Embed Size (px)
Citation preview

doi:10.1016/j.jmb.2008.01.051 J. Mol. Biol. (2008) 377, 1117–1129
Available online at www.sciencedirect.com
SEA Domain Autoproteolysis Accelerated byConformational Strain: Energetic Aspects
Anders Sandberg1,2, Denny G. A. Johansson1, Bertil Macao1
and Torleif Härd1,2⁎
1Department of MedicalBiochemistry, GöteborgUniversity, PO Box 440,SE-405 30 Göteborg, Sweden2The Swedish NMR Centre,Göteborg University,PO Box 465, SE-405 30Göteborg, Sweden
Received 5 November 2007;received in revised form28 December 2007;accepted 18 January 2008Available online30 January 2008
*Corresponding author. DepartmentBiochemistry, Göteborg University, PGöteborg, Sweden. E-mail address:Abbreviations used: GdmCl, guan
GPS, G protein-coupled receptor proinclusion body; IMAC, immobilizedchromatography; NTA, nitrilotriacetterminal nucleophile; SEA, sea urchenterokinase, and agrin; TS, transitio
0022-2836/$ - see front matter © 2008 P
A subclass of proteins with the SEA (sea urchin sperm protein, enterokinase,and agrin) domain fold exists as heterodimers generated by autoproteolyticcleavage within a characteristic G−1S+1VVV sequence. Autoproteolysisoccurs by a nucleophilic attack of the serine hydroxyl on the vicinal glycinecarbonyl followed by anN→Oacyl shift and hydrolysis of the resulting ester.The reaction has been suggested to be accelerated by the straining of thescissile peptide bond upon protein folding. In an accompanying article, wereport the mechanism; in this article, we provide further key evidence andaccount for the energetics of coupled protein folding and autoproteolysis.Cleavage of the GPR116 domain and that of the MUC1 SEA domain occurwith half-life (t½) values of 12 and 18 min, respectively, with lowering of thefree energy of the activation barrier by ∼10 kcal mol−1 compared withuncatalyzed hydrolysis. The free energies of unfolding of the GPR116 andMUC1 SEA domains were measured to ∼11 and ∼15 kcal mol−1, res-pectively, but ∼7 kcal mol−1 of conformational energy is partitioned as strainover the scissile peptide bond in the precursor to catalyze autoproteolysis bysubstrate destabilization. A straining energy of ∼7 kcal mol−1 was measuredby using both a pre-equilibrium model to analyze stability and cleavagekinetics data obtained with the GPR116 SEA domain destabilized by coremutations or urea addition, as well as the difference in thermodynamicstabilities of the MUC1 SEA precursor mutant S1098A (with a G−1A+1VVVmotif) and the wild-type protein. The results imply that cleavage by N→Oacyl shift alone would proceedwith a t½ of∼2.3 years, which is too slow to bebiochemically effective. A subsequent review of structural data on other self-cleaving proteins suggests that conformational strain of the scissile peptidebond may be a common mechanism of autoproteolysis.
© 2008 Published by Elsevier Ltd.
Keywords: autoproteolysis; protein folding; protein stability; conformationalstrain; SEA domain
Edited by C. R. MatthewsIntroduction
Autoproteolysis of the MUC1 SEA (sea urchinsperm protein, enterokinase, and agrin) domain is an
of MedicalO Box 440, SE-405 [email protected] chloride;teolytic site; IB,metal affinityic acid; Ntn, N-in sperm protein,n state.
ublished by Elsevier Ltd.
intramolecular reaction where the protein chain iscleaved between a glycine and a serine at the con-served G−1S+1VVV motif.1,2 This reaction, which isprobably similar in all systems undergoing autopro-teolysis,3 proceeds via an N→O acyl shift where thenucleophilic serine (S+1) hydroxyl group attacks theglycine (G−1) carbonyl atom, which generates anester that is subsequently hydrolyzed into novel N′and C′ termini.4 The result is a heterodimeric yetsingle-domain structure. For the SEA domains, thiscleavage reaction may have evolved as a membrane-protective function2 and/or to serve as a receptor–ligand entity.5 Several other SEA domains with thecharacteristic GSVVV sequence also undergo auto-proteolysis, and although most of these are found

1118 Autoproteolysis by Conformational Strain: Energetics
in the mucin protein family, there is also one homo-logue in the non-mucin GPR116 orphan receptor.2,6
The uncatalyzed hydrolysis of internal peptidebonds is an extremely slow reaction that proceedswith a half-life (t½) of ∼2100 years (at 21 °C andpH 6.8).7 In the SEA domains, this reaction is cata-lyzed not only by the N→O acyl shift but also byconformational strain,2,8 bringing down the t½ towithin minutes.9–11 In an accompanying article,8 weaddress several of the immediate implications of amechanism involving a folding-induced straining(destabilization) of the scissile peptide bond (thesubstrate) as opposed to catalysis by traditionaltransition-state (TS) stabilization. In brief, we showthat the strain provides a mechanistic link betweenthe folding and cleavage reactions. Several experi-mental findings support this interpretation: (i)extensive mutational studies of polar residues nearthe cleavage site do not affect the cleavage; (ii) nativestructure formation and cleavage are concomitant;(iii) the rate of the reaction can be manipulatedby loop insertions that presumably act to alleviatestrain; and (iv) the G−1A+1 precursor mutant (theS1098A mutant), which, despite lacking the nucleo-philic serine, is able to undergo spontaneous cleav-age. Molecular dynamics simulations of an un-cleaved precursor model (with an intact G−1S+1
bond) also demonstrate that there is a transientcleavage-competent conformation with a strainedtrans peptide that positions the catalytic Ser1098 forefficient nucleophilic attack at the Gly1097 carbonyl,thus allowing the N→O acyl shift to proceed.The mechanism implies that the energy required
for autoproteolysis must stem from the conforma-tional free energy, which we here define as the freeenergy of the unfolded state relative to the foldedstate (i.e., ΔGD−N=GD−GN). Based on TS theoryand considerations of the upper limit of the effect ofthe serine hydroxyl acting as a nucleophile, it waspreviously estimated that a ΔGD−N of 4.5–7 kcalmol−1 would be sufficient to drive the cleavagereaction in the MUC1 SEA domain.2 Since theΔGD−N for proteins typically ranges from 5 to15 kcal mol−1, this is not an unrealistic assumpt-ion.12 That the energy released upon SEA proteinfolding is put to practical use by the triggering of achemical reaction is also an attractive novel bio-chemical mechanism.Here we address this final issue concerning SEA
autoproteolysis, namely, if the ΔGD−N can accountfor the observed rates of proteolysis, and just howmuch the scissile bond is strained during catalysis.We do this by measuring the conformational stabi-lities and cleavage rates of the wild type and seve-ral mutants of theMUC1 SEA domain and of the SEAdomain from the human GPR116 receptor. Both pro-teins proved to be very stable, with ΔGD−N valuesexceeding 10 kcal mol−1, and the rates of proteoly-sis were determined to 6.4×10−4 s−1 (t½ ∼18 min) forMUC1 SEA and 9.3×10−4 s−1 (t½ ∼12 min) forGPR116 SEA. These correspond to a lowering of thefree energy of activation barrier (ΔΔG‡) of ∼10 kcalmol−1 when compared with the spontaneous hydro-
lysis of peptide bonds.7 Much of this accelerationstems from the destabilizing strain energy, which wemeasured to 7 kcal mol−1 in two experiments. In thefirst experiment, an evaluation of the cleavage ratesof destabilized GPR116 SEA as a function of ΔGD−Nyielded 7.1 kcal mol−1 of strain energy. The secondexperiment demonstrated that the MUC1 SEA pre-cursor mutant S1098A is destabilized by 7.4 kcalmol−1 compared with wild type. The results alsoimply that the N→O acyl shift mechanism con-tributes with a ΔΔG‡ of 4 kcal mol−1. These twocatalytic mechanisms (i.e., strain-induced destabiliza-tion of the scissile peptide bond and the N→O acylshift) must therefore work in concert to generate thefast cleavage rates observed for the SEA domains.Finally, a survey of the literature data suggests thatcoupled folding and cleavage mechanisms are com-mon among proteins undergoing autoproteolysis.
