, A. (1993) An aggregating
elastin-like pentapeptide. Journal of Biumolecular Structure and Dynamics 1 1,I8 1-
190.
Castiglione-Moreiii, A., Scopa, A. & Tamburro, A. (1990) Spectruscopic studies on elastin-
like synthetic polypeptides. International Journal of Biological Macromolecules 12,
363-368.

Chen, V. (1999) Human elastin peptide coated synthetic materials in blood contacting
applications. ChemicaI Engineering and Applied Chemistry, pp. 119. Toronto:
University of Toronto.
Cleary , E. G. & Cliff, W. J. (1978) The substnicture of elastin. Experimenfal mrd Molecular
Biology 28,227-246.
Cox, B. A., Starcher, B. C. & Urry, D. W. (1973) Coacervation of a-elastin resuIts in fiber
formation. Biochimica et Biophysica Acta 3 17,209- 13.
Cox, B., Starcher, B. & Urry, D. (1974) Coacervation of tropoelastin results in fiber
formation. Jouml ofBiologica1 Chemistry 249,997-998.
Cox, D. L., Mecham, R. P. & Sexton, O. J. (1982) Lysine derived cross-links are present in a
non-elastin, proline-rich protein fraction of I g u m iguana eggshell. Comparative
Biochemistry and Pttysiology 72B, 6 19-623.
Davis, E. C. (1993) Stability ofelastin in the developing mouse aorta: a quantitative
radioautographic shtdy. Histochemistry 100, 17-26.
de Chdain, T., Phillips, J.H. & Hinek, A. (1999) Bioengineering of elastic cartilage with
aggregated porcine and human auricular chondroçytes and hydrogels containing
dginate, collagen, and K-elastin. Journal of Biomedical Matenals Research 44,280-
288.
Debeiie, L. & Alix, A. (1995) Optical spectroscopie determination of bovine tropoeIastin
molecular model. Journal of Molecular Structure 348,321-324.
Debelle, L., Ah, A. J.. Wei, S. M., Jacob, M. P., Huvenne, J., Berjot, M. & Legrand, P.
(1998) The secondary structure and architecture of human elastin. Europem Journal
of Biochemistry 258,533-539.
Debelle, L., Alix, A., Jacob, M., Huvenne, J., Be rjot, M., Sombret, B. & Legrand, P. (1995)
Bovine elastin and K-elastin secondary structure determination by optical
specîroscopies. J o u d of Biological Chemisby 270,26099-26 103.

Debelle, L., Wei, S. M., Jacob, M. P., Hornebeck, W. & Alix, A. 1. (1992) Predictions of the
secondary strucnue and antigenicity of human and bovine tropoelastins. European
Biophysics Journal 21,321-9.
Didisheim, P. & Watson, J. T. (1996) Cardiovascular Applications. In Biomaterials Science;
An Introduction to Materids in Medicine (ed. B. D. Ratner, A, S. Hoffman, F. J.
Schoen & J. E. Lemons), pp. 283-297. San Diego: Academic Press.
Dutoya, S., Vema, A.. Lefebvre, F. & Rabaud, M. (7000) Elastin-denved protein coating
ont0 poly(etby1ene terephthalate). Technical, microstructural and biologicai studies.
Biomateriais 21,1521-1529.
Eyre, D., Paz, M. & Gdlop, P. (1984) Cross-iinking in coliagen and elastin. Annual Review
of Biochemistry 53.7 17-748.
Foster, J. A., Rubin, L., Kagm, H, M., Franzblau, C., Bruenger, E. & Sandberg, L. B. (1974)
Isolation and characterization of crosslinked peptides from elastin. Journal of
Biological Chcmistry 249,6 19 1-6 1%.
Foster, J., Bmenger, E., Rubin, L., Imberman, M., Kagan, H., Mecham, R. & Franzblau, C.
(1976) Circular dichroism studies of an elastin crosslinked peptide. Biopolymers 15,
833-841.
Freifelder, D, (1982) Phvsical Biochemistry. New York: WH. Freeman and Company.
Gerber, G. E. & Anwar, R. A. (1974) Structural studies on cross-linked regions of elastin.
J o u d ofBiological Chemistry 249,5200-5207.
Gerber, G. E. & Anwar, R. A. (1975) Comparative studies of the cross-Iinked regions of
elastin from bovine ligamenturn nuchae and bovine, porcine and human aorta.
Biochemical JoumI 149,685-695.
Gibson, M. A., HatzinikoIas, G., Kumaratilake, J. S., Sandberg, L. B., NichoU, J. K.,
SutherIand, G. R, & Cleary, E. G. (1996) Further characterization of proteins
associated with elastic fibre mim€ibrïls induding the molecular cIoning of MAGP-2
(MP25). J o u d of Biological îhemistry 271.

