Embed Size (px)
Citation preview

CHARACTERIZATION OF HCV GENOTYPE 3A STRUCTURAL
PROTEINS CORE, E1, E2 AND DEVELOPMENT OF PSEUDOTYPE
PARTICLES (HCVPP) AND INTERGENOTYPIC CHIMERA (HCVCC)
By
SADIA ANJUM 2007-NUST-DirPhD- Virology-21
A Dissertation Submitted in the Partial Fulfillment of the Requirement for
the Degree of Doctor of Philosophy (PhD)
IN
VIROLOGY
Supervisor
Dr. Ishtiaq Qadri
Atta-ur Rahman School of Applied Biosciences National University of Sciences & Technology
Islamabad, Pakistan

i
In the Name of God, the Merciful, the Compassionate
Read! In the Name of your Sustainer who created,
created man out of a germ cell.
Read! for your Sustainer is the Most Bountiful
One who has taught mankind the use of the pen taught mankind what he did not know!" –
Surah al'Alaq,

ii
ACKNOWLEDGEMENT
With the grace of God I have completed my PhD work. I highly acknowledge guidance and support of people who have helped me in completion of this task.
My immense gratitude goes to my Supervisor Dr.Ishtiaq Qadri for taking me in his kind supervision, for providing enthusiasm, vision and encouragement. I highly acknowledge his consistent support and guidance that led me in completion of this difficult task.
Being an HEC scholar I highly acknowledge financial support provided by the HEC throughout my PhD. I also highly acknowledge French Embassy for providing me an opportunity and finance to work in one of the well recognized lab in France. I am sincerely thankful to NUST Pakistan and CNRS France for providing me scholarship to complete my research project.
I owe colossal acknowledgment to Jean Dubuisson for accepting me in his lab and providing me opportunity for tremendous learning. I am very grateful for his kind and skilled assistance in my sequence analysis and for providing directions to my research. I am also very Great full to Anna Albecka for helping me with HCVpp system and Gabrielle Vieyres for helping me with my neutralization assay.
I highly gratify very skilled support and guidance of Dr. Czeslaw Wychowski for construction of HCVcc model, working with him was an opportunity of tremendous learning. I would not miss the opportunity to acknowledge consistence guidance and support from Czeslaw’s PhD students, Khaled Alsaleh and Pierre-yves Delavalle.
I sincerely acknowledge Birke Andrea Tews for providing me her construct to take out Goussia luciferase reporter gene for my chimera and Dr. Costin-Ioan Popescu for his help and support in my experiments.
I also earnestly acknowledge Dr. Yves Rouillé for teaching me Immuno staining and for his enormous help in microscopy. I owe very special thanks for a lot of support from Dr. Sandrine Belouzard, Dr. Laurence Cocquerel and Sophana Ung trough out my stay in France.
I am very grateful to Dr. Hajra Sadia, my special committee member for her guidance and support and my GEC members Dr. Najam us Sahar Sadaf Zaidi, Dr. Sadia Andleeb and Dr. Sheeba Murad Mall for helping me in proof reading of the manuscript.
I highly acknowledge contribution and assistance of Muhammad Sohail Afzal for compiling my results and Baber Aslam and Rehan Zafar for their assistance in in slico modelling.
I owe lot of thanks to my lab fellows, Muhammad Sohail Afzal, Kaneez Fatima, Sidra Ali, Talha Shafi, Faiza Rasheed, Mayriam Bibi and Sadia Zahid and for their help in thesis write up.
Very special thanks to my Husband Tahir Ahmad for his continuous support and colossal help in Lab. Special gratitude for my parents, brother and sisters for their support and for taking care of my kids during my PhD, without their support it would have been an impossible task.

