Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/susc
Surface Science 601 (2007) 5332–5339
Segregation at the surface of an Au/Pd alloy exposed to CO
Vıctor Soto-Verdugo a, Horia Metiu b,*
a Department of Physics, University of California, Santa Barbara, CA 93106-9510, United Statesb Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106-9510, United States
Received 18 June 2007; accepted for publication 14 August 2007Available online 8 September 2007
Abstract
We use density functional theory (DFT) with the generalized gradient approximation (GGA) and the revised Perdew–Burke–Ernzer-hoff (rPBE) functional, to study the surface composition of the (111) and (100) dilute Pd/Au alloy. We find that the energy of Pd atomsis lower when they substitute an Au atom in the bulk than when they substitute an Au atom in the surface layer, or when they areadsorbed on the surface. Whether they are in the surface layer or in the bulk, the Pd atoms interact very weakly with each other. COadsorbs on the Pd atom in the surface layer and the energy of this complex is lower than that of CO in gas and Pd atom in the bulk.The interaction between the PdCO complexes formed when CO adsorbs on a Pd atom imbedded in the surface layer, is also negligible.We use these energies, equilibrium thermodynamics, and a simple lattice–gas model to examine the equilibrium composition of the sur-face layer, as a function of temperature, CO pressure and the Pd/Au ratio. We find that the surface Pd concentration for a nanoparticleof an Au/Pd alloy differs from that in a bulk sample. The difference is due mainly to the fact that in a nanoparticle the migration of Pdatoms to the surface depletes the bulk concentration while in a large sample; the bulk provides an infinite source of Pd atoms to populatethe surface sites. This system is of interest because Pd/Au alloys are selective catalysts for vinyl acetate synthesis when the Pd concen-tration on the surface is very low.� 2007 Elsevier B.V. All rights reserved.
Keywords: Catalysis; Chemisorption; Alloys; Surface segregation; Vinyl acetate
1. Introduction
The surface composition of Au/Pd alloys has been stud-ied by a variety of techniques which indicate that the sur-face concentration of Au is greater than its concentrationin the bulk. Auger electron spectroscopy (AES) determined(from the ratios of the Pd (330 eV) to Au (71 eV) peaks)that the surface of polycrystalline alloy foils [1] and of va-por-deposited thin films [2] has a higher concentration ofAu than the bulk. Au enrichment in the surface of poly-crystalline Au/Pd alloys was also found by ion scatteringspectroscopy (ISS) [3]. LEED and ISS measurements [4,5]on Au3Pd(110) single crystal surfaces found that the upperlayer was pure Au, a result confirmed by scanning tunnel-ing microscopy (STM) studies [6]. Moreover, after heating
0039-6028/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.susc.2007.08.022
* Corresponding author. Tel.: +1 805 893 2256; fax: +1 805 893 4120.E-mail address: [email protected] (H. Metiu).
to 875 K, the surface of the Au3Pd(1 00) becomes pure,unreconstructed Au(1 00). AES, LEISS, X-ray photoelec-tron spectroscopy (XPS), and LEED measurements [7] ofultra-thin films of Pd on Au(1 11), found that a Pd/Au al-loy is formed if the substrate temperature is above 300 K.Goodman et al. [8] prepared Au/Pd alloy films on aMo(110) substrate, by using vapor deposition. The Pd sur-face concentration was determined using ISS, XPS, infra-red (IR) spectroscopy and thermal desorption (TPD) ofadsorbed CO. They too found that the surface is rich inAu.
Pd catalyzes the formation of vinyl acetate from aceticacid, ethylene and oxygen and alloying with Au increasesselectivity. Goodman’s group studied this process exten-sively [9–12] and found that selectivity is highest whenthe amount of Pd in the alloy is small. They proposed thatthe reaction center for the formation of the vinyl acetateconsists of two non-contiguous Pd atoms. They explained
