Embed Size (px)
Citation preview

Endocrine Journal Advance Publication
Endocrine Journal 2014Advance Publication
doi: 10.1507/endocrj. EJ14-0021
MULTIPLE ENDOCRINE NEOPLASIA TYPE 1 (MEN1) or Wermer syndrome is a hereditary endo-crine cancer syndrome occurring in approximately one every 30,000 individuals regardless of sex or ethnic group [1]. It is characterized by the development of parathyroid adenomas (95% of cases), gastroentero-pancreatic neuroendocrine tumors (30-80% of cases), and anterior pituitary adenomas (15-90% of cases) [2]. Other endocrine and non-endocrine neoplasms (such as adrenocortical and thyroid tumors, visceral and cutane-ous lipomas, meningiomas, facial angiofibromas and collagenomas, and thymic, gastric, and bronchial car-cinoids) can also occur [3]. MEN1 should be suspect-ed in patients with an endocrinopathy involving two of the three characteristically affected organs, or else with an endocrinopathy of one of these organs plus a first-degree relative affected by MEN1 syndrome.
MEN1 gene consists of ten exons, spanning about 10 kb, and encodes a 610 amino acid protein named menin. MEN1 follows Knudson’s “two-hit” model for tumor suppressor gene carcinogenesis [4]. The first hit is a heterozygous MEN1 germline mutation, inher-ited from one parent (familial cases) or developed at
Sarcomatoid carcinoma of the kidney in a MEN1 patient: Case report and genetic profileTiziana Cavalli, Francesco Giudici, Gabriella Nesi, Francesca Marini, Francesca Giusti, Loredana Cavalli, Maria Luisa Brandi and Francesco Tonelli
Department of Surgery and Translational Medicine, University of Florence, Florence, Italy
Abstract. Renal tumors are exceedingly rare in Multiple Endocrine Neoplasia type 1 (MEN1), a pleyotropic hereditary cancer disorder affecting the endocrine system. Herein we report a unique case of renal sarcomatoid carcinoma with concomitant ipsilateral non-secreting adrenal adenoma occurring in a young male MEN1 patient, previously operated for hyperparathyroidism and multiple pancreatic neuroendocrine neoplasms. Molecular analysis in the MEN1 locus at 11q13 showed loss of heterozygosity in the adrenal lesion, while kidney cancer was unrelated to MEN1 syndrome.
Key words: Multiple endocrine neoplasia type 1, Sarcomatoid carcinoma, Kidney
an early embryonic stage (sporadic cases) and present in all cells at birth. The second hit is a MEN1 somatic mutation, usually a large deletion occurring in the pre-disposed endocrine cells, giving them the survival advantage for tumor development [5].
MEN1 mutations are located along the entire 1830-bp coding region, without mutation hot spots or genotype/phenotype correlations, suggesting that unrecognized genetic and environmental modifiers could play a role in the expression of the MEN1 phenotype [6].
Patient and Methods
Case reportA 36-year-old adopted male patient affected by
MEN1 was admitted in 2010 for a renal tumor and a concomitant ipsilateral non-secreting adrenal mass. In 2000, the patient had undergone duodenopancreatec-tomy extended to the pancreatic body and resection of the second hepatic segment, owing to duodeno-pan-creatic neuroendocrine tumors (NET) metastatic to the regional lymph nodes. Histologic examination of the liver resection specimen gave focal nodular hyperpla-
Submitted Jan. 11, 2014; Accepted May 7, 2014 as EJ14-0021Released online in J-STAGE as advance publication May 30, 2014Correspondence to: Francesco Tonelli, M.D., Department of Surgery and Translational Medicine, University of Florence, Largo Brambilla 3, 50139 Florence, Italy. E-mail: [email protected]
©The Japan Endocrine Society
Original
Authors’ contribution: All authors contributed equally to this work: T.Cavalli, F.Giudici collected the data; G.Nesi, F.Giudici, T.Cavalli, F. Marini analyzed data; T.Cavalli, F.Giudici, F.Tonelli, F.Marini, F.Giusti and L.Cavalli wrote the manuscript, M.L.Brandi supervised the manuscript.