Table of content
iii
Table of contents INTRODUCTION ........................................................................................................................................ 1
LITERATURE REVIEW ............................................................................................................................. 6
2.1 CLASSIFICATION AND GENETIC VARIABILITY sentense case .............................................. 8
2.2 HCV VIRION ASSEMBLY AND RELEASE .................................................................................. 9
2.3 HCV CORE IS A MULTIFUNCTIONAL PLEIOTROPIC PROTEIN .......................................... 12
2.4 HCV GLYCOPROTEINS, ENTRY, INFECTIVITY, HETRODIMERIZATION AND FUSION ...................................................................................................................................................... 16
2.5 P7 AN ION CHANNEL PROTEIN ................................................................................................. 34
2.6 NS2 GENE AND PROTEIN ........................................................................................................... 35
2.7 HCV NEUTRALIZATION ............................................................................................................. 38
MATERIAL AND METHODS .................................................................................................................. 41
3.1 ENROLLMENT OF PATIENTS..................................................................................................... 41
3.2 SAMPLE COLLECTION AND STORAGE ................................................................................. 41
3.3 RNA ISOLATION ........................................................................................................................... 41
3.4 GENOTYPING ................................................................................................................................ 42
3.5 PRIMER DESIGNING .................................................................................................................... 42
3.6 cDNA SYNTHESIS ......................................................................................................................... 45
3.7 CORE - NS2 GENE AMPLIFICATION ......................................................................................... 45
3.8 CLONING OF HCV CORE-NS2 REGION IN pCRII-TOPO VECTOR ....................................... 46
3.8.1 DNA Ligation ...................................................................................................................... 48
3.8.2 Bacterial Transformation ..................................................................................................... 48
3.8.3 Plasmid DNA Isolation and Restriction Endonuclease Analysis ......................................... 48
3.8.4 Sequencing and Phylogenetic Analysis ............................................................................... 49

Table of content
iv
3.9 IN SILICO MODELING AND PROTEIN-PROTEIN INTERACTION FOR HCV CORE AND STAT1 ............................................................................................................................................... 50
3.10 CLONING OF HCV ENVELOPE PROTEINS E1E2 IN MAMMALIAN EXPRESSION VECTOR pcDNA 3.1 ................................................................................................................................. 51
3.11 CHARACTERIZATION OF HCV GLYCOPROTEINS BY USING HCVpp SYSTEM. ........... 53
3.11.1 Transfection ....................................................................................................................... 53
3.11.2 Glycoprotein Analysis........................................................................................................ 54
3.11.3 Western Blot ...................................................................................................................... 55
3.11.4 Immuno Fluorescent Labeling ........................................................................................... 56
3.11.5 Immunoprecipitation (CD81 Pull Down Assay) ................................................................ 56
3.11.6 HCVpp Infectivity Assay ................................................................................................... 57
3.12 PCR BASED MUTAGENESIS IN E2 GENE .............................................................................. 58
3.12.1 Fusion PCR ........................................................................................................................ 59
3.12.2 Characterization of HCVpp Mutants ................................................................................. 60
3.13 NUTRALIZATION OF HCVpp .................................................................................................... 60
3.13.1 Isolation of Antibodies ....................................................................................................... 61
3.13.2 GNA Capture ELISA for HCV E2 .................................................................................... 61
3.13.3 Neutralization Assay .......................................................................................................... 62
3.14 CONSTRUCTION AND CHARACTERIZATION OF INTER- GENOTYPIC CHIMERA FOR STRUCTURAL PROTEINS .......................................................................................... 62
3.14.1 Cloning of Core-NS2 Region in pJFH1 ............................................................................. 62
3.14.2 Cloning of Core-NS2 region in pJFH1del ......................................................................... 63
3.14.3 Cloning of Core-NS2 region in pJFH1VP ......................................................................... 64
3.14.4 Cloning of Gossia luciferase repoter gene in 3a-JFH1VP chimera ................................... 64
3.15 CHARACTERIZATION OF 3APAK-JFH1 (CHIMERA) .......................................................... 65
3.15.1 Preparation of DNA for In vitro Transcription .................................................................. 65
3.15.2 In vitro Transcription of 3a-JFH1 Chimera ....................................................................... 67
3.15.3 Electroprotaion of Huh7 cells with 3aPAK-JFH1 RNA .................................................... 68

