Embed Size (px)
Citation preview

Std
CNNGa
b
c
d
a
ARRAA
KCTRB
1
Iw[orembsmtte
8
0h
Leukemia Research 38 (2014) 236– 242
Contents lists available at ScienceDirect
Leukemia Research
journa l h o me pag e: www.elsev ier .com/ locate / leukres
elective strong synergism of Ruxolitinib and second generationyrosine kinase inhibitors to overcome bone marrow stroma relatedrug resistance in chronic myelogenous leukemia
oncetta Quintarelli a,b,∗,1, Biagio De Angelisa,b,1, Santa Errichielloa,b, Simona Carusoa,b,icola Espositoa,b, Irene Colavitab, Maddalena Raiab, Simona Pagliucaa,b,ovella Pugliesea,b, Antonio M. Risitanoa,b, Marco Picardia,b, Luigia Lucianoa,iuseppe Saglioc, Giovanni Martinelli d, Fabrizio Panea,b
Dipartimento di Medicina Clinica e Chirurgia, University of Naples Federico II, ItalyCEINGE Biotecnologie Avanzate, Napoli, ItalyDepartment of Clinical and Biological Sciences, University of Turin, Orbassano, ItalyDepartment of Haematology/Oncology “L. e A. Seràgnoli”, University of Bologna, Bologna, Italy
r t i c l e i n f o
rticle history:eceived 19 July 2013eceived in revised form 4 November 2013ccepted 7 November 2013
a b s t r a c t
The IC50 of TKIs is significantly increased when BCR-ABL+ K562 cell line is cultured in stroma conditionedmedia produced by BM mesenchymal cells. In particular, while the Imatinib IC50 in the stromal co-cultures was well above the in vivo through levels of the drug, the IC50s of second generation TKIswere still below their through levels. Moreover, we provide a formal comparison of the synergy between
vailable online 15 November 2013
eywords:hronic myelogenous leukemiayrosine kinase inhibitoresistance
first and second generation TKIs with the JAK inhibitor Ruxolitinib to overcome BM stroma related TKIresistance. Taken together, our data provide a rationale for the therapeutic combination of TKIs andRuxolitinib with the aim to eradicate primary BCR-ABL+ cells homed in BM niches.
© 2013 Elsevier Ltd. All rights reserved.
M microenvironment
. Introduction
Most patients with CML in chronic phase (CML-CP) treated withmatinib achieve long term complete cytogenetic response (CCR),
hile only a minority achieve complete molecular response (CMR)1]. Moreover, although second-generation TKIs yield higher ratesf CMR than Imatinib [2,3], there is still no evidence that CMResults in CML stem cell eradication in the majority of patients. Sev-ral experimental evidences and clinical observations indicate thatost patients achieving CCR have BCR-ABL transcripts detectable
y RT-PCR, and that BCR-ABL-expressing leukemia stem cells per-ist in the BM of patients with CML even in sustained undetectableolecular residual disease [4]. In addition, Imatinib discontinua-
ion studies demonstrated that only a minority of CML patients, i.e.hose with more than a 4-log reduction of minimal residual dis-ase, is eligible for discontinuation trials [5], suggesting that new
∗ Corresponding author at: Università di Napoli Federico II, Via S. Pansini, 5, Napoli0131, Italy. Tel.: +39 0813737869; fax: +39 0813737808.
E-mail address: [email protected] (C. Quintarelli).1 These authors contributed equally to this work.
145-2126/$ – see front matter © 2013 Elsevier Ltd. All rights reserved.ttp://dx.doi.org/10.1016/j.leukres.2013.11.006
additional therapeutic strategies are required to eradicate the dis-ease in a larger number of patients. Noteworthy, recent evidencessuggest that upon TKI treatment pressure, CML stem cell survivalis due to BCR-ABL kinase independent mechanisms [6–9]. Thus,the curative treatment strategies for CML must also include theinhibition of survival pathways in addition to those activated bythe hallmark BCR-ABL oncoprotein. Ph+ leukemic stem cell (LSC)viability is supported by an aberrant activation of pro-survivaland self-renewal pathways regulated by both cell-intrinsic factors,including �-catenin and Sonic Hedgehog (Shh) [10,11], and cell-extrinsic stroma related stimulations [12]. Enhanced survival of LSCin protective microenvironments, provided by the BM niche [11], aswell as the physiological dormancy of cells in these niches, also con-tributes to their persistence despite TKI treatment. In this regard,it is important to note that the BM microenvironment is a sourceof both extracellular matrices and high local concentrations ofcytokines and growth factors, all involved in the signal transductionmostly mediated by the activation of JAK–STAT pathways [13]. In
particular, previous studies have shown that stroma-mediated drugresistance is mediated by the increase in the phosphorylation levelof Stat3 (Tyr705) [14], and that JAK inhibitor potentiates Nilotinib-mediated cell death in the context of the BM microenvironment
ia Re
[ttmpwcRewa
2
2
plliptbhf
2
taodCiWcaTtaf
2
mwHfl(Cwwa
2
tmSs
2
ttoTRcIam
C. Quintarelli et al. / Leukem
15,16], although the observed synergistic effect was nullified byoxic effect toward non-leukemic cells. In this paper, we showhat the TKI IC50 modulation related to BM stroma microenviron-
ent may account for the differential level of response observed inatients treated with first or second generation of TKIs. Moreover,e prove that either Nilotinib or Dasatinib (used at in vivo appli-
able concentration) are very strongly synergic with JAK inhibitoruxolitinib to overcome the BM stroma related TKI resistance inither CML cell lines or CD34+ progenitor cells from CML patients,ith minimal toxic effect on progenitor cells from healthy donors
t the applied concentrations.
