Embed Size (px)
Citation preview

1250 Chapter 24 Natural Products (Secondary Metabolites)
Introduction
Natural products have primary ecological functions.
Plants produce a vast and diverse assortment of organic compounds,the great majority of which do not appear to participate directly ingrowth and development. These substances, traditionally referred toas secondary metabolites, often are differentially distributed amonglimited taxonomic groups within the plant kingdom. Their functions,many of which remain unknown, are being elucidated with increas-ing frequency. The primary metabolites, in contrast, such as phyto-sterols, acyl lipids, nucleotides, amino acids, and organic acids, arefound in all plants and perform metabolic roles that are essentialand usually evident.
Although noted for the complexity of their chemical structuresand biosynthetic pathways, natural products have been widely per-ceived as biologically insignificant and have historically received lit-tle attention from most plant biologists. Organic chemists, however,have long been interested in these novel phytochemicals and haveinvestigated their chemical properties extensively since the 1850s.Studies of natural products stimulated development of the separa-tion techniques, spectroscopic approaches to structure elucidation,and synthetic methodologies that now constitute the foundation ofcontemporary organic chemistry. Interest in natural products wasnot purely academic but rather was prompted by their great utilityas dyes, polymers, fibers, glues, oils, waxes, flavoring agents, per-fumes, and drugs. Recognition of the biological properties of myriadnatural products has fueled the current focus of this field, namely,the search for new drugs, antibiotics, insecticides, and herbicides.Importantly, this growing appreciation of the highly diverse biologi-cal effects produced by natural products has prompted a reevalua-tion of the possible roles these compounds play in plants, especiallyin the context of ecological interactions. As illustrated in this chapter,many of these compounds now have been shown to have important
C H A P T E R 24Natural Products (Secondary Metabolites)
Introduction24.1 Terpenoids24.2 Synthesis of IPP24.3 Prenyltransferase and terpene
synthase reactions24.4 Modification of terpenoid
skeletons24.5 Toward transgenic terpenoid
production24.6 Alkaloids24.7 Alkaloid biosynthesis24.8 Biotechnological application
of alkaloid biosynthesis research
24.9 Phenylpropanoid and phenylpropanoid-acetate pathway metabolites
24.10 Phenylpropanoid and phenylpropanoid-acetate biosynthesis
24.11 Biosynthesis of lignans, lignins,and suberization
24.12 Flavonoids24.13 Coumarins, stilbenes,
styrylpyrones, and arylpyrones24.14 Metabolic engineering of
phenylpropanoid production: a possible source of enhanced fibers, pigments, pharmaceuti-cals, and flavoring agents
C H A P T E R O U T L I N E
Biochemistry & Molecular Biology of Plants, B. Buchanan, W. Gruissem, R. Jones, Eds.© 2000, American Society of Plant Physiologists
Rodney CroteauToni M. KutchanNorman G. Lewis

125124.1–Terpenoids
adaptive significance in protection againstherbivory and microbial infection, as attrac-tants for pollinators and seed-dispersing ani-mals, and as allelopathic agents (allelochem-icals that influence competition among plantspecies). These ecological functions affectplant survival profoundly, and we think itreasonable to adopt the less pejorative term“plant natural products” to describe sec-ondary plant metabolites that act primarilyon other species.
The boundary between primary andsecondary metabolism is blurred.
Based on their biosynthetic origins, plantnatural products can be divided into threemajor groups: the terpenoids, the alkaloids,and the phenylpropanoids and allied pheno-lic compounds. All terpenoids, includingboth primary metabolites and more than25,000 secondary compounds, are derivedfrom the five-carbon precursor isopentenyldiphosphate (IPP). The 12,000 or so knownalkaloids, which contain one or more nitro-gen atoms, are biosynthesized principallyfrom amino acids. The 8000 or so phenoliccompounds are formed by way of either theshikimic acid pathway or the malonate/acetate pathway.
Primary and secondary metabolites can-not readily be distinguished on the basis ofprecursor molecules, chemical structures, orbiosynthetic origins. For example, both pri-mary and secondary metabolites are foundamong the diterpenes (C20) and triterpenes(C30). In the diterpene series, both kaurenoicacid and abietic acid are formed by a verysimilar sequence of related enzymatic reac-tions (Fig. 24.1); the former is an essential in-termediate in the synthesis of gibberellins,i.e., growth hormones found in all plants(see Chapter 17), whereas the latter is a resincomponent largely restricted to members ofthe Fabaceae and Pinaceae. Similarly, the es-sential amino acid proline is classified as aprimary metabolite, whereas the C6 analogpipecolic acid is considered an alkaloid and
thus a natural product (Fig. 24.1). Evenlignin, the essential structural polymer ofwood and second only to cellulose as themost abundant organic substance in plants,is considered a natural product rather than a primary metabolite.
In the absence of a valid distinctionbased on either structure or biochemistry, wereturn to a functional definition, with prima-ry products participating in nutrition and es-sential metabolic processes inside the plant,and natural (secondary) products influenc-ing ecological interactions between the plantand its environment. In this chapter, weprovide an overview of the biosynthesis ofthe major classes of plant natural products,emphasizing the origins of their structural diversity, as well as their physiological func-tions, human uses, and potential biotechno-logical applications.
24.1 Terpenoids
Terpenoids perhaps are the most structurallyvaried class of plant natural products. Thename terpenoid, or terpene, derives from thefact that the first members of the class were
Abietic acidCOOH
Pipecolic acid
COOH
H
N
Kaurenoic acidCOOH
COOH
ProlineH
N
Secondary metabolitePrimary metabolite
Figure 24.1Kaurenoic acid and proline are primary metabo-lites, whereas the closely related compounds abieticacid and pipecolic acid are considered secondarymetabolites.

isolated from turpentine (“terpentin” in Ger-man). All terpenoids are derived by repeti-tive fusion of branched five-carbon unitsbased on isopentane skeleton. These mono-mers generally are referred to as isopreneunits because thermal decomposition ofmany terpenoid substances yields the alkenegas isoprene as a product (Fig. 24.2, upperpanel) and because suitable chemical condi-tions can induce isoprene to polymerize inmultiples of five carbons, generating numer-ous terpenoid skeletons. For these reasons,the terpenoids are often called isoprenoids,although researchers have known for wellover 100 years that isoprene itself is not the biological precursor of this family ofmetabolites.
24.1.1 Terpenoids are classified by thenumber of five-carbon units they contain.
The five-carbon (isoprene) units that makeup the terpenoids are often joined in a “head-to-tail” fashion, but head-to-head fusions arealso common, and some products are formedby head-to-middle fusions (Fig. 24.2, lowerpanel). Accordingly, and because extensivestructural modifications with carbon–carbonbond rearrangements can occur, tracing theoriginal pattern of isoprene units is some-times difficult.
The smallest terpenes contain a singleisoprene unit; as a group, they are namedhemiterpenes (half-terpenes). The best
1252 Chapter 24 Natural Products (Secondary Metabolites)
known hemiterpene is isoprene itself, a vol-atile product released from photosynthe-tically active tissues. The enzyme isoprenesynthase is present in the leaf plastids of nu-merous C3 plant species, but the metabolicrationale for the light-dependent productionof isoprene is unknown (acclimation to hightemperatures has been suggested). Estimatedannual foliar emissions of isoprene are quitesubstantial (5 × 108 metric tons of carbon),and the gas is a principal reactant in theNOx radical–induced formation of tropo-spheric ozone (see Chapter 22, Fig. 22.37).
C10 terpenoids, although they consist oftwo isoprene units, are called monoterpenes;as the first terpenoids isolated from turpen-tine in the 1850s, they were considered to be the base unit from which the subsequentnomenclature is derived. The monoterpenesare best known as components of the vol-atile essences of flowers and of the essentialoils of herbs and spices, in which they makeup as much as 5% of plant dry weight. Mono-terpenes are isolated by either distillation orextraction and find considerable industrialuse in flavors and perfumes.
The terpenoids that derive from threeisoprene units contain 15 carbon atoms andare known as sesquiterpenes (i.e., one andone-half terpenes). Like monoterpenes,many sesquiterpenes are found in essentialoils. In addition, numerous sesquiterpenoidsact as phytoalexins, antibiotic compoundsproduced by plants in response to microbialchallenge, and as antifeedants that discour-age opportunistic herbivory. Although theplant hormone abscisic acid is structurally asesquiterpene, its C15 precursor, xanthoxin, isnot synthesized directly from three isopreneunits but rather is produced by asymmetriccleavage of a C40 carotenoid (see Chapter 17).
The diterpenes, which contain 20 car-bons (four C5 units), include phytol (the hy-drophobic side chain of chlorophyll), thegibberellin hormones, the resin acids ofconifer and legume species, phytoalexins,and a host of pharmacologically importantmetabolites, including taxol, an anticanceragent found at very low concentrations(0.01% dry weight) in yew bark, and for-skolin, a compound used to treat glaucoma.Some gibberellins have only 19 carbon atomsand are considered norditerpenoids sincethey have lost 1 carbon through a metaboliccleavage reaction (see Chapter 17).
Figure 24.2Terpenes are synthesized from C5 units. The upper panel shows the structures ofthe isopentane skeleton and isoprene gas. The lower panel shows how differentpatterns of isoprene unit assembly yield a variety of different structures. For ex-ample, the triterpene squalene is formed by head-to-head fusion of two moleculesof farnesyl diphosphate (FPP), which itself is the product of the head-to-tail fu-sion of isopentenyl diphosphate (IPP) and geranyl diphosphate (GPP) (see Fig.24.7). The monoterpene pyrethrin I (see Fig. 24.10) results from a head-to-middlefusion of two C5 units.
Isopentane Isoprene
Head-to-tail Head-to-middleHead-to-head

125324.2–Synthesis of IPP
The triterpenes, which contain 30 car-bon atoms, are generated by the head-to-head joining of two C15 chains, each ofwhich constitutes three isoprene units joinedhead-to-tail. This large class of molecules in-cludes the brassinosteroids (see Chapter 17),the phytosterol membrane components (seeChapter 1), certain phytoalexins, varioustoxins and feeding deterrents, and compo-nents of surface waxes, such as oleanolicacid of grapes.
The most prevalent tetraterpenes (40carbons, eight isoprene units) are the carote-noid accessory pigments which perform essential functions in photosynthesis (seeChapter 12). The polyterpenes, those con-taining more than eight isoprene units, in-clude the prenylated quinone electron carri-ers (plastoquinone and ubiquinone; seeChapters 12 and 14), long-chain polyprenolsinvolved in sugar transfer reactions (e.g.,dolichol; see Chapters 1 and 4), and enor-mously long polymers such as rubber (aver-age molecular mass greater than 106 Da), often found in latex.
Natural products of mixed biosyntheticorigins that are partially derived from ter-penoids are often called meroterpenes. Forexample, both cytokinins (see Chapter 17)and numerous phenylpropanoid compoundscontain C5 isoprenoid side chains. Certain al-kaloids, including the anticancer drugs vin-cristine and vinblastine, contain terpenoidfragments in their structures (see Fig. 24.34).Additionally, some modified proteins in-clude a 15- or 20-carbon terpenoid side chainthat anchors the protein in a membrane (seeChapter 1).
24.1.2 A diverse array of terpenoidcompounds is synthesized by variousconserved reaction mechanisms.
At the turn of the 20th century, structural in-vestigations of many terpenoids led OttoWallach to formulate the “isoprene rule,”which postulated that most terpenoids couldbe constructed hypothetically by repetitivelyjoining isoprene units. This principle provid-ed the first conceptual framework for a com-mon structural relationship among terpenoidnatural products (Box 24.1). Wallach’s ideawas refined in the 1930s, when Leopold Ruzicka formulated the “biogenetic isoprene
rule,” emphasizing mechanistic considera-tions of terpenoid synthesis in terms ofelectrophilic elongations, cyclizations, andrearrangements. This hypothesis ignores theprecise character of the biological precursorsand assumes only that they are “isoprenoid”in structure. As a working model for ter-penoid biosynthesis, the biogenetic isoprenerule has proved essentially correct.
Despite great diversity in form andfunction, the terpenoids are unified in theircommon biosynthetic origin. The biosynthe-sis of all terpenoids from simple, primarymetabolites can be divided into four overallsteps: (a) synthesis of the fundamental pre-cursor IPP; (b) repetitive additions of IPP toform a series of prenyl diphosphate ho-mologs, which serve as the immediate pre-cursors of the different classes of terpenoids;(c) elaboration of these allylic prenyl diphos-phates by specific terpenoid synthases toyield terpenoid skeletons; and (d) secondaryenzymatic modifications to the skeletons(largely redox reactions) to give rise to thefunctional properties and great chemical di-versity of this family of natural products.
24.2 Synthesis of IPP
24.2.1 Biosynthesis of terpenoids iscompartmentalized, as is production of theterpenoid precursor IPP.
Although terpenoid biosynthesis in plants,animals, and microorganisms involves simi-lar classes of enzymes, important differencesexist among these processes. In particular,plants produce a much wider variety of ter-penoids than do either animals or microbes,a difference reflected in the complex organi-zation of plant terpenoid biosynthesis at thetissue, cellular, subcellular, and genetic lev-els. The production of large quantities of ter-penoid natural products as well as their sub-sequent accumulation, emission, or secretionis almost always associated with the pres-ence of anatomically highly specializedstructures. The glandular trichomes (Fig.24.3A, B) and secretory cavities of leaves(Fig. 24.3C) and the glandular epiderms offlower petals generate and store or emit ter-penoid essential oils that are important be-cause they encourage pollination by insects.The resin ducts and blisters of conifer species

1254 Chapter 24 Natural Products (Secondary Metabolites)
Box 24.1 Early investigators formulated rules for identifying and namingisoprenoid structures.
In the late 1800s, chemists struggledto define the structures of the monoter-penes. The mixed results achieved bythese efforts are illustrated by the numer-ous structures proposed for camphor(C10H16O; see structures at left of figure,which include the names of the proposersand the dates proposed). Chromatograph-ic purification techniques and spectro-scopic methods for structure elucidationwere not available to these earlychemists, who relied on the preparationof crystalline derivatives to assess purityand on chemical degradation studies todetermine structures. Systematic study ofthe monoterpenes led the Germanchemist Otto Wallach to recognize thatmany terpenoid compounds might beconstructed by joining isoprene units,generally in a repetitive head-to-tail fashion, as in Bredt’s correct proposedstructure for camphor (see figure). This
concept, known as the isoprene rule, earned Wallach the Nobel Prize in Chem-istry in 1910.
By the 1930s, faced with a bewilder-ing array of terpenoid substances,Leopold Ruzicka and his contemporariessought to develop a unifying principlethat could rationalize the natural occur-rence of all of the known terpenoids,even those that did not strictly fit Wal-lach’s isoprene rule. Ruzicka’s ingenioussolution to the problem was to focus onreaction mechanisms and ignore the pre-cise character of the biological precursor,assuming only that it had a terpenoidstructure during reaction. He hypothe-sized the involvement of electrophilic re-actions that generated carbocationic inter-mediates, which underwent subsequentC5 addition, cyclization, and in some cas-es skeletal rearrangement before elimina-tion of a proton or capture by a nucle-
ophile to yield the observed terpenoidproducts. This proposal, which Ruzickacalled the biogenetic isoprene rule, canbe stated simply: A compound is “iso-prenoid” if it is derived biologically froman “isoprenoid” precursor, with or with-out rearrangements. Ruzicka’s conceptdiffers from Wallach’s in its emphasis onbiochemical origin rather than structure.The great strength of the biogenetic iso-prene rule lay in its use of mechanisticconsiderations to classify the bulk ofknown terpenoids, including structuresthat did not strictly follow Wallach’s iso-prene rule. Application of the biogeneticisoprene rule to the origin of several ofthe common monoterpene skeletons is il-lustrated in the right panel of the figure(note the bornane skeleton from whichcamphor is derived). Ruzicka was award-ed the Nobel Prize in Chemistry in 1939.
O O
O
O
O
O
+
+
+
+
+
++
(Bredt, 1893)
(Perkin, 1898)
(Tiemann, 1895)
(Meyer, 1870) (Hlasiwetz, 1870)
(Bouveault, 1897)
Pinane skeleton Terpinen-4-yl cationα-Terpinyl cation
Fenchane skeleton Bornane skeleton Thujane skeleton
(Fig. 24.3D) produce and accumulate a defen-sive resin consisting of turpentine (monoter-pene olefins) and rosin (diterpenoid resinacids). Triterpenoid surface waxes are formedand excreted from specialized epidermis,and laticifers produce certain triterpenes and
polyterpenes such as rubber. These special-ized structures sequester natural productsaway from sensitive metabolic processes andthereby prevent autotoxicity. Most structuresof this type are nonphotosynthetic and musttherefore rely on adjacent cells to supply

125524.2–Synthesis of IPP
the carbon and energy needed to drive ter-penoid biosynthesis.
A more fundamental, and perhaps uni-versal, feature of the organization of ter-penoid metabolism exists at the subcellularlevel. The sesquiterpenes (C15), triterpenes(C30), and polyterpenes appear to be pro-duced in the cytosolic and endoplasmicreticulum (ER) compartments, whereas iso-prene, the monoterpenes (C10), diterpenes(C20), tetraterpenes (C40), and certain preny-lated quinones originate largely, if not exclu-sively, in the plastids. The evidence now in-dicates that the biosynthetic pathways forthe formation of the fundamental precursorIPP differ markedly in these compartments,with the classical acetate/mevalonate path-way being active in the cytosol and ER andthe glyceraldehyde phosphate/pyruvatepathway operating in the plastids. Regula-tion of these dual pathways may be difficultto assess, given that plastids may supply
IPP to the cytosol for use in biosynthesis,and vice versa. Mitochondria, a third com-partment, may generate the ubiquinoneprenyl group by the acetate/mevalonatepathway, although little is known about thecapability of these organelles for terpenoidbiosynthesis.
24.2.2 Hydroxymethylglutaryl-CoAreductase, an enzyme in theacetate/mevalonate pathway, is highly regulated.
The basic enzymology of IPP biosynthesis byway of the acetate/mevalonate pathway iswidely accepted (Fig. 24.4). This cytosolicIPP pathway involves the two-step conden-sation of three molecules of acetyl-CoAcatalyzed by thiolase and hydroxymethyl-glutaryl-CoA synthase. The resulting product, 3-hydroxy-3-methylglutaryl-CoA(HMG-CoA), is subsequently reduced by
Figure 24.3(A) Scanning electron micrograph of the leaf surface of thyme. The round structures are peltate glandular tri-chomes, in which monoterpenes and sesquiterpenes are synthesized. (B) Light micrograph of a glandular tri-chome from spearmint, shown in longitudinal section. C, subcuticular space; S, secretory cells; St, stalk; B,basal cell; E, epidermal cell. (C) Light micrograph of a secretory cavity in a lemon leaf, shown in cross-section.L, lumen; Sh, sheath cells; P, parenchyma cell. (D) Light micrograph of a resin duct in wood of Jeffrey pine,shown in cross-section. X, secondary xylem.
(A) (B)
(C) (D)
L
S
StB E
Sh
SP
X
Sh
L
S
C
100 µ

1256 Chapter 24 Natural Products (Secondary Metabolites)
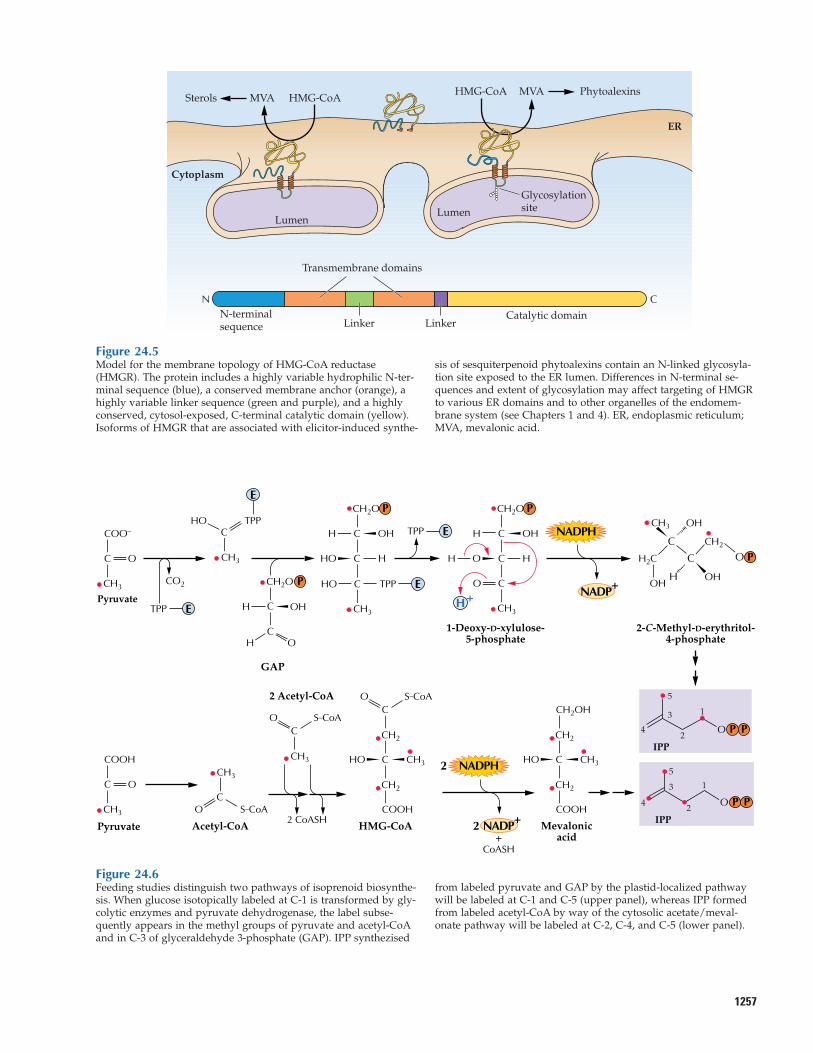
HMG-CoA reductase is one of the mosthighly regulated enzymes in animals, beinglargely responsible for the control of choles-terol biosynthesis. Accumulated evidence indicates that the plant enzyme, which is located in the ER membrane, is also highlyregulated. In many cases, small gene fami-lies, each containing multiple members, en-code this reductase. These gene families areexpressed in complex patterns, with individ-ual genes exhibiting constitutive, tissue- ordevelopment-specific, or hormone-inducibleexpression. Specific HMG-CoA reductasegenes can be induced by wounding or path-ogen infection. The activity of HMG-CoA re-ductase may be subject to posttranslationalregulation, for example, by a protein kinasecascade that phosphorylates and thereby in-activates the enzyme. Allosteric modulationprobably also plays a regulatory role. Prote-olytic degradation of HMG-CoA reductaseprotein and the rate of turnover of the corre-sponding mRNA transcripts may also influ-ence enzyme activity. Researchers have notarrived at a unified scheme that explainshow the various mechanisms that regulateHMG-CoA reductase facilitate the produc-tion of different terpenoid families. The precise biochemical controls that influenceactivity have been difficult to assess in vitrobecause the enzyme is associated with theER membrane. A model proposed to ration-alize the selective participation of HMG-CoAreductase in the biosynthesis of differentmevalonate-derived terpenoids is shown in Figure 24.5.
24.2.3 In plastids, IPP is synthesized frompyruvate and glyceraldehyde 3-phosphate.
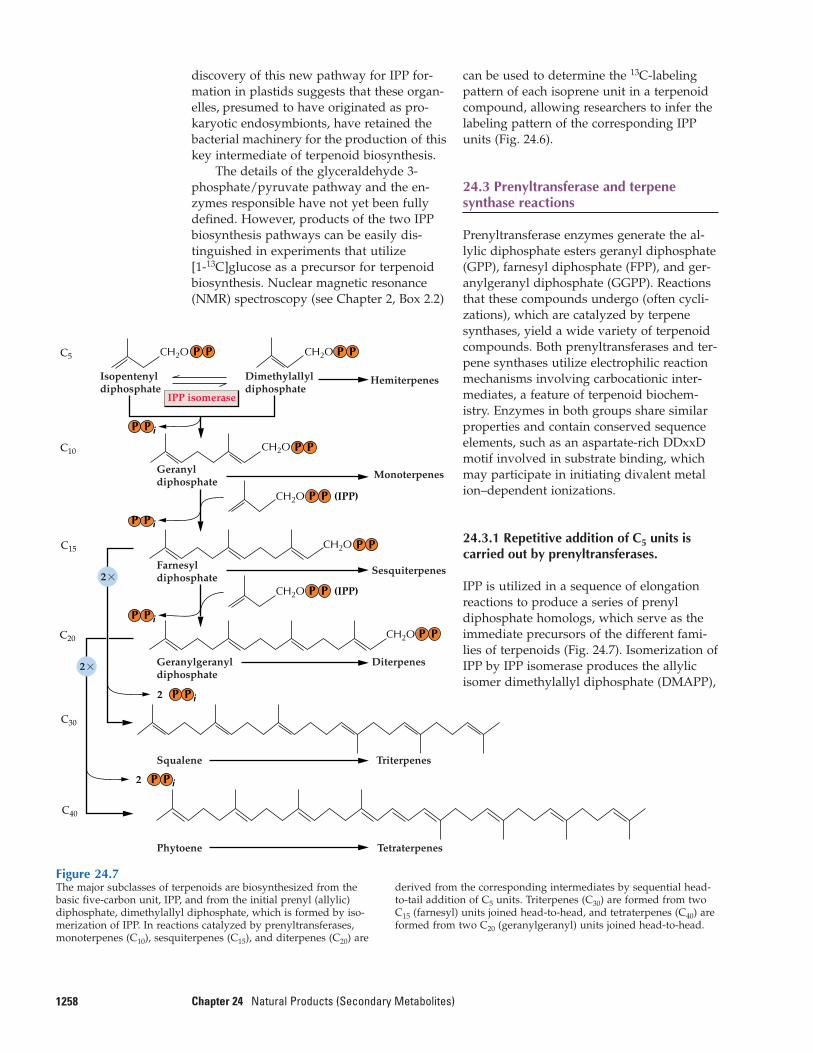
The plastid-localized route to IPP involves a different pathway, demonstrated in greenalgae and many eubacteria as well as plants.In this pathway, pyruvate reacts with thia-mine pyrophosphate (TPP) to yield a two-carbon fragment, hydroxyethyl-TPP,which condenses with glyceraldehyde 3-phosphate (see Chapter 12, Fig. 12.41, forsimilar TPP-mediated C2 transfers catalyzedby transketolase). TPP is released to form afive-carbon intermediate, 1-deoxy-D-xylulose 5-phosphate, which is rearranged and re-duced to form 2-C-methyl-D-erythritol 4-phosphate and subsequently transformed to yield IPP (Fig. 24.6, upper pathway). The
Thiolase
HMG-CoAsynthase
HMG-CoAreductase
MVA kinase
MVAP kinase
MVAPP decarboxylase
NADPH
CCH3
CH2 COOH
CH2
HO
COOH
COOH
COOH
C
CCH3
CH2
CH2
HO
CH2OH
CCH3
CH2
CH2
HO
CCH3
O
Acetyl-CoA
Acetyl-CoA
3-Hydroxy-3-methyl-glutaryl-CoA(HMG-CoA)
Mevalonic acid5-phosphate(MVAP)
Mevalonic acid5-diphosphate(MVAPP)
Acetoacetyl-CoA
CoASH
CoASH
CoASH
O
C CH2
CH3
CO2 H2O
CH2
CH2O
2
NADP+2
+ADP
+
ATP
ADP
ATP
ADP
ATP
CoAS
CCH2
O
CCH3
O
CoAS
CCH3
O
CoAS
Acetyl-CoA
CCH3
O
CoAS
CoAS
P P
CH2O P P
P i
CCH3
CH2
CH2
HO
CH2O P
Mevalonic acid(MVA)
Isopentenyldiphosphate(IPP)
Figure 24.4The acetate/mevalonatepathway for the forma-tion of IPP, the basicfive-carbon unit of ter-penoid biosynthesis.Synthesis of each IPPunit requires threemolecules of acetyl-CoA.
HMG-CoA reductase in two coupled reac-tions that form mevalonic acid. Two sequen-tial ATP-dependent phosphorylations ofmevalonic acid and a subsequent phospho-rylation/elimination-assisted decarboxylationyield IPP.
1257
N-terminalsequence Linker Linker
Catalytic domain
Transmembrane domains
Lumen
SterolsPhytoalexinsMVAHMG-CoA
HMG-CoAMVA
Lumen
Glycosylationsite
ER
Cytoplasm
N C
Figure 24.5Model for the membrane topology of HMG-CoA reductase(HMGR). The protein includes a highly variable hydrophilic N-ter-minal sequence (blue), a conserved membrane anchor (orange), ahighly variable linker sequence (green and purple), and a highlyconserved, cytosol-exposed, C-terminal catalytic domain (yellow).Isoforms of HMGR that are associated with elicitor-induced synthe-
sis of sesquiterpenoid phytoalexins contain an N-linked glycosyla-tion site exposed to the ER lumen. Differences in N-terminal se-quences and extent of glycosylation may affect targeting of HMGRto various ER domains and to other organelles of the endomem-brane system (see Chapters 1 and 4). ER, endoplasmic reticulum;MVA, mevalonic acid.
Acetyl-CoA
2 Acetyl-CoA
HMG-CoA
GAP
1-Deoxy-D-xylulose-5-phosphate
2-C-Methyl-D-erythritol-4-phosphate
Mevalonicacid
IPP
13
42
5
Pyruvate
C
C
C
C
C CH2OH
C
CH3
O
COO–
Pyruvate
C
CH3
O
O
COOH
TPPHO
IPP
O
13
42
5
P P
O P P
TPP
CH3
CO2
CH3
CH2O
CH3 CH3
C
C
HO
OH
H H O
OHH
C
C
C
HO
H OH
H
C
C
O
COH
P
CH2O P CH2O P
H H2C
CH2
OH
S–CoA COOH
CH3 CH3
CH2
O S–CoA
CH3
S–CoA
2 CoASH
O
HO
CH2
C
COOH
CH2
HO
CH2
NADPH
NADP+
NADPH2
NADP+2
C
OH
C PO
H
OHCH3
E
E
TPP E
TPP E
+CoASH
H+
Figure 24.6Feeding studies distinguish two pathways of isoprenoid biosynthe-sis. When glucose isotopically labeled at C-1 is transformed by gly-colytic enzymes and pyruvate dehydrogenase, the label subse-quently appears in the methyl groups of pyruvate and acetyl-CoAand in C-3 of glyceraldehyde 3-phosphate (GAP). IPP synthezised
from labeled pyruvate and GAP by the plastid-localized pathwaywill be labeled at C-1 and C-5 (upper panel), whereas IPP formedfrom labeled acetyl-CoA by way of the cytosolic acetate/meval-onate pathway will be labeled at C-2, C-4, and C-5 (lower panel).
discovery of this new pathway for IPP for-mation in plastids suggests that these organ-elles, presumed to have originated as pro-karyotic endosymbionts, have retained thebacterial machinery for the production of thiskey intermediate of terpenoid biosynthesis.
The details of the glyceraldehyde 3-phosphate/pyruvate pathway and the en-zymes responsible have not yet been fullydefined. However, products of the two IPPbiosynthesis pathways can be easily dis-tinguished in experiments that utilize [1-13C]glucose as a precursor for terpenoidbiosynthesis. Nuclear magnetic resonance(NMR) spectroscopy (see Chapter 2, Box 2.2)
1258 Chapter 24 Natural Products (Secondary Metabolites)
C5
C10
C15
C20
C30
C40
MonoterpenesGeranyldiphosphate
Isopentenyldiphosphate
Dimethylallyldiphosphate
Hemiterpenes
CH2O CH2O
Farnesyldiphosphate
Sesquiterpenes
CH2O
Geranylgeranyldiphosphate
Diterpenes
CH2O
Squalene Triterpenes
Phytoene Tetraterpenes
CH2O (IPP)2�
2�
P P P P
P
IPP isomerase
P i
P P i
P P i
P2 P i
P2 P i
P P
P P
CH2O (IPP)P P
P P
CH2O P P
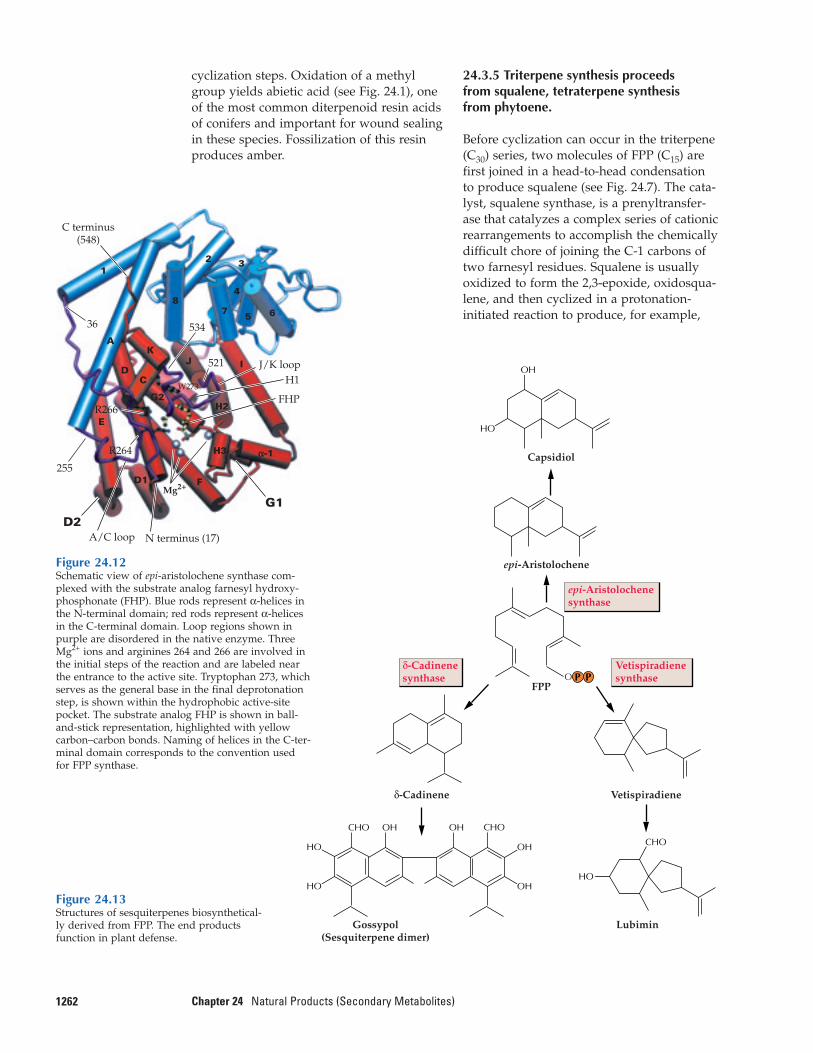
Figure 24.7The major subclasses of terpenoids are biosynthesized from the basic five-carbon unit, IPP, and from the initial prenyl (allylic)diphosphate, dimethylallyl diphosphate, which is formed by iso-merization of IPP. In reactions catalyzed by prenyltransferases,monoterpenes (C10), sesquiterpenes (C15), and diterpenes (C20) are
derived from the corresponding intermediates by sequential head-to-tail addition of C5 units. Triterpenes (C30) are formed from twoC15 (farnesyl) units joined head-to-head, and tetraterpenes (C40) areformed from two C20 (geranylgeranyl) units joined head-to-head.
can be used to determine the 13C-labelingpattern of each isoprene unit in a terpenoidcompound, allowing researchers to infer thelabeling pattern of the corresponding IPPunits (Fig. 24.6).
24.3 Prenyltransferase and terpenesynthase reactions
Prenyltransferase enzymes generate the al-lylic diphosphate esters geranyl diphosphate(GPP), farnesyl diphosphate (FPP), and ger-anylgeranyl diphosphate (GGPP). Reactionsthat these compounds undergo (often cycli-zations), which are catalyzed by terpene synthases, yield a wide variety of terpenoidcompounds. Both prenyltransferases and ter-pene synthases utilize electrophilic reactionmechanisms involving carbocationic inter-mediates, a feature of terpenoid biochem-istry. Enzymes in both groups share similarproperties and contain conserved sequenceelements, such as an aspartate-rich DDxxDmotif involved in substrate binding, whichmay participate in initiating divalent metalion–dependent ionizations.
24.3.1 Repetitive addition of C5 units iscarried out by prenyltransferases.
IPP is utilized in a sequence of elongationreactions to produce a series of prenyldiphosphate homologs, which serve as theimmediate precursors of the different fami-lies of terpenoids (Fig. 24.7). Isomerization ofIPP by IPP isomerase produces the allylicisomer dimethylallyl diphosphate (DMAPP),