Results
Kinetics of cleavage
Previous in vivo measurements of MUC1 SEA do-main cleavage have estimated the t½ of the reactionto 5 min.9–11 In order to get good in vitro data on thisreaction, we produced SEA as insoluble inclusionbodies (IBs) in their uncleaved form (not shown). Therefolding and cleavage were then monitored withsodium dodecyl sulfate (SDS) polyacrylamide gelelectrophoresis (PAGE). The uncleaved precursorproteins were found to be aggregation prone duringthe refolding assays, as is expected for an intercon-verting ensemble of collapsed precursor confor-mations with only partly formed secondary struc-ture and hydrophobic cores.8 In order to preventaggregation, we therefore applied the denatureduncleaved protein to an immobilized metal affinitychromatography (IMAC) column containing Ni2+
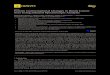
prior to the initiation of folding. Folding was thencarried out by switching to native conditions. Diffe-rent time points were obtained by incubating therefolded protein in the gel for different periods. Therelative intensities of the bands on the SDS-PAGEgel gave a rate of hydrolysis of 9.30±0.50×10−4 s−1
for the GPR116 SEA domain and that of 6.42±0.35×10−4 s−1 for the MUC1 SEA domain, corres-ponding to t½ values of ∼12 and ∼18 min, respec-tively (Fig. 1). Comparing these rates with the un-catalyzed reaction at pH 6.8 (3.6× 10− 11 s− 1)7
suggests that the concerted actions of strain and theN→O acyl shift lower theΔΔG‡ by ∼10 kcal mol−1.During the refolding of these proteins, we noticed
a competing side reaction where folding occurs intodimeric, presumably domain-swapped, non-nativestructures (not shown). These non-native structuresprobably have unstrained G−1S+1 peptide bonds asthey cannot undergo autoproteolysis. Dimerizationis also the main reason that the cleavage shownin Fig. 1 only proceeds to ∼84% for MUC1 SEA and∼43% for GPR116 SEA. Furthermore, the stability of
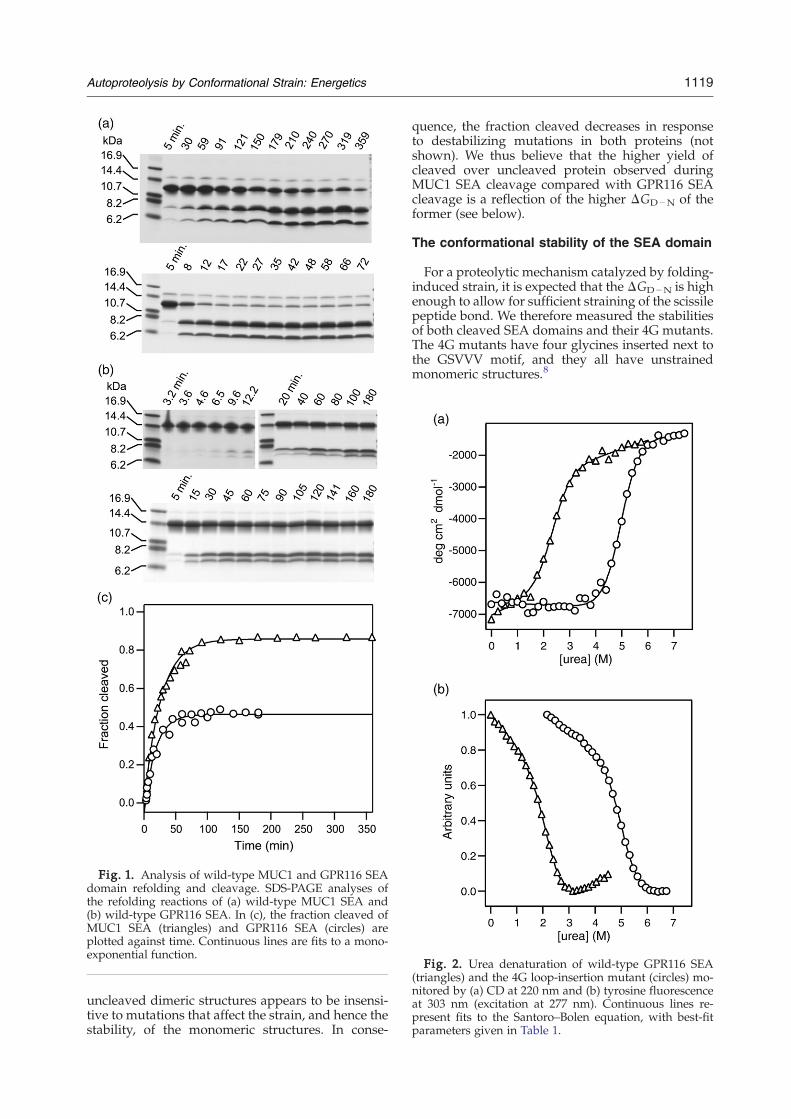
Fig. 2. Urea denaturation of wild-type GPR116 SEA(triangles) and the 4G loop-insertion mutant (circles) mo-nitored by (a) CD at 220 nm and (b) tyrosine fluorescenceat 303 nm (excitation at 277 nm). Continuous lines re-present fits to the Santoro–Bolen equation, with best-fitparameters given in Table 1.
Fig. 1. Analysis of wild-type MUC1 and GPR116 SEAdomain refolding and cleavage. SDS-PAGE analyses ofthe refolding reactions of (a) wild-type MUC1 SEA and(b) wild-type GPR116 SEA. In (c), the fraction cleaved ofMUC1 SEA (triangles) and GPR116 SEA (circles) areplotted against time. Continuous lines are fits to a mono-exponential function.
1119Autoproteolysis by Conformational Strain: Energetics
uncleaved dimeric structures appears to be insensi-tive to mutations that affect the strain, and hence thestability, of the monomeric structures. In conse-
quence, the fraction cleaved decreases in responseto destabilizing mutations in both proteins (notshown). We thus believe that the higher yield ofcleaved over uncleaved protein observed duringMUC1 SEA cleavage compared with GPR116 SEAcleavage is a reflection of the higher ΔGD−N of theformer (see below).
The conformational stability of the SEA domain
For a proteolytic mechanism catalyzed by folding-induced strain, it is expected that the ΔGD−N is highenough to allow for sufficient straining of the scissilepeptide bond. We therefore measured the stabilitiesof both cleaved SEA domains and their 4G mutants.The 4G mutants have four glycines inserted next tothe GSVVV motif, and they all have unstrainedmonomeric structures.8
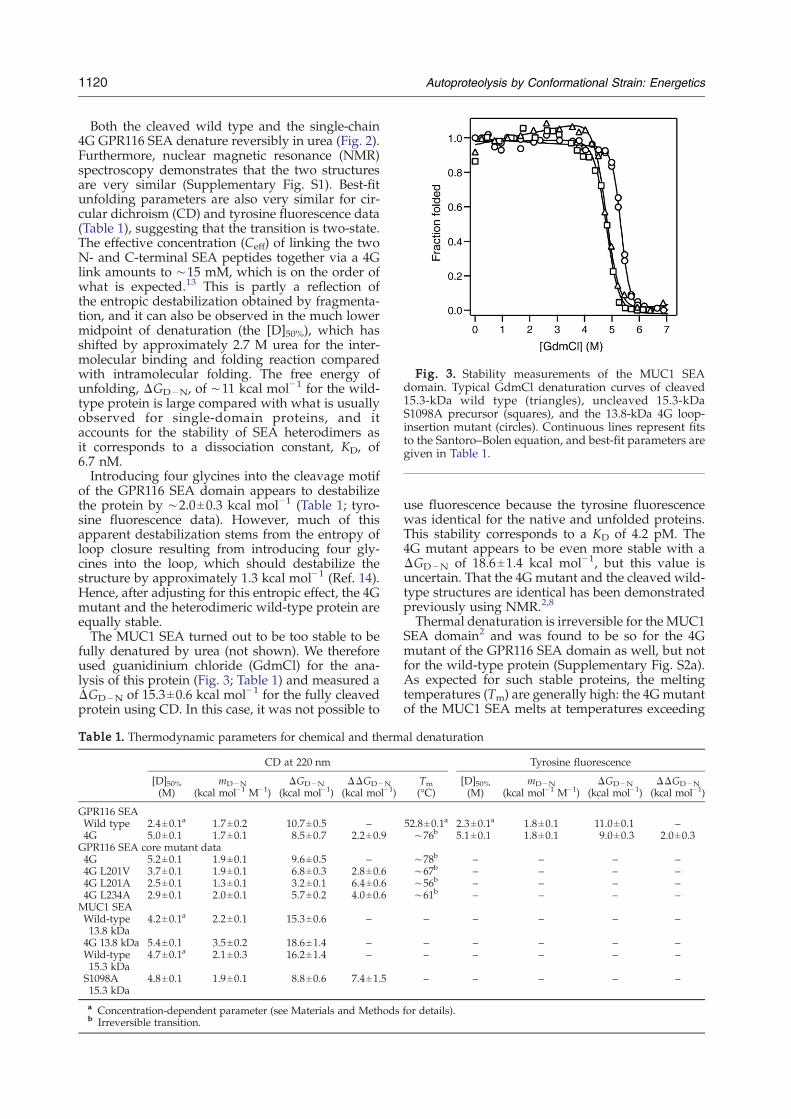
Fig. 3. Stability measurements of the MUC1 SEAdomain. Typical GdmCl denaturation curves of cleaved15.3-kDa wild type (triangles), uncleaved 15.3-kDaS1098A precursor (squares), and the 13.8-kDa 4G loop-insertion mutant (circles). Continuous lines represent fitsto the Santoro–Bolen equation, and best-fit parameters aregiven in Table 1.
1120 Autoproteolysis by Conformational Strain: Energetics
Both the cleaved wild type and the single-chain4G GPR116 SEA denature reversibly in urea (Fig. 2).Furthermore, nuclear magnetic resonance (NMR)spectroscopy demonstrates that the two structuresare very similar (Supplementary Fig. S1). Best-fitunfolding parameters are also very similar for cir-cular dichroism (CD) and tyrosine fluorescence data(Table 1), suggesting that the transition is two-state.The effective concentration (Ceff) of linking the twoN- and C-terminal SEA peptides together via a 4Glink amounts to ∼15 mM, which is on the order ofwhat is expected.13 This is partly a reflection ofthe entropic destabilization obtained by fragmenta-tion, and it can also be observed in the much lowermidpoint of denaturation (the [D]50%), which hasshifted by approximately 2.7 M urea for the inter-molecular binding and folding reaction comparedwith intramolecular folding. The free energy ofunfolding, ΔGD−N, of ∼11 kcal mol−1 for the wild-type protein is large compared with what is usuallyobserved for single-domain proteins, and itaccounts for the stability of SEA heterodimers asit corresponds to a dissociation constant, KD, of6.7 nM.Introducing four glycines into the cleavage motif
of the GPR116 SEA domain appears to destabilizethe protein by ∼2.0±0.3 kcal mol−1 (Table 1; tyro-sine fluorescence data). However, much of thisapparent destabilization stems from the entropy ofloop closure resulting from introducing four gly-cines into the loop, which should destabilize thestructure by approximately 1.3 kcal mol−1 (Ref. 14).Hence, after adjusting for this entropic effect, the 4Gmutant and the heterodimeric wild-type protein areequally stable.The MUC1 SEA turned out to be too stable to be
fully denatured by urea (not shown). We thereforeused guanidinium chloride (GdmCl) for the ana-lysis of this protein (Fig. 3; Table 1) and measured aΔGD−N of 15.3±0.6 kcal mol−1 for the fully cleavedprotein using CD. In this case, it was not possible to
Table 1. Thermodynamic parameters for chemical and therm
CD at 220 nm
[D]50%(M)
mD−N(kcal mol−1 M−1)
ΔGD−N(kcal mol−1)
ΔΔGD−N(kcal mol−1)
GPR116 SEAWild type 2.4±0.1a 1.7±0.2 10.7±0.5 –4G 5.0±0.1 1.7±0.1 8.5±0.7 2.2±0.9GPR116 SEA core mutant data4G 5.2±0.1 1.9±0.1 9.6±0.5 –4G L201V 3.7±0.1 1.9±0.1 6.8±0.3 2.8±0.64G L201A 2.5±0.1 1.3±0.1 3.2±0.1 6.4±0.64G L234A 2.9±0.1 2.0±0.1 5.7±0.2 4.0±0.6MUC1 SEAWild-type13.8 kDa
4.2±0.1a 2.2±0.1 15.3±0.6 –
4G 13.8 kDa 5.4±0.1 3.5±0.2 18.6±1.4 –Wild-type15.3 kDa
4.7±0.1a 2.1±0.3 16.2±1.4 –
S1098A15.3 kDa
4.8±0.1 1.9±0.1 8.8±0.6 7.4±1.5
a Concentration-dependent parameter (see Materials and Methodsb Irreversible transition.