. Materials/subjects and methods
.1. Cell lines and tumor cells
K562 cells (DSMZ, Braunschweig, Germany) were maintained in culture in com-lete medium (defined as Regular Media [RM]), as previously described [17]. Cell
ine viability was assessed by trypan blue exclusion. In selected experiments, cellines were incubated for 72 h in the presence of the required concentrations of Imat-nib, Nilotinib (kindly provided by Novartis, Basel, Switzerland), Dasatinib (kindlyrovided by Bristol–Myers Squibb) or Ruxolitinib (INCB018424, Selleckchem, Hous-on, TX). Apoptotic rate was evaluated by Annexin-V Kit and cell cycle was assessedy BRdU Flow Kits (BD Pharmingen), following the manufacturer’s instructions. Theuman stroma cell line HS-5 was cultured in �-MEM with 10% FBS and sub-cultured
ollowing the manufacturer’s instructions.
.2. Samples from CML patients and healthy donors
BM samples were collected from six newly diagnosed patients with CML-CP,hree CML patients in molecular response (MR4) [18] and three healthy donors (HD)fter approval by the Institutional Review Board (IRB) of the Federico II Universityf Naples. Mononuclear cells were isolated from BM or PB aspirates, as previouslyescribed [19]. CD34+ hematopoietic progenitor cells were selected by the DirectD34 Progenitor Cell Isolator kit (Miltenyi Biotec Inc., Auburn, CA), and cultured
n AIM-V serum-free media (Invitrogen) at a starting density of 1 × 105 cells/ml.hen specified, the following cytokines were included [high growth factor (GF)
ocktail] [20]: 100 ng/ml SCF, 100 ng/ml FLT3 ligand, 20 ng/ml G-CSF, 20 ng/ml IL-3,nd 20 ng/ml IL-6 (all from R&D Systems). Depending on the specific experiment,KIs were added as follows: 1000 nM Imatinib, 400 nM Nilotinib and 2.5 nM Dasa-inib [21]. MSCA-1+ cells were selected from 8 BM samples of patients with CML-CPnd two BM samples of HDs, using a MicroBead kit (Miltenyi) following the manu-acturer’s instructions.
.3. Generation of mesenchymal SCM
HS-5 cell line was plated to achieve 80% confluence. Thereafter, the completeedia was removed, and the adherent cells were washed with PBS. Then, HS-5ere cultured in serum-free media for 24 h. HS-5 serum-free supernatant (namedS-5/SCM) was removed from the cells and filtered through a 0.22 �m filter. SCM
rom HD and patients with CML was obtained with the same procedure after ateast two cell passages of two mesenchymal stroma cell lines obtained from HDsnamed HD/SCM) and eight stroma cell lines obtained each from a BM sample of aML patient (named CML/SCM). After SCM collection, mesenchymal stroma cell linesere analyzed by human MSC Phenotyping Kit (Miltenyi). HS-5/SCM and CML/SCMsere analyzed for the concentration of soluble factors by Bio-Plex assay (Bio-Rad),
ccording to the manufacturer’s instructions.
.4. Colony Forming Cell (CFC) assay
1 × 105 cells CD34+ cells isolated from BM of six CML donors and three HDs werereated with TKIs alone or in combination with Ruxolitinib, plated in triplicate in
ethylcellulose medium supplemented with recombinant cytokines (MethoCult;temCell Technologies), and incubated at 37 ◦C. Colony-forming units (CFU) werecored using a high-quality inverted microscope after 2 weeks of culture [22].
.5. Statistical analysis
All data are presented as mean ± 1 SD. The Student’s t test was used to evaluatehe statistical significance of differences using the non parametric Mann–Whitneyest, with a p value <.05 indicating a significant difference. IC50 was calculated basedn the level of residual viable cells (Annexin-Vneg.ve/PIneg.ve) after treatment withKIs at increasing doses. Data were analyzed by a specific software (MasterPlex
eaderFit). The combination index (CI) was defined as previously specified [23], andalculated by CompuSyn software based on Chou’s median-effect equation [24,25].n particular, based on the CI value, we defined a very strong synergism (CI < 0.1),strong synergism (0.1 < CI > 0.3) and a synergism (0.3 < CI > 0.7), according to theanufacturer’s instructions.
search 38 (2014) 236– 242 237
3. Results
3.1. The IC50 of either first or second generation TKIs significantlyincreases when Ph+ cell line are treated in the presence ofHS-5/SCM
K562 cells were exposed to increasing doses of Imatinib, Nilot-inib or Dasatinib (range 10−1–105 nM) in either the presence orabsence of HS-5/SCM, to quantify the effect of exposure to HS-5/SCM on the modulation of TKI activity. Thereafter, cell viabilitywas monitored by Annexin-V/PI staining after 72 h. As shownin Fig. 1A, we observed that the IC50 of Imatinib, Nilotinib andDasatinib were significantly increased when K562 cells were cul-tured in the presence of HS-5/SCM (IC50HS5-SCM: 7957.16 nM,889.06 nM, 2.85 nM, respectively) respect to the control culturesin RM (IC50RM: 545.46 nM, 13.93 nM and 1.12 nM, respectively).