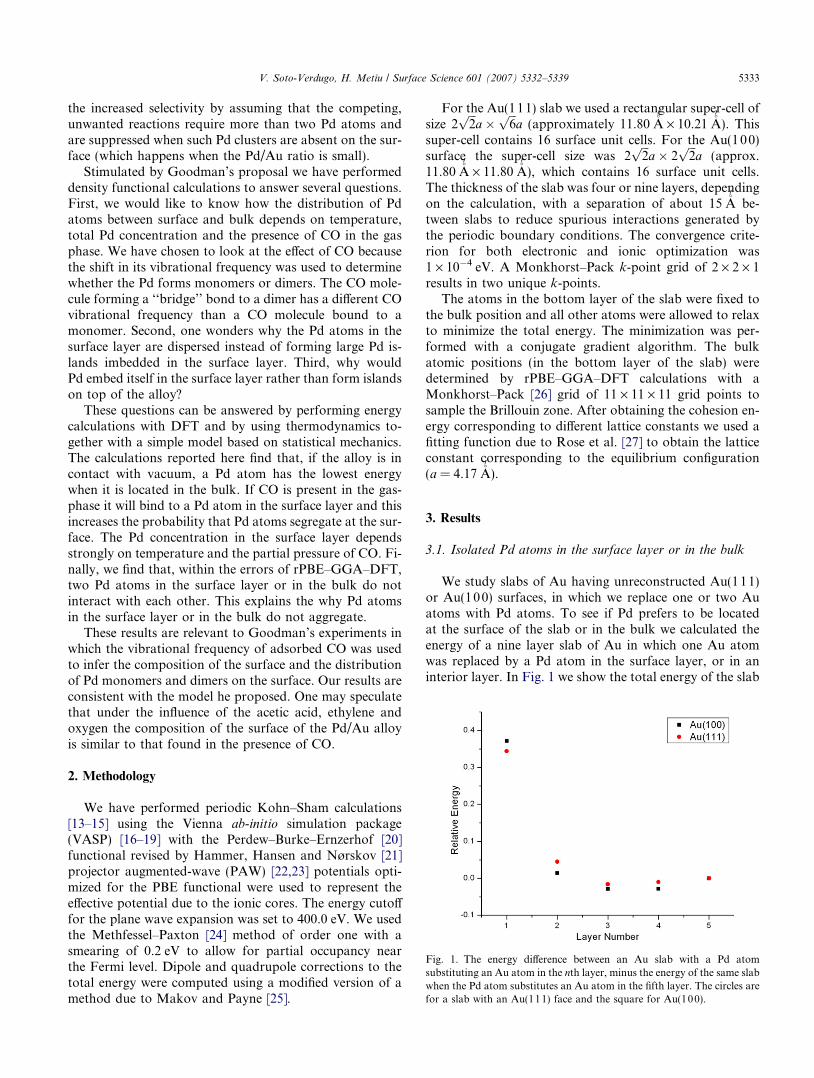
Fig. 1. The energy difference between an Au slab with a Pd atomsubstituting an Au atom in the nth layer, minus the energy of the same slabwhen the Pd atom substitutes an Au atom in the fifth layer. The circles arefor a slab with an Au(111) face and the square for Au(100).
V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339 5333
the increased selectivity by assuming that the competing,unwanted reactions require more than two Pd atoms andare suppressed when such Pd clusters are absent on the sur-face (which happens when the Pd/Au ratio is small).
Stimulated by Goodman’s proposal we have performeddensity functional calculations to answer several questions.First, we would like to know how the distribution of Pdatoms between surface and bulk depends on temperature,total Pd concentration and the presence of CO in the gasphase. We have chosen to look at the effect of CO becausethe shift in its vibrational frequency was used to determinewhether the Pd forms monomers or dimers. The CO mole-cule forming a ‘‘bridge’’ bond to a dimer has a different COvibrational frequency than a CO molecule bound to amonomer. Second, one wonders why the Pd atoms in thesurface layer are dispersed instead of forming large Pd is-lands imbedded in the surface layer. Third, why wouldPd embed itself in the surface layer rather than form islandson top of the alloy?
These questions can be answered by performing energycalculations with DFT and by using thermodynamics to-gether with a simple model based on statistical mechanics.The calculations reported here find that, if the alloy is incontact with vacuum, a Pd atom has the lowest energywhen it is located in the bulk. If CO is present in the gas-phase it will bind to a Pd atom in the surface layer and thisincreases the probability that Pd atoms segregate at the sur-face. The Pd concentration in the surface layer dependsstrongly on temperature and the partial pressure of CO. Fi-nally, we find that, within the errors of rPBE–GGA–DFT,two Pd atoms in the surface layer or in the bulk do notinteract with each other. This explains the why Pd atomsin the surface layer or in the bulk do not aggregate.
These results are relevant to Goodman’s experiments inwhich the vibrational frequency of adsorbed CO was usedto infer the composition of the surface and the distributionof Pd monomers and dimers on the surface. Our results areconsistent with the model he proposed. One may speculatethat under the influence of the acetic acid, ethylene andoxygen the composition of the surface of the Pd/Au alloyis similar to that found in the presence of CO.
2. Methodology
We have performed periodic Kohn–Sham calculations[13–15] using the Vienna ab-initio simulation package(VASP) [16–19] with the Perdew–Burke–Ernzerhof [20]functional revised by Hammer, Hansen and Nørskov [21]projector augmented-wave (PAW) [22,23] potentials opti-mized for the PBE functional were used to represent theeffective potential due to the ionic cores. The energy cutofffor the plane wave expansion was set to 400.0 eV. We usedthe Methfessel–Paxton [24] method of order one with asmearing of 0.2 eV to allow for partial occupancy nearthe Fermi level. Dipole and quadrupole corrections to thetotal energy were computed using a modified version of amethod due to Makov and Payne [25].
For the Au(1 11) slab we used a rectangular super-cell ofsize 2
ffiffiffi2p
a�ffiffiffi6p
a (approximately 11.80 �A0
· 10.21 �A0). This
super-cell contains 16 surface unit cells. For the Au(1 00)surface the super-cell size was 2
ffiffiffi2p
a� 2ffiffiffi2p
a (approx.11.80 �A
0· 11.80 �A
0), which contains 16 surface unit cells.
