Embed Size (px)
Citation preview

Selected papers from Journal of Computational Chemistry
February, 2013 – July, 2013
Copyright © 2013 Wiley Periodicals, Inc., A Wiley Company
Edited By: Charles L. Brooks III, Masahiro Ehara, GernotFrenking, and Peter R. Schreiner
Impact Factor: 4.583
ISI Journal Citation Reports © Ranking: 2011: 26/154 (Chemistry Multidisciplinary)
Tibor, again.
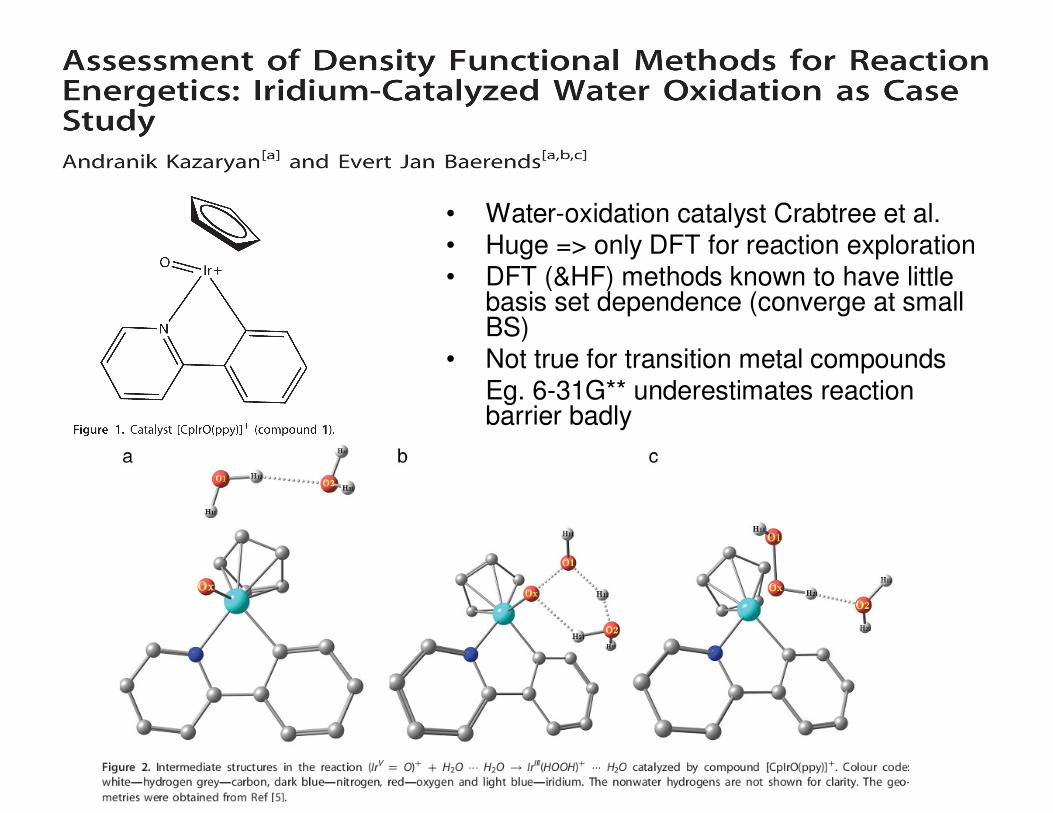
• Water-oxidation catalyst Crabtree et al.• Huge => only DFT for reaction exploration• DFT (&HF) methods known to have little
basis set dependence (converge at small BS)
• Not true for transition metal compoundsEg. 6-31G** underestimates reaction barrier badly

• Reaction
– reactant complex(RC)
– transition state(TS)
– troduct complex(PC)
• Basis set
– Slater type orbital (STO)
– Gaussian type orbital (GTO)
• Methods
– CCSD on a simpler model compound to have a reference
– Various DFT methods: with Grimme’s dispersion correction (-D3) and without
• Codes
– for GTOs:Turbomole, NWChem
– For STOs:ADF2012
• Basis set functions: radial*angular
– radial:spherical harmonics
• Slater type orbitals (STO)
– H-atom like orbitals
– No radial nodes
– Exponential decay at long range
– Kato’s cusp condition at short range
– Computationally difficult->overlap int.