We also investigated whether apoptosis was only delayed in thepresence of HS-5/SCM. To this aim, we cultured K562 cell line withTKIs at the calculated IC50HS5-SCM for an additional 3 days in thepresence of HS-5/SCM or RM. On day 6, cells were re-plated in RMfor a total of 16 days without addition of TKIs. Interestingly, onlythe residual K562 cells cultured in HS-5/SCM (Fig. 1C) kept a sig-nificant ability to proliferate after TKI withdrawal, whereas thoseexposed to TKI without stromal protection (Fig. 1B) failed to re-expand once TKI exposure was suspended. All these effects weredirectly mediated by soluble factors released by BM stroma, sinceHS-5/SCM withdrawal before TKI exposure resulted in the com-plete absence of protection against the TKI effect (data not shown).Moreover, serial dilutions of HS-5/SCM, ranging from 50% to 0.05%,resulted in a dose dependent effect on resistance to TKI exposure(Supplemental Fig. 1).
Supplementary material related to this article can be found,in the online version, at http://dx.doi.org/10.1016/j.leukres.2013.11.006.
3.2. CML derived BM mesenchymal cell lines produce severalSTAT3 activating cytokines
Since it was already reported that BM stroma soluble factorsinduce STAT3 activation [14], we also quantified the amountsof several cytokines by Bioplex ELISA assay, in HS-5/SCM andCML/SCM derived from eight BM mesenchymal stromal cell linesgenerated from untreated CP-CML patients (Supplemental Fig. 2).IL-1a, IL-1b, IL-7, IL-15 and MIP-1a were expressed exclusively byHS-5 cell line, while SDF-1a, TRAIL and HGF cytokines were foundalso in CML derived stroma. Moreover, most of the other cytokinestested, whereas present in significantly higher levels in HS-5/SCM,were also produced in CML/SCM (M-CSF, SCF, PDGF-bb, IL-6, IL-8,IL-10, IL-12, G-CSF, GM-CSF, TNF-a and VEGF).
Supplementary material related to this article can be found,in the online version, at http://dx.doi.org/10.1016/j.leukres.2013.11.006.
3.3. Second generation TKIs differentially synergize withRuxolitinib to overcome stroma related drug resistance in CML cellline
Recent convincing experimental evidences have shown that BMstroma conditioned media activates Jak2–Tyk2–Stat3 pathway inCML cells [14,16] and that turning off the JAK pathway using theJAK inhibitor sensitizes CML cells to Nilotinib treatment [15]. Inthe era of TKI combinatory therapy, we next sought to determine a
potential differential level of synergism with Ruxolitinib betweenfirst and second generation of TKIs.Whereas Ruxolitinib, as single agent, showed only a mod-est effect on overall cell viability after 72 h of treatment, the
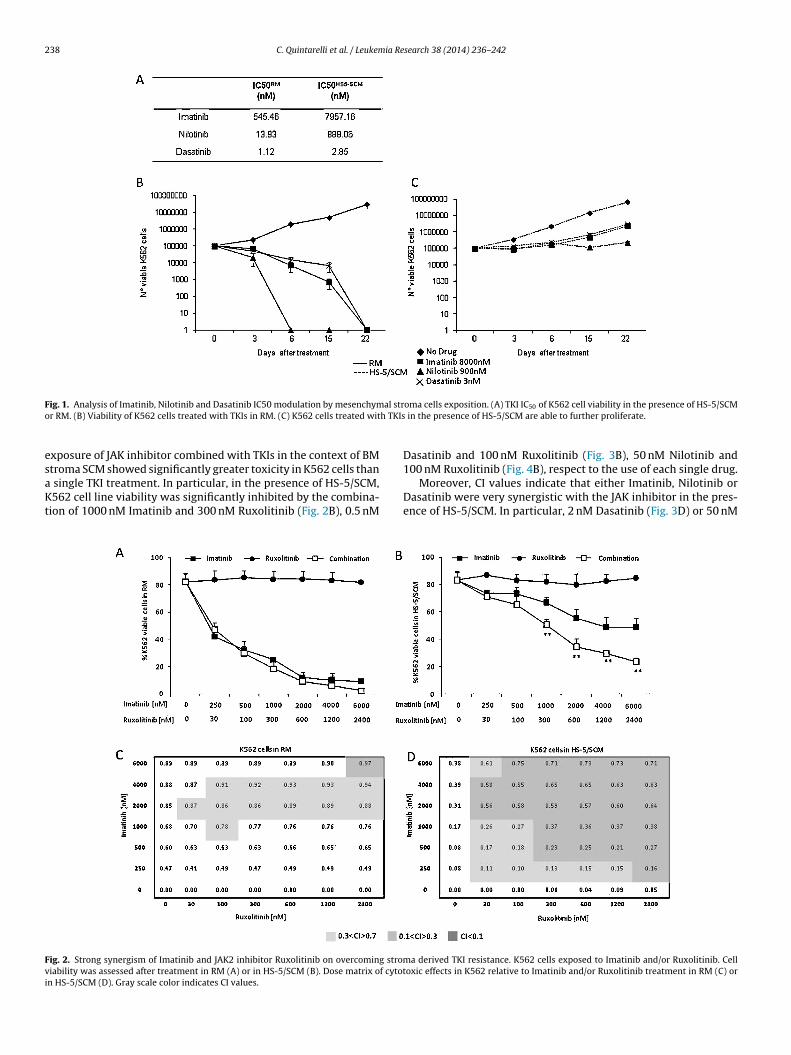
238 C. Quintarelli et al. / Leukemia Research 38 (2014) 236– 242
F al stro h TKIs
esaKt
Fvi
ig. 1. Analysis of Imatinib, Nilotinib and Dasatinib IC50 modulation by mesenchymr RM. (B) Viability of K562 cells treated with TKIs in RM. (C) K562 cells treated wit
xposure of JAK inhibitor combined with TKIs in the context of BM
troma SCM showed significantly greater toxicity in K562 cells thansingle TKI treatment. In particular, in the presence of HS-5/SCM,562 cell line viability was significantly inhibited by the combina-
ion of 1000 nM Imatinib and 300 nM Ruxolitinib (Fig. 2B), 0.5 nM
ig. 2. Strong synergism of Imatinib and JAK2 inhibitor Ruxolitinib on overcoming stromiability was assessed after treatment in RM (A) or in HS-5/SCM (B). Dose matrix of cytotn HS-5/SCM (D). Gray scale color indicates CI values.