125924.3–Prenyltransferase and Terpene Synthase Reactions
which is considered the first prenyl diphos-phate. Because DMAPP and related prenyldiphosphates contain an allylic double bond,these compounds can be ionized to generateresonance-stabilized carbocations. Onceformed, a carbocation intermediate of n car-bons can react with IPP to yield a prenyldiphosphate homolog containing n + 5 car-bons. Thus, the reactive primer DMAPP un-dergoes condensation with IPP to yield theC10 intermediate GPP. Repetition of the reac-tion cycle by addition of one or twomolecules of IPP provides FPP (C15) orGGPP (C20), respectively. Each prenyl ho-molog in the series arises as an allylic diphos-phate ester that can ionize to form a reso-nance-stabilized carbocation and condensewith IPP in another round of elongation(Fig. 24.8).
The electrophilic elongation reactionsthat yield C10, C15, and C20 prenyl diphos-phates are catalyzed by enzymes known col-lectively as prenyltransferases. GPP, FPP, andGGPP are each formed by specific prenyl-transferases named for their products (e.g.,farnesyl diphosphate synthase). The new al-lylic double bond introduced in the courseof the prenyltransferase reaction is common-ly in the trans geometry, although this is notalways the case: The transferase responsiblefor rubber biosynthesis introduces cis-doublebonds, which are responsible for the elastici-ty of that polymer. Prenylation reactions arenot limited to elongations involving IPP; thesame basic carbocationic mechanism permitsthe attachment of prenyl side chains to atomsof carbon, oxygen, nitrogen, or sulfur in a
wide range of nonterpenoid compounds, including proteins.
The most extensively studied prenyl-transferase, farnesyl diphosphate synthase,plays an important role in cholesterol bio-synthesis in humans. Farnesyl diphosphatesynthases from microbes, plants, and ani-mals exhibit high sequence conservation. Thefirst enzyme of the terpenoid pathway to bestructurally defined is recombinant avianfarnesyl diphosphate synthase, the crystalstructure of which has been determined.
24.3.2 The enzyme limonene synthase is amodel for monoterpene synthase action.
The families of enzymes responsible for theformation of terpenoids from GPP, FPP, andGGPP are known as monoterpene, sesquiter-pene, and diterpene synthases, respectively.These synthases use the correspondingprenyl diphosphates as substrates to formthe enormous diversity of carbon skeletonscharacteristic of terpenoids. Most terpenoidsare cyclic, and many contain multiple ringsystems, the basic structures of which aredetermined by the highly specific synthases.Terpenoid synthases that produce cyclicproducts are also referred to as “cyclases,”although examples of synthases producingacyclic products are also known.
A diverse array of monoterpene syn-thases has been isolated from essential oil-producing angiosperm species and resin-producing gymnosperms. These enzymesuse a common mechanism in which ionization
Figure 24.8The prenyltransferase reaction.
A divalent metal cation promotesthe ionization of an allylic diphosphatesubstrate, yielding a charge-delocalizedcation that probably remains paired withthe pyrophosphate anion.
The cation is added to IPP, generatinga tertiary carbocation that correspondsto the next C5 isoprenolog.
Deprotonation of the enzyme-boundintermediate yields an allylic (prenyl)diphosphate five carbons longer thanthe starting substrate.
R O
M2+H
R
H H+
R
OPP
R1
1 2 3
2 3PP P
O P PO P P
P P iAllylic diphosphate ester (n carbons) Allylic diphosphate ester (n + 5 carbons)
IPP
+
+
P i

of GPP leads initially to the tertiary allylicisomer linalyl diphosphate (LPP; Fig. 24.9).This isomerization step is required becauseGPP cannot cyclize directly, given the pres-ence of the trans-double bond. Ionization of the enzyme-bound LPP intermediate pro-motes cyclization to a six-membered ringcarbocation (the α-terpinyl cation), whichmay undergo additional electrophilic cycli-zations, hydride shifts, or other rearrange-ments before the reaction is terminated bydeprotonation of the carbocation or captureby a nucleophile (e.g., water). Variations onthis simple mechanistic scheme, involvingsubsequent reactions of the α-terpinyl carbo-cation, are responsible for the enzymatic for-mation of most monoterpene skeletons (seeBox 24.1).
The simplest monoterpene synthase re-action is catalyzed by limonene synthase, auseful model for all terpenoid cyclizations(Fig. 24.9). The electrophilic mechanism ofaction used by limonene synthase can beviewed as an intramolecular equivalent ofthe prenyltransferase reaction (see Fig. 24.8).Synthases that produce acyclic olefin prod-ucts (e.g., myrcene) and bicyclic products (α-and β-pinene) from GPP are also known, asare enzymes that transform GPP to oxy-genated derivatives such as 1,8-cineole andbornyl diphosphate (Fig. 24.10), the precur-sor of camphor (see Box 24.1).
An interesting feature of the monoter-pene synthases is the ability of these en-zymes to produce more than one product;for example, pinene synthase from severalplant sources produces both α- and β-pinene.The pinenes are among the most commonmonoterpenes produced by plants and areprincipal components of turpentine of the
1260 Chapter 24 Natural Products (Secondary Metabolites)
pines, spruces, and firs. The compounds aretoxic to bark beetles and their pathogenicfungal symbionts, which cause serious dam-age to conifer species worldwide. Many con-ifers respond to bark beetle infestation byup-regulating synthesis of monoterpenes, a process analogous to the production of antimicrobial phytoalexins, when underpathogen attack (Fig. 24.11). Other monoter-penes have quite different functions. Thus,linalool (see Fig. 24.10) and 1,8-cineole emit-ted by flowers serve as attractants for polli-nators, including bees, moths, and bats. 1,8-Cineole and camphor act as foliar feed-ing deterrents to large herbivores such ashares and deer and also may provide a competitive advantage to several angio-sperm species as allelopathic agents that inhibit germination of the seeds of otherspecies.
Exceptions to the general pattern ofhead-to-tail joining of isoprene units seen in limonene, the pinenes, and most othermonoterpenes derived from GPP are the “irregular” monoterpenes. An example ofthis type is the family of insecticidal mono-terpene esters called pyrethrins, found inChrysanthemum and Tanacetum species.These monoterpenoids, which exhibit ahead-to-middle joining of C5 units, havegained wide use as commercial insecticidesbecause of their negligible toxicity to mam-mals and their limited persistence in the environment (see Fig. 24.10).
24.3.3 Sesquiterpene synthases generateseveral compounds that function in plant defense.
The electrophilic mechanisms for the forma-tion of the C15 sesquiterpenes from FPPclosely resemble those used by monoterpenesynthases, although the increased flexibilityof the 15-carbon farnesyl chain eliminatesthe need for the preliminary isomerizationstep except in forming cyclohexanoid-typecompounds. The additional C5 unit and dou-ble bond of FPP also permit formation of agreater number of skeletal structures than in the monoterpene series. The best knownsesquiterpene synthase of plant origin is epi-aristolochene synthase from tobacco, thecrystal structure of which has been deter-mined (Fig. 24.12). This enzyme cyclizes FPP
Figure 24.9(–)-Limonene synthase catalyzes the simplest of allterpenoid cyclizations and serves as a model for thisreaction type. Ionization of GPP, assisted by divalentmetal ions, provides the delocalized carbocation–diphosphate anion pair, which collapses to form theenzyme-bound tertiary allylic intermediate linalyldiphosphate. This required isomerization step, fol-lowed by rotation about the C-2–C-3 single bond,overcomes the original stereochemical impediment todirect cyclization of the geranyl precursor. A subse-quent assisted ionization of the linalyl diphosphateester promotes an anti-endo-cyclization to the α-ter-pinyl cation, which undergoes deprotonation to formlimonene, a compound now thought to be an impor-tant cancer preventive in humans.
(3S)-Linalyl diphosphate(cisoid rotamer)
(3S)-Linalyl diphosphate(transoid rotamer)
α-Terpinyl cation
(–)-Limonene
M2+
M2+
Geranyl diphosphate
O
O
P
P i
P i
P i
P
P P
P
P
P
P iP
O P P
+
+
+

126124.3–Prenyltransferase and Terpene Synthase Reactions
and catalyzes a methyl migration to yieldthe olefin precursor of the phytoalexin capsidiol, which is elicited by pathogen at-tack. Vetispiradiene synthase from potatoprovides the olefin precursor of the phy-toalexin lubimin in this species, whereas δ-cadinene synthase from cotton yields theolefin precursor of the important defensecompound gossypol, the latter being current-ly studied as a possible male contraceptive(Fig. 24.13). Some sesquiterpene synthasesinvolved in the production of conifer resinare capable of individually producing morethan 25 different olefins.
24.3.4 Diterpene synthases catalyze twodistinct types of cyclization reactions.
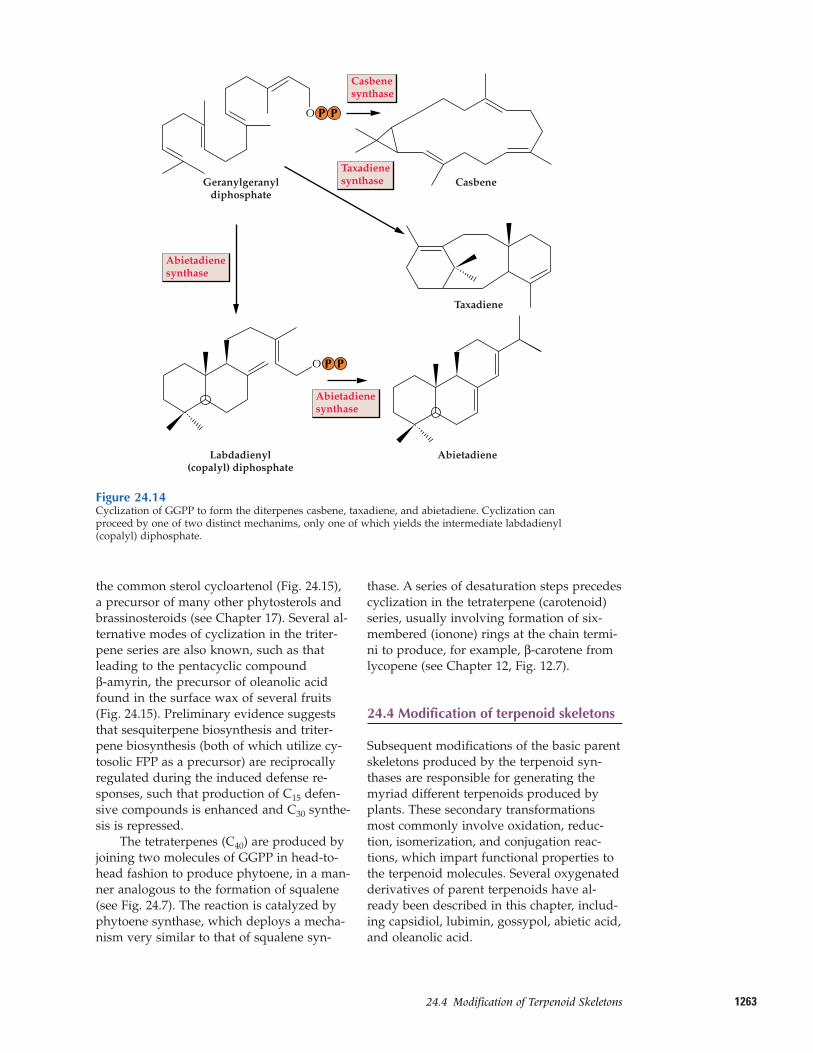
Two fundamentally different types of enzy-matic cyclization reactions occur in thetransformation of GGPP to diterpenes (Fig.24.14). The first resembles the reactions cat-alyzed by monoterpene and sesquiterpenesynthases, in which the cyclization involvesionization of the diphosphate ester and at-tack of the resulting carbocation on an interi-or double bond of the geranylgeranyl sub-strate. An example of this type is casbenesynthase, which is responsible for produc-tion of the phytoalexin casbene in castorbean. Taxadiene synthase from yew speciesuses a mechanistically similar, but morecomplex, cyclization to produce the tricyclicolefin precursor of taxol.
Abietadiene synthase from grand fir ex-emplifies the second type of cyclization, inwhich protonation of the terminal doublebond to generate a carbonium ion initiatesthe first cyclization to a bicyclic intermediate(labdadienyl diphosphate, also known as co-palyl diphosphate). Ionization of the diphos-phate ester promotes the second cyclizationstep to give the tricyclic olefin product, abi-etadiene; a single enzyme catalyzes both
Myrcene α-Pinene
Limonene
1,8-Cineole
Bornyl diphosphate Pyrethrin I
β-Pinene
O
Linalool
OH
COOR
O P P
Figure 24.10Structures of monoterpenes, including insecticidalcompounds (α- and β-pinene, pyrethrin), pollinatorattractants (linalool and 1,8-cineole), and antiher-bivory agents (1,8-cineole).
Figure 24.11Mass attack by mountain pine beetles on a lodgepolepine (Pinus contorta) bole. Each white spot on thetrunk represents a beetle entry point at which resinhas been secreted. This tree has survived the attackbecause turpentine production was sufficient to killall of the bark beetles, which have been “pitchedout” by resin outflow. On evaporation of the turpen-tine and exposure to air, the diterpenoid resin acidsform a solid plug that seals the wound.
cyclization steps. Oxidation of a methylgroup yields abietic acid (see Fig. 24.1), oneof the most common diterpenoid resin acidsof conifers and important for wound sealing in these species. Fossilization of this resinproduces amber.
1262 Chapter 24 Natural Products (Secondary Metabolites)
24.3.5 Triterpene synthesis proceeds from squalene, tetraterpene synthesis from phytoene.
Before cyclization can occur in the triterpene(C30) series, two molecules of FPP (C15) arefirst joined in a head-to-head condensationto produce squalene (see Fig. 24.7). The cata-lyst, squalene synthase, is a prenyltransfer-ase that catalyzes a complex series of cationicrearrangements to accomplish the chemicallydifficult chore of joining the C-1 carbons oftwo farnesyl residues. Squalene is usuallyoxidized to form the 2,3-epoxide, oxidosqua-lene, and then cyclized in a protonation-initiated reaction to produce, for example,
E
DC
KA
1
8
2
7 5 6
αα--1
4
3
J I
H2
H3
FD1
G2
36 534
521
W273
R266
R264
255
Mg2+
J/K loopH1
FHP
N terminus (17)A/C loop
C terminus(548)
G1
D2
Figure 24.12Schematic view of epi-aristolochene synthase com-plexed with the substrate analog farnesyl hydroxy-phosphonate (FHP). Blue rods represent α-helices inthe N-terminal domain; red rods represent α-helicesin the C-terminal domain. Loop regions shown inpurple are disordered in the native enzyme. ThreeMg2+ ions and arginines 264 and 266 are involved inthe initial steps of the reaction and are labeled nearthe entrance to the active site. Tryptophan 273, whichserves as the general base in the final deprotonationstep, is shown within the hydrophobic active-sitepocket. The substrate analog FHP is shown in ball-and-stick representation, highlighted with yellowcarbon–carbon bonds. Naming of helices in the C-ter-minal domain corresponds to the convention usedfor FPP synthase.
Figure 24.13Structures of sesquiterpenes biosynthetical-ly derived from FPP. The end productsfunction in plant defense.
OH
HO
CHO
HO
HO
HO
CHO OH CHOOH
OH
OH
P PO
epi-Aristolochene
δ-Cadinene
Capsidiol
Vetispiradiene
LubiminGossypol(Sesquiterpene dimer)
FPP
epi-Aristolochenesynthase
δ-Cadinenesynthase
Vetispiradienesynthase
126324.4–Modification of Terpenoid Skeletons
the common sterol cycloartenol (Fig. 24.15),a precursor of many other phytosterols andbrassinosteroids (see Chapter 17). Several al-ternative modes of cyclization in the triter-pene series are also known, such as thatleading to the pentacyclic compound β-amyrin, the precursor of oleanolic acidfound in the surface wax of several fruits(Fig. 24.15). Preliminary evidence suggeststhat sesquiterpene biosynthesis and triter-pene biosynthesis (both of which utilize cy-tosolic FPP as a precursor) are reciprocallyregulated during the induced defense re-sponses, such that production of C15 defen-sive compounds is enhanced and C30 synthe-sis is repressed.
The tetraterpenes (C40) are produced byjoining two molecules of GGPP in head-to-head fashion to produce phytoene, in a man-ner analogous to the formation of squalene(see Fig. 24.7). The reaction is catalyzed byphytoene synthase, which deploys a mecha-nism very similar to that of squalene syn-
thase. A series of desaturation steps precedescyclization in the tetraterpene (carotenoid)series, usually involving formation of six-membered (ionone) rings at the chain termi-ni to produce, for example, β-carotene fromlycopene (see Chapter 12, Fig. 12.7).
24.4 Modification of terpenoid skeletons
Subsequent modifications of the basic parentskeletons produced by the terpenoid syn-thases are responsible for generating themyriad different terpenoids produced byplants. These secondary transformationsmost commonly involve oxidation, reduc-tion, isomerization, and conjugation reac-tions, which impart functional properties tothe terpenoid molecules. Several oxygenatedderivatives of parent terpenoids have al-ready been described in this chapter, includ-ing capsidiol, lubimin, gossypol, abietic acid,and oleanolic acid.
Figure 24.14Cyclization of GGPP to form the diterpenes casbene, taxadiene, and abietadiene. Cyclization canproceed by one of two distinct mechanims, only one of which yields the intermediate labdadienyl(copalyl) diphosphate.
CasbeneGeranylgeranyldiphosphate
Labdadienyl(copalyl) diphosphate
Abietadiene
Taxadiene
O
O P P
P P
Casbenesynthase
Taxadienesynthase
Abietadienesynthase
Abietadienesynthase

Many of the hydroxylations or epoxida-tions involved in introducing oxygen atomsinto the terpenoid skeletons are performedby cytochrome P450 mixed-function oxidas-es. Because these reactions are not unique toterpenoid biosynthesis, this section will notfocus on specific enzyme types but rather onthe general role of secondary transforma-tions as the wellspring of diversity in ter-penoid structure and function.
24.4.1 The conversion of (–)-limonene to(–)-menthol in peppermint and carvone inspearmint illustrates the biochemistry ofterpenoid modification.
The principal and characteristic essential oilcomponents of peppermint (Mentha piperita)and spearmint (M. spicata) are produced bysecondary enzymatic transformations of (–)-limonene (Fig. 24.16). In peppermint, a microsomal cytochrome P450 limonene 3-hydroxylase introduces an oxygen atom at an allylic position to produce (–)-trans-isopiperitenol. A soluble NADP+-dependentdehydrogenase oxidizes the alcohol to a ke-tone, (–)-isopiperitenone, thereby activatingthe adjacent double bond for reduction by asoluble, NADPH-dependent, regiospecific
1264 Chapter 24 Natural Products (Secondary Metabolites)
reductase to produce (+)-cis-isopulegone. Anisomerase next moves the remaining doublebond into conjugation with the carbonylgroup, yielding (+)-pulegone. One regiospe-cific, NADPH-dependent, stereoselective reductase converts (–)-pulegone to either (+)-isomenthone or the predominant species,(–)-menthone. Similar reductases producethe menthol isomers from these ketones. (–)-Menthol greatly predominates among thementhol isomers (constituting as much as40% of the essential oil) and is the compo-nent primarily responsible for the character-istic flavor and cooling sensation of pepper-mint. The menthol isomers are often foundas acetate esters, formed by the action of anacetyl CoA-dependent acetyltransferase. Thementhol and menthyl acetate content of pep-permint oil glands increases with leaf matu-rity. Environmental factors greatly influenceoil composition. Water stress and warmnight growth conditions both promote theaccumulation of the more-oxidized pathwayintermediates such as (+)-pulegone.
The pathway in spearmint is muchshorter. In this instance, a cytochrome P450limonene 6-hydroxylase specifically intro-duces oxygen at the alternative allylic posi-tion to produce (–)-trans-carveol, which is oxidized to (–)-carvone by the soluble
Cycloartenol
HO
β-Amyrin
HO
Oleanolic acid
HO
COOH
Brassinolide
HO
HO
HO
OH
OO
Oxidosqualene
O
Figure 24.15Structures of triterpenes.This class of squalene-derived products in-cludes brassinosteroidregulators of plantgrowth and surface waxcomponents.
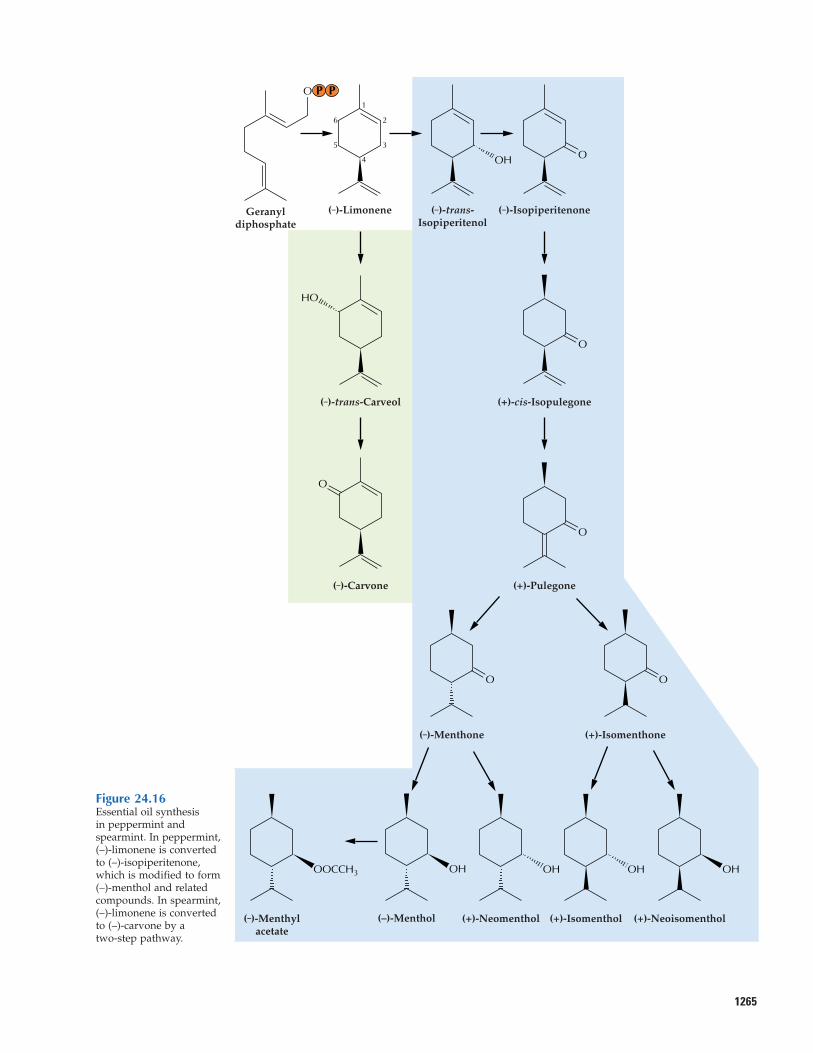
1265
Figure 24.16Essential oil synthesis in peppermint andspearmint. In peppermint,(–)-limonene is convertedto (–)-isopiperitenone,which is modified to form (–)-menthol and relatedcompounds. In spearmint,(–)-limonene is converted to (–)-carvone by a two-step pathway.
(–)-LimoneneGeranyldiphosphate
O
(–)-trans-Isopiperitenol
OH
HO
(–)-trans-Carveol
(–)-Isopiperitenone
(+)-cis-Isopulegone
(+)-Pulegone
(–)-Menthone (+)-Isomenthone
OH
(+)-Neoisomenthol
OH
(+)-Isomenthol
OH
(+)-Neomenthol
OH
(–)-Menthol
OOCCH3
(–)-Menthylacetate
(–)-Carvone
O
O
O
O
O O
1
4
2
35
6
PP

NADP+-dependent dehydrogenase. Al-though most of the enzymatic machinerypresent in peppermint oil glands is also present in spearmint, the specificity of theseenzymes is such that (–)-carvone is a verypoor substrate. Consequently, carvone, thecharacteristic component of spearmint flavor,accumulates as the major essential oil com-ponent (about 70%). Similar reaction se-quences initiated by allylic hydroxylationsand subsequent redox metabolism and con-jugations are very common in the monoter-pene, sesquiterpene, and diterpene classes.
24.4.2 Some terpenoid skeletons areextensively decorated.
Reactions similar to those responsible for essential oil production in mints generatemyriad terpenoid compounds of biologicalor pharmaceutical interest. Such reactionsconvert sesquiterpene olefin precursors tophytoalexins (see Fig. 24.13), allelopathicagents, and pollinator attractants. Additionalsesquiterpenes generated by modifyingolefin precursors include juvabione (Fig.24.17), a compound from fir species that exhibits insect juvenile hormone activity;sirenin, a sperm attractant of the water moldallomyces; and artemisinin, a potent anti-malarial drug from annual wormwood(Artemisia annua, also known as Qinghaosu,a plant used in traditional Chinese medicinesince about 200 B.C.). A related enzymaticreaction sequence converts the parent diter-pene olefin taxadiene to the anticancer drugtaxol in yew species, in which the basic ter-penoid nucleus is modified extensively by acomplex pattern of hydroxylations and acy-lations. Esters of phorbol (another highlyoxygenated diterpene) produced by speciesof the Euphorbiaceae are powerful irritantsand cocarcinogens. After introduction of ahydroxyl group, subsequent oxidation cangenerate a carboxyl function such as thatfound in abietic acid (see Fig. 24.1) andoleanolic acid, and also provide the struc-tural elements for lactone ring formation.Sesquiterpenes bearing such lactone rings,e.g., costunolide, are produced and accumu-lated in the glandular hairs on the leaf sur-faces of members of the Asteraceae, wheresome of these compounds serve as feedingrepellents to herbivorous insects and mam-
1266 Chapter 24 Natural Products (Secondary Metabolites)
mals. Monoterpene lactones include nepeta-lactone (the active principle of catnip as wellas an aphid pheromone), a member of the iridoid family of monoterpenes, which areformed by a cyclization reaction quite differentfrom that of other monoterpenes (Fig. 24.17).
The limonoids are a family of oxygenat-ed nortriterpene antiherbivore compounds.Like the sesquiterpene lactones, these sub-stances taste very bitter to humans and prob-ably to other mammals as well. A powerfulinsect antifeedant compound is azadirachtinA, a highly modified limonoid from the neemtree (Azadirachta indica). Other oxygenatedtriterpenoid natural products with unusualbiological properties include the phytoecdy-sones, a family of plant steroids that act ashormones and stimulate insect molting; thesaponins, so named because of their soap-like, detergent properties; and the cardeno-lides, which, like the saponins, are glyco-sides, in that they bear one or more attachedsugar residues. Ingestion of α-ecdysone byinsects disrupts the molting cycle, usuallywith fatal consequences. The saponins andcardenolides are toxic to many vertebrateherbivores; this family of compounds in-cludes well-known fish poisons and snailpoisons of significance in the control ofschistosomiasis. Many of these products arealso cardioactive and anticholesterolemicagents of pharmacological significance. Digi-toxin, the glycone (glycosylated form) ofdigitoxigenin (Fig. 24.17) extracted from fox-glove (Digitalis), is used widely in carefullyprescribed doses for treatment of congestiveheart disease.
The broad range of insect and higher an-imal toxins and deterrents among the modi-fied triterpenes leaves little doubt as to theirrole in plant defense. Interestingly, some her-bivores have developed the means to cir-cumvent the toxic effects of these terpenoidsand adapt them to their own defense pur-poses. The classical example of this phe-nomenon is the monarch butterfly, a special-ist feeder on milkweeds (Asclepias) whichcontain cardenolides that are toxic to mostherbivores and are even associated with live-stock poisoning. Monarch caterpillars, how-ever, feed on milkweeds and accumulate thecardenolides without apparent ill effects. Asa result, both caterpillars and the adult but-terflies contain enough cardenolides to betoxic to their own predators such as birds.
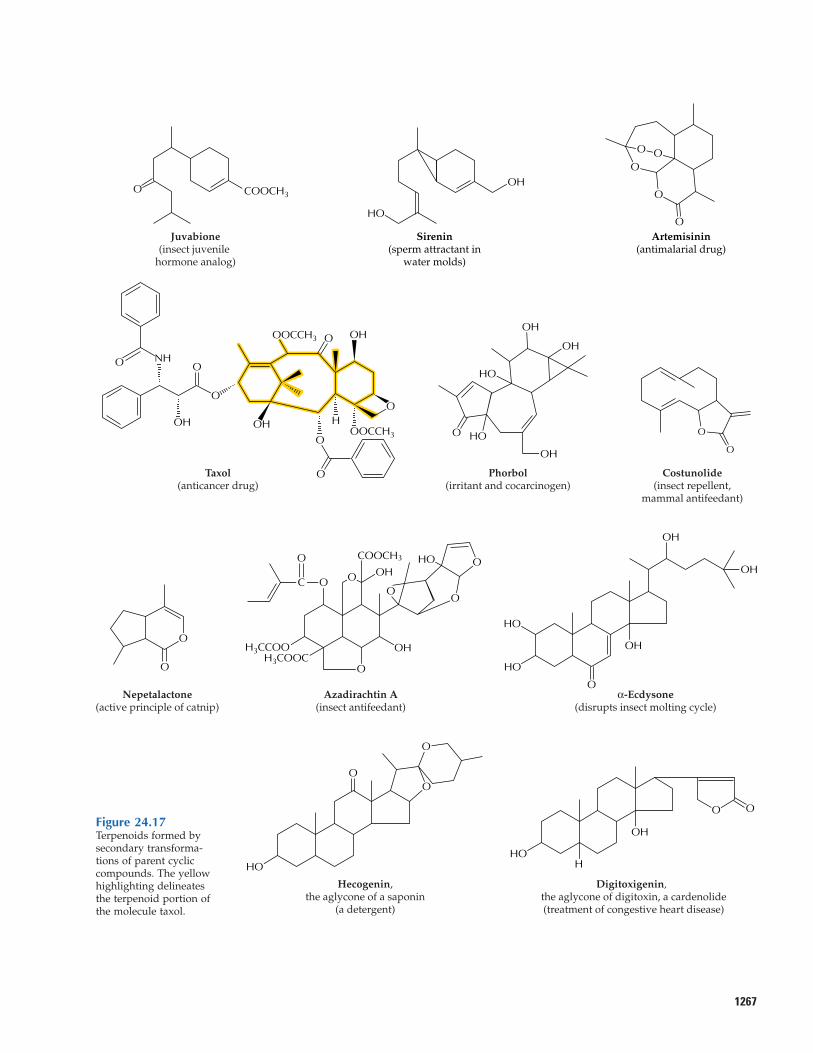
1267
O NH
O
O
Nepetalactone(active principle of catnip)
COOCH3
Sirenin(sperm attractant in
water molds)
Artemisinin(antimalarial drug)
O
Juvabione(insect juvenile
hormone analog)
O COOCH3
O
O
O
O
O
HO
HO
OH
OH
OH
Phorbol(irritant and cocarcinogen)
O
HO
O
O
Hecogenin,the aglycone of a saponin
(a detergent)
O
HOH
O
OH
Digitoxigenin, the aglycone of digitoxin, a cardenolide(treatment of congestive heart disease)
O
O
Costunolide(insect repellent,
mammal antifeedant)
O
HO
OH
OOO
OH3COOC
H3CCOO
O
OH
HO
COH
O
Azadirachtin A(insect antifeedant)
Oα-Ecdysone
(disrupts insect molting cycle)
OH
OH
OH
HO
HO
OO
HOH OHO
OOCCH3
OOCCH3 OH
Taxol(anticancer drug)
O
O
O
Figure 24.17Terpenoids formed bysecondary transforma-tions of parent cycliccompounds. The yellowhighlighting delineatesthe terpenoid portion ofthe molecule taxol.

