Embed Size (px)
Citation preview
Schrodinger Bootcamp ~ Fall 2016

BioLuminate ~ Structure-Based Biologics
• Antibody Design • Model prediction from sequence • Humanization
• Protein Design • Residue Scanning (stability/affinity) • Cysteine scanning • Protein crosslinking • Affinity maturation • Protein FEP
• Liability Identification • Aggregation propensity • Reactive residue (glycosylation, proteolysis, oxidation,
deamidation) • Peptide QSAR,Titration curve/Isoelectric point

• Protein Modeling • Homology model building • Protein-Protein docking • Prime de novo loop modeling • Protein interaction analysis • Peptide docking • Peptide QSAR • Multiple Sequence Viewer • Protein reliability report • Molecular dynamics, energy minimization, etc.
BioLuminate ~ Structure-Based Biologics

• Antibody prediction
• Easily go from sequence to predicted Fv
• Aggregation propensity
• Protein Surface Analysis reveals aggregation hotspots
Topics For Today’s Bootcamp
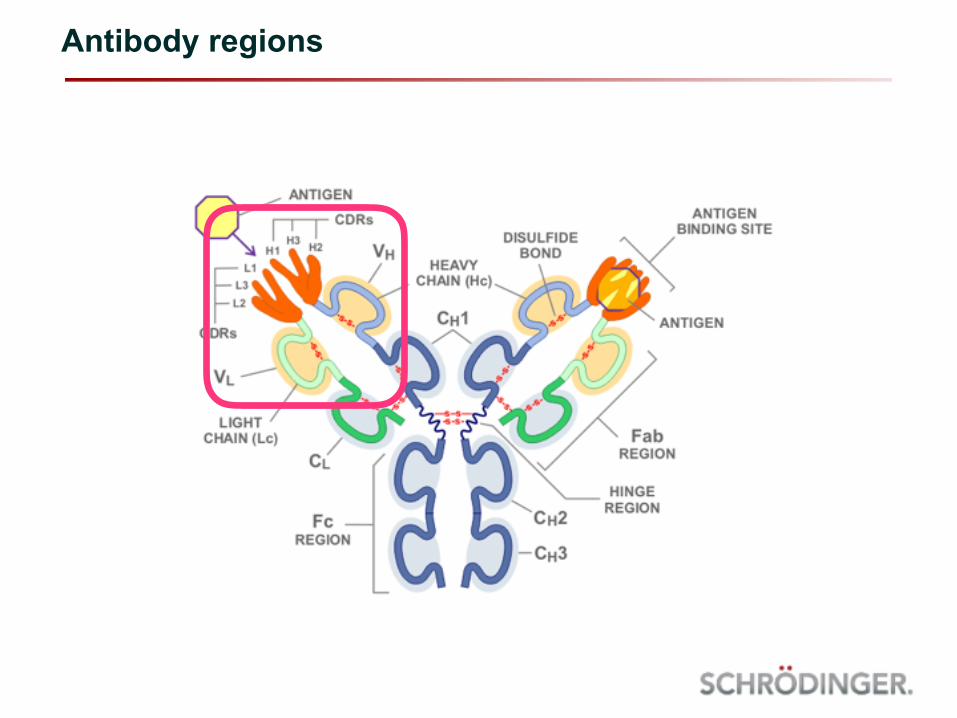
Antibody regions