oma cells exposition. (A) TKI IC50 of K562 cell viability in the presence of HS-5/SCM in the presence of HS-5/SCM are able to further proliferate.
Dasatinib and 100 nM Ruxolitinib (Fig. 3B), 50 nM Nilotinib and
100 nM Ruxolitinib (Fig. 4B), respect to the use of each single drug.Moreover, CI values indicate that either Imatinib, Nilotinib orDasatinib were very synergistic with the JAK inhibitor in the pres-ence of HS-5/SCM. In particular, 2 nM Dasatinib (Fig. 3D) or 50 nM
a derived TKI resistance. K562 cells exposed to Imatinib and/or Ruxolitinib. Celloxic effects in K562 relative to Imatinib and/or Ruxolitinib treatment in RM (C) or
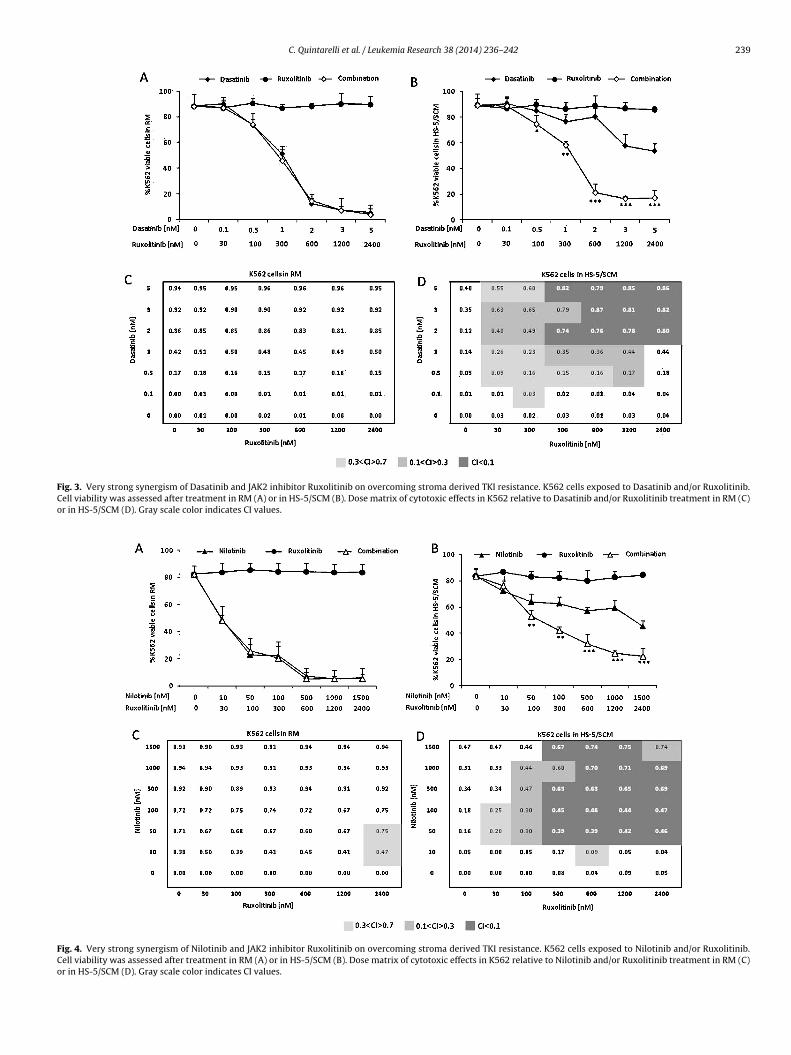
C. Quintarelli et al. / Leukemia Research 38 (2014) 236– 242 239
Fig. 3. Very strong synergism of Dasatinib and JAK2 inhibitor Ruxolitinib on overcoming stroma derived TKI resistance. K562 cells exposed to Dasatinib and/or Ruxolitinib.Cell viability was assessed after treatment in RM (A) or in HS-5/SCM (B). Dose matrix of cytotoxic effects in K562 relative to Dasatinib and/or Ruxolitinib treatment in RM (C)or in HS-5/SCM (D). Gray scale color indicates CI values.