2 Cavalli et al.
Endocrine Journal Advance Publication
low-up, no adjuvant chemotherapy was recommended. A thoraco-abdominal CT scan performed in January 2011 showed no abnormality in the lungs, mediasti-num, spleen, right kidney, or abdominal lymph nodes. In February 2011, secretin test and routine exams showed normal levels of gastrin, insulin, glucagon, PP, VIP, ACTH, GH, cortisol, thyroid hormones, calcium, PTH, gonadotropins, testosterone and prolactin, and normalization of previously elevated NSE.
The patient is free of disease 48 months after surgery.
Genetic analysisThe patient consented to multiple biopsies being
taken from the fresh surgical specimen for genetic analysis. Somatic DNA and total RNA were extracted from tissue samples.
The somatic genotype of tumor biopsies was com-pared with the constitutional DNA for evaluation of loss of heterozygosity (LOH) in the MEN1 locus at 11q13 Microsatellite PCR-based analysis for LOH in the MEN1 locus at 11q13 was performed using both kidney cancer and adrenal adenoma samples. Four specific microsatellite markers (D11S480, PYGM, D11S449, D11S913), flanking the 11q12-13 locus,
sia. One year later, he received total parathyroidec-tomy and thymectomy, with parathyroid graft in the right forearm. The patient was also affected by pitui-tary prolactinoma. Genetic analysis on genomic DNA from blood demonstrated the presence of germinal het-erozygote missense Leu414Pro mutation in exon 9 of the MEN1 gene.
Abdominal magnetic resonance imaging (MRI), performed in August 2009 as part of the scheduled follow-up of MEN1 patients, documented a 2.2-cm inhomogeneous mass of the left kidney, suggestive of malignancy. A 1-cm nodule, slightly impregnated with contrast medium, indicative of metastasis, was seen in the left adrenal gland.
Pancreatic and adrenal biochemical and hormonal profiles (gastrin, glucagon, PP, VIP, cortisol, ACTH) were normal. Tumor markers (alpha-fetoprotein, CA-125, CEA, CA 19-9) and chromogranin A were within normal range, while neuron-specific enolase (NSE) was slightly above the limit (16.7 ng/mL instead of 13). The patient had suffered renal colic due to ure-teral stones, treated with lithotripsy and subsequently surgically removed, but renal function was normal with creatinine at 1.04 mg/dL and urea at 0.39 g/L.
In September 2009 a total body octreoscan showed increased uptake of 111In-octreotide by the left kidney (Fig. 1A). In April 2010 the patient underwent endo-scopic ultrasonography, which raised suspicion of a 3-mm hypoechoic nodule in the pancreatic remnant. Abdominal computed tomography (CT) confirmed a 2.8-cm renal mass with fibro-calcified amorphous areas, showing moderate inhomogeneous contrast enhance-ment. A 1x1.5-cm nodule showing intense contrast enhancement was detected in the left adrenal gland. A stone of 0.5-cm was evident in the lower calyx of the left kidney. There was no evidence of hydronephrosis. In May 2010, total body 18FDG positron emission tom-ography (PET) demonstrated moderate hypermetabolic activity in the left adrenal gland and minimum uptake by the left renal mass.
In July 2010 the patient underwent surgical inter-vention. After a lumbar incision and removal of the left twelfth rib, the kidney with the adipose capsule was isolated. A 3-cm renal mass was identified deep in the middle of the kidney, reaching as far as the pelvis (Fig. 1B) and an adrenal nodule of 1.5 cm in diameter was also seen. Nephro-adrenalectomy was carried out. Intraoperative ultrasonography showed no lesion in the distal residual pancreas. In the subsequent clinical fol-
Fig. 1 Total body octreoscan (SSRS) showing increased uptake of 111In-octreotide by the left kidney (A). Macroscopic view of the specimen demonstrating the renal tumor and the adrenal gland lesion (arrow) (B).