• Gaussian type orbitals(GTO)
– Easier to calculate
– problem: no cusp=> we need more of
them to approximate reality
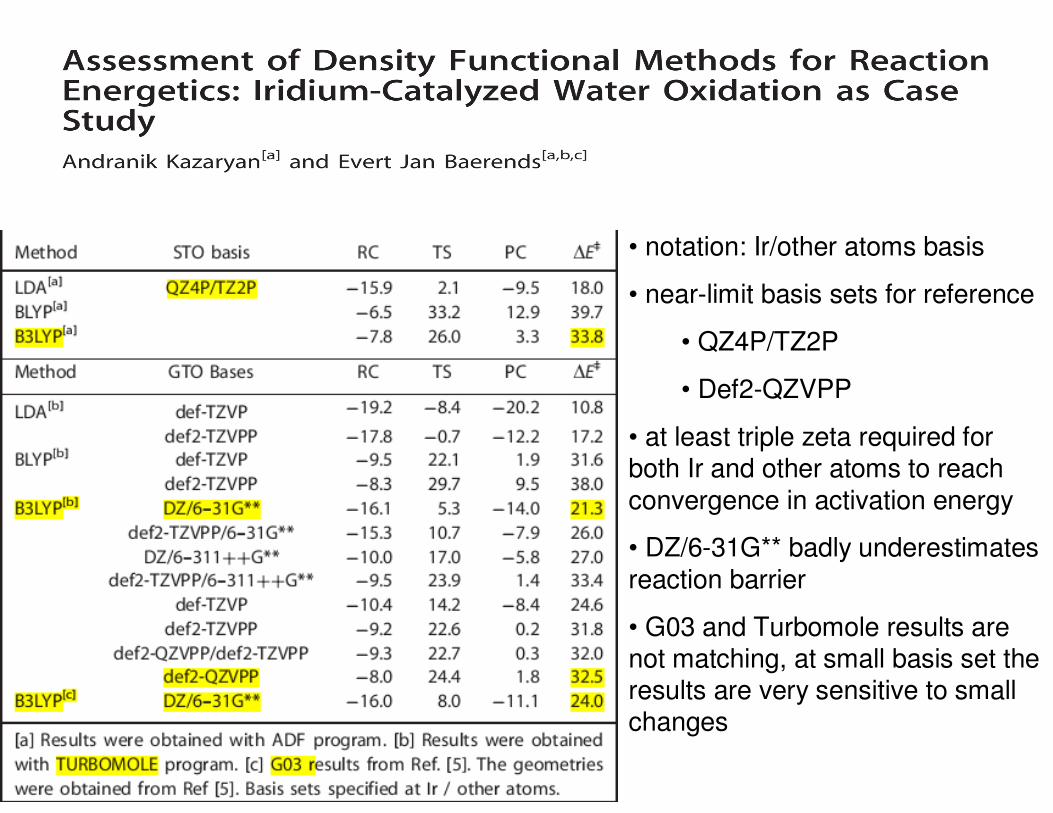
• notation: Ir/other atoms basis
• near-limit basis sets for reference
• QZ4P/TZ2P
• Def2-QZVPP
• at least triple zeta required for both Ir and other atoms to reach
convergence in activation energy
• DZ/6-31G** badly underestimates
reaction barrier
• G03 and Turbomole results are not matching, at small basis set the results are very sensitive to small changes