24.5 Toward transgenic terpenoid production
With recent success in the cloning of genesthat encode enzymes of terpenoid synthesis,the transgenic manipulation of plant ter-penoid metabolism may present a suitableavenue for achieving a number of goals. Sev-eral agriculturally important crop specieshave been bred selectively to produce rela-tively low amounts of unpalatable terpenoiddefense compounds; in the process, thesecultivars have lost not only defense capabili-ties but also, in some cases, quality attributessuch as flavor and color. The selective rein-troduction of terpenoid-based defense chemistry is certainly conceivable, as is theengineering of pathways into fruits and veg-etables to impart desirable flavor properties.The aroma profiles of ornamental plantspecies might be modified by similar ap-proaches. Likewise, transgene expressionmight accelerate the rate of slow biosyntheticsteps and thereby increase the yields of essential oils used in flavors and perfumes,phytopharmaceuticals (e.g., artemisinin and taxol), insecticides (e.g., pyrethrins andazadirachtin), and a wide range of industrialintermediates that are economically inacces-sible by traditional chemical synthesis.
The genetic engineering of terpenoid-based insect defenses is particularly appeal-ing, given the array of available monoterpene,sesquiterpene, diterpene, and triterpenecompounds that are toxic to insects notadapted to them. Attracting predators andparasitoids of the target insect or modifyinghost attractants, oviposition stimulators, andpheromone precursors offers even more so-phisticated strategies for pest control. For ef-fective transgenic manipulation of such ter-penoid biosynthetic pathways, promoters fortissue-specific, developmentally controlled,and inducible expression are required, as arepromoters for targeting production to secre-tory structures of essential oil plants andconifers. The latter are the most likelyspecies for initial manipulation because theyalready are adapted for terpenoid accumula-tion, and the antecedent and subsequentmetabolic steps are largely known.
The engineering of terpenoid biosyn-thetic pathways into plant species that donot ordinarily accumulate these naturalproducts presents a greater opportunity but
1268 Chapter 24 Natural Products (Secondary Metabolites)
an even greater challenge, given that littlemetabolic context exists in these cases. Insuch species, issues of subcellular sites ofsynthesis, requirements for sufficiency ofprecursor flux, and the fate of the desiredproduct might present additional difficulties.Clearly, targeting a terpenoid synthase to thecellular compartment containing the appro-priate C10, C15, C20, or C30 precursor will bean important consideration. Sufficient flux ofIPP at the production site to drive the path-way also will be essential. Because con-straints in precursor flow ultimately willlimit the effectiveness of transgenes for sub-sequent pathway steps, information aboutthe flux controls on IPP biosynthesis in bothcytosol and plastid, and about the interac-tions of these controls, is sorely needed.
Very few published examples of the ge-netic engineering of terpenoid metabolismare currently available, although two notablesuccesses have been achieved in the area ofterpenoid vitamins. The ratio of beneficialtocopherol (vitamin E) isomers in oilseedshas been altered by this means, and an in-creased concentration of β-carotene (a vita-min A precursor) in both rice kernels andrapeseed has been obtained by manipulatingthe carotenoid pathway. In another, caution-ary example, however, overexpression in atransgenic tomato of the enzyme that divertsGGPP to carotenoids resulted in a dwarfphenotype, an unintended consequence ofdepleting the precursor of the gibberellinplant hormones.
24.6 Alkaloids
24.6.1 Alkaloids have a 3000-year historyof human use.
For much of human history, plant extractshave been used as ingredients in potionsand poisons. In the eastern Mediterranean,use of the latex of the opium poppy (Papaversomniferum; Fig. 24.18) can be traced back atleast to 1400 to 1200 B.C. The Sarpagandharoot (Rauwolfia serpentina) has been used inIndia since approximately 1000 B.C. Ancientpeople used medicinal plant extracts aspurgatives, antitussives, sedatives, and treat-ments for a wide range of ailments, includ-ing snakebite, fever, and insanity. As the useof medicinal plants spread westward across

126924.6–Alkaloids
Arabia and Europe, new infusions and de-coctions played a role in famous events. Dur-ing his execution in 399 B.C., the philoso-pher Socrates drank an extract of coniine-containing hemlock (Conium maculatum; Fig. 24.19). In the last century B.C., QueenCleopatra used extracts of henbane (Hyoscya-mus), which contains atropine (Fig. 24.20), todilate her pupils and appear more alluringto her male political rivals.
Over the centuries, the king of all medic-inals has been opium, which was widelyconsumed in the form of Theriak, a concoc-tion consisting mainly of opium, dried snakemeat, and wine (Box 24.2). Analysis of theindividual components of opium led to theidentification of morphine (Fig. 24.21A),named for Morpheus, the god of dreams inGreek mythology. The isolation of morphinein 1806 by German pharmacist FriedrichSertürner gave rise to the study of alkaloids.
The term alkaloid, coined in 1819 inHalle, Germany, by another pharmacist, CarlMeissner, finds its origin in the Arabic nameal-qali, the plant from which soda was firstisolated. Alkaloids were originally definedas pharmacologically active, nitrogen-containing basic compounds of plant origin.
(A) (B)
Figure 24.18(A) Maturing capsule of the opium poppy Pa-paver somniferum. When the capsule is wound-ed, a white, milky latex is exuded. Poppy latexcontains morphine and related alkaloids suchas codeine. When the exuded latex is allowedto dry, a hard, brown substance called opiumis formed. (B) Statuette from Gazi of a goddessof sleep crowned with capsules of the opiumpoppy (1250–1200 B.C.).
Figure 24.19(A). The piperidine alkaloid coniine, the first alkaloid to be synthe-sized, is extremely toxic, causing paralysis of motor nerve endings.(B) In 399 B.C., the philosopher Socrates was executed by consum-
ing an extract of coniine-containing poisonous hemlock. This depic-tion of the event, “The Death of Socrates,” was painted by Jacques-Louis David in 1787.
H
N
Coniine
Conium maculatum
H
(B)

After 190 years of alkaloid research, this def-inition as such is no longer comprehensiveenough to encompass the alkaloid field, butin many cases it is still appropriate. Alka-loids are not unique to plants. They havealso been isolated from numerous animalsources (Fig. 24.21B and Box 24.3), althoughstill to be determined is whether biosynthe-
1270 Chapter 24 Natural Products (Secondary Metabolites)
sis de novo occurs in each organism. Manyof the alkaloids that have been discoveredare not pharmacologically active in mam-mals and some are neutral rather than basicin character, despite the presence of a nitro-gen atom in the molecule.
Alkaloid-containing plants weremankind’s original “materia medica.” Manyare still in use today as prescription drugs(Table 24.1). One of the best-known prescrip-tion alkaloids is the antitussive and anal-gesic codeine from the opium poppy (Fig.24.21A). Plant alkaloids have also served asmodels for modern synthetic drugs, such asthe tropane alkaloid atropine for tropicamideused to dilate the pupil during eye examina-tions and the indole-derived antimalarial al-kaloid quinine for chloroquine (Fig. 24.22).
In addition to having a major impact onmodern medicine, alkaloids have also influ-enced world geopolitics. Notorious examplesinclude the Opium Wars between China andBritain (1839–1859) and the efforts currentlyunderway in various countries to eradicate
N
CH3
CH2OH
OOCCH
Hyoscyamus niger Atropine
Figure 24.20Stucture of the anti-cholinergic tropane alka-loid atropine fromHyoscyamus niger.
Box 24.2 Theriak, an ancient antipoisoning nostrum containing opium,wine, and snake meat, is still used today in rare instances.
One of the oldest and most long-livedmedications in the history of mankind isTheriak. Originating in Greco-Roman cul-ture, Theriak consists of mainly opiumand wine with a variety of plant, animal,and mineral constituents. Panel A of thefigure shows a recipe for Theriak from theFrench Pharmacopée Royale in 1676.
Theriak was developed as an antidoteagainst poisoning, snake bites, spiderbites, and scorpion stings. History has itthat the Roman Emperor Nero contractedthe Greek physician Andromachus to dis-cover a medicine that was effectiveagainst all diseases and poisons. Andro-machus improved the then-existing recipeto include, in addition to opium, five oth-er plant poisons and 64 plant drugs. An-other crucial component was dried snakemeat, believed to act against snake bitesby neutralizing the venom.
Today, Theriak is still prescribed inrare cases in Europe for pain and other ailments. Panel B shows a valuable Theriak-holding vessel made of Nym-phenburg porcelain (in about 1820),which is on display in the Residenz Phar-macy in Munich, Germany.
(A) (B)

127124.6–Alkaloids
illicit production of heroin, a semisyntheticcompound derived by acetylation of mor-phine (Fig. 24.23), and cocaine, a naturallyoccurring alkaloid of the coca plant (Fig.24.24). Because of their various pharmaco-logical activities, alkaloids have influenced
CodeinePapaver somniferum Morphine
(A)
H
H3CO
O O
H
HO
N CH3
HOHO
N CH3
(B)
Box 24.3 Some butterflies and moths use alkaloids for sexual signaling or forprotection against predators.
Alkaloid-bearing species have beenfound in nearly all classes of organisms,including frogs, ants, butterflies, bacteria,sponges, fungi, spiders, beetles, andmammals. Alkaloids of various structureshave been isolated from a variety of ma-rine creatures. Some animals, such as am-phibians, produce an array of either toxicor noxious alkaloids in the skin or the se-cretory glands. Others, such as the insectsdescribed below, use plant alkaloids as asource of attractants, pheromones, anddefense substances.
Some butterflies gather alkaloidal pre-cursors from plants that are not their foodsources and convert these compoundsinto pheromones and defense com-pounds. Larvae of the cinnabar moth, Tyr-ia jacobaea, continuously graze theirplant host Senecio jacobaea until theplant is completely defoliated (see panelA). The alkaloids thus obtained by the lar-vae are retained throughout metamorpho-sis. Male Asian and American arctiidmoths incorporate pyrrolizidine alkaloidsinto their reproductive biology by seques-tering these alkaloids in abdominal scentorgans called coremata, which are evert-ed in the final stages of their courtship torelease the pheromones necessary to gainacceptance by a female. The coremata ofa male Asian arctiid moth (Creatonotostransiens) are directly proportional to thepyrrolizidine alkaloid content of its dietduring the larval stage (see panel B). The
courtship success of these male butterfliestherefore depends on their ingesting alka-loids from higher plants.
The larvae of a second insect group,the Ithomiine butterflies, feed on solana-ceous plants and sequester the plant tox-ins, including tropane alkaloids andsteroidal glycoalkaloids. However, theadult Ithomiinae do not contain theseSolanaceae alkaloids but prefer to ingestplants that produce pyrrolizidine alka-loids, sequestering these bitter substancesas N-oxides and monoesters. The pyr-rolizidine alkaloid derivatives protectIthomiinae butterflies from an abundantpredator, the giant tropical orb spider. Thespider will release a field-caught butterflyfrom its web but will readily eat a freshlyemerged adult that has not yet had an opportunity to feed on the preferred hostplant. When palatable butterflies werepainted externally with a solution ofpyrrolizidine alkaloids, the spider re-leased them from its web. In contrast,palatable butterflies treated the same waywith Solanaceae alkaloids were devoured.In general, mostly male butterflies arefound feeding on the pyrrolizidine alka-loid-accumulating plants; however, asmuch as 50% of the pyrrolizidine alka-loids present in these males is sequesteredin the spermatophores and transferred tofemales at mating. In some butterflyspecies, the protective alkaloids are thentransferred to the eggs.
(A)
(B)
Figure 24.21(A) Structures of the alkaloids codeine and mor-phine from the opium poppy Papaver somnifer-um. Asymmetric (chiral) carbons are highlightedwith red dots. (B) The frog Bufo marinus accu-mulates a considerable amount of morphine inits skin.

human history profoundly, both for good andill. Of interest to plant biologists, however, isthe evolutionary selection process in plantsthat has caused alkaloids to evolve into sucha large number of complex structures and toremain effective over the millennia.
1272 Chapter 24 Natural Products (Secondary Metabolites)
24.6.2 Physiologically active alkaloidsparticipate in plant chemical defenses.
More than 12,000 alkaloids have been isolat-ed since the discovery of morphine. About20% of the species of flowering plants pro-duce alkaloids, and each of these species ac-cumulates the alkaloids in a unique, definedpattern. Some plants, such as the periwinkle(Catharanthus roseus) contain more than 100different monoterpenoid indole alkaloids.Why should a plant invest so much nitrogeninto synthesizing such a large number of al-kaloids of such diverse structure? The role ofalkaloids in plants has been a longstandingquestion, but a picture has begun to emergethat supports an ecochemical function forthese compounds.
The role of chemical defense for alka-loids in plants is supported by their widerange of physiological effects on animals andby the antibiotic activities many alkaloidspossess. Various alkaloids also are toxic toinsects or function as feeding deterrents. For example, nicotine, found in tobacco, wasone of the first insecticides used by humansand remains one of the most effective (Fig.24.25). Herbivory has been found to stimu-late nicotine biosynthesis in wild tobaccoplants. Another effective insect toxin is caf-feine, found in seeds and leaves of cocoa,
Cinchona officinalis
CH3O
N
N
CHCH2
HHO
Quinine
H
H
Table 24.1—Physiologically active alkaloids used in modern medicine
Alkaloid Plant source Use
Ajmaline Rauwolfia serpentina Antiarrythmic that functions by inhibiting glucose uptake by heart tissue mitochondria
Atropine, Hyoscyamus niger Anticholinergic, antidote to nerve gas poisoning-(±)-hyoscyamineCaffeine Coffea arabica Widely used central nervous system stimulantCamptothecin Camptotheca acuminata Potent anticancer agentCocaine Erythroxylon coca Topical anaesthetic, potent central nervous system stimulant, and adrenergic
blocking agent; drug of abuseCodeine Papaver somniferum Relatively nonaddictive analgesic and antitussiveConiine Conium maculatum First alkaloid to be synthesized; extremely toxic, causes paralysis of motor nerve
endings, used in homeopathy in small dosesEmetine Uragoga ipecacuanha Orally active emetic, amoebicideMorphine P. somniferum Powerful narcotic analgesic, addictive drug of abuseNicotine Nicotiana tabacum Highly toxic, causes respiratory paralysis, horticultural insecticide; drug of abusePilocarpine Pilocarpus jaborandi Peripheral stimulant of the parasympathetic system, used to treat glaucomaQuinine Cinchona officinalis Traditional antimalarial, important in treating Plasmodium falciparum strains that
are resistant to other antimalarialsSanguinarine Eschscholzia californica Antibacterial showing antiplaque activity, used in toothpastes and oral rinsesScopolamine H. niger Powerful narcotic, used as a sedative for motion sicknessStrychnine Strychnos nux-vomica Violent tetanic poison, rat poison, used in homeopathy(+)-Tubocurarine Chondrodendron tomentosm Nondepolarizing muscle relaxant producing paralysis, used as an adjuvant
to anaesthesiaVinblastine Catharanthus roseus Antineoplastic used to treat Hodgkin’s disease and other lymphomas.
Figure 24.22Structure of the monoterpenoid indole alkaloid-derived quinine from Cinchona officinalis. An anti-malarial quinine-containing tonic prepared from the bark of C. officinalis greatly facilitated Europeanexploration and inhabitation of the tropics during the past two centuries.
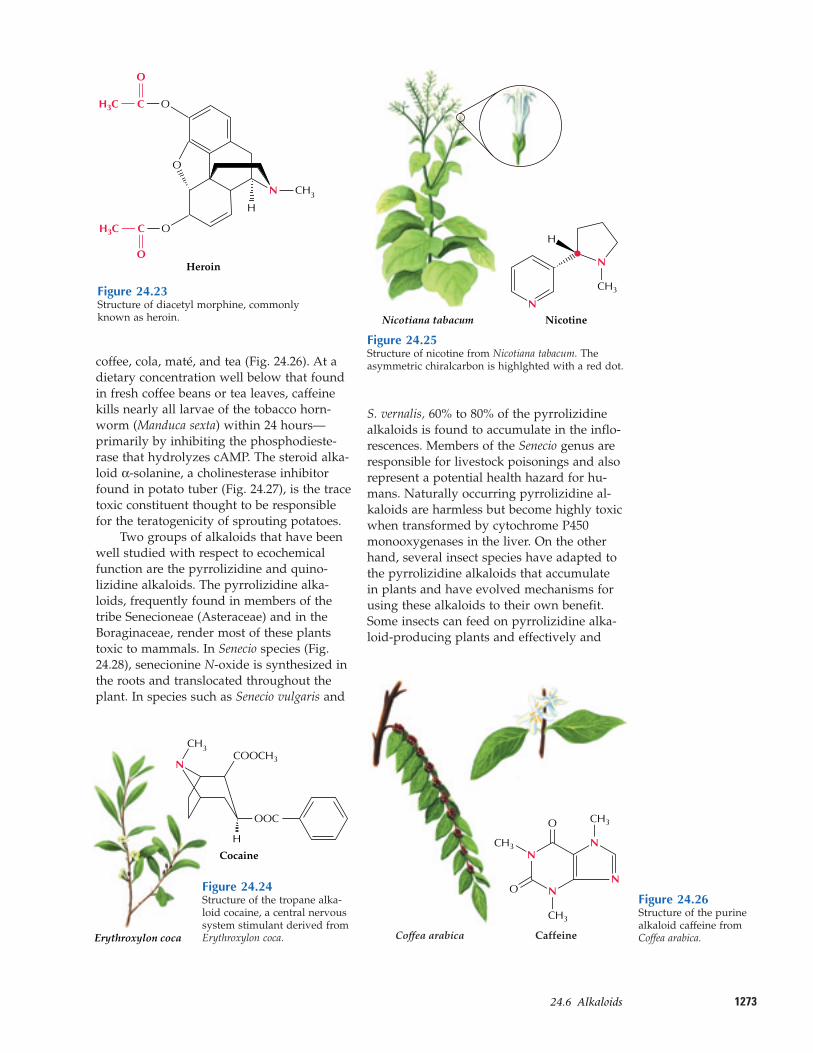
127324.6–Alkaloids
coffee, cola, maté, and tea (Fig. 24.26). At adietary concentration well below that foundin fresh coffee beans or tea leaves, caffeinekills nearly all larvae of the tobacco horn-worm (Manduca sexta) within 24 hours—primarily by inhibiting the phosphodieste-rase that hydrolyzes cAMP. The steroid alka-loid α-solanine, a cholinesterase inhibitorfound in potato tuber (Fig. 24.27), is the tracetoxic constituent thought to be responsiblefor the teratogenicity of sprouting potatoes.
Two groups of alkaloids that have beenwell studied with respect to ecochemicalfunction are the pyrrolizidine and quino-lizidine alkaloids. The pyrrolizidine alka-loids, frequently found in members of thetribe Senecioneae (Asteraceae) and in theBoraginaceae, render most of these plantstoxic to mammals. In Senecio species (Fig.24.28), senecionine N-oxide is synthesized inthe roots and translocated throughout theplant. In species such as Senecio vulgaris and
S. vernalis, 60% to 80% of the pyrrolizidinealkaloids is found to accumulate in the inflo-rescences. Members of the Senecio genus areresponsible for livestock poisonings and alsorepresent a potential health hazard for hu-mans. Naturally occurring pyrrolizidine al-kaloids are harmless but become highly toxicwhen transformed by cytochrome P450monooxygenases in the liver. On the otherhand, several insect species have adapted tothe pyrrolizidine alkaloids that accumulatein plants and have evolved mechanisms forusing these alkaloids to their own benefit.Some insects can feed on pyrrolizidine alka-loid-producing plants and effectively and
O
Heroin
O
OCH3C
O
CH3C
H
O
N CH3
Figure 24.23Structure of diacetyl morphine, commonlyknown as heroin.
Erythroxylon coca
N
CH3
H
OOC
COOCH3
Cocaine
Figure 24.24Structure of the tropane alka-loid cocaine, a central nervoussystem stimulant derived fromErythroxylon coca.
N
N
H
CH3
NicotineNicotiana tabacum
Figure 24.25Structure of nicotine from Nicotiana tabacum. Theasymmetric chiralcarbon is highlghted with a red dot.
Caffeine
O
O
CH3
CH3
CH3
N
NN
N
Coffea arabica
Figure 24.26Structure of the purinealkaloid caffeine fromCoffea arabica.
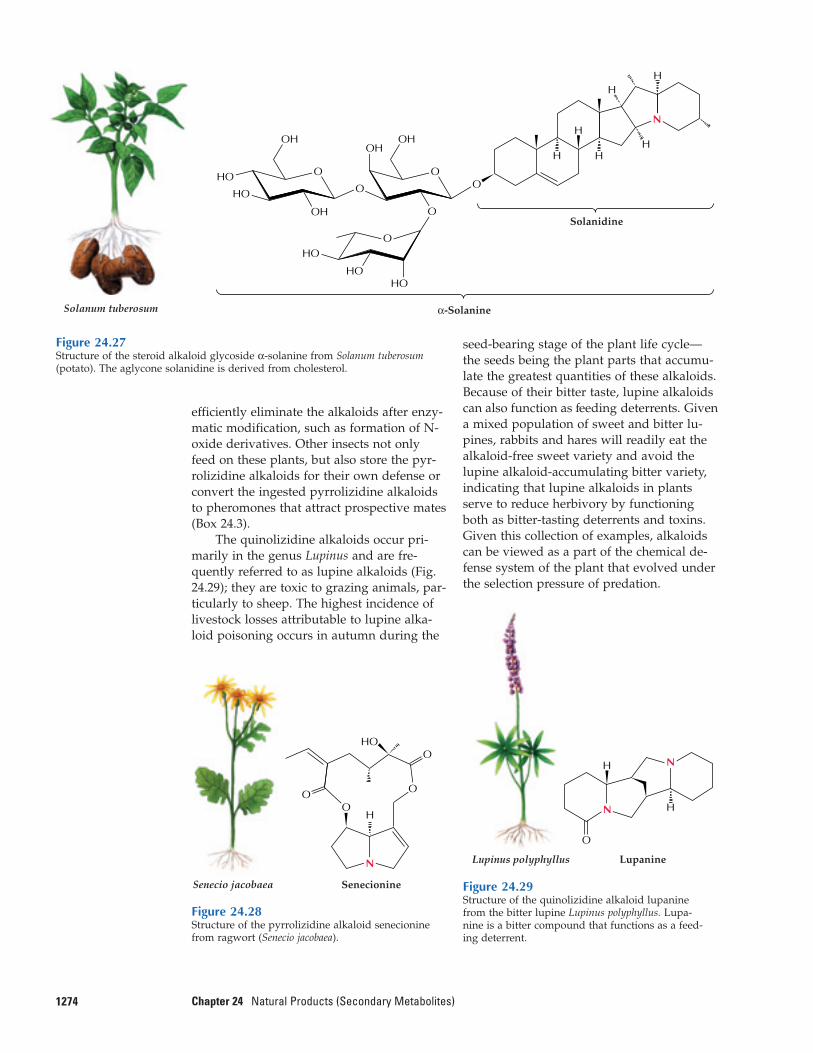
efficiently eliminate the alkaloids after enzy-matic modification, such as formation of N-oxide derivatives. Other insects not onlyfeed on these plants, but also store the pyr-rolizidine alkaloids for their own defense orconvert the ingested pyrrolizidine alkaloidsto pheromones that attract prospective mates(Box 24.3).
The quinolizidine alkaloids occur pri-marily in the genus Lupinus and are fre-quently referred to as lupine alkaloids (Fig.24.29); they are toxic to grazing animals, par-ticularly to sheep. The highest incidence oflivestock losses attributable to lupine alka-loid poisoning occurs in autumn during the
1274 Chapter 24 Natural Products (Secondary Metabolites)
Figure 24.27Structure of the steroid alkaloid glycoside α-solanine from Solanum tuberosum(potato). The aglycone solanidine is derived from cholesterol.
Solanum tuberosum α-Solanine
Solanidine
O
OH
HO
OH
HO
HO
HOHO
O
OH
O
O
O
OH
O
H H
HH
HH
N
O
O
N
O
OH
HO
Senecio jacobaea Senecionine
Figure 24.28Structure of the pyrrolizidine alkaloid senecioninefrom ragwort (Senecio jacobaea).
O
N
N H
H
LupanineLupinus polyphyllus
Figure 24.29Structure of the quinolizidine alkaloid lupaninefrom the bitter lupine Lupinus polyphyllus. Lupa-nine is a bitter compound that functions as a feed-ing deterrent.
seed-bearing stage of the plant life cycle—the seeds being the plant parts that accumu-late the greatest quantities of these alkaloids.Because of their bitter taste, lupine alkaloidscan also function as feeding deterrents. Givena mixed population of sweet and bitter lu-pines, rabbits and hares will readily eat thealkaloid-free sweet variety and avoid thelupine alkaloid-accumulating bitter variety,indicating that lupine alkaloids in plantsserve to reduce herbivory by functioningboth as bitter-tasting deterrents and toxins.Given this collection of examples, alkaloidscan be viewed as a part of the chemical de-fense system of the plant that evolved underthe selection pressure of predation.

127524.6–Alkaloids
24.6.3 Alkaloid biosynthesis research hasbeen greatly aided by the development oftechniques for culturing plant cells.
Many alkaloids have complex chemicalstructures and contain multiple asymmetriccenters, complicating structure elucidationand making study of the biosynthesis of al-kaloids quite difficult until relatively recent-ly. For example, although nicotine (oneasymmetric center; see Fig. 24.25) was dis-covered in 1828, its structure was not knownuntil it was synthesized in 1904, and thestructure of morphine (five asymmetric cen-ters; see Fig. 24.21) was not unequivocallyelucidated until 1952, almost 150 years afterits isolation. Almost all of the enzymes in-volved in the biosynthesis of these two alka-loids have been identified, but 190 years af-ter morphine was first isolated, itsbiosynthetic pathway remains incomplete.
Why has it been so difficult to elucidatealkaloid biosynthetic pathways? Plants syn-thesize natural products at a relatively slug-gish rate, so steady-state concentrations ofthe alkaloid biosynthetic enzymes are low. Inaddition, the large amounts of tannins andother phenolics that accumulate in plants in-terfere with the extraction of active enzymes.Even when plants are treated with radiola-beled precursors and the resulting radioactivealkaloids are chemically degraded to identifythe position of the label, the low rate of nat-ural product metabolism can prevent thehigh rates of incorporation that yield clearlyinterpretable results. The use of polyvinyl-pyrrolidone and Dowex-1 in preparing pro-tein extracts from plant tissues has helpedovercome the enzyme inactivation by pheno-lic compounds, but isolation of the enzymesinvolved in natural product synthesis hashad only limited success because of theirvery low concentrations in the plant.
Not until the 1970s were suspension cul-tures of plant cells established that were ca-pable of producing high concentrations of alkaloids (Fig. 24.30). As an experimentalsystem, cell culture provides several advan-
tages over whole-plant studies, including theyear-round availability of plant material; theundifferentiated, relatively uniform state ofdevelopment of the cells; the absence of in-terfering microorganisms; and most impor-tantly, the compressed vegetative cycle. Plantcell cultures can synthesize large amounts ofsecondary products within a two-week culti-vation period. This is very favorable in com-parison with in planta production, for whichthe time frame for alkaloid accumulationmay vary from one season for annual plantsto several years for some perennial species.In plant cell culture, the rate of alkaloidbiosynthesis can be increased, greatly facili-tating its study (Table 24.2). Moreover, thegreater metabolic rates associated with cellcultures promote the incorporation of la-beled precursors during alkaloid biosynthe-sis. Hormones regulate the accumulation ofalkaloids in culture, and in many cases, al-kaloid biosynthesis can be induced by theaddition of abiotic and biotic elicitor sub-stances to the culture. These advances haveprovided a powerful system with which toanalyze the regulation of alkaloid biosynthe-sis. Since the advent of alkaloid productionin culture, more than 80 new enzymes thatcatalyze steps in the biosynthesis of indole,isoquinoline, tropane, pyrrolizidine, acri-done, and purine classes of alkaloids havebeen discovered and partially characterized.
Figure 24.30Callus cultures estab-lished from plants canbe optimized to producehigh concentrations of awide variety of naturalproducts. In some of the examples shown,metabolite pigmentsgive the calli distinctivecolors.
Table 24.2—Production of selected alkaloids in plant cell culture
Secondary metabolite Species Yield (g/l) % Dry weight
Berberine Coptis japonica 7.0 12Jatrorrhizine Berberis wilsoniae 3.0 12Raucaffricine Rauwolfia serpentina 1.6 3
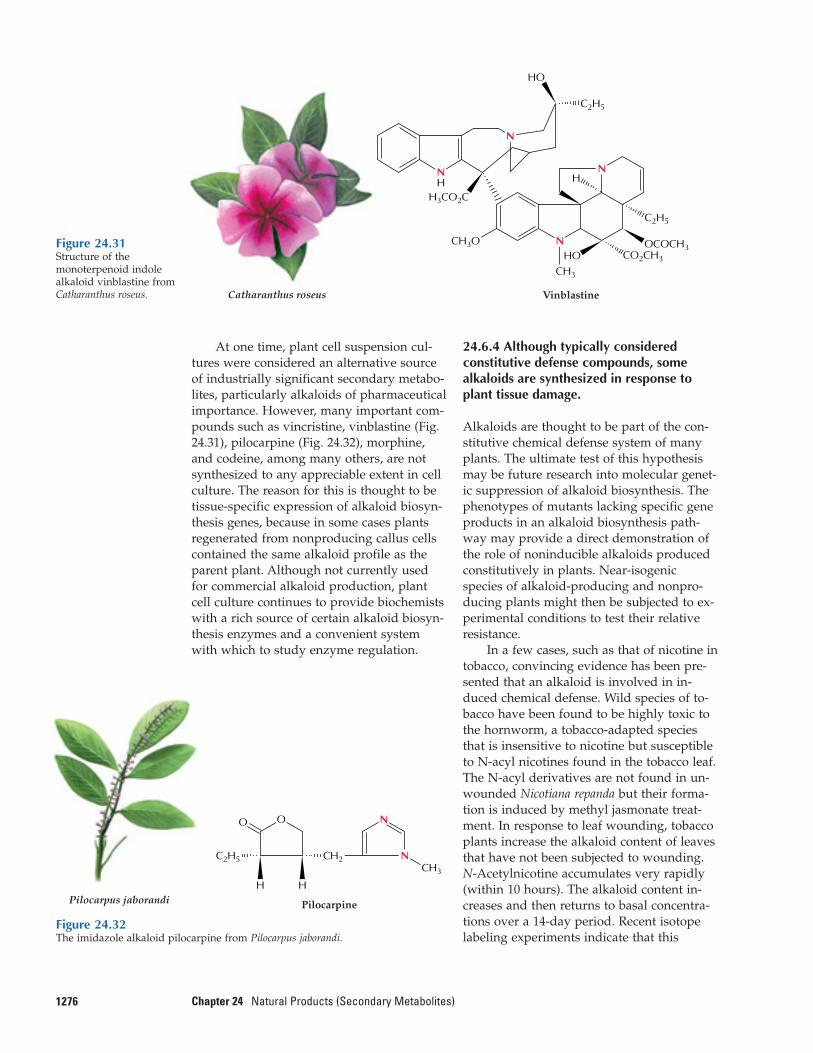
At one time, plant cell suspension cul-tures were considered an alternative sourceof industrially significant secondary metabo-lites, particularly alkaloids of pharmaceuticalimportance. However, many important com-pounds such as vincristine, vinblastine (Fig.24.31), pilocarpine (Fig. 24.32), morphine,and codeine, among many others, are notsynthesized to any appreciable extent in cellculture. The reason for this is thought to betissue-specific expression of alkaloid biosyn-thesis genes, because in some cases plantsregenerated from nonproducing callus cellscontained the same alkaloid profile as theparent plant. Although not currently usedfor commercial alkaloid production, plantcell culture continues to provide biochemistswith a rich source of certain alkaloid biosyn-thesis enzymes and a convenient systemwith which to study enzyme regulation.
1276 Chapter 24 Natural Products (Secondary Metabolites)
24.6.4 Although typically consideredconstitutive defense compounds, somealkaloids are synthesized in response toplant tissue damage.
Alkaloids are thought to be part of the con-stitutive chemical defense system of manyplants. The ultimate test of this hypothesismay be future research into molecular genet-ic suppression of alkaloid biosynthesis. Thephenotypes of mutants lacking specific geneproducts in an alkaloid biosynthesis path-way may provide a direct demonstration ofthe role of noninducible alkaloids producedconstitutively in plants. Near-isogenicspecies of alkaloid-producing and nonpro-ducing plants might then be subjected to ex-perimental conditions to test their relativeresistance.
In a few cases, such as that of nicotine intobacco, convincing evidence has been pre-sented that an alkaloid is involved in in-duced chemical defense. Wild species of to-bacco have been found to be highly toxic tothe hornworm, a tobacco-adapted speciesthat is insensitive to nicotine but susceptibleto N-acyl nicotines found in the tobacco leaf.The N-acyl derivatives are not found in un-wounded Nicotiana repanda but their forma-tion is induced by methyl jasmonate treat-ment. In response to leaf wounding, tobaccoplants increase the alkaloid content of leavesthat have not been subjected to wounding.N-Acetylnicotine accumulates very rapidly(within 10 hours). The alkaloid content in-creases and then returns to basal concentra-tions over a 14-day period. Recent isotopelabeling experiments indicate that this
VinblastineCatharanthus roseus
HO
HOCH3
C2H5
C2H5
H3CO2C
OCOCH3
N
N
N
NH H
CO2CH3
CH3O
Figure 24.32The imidazole alkaloid pilocarpine from Pilocarpus jaborandi.
PilocarpinePilocarpus jaborandi
OO
H
N
NCH3
H
CH2C2H5
Figure 24.31Structure of themonoterpenoid indolealkaloid vinblastine fromCatharanthus roseus.