Table of content
v
3.16 REPLICATION AND INFECTIVITY ASSESSMENT FOR M2-JFH1VP CHIMERA .............. 69
3.16.1 Replication Assay .............................................................................................................. 69
3.16.2 Infectivity Assay ................................................................................................................ 69
3.16.3 Immuno Fluorescent Labeling ........................................................................................... 69
RESULTS ................................................................................................................................................... 71
4.1 AMPLIFICATION AND CLONING OF HCV CORE-NS2 REGION .......................................... 71
4.1.1 Core to NS2 Amplification .................................................................................................. 71
4.1.2 Core-NS2 Cloning ............................................................................................................... 71
4.2. CORE-NS2 DNA SEQUENCING: ................................................................................................ 74
4.2.1 Amino Acid Sequence Analysis .......................................................................................... 75
4.3 IN SILICO CHARACTERIZATION OF STAT1 INTERACTING DOMAIN OF HCV CORE: ......................................................................................................................................................... 83
4.4 HCVpp PRODUCTION AND CHARACTERIZATION ............................................................. 86
4.5 HCVpp MUTATAGENESIS AND THEIR CHARACTERIZATION ......................................... 90
4.5.1 Removal or addition of glycosylation site at position 499. .................................................. 91
4.5.2 Deletion of 5 extra amino acids in intergenotpic variable region ........................................ 91
4.5.3 Switching of intergenotypic region ...................................................................................... 92
4.5.4 Characterization of E2 Mutants for CD81 binding and Infectivity .................................... 95
4.5.5 Characterization of E2 Mutants for infectivity .................................................................. 96
4.5.6 Incorporation of A4 Epitope In Glycoprotein E1 .............................................................. 97
4.6 HCVpp NEUTRALIZATION BY SERUM DERIVED IgG ........................................................ 98
4.7 PHYLOGENETIC ANALYSIS OF HCV STRUCTURAL PROTEINS CORE, E1 AND E2 .............................................................................................................................................................. 103
4.8 CONSTRUCTION AND CHARACTERIZATION OF INTERGENOTYPIC CHIMERA FOR STRUCTURAL PROTEINS ........................................................................................................... 107
4.8.1 Cloning of Core-NS2 region in pJFH1del ......................................................................... 107
4.8.2 Cloning of Core-NS2 region in pJFH1VP. ........................................................................ 108

Table of content
vi
4.8.3 Characterization of M2JFH1VP Chimera .......................................................................... 110
4.9 CLONING OF GAUSSIA LUCIFERASE REPORTER GENE IN M2JFH1VP CHIMERA. ............................................................................................................................................... 113
4.10 CHARACTERIZATION OF 3aPAK -JFH1 (CHIMERA) ......................................................... 115
4.10.1: In vitro Transcription and Electroporation of 3aPAK -JFH1 .......................................... 115
4.10.2 Immuno fluorescent labeling. .......................................................................................... 116
4.10.3 Luciferase Assay for Replication and Infectivity of 3aPAK-JFH1 Chimera ................... 119
DISSCUSSION ......................................................................................................................................... 121
Phylogenetic Analysis for HCV Core and Envelop Protein ..................................................................... 135
HCVcc Model system for Pakistani Isolates ............................................................................................. 139
SUMMARY………………………………………………………………………………………………………………………………139
REFRENCES ............................................................................................................................................ 141