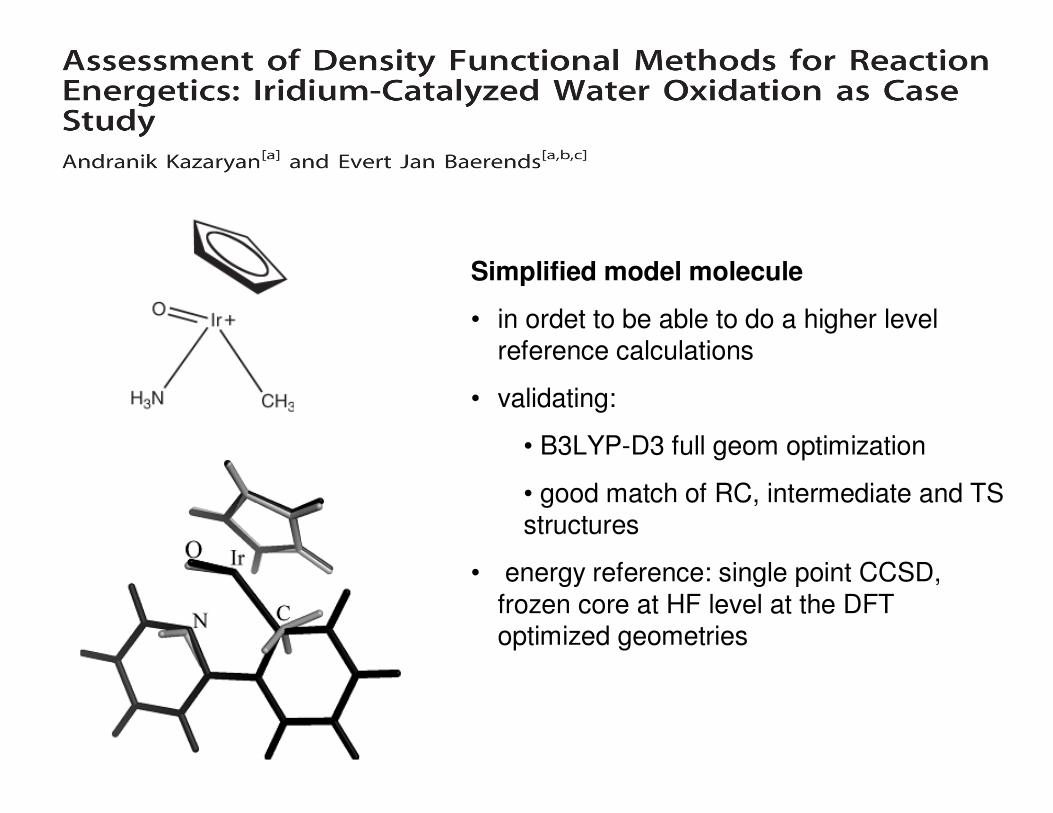
Simplified model molecule
• in ordet to be able to do a higher level reference calculations
• validating:
• B3LYP-D3 full geom optimization
• good match of RC, intermediate and TS structures
• energy reference: single point CCSD,
frozen core at HF level at the DFT optimized geometries
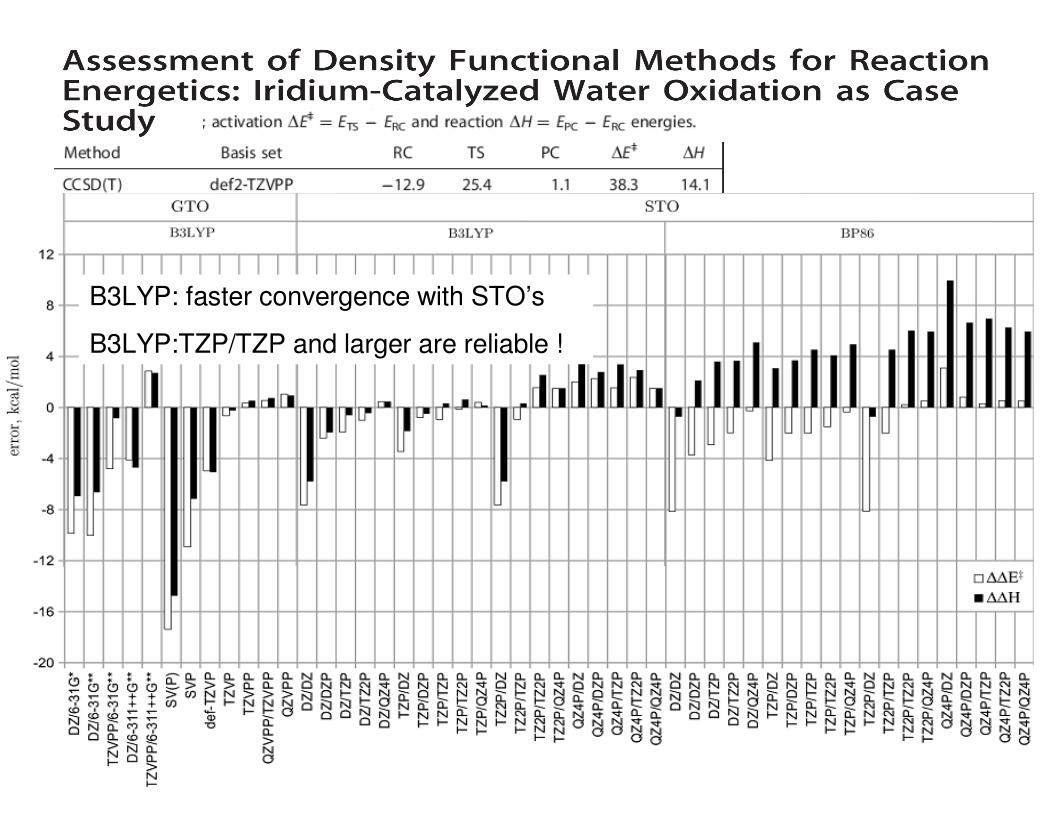
B3LYP: faster convergence with STO’s
B3LYP:TZP/TZP and larger are reliable !