Gibson, M. A., Sandberg, L. B., Grosso, L. E. & Cleary, E. G. (1991) Complementary DNA
cloning estab iishes rnicrofibnl-associated glycoprotein (MAGP) to be a discrete
component of the eIastin-associated rnicrofibrils. Journal of Biological Chemistry
266,7596-7601,
Gosline, J. (1976) The physicai properties of elastic tissue. In International Review of
Connective Tissue Research (ed. D. HaU & D. Jackson), pp. 21 1-249. New York:
Academic Press.
Gosline, 1. (1978) Hydrophobic interaction and a model for the elasticity of elastin.
Biopolymers t7,677495.
Gotte, L., Giro, M., Volpin, D. & Horne, R. (1974) The ~Itrastructural organization of
elastin. J o u d of Ultrastructure Research 46,23-33.
Gotte, L., Mamrni, M. & kzzin, G. (1972) Scanning electron microscope observations of
elastin. Connedve Tissue Research 1,6147.
Gray, W., Sandberg, L. & Foster, 1. (1973) Molecular model for elastin stntcnire and
function. Nature 246,46 1466.
Greenke Jr., T. K., Ross, R. & Hartman, J. L. (1966) The fine stmchire of elastic fibers.
Journal of Ce11 Biology 30-59-7 1.
Greenwaid, S. E. & Berry, C. L. (2000) Impmving vascular grafts: the importance of
mechanical and haemodynamic properties. Journal of Paihology 190,292-299.
Grosso, L E., Whitehouse, L, A. & Mecham, R. P. (1990) Immunohistochernical detection
of intraceiiular trpoelastin: an assay for elastin production and its use in the
detection and assessrnent of elas togenic factors. Amencan Journal of Respirmory
CeIl& MoIeculur Biology 3,45-9.
HamoQakas, S., Etmektzoglou, T. & Kafatos, F. (1985) Amino acid periodicities and their
stmcttnaf implications for the evolutionarily conservative cenaal domain of some
siikmoth chorion proteins. Journal of Molecular Biotogy 186,583-589.

Harkness, R. D. (1968) Mechanicd properties of collagenous tissues. Treatise on Collagen
2A, 247-3 10.
Hinek, A. & Rabinovitch, M. (1994) 67-kD elastin-binding protein is a protective
"companion" of extracellular insoluble elastin and intracellular tropoelastin. Journal
of Ce11 Biology 126,563-574.
Hinek, A., Wrenn, D., Mecham, R. & Barondes, S. (1988) The elastin receptor: a
galactoside-binding protein. Science 239, 1539-1541.
Hoeve, C.A.J. & Flory, P.J. (1958) The elastic properties of elastin. Journal of the American
Chernical Society 80,6523-6526.
Hoeve, C.A.J. & Fiory, P.J. (1974) The elastic properties of elastin. Biopolymers 13,677-
686.
Holiinger, J.O., Schrnitz, J.P., Yaskovich, R., Long, M.M., Prasad, KU. & Urry, D.W.
(1988) A synthetic polypentapeptide of eiastin for initiating calcification. Calcifed
Tissue international 42,23 1-236.
Indik, Z., Abrams, W., Kucich, U., Gibson, C., Mecham, R. & Rosenbloorn, J. (1990)
Production of recombinant human tropoelastin: characterization and demonstration of
immunologie and chemotactic activity. Archives of Biochemisrry and Biophysics 280,
80-86.
Indik, 2, Yeh, H., ûmstein-Goldstein, N., Kucich, U., Abrams, W., Rosenbloom, J. C. &
Rosenbloom, J. (1989) Structure of the elastin gene and alternative splicing of elastin
mRNA: implications for human disease. American Journal of Medical Genetics 34,
8 1-90.
indik, Z., Yeh, EL, Omstein-Goldstein, N., Sheppard, P., Anderson, N., Rosenbloom, J. C.,
Peltonen, L. & Rosenbloom, 1. (1987) Altemative spIicing of human elastin mRNA
indicated by sequence andysis of cloned genornic and complementary DNA.
Proceedings of the National Academy of Sciences USA 84,56804