List of tables
vii
List of tables
Table No. Title Page No.
Table 3.1 HCV patient information enrolled in the current study 43
Table 3.2 Primers used for amplification and sequencing of core-NS2
region
44
Table 3.3 Primers Used for Mutagenesis 59
Table 4.1 Core-NS2 Sequence Accession Number (NCBI) 74
Table 4.2 Amino acid sequence variations of HCV (3a) structural proteins
(core, E1, E2)
79-83
Table 4.3 In vivo Mutations observed in HCV Core and their Effect on STAT1 Interaction
84
Table 4.4 Results of four different models for Core, STAT1 interaction, showing interacting residues of Core and STAT1
84
Table 4.5 Results of cluster model 1 showing the contact points between HCV Core and STAT1 along with bond type and net charge.
85

List of figures
viii
List of figures
Figure No. Title
Page No.
Figure 2.1 HCV Discovery 8
Figure 2.2 Global distribution of HCV genome 10
Figure 2.3 HCV polyprotein processing and topology 11
Figure 2.4 The various steps of HCV entry in hepatocytes 13
Figure 2.5 Schematic representation of the assembly and release of HCV 14
Figure 2.6 Structural and Functional domains of the HCV core protein 16
Figure 2.7 Analysis of the HCV E1 putative fusion domain 22
Figure 2.8 Conserved residues within the putative CD81 binding domains of E2
23
Figure 2.9 Amino acid sequence diversity in HCV E1–E2 envelope glycoproteins
26
Figure 2.10 Conservation of sequences close to glycosylation sites E2N1, E2N6, and E2N11.
28
Figure 2.11 Tertiary structure of HCVE2 showing location of various domains 32
Figure 2.12 Cellular receptors for hepatitis C virus 33
Figure 2.13 HCV life Cycle 34
Figure 3.1 A Schematic presentation of pCRII-TOPO vector. A; linerized plasmid. B; Circular plasmid after Core-NS2 cloning between P lac and lac Z sites
47

List of figures
ix
Figure 3.2 Recombinant pcDNA3.1 showing cloning position of E1E2 genes of HCV along with signal sequence (Sc) from 3ˈend of Core
53
Figure 3.3 3aPAK-JFH1 chimera. 65
Figure 4.1 First round PCR (A). Second round (nested) PCR (B) 71
Figure 4.2 Restriction digestion of recombinant pCRII TOPO vector from patient M with EcoRI.
72
Figure 4.3 Cloning in pCRII TOPO vector from patient S, digested with EcoR1
72
Figure 4.4 Recombinant pCRII TOPO vector from patient k. 73
Figure 4.5 Cloning in pCRII TOPO vector from patient R, digested with EcoR1
73
Figure 4.6 Amino acid sequence Alignment. 75-78
Figure 4.7 HCV Core and STAT1 Interacting domains. 86
Figure 4.8 Western blot analysis showing E2 expression in HEK293T cells from patient M and S.
87
Figure 4.9 Western blot analysis showing E2 expression in HEK293T cells from patients K and R.
87
Figure 4.10 Western blot analysis showing CD81 pull down of E2 incorporated in HCVpp
88
Figure 4.11 Western blot analysis showing CD81 pull down of E2 incorporated in HCVpp. Secreted HCVpp
88
Figure 4.12 Bar graph showing HCV pp infectivity based on luciferase activity
89
Figure 4.13 Bar graph showing HCV pp infectivity based on luciferase activity in presense of 3a as refrence
90