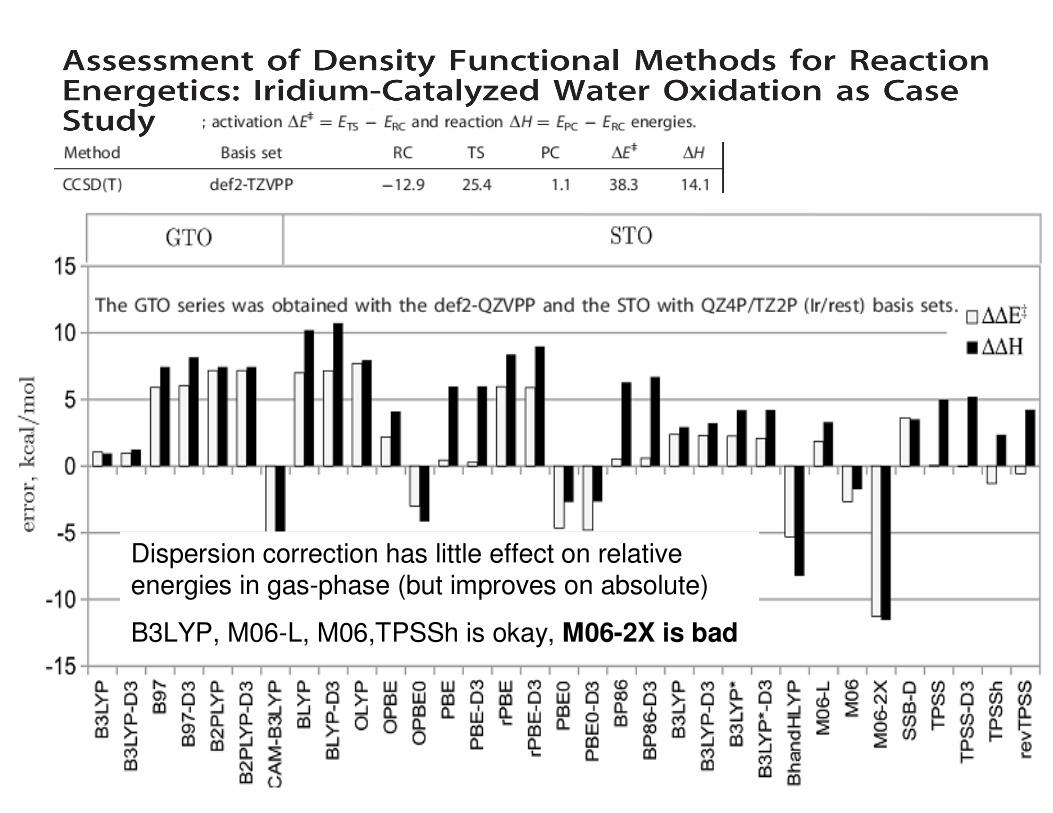
Dispersion correction has little effect on relative energies in gas-phase (but improves on absolute)
B3LYP, M06-L, M06,TPSSh is okay, M06-2X is bad

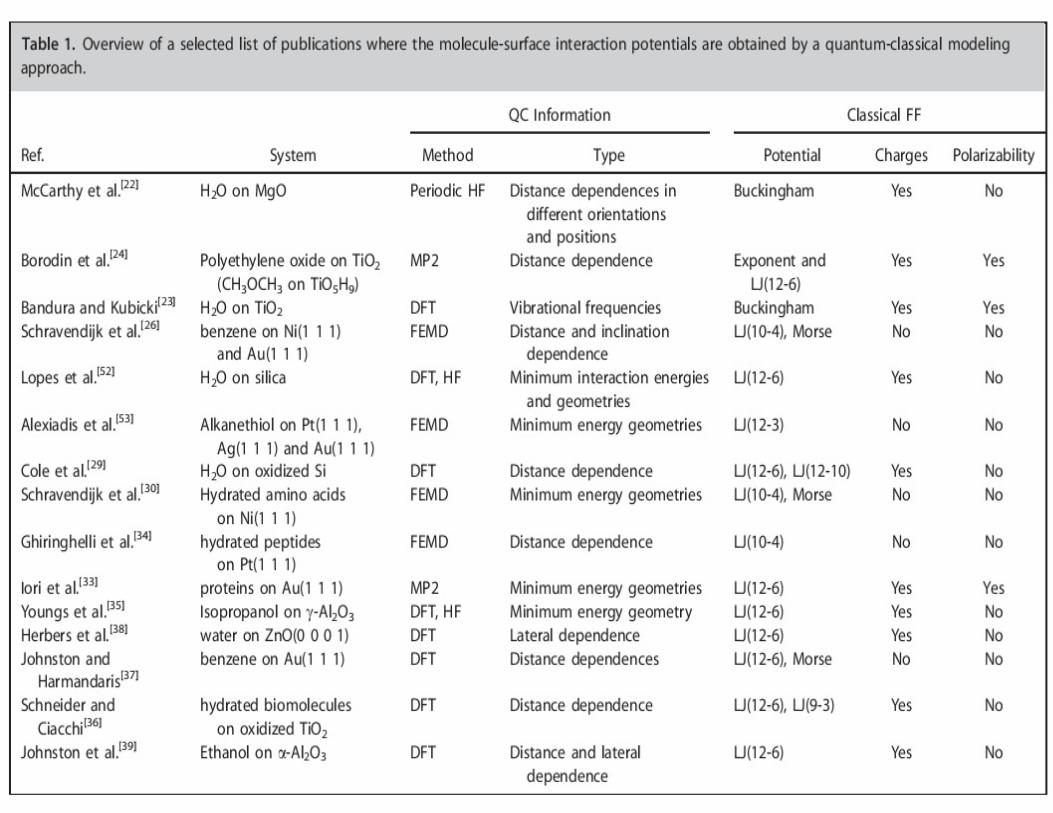
• Absorption of small and macromolecules on surfaces is very important
• Commonly used force-fields are parameterized not forinterfacial systems, but for bulk liquid and solid phases
• Dual-scale modelling approach1.Quantum chemical calculations
2. force field parameterisation and classical MD
– Surface site specific absorption energies
– Extensive sampling of near-surface conformations:
• Not done in the past
• Distance, position, orientation of the molecule sampling=> reliable FF
• Phase-space exploration and fitting strategies to obtain representative and reliable force field are recommended.