127724.7–Alkaloid Biosynthesis
derivative is formed from a preexisting poolof nicotine. De novo nicotine biosynthesisoccurs in roots, followed by transport toleaves, but only after 36 hours. The increasein nicotine biosynthesis results in a 10-foldincrease of the alkaloid in the xylem fluid.
Freshly hatched hornworm larvae fedwounded leaves achieve only half theweight gain obtained by counterparts fedleaf material from unwounded plants. Re-cent studies demonstrate that, given thechoice, hornworms will abandon a woundedplant. Hornworms not permitted to leave awounded plant exhibit much higher mortali-ty rates and much lower growth rates thanthose fed on unwounded plants.
Inducible synthesis of nicotine and otheralkaloids appears to involve methyl jas-monate, a volatile plant growth regulator(see Chapter 17). Endogenous jasmonatepools increase rapidly when plant cells aretreated with an elicitor prepared from yeastcell walls. In turn, jasmonates are known toinduce accumulation of secondary metabo-lites in cell culture. More than 140 differentcultured plant species respond to the addi-tion of methyl jasmonate by increasing theirproduction of natural products. Althoughstudies of this type with intact plants are notas extensive as with cell suspension cultures,clear examples have been demonstrated withtobacco plants, in which leaf wounding pro-duces an increase in endogenous jasmonicacid pools in shoots and roots. Moreover, theapplication of methyl jasmonate to tobaccoleaves increases both endogenous jasmonicacid in roots and de novo nicotine biosyn-thesis. These results imply that jasmonatemay play a role in regulating the defense re-sponses of alkaloid-producing plants.
24.7 Alkaloid biosynthesis
24.7.1 Plants biosynthesize alkaloids from simple precursors, using many unique enzymes.
Until the mid-20th century, our view of howalkaloids are synthesized in plants wasbased on biogenic hypotheses. Pathwayssuggested by illustrious natural productchemists such as Sir Robert Robinson,Clemens Schöpf, Ernst Winterstein, andGeorg Trier were based on projections con-
sidered feasible within the realm of organicchemistry. In the 1950s, however, alkaloidbiosynthesis became an experimental sci-ence, as radioactively labeled organicmolecules became available for testing hy-potheses. These early precursor-feeding ex-periments clearly established that alkaloidsare in most cases formed from L-amino acids(e.g., tryptophan, tyrosine, phenylalanine, ly-sine, and arginine), either alone or in combi-nation with a steroidal, secoiridoid (e.g., sec-ologanin), or other terpenoid-type moiety.One or two transformations can convertthese ubiquitous amino acids from primarymetabolites to substrates for highly species-specific alkaloid metabolism. Although wedo not thoroughly understand how most ofthe 12,000 known alkaloids are made byplants, several well-investigated systems can serve as examples of types of buildingblocks and enzymatic transformations thathave evolved in alkaloid biosynthesis.
The L-tryptophan–derived monoter-penoid indole alkaloid ajmalicine was thefirst alkaloid for which biosynthesis wasclarified at the enzyme level (Fig. 24.33); inthat study plant cell suspension cultures ofthe Madagascar periwinkle C. roseus (see Fig.24.31) were used. In plants, the biosynthesisof ajmalicine and more than 1800 othermonoterpenoid indole alkaloids begins withthe decarboxylation of the amino acid L-tryptophan by tryptophan decarboxylase toform tryptamine. Then tryptamine, by actionof strictosidine synthase, is stereospecificallycondensed with the secoiridoid secologanin(derived in multiple enzymatic steps fromgeraniol) to form 3α-strictosidine. Strictosi-dine can then be enzymatically permutatedin a species-specific manner to form a multi-tude of diverse structures (Fig. 24.34). Theelucidation of the enzymatic formation of ajmalicine by using plant cell cultures laidthe groundwork for analysis of more-complex biosynthetic pathways, such asthose leading to two other L-tryptophan–derived monoterpenoid indole alkaloids, ajmaline (Fig. 24.35) and vindoline.
24.7.2 The berberine synthesis pathway hasbeen defined completely.
The first alkaloid for which each biosynthet-ic enzyme has been identified, isolated, andcharacterized from the primary metabolite
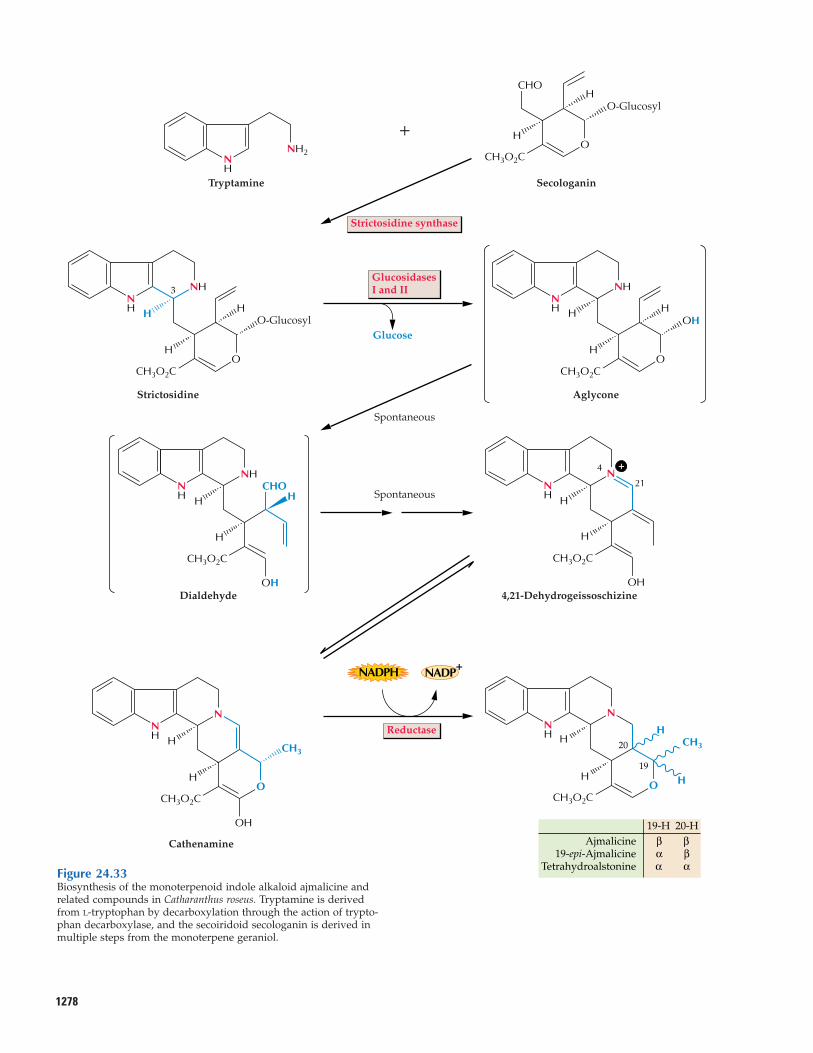
1278
NHNH H
H
HO-Glucosyl
Glucose
OCH3O2C
Strictosidine
NHNH H
O
OH
H
H
CH3O2C
Aglycone
NH H
NH
H
OH
HCHO
CH3O2C
Dialdehyde
NH H
H
OH
CH3O2C
21
4 N
4,21-Dehydrogeissoschizine
NH H
H
HCH3
H
N
OCH3O2C
NH H
N
H
OH
O
CH3
CH3O2C
Cathenamine Ajmalicine19-epi-Ajmalicine
Tetrahydroalstonine
19-H 20-H
20
3
19
β βα βα α
Tryptamine
NH
NH2
Secologanin
H
HO-Glucosyl
O
CHO
CH3O2C
+
Strictosidine synthase
GlucosidasesI and II
Spontaneous
Spontaneous
NADPH NADP+
Reductase
+
Figure 24.33Biosynthesis of the monoterpenoid indole alkaloid ajmalicine andrelated compounds in Catharanthus roseus. Tryptamine is derivedfrom L-tryptophan by decarboxylation through the action of trypto-phan decarboxylase, and the secoiridoid secologanin is derived inmultiple steps from the monoterpene geraniol.
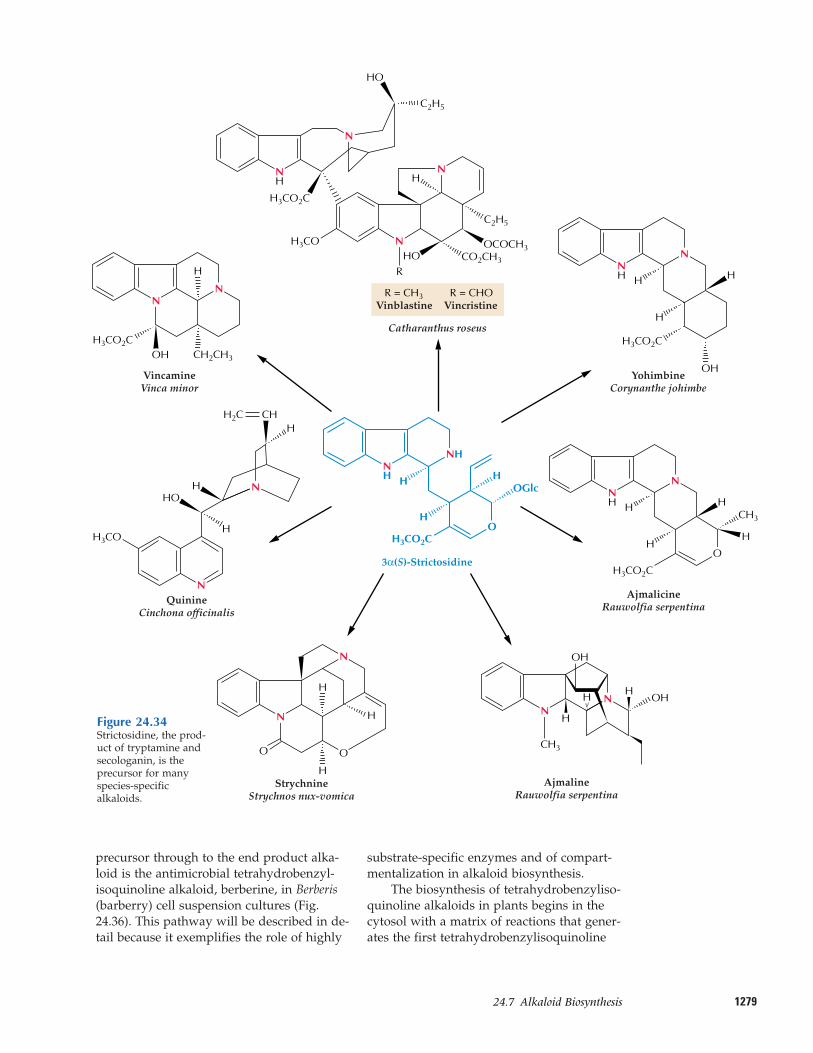
127924.7–Alkaloid Biosynthesis
precursor through to the end product alka-loid is the antimicrobial tetrahydrobenzyl-isoquinoline alkaloid, berberine, in Berberis(barberry) cell suspension cultures (Fig.24.36). This pathway will be described in de-tail because it exemplifies the role of highly
substrate-specific enzymes and of compart-mentalization in alkaloid biosynthesis.
The biosynthesis of tetrahydrobenzyliso-quinoline alkaloids in plants begins in thecytosol with a matrix of reactions that gener-ates the first tetrahydrobenzylisoquinoline
YohimbineCorynanthe johimbe
NH
N
H
H
OH
H
H3CO2C
Vinblastine
Catharanthus roseus
R = CH3
HO
HOR
C2H5
C2H5
H3CO2C
OCOCH3
N
N
N
H
CO2CH3
H3CO
AjmalicineRauwolfia serpentina
NH
N
O
HCH3
HH
H
H3CO2C
QuinineCinchona officinalis
H3CO
N
N
H
HCHH2C
HHO
AjmalineRauwolfia serpentina
StrychnineStrychnos nux-vomica
O O
H
H
H
N
N
VincamineVinca minor
H3CO2COH
H
CH2CH3
NN
3α(S)-Strictosidine
H3CO2CO
H
H
HOGlc
NHNH
H
H H
OH
OHNN
CH3
NH
VincristineR = CHO
Figure 24.34Strictosidine, the prod-uct of tryptamine andsecologanin, is the precursor for manyspecies-specific alkaloids.

alkaloid, (S)-norcoclaurine (Fig. 24.37). Thepathway proceeds from two molecules of L-tyrosine. One is decarboxylated to formtyramine or is acted on by a phenol oxidaseto form L-dopa. Dopamine can then beformed by decarboxylation of L-dopa or bythe action of a phenol oxidase on tyramine.Determining which of these two pathways ispredominant in a given plant is difficult be-cause all of the enzyme activities are presentin protein extracts. The benzyl moiety of (S)-norcoclaurine is formed by transamina-tion of the second L-tyrosine molecule toform p-hydroxyphenylpyruvate, which isnext decarboxylated to p-hydroxyphenyl-acetaldehyde. Dopamine and p-hydroxy-phenylacetaldehyde are then stereoselectively
1280 Chapter 24 Natural Products (Secondary Metabolites)
condensed to form (S)-norcoclaurine. Aseries of methylation and oxidation reac-tions yield the branchpoint intermediate of benzylisoquinoline alkaloid biosynthesis, (S)-reticuline (Fig. 24.38).
In Berberis, the N-methyl group of (S)-reticuline is oxidized to the berberinebridge carbon C-8 of (S)-scoulerine (see Fig. 24.37). The specific pathway from (S)-scoulerine that leads to berberine pro-ceeds with O-methylation to (S)-tetrahydro-columbamine. The 3-O-methyl moiety oftetrahydrocolumbamine is converted to themethylenedioxy bridge of canadine by cana-dine synthase, a microsomal cytochromeP450–dependent oxidase. The final step inthe biosynthesis of berberine is catalyzed by(S)-tetrahydroprotoberberine oxidase, an en-zyme shown to contain a covalently boundflavin. The end product alkaloid berberineaccumulates in the central vacuole of theBerberis cell.
The berberine bridge enzyme and (S)-tetrahydroprotoberberine oxidase arecompartmentalized together in vesicles ap-parently derived from the smooth endoplas-mic reticulum. Each of these enzymes con-sumes 1 mol of O2 and produces 1 mol ofH2O2 per mole of berberine formed. Overall,the course of reactions from 2 mol of L-tyro-sine to 1 mol of berberine consumes 4 mol of S-adenosylmethionine and 2 mol ofNADPH.
24.7.3 Elucidation of other alkaloidbiosynthetic pathways is progressing.
The enzymes that catalyze the biosynthesisof the benzophenanthridine alkaloid ma-carpine in the California poppy Eschscholziacalifornica have also been identified, isolated,and characterized, as have nearly all of theenzymes of morphine biosynthesis in theopium poppy (Fig. 24.39). Good progress has been made toward understanding theenzymatic formation of the tropane alkaloidscopolamine in Hyoscyamus niger and of the acridone alkaloid furofoline-I in Rutagraveolens.
Studies have revealed that the chemicaltransformations required for alkaloid biosyn-thesis are catalyzed by highly stereo-, regio-,and substrate-specific enzymes that are pres-ent only in specific species. These enzymes
Rauwolfia serpentina Ajmaline
H
HH
OH
OHNN
CH3
Figure 24.35Structure of the monoterpenoid indole alkaloid ajmaline from Rauwolfia serpentina.
Figure 24.36The Berberis wilsoniae plant (left) and cell suspension culture (right). The cell sus-pension culture derives its color from optimized production of the highly oxi-dized benzylisoquinoline alkaloid berberine. Plant cell cultures (like this one) thatproduce large quantities of alkaloids have led to the complete elucidation of sev-eral alkaloid biosynthetic pathways.
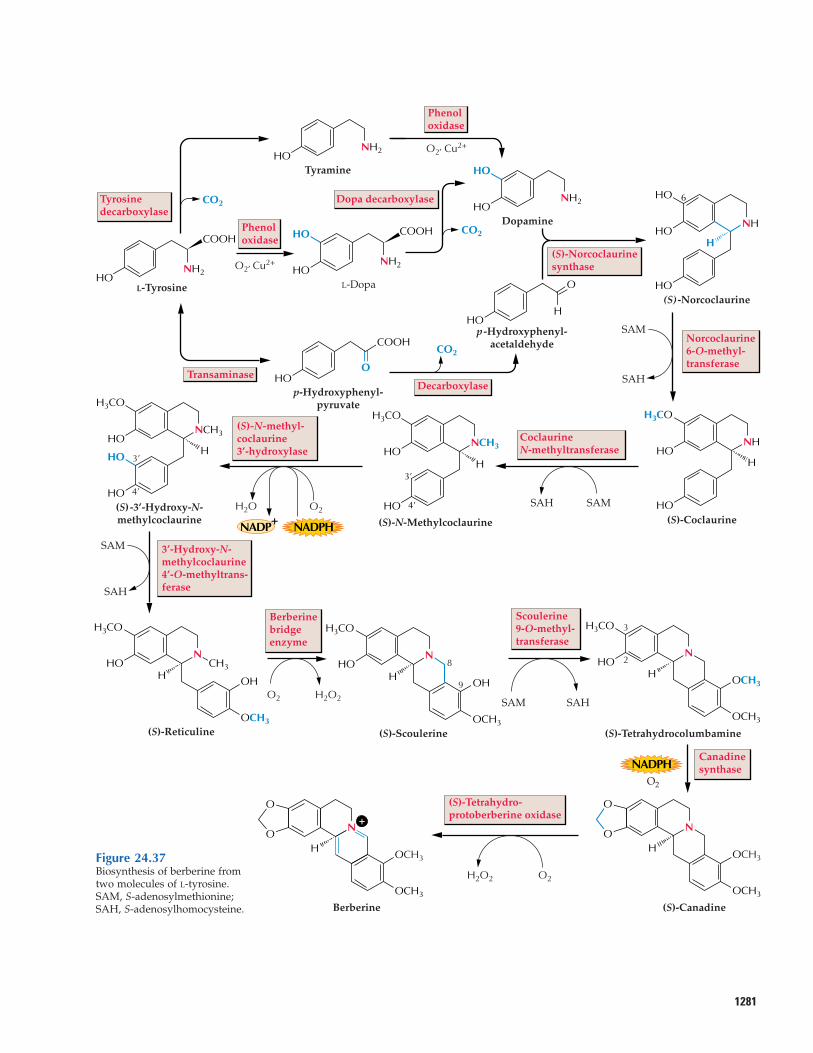
1281
CO2
Transaminase
Tyrosinedecarboxylase
Phenoloxidase
Dopa decarboxylase
Norcoclaurine6-O-methyl-transferase
Canadinesynthase
(S)-Tetrahydro-protoberberine oxidase
3’-Hydroxy-N-methylcoclaurine4’-O-methyltrans-ferase
(S)-Norcoclaurinesynthase
Decarboxylase
O2′ Cu2+
O2′ Cu2+
SAM
SAM
SAM
SAH
SAH
SAH
SAM
SAH
Phenoloxidase
CO2
CO2
CoclaurineN-methyltransferase
Scoulerine9-O-methyl-transferase
Berberinebridge enzyme
O2 H2O2
H
HO 6
HO
HO
NH
L-TyrosineHO
NH2
COOH
p-Hydroxyphenyl-pyruvate
OHO
COOH
HONH2
Tyramine HO
HONH2
Dopamine
O
HHO
p-Hydroxyphenyl-acetaldehyde
(S) -Norcoclaurine
HHO
H3CO
HO
NH
(S)-Coclaurine
3’
4’
8
9
3
2
H3’
4’HO
NCH3HO
H3CO
(S)-N-Methylcoclaurine
HO
H3CO
HO
HO
NCH3
H
(S)-3’-Hydroxy-N-methylcoclaurine
HO CH3
OH
OCH3
H
N
H3CO
(S)-Reticuline
HOH
OH
OCH3
N
H3CO
(S)-Scoulerine
H3CO
HON
H OCH3
OCH3
(S)-Tetrahydrocolumbamine
O2
O2H2O2
HO
O
N
OCH3
OCH3
(S)-Canadine
L-Dopa
HO
HONH2
COOH
Berberine
HO
O
OCH3
OCH3
N
NADPH
NADPH
(S)-N-methyl-coclaurine3’-hydroxylase
O2H2O
NADP+
+
Figure 24.37Biosynthesis of berberine fromtwo molecules of L-tyrosine.SAM, S-adenosylmethionine;SAH, S-adenosylhomocysteine.
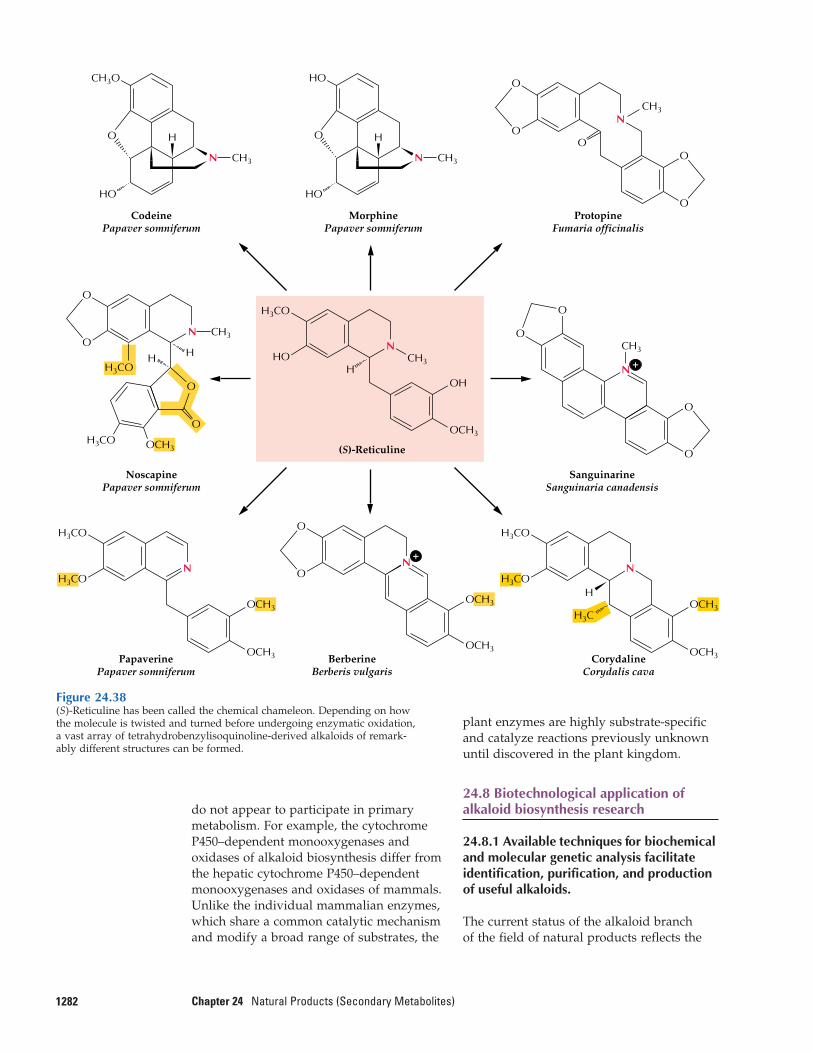
do not appear to participate in primarymetabolism. For example, the cytochromeP450–dependent monooxygenases and oxidases of alkaloid biosynthesis differ from the hepatic cytochrome P450–dependentmonooxygenases and oxidases of mammals.Unlike the individual mammalian enzymes,which share a common catalytic mechanismand modify a broad range of substrates, the
1282 Chapter 24 Natural Products (Secondary Metabolites)
plant enzymes are highly substrate-specificand catalyze reactions previously unknownuntil discovered in the plant kingdom.
24.8 Biotechnological application ofalkaloid biosynthesis research
24.8.1 Available techniques for biochemicaland molecular genetic analysis facilitateidentification, purification, and productionof useful alkaloids.
The current status of the alkaloid branch of the field of natural products reflects the
N
O
O
CH3
ProtopineFumaria officinalis
O
O
CH3
N
O
O
O
O
SanguinarineSanguinaria canadensis
OCH3
H3CO
CorydalineCorydalis cava
N
H
H3C
OCH3
OH
HO
H3CO
(S)-Reticuline
N
H
OCH3
H3CO
H3CO
PapaverinePapaver somniferum
NoscapinePapaver somniferum
N
CH3H3CO
O
O
O
O
H3CO
O
H H
CodeinePapaver somniferum
HO
N CH3
HO
CH3O
MorphinePapaver somniferum
HO
H
HO
O
O
O
BerberineBerberis vulgaris
N
OCH3
OCH3OCH3
OCH3
OCH3
H3CO
N CH3
N CH3
+
+
Figure 24.38(S)-Reticuline has been called the chemical chameleon. Depending on howthe molecule is twisted and turned before undergoing enzymatic oxidation,a vast array of tetrahydrobenzylisoquinoline-derived alkaloids of remark-ably different structures can be formed.
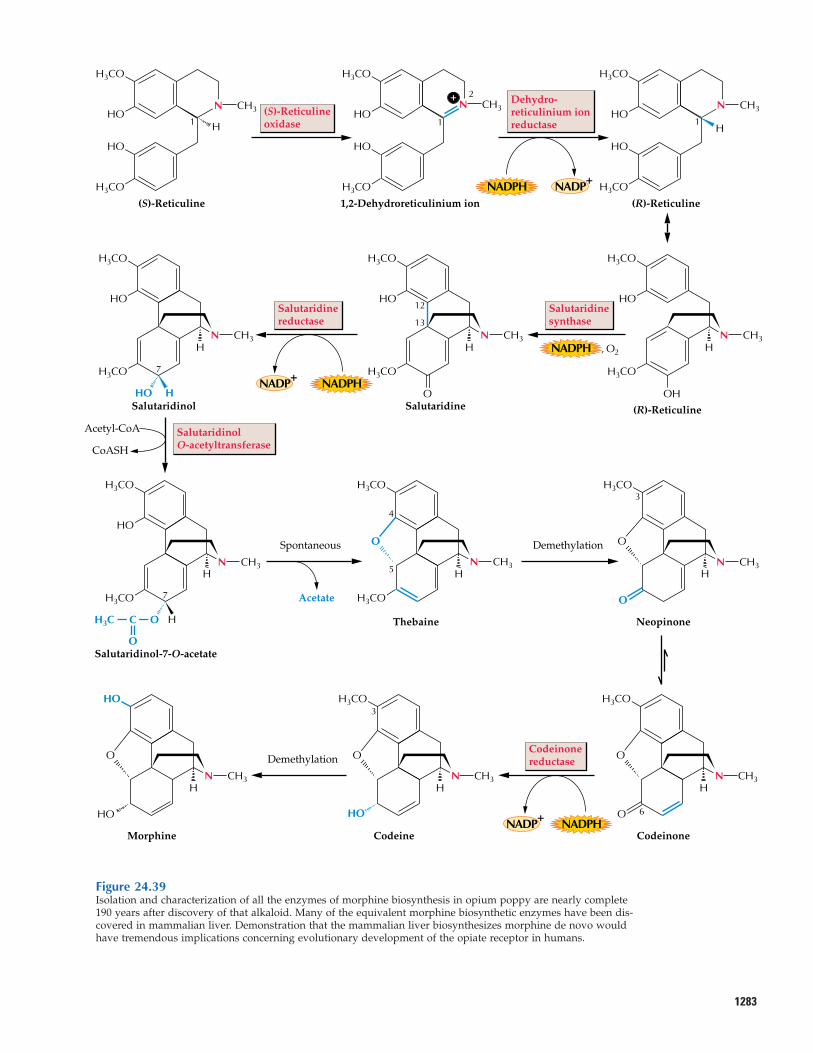
1283
H
H
(S)-Reticuline
H3CO
HO
HO1
H3CO
H
1,2-Dehydroreticulinium ion
H3CO
HO
HO1
2
H3CO
(S)-Reticulineoxidase
Dehydro-reticulinium ionreductase
SalutaridinolO-acetyltransferase
Salutaridinesynthase
(R)-Reticuline
(R)-Reticuline
H3CO
HO
HO1
H3CO
H3CO
HO
OH
H3CO
H
Salutaridinereductase
Salutaridine
H3CO
HO
H3CO
12
13
O
H
Salutaridinol
Acetyl-CoA
CoASH
H3CO
HO
HO H
7
NADPH
NADPH , O2
NADP+
NADPH NADP+
H3CO
H
Neopinone
H3CO
OO
O
H
Spontaneous Demethylation
Demethylation
Thebaine
H3CO
H3CO
3
3
4
5H
Salutaridinol-7-O-acetate
H3CO
H3C C
O
O
Acetate
HO
H
7H3CO
H
Codeinone
H3CO
O
O 6
H
CodeineMorphine
H3CO
OO
HO
H
HO
HO
Codeinonereductase
NADPHNADP+
N CH3
N CH3
N CH3
N CH3 N CH3 N CH3
N CH3 N CH3
N CH3N CH3
N CH3N CH3+
Figure 24.39Isolation and characterization of all the enzymes of morphine biosynthesis in opium poppy are nearly complete190 years after discovery of that alkaloid. Many of the equivalent morphine biosynthetic enzymes have been dis-covered in mammalian liver. Demonstration that the mammalian liver biosynthesizes morphine de novo wouldhave tremendous implications concerning evolutionary development of the opiate receptor in humans.