List of figures
x
Figure 4.14 First round PCR for the introduction of mutations in M2pp clone. 91
Figure 4.15 Second round PCR (fusion PCR) for the introduction of mutations in M2 clone
92
Figure 4.16 Fusion PCR products in PcDNA3.1 Vector 92
Figure 4.17 First round PCR for the introduction of mutation in 3awtpp clone 93
Figure 4.18 Second round PCR (fusion PCR) for the introduction of mutations in 3awt clone
94
Figure 4.19 Cloning of fusion PCR products in pCDNA3.1 vector 94
Figure 4.20 Western blot showing CD81 pull down from cell lysate 95
Figure 4.21 CD81 pull down from HCVpp (3a). 95
Figure 4.22 Bar graph showing HCVpp infection in Huh7 cells measured as luciferase activity for HVR 499 mutant
96
Figure 4.23 Bar graph showing HCVpp infection in Huh7 cells, measured as luciferase activity for HVR575 mutants.
97
Figure 4.24 Bar graph showing HCVpp infection in Huh7 cells 98
Figure 4.25 Graph showing HCVpp neutralization measured as luciferase activity by HCVpp infected cells for M1
99
Figure 4.26 Graph showing HCVpp neutralization measured as luciferase activity by HCVpp infected cells For 3aWt
100
Figure 4.27 Graph showing serum IgG derived neutralization of HCVpp bearing mutations in E2 glycoprotein.
101
Figure 4.28 Graph showing serum IgG derived neutralization of HCVpp bearing mutations in E2 IgHVR
102
Figure 4.29 Phylogenetic relation of HCV core protein, genotype 3a 104
Figure 4.30 Phylogenetic relation of HCV glycoprotein E1E2 for 3a genotype
105-6

List of figures
xi
Figure 4.31 Purified products digested with Bst11071 and BglII 107
Figure 4.32 Colony PCR of potential recombinant clones. 108
Figure 4.33 Purified digestion fragments showing pJFH1VP, M1JFH1del, M2JFH1del
108
Figure 4.34 Colony PCR 109
Figure 4.35 Schematic representation of JFH1VP chimera 109
Figure 4.36 Schematic representation of NS2 topology 110
Figure 4.37 In vitro transcription of JFH1C6S4, M1JFH1VP and M2JFH1VP 110
Figure 4.38 Huh7 cells showing expression of Core 48 hours post electroporation with M2FH1VP RNA.
111
Figure 4.39 Huh7 cells showing expression of core 72 hours post electroporation with M2J-JFH1VP RNA.
111
Figure 4.40 Huh7 cells showing expression of core72 hours post electroporation with M2J-JFH1VP RNA with DPI background
112
Figure 4.41 Huh7 cells showing expression of NS5a 72 hr post electroporation with M2JFH1VP RNA.
112
Figure 4.42 Huh7 cells showing expression of E2 72hr post infection with M2 JFH1VP virus produced in cell culture.
113
Figure 4.43 Bst11071 digested and purified fragments of M2J-JFH1VP and JFH1CS-N6A4-Gluc
114
Figure 4.44 3aPAK-JFH1 recombinant positive PCR 114
Figure 4.45 3aPAK-JFH1 and JFH1C6S4 digested with Xba1 115
Figure 4.46 In vitro transcribed RNA from 3aPAK-JFH1and JFH1C6S4 116

List of figures
xii
Figure 4.47 Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour
after transformation with JFH1C6S4 RNA.
117
Figure 4.48 Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour after transformation with 3a PAK-JFH1 RNA.
117
Figure 4.49 Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour after infection with cell cultured derived JFHI virus
118
Figure 4.50 Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour after infection with cell culture derived 3a PAK-JFH1 recombinant virus.
118
Figure 4.51 3aPAK-JFH1 replication vs JFH1CSA4 119
Figure 4.52 3aPAK-JFH1 infection vs JFH1CSA4 120

List of acronyms
xiii
List of Acronyms
AA Amino Acid
Apo B Apolipoprotein B
Asp Aspartate
CCl2 Cesium chloride
CIDE-B Cell death-Inducing DFFA-like Effectors B
CLDN1 Claudin1
CRD Calcium-dependent carbohydrate Recognition Domain
CT C-Terminal
DAPI 4', 6-Diamidino-2-Phenylindole
dATPs Deoxy Adenoside triphosphate
DC-SIGN Dendritic cell-specific Intercellular molecule-3-Grabbing Non-integrin
dCTPs Deoxycytinoside triphosphate
dGTPs Deoxyguanoside triphosphate
DMEM Dulbecco’s modified Eagle’s medium
dNTPs Deoxyribonucleoside triphosphates
DTT Dithiothreitol
dUTPs Deoxyurinoside triphosphate
E1 Envelop protein1
E2 Envelop protein2
ELISA Enzyme linked immunosorbant assay
ER Endoplasmic reticulum
euHCVdb European HCV database
FCS Fetal calf serum