3Unexpected malignancy in MEN1
Endocrine Journal Advance PublicationEndocrine Journal Advance Publication
were used for the LOH assessment. Four independ-ent PCR amplifications were separately made in a final volume of 12.5μL using PuReTaq Ready-To-Go PCR beads (GE Healthcare, Buckinghamshire, UK). Thermal cycling conditions were 94°C for 3 min, 35 cycles at 94°C for 30 sec, 55°C for 30 sec and 72°C for 2 min, followed by an additional stabilization step at 72°C for 20 min. An aliquot of each amplification product was denatured at 95°C for 5 min in a solution of formamide and GENESCAN 400HD [ROX] stand-ard size (Applied Biosystems, Foster City, CA, USA), and then analyzed on the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) by Genescan® analysis software.
Sequencing of MEN1 coding region (exons 2-10) and intron-exon junctions was also performed on somatic DNA from adrenocortical and kidney tumor samples, as previously described [7].
In addition, total mRNAs from tumor specimens were reversely transcribed to cDNA and then the region of cDNA containing the MEN1 mutation was PCR-amplified, using a specific couple of primers, and then sequenced to verify if MEN1 wild type, or mutated allele, or both were expressed in kidney cancer and adrenal adenoma.
Immunohistochemical analysisRepresentative sections of the lesions were selected
for IHC analysis. As primary antibodies we used rabbit monoclonal Menin Antibody (clone EPR3986, dilution 1:100; Novus Biologicals, Cambridge, UK) and Rabbit polyclonal Anti-Somatostatin Receptor 1 (SSTR1, dilution 1:50; Abcam, Cambridge UK).
Positive controls comprised colon for Menin and pancreas for Somatostatin Receptor. Negative control was performed by substituting the primary antibody with a Rabbit Serum (Dako, Carpinteria, CA). The control sections were treated in parallel with the sam-ples. All sections were then lightly counterstained with Mayer’s hematoxylin and mounted with Permount.
Results
Pathologic analysisGrossly, the renal neoplasm was gray-white in color,
with hemorrhage, necrosis and bony hard areas. The adrenal lesion appeared as a small, encapsulated, gold-en-yellow nodule (Fig. 1B). On histologic examination, the kidney tumor consisted of an epithelial component
with tubulo-papillary morphology and a mesenchymal component with extensive production of osseous and chondroid matrix (Fig. 2). No neoplastic invasion of the perirenal fat and vascular structures was detected. The adrenal cortical tumor was composed of compact cells with eosinophilic cytoplasm, arranged in cords and nests (Fig. 2). There was no evidence of capsular and vascular invasion. Necrosis and atypical mitotic Figs. were not identified. The proliferative fraction was immunohis-tochemically assessed with Ki-67 at less than 1%. These findings resulted in the diagnosis of renal sarcomatoid carcinoma and adrenal cortical adenoma.
Genetic analysisMicrosatellite analysis of 11q12-13 locus showed no
LOH in the kidney tumor, while the adrenal adenoma showed an allelic loss for D11S480 and D11S449 markers when compared to constitutive genomic DNA (Fig. 3). PYGM and D11S913 microsatellites were not informative, due to their homozygous status.
Sequencing of MEN1 gene coding region and intron-exon junctions on somatic DNA confirmed the loss of MEN1 wild type copy in the adrenal adenoma and the retainment of MEN1 wild type copy in the kid-ney tumor (Fig. 4). Sequencing analysis also excluded the presence of missense, nonsense or small deletion/insertion mutations inactivating the wild type allele of MEN1 in the kidney tumor.