many new advances in analytical chemistry,enzymology, and pharmacology. Only mini-mal quantities of a pure alkaloid are nownecessary for a complete structure to be elucidated by mass and NMR spectroscopicanalyses. Absolute stereochemistry can beunambiguously assigned by determining thecrystal structure. The pharmacological activi-ties of crude plant extracts or pure substancesare determined by fully automated systems,such that millions of data points are collectedeach year in industrial screening programs.The factor that limits the number of biologi-cal activities for which we can test is thenumber of available target enzymes and re-ceptors. As more of the underlying biochem-ical bases for diseases continue to be discov-ered, the number of test systems will increase.
What happens when a small quantity ofan alkaloid of complex chemical structurefrom a rare plant is found to be physiologi-cally active? The alkaloid must first pass ani-mal and clinical trials; if these are successful,eventually enough material will be neededto satisfy market demand. Researchers candevelop biomimetic syntheses, which dupli-cate at least part of the biosynthetic pathwaysof plants to yield synthetic compounds; al-ternatively, they can alter the metabolism ofthe plant to change the alkaloid profile (Fig.24.40). The regulation of alkaloid biosynthe-sis in cell culture can also be influenced toproduce a desired alkaloid. The followingsuccessful studies demonstrate the viabilityof these approaches.
24.8.2 Metabolic engineering of medicinalplants may be the pharmaceuticalbiotechnology of the future.
The tropane class of alkaloids, found mainlyin the Solanaceae, contains the anticholiner-gic drugs hyoscyamine and scopolamine.Solanaceous plants have been used tradition-ally for their medicinal, hallucinogenic, andpoisonous properties, which derive, in part,from tropane alkaloids. For obtaining im-proved sources of pharmaceuticals, metabol-ic engineering of the plants that serve ascommercial sources of scopolamine couldaugment classical breeding in the effort todevelop plants with an optimal alkaloid pattern. The current commercial source ofscopolamine is Duboisia, which is cultivated
1284 Chapter 24 Natural Products (Secondary Metabolites)
on plantations in Australia, Indonesia, andBrazil. Certain other tropane alkaloid–pro-ducing species accumulate hyoscyamine in-stead of scopolamine as the major alkaloid.The question arises whether expression of atransgene in a medicinal plant would alterthe alkaloid-producing pattern such thatmore of the pharmaceutically useful alka-loid, scopolamine, is obtained. To this end, a
Figure 24.40Using antisense/cosuppression technologies (seeChapter 7) or overexpression, medicinal plants canbe tailored to produce pharmaceutically importantalkaloids by eliminating interfering metabolic stepsor by introducing desired metabolic steps. Express-ing an entire alkaloid biosynthesis pathway of 20 to30 enzymes in a single microorganism is currentlybeyond our technical capability. However, alteringthe pathway in a plant and producing the desired al-kaloid either in culture or in the field may now bepossible. For example, to accumulate more of the endproduct alkaloid, a side pathway that also uses thesame precursor may have to be blocked (A). To accu-mulate an alkaloid not normally produced in a par-ticular plant species, a transgene (from another plantor a microorganism) may be introduced (B). If theend product alkaloid would be more useful as a par-ticular derivative, for example, as a more soluble gly-coside, a gene that encodes a glycosyl transferasecould be introduced (C).
Precursor
Product 3
Product 4
Introducetransgene
Product 2
Introducetransgene
Product 1
(A)
(B)
(C)
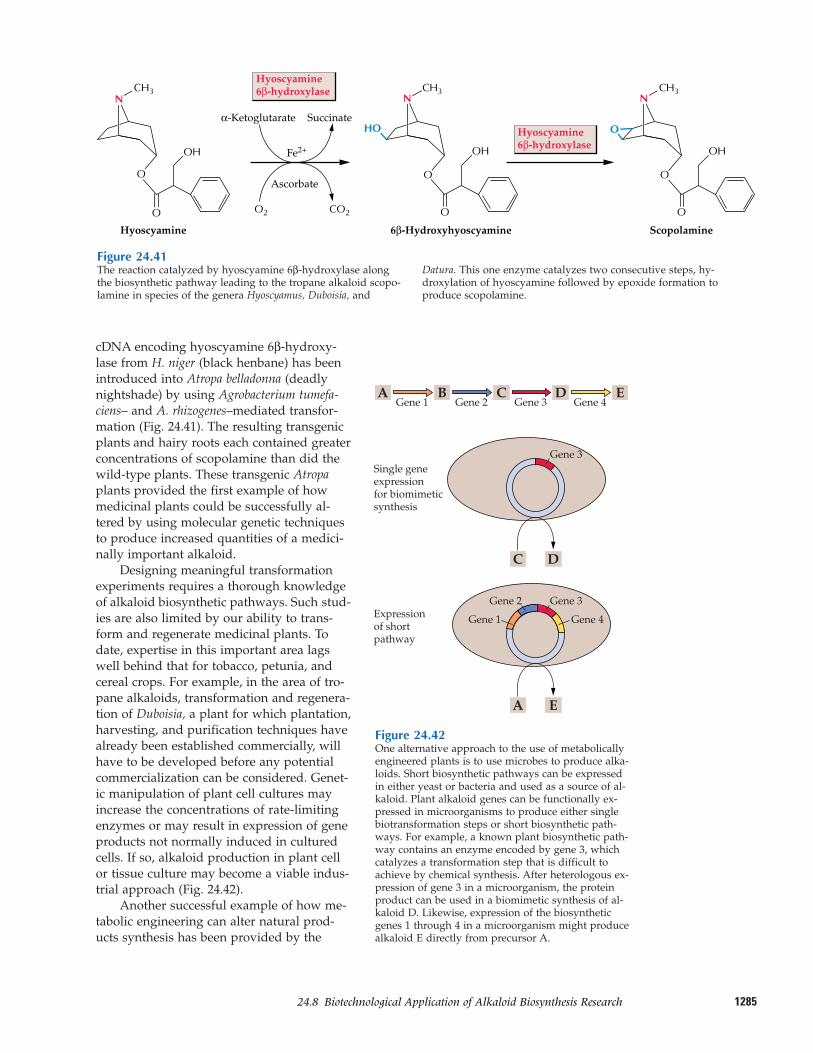
128524.8–Biotechnological Application of Alkaloid Biosynthesis Research
cDNA encoding hyoscyamine 6β-hydroxy-lase from H. niger (black henbane) has beenintroduced into Atropa belladonna (deadlynightshade) by using Agrobacterium tumefa-ciens– and A. rhizogenes–mediated transfor-mation (Fig. 24.41). The resulting transgenicplants and hairy roots each contained greaterconcentrations of scopolamine than did thewild-type plants. These transgenic Atropaplants provided the first example of howmedicinal plants could be successfully al-tered by using molecular genetic techniquesto produce increased quantities of a medici-nally important alkaloid.
Designing meaningful transformationexperiments requires a thorough knowledgeof alkaloid biosynthetic pathways. Such stud-ies are also limited by our ability to trans-form and regenerate medicinal plants. Todate, expertise in this important area lagswell behind that for tobacco, petunia, andcereal crops. For example, in the area of tro-pane alkaloids, transformation and regenera-tion of Duboisia, a plant for which plantation,harvesting, and purification techniques havealready been established commercially, willhave to be developed before any potentialcommercialization can be considered. Genet-ic manipulation of plant cell cultures mayincrease the concentrations of rate-limitingenzymes or may result in expression of geneproducts not normally induced in culturedcells. If so, alkaloid production in plant cellor tissue culture may become a viable indus-trial approach (Fig. 24.42).
Another successful example of how me-tabolic engineering can alter natural prod-ucts synthesis has been provided by the
CO2O2
OH
Hyoscyamine6β-hydroxylase
Hyoscyamine6β-hydroxylase
α-Ketoglutarate Succinate
Ascorbate
Hyoscyamine 6β-Hydroxyhyoscyamine Scopolamine
Fe2+
NCH3
NCH3
NCH3
O
O
OH
O
O
HO
OH
O
O
O
Figure 24.41The reaction catalyzed by hyoscyamine 6β-hydroxylase alongthe biosynthetic pathway leading to the tropane alkaloid scopo-lamine in species of the genera Hyoscyamus, Duboisia, and
Datura. This one enzyme catalyzes two consecutive steps, hy-droxylation of hyoscyamine followed by epoxide formation toproduce scopolamine.
Figure 24.42One alternative approach to the use of metabolicallyengineered plants is to use microbes to produce alka-loids. Short biosynthetic pathways can be expressedin either yeast or bacteria and used as a source of al-kaloid. Plant alkaloid genes can be functionally ex-pressed in microorganisms to produce either singlebiotransformation steps or short biosynthetic path-ways. For example, a known plant biosynthetic path-way contains an enzyme encoded by gene 3, whichcatalyzes a transformation step that is difficult toachieve by chemical synthesis. After heterologous ex-pression of gene 3 in a microorganism, the proteinproduct can be used in a biomimetic synthesis of al-kaloid D. Likewise, expression of the biosyntheticgenes 1 through 4 in a microorganism might producealkaloid E directly from precursor A.
AGene 1
BGene 2
Single geneexpression for biomimeticsynthesis
CGene 3
Gene 1
Gene 2
Gene 4
Gene 3
DGene 4
E
A
C D
E
Expression of shortpathway
Gene 3

transformation of Brassica napus (canola)with the cDNA encoding the C. roseus tryp-tophan decarboxylase used in biosynthesisof monoterpenoid indole alkaloids. Useful-ness of seed from this oil-producing crop as animal feed has been limited in part bythe presence of indole glucosinolates (seeChapters 8 and 16), sulfur-containing com-pounds that make the protein meal lesspalatable. The tryptophan decarboxylasetransgene in canola redirects tryptophanpools away from indole glucosinolatebiosynthesis and into tryptamine (Fig. 24.43).The mature seed of the transgenic canolaplants contains less of the indole glucosi-nates and does not accumulate tryptamine,making it more suitable for use as animalfeed and achieving a potentially economical-ly useful product.
To date, the elucidation of enzymaticsyntheses of at least eight alkaloids is eithercomplete or nearly complete: ajmaline, vin-doline, berberine, corydaline, macarpine,morphine, berbamunine, and scopolamine.Of these alkaloids, those in current industrialuse, such as morphine and scoploamine, arestill being isolated from the plants that pro-duce them rather than synthesized. The fu-ture for research on these alkaloids lies in
1286 Chapter 24 Natural Products (Secondary Metabolites)
the development of alternative systems ofproduction, such as plant cell or microbialcultures, and in the development of plantswith an improved spectrum of alkaloids fora more efficient production of the pharma-ceuticals currently isolated from field-grownplants. The design of these alternative sys-tems and optimized plants requires molecu-lar manipulation, which in turn requiresknowledge of alkaloid biosynthetic path-ways at the enzyme level. Much progresshas been made with select alkaloids, butmuch remains to be discovered about the enzymatic synthesis of pharmaceutically important alkaloids such as camptothecin,quinine, and emetine, to name only a fewexamples. cDNAs have now been isolatedfor approximately 20 enzymes of alkaloidbiosynthesis, and the rate at which newclones are identified is certain to increase inthe coming years. As genes are isolated, wecan anticipate that heterologous expressionsystems will be developed in bacterial, yeast,and insect cell culture systems to allow pro-duction of single enzymes, and perhapseven short pathways, for biomimetic synthe-ses of alkaloids. Our understanding of howthe expression of alkaloid biosynthesis genesis regulated by elicitors or in specific tissueswill also improve as the promoters of alka-loid biosynthetic genes are analyzed. The fu-ture will almost certainly bring geneticallyengineered microorganisms and eukaryoticcell cultures that produce alkaloids, metabol-ically engineered medicinal plants with tai-lored alkaloid spectra, pharmaceutically im-portant alkaloids in plant cell culture, andeven enzymatic synthesis of as yet unknownalkaloids through combinatorial biochemistry.
24.9 Phenylpropanoid andphenylpropanoid-acetate pathway metabolites
24.9.1 Plants contain a remarkably diversearray of phenolic compounds.
Plants originated in an aquatic environment.Their successful evolutionary adaptation toland was achieved largely by massive forma-tion of “plant phenolic” compounds. Al-though the bulk of these substances as-sumed cell wall structural roles, a vast array of nonstructural constituents was
Figure 24.43Metabolic engineering to improve the quality of canola oil. A canola cultivaris transformed with a gene from Catharanthus roseus that encodes tryptophandecarboxylase, an enzyme involved in biosynthesis of monoterpenoid indolealkaloids. The transgene effectively directs the L-tryptophan pool away fromuse in biosynthesis of the bitter indole glucosinolate and into the productionof tryptamine. WT, wild type.
L-Tryptophan
NH2
COOH
CO2
NH
Indole glucosinolate
S Glc
NH
Tryptamine
Transgeniccanola
WT canola
NH
NH2
Tryptophandecarboxylase
N O SO3–

128724.9–Phenylpropanoid and Phenylpropanoid-Acetate Pathway Metabolites
also formed, having such various roles asdefending plants, determining certain distin-guishing features of different woods andbarks (e.g., durability), establishing flowercolor, and contributing substantially to cer-tain flavors (tastes and odors). These func-tions and others performed by plant pheno-lics are essential for the continued survivalof all types of vascular plants. Accountingfor about 40% of organic carbon circulatingin the biosphere, these phenolic compoundsare primarily derived from phenylpropanoid,phenylpropanoid-acetate, and related bio-chemical pathways such as those leading to“hydrolyzable” tannins. Furthermore, it istheir reassimilation back to carbon dioxideduring biodegradation (mineralization) thatpresents the rate-limiting step in recyclingbiological carbon.
Plant phenolics are generally character-ized as aromatic metabolites that possess, orformerly possessed, one or more “acidic”hydroxyl groups attached to the aromaticarene (phenyl) ring (Fig. 24.44). These com-pounds plagued plant scientists for years byinterfering with experimental methods. Forexample, when exposed to air, plant pheno-lics readily oxidize and turn brown, generat-ing products that form complexes with pro-teins and inhibit enzyme activity. Manyprotocols now used to isolate plant proteinsand nucleic acids include special precautionsdesigned to minimize interference by pheno-lic compounds. Cultured plant tissues canalso release phenolics that inhibit growth ofcallus and regeneration of plantlets. At thesame time, phenolic compounds are increas-ingly being recognized for their profoundimpact on plant growth, development, re-production, and defense; indeed, scientistshave come to appreciate their significancemore fully, particularly over the past fewdecades.
The discussion of plant phenolic sub-stances is a discussion of plant diversity it-self. Characteristics unique to each of theroughly 250,000 species of vascular plantsarise, at least in part, through differential deposition of highly specialized phenyl-propanoid and phenylpropanoid-acetatederivatives. No single species can be used toillustrate the extraordinary diversity of “sec-ondary” metabolites that exists within theplant kingdom, because many branches ofthe pathways are found or are amplified
only in specific plant families. Placing undueemphasis on any single plant species can ob-scure the extremely broad variation in bio-synthetic capabilities that has yielded thisspectrum of different plant types.
24.9.2 Most, but not all, plant phenoliccompounds are products ofphenylpropanoid metabolism.
Most plant phenolics are derived from thephenylpropanoid and phenylpropanoid-acetate pathways (Fig. 24.45) and fulfill avery broad range of physiological roles in planta. In ferns, fern allies, and seedplants, polymeric lignins reinforce special-ized cell walls, enabling them to supporttheir massive weights on land and to trans-port water and minerals from roots toleaves. Closely related to lignins, the lignanscan vary from dimers to higher oligomers.Widespread throughout the plant kingdom,lignans can, for example, either help defend
OH
Phenylring
Phenol
“Acidic”hydroxylorphenolicgroup
Figure 24.44Structure of phenol.
C
C
C
C3
Phenylpropanoidskeleton (C6C3)
C6
Phenylpropanoid–acetate skeleton (C6C3–C6), with phenylpropanoid-derived (C6C3) and acetate-derived (3 � C2) rings
3 � C2C6C3
Coniferyl alcohol,a component oflignins and many lignans
Quercetin, a flavonoid(C6C3–C6)
Phenylpropanoid skeletonAcetate-derived rings
O
O
HOOH
HO
OH
HO
OH
OH
H3CO
Figure 24.45Phenylpropanoid and phenylpropanoid-acetate skeletons and representative plantcompounds based on those structures.
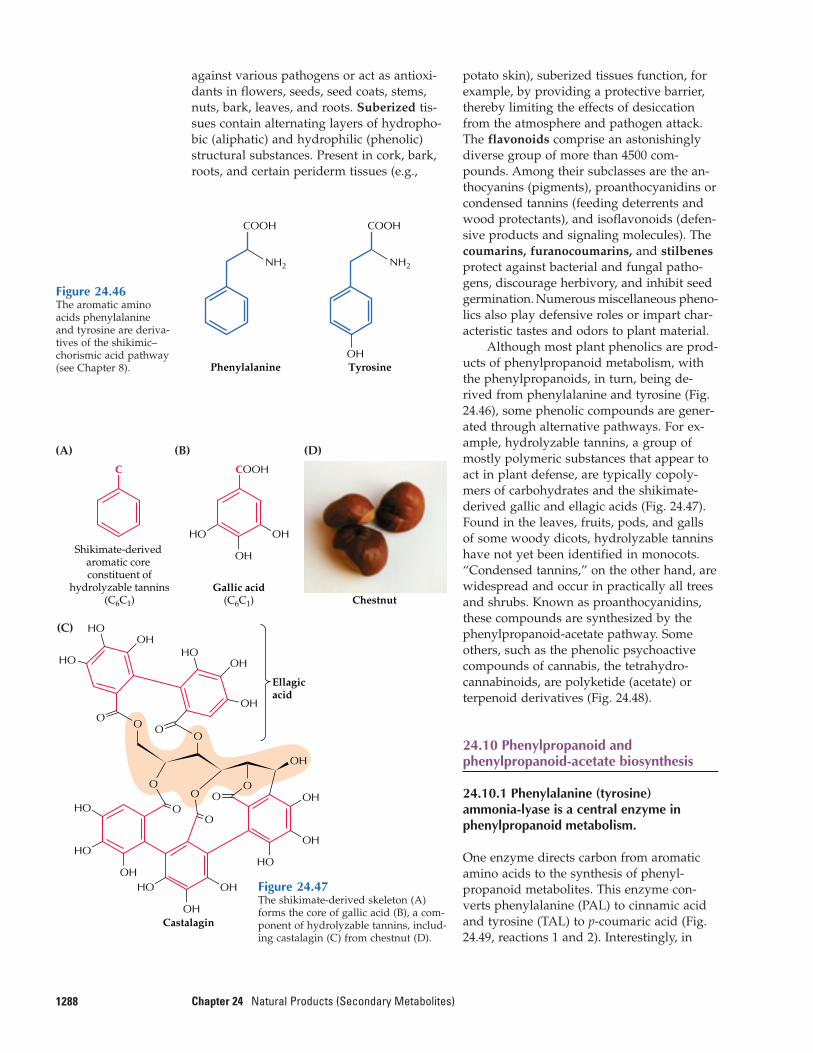
against various pathogens or act as antioxi-dants in flowers, seeds, seed coats, stems,nuts, bark, leaves, and roots. Suberized tis-sues contain alternating layers of hydropho-bic (aliphatic) and hydrophilic (phenolic)structural substances. Present in cork, bark,roots, and certain periderm tissues (e.g.,
1288 Chapter 24 Natural Products (Secondary Metabolites)
potato skin), suberized tissues function, forexample, by providing a protective barrier,thereby limiting the effects of desiccationfrom the atmosphere and pathogen attack.The flavonoids comprise an astonishinglydiverse group of more than 4500 com-pounds. Among their subclasses are the an-thocyanins (pigments), proanthocyanidins orcondensed tannins (feeding deterrents andwood protectants), and isoflavonoids (defen-sive products and signaling molecules). Thecoumarins, furanocoumarins, and stilbenesprotect against bacterial and fungal patho-gens, discourage herbivory, and inhibit seedgermination. Numerous miscellaneous pheno-lics also play defensive roles or impart char-acteristic tastes and odors to plant material.
Although most plant phenolics are prod-ucts of phenylpropanoid metabolism, withthe phenylpropanoids, in turn, being de-rived from phenylalanine and tyrosine (Fig.24.46), some phenolic compounds are gener-ated through alternative pathways. For ex-ample, hydrolyzable tannins, a group ofmostly polymeric substances that appear toact in plant defense, are typically copoly-mers of carbohydrates and the shikimate-derived gallic and ellagic acids (Fig. 24.47).Found in the leaves, fruits, pods, and gallsof some woody dicots, hydrolyzable tanninshave not yet been identified in monocots.“Condensed tannins,” on the other hand, arewidespread and occur in practically all treesand shrubs. Known as proanthocyanidins,these compounds are synthesized by thephenylpropanoid-acetate pathway. Someothers, such as the phenolic psychoactivecompounds of cannabis, the tetrahydro-cannabinoids, are polyketide (acetate) or terpenoid derivatives (Fig. 24.48).
24.10 Phenylpropanoid andphenylpropanoid-acetate biosynthesis
24.10.1 Phenylalanine (tyrosine) ammonia-lyase is a central enzyme inphenylpropanoid metabolism.
One enzyme directs carbon from aromaticamino acids to the synthesis of phenyl-propanoid metabolites. This enzyme con-verts phenylalanine (PAL) to cinnamic acidand tyrosine (TAL) to p-coumaric acid (Fig.24.49, reactions 1 and 2). Interestingly, in
Phenylalanine
NH2
COOH
TyrosineOH
NH2
COOH
Figure 24.46The aromatic aminoacids phenylalanine and tyrosine are deriva-tives of the shikimic–chorismic acid pathway(see Chapter 8).
(A)
(C)
(B)
Shikimate-derived aromatic core constituent of
hydrolyzable tannins(C6C1)
Ellagicacid
OHHO
OH
C
Gallic acid(C6C1) Chestnut
Castalagin
COOH
OHHO
OH
HO
HO
OH
OH
OH
OH
O
O
O
O
O
O
O
HO
OHHO
HO
OHHO
O
O
(D)
O
OH
Figure 24.47The shikimate-derived skeleton (A)forms the core of gallic acid (B), a com-ponent of hydrolyzable tannins, includ-ing castalagin (C) from chestnut (D).

128924.10–Phenylpropanoid and Phenylpropanoid-Acetate Biosynthesis
most vascular plants, Phe is the highly pre-ferred substrate, but the monocot enzymecan utilize both Phe and Tyr. PAL has beendetected in a few aquatic plants, where itprobably functions in formation of simpleflavonoids, such as the C-glucosyl–linkedlucenin and vicenin of Nitella species (Fig.24.50). Lignins, however, are not present inaquatic plants. Thus, this PAL (TAL) enzy-matic step and the products of the variousphenylpropanoid and phenylpropanoid-ac-etate pathways appear to have been key tothe plant colonization of land.
PAL is the most extensively studied en-zyme in the phenylpropanoid pathway, per-haps in all secondary metabolism. In someplants, PAL appears to be encoded by a sin-gle gene, whereas in others it is the productof a multigene family. The enzyme requiresno cofactor for activity. The ammonium ionliberated by the PAL reaction is recycled byway of glutamine synthetase and glutamatesynthetase (GS-GOGAT; see Chapter 8).Once assimilated into glutamate, the aminogroup can be donated to prephenate, form-ing arogenate, a precursor of both phenylala-nine and tyrosine (Fig. 24.51). This nitrogen-cycling process ensures a steady supply ofthe aromatic amino acids from which plantphenolics are derived.
24.10.2 Biochemical pathways to distinct phenolic classes share manycommon features.
During the 1960s and early 1970s, impressiveprogress was made in defining many of thesalient features of the pathway that converts
cinnamic acid to the monolignols (see Fig.24.49). This pathway essentially comprisesfour types of enzymatic reactions: aromatichydroxylations, O-methylations, CoA liga-tions, and NADPH-dependent reductions.More recently, the precise enzymology in-volved in earlier parts of the pathway hascome under renewed attention, focusing par-ticularly on aromatic hydroxylations and onwhether the O-methylation steps utilize thefree acids or CoA esters.
Aromatic ring hydroxylation involvesthree distinct hydroxylation conversions, allof which are believed to be microsomal. Thebest studied of these enzymes, cinnamate-4-hydroxylase, is an oxygen-requiring,NADPH-dependent, cytochrome P450 en-zyme that catalyzes the regiospecific hydrox-ylation at the para-position of cinnamic acidto give p-coumaric acid (see Fig. 24.49, reac-tion 3). The other two hydroxylases original-ly were thought to introduce hydroxylgroups into the free acids p-coumarate orferulate (or their CoA ester forms), yieldingthe diphenol (catechol) products caffeic acidor 5-hydroxyferulic acid (or their CoAderivatives), respectively (see Fig. 24.49, re-action 4). At this time, however, substantialconfusion remains as to how the caffeoylmoiety of caffeic acid or caffeoyl-CoA isformed. Whether this biosynthesis involves anonspecific phenolase-catalyzed conversionor whether some other enzymatic step (e.g.,one involving an NADPH-dependent cy-tochrome P450) is used is still not known.Additionally, although ferulate-5-hydroxy-lase has been established as an NADPH-dependent cytochrome P450 enzyme, there
Figure 24.48Not all plant phenolic compounds are derived from phenyl-propanoid substrates. Whereas mescaline, the psychoactive compo-nent of peyote, is a phenylpropanoid derivative (A), the phenolic
compound ∆1-3,4-cis-tetrahydrocannabinol (B), a psychoactive com-ponent of cannabis, is a product of polyketide synthesis, the repeat-ed condensation of acetyl-CoA units derived from malonyl-CoA.
Mescaline Peyote cactus Cannabis/Hemp
(A)
OCH3
NH2
H3CO OCH3
OH
(B)
∆1-3,4-cis-Tetrahydrocannabinol
O
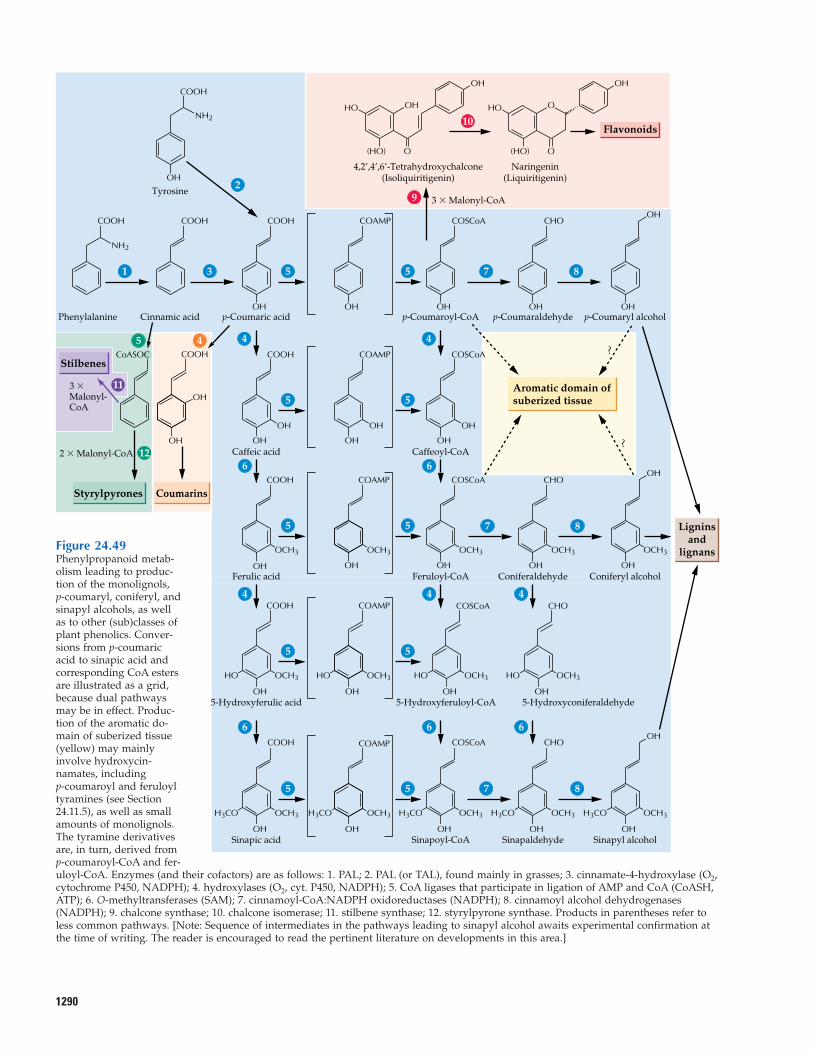
1290
Phenylalanine
Stilbenes
Styrylpyrones Coumarins
3 �Malonyl- CoA
2 � Malonyl-CoA
COOH
Cinnamic acid
COOH
10
87
OH
Tyrosine
COOH
2
OHp-Coumaric acid
COOH
4
11
12
5 4 4
OHCaffeic acid
COOH
OH
COOH
OH
OH
CoASOC
OHFerulic acid
COOH
OH5-Hydroxyferulic acid
COOH
OHSinapic acid
COOH
OH
COAMP
OH
COAMP
OH
OH
OH
COAMP
OHp-Coumaroyl-CoA
3 � Malonyl-CoA
4,2’,4’,6’-Tetrahydroxychalcone(Isoliquiritigenin)
COSCoA
(HO) O
9
OHCaffeoyl-CoA
COSCoA
OH
66
4 4
6 6
OHFeruloyl-CoA
OH5-Hydroxyferuloyl-CoA
OHSinapoyl-CoA
COSCoA
5
7 8
5
55
7 8
31
OHp-Coumaraldehyde
CHO
OHConiferaldehyde
CHO
OHSinapaldehyde
CHO
OHp-Coumaryl alcohol
OHConiferyl alcohol
OH
?
?
OHSinapyl alcohol
H3CO OCH3H3CO OCH3
Aromatic domain ofsuberized tissue
Ligninsand
lignans
Flavonoids
OH
HO
Naringenin(Liquiritigenin)
(HO) O
O
OH
HOOH
H3CO OCH3H3CO OCH3
HO OCH3
H3CO
HO OCH3 HO OCH3
OCH3 OCH3 OCH3 OCH3 OCH3
COAMP COSCoA
55
OH
COAMP
OCH3
55
55
COSCoA4
6
OH5-Hydroxyconiferaldehyde
HO OCH3
CHO
NH2
NH2
OH
OH
Figure 24.49Phenylpropanoid metab-olism leading to produc-tion of the monolignols, p-coumaryl, coniferyl, andsinapyl alcohols, as wellas to other (sub)classes ofplant phenolics. Conver-sions from p-coumaricacid to sinapic acid andcorresponding CoA estersare illustrated as a grid,because dual pathwaysmay be in effect. Produc-tion of the aromatic do-main of suberized tissue(yellow) may mainly involve hydroxycin-namates, including p-coumaroyl and feruloyltyramines (see Section24.11.5), as well as smallamounts of monolignols.The tyramine derivativesare, in turn, derived fromp-coumaroyl-CoA and fer-uloyl-CoA. Enzymes (and their cofactors) are as follows: 1. PAL; 2. PAL (or TAL), found mainly in grasses; 3. cinnamate-4-hydroxylase (O2,cytochrome P450, NADPH); 4. hydroxylases (O2, cyt. P450, NADPH); 5. CoA ligases that participate in ligation of AMP and CoA (CoASH,ATP); 6. O-methyltransferases (SAM); 7. cinnamoyl-CoA:NADPH oxidoreductases (NADPH); 8. cinnamoyl alcohol dehydrogenases(NADPH); 9. chalcone synthase; 10. chalcone isomerase; 11. stilbene synthase; 12. styrylpyrone synthase. Products in parentheses refer toless common pathways. [Note: Sequence of intermediates in the pathways leading to sinapyl alcohol awaits experimental confirmation atthe time of writing. The reader is encouraged to read the pertinent literature on developments in this area.]
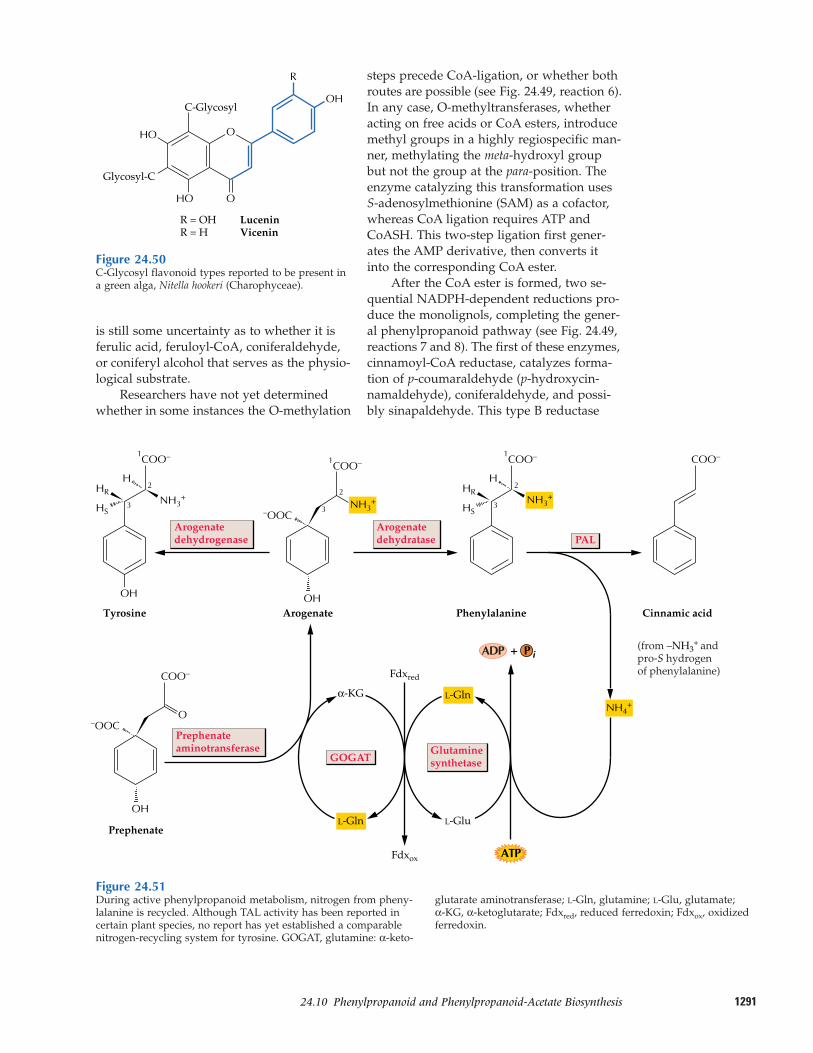
129124.10–Phenylpropanoid and Phenylpropanoid-Acetate Biosynthesis
is still some uncertainty as to whether it isferulic acid, feruloyl-CoA, coniferaldehyde,or coniferyl alcohol that serves as the physio-logical substrate.
Researchers have not yet determinedwhether in some instances the O-methylation
steps precede CoA-ligation, or whether bothroutes are possible (see Fig. 24.49, reaction 6).In any case, O-methyltransferases, whetheracting on free acids or CoA esters, introducemethyl groups in a highly regiospecific man-ner, methylating the meta-hydroxyl groupbut not the group at the para-position. Theenzyme catalyzing this transformation usesS-adenosylmethionine (SAM) as a cofactor,whereas CoA ligation requires ATP andCoASH. This two-step ligation first gener-ates the AMP derivative, then converts itinto the corresponding CoA ester.
After the CoA ester is formed, two se-quential NADPH-dependent reductions pro-duce the monolignols, completing the gener-al phenylpropanoid pathway (see Fig. 24.49,reactions 7 and 8). The first of these enzymes,cinnamoyl-CoA reductase, catalyzes forma-tion of p-coumaraldehyde (p-hydroxycin-namaldehyde), coniferaldehyde, and possi-bly sinapaldehyde. This type B reductase
Figure 24.50C-Glycosyl flavonoid types reported to be present ina green alga, Nitella hookeri (Charophyceae).
O
O
HO
R
HO
Glycosyl-C
R = OH LuceninR = H Vicenin
C-GlycosylOH
OH
3
2
1
NH3+
H
HS
HR
COO–
TyrosineOH
3
2
1
NH3+ NH3
+
–OOC
COO–
Arogenate
3
2
1
NH4+
H
HS
HR
COO–
Phenylalanine
COO–
Cinnamic acid
Arogenatedehydrogenase
Arogenatedehydratase
OH
O–OOC
COO–
Prephenate
α-KG
GOGAT
PAL
ATP
ADP + P i
Fdxox
Fdxred
Glutaminesynthetase
L-Glu
L-Gln
L-Gln
Prephenateaminotransferase
(from –NH3+ and
pro-S hydrogenof phenylalanine)
Figure 24.51During active phenylpropanoid metabolism, nitrogen from pheny-lalanine is recycled. Although TAL activity has been reported incertain plant species, no report has yet established a comparable nitrogen-recycling system for tyrosine. GOGAT, glutamine: α-keto-
glutarate aminotransferase; L-Gln, glutamine; L-Glu, glutamate; α-KG, α-ketoglutarate; Fdxred, reduced ferredoxin; Fdxox, oxidizedferredoxin.