use fluorescence because the tyrosine fluorescencewas identical for the native and unfolded proteins.This stability corresponds to a KD of 4.2 pM. The4G mutant appears to be even more stable with aΔGD−N of 18.6±1.4 kcal mol−1, but this value isuncertain. That the 4G mutant and the cleaved wild-type structures are identical has been demonstratedpreviously using NMR.2,8
Thermal denaturation is irreversible for the MUC1SEA domain2 and was found to be so for the 4Gmutant of the GPR116 SEA domain as well, but notfor the wild-type protein (Supplementary Fig. S2a).As expected for such stable proteins, the meltingtemperatures (Tm) are generally high: the 4G mutantof the MUC1 SEA melts at temperatures exceeding
al denaturation
Tyrosine fluorescence
Tm(°C)
[D]50%(M)
mD−N(kcal mol−1 M−1)
ΔGD−N(kcal mol−1)
ΔΔGD−N(kcal mol−1)
52.8±0.1a 2.3±0.1a 1.8±0.1 11.0±0.1 –∼76b 5.1±0.1 1.8±0.1 9.0±0.3 2.0±0.3
∼78b – – – –∼67b – – – –∼56b – – – –∼61b – – – –
– – – – –
– – – – –– – – – –
– – – – –
for details).
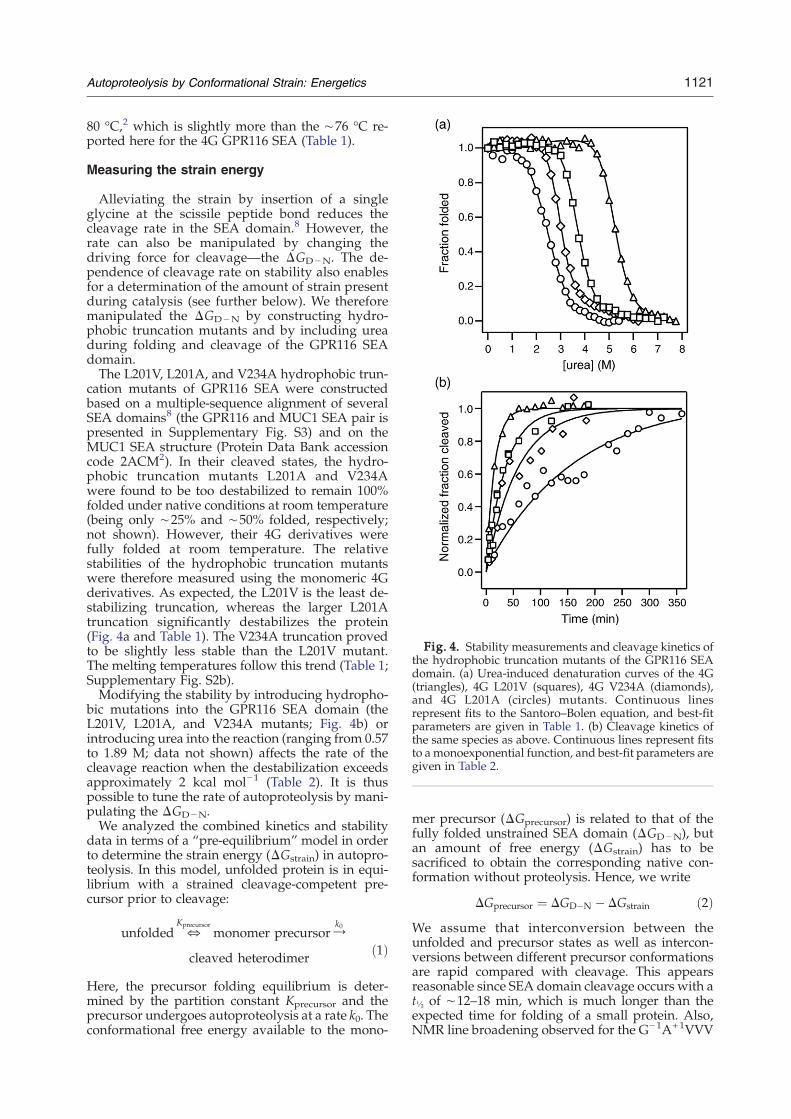
Fig. 4. Stability measurements and cleavage kinetics ofthe hydrophobic truncation mutants of the GPR116 SEAdomain. (a) Urea-induced denaturation curves of the 4G(triangles), 4G L201V (squares), 4G V234A (diamonds),and 4G L201A (circles) mutants. Continuous linesrepresent fits to the Santoro–Bolen equation, and best-fitparameters are given in Table 1. (b) Cleavage kinetics ofthe same species as above. Continuous lines represent fitsto amonoexponential function, and best-fit parameters aregiven in Table 2.
1121Autoproteolysis by Conformational Strain: Energetics
80 °C,2 which is slightly more than the ∼76 °C re-ported here for the 4G GPR116 SEA (Table 1).
Measuring the strain energy
Alleviating the strain by insertion of a singleglycine at the scissile peptide bond reduces thecleavage rate in the SEA domain.8 However, therate can also be manipulated by changing thedriving force for cleavage—the ΔGD−N. The de-pendence of cleavage rate on stability also enablesfor a determination of the amount of strain presentduring catalysis (see further below). We thereforemanipulated the ΔGD−N by constructing hydro-phobic truncation mutants and by including ureaduring folding and cleavage of the GPR116 SEAdomain.The L201V, L201A, and V234A hydrophobic trun-
cation mutants of GPR116 SEA were constructedbased on a multiple-sequence alignment of severalSEA domains8 (the GPR116 and MUC1 SEA pair ispresented in Supplementary Fig. S3) and on theMUC1 SEA structure (Protein Data Bank accessioncode 2ACM2). In their cleaved states, the hydro-phobic truncation mutants L201A and V234Awere found to be too destabilized to remain 100%folded under native conditions at room temperature(being only ∼25% and ∼50% folded, respectively;not shown). However, their 4G derivatives werefully folded at room temperature. The relativestabilities of the hydrophobic truncation mutantswere therefore measured using the monomeric 4Gderivatives. As expected, the L201V is the least de-stabilizing truncation, whereas the larger L201Atruncation significantly destabilizes the protein(Fig. 4a and Table 1). The V234A truncation provedto be slightly less stable than the L201V mutant.The melting temperatures follow this trend (Table 1;Supplementary Fig. S2b).Modifying the stability by introducing hydropho-
bic mutations into the GPR116 SEA domain (theL201V, L201A, and V234A mutants; Fig. 4b) orintroducing urea into the reaction (ranging from 0.57to 1.89 M; data not shown) affects the rate of thecleavage reaction when the destabilization exceedsapproximately 2 kcal mol−1 (Table 2). It is thuspossible to tune the rate of autoproteolysis by mani-pulating the ΔGD−N.We analyzed the combined kinetics and stability
data in terms of a “pre-equilibrium” model in orderto determine the strain energy (ΔGstrain) in autopro-teolysis. In this model, unfolded protein is in equi-librium with a strained cleavage-competent pre-cursor prior to cleavage:
unfolded fKprecursor
monomer precursorYk0
cleaved heterodimer ð1Þ
Here, the precursor folding equilibrium is deter-mined by the partition constant Kprecursor and theprecursor undergoes autoproteolysis at a rate k0. Theconformational free energy available to the mono-
mer precursor (ΔGprecursor) is related to that of thefully folded unstrained SEA domain (ΔGD−N), butan amount of free energy (ΔGstrain) has to besacrificed to obtain the corresponding native con-formation without proteolysis. Hence, we write
DGprecursor ¼ DGD�N � DGstrain ð2ÞWe assume that interconversion between theunfolded and precursor states as well as intercon-versions between different precursor conformationsare rapid compared with cleavage. This appearsreasonable since SEA domain cleavage occurs with at½ of ∼12–18 min, which is much longer than theexpected time for folding of a small protein. Also,NMR line broadening observed for the G−1A+1VVV
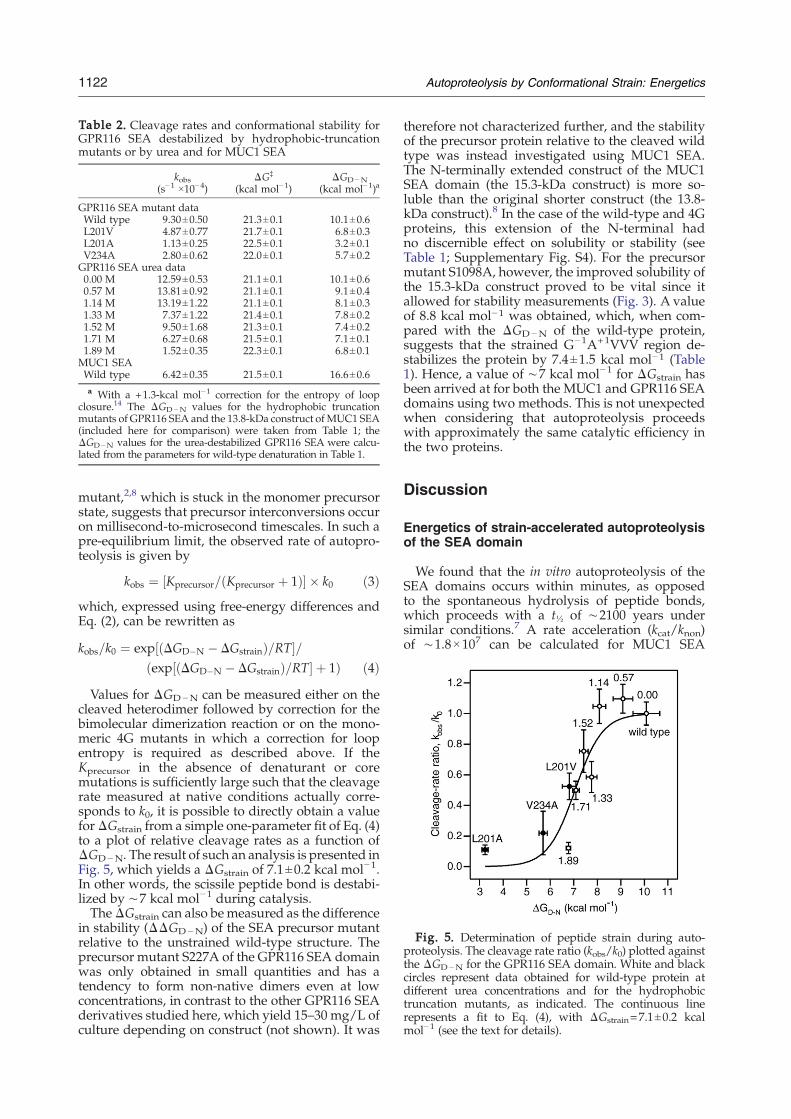
Table 2. Cleavage rates and conformational stability forGPR116 SEA destabilized by hydrophobic-truncationmutants or by urea and for MUC1 SEA
kobs(s−1 ×10−4)
ΔG‡
(kcal mol−1)ΔGD−N
(kcal mol−1)a
GPR116 SEA mutant dataWild type 9.30±0.50 21.3±0.1 10.1±0.6L201V 4.87±0.77 21.7±0.1 6.8±0.3L201A 1.13±0.25 22.5±0.1 3.2±0.1V234A 2.80±0.62 22.0±0.1 5.7±0.2GPR116 SEA urea data0.00 M 12.59±0.53 21.1±0.1 10.1±0.60.57 M 13.81±0.92 21.1±0.1 9.1±0.41.14 M 13.19±1.22 21.1±0.1 8.1±0.31.33 M 7.37±1.22 21.4±0.1 7.8±0.21.52 M 9.50±1.68 21.3±0.1 7.4±0.21.71 M 6.27±0.68 21.5±0.1 7.1±0.11.89 M 1.52±0.35 22.3±0.1 6.8±0.1MUC1 SEAWild type 6.42±0.35 21.5±0.1 16.6±0.6a With a +1.3-kcal mol−1 correction for the entropy of loop
closure.14 The ΔGD−N values for the hydrophobic truncationmutants of GPR116 SEA and the 13.8-kDa construct of MUC1 SEA(included here for comparison) were taken from Table 1; theΔGD−N values for the urea-destabilized GPR116 SEA were calcu-lated from the parameters for wild-type denaturation in Table 1.