Fig. 4. Very strong synergism of Nilotinib and JAK2 inhibitor Ruxolitinib on overcoming stroma derived TKI resistance. K562 cells exposed to Nilotinib and/or Ruxolitinib.Cell viability was assessed after treatment in RM (A) or in HS-5/SCM (B). Dose matrix of cytotoxic effects in K562 relative to Nilotinib and/or Ruxolitinib treatment in RM (C)or in HS-5/SCM (D). Gray scale color indicates CI values.
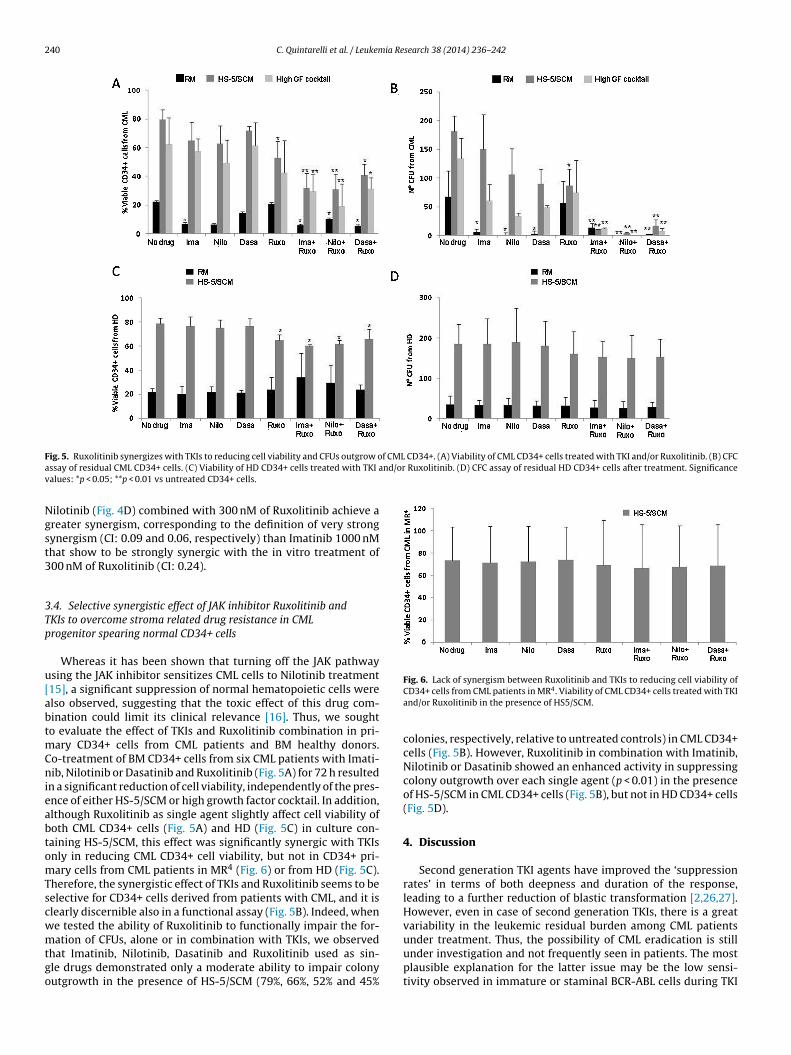
240 C. Quintarelli et al. / Leukemia Research 38 (2014) 236– 242
F f CML CD34+. (A) Viability of CML CD34+ cells treated with TKI and/or Ruxolitinib. (B) CFCa nd/or Ruxolitinib. (D) CFC assay of residual HD CD34+ cells after treatment. Significancev
Ngst3
3Tp
u[abtmCnieabtomTscwmtgo
Fig. 6. Lack of synergism between Ruxolitinib and TKIs to reducing cell viability of
ig. 5. Ruxolitinib synergizes with TKIs to reducing cell viability and CFUs outgrow ossay of residual CML CD34+ cells. (C) Viability of HD CD34+ cells treated with TKI aalues: *p < 0.05; **p < 0.01 vs untreated CD34+ cells.
ilotinib (Fig. 4D) combined with 300 nM of Ruxolitinib achieve areater synergism, corresponding to the definition of very strongynergism (CI: 0.09 and 0.06, respectively) than Imatinib 1000 nMhat show to be strongly synergic with the in vitro treatment of00 nM of Ruxolitinib (CI: 0.24).