Sequencing of the PCR-amplified cDNA region, containing the MEN1 mutation, proved that in the kid-ney cancer both wild type and mutated MEN1 mRNAs were expressed, while in the adrenal adenoma, only mutated mRNA was expressed, confirming the loss of the wild type allele at genetic level (Fig. 5).
Immunohistochemical analysis of expression of menin and SSTR
As shown in Fig. 6, menin immunostaining was absent in the adrenal adenoma whereas it displayed strong nuclear signal in the adjacent normal cortex. Immunohistochemistry also demonstrated preser-vation of wild-type menin expression in the kidney tumor (Fig. 7).
Antibody reactivity for STTR1 was seen in limited tumor clusters or single cells of the renal carcinoma. Immunostaining was predominantly localized to the plasma membrane but was also variably observed in the cytoplasm (Fig. 7).
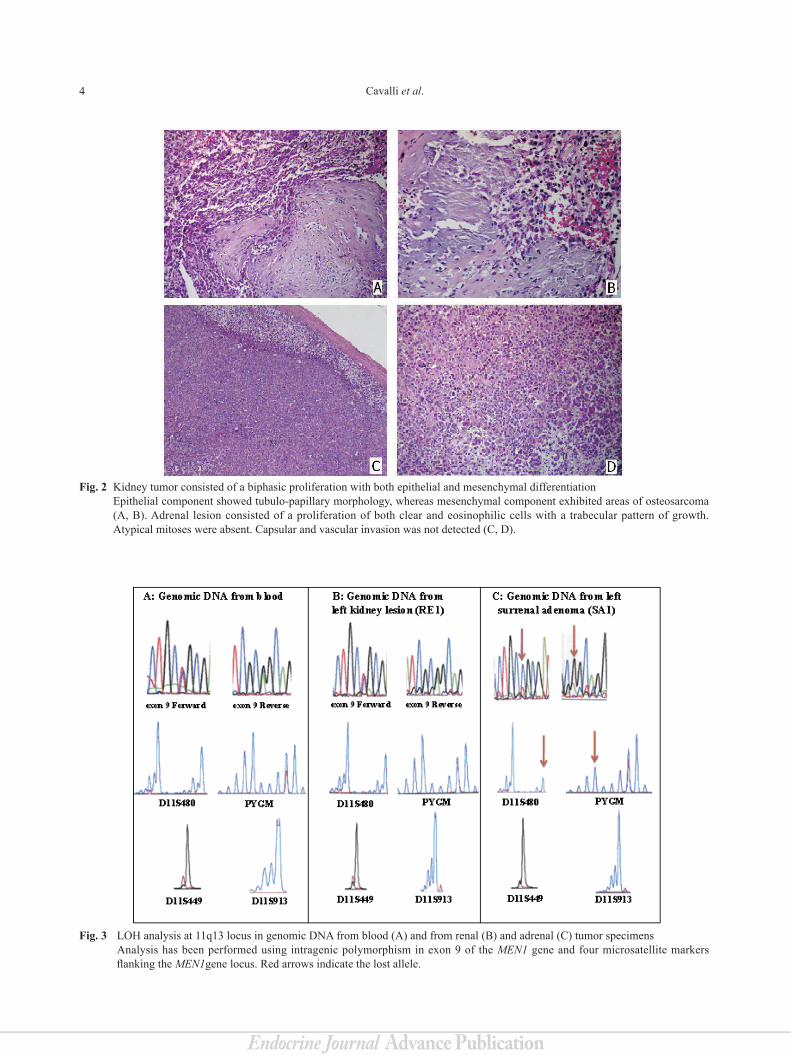
4 Cavalli et al.
Endocrine Journal Advance Publication
Fig. 2 Kidney tumor consisted of a biphasic proliferation with both epithelial and mesenchymal differentiation Epithelial component showed tubulo-papillary morphology, whereas mesenchymal component exhibited areas of osteosarcoma (A, B). Adrenal lesion consisted of a proliferation of both clear and eosinophilic cells with a trabecular pattern of growth. Atypical mitoses were absent. Capsular and vascular invasion was not detected (C, D).