Antibody Loop Prediction
For antibodies, L1-L3, H1, H2 are typically “pretty good” in homology models
H3 is a problem:
From blind prediction on 9 structures; four different approaches, as described in Almagro et al (2011) Proteins 79 3050-3066
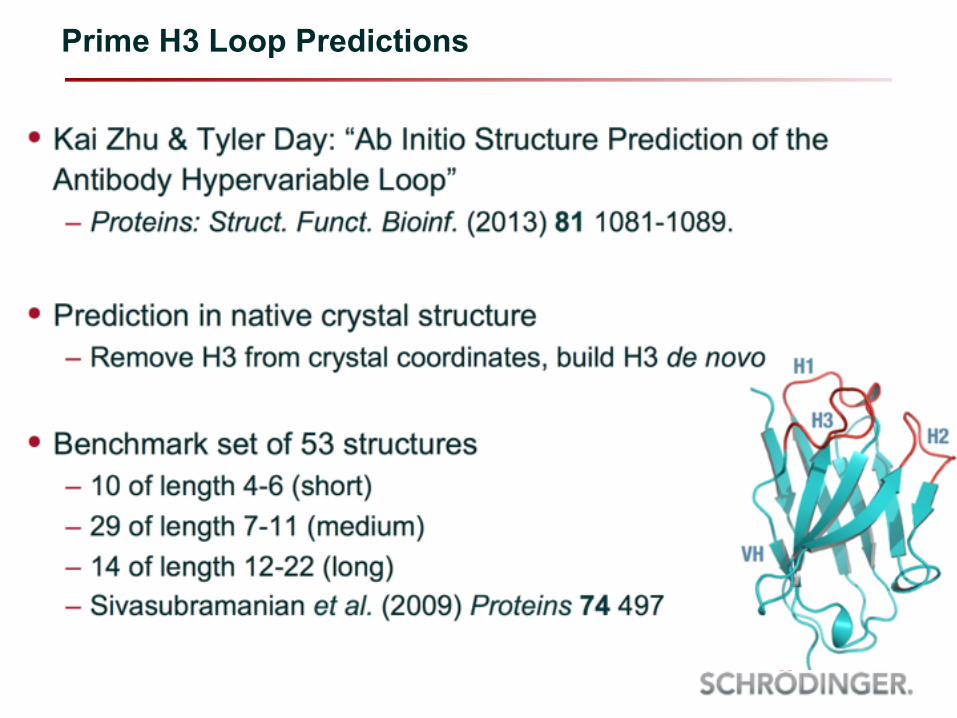
Prime H3 Loop Predictions

Prime H3 Loop Predictions
Average RMS deviation from x-ray for H3 loop (A)
• Possible reasons for poorer results for long loops • Sampling • Methodology • Environment

Prime H3 Loop Predictions with Symmetry
Average RMS deviation from x-ray for H3 loop (A)

• Fully automated (options for advanced users) • Best performance in 2nd blinded antibody modeling assessment (AMA-II)
Antibody prediction in BioLuminate
Volume 82, Issue 8 August 2014
• 7 Participants: – Schödinger; CCG; Accelrys; Rosetta (Jeff Grey @John Hopkins)),
Macromoltek; Astellas Pharma + Osaka U; PIGS server
• Predict 10 unpublished structures (4 human Ab, 6 mouse Ab) • Two stages:
– Stage 1: Predict full Fv from sequence – Stage 2: Predict H3 given xray coordinates of remainder of structure