.4. Selective synergistic effect of JAK inhibitor Ruxolitinib andKIs to overcome stroma related drug resistance in CMLrogenitor spearing normal CD34+ cells
Whereas it has been shown that turning off the JAK pathwaysing the JAK inhibitor sensitizes CML cells to Nilotinib treatment15], a significant suppression of normal hematopoietic cells werelso observed, suggesting that the toxic effect of this drug com-ination could limit its clinical relevance [16]. Thus, we soughto evaluate the effect of TKIs and Ruxolitinib combination in pri-
ary CD34+ cells from CML patients and BM healthy donors.o-treatment of BM CD34+ cells from six CML patients with Imati-ib, Nilotinib or Dasatinib and Ruxolitinib (Fig. 5A) for 72 h resulted
n a significant reduction of cell viability, independently of the pres-nce of either HS-5/SCM or high growth factor cocktail. In addition,lthough Ruxolitinib as single agent slightly affect cell viability ofoth CML CD34+ cells (Fig. 5A) and HD (Fig. 5C) in culture con-aining HS-5/SCM, this effect was significantly synergic with TKIsnly in reducing CML CD34+ cell viability, but not in CD34+ pri-ary cells from CML patients in MR4 (Fig. 6) or from HD (Fig. 5C).
herefore, the synergistic effect of TKIs and Ruxolitinib seems to beelective for CD34+ cells derived from patients with CML, and it islearly discernible also in a functional assay (Fig. 5B). Indeed, whene tested the ability of Ruxolitinib to functionally impair the for-
ation of CFUs, alone or in combination with TKIs, we observedhat Imatinib, Nilotinib, Dasatinib and Ruxolitinib used as sin-le drugs demonstrated only a moderate ability to impair colonyutgrowth in the presence of HS-5/SCM (79%, 66%, 52% and 45%
CD34+ cells from CML patients in MR4. Viability of CML CD34+ cells treated with TKIand/or Ruxolitinib in the presence of HS5/SCM.
colonies, respectively, relative to untreated controls) in CML CD34+cells (Fig. 5B). However, Ruxolitinib in combination with Imatinib,Nilotinib or Dasatinib showed an enhanced activity in suppressingcolony outgrowth over each single agent (p < 0.01) in the presenceof HS-5/SCM in CML CD34+ cells (Fig. 5B), but not in HD CD34+ cells(Fig. 5D).
4. Discussion
Second generation TKI agents have improved the ‘suppressionrates’ in terms of both deepness and duration of the response,leading to a further reduction of blastic transformation [2,26,27].However, even in case of second generation TKIs, there is a greatvariability in the leukemic residual burden among CML patients
under treatment. Thus, the possibility of CML eradication is stillunder investigation and not frequently seen in patients. The mostplausible explanation for the latter issue may be the low sensi-tivity observed in immature or staminal BCR-ABL cells during TKI
ia Re
timsrtcabp
htosBamioHwtot11iartI
ptttstsTassrwilqsCaiC
dopwatcvdop
C. Quintarelli et al. / Leukem
reatment [28]. Despite several mechanisms have been proposed,ncluding increased BCR–ABL levels and activity [29], so far the
olecular basis of this clinical observation is still poorly under-tood. However, both clinical and in vitro preclinical data, in whichesidual BCR-ABL immature cells were isolated despite successfulreatment with Imatinib mesylate [4], have shown that CML stemell survival is not dependent on BCR-ABL kinase activity [6,8],nd that, consequently, BCR-ABL independent mechanisms maye responsible for primary and secondary TKI resistance in CMLatients.
In this regard, since BM niches represent a rich milieu regulatingematopoietic stem cell self-renewal, proliferation and differen-iation, we investigated its role in the modulation of TKI effectsn CML cells. The results of our study confirm that mesenchymaltromal microenvironment holds also a relevant role in regulatingCR-ABL leukemia cell fate under TKI treatment. Indeed, applyingn in vitro model based on the co-culture of CML cells and BM stro-al derived HS-5 cell line, we demonstrated that IC50s significantly
ncrease for the three clinically available TKIs. In particular, in casef Imatinib and Nilotinib, IC50 increased by approximately one log.owever, while the Imatinib IC50 in the stromal co-cultures wasell above the through levels of the drug when administered at
he standard dose of 400 mg OD (7957 nM vs 1600 nM), the IC50sf second generation TKIs in the same conditions were still belowheir corresponding through levels at the steady-state (889 nM vs700 nM for Nilotinib 400 mg BID; 2.85 nM vs 5.5 nM for Dasatinib00 mg OD). These observations may account for the higher activ-
ty of these drugs against the BCR-ABL immature cell compartment,nd hence provide a plausible explanation for the deeper molecularesponses observed in CML patients treated with second genera-ion TKIs compared to those observed in CML patients treated withmatinib.
Importantly, our data also show that stromal soluble factorsreserve viability of CML cells under treatment with TKIs, sincehey maintain a good proliferating capability in subsequent longerm sub-cultures. Indeed, despite TKI treatment at concentra-ions corresponding to their stromal co-culture IC50s, BCR-ABL cellshow an initial proliferation arrest and then, only those treated inhe presence of stromal soluble factors retain their ability to re-tart proliferation in long term sub-cultures after TKI withdrawal.hus, stromal cells provide long lasting protection to CML cellsgainst apoptosis, resembling the clinical situation in which Ph+tem cells homed in BM niches under TKI treatment survive inmall numbers and in a sort of quiescent status, but are ready toesume proliferation upon TKI discontinuation. Moreover, sincee demonstrated that CML cells are protected from TKI activ-
ty by soluble factors either derived from immortalized HS-5 celline or stroma CML cell lines (data not shown), we performed auantitative evaluation of cytokines present in this two cellularettings. The majority of the investigated cytokines, such as M-SF, SCF, PDGF-bb, IL-6, IL-8, IL-10, IL-12, G-CSF, GM-CSF, TNF-and VEGF but not the chemotactic factor SDF-1, whereas presentn significantly higher levels in HS-5/SCM, were also found inML/SCM.