The thickness of the slab was four or nine layers, dependingon the calculation, with a separation of about 15 �A
0be-
tween slabs to reduce spurious interactions generated bythe periodic boundary conditions. The convergence crite-rion for both electronic and ionic optimization was1 · 10�4 eV. A Monkhorst–Pack k-point grid of 2 · 2 · 1results in two unique k-points.
The atoms in the bottom layer of the slab were fixed tothe bulk position and all other atoms were allowed to relaxto minimize the total energy. The minimization was per-formed with a conjugate gradient algorithm. The bulkatomic positions (in the bottom layer of the slab) weredetermined by rPBE–GGA–DFT calculations with aMonkhorst–Pack [26] grid of 11 · 11 · 11 grid points tosample the Brillouin zone. After obtaining the cohesion en-ergy corresponding to different lattice constants we used afitting function due to Rose et al. [27] to obtain the latticeconstant corresponding to the equilibrium configuration(a = 4.17 �A
0).
3. Results
3.1. Isolated Pd atoms in the surface layer or in the bulk
We study slabs of Au having unreconstructed Au(1 11)or Au(10 0) surfaces, in which we replace one or two Auatoms with Pd atoms. To see if Pd prefers to be locatedat the surface of the slab or in the bulk we calculated theenergy of a nine layer slab of Au in which one Au atomwas replaced by a Pd atom in the surface layer, or in aninterior layer. In Fig. 1 we show the total energy of the slab

Fig. 2. Relative energies of different configurations of two Pd atoms substituting Au atoms in the two top layers of the Au(111) surface. The energies arewith respect to the lowest energy configuration, configuration k where the Pd atoms are furthest apart.
5334 V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339
with the Pd atom in the nth layer minus the total energy ofthe slab when the Pd atom was in the fifth layer below thesurface, as a function of n. The energy is highest when thePd atom is in the surface layer; as far as energy is con-cerned, the Pd atoms prefer to be located in the bulk. En-tropy will enhance this tendency since our ability to guessthe position of the Pd atom when it is in the bulk is lowerthan when it is on the surface; thus there is less informationabout the location of Pd when it is in the bulk, hence more‘‘disorder’’, higher entropy, lower free energy [28].
Besides replacing an Au atom in the surface layer or inthe bulk, the Pd atom may also sit on top of the Au surface.The energy required for removing the Pd atom from thesurface layer onto the surface and leaving a gold vacancybehind is 1.33 eV for the (111) surface. Within the errorsof rPBE–GGA–DFT this energy is independent of the dis-tance between the Pd atom on top of the surface and theAu vacancy. The probability that Pd will leave a substitu-tional site in the surface layer to move onto the surface isvery low.
3.2. Pd–Pd interaction
To find whether the Pd atoms in the alloy have a ten-dency to form Pd clusters we have calculated the energyof two Pd atoms, each replacing a gold atom at differentlattice sites. These sites can be in the top surface layer orin the layer immediately beneath. The position of the Pd
atoms in the slab and the energies of the slabs are shownin Fig. 2 (for the (111) surface) and Fig. 3 (for the (100)surface). The zero of energy is the energy of the slab withthe two Pd atoms in the bulk, separated by a large distance(Fig. 2k, for the (111) slab, and Fig. 3m, for the (100)slab). It is difficult to estimate the errors of rPBE–GGA–DFT but they are certainly larger than the interactionenergies shown in Fig. 2. Therefore, within the errors ofrPBE–GGA–DFT, there is no interaction between thetwo Pd atoms. For example, Fig. 2a–d show that the en-ergy of two Pd atoms in the surface layer is independentof the distance between them. The same story is told byFig. 2e–g in which one atom is in the surface layer andthe other in the layer underneath. The interaction energyis negligible for all pairings of two Pd atoms.
The lack of interaction between the Pd atoms explainswhy they do not form large Pd clusters, except perhapsby accident. This result supports Goodman’s conjecturethat the system he was working had with monomers ornon-contiguous dimers (formed accidentally) and give cre-dence to the assumption that this may be responsible forthe increased selectivity of the alloys having a low Pdconcentration.