List of acronyms
xiv
FP Fusion peptide
FRET Fluorescence resonant energy transfer
GFP Green fluorescent protein
Gly Glycine
GNA Galanthus nivialis lectin
GST Glutathione-S-Transferase
GT Genotype
HAV Hepatitis A virus
HBV Hepatitis B virus
HCC Hepatocellular carcinoma
HCV Hepatitis C virus
HCVcc Hepatitis C virus chimera of core
HCV-LP Hepatitis C virus like Particles
HCVpp Hepatitis C virus pseudoparticles
HEK cells Human epithelial kidney 293T cells
HIV Human immunodeficiency virus
HSDL High density lipoprotein
Hsp 90 Heat shock protein 90
Huh7 Human hepatoma cell line
HVR II Hypervariable region II
IgG Immunoglobulin G
IPTG Isopropyl beta-D-1-thiogalactopyranoside
IRES Internal ribosome entry site
JFH1 Japenese fulminant hepatitis isolate1
LB Luria-Bertani

List of acronyms
xv
LD Lipid droplets
LDL Low density lipoproteins
LDLR Low density lipoprotein receptors
LEL Large exracellular loop
L-SIGN Liver/lymph node- specific Intercellular molecule-3-Grabbing Non-integrin
Lys Lysine
MAbs Monoclonal antibodies
MEM Minimal essential medium
ml Millilitre
MLV Murine leukaemia virus
MTT Microsomal triglyceride transfer protein
NANBH Non-A Non-B Hepatitis
NCBI National Commission on Biotechnology Information
NCVI NUST Center of Virology & Immunology
NMR Nuclear Magnetic Resonance
NS2 Non Structural protein 2
NS3 Non Structural protein 3
NS4A Non Structural protein 4A
NS4B Non Structural protein 4B
NS5A Non Structural protein 5A
NS5B Non Structural protein 5B
OCLN Occluding
PBS Phosphate buffer saline
PCR Polymerase chain reaction

List of acronyms
xvi
PFA Paraformaldehyde in PBS
RdRp RNA-dependent RNA polymerase
rpm Rotations per minute
RQA Recurrence Quantification Analysis
RT PCR Reverse transcriptase
SDS-PAGE Sodium dodecyl-polyacrlyamide gel electrophoresis
SEL Small extracellular loop
SR-BI Scavenger Receptor BI
SRP Signal recognition particle
TBEV Tick Borne encephalitis Virus
TE buffer Tris EDTA buffer
TMDs Transmembrane Domains
UTRs Untranslated Regions
VLDL Very Low density lipoprotein
VLPs Virus like particles or Lipoviroparticles
X-gal Bromo-chloro-indolyl-galactopyranoside
μl micro litre