Fig. 3 LOH analysis at 11q13 locus in genomic DNA from blood (A) and from renal (B) and adrenal (C) tumor specimens Analysis has been performed using intragenic polymorphism in exon 9 of the MEN1 gene and four microsatellite markers
flanking the MEN1gene locus. Red arrows indicate the lost allele.
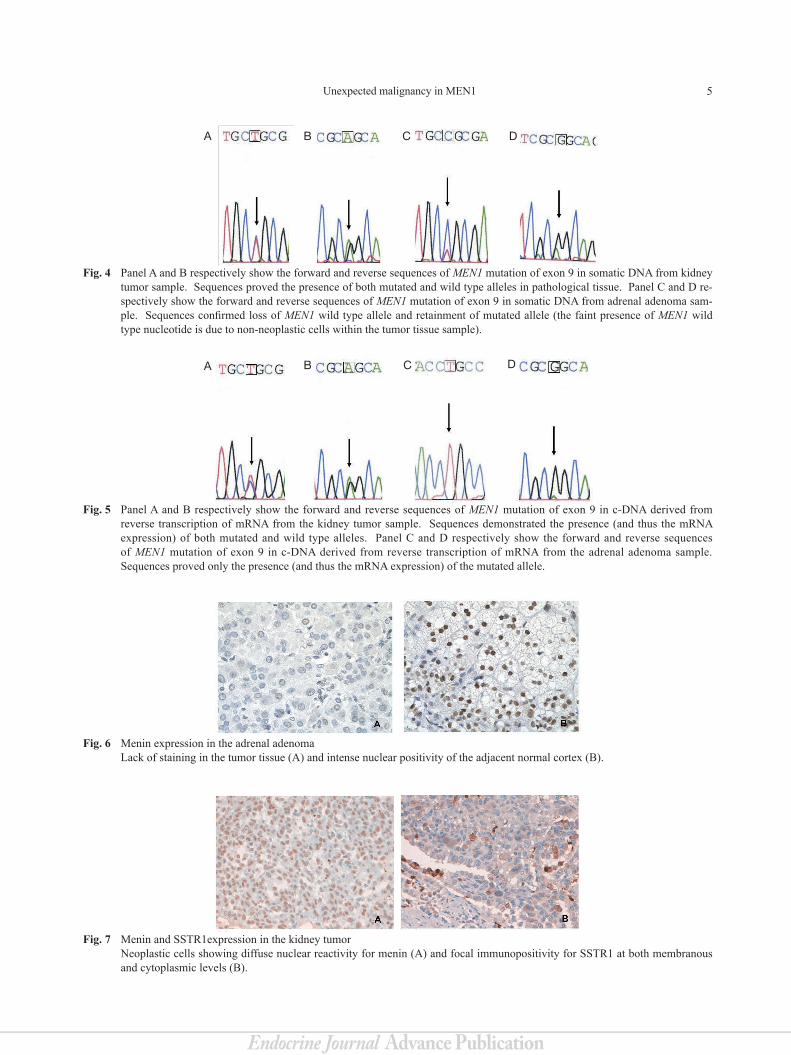
5Unexpected malignancy in MEN1
Endocrine Journal Advance PublicationEndocrine Journal Advance Publication
Fig. 4 Panel A and B respectively show the forward and reverse sequences of MEN1 mutation of exon 9 in somatic DNA from kidney tumor sample. Sequences proved the presence of both mutated and wild type alleles in pathological tissue. Panel C and D re-spectively show the forward and reverse sequences of MEN1 mutation of exon 9 in somatic DNA from adrenal adenoma sam-ple. Sequences confirmed loss of MEN1 wild type allele and retainment of mutated allele (the faint presence of MEN1 wild type nucleotide is due to non-neoplastic cells within the tumor tissue sample).