3.3. The effect of CO on Pd location
We have calculated the adsorption energy of CO on avariety of Pd/Au alloy slabs (see Figs. 4–7). In Fig. 4 we
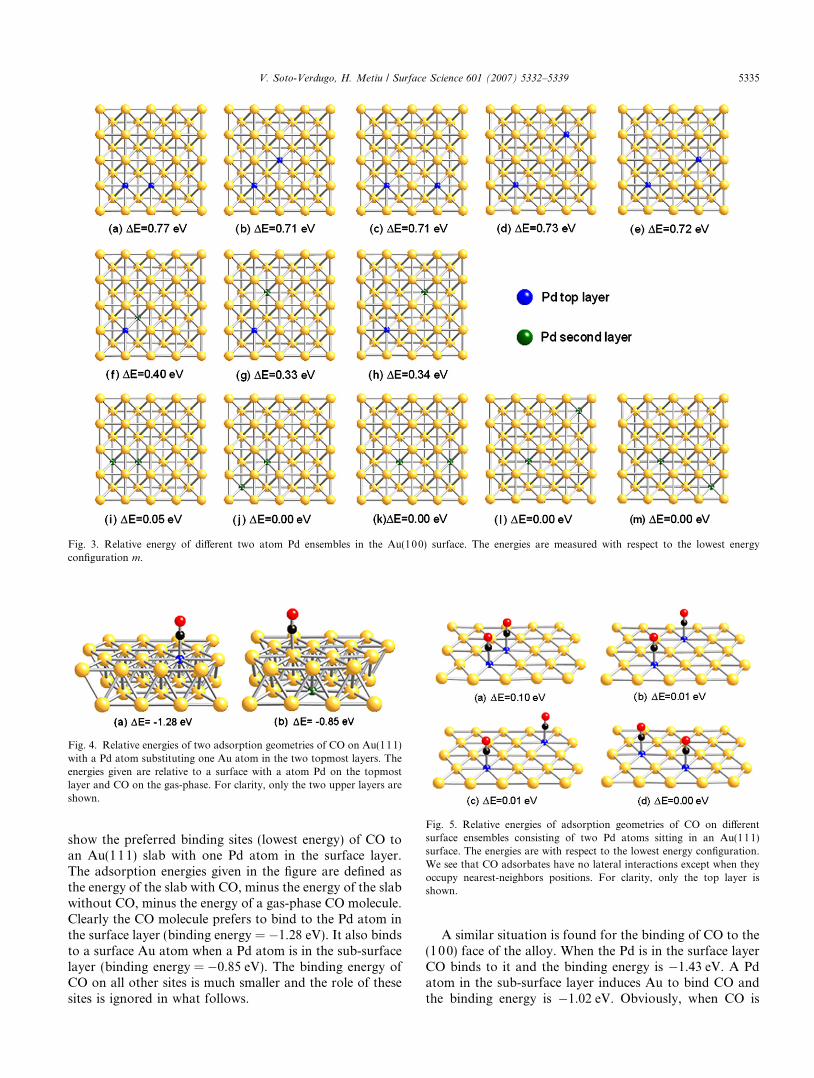
Fig. 3. Relative energy of different two atom Pd ensembles in the Au(100) surface. The energies are measured with respect to the lowest energyconfiguration m.
Fig. 4. Relative energies of two adsorption geometries of CO on Au(111)with a Pd atom substituting one Au atom in the two topmost layers. Theenergies given are relative to a surface with a atom Pd on the topmostlayer and CO on the gas-phase. For clarity, only the two upper layers areshown.
Fig. 5. Relative energies of adsorption geometries of CO on differentsurface ensembles consisting of two Pd atoms sitting in an Au(111)surface. The energies are with respect to the lowest energy configuration.We see that CO adsorbates have no lateral interactions except when theyoccupy nearest-neighbors positions. For clarity, only the top layer isshown.
V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339 5335
show the preferred binding sites (lowest energy) of CO toan Au(111) slab with one Pd atom in the surface layer.The adsorption energies given in the figure are defined asthe energy of the slab with CO, minus the energy of the slabwithout CO, minus the energy of a gas-phase CO molecule.Clearly the CO molecule prefers to bind to the Pd atom inthe surface layer (binding energy = �1.28 eV). It also bindsto a surface Au atom when a Pd atom is in the sub-surfacelayer (binding energy = �0.85 eV). The binding energy ofCO on all other sites is much smaller and the role of thesesites is ignored in what follows.
A similar situation is found for the binding of CO to the(100) face of the alloy. When the Pd is in the surface layerCO binds to it and the binding energy is �1.43 eV. A Pdatom in the sub-surface layer induces Au to bind CO andthe binding energy is �1.02 eV. Obviously, when CO is

Fig. 6. Relative energies of two adsorption geometries of CO on Au(100)with a Pd atom substituting one Au atom in the two topmost layers. Theenergies given are relative to a surface with a atom Pd on the topmostlayer and CO on the gas-phase. For clarity, only the two topmost layersare shown.
5336 V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339
present in the gas-phase it will induce an accumulation ofPd atoms in the surface layer. CO can also adsorb in abridge configuration between two neighboring Pd atoms.This configuration has an adsorption energy of �1.34 eV.
Figs. 5 and 7 show that the energy of two Pd atoms,each having a CO molecule adsorbed on it, is independentof the Pd–Pd distance. This lack of interaction means thatthe Pd atoms with CO on them are randomly distributedon the surface lattice sites. CO adsorption will induce anenrichment of Pd in the surface layer but it will not causethe Pd atoms to form clusters.
We have also calculated the energy of CO adsorbed on aPd atom located on the top of an Au slab which has a goldvacancy on the surface, and found it to be 0.97 eV higherthan the energy of CO adsorbed on a Pd atom imbeddedin the surface layer. We conclude that the presence of COin the gas-phase will not induce the Pd atoms to leave theirposition in the surface layer and move to the surface.