Method Fv RMSD Framework RMSD
All loops RMSD –H3
H3 RMSD
Schrödinger 1.1 ± 0.2Å 0.8 ± 0.2Å 1.1 ± 0.4Å 2.7 ± 0.8ÅAccelrys 1.1 ± 0.3Å 0.9 ± 0.3Å 1.1 ± 0.5Å 3.0 ± 1.1ÅCCG 1.1 ± 0.2Å 0.9 ± 0.3Å 1.0 ± 0.3Å 3.3 ± 0.9ÅRosetta (Jeff Grey) 1.1 ± 0.2Å 0.8 ± 0.2Å 1.1 ± 0.4Å 2.6 ± 0.9Å
Macromoltek 1.4 ± 0.2Å 1.2 ± 0.2Å 1.2 ± 0.3Å 3.0 ± 1.0ÅAstellas + Osaka U 1.1 ± 0.2Å 0.8 ± 0.2Å 1.0 ± 0.2Å 2.3 ± 0.6ÅPIGS server 1.2 ± 0.1Å 0.9 ± 0.2Å 0.9 ± 0.4Å 3.1 ± 1.1Å
Average 1.1 ± 0.2Å 0.9 ± 0.2A 1.1 + 0.4Å 2.8 ± 0.9Å
AMA-II : Overall results for Round 1: Full Fv from sequence
• All methods are generally producing decent models • H3 is the recurrent problem
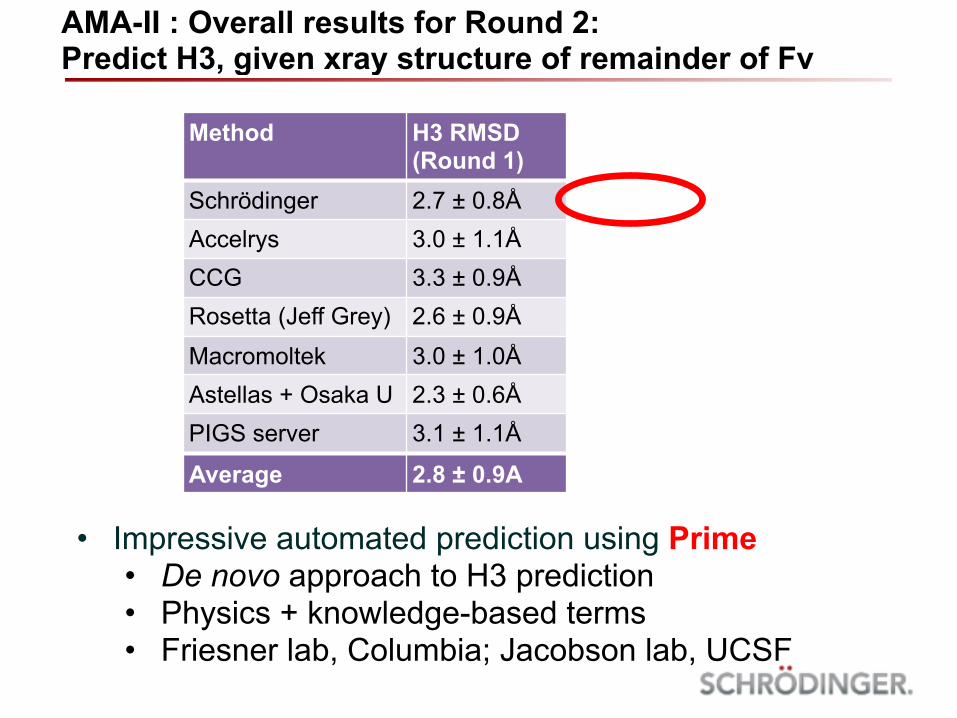
Method H3 RMSD (Round 1)
H3 RMSD (Round 2)
Schrödinger 2.7 ± 0.8Å 1.4 ± 1.1ÅAccelrys 3.0 ± 1.1Å 2.3 ± 1.0ÅCCG 3.3 ± 0.9Å 2.5 ± 1.6ÅRosetta (Jeff Grey) 2.6 ± 0.9Å 2.1 ± 1.1Å
Macromoltek 3.0 ± 1.0Å 3.3 ± 1.2ÅAstellas + Osaka U 2.3 ± 0.6Å 1.4 ± 1.9ÅPIGS server 3.1 ± 1.1Å
Average 2.8 ± 0.9A 2.2 ± 0.9Å