Ito, S. H-, Ishimaru, S. & Wilson, S. E. (1998) Effect of coacervated a-elastin on
proliferation of vascular smooth muscle and endothelid cells. Angiology 49,289-
297.
Jacob, M. & Robert, L. (1 989) Isolation, characterization, and biochemical properties of
elastin. In Elastin and Elastase, vol. 1 (ed. L. Robert & W. Hornebeck), pp. 49-65.
Florida: CRC Press.
Jarnieson, A., Downs, C. & Waiton, A. (1972) Studies of eIastin coacervation by quasielastic
light scattenng. Biochimica et Biophysica Acta 27 1,3447.
Jensen, S., Vrhovski, B. & Weiss, A. (2000) Domain 36 of tropoelastin plays a dominant role
in association by coacervation. Journal of Biological Chemistry 275,2849-28454.
Johnson, D. f., LaBomene, J., Rabinovitch, M. & Keeley, F. W. (1993) Relative efficiency of
incorporation of newly synthesized elastin and collagen into aorta, pulmonary artery
and pulmonary vein of growing pigs. Connecrive Tissue Research 29,213-221.
Kagan, H. M. & Trackman, P. C. (1991) Properties and function of lysyl oxidase. Amencan
Joumal of Respiration and Cellular Molecular Biology 5,206-10.
Kaibara, K., Sakai, K, Okamoto, K., Uemura, Y., Miyakawa, K. & Kondo, M. (1992) a-
elastin coacervate as a protein iiquid membrane: effect of pH on transmembrane
potential responses. Biopolymers 32,1173-1 180.
Kondo, M., Nakashima, Y., Kodama, H. & Okamoto, K. (1987) Study on coacervation of the
repeat pentapeptide mode1 of tropaelastin: effect of cations. Journal of Biochemisrry
10 1,89-94.
Kyte, f. & Doolittle, R. (1982) A simple method for displaying the hydrophobie character of
a protein. Journal of Molecular Biology 157, 105-132.
Lansing, A., Rosenthal, T. B., AIex, M. & Dempsey, E. W. (1952) The structure and
chemical characterization of elastic fibers as reveaied by elastase and by electron
mimscopy. Anarornical Record 1 14,555-575.

Lefebvre, F.. Drouillet, F., Savin de Larclause, AM., Aprahamian, M., Midy, D., Bordenave,
L. & Rabaud, M. (1989) Repair of experimental arteriotomy in rabbit aorta using a
new resorbable eIastin-fibrin biomaieriai. Journal of Biomedical Materials Research
23,1423-1432.
Legrand, Y. & Fauvel, L. (1992) MolecuIar mechanism of the interaction of subendothelial
microfibrils with blood platelets. [Review]. Nouvelle Revue Francaise d'Hematologie
34, 17-25.
Li, D.Y., Faury, G., Taylor, D.G., Davis, E.C., Boyle, W.A., Mecharn, RP., Stenzel, P.,
Boak, B. & Keating, M.T. (1998) Novel arterial pathology in mice and hurnans
hernizygous for elastin. Journal of Clinical Investigation 102, 1783- 1787.
Luan, C., Hanis, R., Prasad, K. & Urry, D. (1990) Differentiai scanning calorimetry studies
of the inverse temperature transition of the polypentapeptide of elastin and its
analogues. Biopolymers 29, 1699-1706.
Mammi, M., Gotte, L. & Pezzin, G. (1%8) Evidence for order in the structure of a-elastin.
Nature 220,37 1-373.
Manavalan, P. & Johnson Jr., W. C. (1983) Sensitivity to circular dichroism to protein
tertiary structure c Iass. Nanrre 305,83 1-832.
Martin, S., Vrhovski, B. & Weiss, A. (1995) Total synthesis and expression in Escherichia
coli of a gene encoding human tropelastin. Gene 154,159-166.
Mecham, R. (1999) Personal Communication. Gordon Research Conference.
Mecham, R. P. & Ramirez, F. (1994) Structure and expression of fibruin-2, a novel
microfibrillar component preferentially located in elastic matrices. Journal of Ce11
Biology 124,855-863.
Mecham, R.P. & Heuser, JE. (1991) The elastic fiber. In Cell Bioi~ev of Extracelluia.
Maaix (ed, ED. Hay), pp. 79-109. New York: Plenum Press.