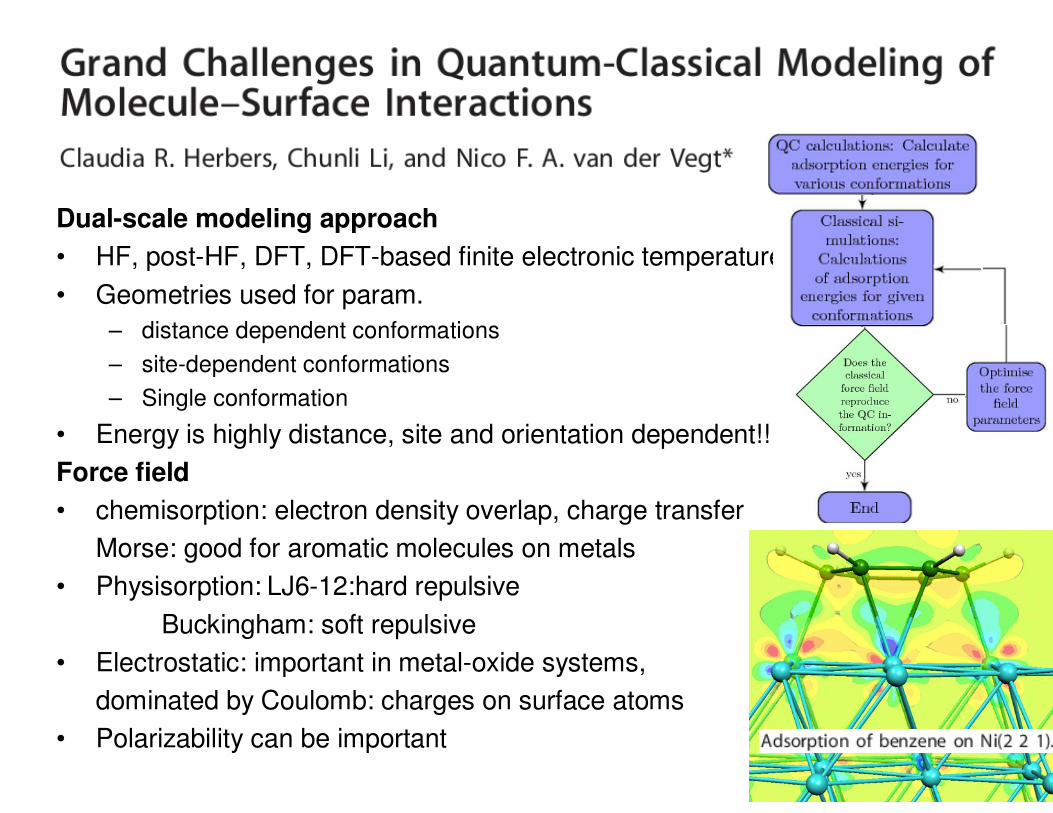
Dual-scale modeling approach
• HF, post-HF, DFT, DFT-based finite electronic temperature MD
• Geometries used for param.
– distance dependent conformations
– site-dependent conformations
– Single conformation
• Energy is highly distance, site and orientation dependent!!