AMA-II : Overall results for Round 2: Predict H3, given xray structure of remainder of Fv
• Impressive automated prediction using Prime • De novo approach to H3 prediction • Physics + knowledge-based terms • Friesner lab, Columbia; Jacobson lab, UCSF
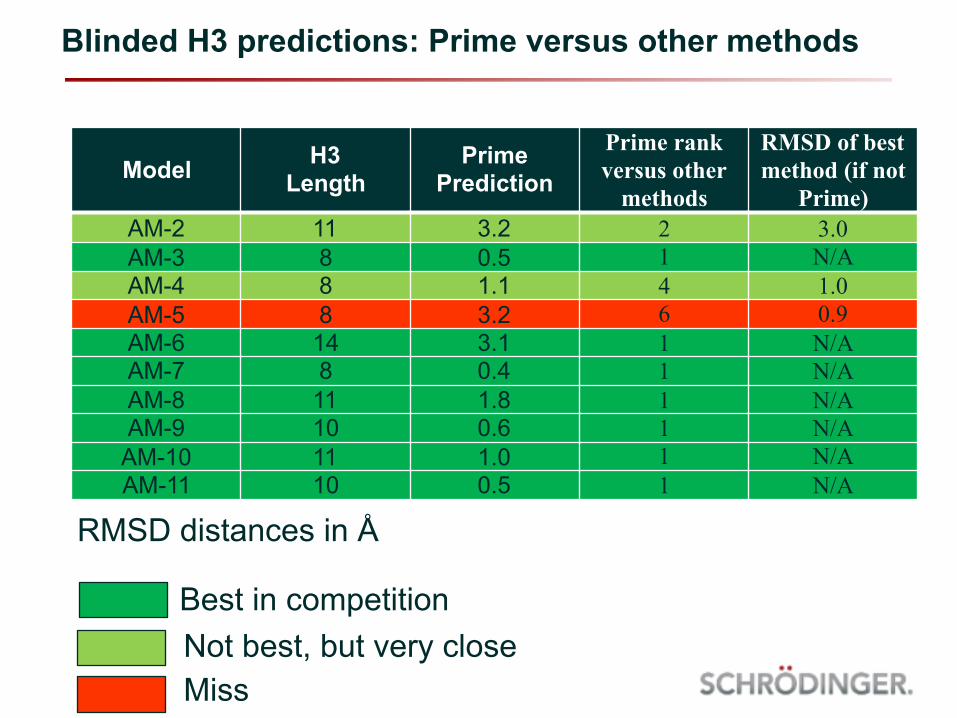
Blinded H3 predictions: Prime versus other methods
Model H3 Length
Prime Prediction
Prime rank versus other
methods
RMSD of best method (if not
Prime)AM-2 11 3.2 2 3.0AM-3 8 0.5 1 N/AAM-4 8 1.1 4 1.0AM-5 8 3.2 6 0.9AM-6 14 3.1 1 N/AAM-7 8 0.4 1 N/AAM-8 11 1.8 1 N/AAM-9 10 0.6 1 N/AAM-10 11 1.0 1 N/AAM-11 10 0.5 1 N/A
RMSD distances in Å
Best in competitionNot best, but very closeMiss
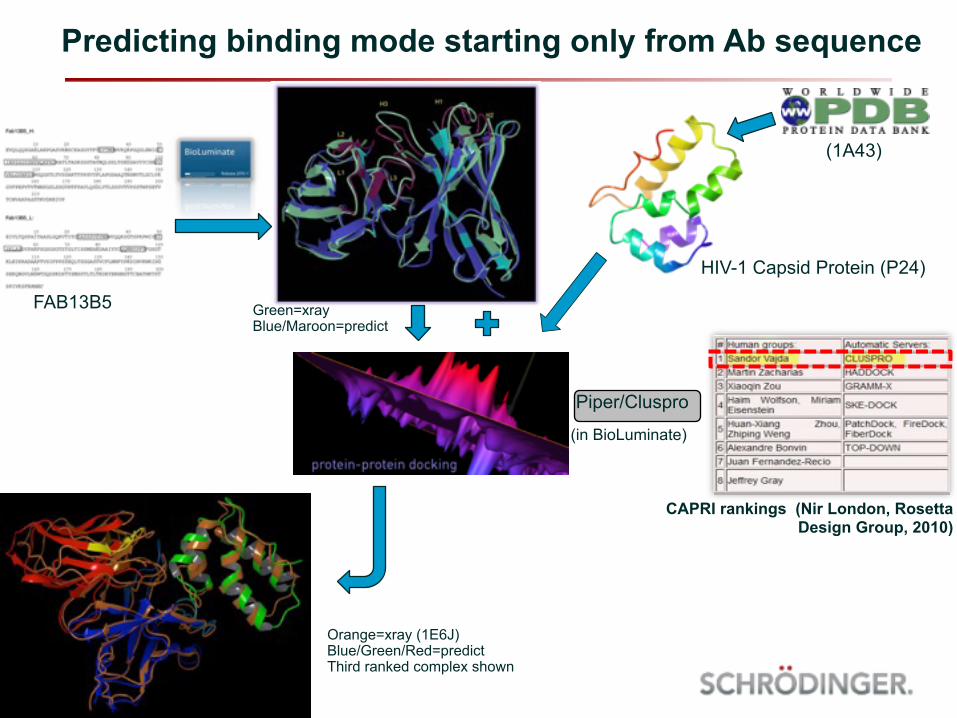
Predicting binding mode starting only from Ab sequence
Piper/Cluspro
FAB13B5
HIV-1 Capsid Protein (P24)
Green=xray Blue/Maroon=predict
Orange=xray (1E6J) Blue/Green/Red=predict Third ranked complex shown
CAPRI rankings (Nir London, Rosetta Design Group, 2010)
(1A43)
(in BioLuminate)