Fig. 5. Determination of peptide strain during auto-proteolysis. The cleavage rate ratio (kobs/k0) plotted againstthe ΔGD−N for the GPR116 SEA domain. White and blackcircles represent data obtained for wild-type protein atdifferent urea concentrations and for the hydrophobictruncation mutants, as indicated. The continuous linerepresents a fit to Eq. (4), with ΔGstrain=7.1±0.2 kcalmol−1 (see the text for details).
1122 Autoproteolysis by Conformational Strain: Energetics
mutant,2,8 which is stuck in the monomer precursorstate, suggests that precursor interconversions occuron millisecond-to-microsecond timescales. In such apre-equilibrium limit, the observed rate of autopro-teolysis is given by
kobs ¼ ½Kprecursor=ðKprecursor þ 1Þ� � k0 ð3Þwhich, expressed using free-energy differences andEq. (2), can be rewritten as
kobs=k0 ¼ exp½ðDGD�N � DGstrainÞ=RT�=ðexp½ðDGD�N � DGstrainÞ=RT� þ 1Þ ð4Þ
Values for ΔGD−N can be measured either on thecleaved heterodimer followed by correction for thebimolecular dimerization reaction or on the mono-meric 4G mutants in which a correction for loopentropy is required as described above. If theKprecursor in the absence of denaturant or coremutations is sufficiently large such that the cleavagerate measured at native conditions actually corre-sponds to k0, it is possible to directly obtain a valueforΔGstrain from a simple one-parameter fit of Eq. (4)to a plot of relative cleavage rates as a function ofΔGD−N. The result of such an analysis is presented inFig. 5, which yields a ΔGstrain of 7.1±0.2 kcal mol−1.In other words, the scissile peptide bond is destabi-lized by ∼7 kcal mol−1 during catalysis.TheΔGstrain can also bemeasured as the difference
in stability (ΔΔGD−N) of the SEA precursor mutantrelative to the unstrained wild-type structure. Theprecursor mutant S227A of the GPR116 SEA domainwas only obtained in small quantities and has atendency to form non-native dimers even at lowconcentrations, in contrast to the other GPR116 SEAderivatives studied here, which yield 15–30 mg/L ofculture depending on construct (not shown). It was
therefore not characterized further, and the stabilityof the precursor protein relative to the cleaved wildtype was instead investigated using MUC1 SEA.The N-terminally extended construct of the MUC1SEA domain (the 15.3-kDa construct) is more so-luble than the original shorter construct (the 13.8-kDa construct).8 In the case of the wild-type and 4Gproteins, this extension of the N-terminal hadno discernible effect on solubility or stability (seeTable 1; Supplementary Fig. S4). For the precursormutant S1098A, however, the improved solubility ofthe 15.3-kDa construct proved to be vital since itallowed for stability measurements (Fig. 3). A valueof 8.8 kcal mol−1 was obtained, which, when com-pared with the ΔGD−N of the wild-type protein,suggests that the strained G−1A+1VVV region de-stabilizes the protein by 7.4±1.5 kcal mol−1 (Table1). Hence, a value of ∼7 kcal mol−1 for ΔGstrain hasbeen arrived at for both the MUC1 and GPR116 SEAdomains using two methods. This is not unexpectedwhen considering that autoproteolysis proceedswith approximately the same catalytic efficiency inthe two proteins.
Discussion
Energetics of strain-accelerated autoproteolysisof the SEA domain
We found that the in vitro autoproteolysis of theSEA domains occurs within minutes, as opposedto the spontaneous hydrolysis of peptide bonds,which proceeds with a t½ of ∼2100 years undersimilar conditions.7 A rate acceleration (kcat/knon)of ∼1.8×107 can be calculated for MUC1 SEA

1123Autoproteolysis by Conformational Strain: Energetics
autoproteolysis based on its observed cleavage rateof ∼6.4×10−4 s−1 (Fig. 1; Table 2) and the rate ofspontaneous hydrolysis of the model compoundacetylglycylglycine N-methylamide in H2O at 25 °Cand pH 6.8 (3.6×10−11 s−1).7 This is equivalent to aΔΔG‡ of ∼9.8 kcal mol−1. The rate of cleavage of theGPR116 SEA domain is very similar, amounting to∼9.30×10−4 s−1 (Fig. 1; Table 2), corresponding to arate acceleration of ∼2.6×107 and a ΔΔG‡ of ∼10.0kcal mol−1. But is this rate enhancement realistic forstrained amides? It has been known for some timethat any distortion of the amide interferes with re-sonance stabilization and thus increases the amide'sspontaneous rate of hydrolysis.15 Indeed, the rateof hydrolysis of a strained cyclic amide (benzoqui-nuclidin-2-one) was demonstrated to be 107 timesfaster than the unstrained counterpart (1-phenyl-2-piperidone), corresponding to a ΔΔG‡ of ∼10 kcalmol−1 (Ref. 16), and a study on a set of anilidesdiffering in the extent of amide distortion foundan acceleration in the rate of hydrolysis of 107 foralkaline-catalyzed hydrolysis and that of 1011 foracid-catalyzed hydrolysis.17If the conformational stability of the SEA domains
is to be viewed as one of the catalysts in the auto-proteolytic reaction, these proteins must be suffi-ciently stable to be able to generate the requiredstrain. Chemical denaturation assays using ureagave a ΔGD−N of ∼11 kcal mol−1 for the cleavedGPR116 SEA protein (Fig. 2; Table 1). The 4Gmutantappears to be destabilized by ∼2 kcal mol−1 (Fig. 2;Table 1), but much of this destabilization stems fromthe increased entropy of loop closure. The MUC1SEA domain proved to be even more stable, re-quiring GdmCl to be fully denatured. A ΔGD−N of15.3 kcal mol−1 wasmeasured for the 13.8-kDawild-type protein, and the 4G mutant of the same con-struct exhibited a similar stability: 18.6 kcal mol−1
(Fig. 3; Table 1). The stabilizing free energies of bothSEA domains are therefore sufficiently large toprovide substantial straining energy, ΔGstrain, in theprecursor.Autoproteolysis of the SEA domains studied
here thus proceeds with a t½ of 12–18 min, which,according to TS theory, corresponds to a ΔΔG‡ of∼10 kcal mol−1, as noted above. But how muchof this value stems from a destabilized bond? Diffe-rent experiments on two SEA domains both indi-cate that the scissile peptide bond is destabilized byΔGstrain≈7 kcal mol−1 during catalysis. In oneexperiment, we quantified the strain by manipulat-ing the driving force of the reaction, the ΔGD−N. Tothis end, we introduced destabilizing hydrophobictruncation mutations into GPR116 SEA, distant fromthe cleavage site. Cleavage kinetics was measuredby using the single mutants L201V, L201A, andV234A, which all expressed as IBs in their uncleavedform. The ΔGD−N was instead measured using thesame mutations introduced into the 4G protein (Fig.4; Tables 1 and 2). Furthermore, we also measuredthe cleavage rates in the presence of different con-centrations of urea and compared these with thecalculated ΔGD−N at each concentration (Table 2). A
plot of the cleavage rate ratio against ΔGD−N clearlydemonstrates their mutual dependence (Fig. 5).Treating these data according to a pre-equilibriummodel where the interconversion between differentprecursor states is rapid compared with cleavagemakes it possible to obtain the ΔGstrain from Eq. (4)fitted to the data in Table 2. The ΔGstrain ob-tained from this analysis is on the order of 7.1±0.2 kcal mol−1.In the other experiment, we used the more tra-
ditional approach of constructing G−1A+1 precursormutants lacking the nucleophilic serine. These SEAmutants are unable to undergo cleavage via theN→O acyl shift and therefore only autoproteolyzeas a result of the strained bond.8 However, the rateof spontaneous cleavage is very slow and thereforedoes not affect the chemical denaturation experi-ments. The 15.3-kDa S1098A MUC1 SEA precursormutant turned out to be 7.4±1.5 kcal mol−1 lessstable than wild type, suggesting that the ΔGstrainis of the same order (Fig. 3; Table 1). The S227AGPR116 SEA precursor mutant is too unstable to becharacterized.Considering that the ΔGstrain is ∼7 kcal mol−1 and
that the spontaneous hydrolysis of an unstrainedamide bond requires a ΔG‡ of ∼32 kcal mol−1 (t½ of∼2100 years at 21 °C)7, and furthermore that theSEA domain cleavage rates measured here require aΔG‡ of ∼21 kcal mol−1, this leaves a ΔΔG‡ con-tribution of ∼4 kcal mol−1 from the N→O acyl shiftmechanism per se after correcting for the ΔGstrain(Fig. 6). Despite the fact that the N→O acyl shift isconserved in all autoproteolytic systems,3 it isprobably not in itself sufficient to catalyze peptidebond cleavage since our treatment predicts anunstrained reaction to proceed with a t½ of ∼2.3years. For the SEA domains, which appear to lack anactive deprotonation mechanism and an oxyanionhole,2,8 much of the required energy for catalysiscomes instead from the destabilization of the scissilebond. Autoproteolysis by strain alone is predicted toproceed with a t½ of ∼5 days (Fig. 6), which is inreasonable agreement with what is actually ob-served experimentally for the MUC1 precursormutant S1098A.8