Force field
• chemisorption: electron density overlap, charge transfer
Morse: good for aromatic molecules on metals
• Physisorption: LJ6-12:hard repulsive
Buckingham: soft repulsive
• Electrostatic: important in metal-oxide systems,
dominated by Coulomb: charges on surface atoms
• Polarizability can be important

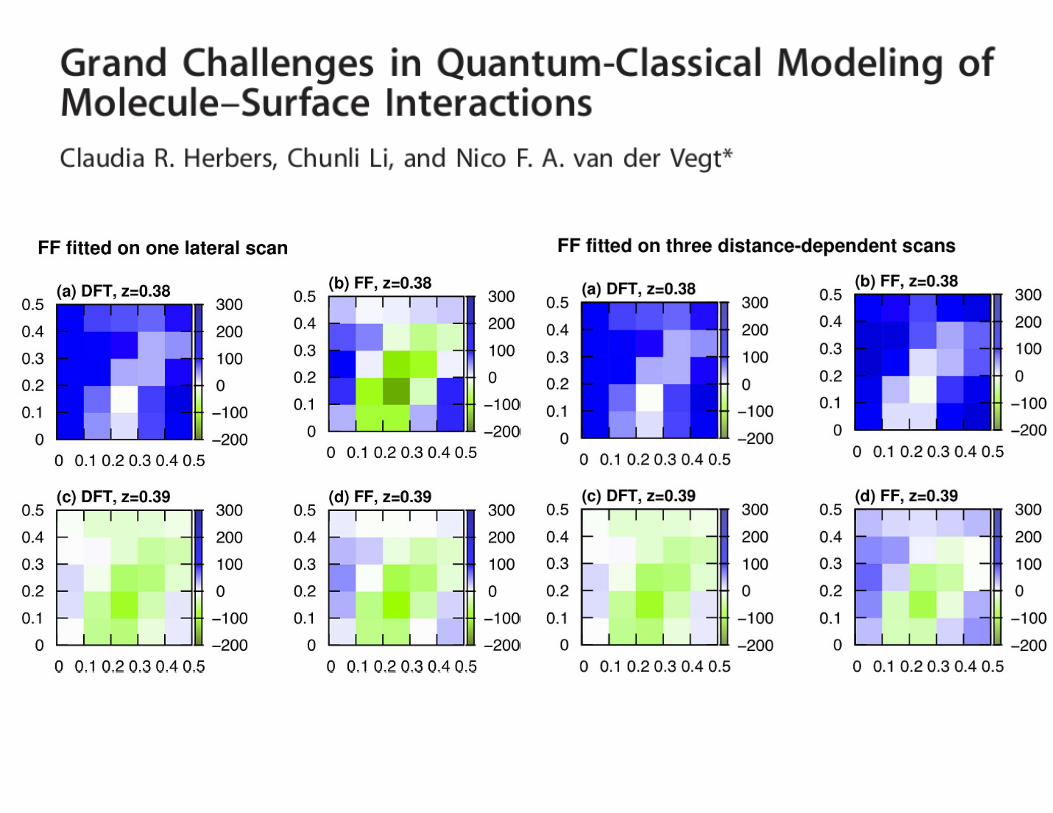
• QC calculation is expensive, we need small, but represen-tative sampling to reconstruct the adsorption landscape
• Vertical, lateral scans
• which scan is more important depends on the system and aim.
• balanced mixture of low- and high-energy conformations are needed
• distance dependence of the adsorption energy, and the molecule’s position and orientation with respect to the surface are also important.
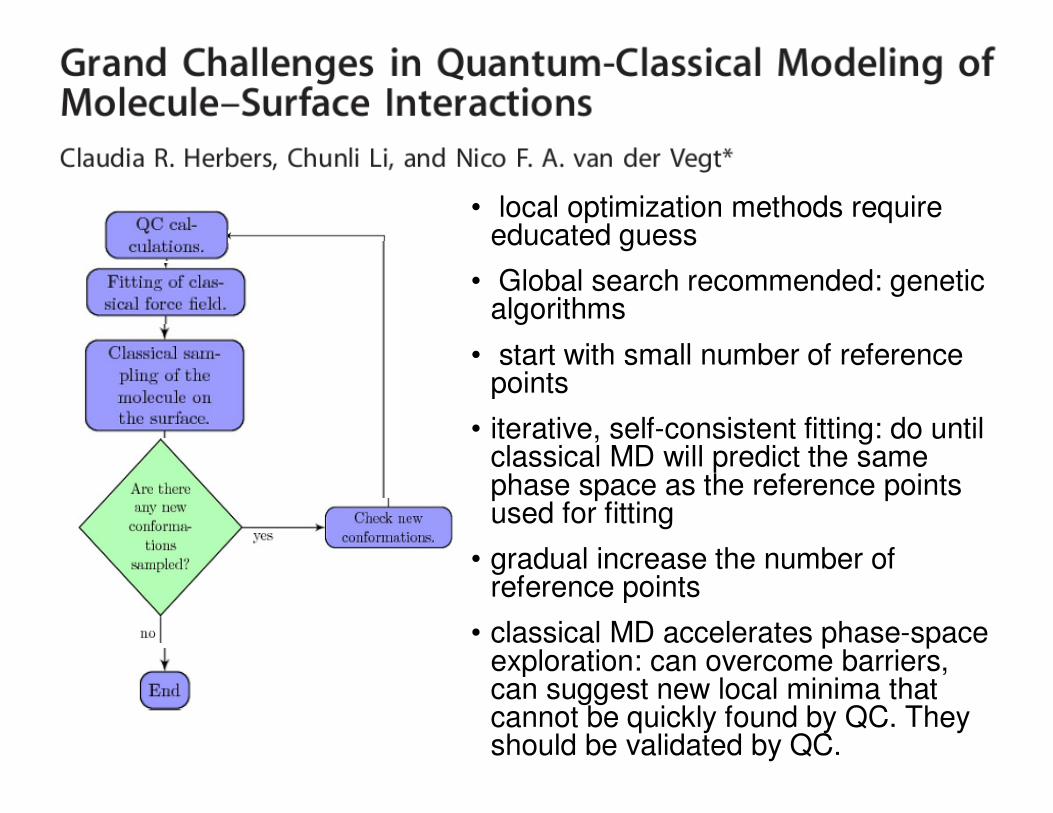
• local optimization methods require educated guess
• Global search recommended: genetic algorithms
• start with small number of reference points
• iterative, self-consistent fitting: do until classical MD will predict the same phase space as the reference points used for fitting