Protein Liability ~ Aggregation Propensity
Aggregation surface from original aggregation prediction tool
Surface analysis using the new Protein Surface Analysis panel

• Correlation between aggregation and sizes and locations of hydrophobic residue clusters on the protein surface
• More than just hydrophobicity?
• Hydrophobic patches • Smoothed hydrophobic potential for each atom, based on
Wildman and Crippen* slogP parameter
• Hydrophilic patches • Positive and negative patched based on atomic partial
charges
Protein Liability ~ Aggregation Propensity
* Wildman, S.A et l.; J Chem Inf Comput (1999) 39; 868-873
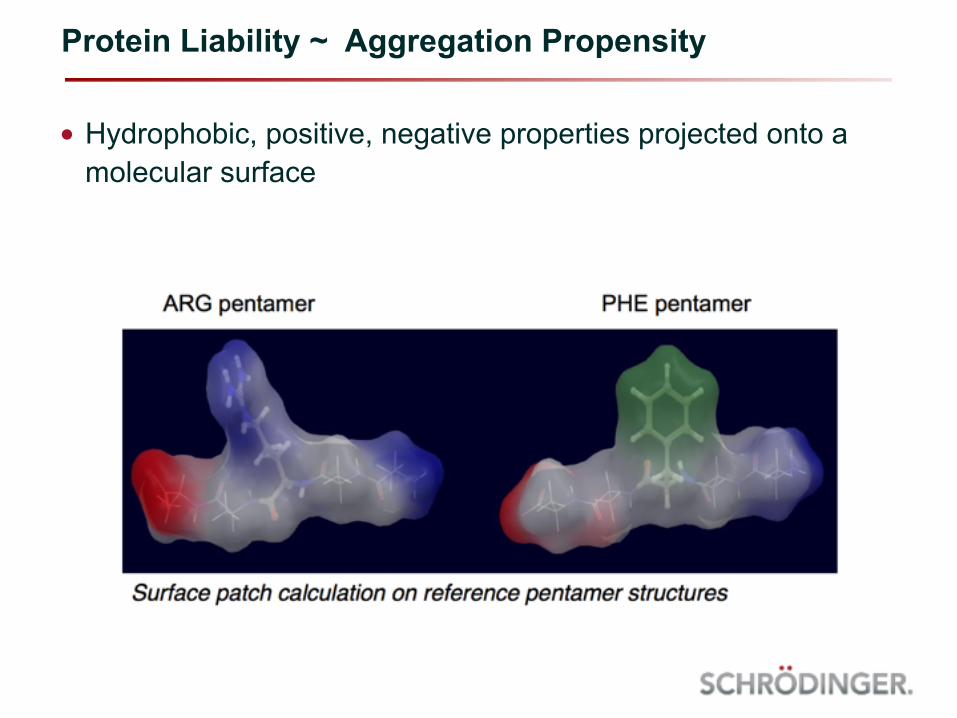
• Hydrophobic, positive, negative properties projected onto a molecular surface
Protein Liability ~ Aggregation Propensity

• From model to full system • Human GH - no aggregation • Bovine GH - aggregates at concentrations > 10um • Chimeric construct - human APR helix grafted onto bGH
Protein Liability ~ Aggregation Propensity

• Use aggregation benchmark datasets to identify driving factor behind aggregation • Adnectins: engineered antibody-like proteins. binding loop
similar to CDRs; loop instability => formation of inclusion bodies • Solubility correlates well with hydrophobic patch score
• VH-CDR3 amyloid: Beta-amyloid fragments cloned into H3 of VH chains • Introduction of charged residues at either end of the
beta-amyloid fragment improves solubility • Correlates well with positive patch score
Protein Liability ~ Aggregation Propensity

• Single model to describe both charge-driven and hydrophobicity-driven aggregation? • Patch descriptors + structure specific properties (pI,
hydrodynamic radius, etc) used to construct overall model • Worked well for the two original datasets (R^2 of 0.54 and
0.85, for adnectin and VH-CDR3-amyloid, respectively • Other external tests sets (antibody structures) yielded
inconclusive results
• Current work ongoing using new and improved descriptor set and AutoQSAR • Encouraging results in addressing solubility, viscosity,
aggregation issues
Protein Liability ~ Aggregation Propensity
• On-line manuals and tutorial files - available from the “Help” menu in BioLuminate and Maestro • On our web site: https://www.schrodinger.com/training • Science and Technology Support Team:
• Maestro 11 specific page: https://www.schrodinger.com/training/maestro11/home
Where to get help…

Acknowledgements
• David A. Pearlman ~ BioLuminate Product Manager
• Kai Zhu ~ Antibody Prediction
• Johannes Maier ~ Protein Surface Analyzer





