Momson, SL., Campbell, DX. & Wright, GM. (2000) Chondrogenesis of the branchial
skeleton in embryonic sea lamprey, Petromyzon marinus. Anatomical Record 260,
252-267.
Narayanan, A. S., Page, R. C., Kuzan, F. & Cooper, C. G. (1978) Elastin cross-linking in
vitro, Studies on factors infl uencing the formation of desmosines by lysyi oxidase
action on tropoelastin. Biochemical Journal 173,857-62.
Ooyama, T., Fukuda, K., O&, H., Nakamura, H. & Hikita, Y. (1987) Substratum-bound
elastin peptide inhibits aortic smooth muscle ce11 migration in vitro. Arteriosclerosis
7,593-598.
Ordinas, A., Homebeck, W., Robert, L. & Caen, J. (1975) Interaction of platelets with
purified macromolecuIes of the artenai wall. Pathologie Biologie 23,44-48.
Partridge, S. M. (1966) Elastin. In *d.
(ed. E. I. Briskey, R. G. Cassens & J. C. Trautman), pp. 327-350. Wisconsin: The
University of Wisconsin Press.
Partridge, S., Davis, H. & Adair, G. (1955) The chemisuy of connective tissues. 2. Soluble
proteins derived h m partial hydrolysis of elastin. Biochemical Journal 6 1,ll-2 1.
PasquaJi Ronchetti, I., Messandrini, A., Baccarani Contri, M., Fornieri, C., Mori, G.,
Quagiino Jr., D. & Valcire, U. (1998) Study of elastic fiber organization by scanning
force rnicroscopy. Matrix Biology 17,75-83.
Pasquali Ronchetti, I., Focnieri, C., Baccarani Contri, M. & Volpin, D. (1979) The
~Itrasûucture of elastin reveaied by freeze-fracture electron rnicroscopy. Micron 10,
89-99.
Podrazky, V. & Jackson, D. (1976) Observations on coacewation of soluble elastins.
Collection Czechoslova Chemicat Communication 41,2296-2302.
Qin, X, Coyne, K. & Waite, J. (1997) Tough tendons: mussel byssus has coilagen with siik-
Hic domains. Journul ofBiologica1 Chemisny 272,32623-32627.

QuintareK, G., Bellocci, M. & Zito, R. (1973) Structural features of insolubie elastin.
Histochenzie 37,494.
Reiersen, H., Clarke, A. & Rees, A. (1998) Short elastin-Iike peptides exhibit the same
temperature-induced structural transitions as elastin polymers: implications for
protein engineering. Journal of Molecular Biology 283,255-264.
Reiser, K., McCormick, R. & Rucker, R. (1992) Enzymatic and nonenzymatic cross-linking
of coIIagen and elastin. FASEB Journal 6,2439-2449.
Roach, M. & Burton, A. (1957) The reason for the shape of the distensibility curves of
artenes. Canadian Journal of Biochemistry and Physiology 35,681-690.
Robson, P., Wright, G. & Keeley, F. (2000) Distinct non-coliagen based cartilages
comprising the endoskeleton of the Atlantic ha&sh, Myxine glutinosa. Anatomy and
Embryology (Berlin) 202,28 1-290.
Robson, P.? Wright, G., Sitan, E., Maiti, A., Rawat, M., Youson, J. & Keeley, F. (1993)
Characterization of lamprin, an unusuai ma& protein from lmprey cartilage.
Journal of Biological Chemistry 268, 1440-1447.
Rosen, S. L. (1971) Fundamental Princi~les of Polyneric Materials for Practicino Enginers.
New York: Barnes & Noble, Inc.
Rosenbloom, J., Abram, W. & Mecham, R. (1993) Extracellular rnatrix 4: the elastic fiber.
FASEB Journal 7,1208-1218.
Ross, R. & Bomstein, P. (1969) The elastic fiber: 1. The separation and partial
characterization of its macromoIecular components. Journal of Ce11 Biology 40,366-
38 1,
Sakai, L. Y., Keene, D. R. & Engvail, E. (1986) Fibrillin, a new 350-kD glycoprotein is a
component of extracellular microfibrils. Joumal of Ce11 Biology 103,2499-2509.