Intramolecular autoproteolysis in other proteins
There are, to our knowledge, six protein familiesand one unique protein—monellin—that containnaturally fragmented polypeptide chains. However,it is only in monellin that the two peptides arebelieved to stem from cleavage by an externalprotease,18 whereas the other proteins obtain theirfragmented structures by an autoproteolytic intra-molecular mechanism. The autoproteolytically frag-mented protein families are (i) the pyruvoyl-dependent enzymes,19 (ii) the hedgehog domain20
(evolutionarily related to the intein domain,21 whichalso has autoproteolytic activity22), (iii) the N-terminal nucleophile (Ntn) hydrolases,23 (iv) theautocleaved G protein-coupled receptor proteolyticsite (GPS) domain,24–26 (v) the nucleoporins,27 and,
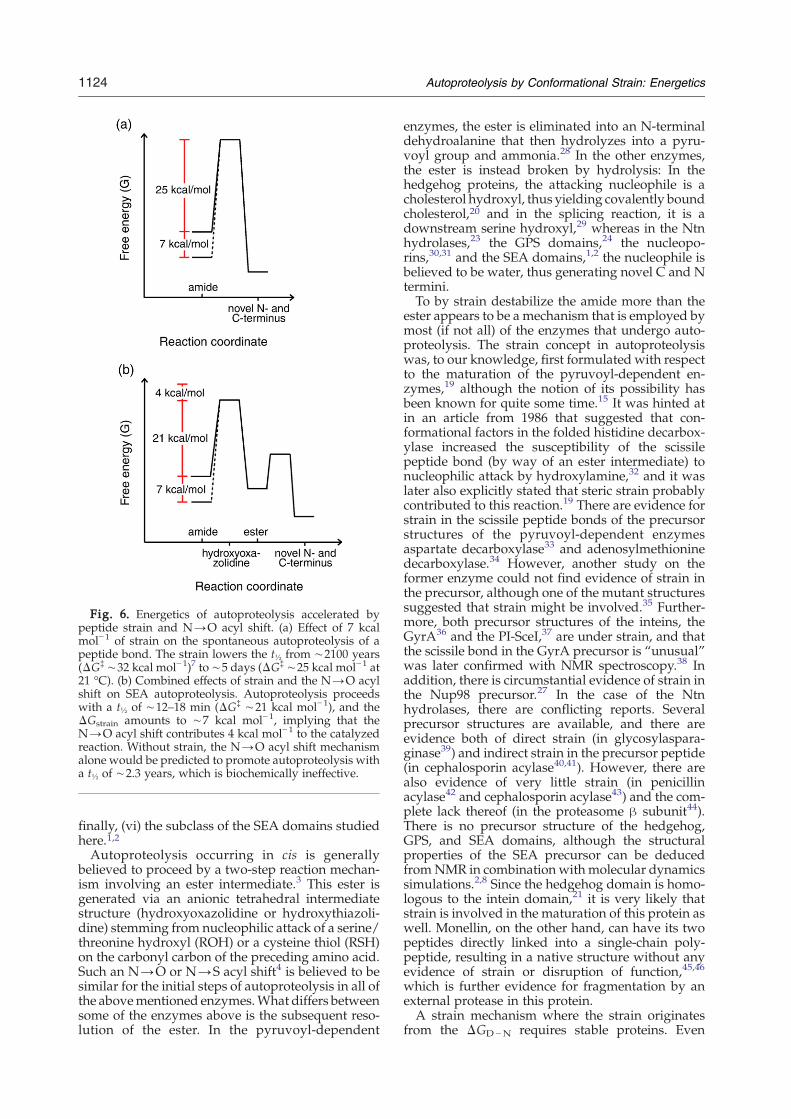
Fig. 6. Energetics of autoproteolysis accelerated bypeptide strain and N→O acyl shift. (a) Effect of 7 kcalmol−1 of strain on the spontaneous autoproteolysis of apeptide bond. The strain lowers the t½ from ∼2100 years(ΔG‡ ∼32 kcal mol−1)7 to ∼5 days (ΔG‡ ∼25 kcal mol−1 at21 °C). (b) Combined effects of strain and the N→O acylshift on SEA autoproteolysis. Autoproteolysis proceedswith a t½ of ∼12–18 min (ΔG‡ ∼21 kcal mol−1), and theΔGstrain amounts to ∼7 kcal mol−1, implying that theN→O acyl shift contributes 4 kcal mol−1 to the catalyzedreaction. Without strain, the N→O acyl shift mechanismalone would be predicted to promote autoproteolysis witha t½ of ∼2.3 years, which is biochemically ineffective.
1124 Autoproteolysis by Conformational Strain: Energetics
finally, (vi) the subclass of the SEA domains studiedhere.1,2Autoproteolysis occurring in cis is generally
believed to proceed by a two-step reaction mechan-ism involving an ester intermediate.3 This ester isgenerated via an anionic tetrahedral intermediatestructure (hydroxyoxazolidine or hydroxythiazoli-dine) stemming from nucleophilic attack of a serine/threonine hydroxyl (ROH) or a cysteine thiol (RSH)on the carbonyl carbon of the preceding amino acid.Such an N→O or N→S acyl shift4 is believed to besimilar for the initial steps of autoproteolysis in all ofthe abovementioned enzymes.What differs betweensome of the enzymes above is the subsequent reso-lution of the ester. In the pyruvoyl-dependent
enzymes, the ester is eliminated into an N-terminaldehydroalanine that then hydrolyzes into a pyru-voyl group and ammonia.28 In the other enzymes,the ester is instead broken by hydrolysis: In thehedgehog proteins, the attacking nucleophile is acholesterol hydroxyl, thus yielding covalently boundcholesterol,20 and in the splicing reaction, it is adownstream serine hydroxyl,29 whereas in the Ntnhydrolases,23 the GPS domains,24 the nucleopo-rins,30,31 and the SEA domains,1,2 the nucleophile isbelieved to be water, thus generating novel C and Ntermini.To by strain destabilize the amide more than the
ester appears to be a mechanism that is employed bymost (if not all) of the enzymes that undergo auto-proteolysis. The strain concept in autoproteolysiswas, to our knowledge, first formulated with respectto the maturation of the pyruvoyl-dependent en-zymes,19 although the notion of its possibility hasbeen known for quite some time.15 It was hinted atin an article from 1986 that suggested that con-formational factors in the folded histidine decarbox-ylase increased the susceptibility of the scissilepeptide bond (by way of an ester intermediate) tonucleophilic attack by hydroxylamine,32 and it waslater also explicitly stated that steric strain probablycontributed to this reaction.19 There are evidence forstrain in the scissile peptide bonds of the precursorstructures of the pyruvoyl-dependent enzymesaspartate decarboxylase33 and adenosylmethioninedecarboxylase.34 However, another study on theformer enzyme could not find evidence of strain inthe precursor, although one of the mutant structuressuggested that strain might be involved.35 Further-more, both precursor structures of the inteins, theGyrA36 and the PI-SceI,37 are under strain, and thatthe scissile bond in the GyrA precursor is “unusual”was later confirmed with NMR spectroscopy.38 Inaddition, there is circumstantial evidence of strain inthe Nup98 precursor.27 In the case of the Ntnhydrolases, there are conflicting reports. Severalprecursor structures are available, and there areevidence both of direct strain (in glycosylaspara-ginase39) and indirect strain in the precursor peptide(in cephalosporin acylase40,41). However, there arealso evidence of very little strain (in penicillinacylase42 and cephalosporin acylase43) and the com-plete lack thereof (in the proteasome β subunit44).There is no precursor structure of the hedgehog,GPS, and SEA domains, although the structuralproperties of the SEA precursor can be deducedfromNMR in combination with molecular dynamicssimulations.2,8 Since the hedgehog domain is homo-logous to the intein domain,21 it is very likely thatstrain is involved in the maturation of this protein aswell. Monellin, on the other hand, can have its twopeptides directly linked into a single-chain poly-peptide, resulting in a native structure without anyevidence of strain or disruption of function,45,46
which is further evidence for fragmentation by anexternal protease in this protein.A strain mechanism where the strain originates
from the ΔGD−N requires stable proteins. Even

1125Autoproteolysis by Conformational Strain: Energetics
though the ΔGD−N appears not to have been de-termined for any of the other enzymes undergoingautoproteolysis, proteins do frequently have stabi-lities on the order of 5–15 kcal mol−1 (Ref. 12).Histidine decarboxylase seems to be a very stableprotein—it is resistant towards denaturation by heat(utilized in a purification step at 70 °C for 2 min)47
and chemicals (5 M GdmCl is required to separatethe two peptides).48 There are also indications in theliterature that the autoproteolytic protein penicillinacylase49 (an Ntn hydrolase) and the inteins50 arevery stable. A high stability in these fragmentedenzymes might therefore be a requirement for main-taining the native state after cleavage, becausefragmentation is inherently destabilizing due to itseffect on the translational and rotational entropy.That the ability of fragmented proteins to recon-stitute into native structure might partly depend onstability has been noted before,51 where fragmenteddihydrofolate reductase (with a ΔGD−N of ∼6 kcalmol−1) has a very unstable fragment-complementedstructure, whereas proteins with higher stabilitieshave native-like complemented structures. Support-ing this is the ΔGD−N of the fragmented proteinmonellin, which amounts to ∼10 kcal mol−1 (Ref.52), a value very similar to the ∼11 kcal mol−1
determined here for the GPR116 SEA domain.Perhaps the most convincing evidence that strain
plays an active part in the autoproteolytic mechan-ism of SEA fragmentation comes from the MUC1SEA precursor mutant S1098A. Although this mu-tant has no nucleophilic residue immediately down-stream of the G1097 in the G−1S+1VVV motif, it stillundergoes autocatalysis at a slow rate.8 This hasalso been observed for a histidine decarboxylaseprecursor mutant, albeit at the bond preceding thescissile peptide bond.19 Since this mechanismprobably involves uncatalyzed peptide bond hydro-lysis by bulk solvent, an anionic TS state53 thatprobably differs from the TS generated via theintramolecular N→O acyl shift is involved. Thisargues against the absolute requirement for anoxyanion hole during peptide bond cleavage inthe SEA domain. Such an oxyanion hole is widelyspeculated to exist in all of the other enzymesundergoing autoproteolysis. However, it is not asclearly defined as it is in the serine proteases, whereit accounts for an acceleration of ∼103 (Ref. 54). Ithas in the case of the pyruvoyl-dependent enzymestherefore been the origin of debate—the initial re-ports found no evidence of an oxyanion hole,33,34,55
but later research argued for the existence of aconserved oxyanion hole in the pyruvate-dependentenzymes as well as in the inteins, hedgehogproteins, and Ntn glycosylasparaginases.35 Workon the precursor structures of the inteins36,37 andNtn hydrolases39–44 do indeed also argue for thepresence of an oxyanion hole, as does the workdone on the Nup98 precursor.27 However, asmentioned above, in the fully cleaved MUC1 SEAstructure, no such stabilization of the tetrahedralintermediate is evident. In fact, the only conservedpolar residue in the vicinity of the cleavage site in all
SEA domains undergoing cleavage is the catalyti-cally active serine.2