4. A lattice–gas model for the distribution of the Pd atoms
between the surface layer and the interior of the slab
One often defines the probability that the Pd atom is atthe surface or in the bulk by using a Boltzmann factor. This
Fig. 7. Relative energies of adsorption geometries of CO on different surfaceenergies are with respect to the lowest energy configuration. For clarity, only
is correct at zero temperature and might be a useful quali-tative criterion at higher temperature. However, thisapproximation does not take into account the effect of con-figurational entropy and the effect of the partial pressure ofCO in the gas. Obviously the latter is an important factor,if there is no CO in the gas Pd prefers to be in the bulk,while in the presence of the gas it will tend to accumulatein the surface layer.
Given the fact that we can neglect Pd–Pd interactions itis easy to use thermodynamics and statistical mechanics tocalculate the concentration of Pd in the surface layer and inthe bulk and its dependence on temperature and CO pres-sure, for a system in thermal equilibrium.
To do this we consider three competing equilibria:
PdB $ PdS ð1ÞPdS þ COg $ PdCOS ð2Þ2PdS þ COg $ Pd2CO ð3Þ
Here PdB is a Pd atom in the bulk and PdS is a Pd atom inthe surface layer. PdCOS is CO adsorbed on a Pd atom inthe surface layer, and Pd2CO is CO adsorbed in the bridgeconfiguration on two Pd atoms. Pd2CO is not the ensembleproposed by Goodman as the active site for the vinyl ace-tate synthesis. Pd2CO involves two nearest-neighbors Pdatoms whereas in the ensemble proposed by Goodmanthe two Pd atoms are next-nearest-neighbors. The equilib-rium PdCOs + COg M Pd2CO is not independent of theones considered above, because of material and lattice siteconservation conditions.
The equilibrium conditions are [29]
lB ¼ lS ð4ÞlCO þ lS ¼ lPdCO ð5ÞlCO þ 2lS ¼ lPd2CO ð6Þ
Here lS, lCO, lB, lPdCO and lPd2CO are the chemical poten-tials of the species PdS, gaseous CO, PdB, PdCOS andPd2CO, respectively. We use the following notation:
ensembles consisting of two Pd atoms sitting in an Au(100) surface. Thethe top layer is shown.
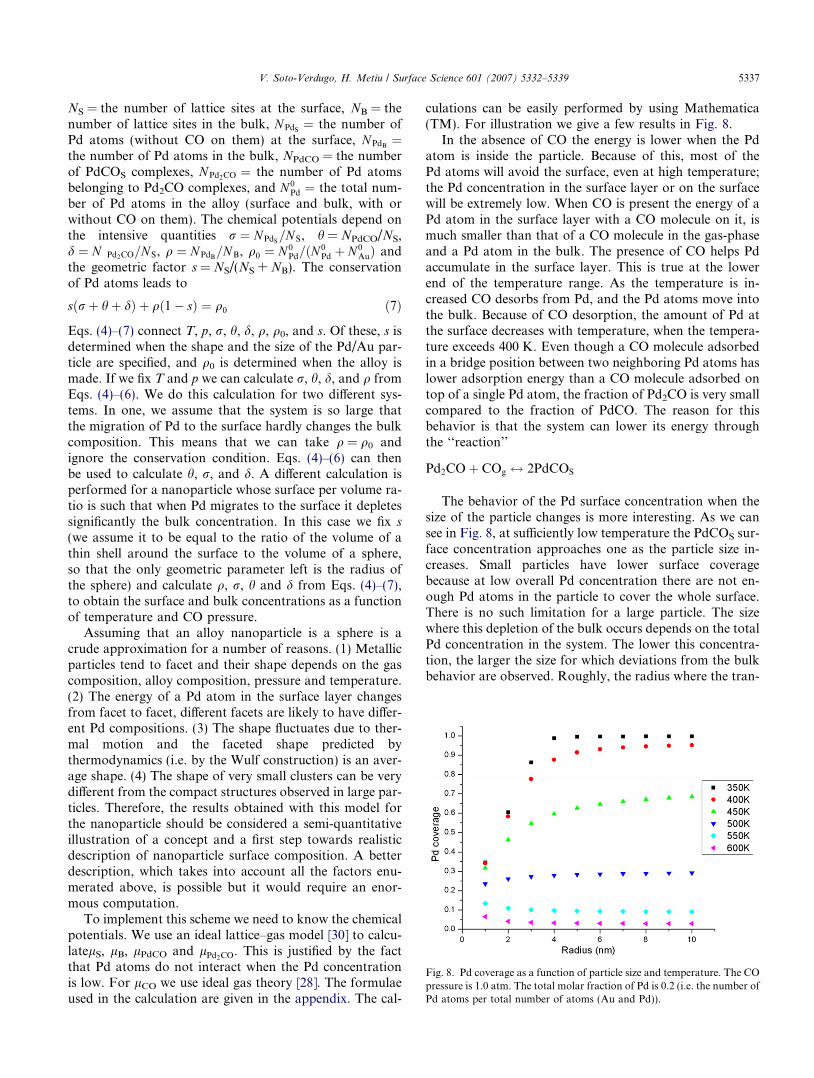
Fig. 8. Pd coverage as a function of particle size and temperature. The COpressure is 1.0 atm. The total molar fraction of Pd is 0.2 (i.e. the number ofPd atoms per total number of atoms (Au and Pd)).