abstracts the pro-S hydride (Fig. 24.52) frombehind the nicotinamide plane of NADPHduring reduction. The second enzyme, cin-namyl alcohol dehydrogenase, is a type Areductase that abstracts the pro-R hydridefrom in front of the nicotinamide plane toyield the monolignols p-coumaryl, coniferyl,and sinapyl alcohols (Fig. 24.52).
The above description is a brief accountof the overall biochemical steps that culmi-nate in monolignol formation. However, thepathway shown in Figure 24.49 is deceptive.Not all cells, tissues, or species of plants uti-lize the entire pathway. In many instances,plants utilize only a small pathway segmentthat directs substrates to one or more of themain metabolic branchpoints; moreover,they may express that truncated pathwayonly in specific tissues. Researchers do notyet fully understand metabolic flux andcompartmentalization in the phenyl-propanoid pathway. Elucidation of theseprocesses will be a necessary step towarddefining or identifying the control points in the pathway.
24.11 Biosynthesis of lignans, lignins, and suberization
The monolignols are primarily convertedinto two distinct classes of plant metabolites:the lignans and the lignins. Most metabolicflux through the phenylpropanoid biosyn-
1292 Chapter 24 Natural Products (Secondary Metabolites)
thetic pathway is directed to the productionof the lignins, which are structural compo-nents of cell walls. Free radicals participatein the reactions that produce both dimeric/oligomeric lignans and lignins as well as related complex plant polymers such asthose in suberized tissue.
24.11.1 Dimeric and oligomeric lignans areformed primarily from coniferyl alcohol.
The term lignan was initially coined byRobert Downs Haworth in 1936 to describe aclass of dimeric phenylpropanoid (C6C3)metabolites linked by way of their 8–8’bonds. More recently, another term, neolig-nan, was used to define all of the othertypes of linkages (e.g., 8–1’-linked dimers),but has since been modified to encompasssubstances derived from allylphenol com-pounds, such as isoeugenol (Fig. 24.53). Inthis chapter, however, we have chosen to use the more convenient name lignan to de-scribe all possible phenylpropanoid (C6C3)coupling products, so long as the couplingmode (e.g., 8–8’, 8–5’) is specified (Fig.24.54). Interestingly, although several thou-sand lignans are now known in nature, relatively few coupling modes have been encountered.
Lignan dimers are found in ferns, gym-nosperms, and angiosperms, but higher
Figure 24.52The stereospecificity of a type B reductase, NADPH-dependent cinnamoyl-CoA reductase (CCR), and a type A reductase, cinnamylalcohol dehydrogenase (CAD). HA, pro-R (the hydrogen projectingupward from the A-face of the nicotinamide ring, i.e., out from the
plane of the page); HB, pro-S (the hydrogen projecting upwardfrom the B-face of the nicotinamide ring, i.e., behind the plane of the page). R, adenine nucleotide diphosphate.
Type B reductase
CCR CAD
Type A reductaseOH
OCH3
CHBO
Coniferyl aldehyde
OH
OCH3
Coniferyl alcohol
CHAHB
OH
OH
OCH3
Feruloyl-CoA ester
NADPH NADP+ NADP+NADPH
COSCoA
C
O
NH2
HA
N
R
+
C
O
NH2
HAHB
N
R
C
O
NH2
HB
N
R
+
C
O
NH2
HAHB
N
R
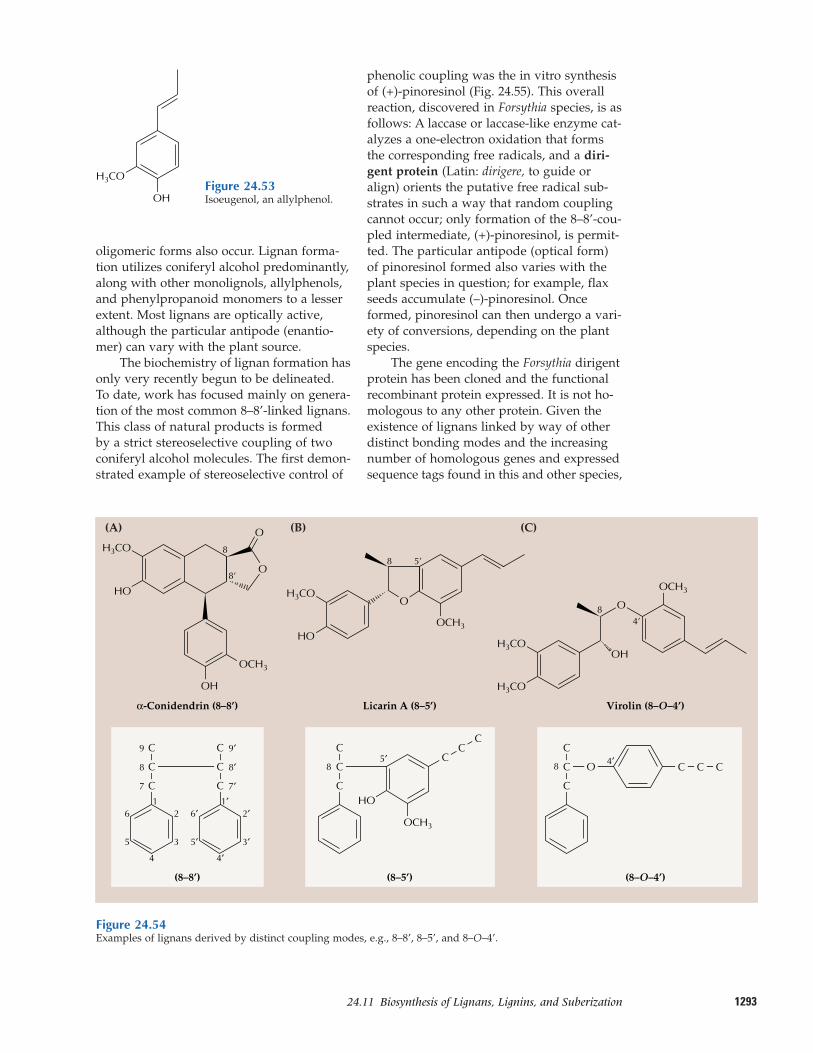
129324.11–Biosynthesis of Lignans, Lignins, and Suberization
oligomeric forms also occur. Lignan forma-tion utilizes coniferyl alcohol predominantly,along with other monolignols, allylphenols,and phenylpropanoid monomers to a lesserextent. Most lignans are optically active, although the particular antipode (enantio-mer) can vary with the plant source.
The biochemistry of lignan formation hasonly very recently begun to be delineated.To date, work has focused mainly on genera-tion of the most common 8–8’-linked lignans.This class of natural products is formed by a strict stereoselective coupling of twoconiferyl alcohol molecules. The first demon-strated example of stereoselective control of
phenolic coupling was the in vitro synthesisof (+)-pinoresinol (Fig. 24.55). This overallreaction, discovered in Forsythia species, is asfollows: A laccase or laccase-like enzyme cat-alyzes a one-electron oxidation that formsthe corresponding free radicals, and a diri-gent protein (Latin: dirigere, to guide oralign) orients the putative free radical sub-strates in such a way that random couplingcannot occur; only formation of the 8–8’-cou-pled intermediate, (+)-pinoresinol, is permit-ted. The particular antipode (optical form)of pinoresinol formed also varies with theplant species in question; for example, flaxseeds accumulate (–)-pinoresinol. Onceformed, pinoresinol can then undergo a vari-ety of conversions, depending on the plantspecies.
The gene encoding the Forsythia dirigentprotein has been cloned and the functionalrecombinant protein expressed. It is not ho-mologous to any other protein. Given the existence of lignans linked by way of otherdistinct bonding modes and the increasingnumber of homologous genes and expressedsequence tags found in this and other species,
Figure 24.53Isoeugenol, an allylphenol.
Figure 24.54Examples of lignans derived by distinct coupling modes, e.g., 8–8’, 8–5’, and 8–O–4’.
OH
H3CO
(A)
OH
OCH3
HO
α-Conidendrin (8–8’)
O
O
9
(8–8’)
OCH3
8
8’
Licarin A (8–5’)
H3CO
H3CO
OCH3
84’
Virolin (8–O–4’)
(8–5’) (8–O–4’)
OH
(B) (C)
C
C
8
7
12
3
6
5
4
9′
8′
7′1′
2′
3′
6′
5′
4′
C
C
C
C
C
C
85′
C
C
CC
C O
C
8 4′
C
C CC
H3CO
HOO
HO
H3CO
OCH3
8 5’
O

1294 Chapter 24 Natural Products (Secondary Metabolites)
Oxidase
or oxidant
E-Coniferyl alcohol
Forsythia intermedia
OH
OCH3
OH
OCH3
+
+
Dirigentprotein
OH
OCH3
•8
OH
• 8’
OCH3
O O
H3CO
:O
88’
H
:O
H
OCH3
H3O+
H3O+
O
O
(+)-Pinoresinol
O
O
8’8 HH
HO
OCH3
OCH3
OH
OH OH
Figure 24.55Proposed biochemicalmechanism accountingfor stereoselective con-trol (regio- and stereo-chemistry) of E-coniferylalcohol coupling inForsythia species. Theparticular enantiomer ofpinoresinol formed canvary with plant species.
we can easily assume that the dirigent pro-tein represents a new class of proteins. Addi-tionally, the mode of action of this protein is of particular interest and may providenew and definitive insight into the macro-molecular assembly processes that lead tolignins and suberins (see Sections 24.11.3and 24.11.5).
Pinoresinol can be enantiospecificallyconverted into lariciresinol and secoisolari-ciresinol, followed by dehydrogenation togive matairesinol (Fig. 24.56 and Box 24.4).This last is the presumed precursor of plicat-ic acid (Figs. 24.56 and 24.57A) and itsanalogs in western red cedar (Thuja plicata),as well as of podophyllotoxin (Figs. 24.56and Fig. 24.57B) in the Indian plant(Podophyllum hexandrum) and may apple (P. peltatum). Podophyllotoxin is used to treatvenereal warts, whereas its semisyntheticderivative, teniposide, is widely used in can-cer treatment. Interestingly, pinoresinol/lariciresinol reductase, which convertspinoresinol into lariciresinol and secoisolari-ciresinol, shows considerable homology tothe phytoalexin-forming isoflavonoid reduc-tases, indicative perhaps of a common evo-lutionary thread in plant defense for boththe lignans and isoflavonoids. Pinoresinol isalso the precursor of the antioxidant sesamin(Fig. 24.57C) in the seeds of sesame (Sesa-mum indicum).
24.11.2 Lignin biosynthesis has beendescribed as a largely nonenzymaticprocess, but differences between syntheticand biologically derived lignins cast doubton this premise.
Derived from the Latin lignum (wood), theterm lignin initially was coined to describe
the noncellulosic encrusting substance pre-sent in woody tissue. After cellulose, ligninsare the most abundant organic natural prod-ucts known, accounting for as much as 20%to 30% of all vascular plant tissue. Deposi-tion of lignins in plants results in the forma-tion of woody secondary xylem tissues intrees, as well as reinforcement of vasculartissues in herbaceous plants and grasses.There are still no methods available for
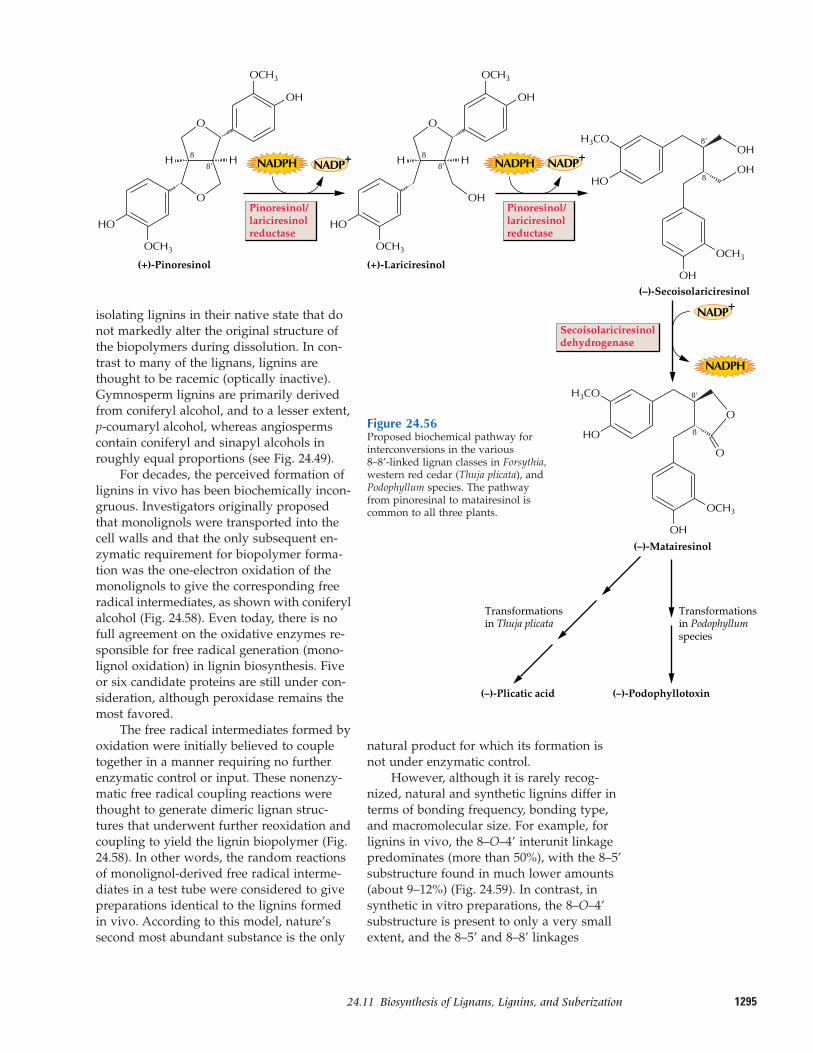
129524.11–Biosynthesis of Lignans, Lignins, and Suberization
Secoisolariciresinoldehydrogenase
Pinoresinol/lariciresinolreductase
(+)-Pinoresinol
O
O
8’8 HH
HO
OCH3
OCH3
OH
Pinoresinol/lariciresinolreductase
(+)-Lariciresinol
O
OH
8’8
8’
8 HH
OCH3
OCH3
OH
(–)-Secoisolariciresinol
Transformationsin Podophyllumspecies
Transformations in Thuja plicata
OCH3
OH
OH
8
8’
(–)-Matairesinol
(–)-Podophyllotoxin(–)-Plicatic acid
OCH3
OH
H3CO
HO
O
O
H3CO
HO
HO
NADPH NADP+ NADPH
NADPH
NADP+
NADP+
OH
isolating lignins in their native state that donot markedly alter the original structure ofthe biopolymers during dissolution. In con-trast to many of the lignans, lignins arethought to be racemic (optically inactive).Gymnosperm lignins are primarily derivedfrom coniferyl alcohol, and to a lesser extent, p-coumaryl alcohol, whereas angiospermscontain coniferyl and sinapyl alcohols inroughly equal proportions (see Fig. 24.49).
For decades, the perceived formation oflignins in vivo has been biochemically incon-gruous. Investigators originally proposedthat monolignols were transported into thecell walls and that the only subsequent en-zymatic requirement for biopolymer forma-tion was the one-electron oxidation of themonolignols to give the corresponding freeradical intermediates, as shown with coniferylalcohol (Fig. 24.58). Even today, there is nofull agreement on the oxidative enzymes re-sponsible for free radical generation (mono-lignol oxidation) in lignin biosynthesis. Fiveor six candidate proteins are still under con-sideration, although peroxidase remains themost favored.
The free radical intermediates formed byoxidation were initially believed to coupletogether in a manner requiring no furtherenzymatic control or input. These nonenzy-matic free radical coupling reactions werethought to generate dimeric lignan struc-tures that underwent further reoxidation andcoupling to yield the lignin biopolymer (Fig.24.58). In other words, the random reactionsof monolignol-derived free radical interme-diates in a test tube were considered to givepreparations identical to the lignins formedin vivo. According to this model, nature’ssecond most abundant substance is the only
natural product for which its formation isnot under enzymatic control.
However, although it is rarely recog-nized, natural and synthetic lignins differ interms of bonding frequency, bonding type,and macromolecular size. For example, forlignins in vivo, the 8–O–4’ interunit linkagepredominates (more than 50%), with the 8–5’substructure found in much lower amounts(about 9–12%) (Fig. 24.59). In contrast, insynthetic in vitro preparations, the 8–O–4’substructure is present to only a very smallextent, and the 8–5’ and 8–8’ linkages
Figure 24.56Proposed biochemical pathway for interconversions in the various 8–8’-linked lignan classes in Forsythia,western red cedar (Thuja plicata), andPodophyllum species. The pathwayfrom pinoresinal to matairesinol iscommon to all three plants.

1296 Chapter 24 Natural Products (Secondary Metabolites)
ly, more details will emerge as this impor-tant process is investigated systematically.
24.11.3 Lignin biosynthesis is controlledspatially and temporally and may involve aproteinaceous template.
Before lignin biosynthesis is initiated, thecells destined to form secondary xylem(i.e., wood; Fig. 24.60) undergo specific
predominate. This disparity suggests thatwithin woody tissues some mechanism inthe lignifying cell wall regulates or man-dates the interunit linkage pattern within thenative biopolymer.
As is becoming increasingly clear, thelignification process in situ is under verytight biochemical control as part of a cell-specific programmed process. In the follow-ing section, we describe known elementsthat control lignification in vivo. Undoubted-
Box 24.4 Dietary lignans have health-protecting functions.
Secoisolariciresinol and matairesinolare common constituents of variousplants, including Forsythia intermedia,flax, and certain vegetables and grains(e.g., green beans and rye). These lignanshave important nutritional functions inhealth protection. During digestion, in-testinal bacteria convert secoisolari-ciresinol and matairesinol to enterodioland enterolactone, respectively (see fig-
ure). These “mammalian” lignans under-go enterohepatic circulation, in whichthey are conjugated in the liver, excretedin the bile, deconjugated in the intestineby bacterial enzymes, absorbed acrossthe intestinal mucosa, and returned to theliver in the portal circulation (see figure).
Enterodiol and enterolactone are be-lieved to be responsible for preventing theonset of and substantially reducing the
rate of incidence of prostate and breastcancers. The protection accrues to indi-viduals on diets rich in grains, vegeta-bles, and berries that contain high con-centrations of secoisolariciresinol andmatairesinol. In contrast, typical Westerndiets tend to be poor in these foods anddo not afford comparable protection.
Renal clearance(urine)
EnterodiolEnterolactone
GlucuronidesSecoisolariciresinol,matairesinol
Absorption
Fecal loss, unconjugated lignans
Lignan,glucuronides
Facultativeaerobes
Enterodiol,enterolactone
(–)-Matairesinol
Enterolactone
OH
O
HO
O
OCH3
OH
O
H3CO
HOO
Enterodiol
OH
OH
OH
HO
(–)-Secoisolariciresinol
OCH3
H3CO
OH
HOOH
OH
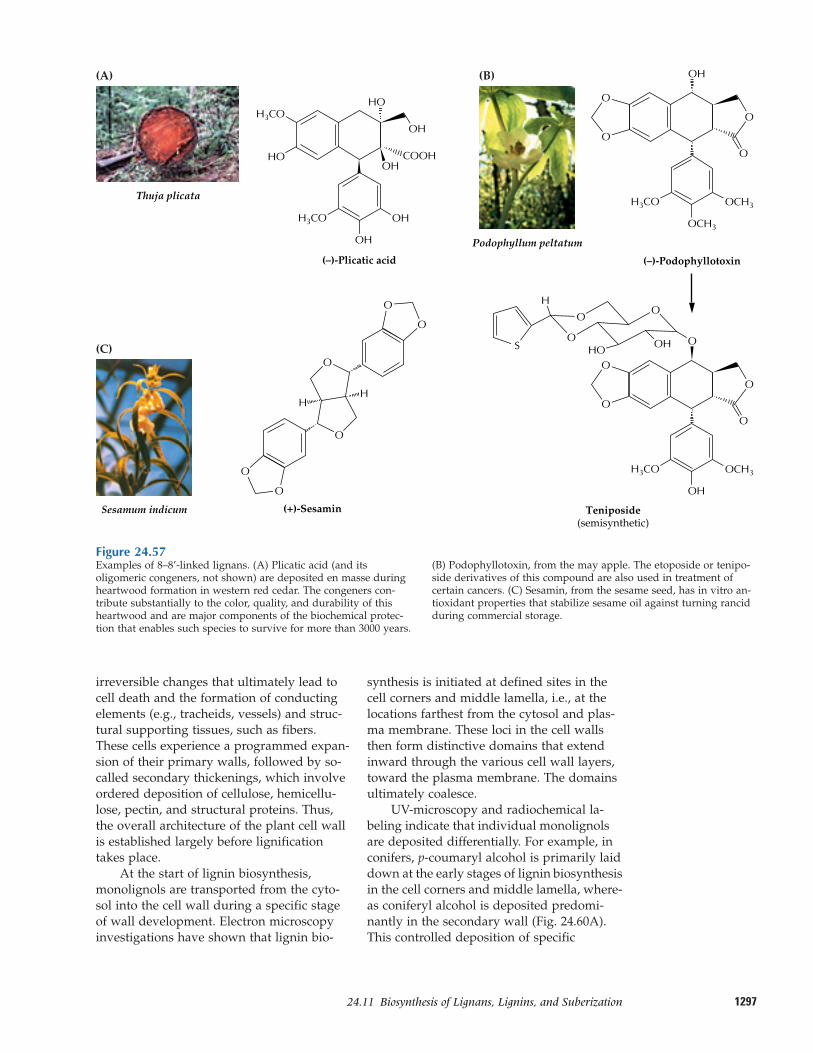
129724.11–Biosynthesis of Lignans, Lignins, and Suberization
irreversible changes that ultimately lead tocell death and the formation of conductingelements (e.g., tracheids, vessels) and struc-tural supporting tissues, such as fibers.These cells experience a programmed expan-sion of their primary walls, followed by so-called secondary thickenings, which involveordered deposition of cellulose, hemicellu-lose, pectin, and structural proteins. Thus,the overall architecture of the plant cell wallis established largely before lignificationtakes place.
At the start of lignin biosynthesis,monolignols are transported from the cyto-sol into the cell wall during a specific stageof wall development. Electron microscopyinvestigations have shown that lignin bio-
synthesis is initiated at defined sites in thecell corners and middle lamella, i.e., at thelocations farthest from the cytosol and plas-ma membrane. These loci in the cell wallsthen form distinctive domains that extendinward through the various cell wall layers,toward the plasma membrane. The domainsultimately coalesce.
UV-microscopy and radiochemical la-beling indicate that individual monolignolsare deposited differentially. For example, inconifers, p-coumaryl alcohol is primarily laiddown at the early stages of lignin biosynthesisin the cell corners and middle lamella, where-as coniferyl alcohol is deposited predomi-nantly in the secondary wall (Fig. 24.60A).This controlled deposition of specific
Figure 24.57Examples of 8–8’-linked lignans. (A) Plicatic acid (and itsoligomeric congeners, not shown) are deposited en masse duringheartwood formation in western red cedar. The congeners con-tribute substantially to the color, quality, and durability of thisheartwood and are major components of the biochemical protec-tion that enables such species to survive for more than 3000 years.
(B) Podophyllotoxin, from the may apple. The etoposide or tenipo-side derivatives of this compound are also used in treatment ofcertain cancers. (C) Sesamin, from the sesame seed, has in vitro an-tioxidant properties that stabilize sesame oil against turning rancidduring commercial storage.
(C)
(–)-Podophyllotoxin
Sesamum indicum
O
O
H3CO OCH3
OCH3
OH
(+)-Sesamin
O
O
HH
O
O
O
O
(B)
Podophyllum peltatum
O
O
H3CO OCH3
OH
O
O
O
O
O
OO
O
Teniposide(semisynthetic)
(A)
Thuja plicata
S OH
H
OH
(–)-Plicatic acid
OH
H3CO
HO
H3CO
OH
OHCOOH
OH
HO
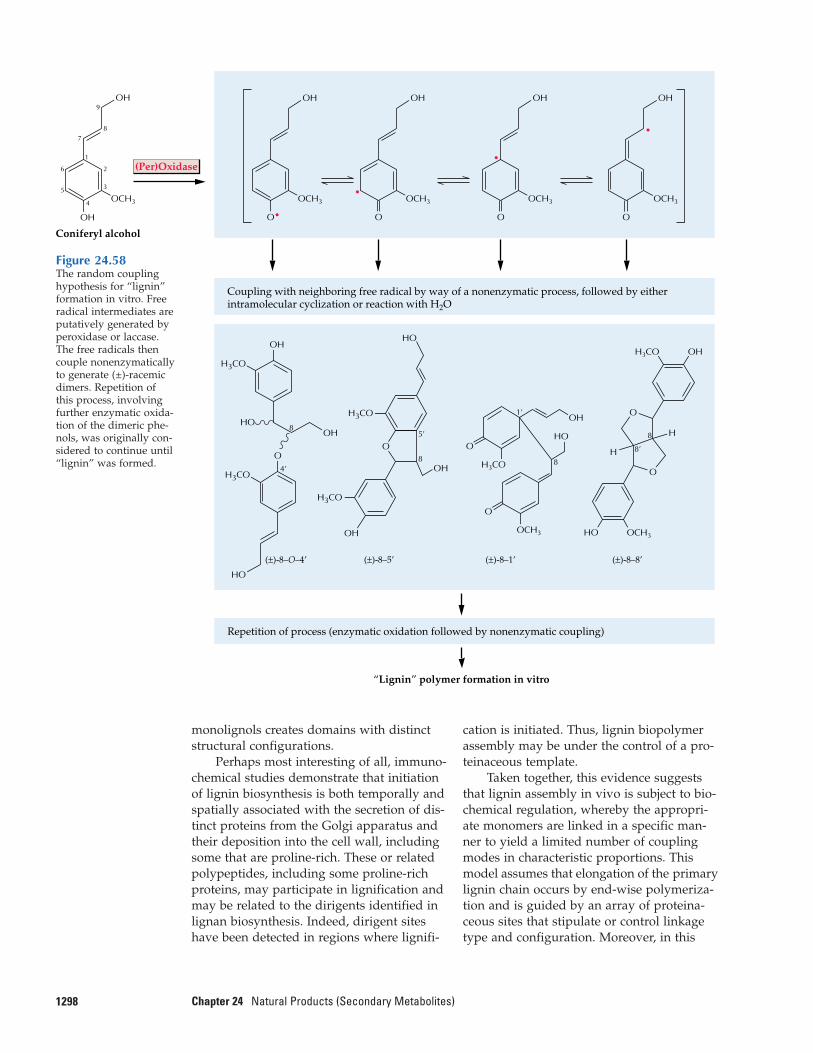
monolignols creates domains with distinctstructural configurations.
Perhaps most interesting of all, immuno-chemical studies demonstrate that initiationof lignin biosynthesis is both temporally andspatially associated with the secretion of dis-tinct proteins from the Golgi apparatus andtheir deposition into the cell wall, includingsome that are proline-rich. These or relatedpolypeptides, including some proline-richproteins, may participate in lignification andmay be related to the dirigents identified inlignan biosynthesis. Indeed, dirigent siteshave been detected in regions where lignifi-
1298 Chapter 24 Natural Products (Secondary Metabolites)
cation is initiated. Thus, lignin biopolymerassembly may be under the control of a pro-teinaceous template.
Taken together, this evidence suggeststhat lignin assembly in vivo is subject to bio-chemical regulation, whereby the appropri-ate monomers are linked in a specific man-ner to yield a limited number of couplingmodes in characteristic proportions. Thismodel assumes that elongation of the primarylignin chain occurs by end-wise polymeriza-tion and is guided by an array of proteina-ceous sites that stipulate or control linkagetype and configuration. Moreover, in this
OH
OCH3
OH9
87
1
2
3
6
5
4
Coniferyl alcohol
Coupling with neighboring free radical by way of a nonenzymatic process, followed by eitherintramolecular cyclization or reaction with H2O
Repetition of process (enzymatic oxidation followed by nonenzymatic coupling)
“Lignin” polymer formation in vitro
O•
OCH3
OH
(Per)Oxidase
OCH3
OH
O
•OCH3
OH
O
•
O
OCH3
OH
•
(±)-8–O–4’
O4’
8
H3CO
HO
OH
OHHO
H3CO
8
1’
(±)-8–1’
O
OHO
OH
H3CO
OCH3
(±)-8–8’
OCH3
H3CO
O
8
8’
O
OH
HO
H3CO
8
5’
H3CO
(±)-8–5’
HO
O
OH
OH
H
H
Figure 24.58The random couplinghypothesis for “lignin”formation in vitro. Freeradical intermediates areputatively generated byperoxidase or laccase.The free radicals thencouple nonenzymaticallyto generate (±)-racemicdimers. Repetition ofthis process, involvingfurther enzymatic oxida-tion of the dimeric phe-nols, was originally con-sidered to continue until“lignin” was formed.
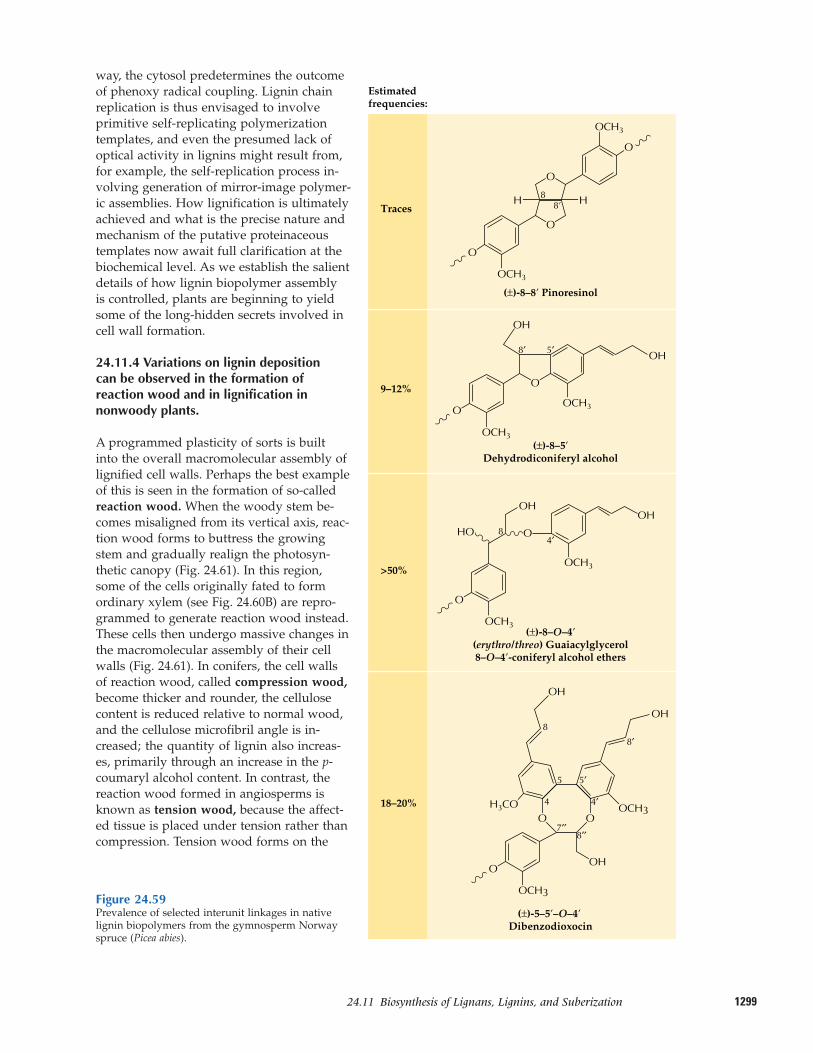
129924.11–Biosynthesis of Lignans, Lignins, and Suberization
way, the cytosol predetermines the outcomeof phenoxy radical coupling. Lignin chainreplication is thus envisaged to involveprimitive self-replicating polymerizationtemplates, and even the presumed lack ofoptical activity in lignins might result from,for example, the self-replication process in-volving generation of mirror-image polymer-ic assemblies. How lignification is ultimatelyachieved and what is the precise nature andmechanism of the putative proteinaceoustemplates now await full clarification at thebiochemical level. As we establish the salientdetails of how lignin biopolymer assembly is controlled, plants are beginning to yieldsome of the long-hidden secrets involved incell wall formation.
24.11.4 Variations on lignin deposition can be observed in the formation ofreaction wood and in lignification innonwoody plants.
A programmed plasticity of sorts is builtinto the overall macromolecular assembly oflignified cell walls. Perhaps the best exampleof this is seen in the formation of so-calledreaction wood. When the woody stem be-comes misaligned from its vertical axis, reac-tion wood forms to buttress the growingstem and gradually realign the photosyn-thetic canopy (Fig. 24.61). In this region,some of the cells originally fated to form ordinary xylem (see Fig. 24.60B) are repro-grammed to generate reaction wood instead.These cells then undergo massive changes inthe macromolecular assembly of their cellwalls (Fig. 24.61). In conifers, the cell wallsof reaction wood, called compression wood,become thicker and rounder, the cellulosecontent is reduced relative to normal wood,and the cellulose microfibril angle is in-creased; the quantity of lignin also increas-es, primarily through an increase in the p-coumaryl alcohol content. In contrast, thereaction wood formed in angiosperms isknown as tension wood, because the affect-ed tissue is placed under tension rather thancompression. Tension wood forms on the
Figure 24.59Prevalence of selected interunit linkages in nativelignin biopolymers from the gymnosperm Norwayspruce (Picea abies).
Traces
Estimatedfrequencies:
9–12%
>50%
18–20%
(±)-8–8’ Pinoresinol
88’
O
O
(±)-8–5’Dehydrodiconiferyl alcohol
8′ 5′
O
O
O
O
OH
OH
O
HO
OH
O
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3(±)-8–O–4’
(erythro/threo) Guaiacylglycerol8–O–4’-coniferyl alcohol ethers
4′8
OH
OCH3
OH
OCH3
OH
OH
(±)-5–5’–O–4’ Dibenzodioxocin
8′′7′′
4′
8′
4
5′5
8
OOH3CO
O
H H
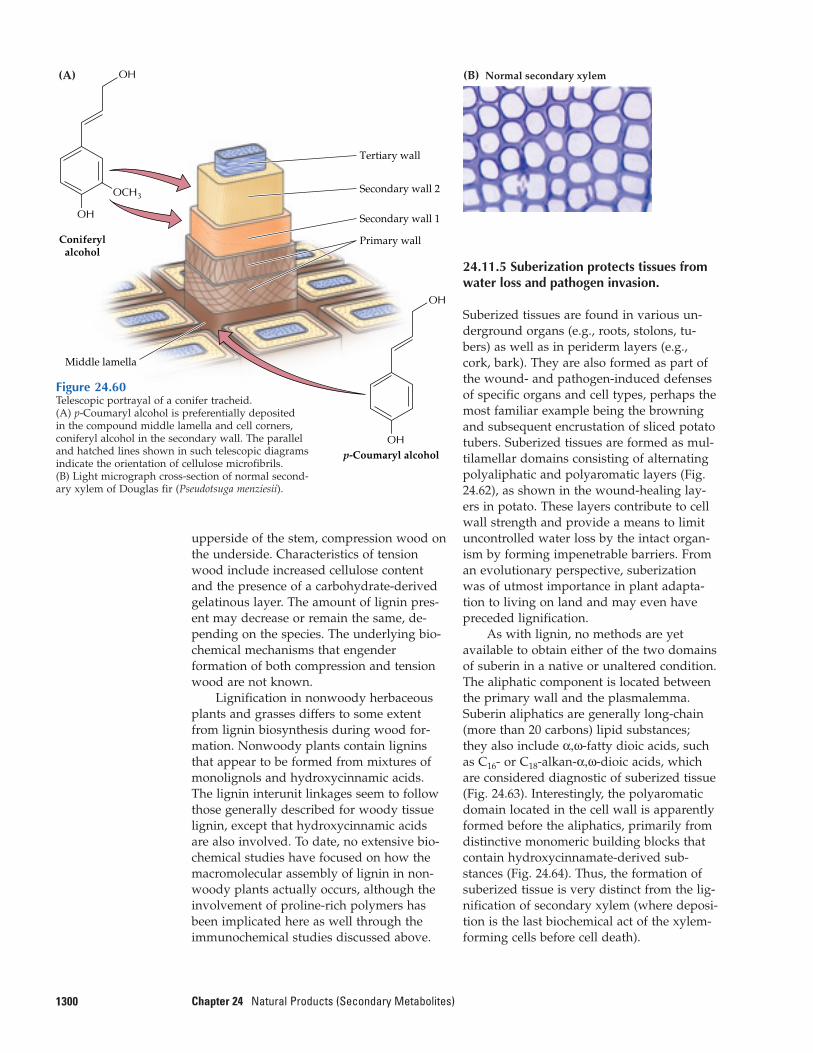
upperside of the stem, compression wood onthe underside. Characteristics of tensionwood include increased cellulose contentand the presence of a carbohydrate-derivedgelatinous layer. The amount of lignin pres-ent may decrease or remain the same, de-pending on the species. The underlying bio-chemical mechanisms that engenderformation of both compression and tensionwood are not known.
Lignification in nonwoody herbaceousplants and grasses differs to some extentfrom lignin biosynthesis during wood for-mation. Nonwoody plants contain ligninsthat appear to be formed from mixtures ofmonolignols and hydroxycinnamic acids.The lignin interunit linkages seem to followthose generally described for woody tissuelignin, except that hydroxycinnamic acidsare also involved. To date, no extensive bio-chemical studies have focused on how themacromolecular assembly of lignin in non-woody plants actually occurs, although theinvolvement of proline-rich polymers hasbeen implicated here as well through the immunochemical studies discussed above.
1300 Chapter 24 Natural Products (Secondary Metabolites)
24.11.5 Suberization protects tissues fromwater loss and pathogen invasion.
Suberized tissues are found in various un-derground organs (e.g., roots, stolons, tu-bers) as well as in periderm layers (e.g.,cork, bark). They are also formed as part ofthe wound- and pathogen-induced defensesof specific organs and cell types, perhaps themost familiar example being the browningand subsequent encrustation of sliced potatotubers. Suberized tissues are formed as mul-tilamellar domains consisting of alternatingpolyaliphatic and polyaromatic layers (Fig.24.62), as shown in the wound-healing lay-ers in potato. These layers contribute to cellwall strength and provide a means to limituncontrolled water loss by the intact organ-ism by forming impenetrable barriers. Froman evolutionary perspective, suberizationwas of utmost importance in plant adapta-tion to living on land and may even havepreceded lignification.
As with lignin, no methods are yetavailable to obtain either of the two domainsof suberin in a native or unaltered condition.The aliphatic component is located betweenthe primary wall and the plasmalemma.Suberin aliphatics are generally long-chain(more than 20 carbons) lipid substances;they also include α,ω-fatty dioic acids, suchas C16- or C18-alkan-α,ω-dioic acids, whichare considered diagnostic of suberized tissue(Fig. 24.63). Interestingly, the polyaromaticdomain located in the cell wall is apparentlyformed before the aliphatics, primarily fromdistinctive monomeric building blocks thatcontain hydroxycinnamate-derived sub-stances (Fig. 24.64). Thus, the formation ofsuberized tissue is very distinct from the lig-nification of secondary xylem (where deposi-tion is the last biochemical act of the xylem-forming cells before cell death).
(A) (B) Normal secondary xylem
Middle lamella
Tertiary wall
Coniferylalcohol
OH
p-Coumaryl alcohol
OCH3
OH
Secondary wall 2
Secondary wall 1
Primary wall
OH
OH
Figure 24.60Telescopic portrayal of a conifer tracheid. (A) p-Coumaryl alcohol is preferentially deposited in the compound middle lamella and cell corners,coniferyl alcohol in the secondary wall. The paralleland hatched lines shown in such telescopic diagrams indicate the orientation of cellulose microfibrils. (B) Light micrograph cross-section of normal second-ary xylem of Douglas fir (Pseudotsuga menziesii).