Fig. 7 Menin and SSTR1expression in the kidney tumorNeoplastic cells showing diffuse nuclear reactivity for menin (A) and focal immunopositivity for SSTR1 at both membranous and cytoplasmic levels (B).
A B C D
A B C D
Fig. 6 Menin expression in the adrenal adenoma Lack of staining in the tumor tissue (A) and intense nuclear positivity of the adjacent normal cortex (B).
Fig. 5 Panel A and B respectively show the forward and reverse sequences of MEN1 mutation of exon 9 in c-DNA derived from reverse transcription of mRNA from the kidney tumor sample. Sequences demonstrated the presence (and thus the mRNA expression) of both mutated and wild type alleles. Panel C and D respectively show the forward and reverse sequences of MEN1 mutation of exon 9 in c-DNA derived from reverse transcription of mRNA from the adrenal adenoma sample. Sequences proved only the presence (and thus the mRNA expression) of the mutated allele.

6 Cavalli et al.
Endocrine Journal Advance Publication
The pathogenesis of the mesenchymal cells is not clearly understood, but it is assumed that the sarcoma-tous component undergoes a metaplastic process in which the tumor cells lose their epithelial character-istics and gain mesenchymal phenotypic qualities via epithelial-mesenchymal transition.
Menin is known to be important for suppressing tumorigenesis in various endocrine and non-endo-crine tissues, via the inhibition of cell proliferation, the positive regulation of cell apoptosis and the con-trol of repair of DNA damages. Menin inhibits cell growth by directly and indirectly regulating numerous and different cell pathways, by associating with pro-moters of various genes as well as by recruiting his-tone-modifying enzymes [17]. Nevertheless, to date, none of menin-regulated pathways or menin protein partners has shown to be alone crucial for the develop-ment of MEN1-related tumors. The menin tissue-spe-cific mechanisms of action and its role in MEN1 tum-origenesis are far to be completely understood. And this is even more true for the rarely described tumors in MEN1 patients, including the sarcomatoid kidney carcinoma, for which the potential tissue-specific role of the MEN1 gene in their tumorigenesis needs to be largely investigated.
The potential role of the MEN1 gene in the devel-opment of rarely described tumors in MEN1 patients needs to be investigated. From our results, obtained through LOH and MEN1 sequencing analyses and MEN1 expressed mRNA sequencing in tumor tissue samples, one may conclude that the MEN1 oncosup-pressor does not appear to have a pathogenetic role in sarcomatoid kidney carcinoma, while its loss has a central role in the tumorigenesis of adrenal carcinoma. However, this finding awaits confirmation in a large series of sporadic sarcomatoid kidney tumors.
Conflict of Interest
None declared.
Discussion
MEN1 phenotype is wide-ranging, and over 20 dif-ferent combinations of endocrine and non-endocrine manifestations have been described [8-12].
Only a few cases of kidney tumors associated with MEN have been reported so far. In 1986 Denker et al. reported a case of hypernephroma in MEN1 [13], while in 1996 Jeddi et al. detailed a case of Wermer syndrome associated with a bilateral renal tumor [14]. To our knowledge, the present case is the first instance of sar-comatoid carcinoma of the kidney in a MEN1 patient. This asymptomatic renal cancer was detected at young age and at an early stage on the follow-up imaging per-formed as control of a MEN1-related adrenal neoplasm.
The different imaging techniques used to monitor and characterize the adrenal lesion of our patient did not tally. MRI labeled the adrenal tumor as a prob-able metastasis, while the kidney uptake of Indium-111-labeled octreotide during octreoscan might have suggested a metastatic neuroendocrine lesion. In lit-erature, somatostatin receptors have been found in certain non-neuroendocrine tumors, including men-ingiomas, well-differentiated brain tumors, malignant lymphomas, renal cell carcinoma, and some breast and lung carcinomas [15].