Sandberg, L., Hackett, T.N., & Carnes, WH. (1969) The solubiiization of an elastin-like
protein From copper-deficient porcine aorta. Biochimica et Biophysica Acta 18 1.20 1-
207.
Sandberg, L., Weissman, N. & Smith, D. (1969) The purification and partial characterization
of a soiubie elastin-iike protein firom copper-deficient porcine aorta. Gbcr'wrrii;uy O,
2940-2945.
Senior, R., Griffin, G. & Mecham, R. (1982) Chemotactic responses of fibroblasts to
tropoelastin and elastin-denved peptides. Journal of Clinical lnvesrigation 70,614-
618.
Serafini-Fracassini, A., Field, J. M., Spina, M., Stephens, W. G. & DeIf, B. (1976) The
macromoIecular organization of the elastin fibriI. Journal of Molecular Biology 100,
73-84.
Shah, M. A., Bergethon, P. R., Boak, A. M., Gdlop, P. M. & Kagan, H. M. (1992) Oxidation
of peptidyt lysine by copper complexes of pyrroIoquinoiine quinone and other
quinones. A mode1 for oxidative pathochemistry. Biochimica et Bioplrysica Acta
1159,311-8.
Starcher, B. & Galione, M. J. (1976) Purification and cornparison of elastins From different
animal species. Analyn'cal Biochemistry 74,44147.
Starcher, B. C. & King, G. S. (1980) The presence of desmosine and isodesmosine in
eggshell membrane protein. Connective Tissue Research 8,53-55.
Starcher, B. C. (1977) Determination of the elastin content of tissues by measuring
desmosine and isodesmosine. Analytical Biochemisny 79, 1 1-5.
Starcher, B. C., Saccomani, G. & Urry, D. W. (1973) Coacervzttion and ion-binding studies
on aoriic elastin. Biochimica et Biophysica Acta 310,481-6,
Szabo, Z., Levi-Mimi, S., Christiano, A., Struminger, C., Stoneking, M., Batzer, M. & Boyd,
C. (1999) Sequential Loss of two neighboring exons of the tropoelastin gene during
primate evohtion. J o u d of Molecular Biology 49,664-671.

Tamburro, A. M., De Stradis, A. & D'Aiessio, L. (1995) Fractal aspects of elastin
supramolecular organization. Journal ofBiomolecular Smccture anà Dynamics 12,
1161-72.
Tambwro, A., Guantieri, V., Daga-Gordini, D. & Abatangelo, G. (1977) Conformational
transitions of u-elastin. Biochimica et Biophysica Acta 492,370-376.
Urry D.W. (1976) On the molecular mechanisms of elastin ~u~c.cpllr~!ieri and coacervate
calcification. Faraday Discussions of the Chernical Sociery 6 1,205-216.
Urry, D.W. (1982) Characterization of solubIe peptides uf eixtir. by- physicd techniques.
Methodr in Enrymology 82,673-7 16.
Urry, D.W. (1983) Whai is elastin; what is not. I/lrrastructural Pathology 4,227-251.
Urry, D.W. (1984) Procein elasticity based on conformations of sequential poiypeptides: the
biological elastic fiber, Jounrol of Prorein Chemistry 3,403436.
Uny, D.W. (1995) Elastic biomoiecular machines. Scientific American, 272,4449.
Uny, D.W., Long, M. M., Cox, B. A., Oh~stii, T., Mitchell, L. W. & Jacobs, M. (1974) The
synthetic polypentapeptide of elastin coacervates and forms filamentous aggregates.
Biochimica et Biophysica Acta 37 1,597-602.
Urry, D.W., Hendrix, CF. & Long, M.M. (1976a) Calcifiçation of a-elristin coacervates: a
bulk property of eIastin. Calcij7ed Essue Research 21,57-65.
Urry, D.W., Long, M M , Hendrix, C I . & Okamoto, K. (1976b) Cross-linked
polypentapeptide of tropoelastin: and insoluble, se- calcifiable matrix.
B i o c h & y 15,4089494
Urry, D.W. & Long, M. M. (1977) On the conformation, coacervation and hnction of
polymeric models of elastin. Advances iit Ekperimental Medicine & Biology 79,685-
714.