Furthermore, an oxyanion hole will only speedthe establishment of the N→O (or N→S) acyl shiftequilibrium, but in this mechanism, the freeprimary amino group (the leaving group) generatedin the ester intermediate may readily attack theester carbonyl, thus reversing the shift. Unless theleaving group is protonated, the equilibrium islikely to be on the amide side. In neutral to alkalinesolvent, the O-acyl derivatives (the esters) thereforereadily convert back to the N-acyl compounds (theamides).4 In non-exposed autoproteolytic sites, anactive protonation mechanism therefore appearsnecessary. A survey of the structures available indi-cates that such a protonation mechanism has beenproposed for all enzymes (i.e., for the pyruvoyl-dependent enzymes,33–35 the inteins,36,37 the hedge-hog domain,21 and the Ntn hydrolases39–44) exceptfor the SEA domain2,8 and the Nup9827 structures,which both have autoproteolytic sites quite exposedto solvent. Yet, autoproteolysis in the wild-typeMUC1 SEA domain still occurs readily at pH 10(data not shown), thus providing further supportfor a shifting of the N→O acyl equilibrium towardsthe ester by conformational destabilization of theamide.In addition to the oxyanion holes, an active de-
protonation mechanism of the nucleophile has beenimplicated from the structural biology of self-cleaving proteins. Judging from their structures, itappears that the nucleophile is ideally poised forattack at the carbonyl. However, as in the case ofMUC1 SEA autoproteolysis, active deprotonation ofthis nucleophile analogous to the catalytic triad inthe serine proteases appears not to be necessary forcleavage to proceed. In the precursor structures forpyruvoyl-dependent enzymes, base-assisted depro-tonation of the catalytic serine was first suggested toexist in aspartate decarboxylase,33 but a laterstructural study could not find evidence for such amechanism in the same protein,35 and the precursorstructures of adenosylmethionine decarboxylasecould not find such a base either.34,55 Neither wasthere any clear evidence of deprotonation of thecysteine nucleophile in the intein precursors,36,37 inanalogy to the fully processed homologous hedge-hog structure.21 For the Ntn hydrolases, there areconflicting reports where deprotonation of thethreonine is suggested to occur in the glycosylaspar-aginase precursor,39 but not for threonine in theproteasome β subunit,44 although the serines inpenicillin acylase42 and cephalosporin40,41,43
appeared to be actively deprotonated, as was theserine in Nup98.27 Consequently, although depro-tonation of the nucleophile might be a contributor insome autoproteolytic proteins, it appears to be ofminor importance in other such proteins.
Conclusions
In an accompanying article, we provide mechan-istic evidence that SEA domain autoproteolysis is

1126 Autoproteolysis by Conformational Strain: Energetics
catalyzed not only by the N→O acyl shift but alsoby the destabilizing strain that is generated over thescissile peptide bond in the uncleaved precursorprotein.8 The native state appears to have difficultiesin accommodating an uncleaved G−1S+1 peptidebond, and autoproteolysis therefore occurs conco-mitantly with folding. Here we demonstrate that theenergy required to strain the peptide bond can befully accounted for by the high conformationalstabilities of the SEA domains—ΔGD−N ∼11 kcalmol−1 for GPR116 SEA and ∼16 kcal mol−1 for theMUC1 SEA domain—which are more than sufficientto give rate enhancements on the order of what hasbeen observed for twisted amides (∼107, corre-sponding to a ΔΔG‡ of ∼10 kcal mol−1).16 Further-more, the observed rates of proteolysis in these twodomains (t½ of ∼12–18 min) are in agreement with aΔΔG‡ of ∼10 kcal mol− 1 compared with thehydrolysis of unstrained amides. A quantitativeanalysis of GPR116 SEA domain cleavage kineticsand stability data obtained using core truncationmutants or destabilizing conditions yield a value forthe strain energy of ΔGstrain=7.1±0.2 kcal mol−1,and a very similar value (7.4±1.5 kcal mol−1) isobtained as the difference in stability of MUC1 SEAwild type and the strained precursor mutantS1098A. This amount of strain and the measuredreaction rates suggest that the N→O acyl shift aloneis not effective in the absence of strain, as it would betoo slow to be compatible with any in vivo bio-chemical requirements (t½ of N2 years for theautoproteolysis of an unstrained G−1S+1VVV motifin the SEA domains; see Fig. 6). Furthermore, wehave reviewed the structural data available forseveral of the autoproteolytic proteins and concludethat the only common denominator in these systemsappears to be the possibility that strain plays anactive part in the cleavage reaction. A mechanisminvolving strain was in fact suggested to play a rolealso in enzymatic hydrolysis of amides,15 before itwas realized that it was the catalytic triad andenzyme–TS complementarity working in concertthat generated the catalytic efficiency of the serineproteases (with a rate acceleration on the orders of∼106 and ∼103, respectively).54 Now it appears as ifpeptide strain indeed might be a general feature ofautoproteolysis, where it works in concert with theN→O acyl shift.
Materials and Methods
Protein production and purification
Expression and purification of the 13.8- and 15.3-kDaconstructs of the MUC1 SEA domain and their precursor(S1098A) and 4G loop-insertion mutants are described inthe accompanying article.8 The longer N-terminallyextended construct of 15.3 kDa was made primarily forthe analysis of the G−1A+1 precursor mutant (S1098A)because of its higher solubility compared with the shorterconstruct. All experiments on MUC1 SEA in this articlewere carried out on the 13.8-kDa protein unless specifi-cally stated otherwise.
The SEA domain corresponding to amino acids 165–282of the human GPR116 protein (Swiss-Prot accession no.Q8IZF2; Supplementary Fig. S3) was cloned with an addi-tional N-terminal GSS-His6 sequence. This protein has amolecular mass of 14,364 Da and a theoretical extinctioncoefficient of 8400 M−1 cm−1 at 278 nm under denaturingconditions (calculated by the method of Gill and vonHippel56). Under native conditions, the extinction coeffi-cient at the same wavelength was determined to 9200 M−1
cm−1, which is the value used for the concentrationdetermination of all GPR116 SEA species in this study.The precursor mutant S227A (with a G−1A+1VVV
cleavage site sequence) and the 4G loop-insertion mutant(GGGGG−1S+1VVV), as well as all hydrophobic trunca-tion mutants (the L201A, L201V, V234A, 4G L201A, 4GL201V, and 4G V234A mutants) were constructed using aQuikChange™ mutagenesis kit (Stratagene). Constructswere confirmed by sequencing, and the identity of theexpressed wild-type protein was also confirmed by massspectrometry (not shown).GPR116 SEA was expressed in Escherichia coli Rosetta
pLysS as soluble protein or IB (see below) at 37 °C underthe control of a T7 promoter. The T7 promoter wasinduced with 0.5 mM IPTG. Expression was continued for4 h, and the cells were washed once with 20 mM Tris–HCland 50 mMNaCl, pH 8.5. After repeated cycles of freezingand thawing (typically three), the cells were resuspendedin the same buffer supplemented with 5% glycerol, 0.5%Triton X-100, 1 mM PMSF, and approximately 5 U ofDNase. Centrifugation at 11,000g for 20 min at 4 °Cseparated the soluble fraction from the IBs.A two-step purification procedure of soluble GPR116
SEA protein (the 4G, 4G L201A, 4G L201V, and 4G V234Amutants were all obtained in the soluble fraction) wascarried out using IMAC [Ni2+–nitrilotriacetic acid (NTA)resin from Sigma] followed by size-exclusion chromato-graphy. The cell lysate was bound to equilibrated Ni2+–NTA resin and washed with 20 mM Tris–HCl, 50 mMNaCl, 5% glycerol, and 10 mM imidazole, pH 8.5. Thecolumn was eluted with the same buffer with 150 mMimidazole and gel filtrated. All size-exclusion chromato-graphy runs were carried out using either a Superdex 75preparative (16/60) or analytical (10/300) column (GEHealthcare) pre-equilibrated with 50 mM K+ phosphateand 50 mM NaCl, pH 7.0. Purified protein was analyzedwith SDS-PAGE with Coomassie staining of the gels.The insoluble fraction containing the GPR116 SEA IBs
(the wild-type protein and the S227A, L201A, L201V, andV234A mutants all expressed as IBs) was refolded andpurified by a two-step procedure similar to the solublefraction. Briefly, the IB deposits were first washed in20 mM Tris–HCl, 50 mM NaCl, 30% glycerol, 0.5% TritonX-100, and 1 mM PMSF, pH 8.5. After centrifugation asabove, they were solubilized in 20 mM Tris–HCl and50 mM NaCl, pH 8.5, supplemented with 4–6 M GdmCl.Refolding was carried out by dilution into a large volume(typically more than a 1:60 dilution) of 20 mM Tris–HCl,50 mM NaCl, and 30% glycerol, pH 8.5, followed byincubation at room temperature for 1–3 h. After centrifu-gation to remove precipitated protein, the soluble refoldedprotein was applied to a pre-equilibrated Ni2+–NTAcolumn and purified as above.