In particular, previous studies showed that stroma-mediatedrug resistance is due to the increase in the phosphorylation levelf Stat3 (Tyr705) [14], thus indicating that the stromal protectionasses through the activation of this clinically relevant pathway,hich is known to sustain cell viability in vivo, and to trigger the
ctivation of Jak2–Tyk2 signaling. Moreover, it was observed thaturning off the JAK pathway using a JAK inhibitor sensitizes CMLells to Nilotinib treatment in the context of the BM microen-
ironment [15,16]. Thus, we investigated whether Ruxolitinibifferentially synergizes with Imatinib, Nilotinib and Dasatinib tovercome stroma related TKI resistance in CML cell line or CD34+rogenitor cells from CML patients.search 38 (2014) 236– 242 241
Our data indicate that Ruxolitinib effectively strongly synergizeswith Imatinib, and achieves a very strong synergism with secondgeneration TKIs, either Dasatinib or Nilotinib, being able to induceapoptosis in both K562 cell line and CD34+ progenitor CML cellsresistant to the elimination by the single TKI agent, in the presenceof BM stroma signaling. Indeed, co-treatment of Ph+ CD34+ cellswith Ruxolitinib and Imatinib, Nilotinib or Dasatinib significantlydecreased CFU outgrowth, providing evidence that it is able to over-come stromal protection. In contrast with Traer et al. [16], we provethat the JAK inhibitor Ruxolitinib, instead the TG101209, used byTraer et al., allows to achieve a therapeutic window of drug com-bination that spear progenitor cells from healthy donors or CMLpatients in MR4.
From a clinical point of view, although second-generationTKIs achieve a deeper and prolonged cytogenetic and molecularresponse rate than Imatinib in patients with CML-CP [3,30,31], theuse of TKIs as a single agent seems unable to eradicate the disease,despite the increase in MR4.0 rate.
Our data strongly support the hypothesis that a drug combina-tion of Ruxolitinib and second generation TKIs (either Nilotinib orDasatinib) may be more effective in eradicating leukemia ratherthan the single agent TKI, with minimal toxic effect on normalhematopoietic cells. However, the in vivo side effects related to theproposed drug combination, need to be determined by a specificclinical trial.
The combined deep down-regulation of BCR/ABL oncogene plusthe down-regulation of the signaling induced by the soluble factorspresent in the stroma BM microenvironment through the JAK/STATpathway may be likely relevant in vivo for the treatment of CMLpatients and may significantly increase the rate of MR4.0, whichis the only condition that may prelude the decision of treatmentinterruption.
Conflict of interest statement
All authors declare no conflicts of interest.
Acknowledgements
This work was supported by grants from Regione Campania(DGRC 2362/07), MIUR (PS 35-126/Ind). Ms Rosanna Scala provideddrafts and editorial assistance to the authors during preparation ofthis manuscript. The authors would like to thank Prof L. Del Vec-chio as the Director of Cytometry Facility at CEINGE BiotecnologieAvanzate.
Contributions. C.Q., B.D.A., S.E., S.C., M.R., I.C. and N.E. performedexperiments. C.Q., B.D.A., S.E., S.C. and F.P. designed the researchand analyzed the data. N.P., A.M.R., S.P., M.P., L.L. and F.P. providedpatient’s samples. C.Q., B.D.A., G.M., G.S. and F.P. provided expertisein Molecular Biology and analyzed the data. C.Q., B.D.A. and F.P.wrote the manuscript. All authors reviewed the manuscript.
References
[1] Deininger M, Goldman JM, Hochhaus A, et al. International randomized studyof interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low riskfor progression or events in patients with newly diagnosed chronic myeloidleukemia in chronic phase (CML-CP) treated with imatinib. Blood 2009:1126.
[2] Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS,et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloidleukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood2012;119(February (5)):1123–9.
[3] Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz J, Larson RA, Gattermann N,
et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronicphase after imatinib resistance or intolerance: 24-month follow-up results.Blood 2011;117(January (4)):1141–5.[4] Chomel JC, Bonnet ML, Sorel N, Bertrand A, Meunier MC, Fichelson S, et al.Leukemic stem cell persistence in chronic myeloid leukemia patients with

2 ia Re
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
42 C. Quintarelli et al. / Leukem
sustained undetectable molecular residual disease. Blood 2011;118(September(13)):3657–60.
[5] Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discon-tinuation of imatinib in patients with chronic myeloid leukaemia who havemaintained complete molecular remission for at least 2 years: the prospective,multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010;11(November (11)):1029–35.
[6] Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al.Chronic myeloid leukemia stem cells are not dependent on BCR-ABL kinaseactivity for their survival. Blood 2011;119(February (6)):1501–10.
[7] Esposito N, Colavita I, Quintarelli C, Sica AR, Peluso AL, Luciano L,et al. SHP-1 expression accounts for resistance to imatinib treatmentin Philadelphia chromosome-positive cells derived from patients withchronic myeloid leukemia. Blood 2011;118(September (13)):3634–44.
[8] Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Humanchronic myeloid leukemia stem cells are insensitive to imatinib despite inhibi-tion of BCR-ABL activity. J Clin Invest 2011;121(January (1)):396–409.