• gradual increase the number of reference points
• classical MD accelerates phase-space exploration: can overcome barriers, can suggest new local minima that cannot be quickly found by QC. They should be validated by QC.

• ReaxFF force field
– Bond, lone-pair, over and under-coordination of atoms with respect to their valency, valence angle (3body), torsion, penalty energy to stabilize 3body system with center atoms with 2 double bonds, conjugated double bonds( 1=2-3=4) 3 and 4 body conjugation terms,H-bond.
• vdW interaction=distance corrected Morse
– Dij:interaction E, rvdw::vdW radius
+empirical parameters
– f13: Shielding to reduce 1-3 repulsion
• Shielded Coulomb interaction, polarization
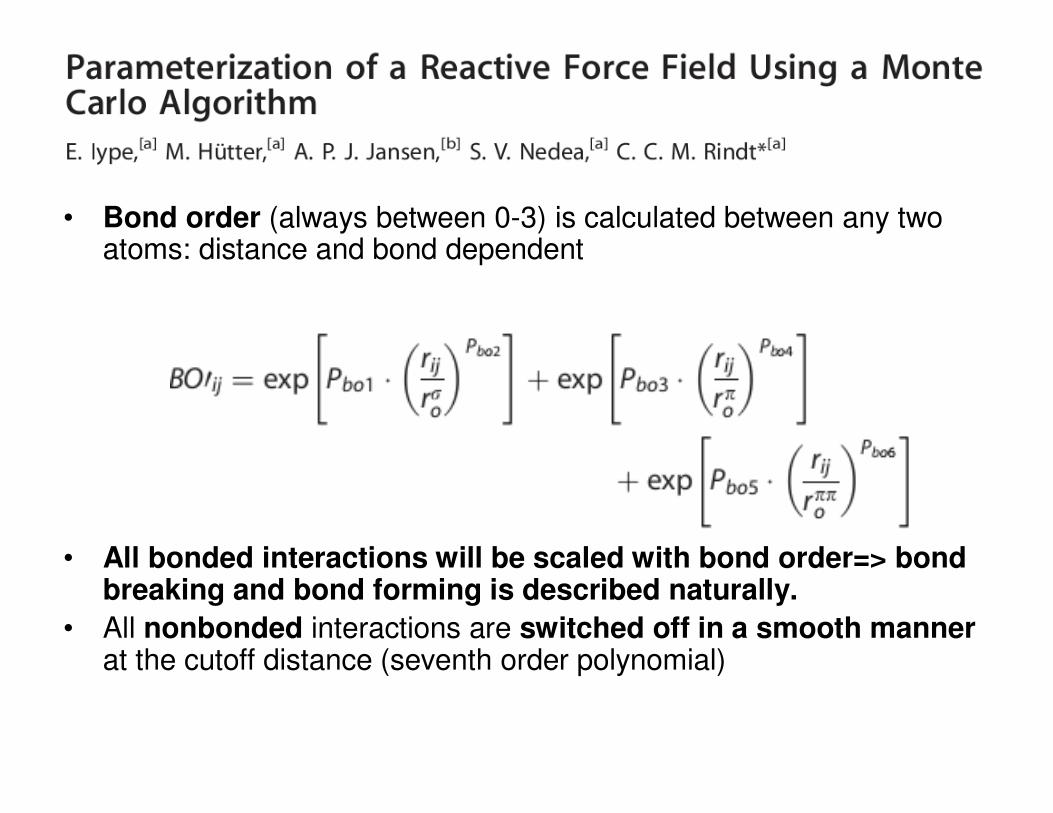
• Bond order (always between 0-3) is calculated between any two atoms: distance and bond dependent
• All bonded interactions will be scaled with bond order=> bond breaking and bond forming is described naturally.