Histologically, sarcomatoid renal cell carcinomas consist of two cell populations, i.e. sarcomatous and carcinomatous. This phenotype can occur in all sub-types of renal cancers with a frequency of 1-8%, but are most common in chromophobe and clear cell carcino-mas. Generally, the sarcomatoid components display features of malignant fibrous histiocytoma or fibrosar-coma. Heterologous differentiation, such as chondro-sarcoma, osteosarcoma and rhabdosarcoma-like differ-entiation, is rare.
Sarcomatoid renal cell cancer occurs in a wide age range (29-81 years) and carries a dismal progression, often presenting at an advanced stage, with a high pro-pensity to metastasize [16].
References
1. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, et al (2001) Guidelines for diagnosis and ther-apy of MEN type 1 and type 2. J Clin Endocrinol Metab 86: 5658-5671.
2. Gibril F, Schumann M, Pace A, Jensen RT (2004)
Multiple Endocrine Neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 83: 43-83.
3. Burgess JR, Harle RA, Tucker P, Parameswaran V,

7Unexpected malignancy in MEN1
Endocrine Journal Advance PublicationEndocrine Journal Advance Publication
Davies P, et al (1996) Adrenal lesions in a large kindred with Multiple Endocrine Neoplasia type 1. Arch Surg 131: 699-702.
4. Knudson AG (1993) Antioncogenes and human cancer. Proc Natl Acad Sci USA 90: 10914-10921.
5. Marx SJ, Agarwal SK, Kester MB, Heppner C, Kim YS, et al (1998) Germline and somatic mutation of the gene for Multiple Endocrine Neoplasia type 1 (MEN1). J Intern Med 243: 447-453.
6. Thakker RV (2000) Multiple Endocrine Neoplasia type 1. Endocrinol Metab Clin North Am 29: 541-567.
7. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, et al (2005) Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. J Clin Endocrinol Metab 90: 2603-2609.
8. Agarwal SK, Lee Burns A, Sukhodolets KE, Kennedy PA, Obungu VH, et al (2004) Molecular pathology of the MEN1 gene. Ann N Y Acad Sci 1014: 189-198.
9. Marx SJ (2005) Molecular genetics of Multiple Endocrine Neoplasia types 1 and 2. Nat Rev Cancer 5: 367-375.
10. Doherty GM (2005) Multiple Endocrine Neoplasia type 1. J Surg Oncol 89: 143-150.
11. Carrasco CA, González AA, Carvajal CA, Campusano C, Oestreicher E, et al (2004) Novel intronic mutation of MEN1 gene causing familial isolated primary hyper-parathyroidism. J Clin Endocrinol Metab 89: 4124-4129.
12. Hao W, Skarulis MC, Simonds WF, Weinstein LS, Agarwal SK, et al (2004) Multiple Endocrine Neoplasia type 1 variant with frequent prolactinoma and rare gas-trinoma. J Clin Endocrinol Metab 89: 3776-3784.
13. Denker PS, Wright D, Hilscher JR, Saba SR, Ramirez G (1986) Hypernephroma associated with Multiple Endocrine Neoplasia type I: a case report. J Urol 136: 896-898.
14. Jeddi A, Vasse N, Prunet D, Buzelin F, Bouchot O, et al (1996) Wermer syndrome associated with a bilateral renal tumor. Prog Urol 6: 103-106.
15. Hanson MW (2001) Scintigraphic evaluation of neu-roendocrine tumors. Appl Radiol 30: 6.
16. Reiter M, Schwope R, Clarkson A (2012) Sarcomatoid renal cell carcinoma: a case report and literature review. J Radiol Case Rep 6: 11-16.
17. Wu T, Hua X (2011) Menin represses tumorigenesis via repressing cell proliferation. Am J Cancer Res 1: 726-739.





