V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339 5337
NS = the number of lattice sites at the surface, NB = thenumber of lattice sites in the bulk, N PdS
¼ the number ofPd atoms (without CO on them) at the surface, NPdB
¼the number of Pd atoms in the bulk, NPdCO = the numberof PdCOS complexes, N Pd2CO ¼ the number of Pd atomsbelonging to Pd2CO complexes, and N 0
Pd ¼ the total num-ber of Pd atoms in the alloy (surface and bulk, with orwithout CO on them). The chemical potentials depend onthe intensive quantities r ¼ NPdS
=NS, h = NPdCO/NS,d ¼ N Pd2CO=NS, q ¼ N PdB
=N B, q0 ¼ N 0Pd=ðN 0
Pd þ N 0AuÞ and
the geometric factor s = NS/(NS + NB). The conservationof Pd atoms leads to
sðrþ hþ dÞ þ qð1� sÞ ¼ q0 ð7Þ
Eqs. (4)–(7) connect T, p, r, h, d, q, q0, and s. Of these, s isdetermined when the shape and the size of the Pd/Au par-ticle are specified, and q0 is determined when the alloy ismade. If we fix T and p we can calculate r, h, d, and q fromEqs. (4)–(6). We do this calculation for two different sys-tems. In one, we assume that the system is so large thatthe migration of Pd to the surface hardly changes the bulkcomposition. This means that we can take q = q0 andignore the conservation condition. Eqs. (4)–(6) can thenbe used to calculate h, r, and d. A different calculation isperformed for a nanoparticle whose surface per volume ra-tio is such that when Pd migrates to the surface it depletessignificantly the bulk concentration. In this case we fix s
(we assume it to be equal to the ratio of the volume of athin shell around the surface to the volume of a sphere,so that the only geometric parameter left is the radius ofthe sphere) and calculate q, r, h and d from Eqs. (4)–(7),to obtain the surface and bulk concentrations as a functionof temperature and CO pressure.
Assuming that an alloy nanoparticle is a sphere is acrude approximation for a number of reasons. (1) Metallicparticles tend to facet and their shape depends on the gascomposition, alloy composition, pressure and temperature.(2) The energy of a Pd atom in the surface layer changesfrom facet to facet, different facets are likely to have differ-ent Pd compositions. (3) The shape fluctuates due to ther-mal motion and the faceted shape predicted bythermodynamics (i.e. by the Wulf construction) is an aver-age shape. (4) The shape of very small clusters can be verydifferent from the compact structures observed in large par-ticles. Therefore, the results obtained with this model forthe nanoparticle should be considered a semi-quantitativeillustration of a concept and a first step towards realisticdescription of nanoparticle surface composition. A betterdescription, which takes into account all the factors enu-merated above, is possible but it would require an enor-mous computation.
To implement this scheme we need to know the chemicalpotentials. We use an ideal lattice–gas model [30] to calcu-latelS, lB, lPdCO and lPd2CO. This is justified by the factthat Pd atoms do not interact when the Pd concentrationis low. For lCO we use ideal gas theory [28]. The formulaeused in the calculation are given in the appendix. The cal-
culations can be easily performed by using Mathematica(TM). For illustration we give a few results in Fig. 8.
In the absence of CO the energy is lower when the Pdatom is inside the particle. Because of this, most of thePd atoms will avoid the surface, even at high temperature;the Pd concentration in the surface layer or on the surfacewill be extremely low. When CO is present the energy of aPd atom in the surface layer with a CO molecule on it, ismuch smaller than that of a CO molecule in the gas-phaseand a Pd atom in the bulk. The presence of CO helps Pdaccumulate in the surface layer. This is true at the lowerend of the temperature range. As the temperature is in-creased CO desorbs from Pd, and the Pd atoms move intothe bulk. Because of CO desorption, the amount of Pd atthe surface decreases with temperature, when the tempera-ture exceeds 400 K. Even though a CO molecule adsorbedin a bridge position between two neighboring Pd atoms haslower adsorption energy than a CO molecule adsorbed ontop of a single Pd atom, the fraction of Pd2CO is very smallcompared to the fraction of PdCO. The reason for thisbehavior is that the system can lower its energy throughthe ‘‘reaction’’
Pd2COþ COg $ 2PdCOS
The behavior of the Pd surface concentration when thesize of the particle changes is more interesting. As we cansee in Fig. 8, at sufficiently low temperature the PdCOS sur-face concentration approaches one as the particle size in-creases. Small particles have lower surface coveragebecause at low overall Pd concentration there are not en-ough Pd atoms in the particle to cover the whole surface.There is no such limitation for a large particle. The sizewhere this depletion of the bulk occurs depends on the totalPd concentration in the system. The lower this concentra-tion, the larger the size for which deviations from the bulkbehavior are observed. Roughly, the radius where the tran-

5338 V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339
sition occurs is given by the condition that the total numberof Pd atoms in the system is equal to the number of latticesites on the surface.