• All nonbonded interactions are switched off in a smooth mannerat the cutoff distance (seventh order polynomial)

• ReaxFF: ~100 parameters/atom: • many of them with no physical meaning: no educated guess can be
made on initial values=> local search is bad• Training set: bond lengths, angles, torsion angles, potential energy
changes, heat of formations, etc
• The σ values used to scale different types of errors.• Monte Carlo Metropolis (MMC) – Simulated annealing: GLOBAL
– random changes in parameters with probabilistic acceptance (Pa)– starting at high temperature and slowly reducing it
• Comparing with parabolic search algorithm: LOCAL– One parameter optimized at a time by taking 3 values of it and fitting a
parabola to the errors.– Parabola min position is estimated, or if it is ∩ than the lowest of three
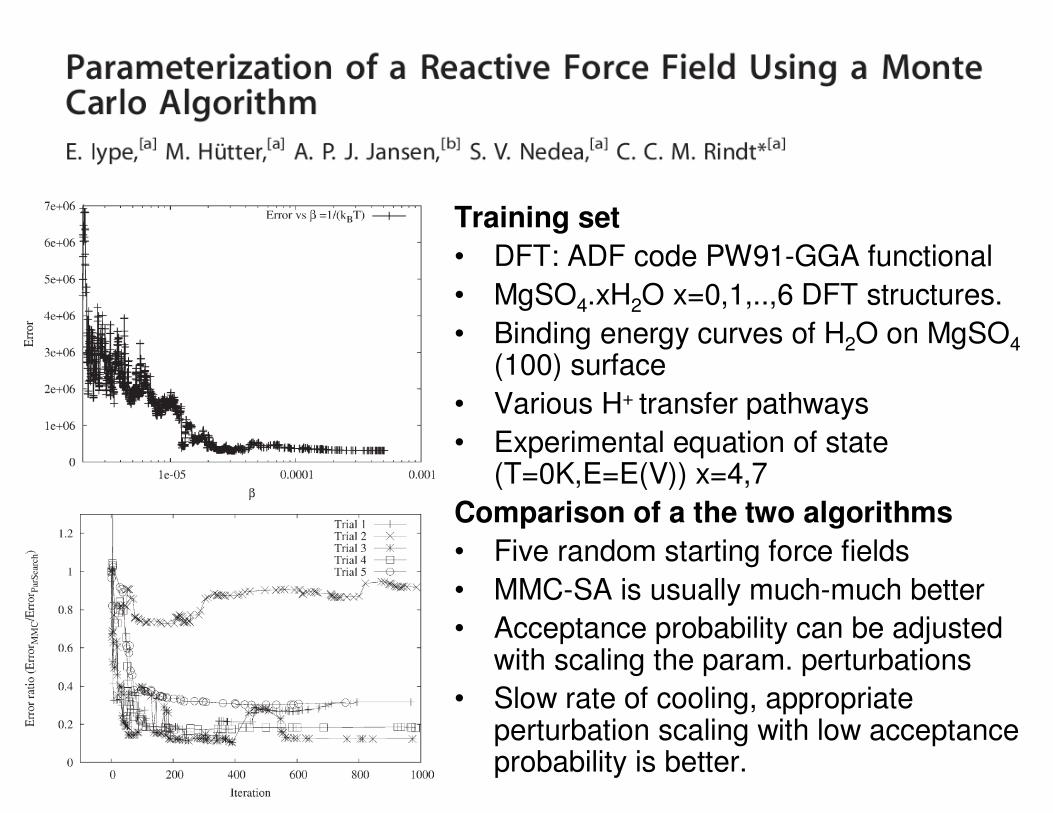
Training set
• DFT: ADF code PW91-GGA functional
• MgSO4.xH2O x=0,1,..,6 DFT structures.
• Binding energy curves of H2O on MgSO4
(100) surface
• Various H+ transfer pathways
• Experimental equation of state(T=0K,E=E(V)) x=4,7
Comparison of a the two algorithms
• Five random starting force fields
• MMC-SA is usually much-much better
• Acceptance probability can be adjustedwith scaling the param. perturbations
• Slow rate of cooling, appropriateperturbation scaling with low acceptanceprobability is better.

Other interesting papers





