130124.11–Biosynthesis of Lignans, Lignins, and Suberization
A further complication to the study ofthe aromatic domain in suberization is thepresence of related phenolic substances. Forexample, in wound-healing suberizing pota-to periderm tissues, chlorogenic acid andmiscellaneous other phenolics are also pres-ent (Fig. 24.65). These compounds do not ap-pear to function in suberization per se butrather may provide a means for topical dis-infection of the exposed cell surfaces, there-by preventing or limiting infection/contami-nation. Some evidence also suggests thepresence of low amounts of monolignols insuberized tissues, but these may be fromsmall amounts of lignin.
Although the polymeric suberin pheno-lic constituents are predominantly derivedfrom hydroxycinnamate, how this aromaticdomain is assembled is unknown. Recentstudies demonstrated that potato tuberwound-healing suberizing tissues containtwo hydroxycinnamoyl-CoA transferases,which catalyze formation of various alkylferulates and (p-coumaroyl) feruloyl tyra-mine derivatives, respectively. How, or if,
(A)
Compression wood
(B) Sequoia sempervirens stem
(D) Compression (reaction)wood tracheid
Pith
Compressionwood
(C) Compression wood xylem
Secondary wall 2 (S2)
Secondary wall 1 (S1)
Primary wall
Intercellular material
Intercellular material
Intercellularspace
Figure 24.61Compression wood (reaction wood). (A) Gym-nosperm showing region of compression wood tis-sue. (B) Cross-section of Sequoia sempervirensshowing pith and compression wood. (C) Lightmicrograph cross-section of compression woodxylem of Douglas fir (Pseudotsuga menziesii). (D) Telescopic portrayal of a tracheid in compres-sion wood.
0.5 µµm
Figure 24.62Suberized tissue consists of layered polyaliphatic andpolyaromatic domains, as shown for wound-healingpotato tuber slices exposed to air. A suberized layer(see arrows) forms five days after exposure of potatotuber slices to air.
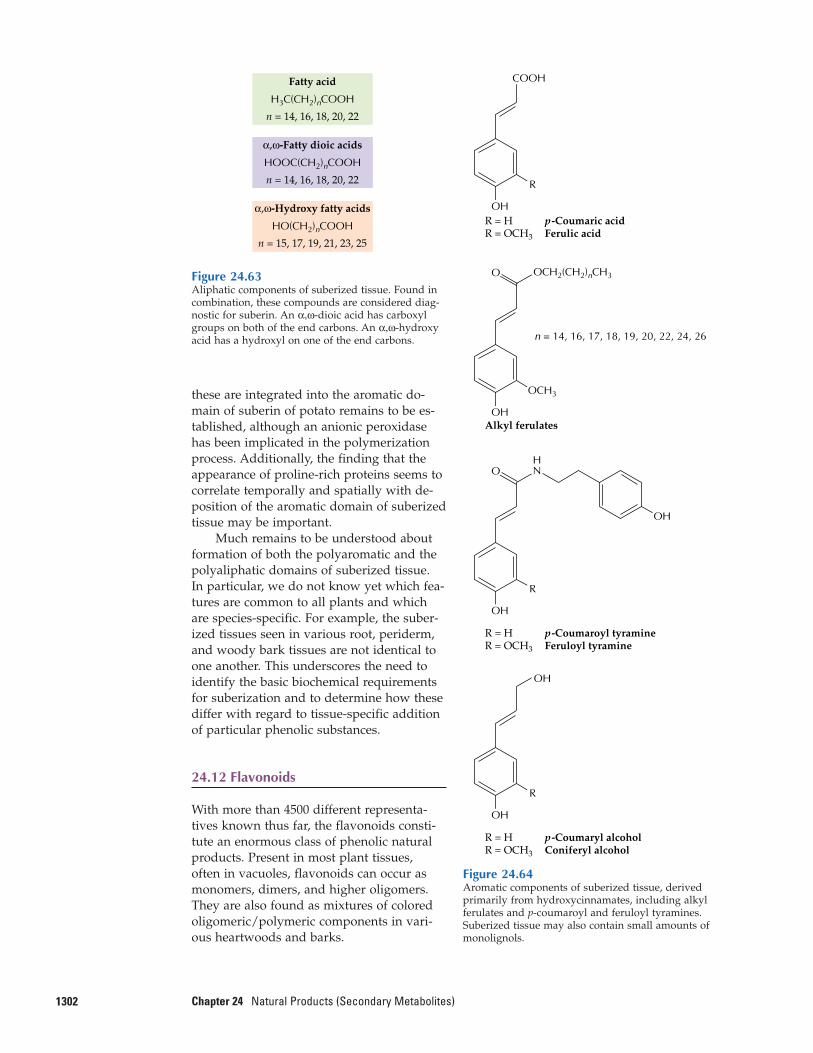
these are integrated into the aromatic do-main of suberin of potato remains to be es-tablished, although an anionic peroxidasehas been implicated in the polymerizationprocess. Additionally, the finding that theappearance of proline-rich proteins seems tocorrelate temporally and spatially with de-position of the aromatic domain of suberizedtissue may be important.
Much remains to be understood aboutformation of both the polyaromatic and thepolyaliphatic domains of suberized tissue. In particular, we do not know yet which fea-tures are common to all plants and whichare species-specific. For example, the suber-ized tissues seen in various root, periderm,and woody bark tissues are not identical toone another. This underscores the need toidentify the basic biochemical requirementsfor suberization and to determine how thesediffer with regard to tissue-specific additionof particular phenolic substances.
24.12 Flavonoids
With more than 4500 different representa-tives known thus far, the flavonoids consti-tute an enormous class of phenolic naturalproducts. Present in most plant tissues, often in vacuoles, flavonoids can occur asmonomers, dimers, and higher oligomers. They are also found as mixtures of coloredoligomeric/polymeric components in vari-ous heartwoods and barks.
1302 Chapter 24 Natural Products (Secondary Metabolites)
Figure 24.63Aliphatic components of suberized tissue. Found incombination, these compounds are considered diag-nostic for suberin. An α,ω-dioic acid has carboxylgroups on both of the end carbons. An α,ω-hydroxyacid has a hydroxyl on one of the end carbons.
H3C(CH2)nCOOH
n = 14, 16, 18, 20, 22
Fatty acid
HOOC(CH2)nCOOH
n = 14, 16, 18, 20, 22
α,ω-Fatty dioic acids
HO(CH2)nCOOH
n = 15, 17, 19, 21, 23, 25
α,ω-Hydroxy fatty acids
Figure 24.64Aromatic components of suberized tissue, derivedprimarily from hydroxycinnamates, including alkylferulates and p-coumaroyl and feruloyl tyramines.Suberized tissue may also contain small amounts ofmonolignols.
OH
OCH3
O OCH2(CH2)nCH3
n = 14, 16, 17, 18, 19, 20, 22, 24, 26
Alkyl ferulates
OH
R
COOH
R = H p-Coumaric acidR = OCH3 Ferulic acid
OH
R
HN
OH
O
R = H p-Coumaroyl tyramineR = OCH3 Feruloyl tyramine
R = H p-Coumaryl alcoholR = OCH3 Coniferyl alcohol
OH
R
OH

130324.12–Flavonoids
(purple, mauve, and blue). Related flavo-noids, such as flavonols, flavones, chalcones,and aurones, also contribute to color defini-tion. Manipulating flower color by targetingvarious enzymatic steps and genes in flavo-noid biosynthesis has been quite successful,particularly in petunia.
Specific flavonoids can also function toprotect plants against UV-B irradiation, arole sometimes ascribed to kaempferol (Fig.24.67). Others can act as insect feeding at-tractants, such as isoquercetin in mulberry, afactor involved in silkworm recognition ofits host species. In contrast, condensed tan-nins such as the proanthocyanidins add adistinct bitterness or astringency to the tasteof certain plant tissues and function as anti-feedants (Fig. 24.68). The flavonoids api-genin and luteolin serve as signal moleculesin legume–rhizobium bacteria interactions,facilitating nitrogen fixation (Fig. 24.69). In arelated function, isoflavonoids are involvedin inducible defense against fungal attack inalfalfa (e.g., medicarpin; Fig. 24.69) and oth-er plant species. Perhaps the most poorlystudied and least understood classes of theflavonoids are the oligomeric and polymericsubstances associated with formation of certain heartwood and bark tissues. These
24.12.1 Flavonoids comprise a diverse set of compounds and perform a widerange of functions.
Many plant–animal interactions are influ-enced by flavonoids. The colors of flowersand fruits, which often function to attractpollinators and seed dispersers, result pri-marily from vacuolar anthocyanins (Fig.24.66) such as the pelargonidins (orange,salmon, pink, and red), the cyanidins (ma-genta and crimson), and the delphinidins
Figure 24.65Suberin deposition has been studied in woundedpotato tubers. In these tissues, suberin formation isaccompanied by the production of an unrelatedphenolic compound, chlorogenic acid.
COOH
OH
OH
OH
OH
O O
HO
Chlorogenic acidSuberized tissue
Figure 24.66Selected anthocyanin pigments: pelargonidin, cyanidin, and delphinidin from geranium,rose, and larkspur, respectively.
OH
OH
HO O
OH
Rosa(Rose)
OH
OH
HO
Delphinium(Larkspur)
Pelargonidin Cyanidin
HO
OH
OH
OH
HO+
O
OH
OH
OH
+O
+
Delphinidin
Pelargonium(Geranium)

compounds include proanthocyanidins andtheir congeners in woody gymnosperms andisoflavonoids in woody legumes from thetropics. In both cases, their massive deposi-tion during heartwood formation contributessignificantly and characteristically to theoverall color, quality, and rot resistance ofwood. These metabolites can be misidenti-fied as lignins because some constituents arenot readily solubilized and are frequentlydissolved only under the same conditionsthat effect lignin dissolution.
1304 Chapter 24 Natural Products (Secondary Metabolites)
Various flavonoids have also been stud-ied extensively from the perspectives ofhealth protection and pharmacological utility,for which mammalian enzyme systems havebeen used to assess flavonoid activity. Flavonoids have been analyzed as modula-tors of immune and inflammatory responses,for their impact on smooth muscle function,and as anticancer, antiviral, antitoxic, andhepatoprotective agents. There is consid-erable current interest in the use of isoflavonoids in cancer prevention. Dietaryconsumption of the isoflavonoids daidzeinand genistein (Fig. 24.70), which are presentin soybeans, is thought to reduce substan-tially the incidence of breast and prostatecancers in humans.
24.12.2 The flavonoid biosynthesis pathwayhas several important branchpoints.
The flavonoids consist of various groups ofplant metabolites, which include chalcones,aurones, flavonones, isoflavonoids, flavones,flavonols, leucoanthocyanidins (flavan-3,4-diols), catechins, and anthocyanins (Figs.24.70 and 24.71).
The first committed step of the flavo-noid pathway is catalyzed by chalcone syn-thase (CHS; see Fig. 24.70). Three moleculesof acetate-derived malonyl-CoA and onemolecule of p-coumaryl-CoA are condensedto generate a tetrahydroxychalcone (see Fig.24.49, reaction 9). CHS, a dimeric polyketidesynthase with each subunit at about 42 kDa,has no cofactor requirements. In certainspecies, the coordinated action of CHS andan NADPH-dependent reductase generates a 6-deoxychalcone (isoliquiritigenin). Bothchalcones can then be converted into au-rones, a subclass of flavonoids found in cer-tain plant species. Beyond CHS, the nextstep shared by most of the flavonoid biosyn-thesis pathways is catalyzed by chalcone isomerase (CHI), which catalyzes a stere-ospecific ring closure isomerization step to form the 2S-flavanones, naringenin, and(less commonly) liquiritigenin (see Fig.24.70). The flavanones may represent the most important branching point inflavonoid metabolism, because isomerizationof these compounds yields the phytoalexinisoflavonoids (Fig. 24.70), introduction of aC-2–C-3 double bond affords flavones and
Figure 24.67Kaempferol, a UV-B protectant, ispresent in many plants such assoybean (Glycine max). Kaempferol
HO
OH
OH
OH
O
O
Soybean
Figure 24.68Red sorghum produces proanthocyanidin antifeedant compounds—condensedtannins, which deter birds from feeding on the seed. White sorghum, which isdeficient in these compounds, is rapidly consumed by birds. Similar compoundsare present in the heartwood of Douglas fir (not shown).
Red sorghum Proanthocyanidin (n = 1–30)
OH
OH
HO O
OH
OH
OH
OH
HO O
OH
OH
OH
OH
HO O
OH n
OH

130524.12–Flavonoids
flavonols (Fig. 24.71), and hydroxylation ofthe 3-position generates dihydroflavonols(Fig. 24.71).
Entry into the isoflavonoid branchpointoccurs by way of two enzymes (see Fig. 24.70).The first, isoflavone synthase (IFS), catalyzesan unusual C-2 to C-3 aryl migration andhydroxylation to give the 2-hydroxyisofla-vanones and has recently been shown to be an NADPH-dependent cytochrome P450enzyme. Dehydration of the 2-hydroxyiso-flavanones, catalyzed by 2-hydroxyisoflava-none dehydratase (IFD), forms the isofla-vonoids genistein and daidzein. Theisoflavonoids can be further metabolized,primarily in the Fabaceae, to yield phy-toalexins (e.g., medicarpin in alfalfa; see Fig.24.70) or to generate isoflavonoid-derivedsubstances known as rotenoids in tropicallegumes (e.g., 9-demethylmunduserone from Amorpha fruticosa; see Fig. 24.70). Therotenoids, which are isolated mainly fromDerris elliptica and related species, are usedextensively as insecticidal agents but haveother applications as well. For example,rotenone is used as a rat poison and an in-hibitor of NADH dehydrogenase. Interest-ingly, the NADPH-dependent isoflavone re-ductase (IFR) step involved in isoflavonoidformation shows considerable homology to
pinoresinol/lariciresinol reductase (see Fig.24.56 and Section 24.11.1), suggesting a phy-logenetic link between both lignan andisoflavonoid pathways for plant defense.
The second branching point in generalflavonoid metabolism involves that of dehy-dration of naringenin at the C-2/C-3 posi-tions to give such abundant flavones as apigenin (Fig. 24.71). This conversion is cat-alyzed by flavone synthase (FNS), which var-ies in enzymatic type depending on the plantspecies. For example, in parsley cell cultures,flavone formation is catalyzed by an α-ketog-lutarate–dependent dioxygenase (FNS I inFig. 24.71), whereas an NADPH-dependentmicrosomal preparation engenders thisreaction in Antirrhinum flowers (FNS II inFig. 24.71).
The third major branchpoint in flavonoidmetabolism is stereospecific 3-hydroxylationof naringenin (or its 3’-hydroxylated analog)to give dihydroflavonols (Fig. 24.71) such asdihydrokaempferol (or dihydroquercetin).The enzyme involved, flavanone 3-hydroxy-lase, is an Fe2+-requiring, α-ketoglutarate–dependent dioxygenase. Specific hydroxyla-tion involving an NADPH-dependent cyto-chrome P450 monooxygenase of naringenincan also directly give dihydroquercetin, whichcan be converted to quercetin (a flavanol) by
Figure 24.69Flavonoids perform diverse functions in alfalfa (Medicago sativa). The flavonoids apigenin and luteolin func-tion as signaling molecules that induce Nod gene expression in compatible Rhizobium bacteria, facilitating thedevelopment of nitrogen-fixing root nodules. The phytoalexin isoflavonoid medicarpin participates in in-ducible plant defense.
(–)-Medicarpin
O
OHO
H
H
OCH3
Nodule
Alfalfa
R = H ApigeninR = OH Luteolin
O
OH O
R
OH
HO
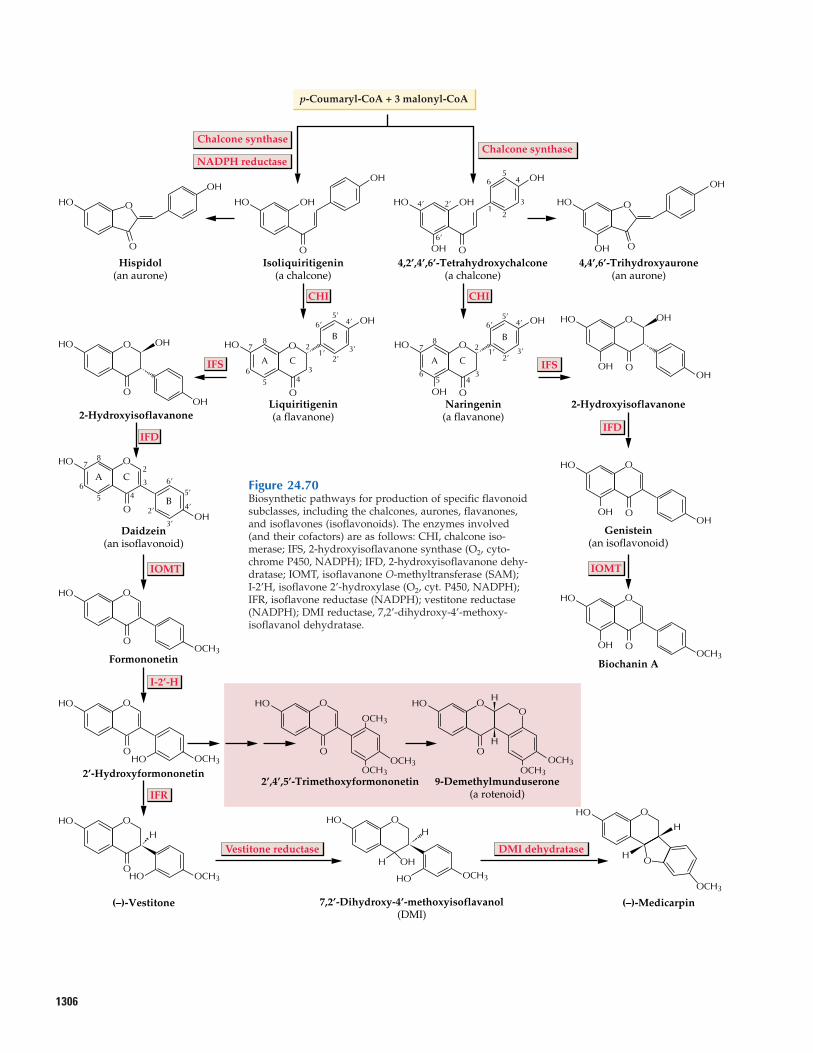
1306
O
O
O
O
O
O
O
O
O O
OO
O
OH
OH
Hispidol(an aurone)
O OH
OH
4,4’,6’-Trihydroxyaurone(an aurone)
O
Chalcone synthaseChalcone synthase
p-Coumaryl-CoA + 3 malonyl-CoA
OH OH
OH OH5
2
6
1
6’
82
5 4
7
6 3
1’ 3’
4’6’5’
2’
4’ 2’ 3
4
Isoliquiritigenin(a chalcone)
OH OH
OH
4,2’,4’,6’-Tetrahydroxychalcone(a chalcone)
OH
Formononetin
OH
OH
A C
B
O
6’
3’
4’2’
4
3
28
56
7
5’
2-Hydroxyisoflavanone
IFSIFS
CHI CHI
IFDIFD
OH
OCH3
O
OH
OH
O
OH
OCH3
O
2’-Hydroxyformononetin
OH
OCH3
O
2’,4’,5’-Trimethoxyformononetin
OH O
9-Demethylmunduserone(a rotenoid)
OH
OCH3
OO
OCH3
OCH3
OCH3
(–)-Vestitone
OH
OCH3HO
H
O
OH O
OH
B
CA C
B
A
OH5’
2’
6’
1’
5 4
78
2 3’
36
4’
Liquiritigenin(a flavanone)
OH O
OHNaringenin
(a flavanone)
Genistein(an isoflavonoid)
Biochanin A
OH
OH OHO
OH
H
H
OCH3
7,2’-Dihydroxy-4’-methoxyisoflavanol(DMI)
OHH H
H
O
OH
HOOCH3
(–)-Medicarpin
OH
H
O
O
OH
OH
IOMT IOMT
I-2’-H
IFR
Daidzein(an isoflavonoid)
HOOCH3
NADPH reductase
Vestitone reductase DMI dehydratase
OH
2-Hydroxyisoflavanone
O
O O
O
OH
OH
Figure 24.70Biosynthetic pathways for production of specific flavonoidsubclasses, including the chalcones, aurones, flavanones,and isoflavones (isoflavonoids). The enzymes involved(and their cofactors) are as follows: CHI, chalcone iso-merase; IFS, 2-hydroxyisoflavanone synthase (O2, cyto-chrome P450, NADPH); IFD, 2-hydroxyisoflavanone dehy-dratase; IOMT, isoflavanone O-methyltransferase (SAM);I-2’H, isoflavone 2’-hydroxylase (O2, cyt. P450, NADPH);IFR, isoflavone reductase (NADPH); vestitone reductase(NADPH); DMI reductase, 7,2’-dihydroxy-4’-methoxy-isoflavanol dehydratase.
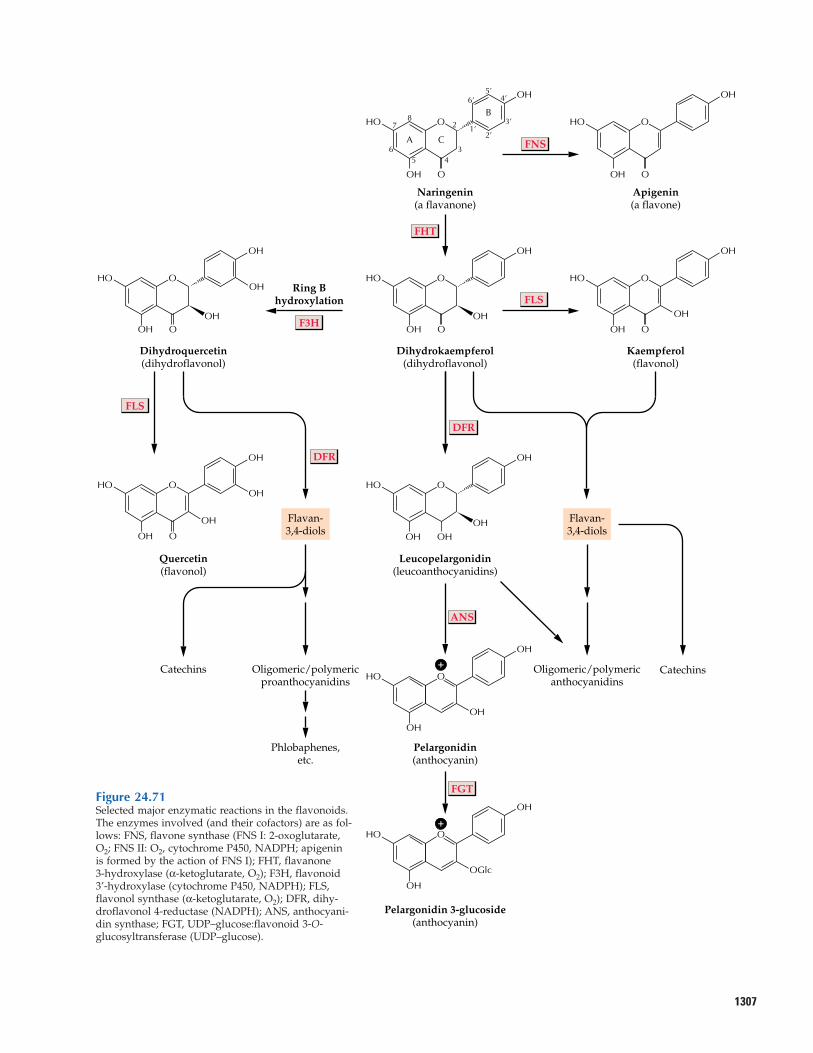
1307
OH5’
2’
B6’
1’
6 3
2
5 4
87 3’
4’
CA
OH O
OH
Naringenin(a flavanone)
O
OH
OH O
OH
Apigenin(a flavone)
O
ANS
FLS
DFR
DFR
OH
OH
OH O
OH
Dihydrokaempferol(dihydroflavonol)
O
FLS
FHT
FNS
Ring Bhydroxylation
OH
OH O
OH
OH
Kaempferol(flavonol)
O
Dihydroquercetin(dihydroflavonol)
OH
OH
OHOH
O
OH O
Catechins
OH
OH
OH O
OH
Leucopelargonidin(leucoanthocyanidins)
Oligomeric/polymericproanthocyanidins
Oligomeric/polymericanthocyanidins
Flavan-3,4-diols
Flavan-3,4-diols
HO
Quercetin(flavonol)
FGT
OH
OH O
OH
Pelargonidin(anthocyanin)
Phlobaphenes,etc.
Pelargonidin 3-glucoside(anthocyanin)
OH
OH O
OH
OH
OH
OH
OH O
OH
OGlc
OH
O
Catechins+
+
F3H
Figure 24.71Selected major enzymatic reactions in the flavonoids.The enzymes involved (and their cofactors) are as fol-lows: FNS, flavone synthase (FNS I: 2-oxoglutarate,O2; FNS II: O2, cytochrome P450, NADPH; apigeninis formed by the action of FNS I); FHT, flavanone3-hydroxylase (α-ketoglutarate, O2); F3H, flavonoid3’-hydroxylase (cytochrome P450, NADPH); FLS,flavonol synthase (α-ketoglutarate, O2); DFR, dihy-droflavonol 4-reductase (NADPH); ANS, anthocyani-din synthase; FGT, UDP–glucose:flavonoid 3-O-glucosyltransferase (UDP–glucose).

flavonol synthase (FLS)–catalyzed C-2–C-3double bond formation; FLS is an α-ketoglu-tarate–dependent dioxygenase. Alternatively,dihydroquercetin can be reduced by anNADPH-dependent dihydroflavonolreductase (DFR) to give the corresponding flavan-3,4-diols (Fig. 24.71).
Subsequent species- and tissue-specificenzymatic conversions can create vast arraysof structurally diverse groups of flavonoids.For example, in flower petals, the leucoan-thocyanidins (e.g., leucopelargonidin) can beconverted to the colored anthocyanins (e.g.,pelargonidin) through the action of a dehy-dratase, anthocyanidin synthase (ANS),which is thought to be an α-ketoglutarate–dependent dioxygenase (Fig. 24.71). Leu-coanthocyanidins can also serve as precur-sors of the (epi-)catechins and condensedtannins. The enzymology associated withthose coupling processes, chain extensionmechanisms, and oxidative modifications,however, is not yet established.
24.13 Coumarins, stilbenes,styrylpyrones, and arylpyrones
24.13.1 Some coumarins, a class of plantdefense compounds, can cause internalbleeding or dermatitis.
Coumarins (e.g., coumarin; Fig. 24.72A) belong to a widespread family of plantmetabolites called the benzopyranones, withmore than 1500 representatives in more than800 species. In plants, these compounds canoccur in seed coats, fruits, flowers, roots,leaves, and stems, although in general thegreatest concentrations are found in fruitsand flowers. Their roles in plants appear tobe mainly defense-related, given their an-timicrobial, antifeedant, UV-screening, andgermination inhibitor properties.
The best known properties of coumarinsindirectly highlight their roles in plant de-fense. Ingesting coumarins from plants suchas clover can cause massive internal bleed-ing in mammals. This discovery ultimatelyled to the development of the rodenticideWarfarin (Fig. 24.72B) and to the use of relat-ed compounds to treat and prevent stroke.Likewise, the photosensitizing compound 8-methoxypsoralen, present in leaf tissue
1308 Chapter 24 Natural Products (Secondary Metabolites)
of Heracleum mantegazzianum (giant hog-weed), can cause photophytodermititis onskin contact and subsequent exposure to UV-A radiation (Fig. 24.73). A comparableform of coumarin-induced dermatitis canalso occur during celery handling. Psoralen(Fig. 24.74), however, is now successfullyused to treat various skin disorders (eczema,psoriasis) by means of a combination of oralingestion and UV-A treatment.
The structure of a representative simplecoumarin, 7-hydroxycoumarin, is shown inFigure 24.75. Additional families of plantcoumarins (see Fig. 24.74) include linear furanocoumarins (e.g., psoralen), angular furanocoumarins (e.g., angelicin), pyra-nocoumarins (e.g., seselin), and pyrone-substituted coumarins (e.g., 4-hydroxy-coumarin).
24.13.2 Coumarin biosynthesis pathwayshave not yet been fully elucidated.
The biosynthetic pathways to the coumarinsare only partially determined at this point;they mainly involve aromatic hydroxyla-tions and additional reactions catalyzed bytrans/cis-hydroxycinnamic acid isomerases,dimethylallyltransferases, various P450/NADPH/O2–dependent synthases and O-methyltransferases (Fig. 24.75). The simplest
Figure 24.72Structures of (A) coumarin (from clover), and (B) asynthetic coumarin, the rodenticide Warfarin.
(A)
Coumarin
Warfarin(synthetic coumarin)
OH
O
O
O
(B)
OO