5. Summary and discussion
In the absence of gases Pd atoms have a strong tendencyto substitute Au atoms in the bulk. The presence of CO inthe gas-phase favors the presence of Pd in the top surfacelayer. The interaction between the Pd atoms, with or with-out CO on them, is very weak and they will not aggregateto form Pd islands within the surface layer, nor will theyform Pd domains in the bulk. The lack of interaction be-tween the Pd atoms makes it easy to provide simple equa-tions that give the equilibrium composition at the surfaceof a spherical nanoparticle, as a function of particle size,net alloy composition, temperature, and CO pressure. Cal-culations with these equations suggest that the ability of thePd/Au alloy to work as a selective catalyst is controlled bythe binding energy of the reactants to the Pd surface atom.If these are too low to overcome the natural tendency ofthe Pd atoms to move into the bulk, then there is hardlyany Pd on the surface and there is little Pd derived catalyticactivity. According to Goodman’s model the best selectiv-ity is achieved when the surface has many clusters contain-ing two next-nearest-neighbor Pd atoms and very fewlarger clusters. Our model suggests that such clusters aremade at random, since the Pd–Pd interaction is too smallto overcome the tendency of the entropy to create a ran-dom distribution. The reaction temperature also plays animportant role, if it is too high and the reactants desorbfrom the Pd atom, the latter will have a tendency to go in-side the bulk under reaction conditions. In this case the cat-alyst will fail since Au does not catalyze vinyl acetateformation.
Acknowledgements
V.S.V. thanks CONACYT and UCMEXUS for sup-port. We are grateful to Steeve Chretien, Greg Mills, RajPala, and Vladimir Shapovalov for useful discussions. Thiswork was partially supported by the Air Force Office ofScientific Research under Grants F49620-01-1-0459 andFAA9550-06-1-0167.
Appendix
The chemical potential of Pd atoms in the bulk of thePd/Au alloy is
lPdB
kT¼ ln
qð1� qÞqB
� �
where qB is the partition function of one Pd atom in thebulk. q is the molar fraction of Pd in the bulk.
The chemical potential of a Pd atom in the surface layer(no CO adsorbed on it) is
lPdS
kT¼ ln
rð1� r� h� dÞqS
� �
Similarly, qS is the partition function for a Pd atom locatedat the surface. The coverages h, r, and d correspond toPdCOS, PdS, and Pd2CO, respectively.
The chemical potential of PdCOS is
lPdCO
kT¼ ln
hð1� r� h� dÞqPdCO
� �
To calculate the chemical potential of Pd2CO we needthe configurational contribution, the number of arrange-ments of two Pd atoms into dimers. An exact solution ofthis problem is not yet known [31,32]. For our purpose itwill be enough to use the Bethe approximation [33] sincethe Pd coverage is low. In this approximation, the chemicalpotential of the Pd2CO is
lPd2CO
kT¼ ln
d dð1� h� rÞ � d½ �d2ð1� r� h� dÞ2qPd2CO
!
For CO on the gas-phase we have the usual result
lCO
kT¼ ln
pK3
kTqCO
� �
In this formulae, p is the pressure of the CO gas, K is thethermal wavelength of a CO molecule,
K ¼ hffiffiffiffiffiffiffiffiffiffiffiffiffiffi2pmkTp
qPdCO, qPd2CO are the partition functions of the PdCOS andPd2CO, and qCO is the rotational and vibrational partitionfunction of gas-phase CO, d is the coordination number ofthe corresponding surface lattice, which is four for the(10 0) surface and six for the (111) surface. With these for-mulas for the chemical potential, the equilibrium condi-tions become
h ¼ rK3pkT
� �qPdCO
qCOqS
� �
ð1� qÞr ¼ qð1� r� h� dÞ qS
qB
� �
d dð1� r� hÞ � d½ � ¼ r2 K3pkT
� �qPd2CO
qCOq2S
� �
In this calculation we made sure that all energies enteringin the partition functions were calculated with the same en-ergy scale. We assume that the vibrational partition func-tions of the slabs cancel in the equilibrium conditions.For example, in the equilibrium PdS + COgas M PdCO itis assumed that the vibrational contribution to the parti-tion function of the PdS slab cancels the vibrational contri-bution from the PdCO slab. However, the vibrationalcontribution from the bending motion of PdCO is includedexplicitly. For the configurational contribution to the par-tition function of Pd2CO we use the Bethe approximationdiscussed in references [31–33].

V. Soto-Verdugo, H. Metiu / Surface Science 601 (2007) 5332–5339 5339
The resulting equations are
h ¼ rExp �b ePdCO � eS � eCOð Þ½ � K3pkT
� �q0PdCO
q0CO
� �ð1� qÞr ¼ qð1� r� h� dÞExp �b eS � eBð Þ½ �d dð1� r� hÞ � d½ � ¼ r2Exp �b ePd2CO � 2eS � eCOð Þ½ �
� K3pkT
� �q0Pd2CO
q0CO
� �
The function q0PdCO is the vibrational partition function ofPdCO when we have the lattice fixed. The function q0CO isthe rotational and vibrational contribution of gas-phaseCO. The function q0Pd2CO is the vibrational partition func-tion of Pd2CO with the surface fixed.