Kinetics of autoproteolysis
The IBs of uncleaved protein were dissolved in 50 mMK+ phosphate, 50 mM NaCl, and 6 M GdmCl, pH 7.5,and purified by IMAC (see above) under denaturing

1127Autoproteolysis by Conformational Strain: Energetics
conditions. Eluted protein was then reapplied to Ni2+
resin equilibrated with 50 mM K+ phosphate, 50 mMNaCl, and 6 M GdmCl, pH 7.5, and washed with threetimes the column volume of the same buffer. Refoldingwas initiated by extensive washing with 50 mM K+
phosphate and 50 mM NaCl, pH 7.5, and the proteineluted with the same buffer made 150 mM imidazole.Eluted protein was then rapidly quenched with standardSDS-PAGE loading buffer, which denatures the proteinand thus marks the end point of the cleavage reactions,and heated at 95 °C for 5 min. For the cleavage kineticsdata obtained at different urea concentrations, only onesample was prepared for each urea concentration (0.00,0.57, 1.14, 1.33, 1.52, 1.71, and 1.89 M). After elution fromthe IMAC column, cleavage thus proceeded in solution.Aliquots from the solutions containing urea were mixedwith SDS-PAGE loading buffer and boiled as above.Cleavage was analyzed with SDS-PAGE, and the gelswere stained with Coomassie and analyzed with theQuantity One software (BioRad) to obtain the pixeldensities of each lane on the SDS-PAGE gel. Rateconstants were obtained by fitting the relative intensitiesof cleaved fragments and uncleaved full-length protein toa monoexponential function using the program IGOR(Wavemetrics). These experiments were all carried out atroom temperature (21±0.5 °C).
Thermal denaturation
CD at 220 nm was monitored as a function oftemperature using a Jasco J-810 spectropolarimeter (JascoCorporation). A scan rate of 1 °C min−1 between 4 and95 °C was used, and the solutions were also subjected to areverse scan after a 1-min incubation time at the highertemperature. Protein concentration was 14 μM in 25 mMK+ phosphate and 25 mM NaCl, pH 7.0. A two-statetransition model was fitted to the experimental data by aleast-squares fitting procedure using the program IGOR inorder to obtain the melting temperature (Tm) as describedpreviously.57,58 The thermal data are provided as supple-mentary material (Supplementary Fig. S2).
Chemical denaturation
Isothermal denaturation curves using urea or GdmCl asdenaturant were obtained at 25 °C (data in Fig. 2) or 21 °C(data in Figs. 3 and 4). Urea concentration was determinedvolumetrically, and GdmCl concentration was determinedby refractive index.59 CD was measured at 220 nm.Separate solutions were prepared for each point andincubated at room temperature for more than 2 h prior tomeasurement. Control experiments indicated that equili-brium was fully established within this period. Proteinconcentrations were 8–28 μM in 25 mM K+ phosphate and25 mM NaCl, pH 7.0 (data in Fig. 2), or 50 mM K+
phosphate and 50 mMNaCl, pH 7.5 (data in Figs. 3 and 4).Fluorescence was measured on 14 μM protein samples
in 25 mMK+ phosphate and 25 mMNaCl, pH 7.0, at 21 °Cusing a Cary Eclipse fluorescence spectrophotometer(Varian) with excitation set at 277 nm and emission at303 nm. In this set of experiments, a solution of nativeprotein was titrated with a solution of denatured protein(in the same buffer with 6 M GdmCl).A two-state model that assumes a linear dependence of
ΔGD −N on denaturant concentration ([D]) over thetransition region was used for quantitative analysis.58,60
The concentration dependence in values of ΔGD−N mea-
sured for the wild-type SEA heterodimer was corrected forby the following equation:
DGD�N ¼ mD�N � ½D�50% � RTlnðC=2Þ ð5Þwhere mD−N is the slope of the transition (−δ(ΔGD−N)/δ[D]), [D]50% is the concentration at the midpoint of de-naturation, R is the gas constant, T is the temperature(in Kelvin), and C is the molar concentration of theheterodimer.61
The effective concentration (Ceff) of linking the twopeptides in the wild-type GPR116 SEA heterodimer toge-ther was taken as:
Ceff ZKS=KD ð6Þwhere KS and KD are the folding equilibrium constants ofthe single chain (4G mutant) and heterodimeric complex(wild-type protein), respectively.13
Acknowledgements
This work was supported by the Swedish Re-search Council (VR), the Hasselblad Foundation, theSwedish Foundation for Strategic Research - TheMucosal Immunobiology and Vaccine Center(MIVAC), and the Knut and Alice WallenbergFoundation. We thank Prof. Mikael Akke and Dr.Wolfgang Hoyer for discussions and valuablesuggestions concerning the pre-equilibrium modelfor autoproteolysis.
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2008.01.051
References
1. Levitin, F., Stern, O., Weiss, M., Gil-Henn, C., Ziv, R.,Prokocimer, Z. et al. (2005). The MUC1 SEA module isa self-cleaving domain. J. Biol. Chem. 280, 33374–33386.
2. Macao, B., Johansson, D. G., Hansson, G. C. & Härd, T.(2006). Autoproteolysis coupled to protein folding inthe SEA domain of the membrane-bound MUC1mucin. Nat. Struct. Mol. Biol. 13, 71–76.
3. Perler, F. B. (1998). Breaking up is easy with esters.Nat. Struct. Biol. 5, 249–252.
4. Iwai, K. & Ando, T. (1967). N→O acyl rearrangement.Methods Enzymol. 11, 263–282.
5. Wreschner, D. H., McGuckin, M. A., Williams, S. J.,Baruch, A., Yoeli, M., Ziv, R. et al. (2002). Generation ofligand–receptor alliances by “SEA” module-mediatedcleavage of membrane-associated mucin proteins.Protein Sci. 11, 698–706.
6. Fredriksson, R., Lagerström, M. C., Höglund, P. J. &Schiöth, H. B. (2002). Novel human G protein-coupledreceptors with long N-terminals containing GPSdomains and Ser/Thr-rich regions. FEBS Lett. 531,407–414.
7. Radzicka, A. & Wolfenden, R. (1996). Rates of un-catalyzed peptide bond hydrolysis in neutral solution

1128 Autoproteolysis by Conformational Strain: Energetics
and the transition state affinities of proteases. J. Am.Chem. Soc. 118, 6105–6109.
8. Johansson, D. G. A., Macao, B., Sandberg, A. & Härd,T. (2008). SEA domain autoproteolysis accelerated byconformational strain: mechanistic aspects. J. Biol.Chem. 377, 1130–1143.
9. Hilkens, J. & Buijs, F. (1988). Biosynthesis of MAM-6,an epithelial sialomucin. Evidence for involvement ofa rare proteolytic cleavage step in the endoplasmicreticulum. J. Biol. Chem. 263, 4215–4222.
10. Linsley, P. S., Kallestad, J. C. & Horn, D. (1988).Biosynthesis of high molecular weight breast carci-noma associated mucin glycoproteins. J. Biol. Chem.263, 8390–8397.
11. Ligtenberg, M. J., Kruijshaar, L., Buijs, F., van Meijer,M., Litvinov, S. V. & Hilkens, J. (1992). Cell-associatedepisialin is a complex containing two proteinsderived from a common precursor. J. Biol. Chem. 267,6171–6177.
12. Fersht, A. (1999). Structure and Mechanism in ProteinScience: A Guide to Enzyme Catalysis and Protein Folding,W.H. Freeman, New York, NY.
13. Zhou, H. X. (2004). Loops, linkages, rings, catenanes,cages, and crowders: entropy-based strategies for sta-bilizing proteins. Acc. Chem. Res. 37, 123–130.
14. Nagi, A. D. & Regan, L. (1997). An inverse correlationbetween loop length and stability in a four-helix-bundle protein. Folding Des. 2, 67–75.
15. Holley, R. W. (1953). Steric inhibition of amide reso-nance and its possible significance in enzyme action.Science, 117, 23–25.
16. Blackburn, G. M., Skaife, C. J. & Kay, I. T. (1980). Straineffects in acyl transfer reactions: Part 5. The kinetics ofhydrolysis of benzoquinuclidin-2-one: a torsionallydistorted amide. J. Chem. Res., Synop., 294–295.
17. Wang, Q. P., Bennet, A. J., Brown, R. S. & Santarsiero,B. D. (1991). Distorted amides as models for activatedpeptide N–C(O) units: 3. Synthesis, hydrolytic profile,and molecular-structure of 2,3,4,5-tetrahydro-2-oxo-1,5-propanobenzazepine. J. Am. Chem. Soc. 113,5757–5765.
18. Murzin, A. G. (1993). Sweet-tasting protein monellinis related to the cystatin family of thiol proteinaseinhibitors. J. Mol. Biol. 230, 689–694.
19. van Poelje, P. D. & Snell, E. E. (1990). Pyruvoyl-dependent enzymes. Annu. Rev. Biochem. 59, 29–59.