UT, D.W., Harris, R. & Long, M. (1982) Irradiation crosslinking of the polytetrapeptide of
elastin and compounding to dacron to produce a potentiai prosthetic materiai with
elasticity and strength. Journal of Biomedicd Mareriais Research 16,11-16.
Urry, D.W., Venkatachaiam, C., Long, M. & Prasad, K. (1983) Dynamic ~ s p k a l s and a
librationd entropy mechanism of elasticity. In Conformation in Bioloev (ed R.
Srinivasan & R. Sana), pp, 11-27, New York: Adenine Press.
Urry, D.W., Wood, S., Harris, R. & Prasad, K. (1984) Polypentapeptide of elastin as an
ehstomeric biomateriai. In Folpers as Biomaterials (ed. S. Shalaby, A. Hohan, B.
Ratner & T. Hohtt), pp. 17-32. New York: Plenum Press.
Urry, D.W., Shaw, R. G. & Prasad, K. U. (1985a) Polypentapeptide of elastin: Temperature
dependence of ellipticity and correlation with elastomeric force. Biochemiccll and
Biophysicai Research Cummunicarions I30,SO-57.
Uny, D.W., Tnpane, T. L. & Prasad, K. U. (198%) Phase-structure transitions of the elastin
polypentapeptide-water system within the frarnework of composition-temperature
studies. Biopoiymers 24,23452356,
Urry, D.W., Prasad, K., Long, M. & Harris, R. (1988) Elastomeric pdypeptides as potential
vascular prosthetic materials. Polymeric Materials Science and Engineering 59,684-
689.
Urry, D.W., Jaggard, J., Harris, R., Chang, D. & Prasad, K. (1990) The poly(nonapeptide) of
elastin: a new elastomeric polypeptide biomateriai. In R.opaess in Biomedical
PoIyners (ed. C. Gebelein & R. Dunn), pp. 171-178, New York: Plenum Press.
Urry, D.W., Luan, C., Parker, T., Gowda, D., Prasad, IL, Reid, M. & Safavy, A. (1991)
Temperature of polypeptide inverse temperature transition depends on niean midue
hydrophobicity. Jorrmal of the Amencan Chernical Society t 13,4346-4348.
Urry, D.W., Gowda, D. C., Parker, T. M., Luan, C. IL, Reid, M. C., Harris, C. M., Pattanaik,
A. & Harris, R. D. (1992) Hyhphobicity scde for proteins based on inverse
temperature transitions. Biopolymers 32,1243-1250-

Volpin, D., Urry, D., Cox, B. & Gotte, L. (1976) Optical diffraction of tropoelastin and
a-elastin coacervates. Biochimica et Biophysica Acta 439,253-258.
Vrhovski, B. & Weiss, A. S. (1998) Biochemistry of tropoelastin. European Journal of
Biochemistry 258.1-18.
Vrhovski, B., Jensen, S. & Weiss, A. (1997) Coacervation characteristics of recombinant
hurnan tropoelastin. European Journal of Biochemistry 250.92-98.
Vyavahare, N., Ogle, M., Schoen, F.J. & Levy, R.J. (1999) Elastin calcification and its
prevention with aiuminum chloride pretreatment. Amencan Jounral of Pathology
155.973-982.
Wad, 1. M. (197 1) Mechanical uroperties of solid p l p e r s . London: John Wiley & Sons,
Ltd.
Weis-Fogh, T. & Andersen, S. (1970) New moIecuIar mode1 for the long-range elasticity of
elasth. Nature 227,718-721.
Wendel, D.P., Taylor, D.G., Alberrine, M., Keating, M.T. & Li, D.Y. (2000) impaired
distal airway development in rnice lacking eIastiri. Americmt Journal of Respiratory
Cell% Molecular Biology 23,320-326.
Xu, M. & Lewis, R. (1990) Structure of a protein superfiber. spider dragline siIk.
Proceedings of the National Academy of Science USA 87,7 12û-7 124.
Yeh, H., Chnstem-Goldstein, N., indik, Z., Sheppard, P., Anderson, N., Rosenblwm, J. C.,
Cicila, G., Ywn, K. & Rosenblwm, J. (1987) Sequence variation of bovine elastin
mRNA due to alternative spiicing. Collagen Related Research 7,235-47.
Zhang, H., Apfelroth, S. D., Hu, W., Davis, E. C., Sanguineti, C., Bonadio, J., Mecham, R. P.
& Ramirez, F. (1994) Structure and expression of firiIliin-2, a novel microfibriliar
component preferentiaIIy located in elastic matrices. J o u d of CeIl Biolugy 124,
855-863