In addition to the equilibrium conditions, it is necessaryto conserve the number of Pd atoms as they distribute be-tween the bulk and surface layer. This condition is given byEq. (7). Numerical solution of these four equations give usthe coverages and bulk Pd concentration in terms of theCO pressure, temperature, molar fraction of Pd and parti-cle size.
References
[1] A. Jablonski, S.H. Overbury, G.A. Somorjai, Surface Science 65(1977) 578.
[2] R. Anton, H. Eggers, J. Veletas, Thin Solid Films 226 (1993) 39.[3] D.G. Swartzfager, S.B. Ziemecki, M.J. Kelley, Journal of Vacuum
Science Technology 19 (1981) 185.[4] J. Kuntze, S. Speller, W. Heiland, P. Deurinck, C. Creemers, A. Atrei,
U. Bardi, Physical Review B 60 (1999) 9010.[5] J. Kuntze, S. Speller, W. Heiland, A. Atrei, G. Rovida, U. Bardi,
Physical Review B 60 (1999) 1535.[6] M. Aschoff, S. Speller, J. Kuntze, W. Heiland, E. Platzgummer, M.
Schmid, P. Varga, B. Baretzky, Surface Science 415 (1998) L1051.
[7] B.E. Koel, A. Sellidj, M.T. Paffett, Physical Review B 46 (1992) 7846.[8] C.-W. Yi, K. Luo, T. Wei, D.W. Goodman, Journal of Physical
Chemistry B 109 (2005) 18535.[9] Y.-F. Han, D. Kumar, C. Sivadinarayana, A. Clearfield, D.W.
Goodman, Catalysis Letters 94 (2004) 131.[10] Y.-F. Han, J.-H. Wang, D. Kumar, Z. Yan, D.W. Goodman, Journal
of Catalysis 232 (2005) 467.[11] M.S. Chen, K. Luo, T. Wei, Z. Yan, D. Kumar, C.W. Yi, D.W.
Goodman, Catalysis Today 117 (2006) 37.[12] M. Chen, D. Kumar, C.-W. Yi, D.W. Goodman, Science 310 (2005)
291.[13] P. Hohenberg, W. Kohn, Physical Review 136 (1964) B864.[14] W. Kohn, L.J. Sham, Physical Review 140 (1965) A1133.[15] M.C. Payne, M.P. Teter, D.C. Allan, T.A. Arias, J.D. Joannopoulos,
Reviews of Modern Physics 64 (1992) 1045.[16] G. Kresse, J. Furthmuller, Physical Review B 54 (1996) 11169.[17] G.Kresse,J.Furthmuller,ComputationalMaterialsScience6(1996)15.[18] G. Kresse, J. Hafner, Physical Review B 47 (1993) 558.[19] G. Kresse, J. Hafner, Physical Review B 49 (1994) 14251.[20] J.P. Perdew, K. Burke, M. Ernzerhof, Physical Review Letters 77
(1996) 3865.[21] B. Hammer, L.B. Hansen, J.K. Nørskov, Physical Review B 59 (1999)
7413.[22] P.E. Blochl, Physical Review B 50 (1994) 17953.[23] G. Kresse, D. Joubert, Physical Review B 59 (1999) 1758.[24] M. Methfessel, A.T. Paxton, Physical Review B 40 (1989) 3616.[25] G. Makov, M.C. Payne, Physical Review B 51 (1995) 4014.[26] H.J. Monkhorst, J.D. Pack, Physical Review B 13 (1976) 5188.[27] J.H. Rose, J.R. Smith, F. Guinea, J. Ferrante, Physical Review B 29
(1984) 2963.[28] H. Metiu, Physical Chemistry: Statistical Physics, Taylor and Francis
Group, NY, 2006.[29] H. Metiu, Physical Chemistry: Thermodynamics, Taylor and Francis
Group, 2006.[30] T.L. Hill, An Introduction to Statistical Thermodynamics, Dover,
Mineola, 1986.[31] J.F. Nagle, Physical Review 152 (1966) 190.[32] D.S. Gaunt, Physical Review 179 (1969) 174.[33] T.S. Chang, Proceedings of the Royal Society of London Series A –
Mathematical and Physical Sciences (1934–1990) 169 (1939) 512.












![Segregation and Solid Evolution during the Solidification ... · constituents?‘]. ... Another approach is through alloy composition modification or ... Bulk chemical composition](https://img.pdfslide.us/doc/110x75/5b8b992a09d3f245638b9d28/segregation-and-solid-evolution-during-the-solidification-constituents.jpg)

![Electrochemical Study of Carbon Steel and 304 Stainless ... · Erosion is a key cause of metal-alloy degradation in many industrial fields [1]. When an alloy is exposed to a flowing](https://img.pdfslide.us/doc/110x75/6000d4a0f928516f14228cd3/electrochemical-study-of-carbon-steel-and-304-stainless-erosion-is-a-key-cause.jpg)