20. Porter, J. A., Young, K. E. & Beachy, P. A. (1996).Cholesterol modification of hedgehog signaling pro-teins in animal development. Science, 274, 255–259.
21. Hall, T. M., Porter, J. A., Young, K. E., Koonin, E. V.,Beachy, P. A. & Leahy, D. J. (1997). Crystal structure ofa Hedgehog autoprocessing domain: homologybetween Hedgehog and self-splicing proteins. Cell,91, 85–97.
22. Noren, C. J., Wang, J. & Perler, F. B. (2000). Dissectingthe chemistry of protein splicing and its applications.Angew. Chem., Int. Ed. Engl. 39, 450–466.
23. Brannigan, J. A., Dodson, G., Duggleby, H. J., Moody,P. C., Smith, J. L., Tomchick, D. R. & Murzin, A. G.(1995). A protein catalytic framework with an N-terminal nucleophile is capable of self-activation.Nature,378, 416–419.
24. Lin, H. H., Chang, G. W., Davies, J. Q., Stacey, M.,Harris, J. & Gordon, S. (2004). Autocatalytic cleavageof the EMR2 receptor occurs at a conserved G protein-coupled receptor proteolytic site motif. J. Biol. Chem.279, 31823–31832.
25. Stacey, M., Lin, H. H., Gordon, S. & McKnight, A. J.(2000). LNB-TM7, a group of seven-transmembrane
proteins related to family-B G-protein-coupled recep-tors. Trends Biochem. Sci. 25, 284–289.
26. Harmar, A. J. (2001). Family-B G-protein-coupled re-ceptors.Genome Biol. 2, reviews 3013.1–reviews 3013.10.
27. Hodel, A. E., Hodel, M. R., Griffis, E. R., Hennig, K. A.,Ratner, G. A., Xu, S. & Powers, M. A. (2002). The three-dimensional structure of the autoproteolytic, nuclearpore-targeting domain of the human nucleoporinNup98. Mol. Cell, 10, 347–358.
28. Recsei, P. A., Huynh, Q. K. & Snell, E. E. (1983).Conversion of prohistidine decarboxylase to histidinedecarboxylase: peptide chain cleavage by nonhydro-lytic serinolysis. Proc. Natl Acad. Sci. USA, 80, 973–977.
29. Xu, M. Q. & Perler, F. B. (1996). The mechanism ofprotein splicing and its modulation by mutation.EMBO J. 15, 5146–5153.
30. Rosenblum, J. S. & Blobel, G. (1999). Autoproteolysisin nucleoporin biogenesis. Proc. Natl Acad. Sci. USA,96, 11370–11375.
31. Teixeira, M. T., Fabre, E. & Dujon, B. (1999). Self-catalyzed cleavage of the yeast nucleoporin Nup145pprecursor. J. Biol. Chem. 274, 32439–32444.
32. Huynh, Q. K. & Snell, E. E. (1986). Histidine decar-boxylase of Lactobacillus 30a. Hydroxylamine cleavageof the –seryl–seryl– bond at the activation site of pro-histidine decarboxylase. J. Biol. Chem. 261, 1521–1524.
33. Albert, A., Dhanaraj, V., Genschel, U., Khan, G.,Ramjee, M. K., Pulido, R. et al. (1998). Crystal structureof aspartate decarboxylase at 2.2 Å resolution pro-vides evidence for an ester in protein self-processing.Nat. Struct. Biol. 5, 289–293.
34. Tolbert, W. D., Zhang, Y., Cottet, S. E., Bennett, E. M.,Ekstrom, J. L., Pegg, A. E. & Ealick, S. E. (2003).Mechanism of human S-adenosylmethionine decarbo-xylase proenzyme processing as revealed by the struc-ture of the S68A mutant. Biochemistry, 42, 2386–2395.
35. Schmitzberger, F., Kilkenny, M. L., Lobley, C. M.,Webb, M. E., Vinkovic, M., Matak-Vinkovic, D. et al.(2003). Structural constraints on protein self-proces-sing in L-aspartate-alpha-decarboxylase. EMBO J. 22,6193–6204.
36. Klabunde, T., Sharma, S., Telenti, A., Jacobs, W. R., Jr.& Sacchettini, J. C. (1998). Crystal structure of GyrAintein from Mycobacterium xenopi reveals structuralbasis of protein splicing. Nat. Struct. Biol. 5, 31–36.
37. Poland, B. W., Xu, M. Q. & Quiocho, F. A. (2000).Structural insights into the protein splicing mechan-ism of PI-SceI. J. Biol. Chem. 275, 16408–16413.
38. Romanelli, A., Shekhtman, A., Cowburn, D. & Muir,T. W. (2004). Semisynthesis of a segmental isotopicallylabeled protein splicing precursor: NMR evidence foran unusual peptide bond at the N-extein–inteinjunction. Proc. Natl Acad. Sci. USA, 101, 6397–6402.
39. Xu, Q., Buckley, D., Guan, C. & Guo, H. C. (1999).Structural insights into the mechanism of intramole-cular proteolysis. Cell, 98, 651–661.
40. Kim, J. K., Yang, I. S., Rhee, S., Dauter, Z., Lee, Y. S.,Park, S. S. & Kim, K. H. (2003). Crystal structuresof glutaryl 7-aminocephalosporanic acid acylase: in-sight into autoproteolytic activation. Biochemistry, 42,4084–4093.
41. Kim, J. K., Yang, I. S., Shin, H. J., Cho, K. J., Ryu, E. K.,Kim, S. H. et al. (2006). Insight into autoproteolyticactivation from the structure of cephalosporin acylase:a protein with two proteolytic chemistries. Proc. NatlAcad. Sci. USA, 103, 1732–1737.
42. Hewitt, L., Kasche, V., Lummer, K., Lewis, R. J.,Murshudov, G. N., Verma, C. S. et al. (2000). Structureof a slow processing precursor penicillin acylase from

1129Autoproteolysis by Conformational Strain: Energetics
Escherichia coli reveals the linker peptide blocking theactive-site cleft. J. Mol. Biol. 302, 887–898.
43. Kim, Y., Kim, S., Earnest, T. N. & Hol, W. G. (2002).Precursor structure of cephalosporin acylase.Insights into autoproteolytic activation in a newN-terminal hydrolase family. J. Biol. Chem. 277,2823–2829.
44. Ditzel, L., Huber, R., Mann, K., Heinemeyer, W., Wolf,D. H. & Groll, M. (1998). Conformational constraintsfor protein self-cleavage in the proteasome. J. Mol.Biol. 279, 1187–1191.
45. Kim, S. H., Kang, C. H., Kim, R., Cho, J. M., Lee, Y. B. &Lee, T. K. (1989). Redesigning a sweet protein: inc-reased stability and renaturability. Protein Eng. 2,571–575.
46. Somoza, J. R., Jiang, F., Tong, L., Kang, C. H., Cho,J. M. & Kim, S. H. (1993). Two crystal structures of apotently sweet protein. Natural monellin at 2.75 Åresolution and single-chain monellin at 1.7 Å resolu-tion. J. Mol. Biol. 234, 390–404.
47. Rosenthaler, J., Guirard, B. M., Chang, G. W. & Snell,E. E. (1965). Purification and properties of histidinedecarboxylase from Lactobacillus 30a. Proc. Natl Acad.Sci. USA, 54, 152–158.
48. Yamagata, S. & Snell, E. E. (1979). Histidine decarbox-ylase from Lactobacillus 30a: reconstitution fromseparated subunits. Biochemistry, 18, 2964–2967.
49. Lindsay, C. D. & Pain, R. H. (1990). The folding andsolution conformation of penicillin G acylase. Eur. J.Biochem. 192, 133–141.
50. Hiraga, K., Derbyshire, V., Dansereau, J. T., Van Roey,P. & Belfort, M. (2005). Minimization and stabilizationof the Mycobacterium tuberculosis recA intein. J. Mol.Biol. 354, 916–926.
51. Smith, V. F. & Matthews, C. R. (2001). Testing the roleof chain connectivity on the stability and structure ofdihydrofolate reductase from E. coli: fragmentcomplementation and circular permutation reveal
stable, alternatively folded forms. Protein Sci. 10,116–128.
52. Xue, W. F., Carey, J. & Linse, S. (2004). Multi-methodglobal analysis of thermodynamics and kinetics inreconstitution of monellin. Proteins, 57, 586–595.
53. Brown, R. S., Bennet, A. J. & Slebockatilk, H. (1992).Recent perspectives concerning the mechanism ofH3O
+-promoted and OH−-promoted amide hydro-lysis. Acc. Chem. Res. 25, 481–488.
54. Carter, P. & Wells, J. A. (1988). Dissecting the catalytictriad of a serine protease. Nature, 332, 564–568.
55. Ekstrom, J. L., Tolbert, W. D., Xiong, H., Pegg, A. E. &Ealick, S. E. (2001). Structure of a human S-adeno-sylmethionine decarboxylase self-processing esterintermediate and mechanism of putrescine stimula-tion of processing as revealed by the H243A mutant.Biochemistry, 40, 9495–9504.
56. Gill, S. C. & von Hippel, P. H. (1989). Calculation ofprotein extinction coefficients from amino acid se-quence data. Anal. Biochem. 182, 319–326.
57. Pace, C. N. & Scholtz, J. M. (1996). Measuring theconformational stability of a protein. In ProteinStructure: A Practical Approach (Creighton, T. E., ed),pp. 299–321, IRL Press, Oxford, UK.
58. Santoro, M. M. & Bolen, D. W. (1988). Unfolding freeenergy changes determined by the linear extrapolationmethod: 1. Unfolding of phenylmethanesulfonylalpha-chymotrypsin using different denaturants. Bio-chemistry, 27, 8063–8068.
59. Nozaki, Y. (1972). The preparation of guanidinehydrochloride. Methods Enzymol. 26, 43–50.
60. Clarke, J. & Fersht, A. R. (1993). Engineered disulfidebonds as probes of the folding pathway of barnase:increasing the stability of proteins against the rate ofdenaturation. Biochemistry, 32, 4322–4329.
61. Neira, J. L., Vazquez, E. & Fersht, A. R. (2000). Stabilityand folding of the protein complexes of barnase. Eur. J.Biochem. 267, 2859–2870.