130924.13–Coumarins, Stilbenes, Styrylpyrones, and Arylpyrones
examples, coumarin and 7-hydroxycoumarin(umbelliferone), are believed to be formedby O-hydroxylation of cinnamic and p-coumaric acids, respectively, followed bytrans/cis-isomerization and ring closure.However, neither the enzymes nor their en-coding genes have yet been obtained.
On the other hand, much more is knownabout the biosynthesis of both linear and an-gular furanocoumarins. These involve re-giospecific prenylation through the action of the corresponding tranferases to yielddemethylsuberosin and osthenol, respective-ly. The subsequent transformations are be-lieved to involve various NADPH-depen-dent, cytochrome P450 oxidase–catalyzedconversions and O-methylations (Fig. 24.75).
Various fungi and yeasts also biosynthe-size coumarins, e.g., the toxic aflatoxins.However, these metabolites are polyketidederivatives and hence are biochemically distinct from their plant analogs.
24.13.3 Stilbenes, styrylpyrones, andarylpyrones constitute another class ofchemical defense compounds.
In addition to the products of the flavonoidpathway, cinnamoyl-CoA and malonyl-CoA(acetate-derived) pathways can in certainplant species also undergo condensation re-actions to yield the corresponding stilbenes,styrylpyrones, and arylpyrones (Fig. 24.76).Comparison of gene sequences for each entry-point enzyme (CHS and stilbene syn-thase) reveals significant homology, aswould be expected for similar enzymatic
systems. Beyond the initial synthases, how-ever, little has yet been described about sub-sequent transformations.
Stilbenes are present in bryophytes,pteridophytes, gymnosperms, and angio-sperms, with more than 300 different stil-benoids known today. The stilbenes play important roles in plants, particularly inheartwood protection, and also have signifi-cance in pharmacology and human health.In plants, they can function as both constitu-tive and inducible defense mechanisms. Stil-benes display weak antibacterial properties
Figure 24.73A linear furanocoumarin, 8-methoxypsoralen, sensitizes human skin to UV-A light. This compound,present in external tissues of Heracleum species, causes severe blistering on skin contact followed byexposure to UV-irradiation.
O OO
8-Methoxypsoralen(a furanocoumarin)
OCH3
Heracleum
Figure 24.74Structures of the linear furanocoumarin psoralen, the angular fura-nocoumarin angelicin, the pyranocoumarin seselin, and the pyrone-substituted coumarin 4-hydroxycoumarin.
OO
Psoralen(a linear furanocoumarin)
O O
O O
OO
Angelicin(an angular furanocoumarin)
4-Hydroxycoumarin(a pyrone-substituted
coumarin)
O
OH
Seselin(a pyranocoumarin)
OO
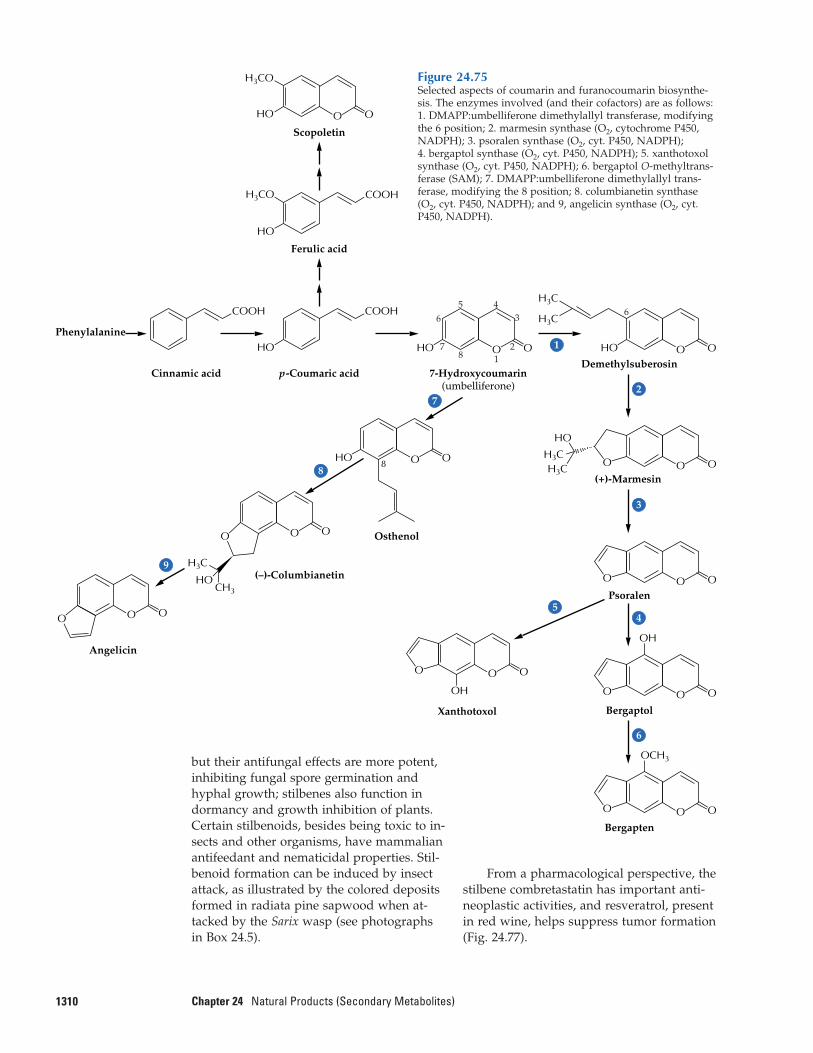
1310 Chapter 24 Natural Products (Secondary Metabolites)
From a pharmacological perspective, thestilbene combretastatin has important anti-neoplastic activities, and resveratrol, presentin red wine, helps suppress tumor formation(Fig. 24.77).
Scopoletin
H3CO
HO OO
7-Hydroxycoumarin(umbelliferone)
Ferulic acid
H3CO
HO HO
HO
HO
COOH
p-Coumaric acid
HO
Cinnamic acid
Phenylalanine
COOH COOH
Demethylsuberosin
Psoralen
(+)-Marmesin
Osthenol
(–)-Columbianetin
8 O
2
1
OO
OO
O
3
BergaptolXanthotoxol
Bergapten
OOO
45
OH
OOO
OH
OOO
O
Angelicin
OOO
6
OCH3
H3C
CH3HO
OOOO
OO
OO
HO
H3CH3C
645
3
28
7
6
1
H3C
H3C
7
8
9
Figure 24.75Selected aspects of coumarin and furanocoumarin biosynthe-sis. The enzymes involved (and their cofactors) are as follows:1. DMAPP:umbelliferone dimethylallyl transferase, modifyingthe 6 position; 2. marmesin synthase (O2, cytochrome P450,NADPH); 3. psoralen synthase (O2, cyt. P450, NADPH); 4. bergaptol synthase (O2, cyt. P450, NADPH); 5. xanthotoxolsynthase (O2, cyt. P450, NADPH); 6. bergaptol O-methyltrans-ferase (SAM); 7. DMAPP:umbelliferone dimethylallyl trans-ferase, modifying the 8 position; 8. columbianetin synthase(O2, cyt. P450, NADPH); and 9, angelicin synthase (O2, cyt.P450, NADPH).
but their antifungal effects are more potent,inhibiting fungal spore germination and hyphal growth; stilbenes also function indormancy and growth inhibition of plants. Certain stilbenoids, besides being toxic to in-sects and other organisms, have mammalianantifeedant and nematicidal properties. Stil-benoid formation can be induced by insectattack, as illustrated by the colored depositsformed in radiata pine sapwood when at-tacked by the Sarix wasp (see photographsin Box 24.5).
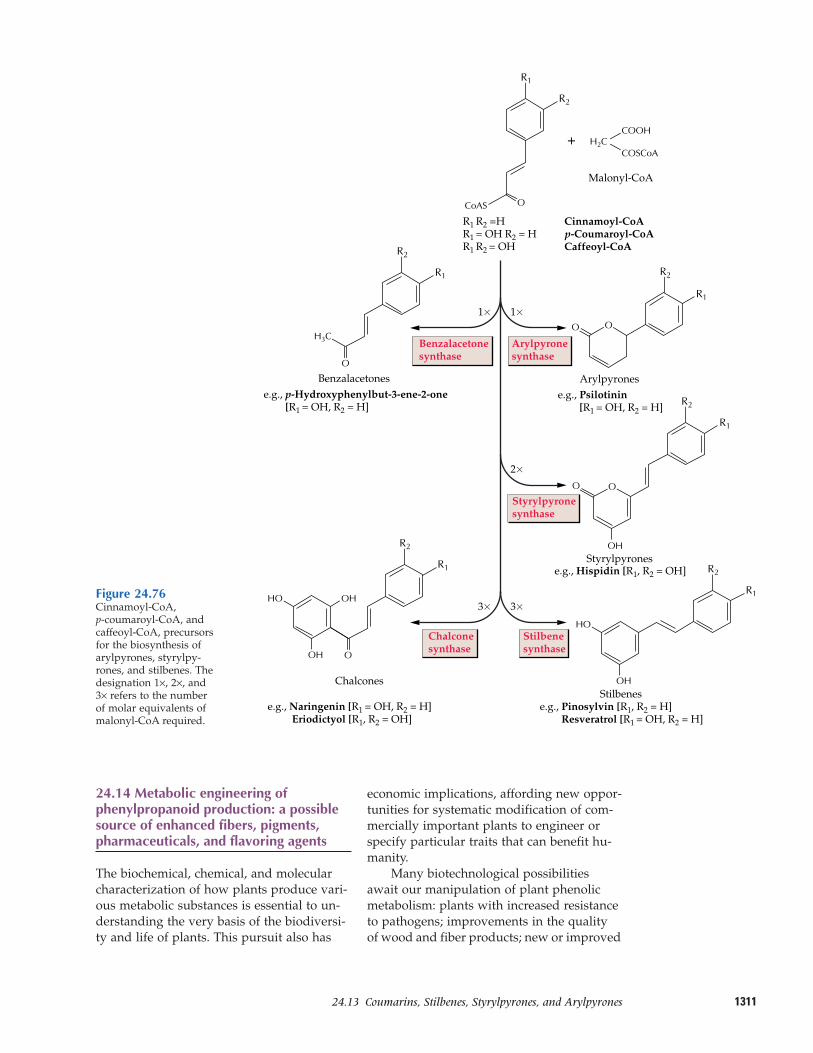
131124.13–Coumarins, Stilbenes, Styrylpyrones, and Arylpyrones
24.14 Metabolic engineering ofphenylpropanoid production: a possiblesource of enhanced fibers, pigments,pharmaceuticals, and flavoring agents
The biochemical, chemical, and molecularcharacterization of how plants produce vari-ous metabolic substances is essential to un-derstanding the very basis of the biodiversi-ty and life of plants. This pursuit also has
economic implications, affording new oppor-tunities for systematic modification of com-mercially important plants to engineer orspecify particular traits that can benefit hu-manity.
Many biotechnological possibilitiesawait our manipulation of plant phenolicmetabolism: plants with increased resistanceto pathogens; improvements in the qualityof wood and fiber products; new or improved
O
H2C
H3C
COOH
COSCoA+
1�1�
Malonyl-CoA
OCoAS
O
Arylpyronese.g., p-Hydroxyphenylbut-3-ene-2-one
[R1 = OH, R2 = H]
O
2�
O
Styrylpyrones
O
OH
3�3�
OH
OH OH
e.g., Naringenin [R1 = OH, R2 = H] Eriodictyol [R1, R2 = OH]
Stilbenes
O
OH
OH
R2
Benzalacetonesynthase
Arylpyronesynthase
Styrylpyronesynthase
Stilbenesynthase
Chalconesynthase
R1 R2 =H Cinnamoyl-CoAR1 = OH R2 = H p-Coumaroyl-CoAR1 R2 = OH Caffeoyl-CoA
e.g., Psilotinin [R1 = OH, R2 = H]
e.g., Pinosylvin [R1, R2 = H] Resveratrol [R1 = OH, R2 = H]
e.g., Hispidin [R1, R2 = OH]
Benzalacetones
Chalcones
R1
R2
R1
R2
R1
R2
R1
R2
R1
R1
R2
Figure 24.76Cinnamoyl-CoA, p-coumaroyl-CoA, andcaffeoyl-CoA, precursorsfor the biosynthesis ofarylpyrones, styrylpy-rones, and stilbenes. Thedesignation 1×, 2×, and3× refers to the numberof molar equivalents ofmalonyl-CoA required.

1312 Chapter 24 Natural Products (Secondary Metabolites)
Box 24.5 Postlignification metabolism and heartwood formation requirenonstructural plant phenolic compounds.
Heartwood represents more than 95%of the merchantable bole of harvestedwood. The heartwood of commerciallyimportant woody plants accounts formore than 60% of the revenues generatedfrom harvesting plant materials before fur-ther factory processing. Heartwood servesas the main source of raw material forlumber, solid wood products, fine furni-ture, paper, and many miscellaneous ap-plications. Despite its economic signifi-cance, however, the general mechanismresponsible for its formation is one of themost poorly studied and poorly under-stood areas of plant science today.
Heartwood is formed by the species-specific deposition of distinct and variedmetabolites that frequently alter the color,durability, texture, and odor of particularwoods relative to that of sapwood. Heart-woods contain strikingly distinctive col-ored metabolites that can readily be ob-served by inspecting cross-sections ofwoody stems of plants, such as tamarack(see panel A of figure), western red cedar (reddish- or pinkish-brown to darkbrown), ebony (jet black), and southernpine (yellow-orange). In contrast, sprucewood, highly valued for pulp and papermanufacture, contains less of the highlycolored heartwood metabolites and hencehas a pale whitish-yellow color. Indeed,the pale color and the very high lignin
content of this wood (about 28%) indicatethat lignin biopolymers themselves arenearly colorless.
Heartwood production is a postsec-ondary xylem-forming process, wherebynonstructural highly colored phenolics(primarily lignan, stilbene, and flavonoid-derived compounds) and other character-istic substances (e.g., terpenes or alka-loids) are infused into wood that hasalready been lignified. Substances similar(if not identical, in some cases) to those inheartwood can also be formed in regionswhere insects or pathogens have attackedsapwood, but these are manifested as amore-localized containment response. Forexample, panel B of the figure shows sap-wood of radiata pine into which a Sirexnoctilio wasp has bored, forming two tun-nels, one for the wasp’s eggs and one fora fungus, Amylostereum noctilio, thatserves as a foodstuff for the larvae. The at-tacked plant responds by increasing thedeposition of various phenolic sub-stances, in this case stilbenes, which areprimarily localized in the affected regions,making them appear lighter-colored thanthe background in the stained wood sec-tion shown in panel C of the figure.
Constitutive heartwood formation, onthe other hand, follows several years ordecades of sapwood growth and develop-ment. According to biochemical details
only now becoming known, heartwoodmetabolites are first deposited in the cen-tral (pith) region of the lignified woodystem tissues, which primarily consist ofdead, lignified cells. Over years of subse-quent growth, heartwood formation grad-ually extends radially, until almost all ofthe woody xylem tissue is encompassed.A transition zone sometimes visible be-tween the heartwood and sapwood is pre-sumed to be involved in the final stages ofbiosynthesis of heartwood metabolitespreceding cell death. The composition ofheartwood metabolites varies extensivelyamong species. For example, Douglas firaccumulates flavonoids and lignans in itsheartwood, whereas yellow poplar de-posits lignans, terpenoids, and alkaloids.
Given that wood is composed largelyof dead cells, how are heartwoodmetabolites deposited? Investigators rec-ognized 50 years ago that ray parenchy-ma cells remain living in lignified sap-wood. As their last function before death,these cells accumulate or biosynthesizesubstances (often in complex species-spe-cific mixtures) that are then infused intolignified woody secondary xylem by wayof pit apertures (see figure, panel D). Thisinfusion process may explain why manyof the heartwood substances also occur at much lower concentrations in sap-wood, where the ray parenchyma cells
Figure 24.77(A) The stilbene, combretastatin, has antineoplastic activity. (B) Another stilbene, resveratrol,present in red grapes and red wine, has potent antitumor properties.
(A)
Resveratrol(a stilbene)
HO
OH
OH
(B)
Combretastatin
H3CO
OHHO
OCH3
OCH3
OCH3

131324.14–Metabolic Engineering of Phenylpropanoid Production
sources of pharmaceuticals, nutriceuticals,pigments, flavors, and fragrances; and selec-tive adjustments to the taste and odor of se-lected plant species. Indeed, a biotechnologicalrevolution is now being witnessed in theplant sciences. The combined use of molecu-lar genetic techniques and conventionalplant breeding approaches is expected toproduce a new generation of plants that areeven further optimized for human use.
By far the largest and economically mostsignificant deployment of plant materials isas a fiber source, whether for pulp/paper,lumber for housing and shelter, wood forfurniture, or other applications. Accordingly,many biotechnological strategies are directedtoward improving fiber and wood propertiesby manipulating the biochemical processesresponsible for cell wall biosynthesis and as-
sociated metabolic functions. This approachcould involve modification of lignin, eitherto render it more susceptible to removal, orto increase its content, thereby increasing thestrength and rigidity of certain fragile crops.Modifying heartwood metabolite formationmay allow researchers to tailor traits such asrot resistance, texture, color, and durabilityin various commercially important woodyplant heartwoods. These goals require fur-ther study of the fundamental mechanismscontrolling both macromolecular assemblypatterns involved in the biosynthesis ofplant cell wall polymers and exploration ofhow and where the heartwood-formingmetabolites are generated.
To this point, most of the biotechnologicalemphasis placed on attempting to engineerlignin content and composition has involved
Bark Sapwood Heartwood
(A) (B)
(D)
Ray parenchyma
Substances secretedthrough pitapertures
Neighboringcell lumen
(C)
are located. At some point during heart-wood formation, the infused phenolicsubstances apparently undergo oxidationto form nonstructural oligomeric andpolymeric components, some of whichcan be removed only under the conditionstypically used for lignin extraction. Withthe further sealing of bordered pits, heart-wood ceases to function in conductingnutrients and water and essentially be-comes a protective structural tissue, highlydurable and resistant to rot.

1314 Chapter 24 Natural Products (Secondary Metabolites)
pursue various interesting questions, includ-ing how heartwood is formed.
Advances in lignan and (iso)flavonoidbiochemistry and molecular biology offer the opportunity to modify concentrations of health protectants and pharmacologicallyactive species in particular plants of choice.Eventually, we should be able to engineerthe formation of secoisolariciresinol, matai-resinol, daidzein, genistein, and similar com-pounds in staple crops that do not ordinarilyproduce them in significant quantities. Thecorresponding transgenic plants thus wouldprovide long-term health benefits as sourcesof cancer preventives. A similar target forenhanced production might be podophyllo-toxin, one of a handful of plant anticancercompounds already in use today.
The potential being unleashed is perhapsmost vividly demonstrated by the impres-sive advances in plant metabolic engineeringseen in the manipulation of flower color byapplication of sense/antisense technologies.Several laboratories in Europe and NewZealand have successfully transformed vari-ous plants such as petunia to alter petal color.
Lastly, knowledge of these pathwayswill eventually lead to the systematic modi-fication and improvement of plant flavorsand fragrances, the properties of which de-fine the very essence of many of our food-stuffs, such as pepper, ginger, and vanilla.
using antisense and sense strategies to targetthe genes that encode various enzymaticsteps in the pathway from phenylalanine tothe monolignols (see Fig. 24.49). This workhas focused primarily on cinnamyl alcoholdehydrogenase and cinnamoyl-CoA reduc-tase and has targeted such plants as tobacco,poplar, and eucalyptus. Although the effectson lignin formation per se have often beenquite small, the transgenic plant tissues pro-duced were highly colored, unlike the origi-nal wild-type plants. Whether these trans-genic plants will have any beneficialproperties, for example, greater ease oflignin removal for pulp/paper applications,is unclear. The pigmentation effects observedwere not anticipated by the researchers in-volved and point to the fact that attempts toalter lignin-forming processes must also takeinto account the related biochemical path-ways that utilize the same substrates.
The finding that lignin formation properis somehow temporally and spatially associ-ated with various presumedly proline-richproteins and dirigent sites holds muchpromise. Full details of the influence of theseproteins on lignin structure may result in the design of new strategies for modifyingboth lignin deposition and structure. Thediscovery of dirigent proteins, pinoresinol/lariciresinol reductases, and their correspond-ing genes also affords the opportunity to
Box 24.6 Phenolics flavor our world.
Phenylpropanoid-derived plant phe-nolics contribute significantly to impart-ing specific fragrances/odors, flavors, andtastes to various plants widely utilized inthe food and beverage industries today(see figure). Although the biochemistry oftheir formation is scarcely addressed inthis brief chapter, their importance cannotbe discounted.
The capsaicinoids, such as capsaicin,are responsible for the pungent propertiesof the red peppers, whereas the piperi-noids flavor black pepper. The delightfultastes of cinnamon and ginger are impart-ed by various cinnamate and gingerolderivatives, respectively; and allylphenols
establish the characteristic tastes andodors of oil of cloves, widely used intoothache treatment, and of the spicesnutmeg and mace. Vanillin, from thevanilla bean, is used extensively in bothbaking and confectionery. In most in-stances, precise biochemical pathways tothese compounds are not yet establishedat the levels of either the enzymes or thegenes.
Plant phenolics are important compo-nents of the characteristic aromas, flavors,and colors of many beverages, whetherfor alcoholic or nonalcoholic consump-tion. Chlorogenic acid, for example, con-stitutes about 4% of the coffee bean and
is thus ingested daily by millions. Greenand black tea leaves contain other plantphenolics, such as (epi-)catechins andvarious other tannins that impart charac-teristic tastes to these popular beverages.Most drinks consumed today would bewatery indeed if not for various pheno-lics, such as vanillin, ferulic acid, certainflavonoids, tannins, and others. An impor-tant endeavor of the flavor and fragranceindustry is to define or identify the mix-tures of various phenolic substances thatcreate pleasing flavors ranging frommaple syrup to whisky.
131524.14–Metabolic Engineering of Phenylpropanoid Production
Vanillin
OH
CHO
OCH3
Phenylethyl alcohol
Orchid
CH2CH2OH
R = H SafroleR = OCH3 Myristicin
Nutmeg
R
O
O
R = H ChavicolR = OCH3 Eugenol
Cloves
OH
R
R = H (–)-EpicatechinR = OH (–)-Epigallocatechin
Green tea
OH
OH
OOH
OH
HO
Red and black peppers
O
Gingerols
n=4,6,8
Ginger rhizome
OH
OH
(CH2)nCH3
OCH3
Cinnamaldehyde
Cinnamon barkCHO
Coffee beans
Chlorogenic acid
OH
OH
HO OH
COOH
Piperine
O
O
N
H3CO
HO
O
RN
H
Capsaicin
NordihydrocapsaicinR =
R =
O
O
OH
Vanilla
O
R

These modifications will ultimately impactthe quality of many of our alcoholic andnonalcoholic beverages, which in turn are often largely determined by their aromaticphenolic constituents (Box 24.6).
Summary
Plants produce a great variety of organiccompounds that are not directly involved inprimary metabolic processes of growth anddevelopment. The roles these natural prod-ucts or secondary metabolites play in plantshave only recently come to be appreciated inan analytical context. Natural products ap-pear to function primarily in defense againstpredators and pathogens and in providingreproductive advantage as attractants of pol-linators and seed dispersers. They may alsoact to create competitive advantage as poi-sons of rival species.
Most natural products can be classifiedinto three major groups: terpenoids, alka-loids, and phenolic compounds (mostlyphenylpropanoids). Terpenoids are com-posed of five-carbon units synthesized byway of the acetate/mevalonate pathway orthe glyceraldehyde 3-phosphate/pyruvatepathway. Many plant terpenoids are toxinsand feeding deterrents to herbivores or areattractants of various sorts. Alkaloids aresynthesized principally from amino acids.These nitrogen-containing compounds pro-tect plants from a variety of herbivorous ani-mals, and many possess pharmacologicallyimportant activity. Phenolic compounds,which are synthesized primarily from products of the shikimic acid pathway, haveseveral important roles in plants. Tannins,lignans, flavonoids, and some simple pheno-lic compounds serve as defenses against her-bivores and pathogens. In addition, ligninsstrengthen cell walls mechanically, andmany flavonoid pigments are important at-tractants for pollinators and seed dispersers.Some phenolic compounds have allelopathicactivity and may adversely influence thegrowth of neighboring plants.
Throughout the course of evolution,plants have developed defenses against her-bivory and microbial attack and producedother natural products to aid competitive-ness. The better-defended, more-competitiveplants have generated more progeny, and so
1316 Chapter 24 Natural Products (Secondary Metabolites)
the capacity to produce and safely store suchecologically useful metabolites has becomewidely established in the plant kingdom.Pressures from herbivores and pathogens, aswell as constant competition, continue to se-lect for new natural products. In cultivatedspecies, however, such chemical defenseshave often been artificially selected against.
Study of the biochemistry of plant natu-ral products has many practical applications.Biotechnological approaches can selectivelyincrease the amounts of defense compoundsin crop plants, thereby reducing the need forcostly and potentially toxic pesticides. Simi-larly, genetic engineering can be utilized toincrease the yields of pharmaceuticals, flavorand perfumery materials, insecticides, fungi-cides, and other natural products of com-mercial value. Although many natural prod-ucts and their functions have been describedin this chapter, the metabolism of naturalproducts in most plant species remains to beelucidated. A great deal of fascinating bio-chemistry remains to be discovered.
Further Reading
Terpenoids
Cane, D. E., ed. (1999) Comprehensive NaturalProducts Chemistry, Vol. 2, Isoprenoids In-cluding Carotenoids and Steroids. Perga-mon/Elsevier, Amsterdam.
Chappell, J. (1995) Biochemistry and molecu-lar biology of the isoprenoid biosyntheticpathway in plants. Annu. Rev. Plant Physi-ol. Plant Mol. Biol. 46: 521–547.
Eisenreich, W., Schwarz, M., Cartayrade, A.,Arigoin, D., Zenk, M. H., Bacher, A. (1998)The deoxylulose phosphate pathway ofterpenoid biosynthesis in plants and mi-croorganisms. Chem. Biol. 5: R221–R233.
Gershenzon, J., Croteau, R. (1993) Terpenoidbiosynthesis: the basic pathway and for-mation of monoterpenes, sesquiterpenesand diterpenes. In Lipid Metabolism inPlants, T. S. Moore, Jr., ed. CRC Press, BocaRaton, FL, pp. 339–388.
Harborne, J. B., and Tomas-Barberan, F. A.,eds. (1991) Ecological Chemistry and Bio-chemistry of Plant Terpenoids. ClarendonPress, Oxford, UK.
Langenheim, J. H. (1994) Higher plant terpenoids: a phytocentric overview of

1317Further Reading
their ecological roles. J. Chem. Ecol. 20:1223–1280.
Lichtenthaler, H. K. (1999) The 1-deoxy-D-xylulose-5-phosphate pathway of iso-prenoid biosynthesis in plants. Annu. Rev.Plant Physiol. Plant Mol. Biol. 50: 47–65.
McGarvey, D., Croteau, R. (1995) Terpenoidbiosynthesis. Plant Cell 7: 1015–1026.
Alkaloids
Cordell, G., ed. (1997) The Alkaloids, Vol. 50.Academic Press, San Diego.
Rosenthal, G. A., and Berenbaum, M. R., eds.(1991) Herbivores: Their Interactions with Sec-ondary Plant Metabolites, Vol. 1: The Chemi-cal Participants, 2nd ed. Academic Press, San Diego.
Southon, I. W., and Buckingham, J., eds.(1989) Dictionary of Alkaloids. Chapman and Hall, London.
Suberin
Bernards, M. A., Lewis, N. G. (1998) Themacromolecular aromatic domain in suber-ized tissues: a changing paradigm. Phyto-chemistry 47: 583–591.
Lignins and lignans
Gang, D. R., Costa, M. A., Fujita, M., Dinkova-Kostova, A. T., Wang, H.-B.,Burlat, V., Martin, W., Sarkanen, S., Davin,L. B., Lewis, N. G. (1999) Regiochemicalcontrol of monolignol radical coupling: anew paradigm for lignin and lignanbiosynthesis. Chem. Biol. 6: 143–151.
Lewis, N. G., Davin, L. B. (1999) Lignans:biosynthesis and function. In ComprehensiveNatural Products Chemistry, Vol. 1, Polyke-tides and Other Secondary Metabolites Includ-ing Fatty Acids and Their Derivatives, D.H.R.Barton, K. Nakanishi, and O. Meth-Cohn,eds.-in-chief. Elsevier, Amsterdam, pp.639–712.
Lewis, N. G., Davin, L. B., Sarkanen, S.(1999) The nature and functions of lignins.In Comprehensive Natural Products Chemistry,Vol. 3, Carbohydrates and Their Derivatives In-cluding Tannins, Cellulose and Related Lignins,D.H.R. Barton, K. Nakanishi, and O. Meth-Cohn, eds.-in-chief. Elsevier, Amsterdam,pp. 617–745.
Sarkanen, S., Lewis, N. G. (1998) Biosynthe-sis of lignins and lignans. ACS Symp. Ser.697: 1–421.
Timell, T. E. (1986) Compression Wood in Gymnosperms, Vols. 1–3. Springer-Verlag, Berlin.
Flavonoids
Dixon, R. A. (1999) Isoflavonoids: biochem-istry, molecular biology and biologicalfunctions. In Comprehensive Natural Prod-ucts Chemistry, Vol. 1, Polyketides and OtherSecondary Metabolites Including Fatty Acidsand Their Derivatives, D.H.R. Barton, K.Nakanishi, and O. Meth-Cohn, eds.-in-chief. Elsevier, Amsterdam, pp. 773–824.
Forkmann, G. (1991) Flavonoids as flowerpigments: the formation of the naturalspectrum and its extension by genetic en-gineering. Plant Breed. 106: 1–26.
Forkmann, G., Heller, W. (1999) Biosynthesisof flavonoids. In Comprehensive NaturalProducts Chemistry, Vol. 1, Polyketides andOther Secondary Metabolites Including FattyAcids and Their Derivatives, D.H.R. Barton,K. Nakanishi, and O. Meth-Cohn, eds.-in-chief. Elsevier, Amsterdam, pp. 713–748.
Harborne, J. B., ed. (1994) The Flavonoids: Ad-vances in Research Since 1986. Chapman andHall, London.
Coumarins and furanocoumarins
Berenbaum, M. R., and Zangerl, A. R. (1996)Phytochemical diversity: adaptation or ran-dom variation. Rec. Adv. Phytochem. 30: 1–24.
Keating, G. J., O’Kennedy, R. (1997) The chem-istry and occurrence of coumarins. In Cou-marins: Biology, Application and Mode of Ac-tion, R. O’Kennedy and R. D. Thornes, eds.John Wiley & Sons, Chichester, pp. 23–66.
Matern, U., Lüer, P., Kreusch, D. (1999)Biosynthesis of coumarins. In Comprehen-sive Natural Products Chemistry, Vol. 1,Polyketides and Other Secondary MetabolitesIncluding Fatty Acids and Their Derivatives,D.H.R. Barton, K. Nakanishi, and O. Meth-Cohn, eds.-in-chief. Elsevier, Amsterdam,pp. 623–638.
Matern, U., Strasser, H., Wendorff, H.,Hamerski, D. (1988) Coumarins and fura-nocoumarins. In Cell Culture and SomaticCell Genetics of Plants, Vol. 5, I. K. Vasil, ed.Academic Press, Orlando, FL, pp. 3–21.

Zobel, A. M. (1997) Coumarins in fruits andvegetables. Proc. Phytochem. Soc. Eur. 41:173–203.
Stilbenes, styrylpyrones, and arylpyrones
Berkert, C., Horn, C., Schnitzler, J.-P., Lehning,A., Heller, W., Veit, M. (1997) Styrylpyronebiosynthesis in Equisetum arvense. Phyto-chemistry 44: 275–283.
Gorham, J., Tori, M., Asakawa, Y. (1995) The
1318 Chapter 24 Natural Products (Secondary Metabolites)
Biochemistry of the Stilbenoids, Vol. 1, Bio-chemistry of Natural Products Series, J. B.Harborne, ed. Chapman and Hall, London.
Schröder, J. (1999) The chalcone/stilbenesynthase-type family of condensing enzymes. In Comprehensive Natural ProductsChemistry, Vol. 1, Polyketides and Other Sec-ondary Metabolites Including Fatty Acids andTheir Derivatives, D.H.R. Barton, K. Naka-nishi, and O. Meth-Cohn, eds.-in-chief. El-sevier, Amsterdam, pp. 749–772.





