[9] Colavita I, Esposito N, Quintarelli C, Nigro E, Pane F, Ruoppolo M, et al. Identifi-cation of Annexin A1 interacting proteins in chronic myeloid leukemia KCL22cells. Proteomics 2013;16(August (13)):2414–8.
10] Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al.Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhi-bition of BCR-ABL. Cancer Cell 2010;18(July (1)):74–87.
11] Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, et al.Microenvironmental protection of CML stem and progenitor cells from tyrosinekinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood2012;121(March (10)):1824–38.
12] Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: hitting amoving target. Cancer Lett 2013;1(September (10)):15–22.
13] Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironmentto leukemogenesis and leukemia progression. Leukemia 2009;23(December(12)):2233–41.
14] Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA.Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrowmicroenvironment model of drug resistance. Mol Cancer Ther 2008;7(October(10)):3169–75.
15] Nair RR, Tolentino JH, Argilagos RF, Zhang L, Pinilla-Ibarz J, Hazlehurst LA.Potentiation of nilotinib-mediated cell death in the context of the bone mar-row microenvironment requires a promiscuous JAK inhibitor in CML. Leuk Res2012;36(June (6)):756–63.
16] Traer E, MacKenzie R, Snead J, Agarwal A, Eiring AM, O’Hare T, et al. Blockadeof JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to
ABL inhibitors. Leukemia 2012;26(May (5)):1140–3.17] Quintarelli C, Dotti G, De Angelis B, Hoyos V, Mims M, Luciano L, et al. CytotoxicT lymphocytes directed to the preferentially expressed antigen of melanoma(PRAME) target chronic myeloid leukemia. Blood 2008;112(September(5)):1876–85.
[
search 38 (2014) 236– 242
18] Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF,et al. European LeukemiaNet recommendations for the management of chronicmyeloid leukemia. Blood 2013;122(August (6)):872–84.
19] Pane F, Cimino G, Izzo B, Camera A, Vitale A, Quintarelli C, et al. Significantreduction of the hybrid BCR/ABL transcripts after induction and consolidationtherapy is a powerful predictor of treatment response in adult Philadelphia-positive acute lymphoblastic leukemia. Leukemia 2005;19(April (4)):628–35.
20] Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L,et al. Primitive, quiescent, Philadelphia-positive stem cells from patientswith chronic myeloid leukemia are insensitive to STI571 in vitro. Blood2002;99(January (1)):319–25.
21] Soverini S, Rosti G, Iacobucci I, Baccarani M, Martinelli G. Choosing the bestsecond-line tyrosine kinase inhibitor in imatinib-resistant chronic myeloidleukemia patients harboring BCR-ABL kinase domain mutations: how reliableis the IC(5)(0)? Oncologist 2011;16(6):868–76.
22] Quintarelli C, Dotti G, Hasan ST, De Angelis B, Hoyos V, Errichiello S, et al.High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived pep-tide can target leukemic and leukemic-precursor cells. Blood 2011;117(March(12)):3353–62.
23] Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, et al. Syner-gistic drug combinations tend to improve therapeutically relevant selectivity.Nat Biotechnol 2009;27(July (7)):659–66.
24] Chou TC, Talalay P. Quantitative analysis of dose–effect relationships: thecombined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul1984;22:27–55.
25] Chou TC. Drug combination studies and their synergy quantification using theChou–Talalay method. Cancer Res 2010;70(January (2)):440–6.
26] Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinibversus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med2010;362(June (24)):2251–9.
27] Larson RA, Hochhaus A, Hughes TP, Clark RE, Etienne G, Kim DW, et al. Nilotinibvs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-yearfollow-up. Leukemia 2012;26(October (10)):2197–203.
28] Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, et al. Persistenceof leukemia stem cells in chronic myelogenous leukemia patients in prolongedremission with imatinib treatment. Blood 2011;118(November (20)):5565–72.
29] Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, et al. Molecu-lar and chromosomal mechanisms of resistance to imatinib (STI571) therapy.Leukemia 2002;16(November (11)):2190–6.
30] Cortes JE, Kim DW, Kantarjian HM, Brummendorf TH, Dyagil I, GriskeviciusL, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic
myeloid leukemia: results from the BELA trial. J Clin Oncol 2012;30(October(28)):3486–92.31] Cortes JE, Jones D, O’Brien S, Jabbour E, Ravandi F, Koller C, et al. Results of dasa-tinib therapy in patients with early chronic-phase chronic myeloid leukemia. JClin Oncol 2010;28(January (3)):398–404.


















![Ruxolitinib - Elsevier...ruxolitinib arm occurred in 85.7% of patients in the best standard therapy arm at or after week 32.[58808] In an extended analysis at week 80, 76% of patients](https://img.pdfslide.us/doc/110x75/6112927320cf746dca4de4fd/ruxolitinib-elsevier-ruxolitinib-arm-occurred-in-857-of-patients-in-the.jpg)




![[alumni]SG InTouch Aug2016 - deloitte.com quality time with our loved ... us with his Chinese rendition of Jay Chou’s Jian Dan Ai and Nathan, ... keeping the score close](https://img.pdfslide.us/doc/110x75/5aa3d9fd7f8b9a07758eccb3/alumnisg-intouch-aug2016-quality-time-with-our-loved-us-with-his-chinese.jpg)