Chapter 1
1
INTRODUCTION Hepatitis C virus (HCV) is a major cause of liver cirrhosis and hepatocellular
carcinoma (HCC). Preventive modalities are absent and the current antiviral treatment is
limited by resistance, toxicity, and high costs (Zeisel et al., 2011). According to the latest
report by Centre of Disease Control (CDC), United States, approximately 200 million
individuals around the globe are suffering from this viral infection constituting roughly
3.3% of the total world population today (Wands et al., 2004). In Pakistan alone, about
10 million people are suffering from this horrendous infection, just about 6% of the total
inhabitants of the country (Raja et al., 2008; Idrees et al., 2008) and the number is still on
rise. Organ transplantation, transfusion of blood or blood products and insecure salutary
injections are the risk factors for HCV transmission. Injectable drug user and
nosocomialy exposed people to infected blood are the high risk population.
According to recent report by Alter et al. (2007) needle stick injury from HCV
positive blood is about 0 to 10 % while rate of prenatal transmission from an infected
mother is 4-7% and occurs only when mother serum is HCV RNA positive at the time of
delivery and the risk increases 4-5 fold when mother is HIV co infected.
Majority of HCV infection leads to persistent chronic infection and only in 10-40
% cases the acute infection is resolved spontaneously. Chronic HCV infection is defined
by elevated aminotransferase level and detection of HCV RNA in the serum. Most of the
time chronic infection remain asymptomatic over a long period of time, however,
sustained chronic infection is distinguish by inflammatory lesions along with intrahepatic
lipid accretion termed as steatosis. Futher disease succession leads to fibrosis progressing
into cirrhosis and terminating into HCC.

Chapter 1
2
Development of HCC has shown a sharp increase in the recent years and is
mostly ascribed to HCV infection (Zoulim et al., 2003; Moradpour et al., 2007). HCV is
hepatotropic and only human and chimpanzees is the natural host.
HCV genome primarily replicates in hepatocytes, other cells such as peripheral
blood mononuclear cells (Cribier et al., 1995), billary cells (loriot et al., 1999) and
enterocytes (Deforges et al., 2004) are also reported to be permissive, though to lower
extent. Huh-7 hepatoma cells and their derivatives are more commonly implicated in
HCV study. Several surrogate forms of HCV have been developed to investigate various
aspects of HCV life cycle. System employed for early studies included production of
HCV like particles (HCV-LP), pseudotype particle (HCVpp) and sub genomic replicon
system. HCV-LP was first reported by Baumert et al. (1998) using recombinant
baculovirus expressing HCV structural proteins of genotype 1b (HCV J strain). Author
demonstrated that HCV structural proteins E1E2 and Core were sufficient to assemble
virus particles presumably from ER while the HCV RNA was selectively incorporated in
the particles. Although these particles were non infectious, HCV-LPs were implicated to
study the signal peptide peptidase processing of Core protein, ER interaction with the
structural proteins, and the role of structural proteins in HCV particle assembly and
morphogenesis (Blanchard et al., 2003b; Ait-Goughoulte et al., 2006; Hourioux et al.,
2007).
Later Bartosh et al. (2003) assembled HCV full length E1E2 glycoproteins on to
retroviral Core proteins from mouse leukemia virus (MLV) and used them to generate
pseudotype particles by cotransfection of 293T cells with three vectors containing HCV
E1E2 glycoproteins, MLV gag-pol Core protein and packaging component from MLV

Chapter 1
3
genome encoding green fluorescent protein (GFP) as a marker. These particles were not
only retained in ER but were also secreted and found to be infectious particle mimicking
the greater homology to HCV, the infectivity of these particles however was different for
different genotypes, the mechanism of which is still not clear (Lavillette et al., 2005).
HCVpp system has been proved to be a great help to understand several aspects of HCV
infection. HCVpp system was introduction must be in present tense intensively employed
to investigate various cellular receptors that interact with envelop proteins. Using this
system E2 has been shown to bind members of the cellular receptor complex, including
CD81 and SR-BI, LDL and members of the tight junction proteins, where as HCV E1 is
proposed to mediate fusion. In E2 the Huh-7 HCVpp model has also been employed to
evaluate the neutralizing activity of the antibodies raised in the serum of chronically
infected Patients (Laville et al., 2005). Blanchard et al. (2006) used HCVpp system to
study cellular mechanism of HCV endocytosis and suggested that HCV employ a
clathrine coated vesicles system to gain entry into susceptible cells. The list of
discoveries contributed by HCVpp system is quite long and is thoroughly reviewed in
literature section. HCVpp system has also been employed to test small molecule
inhibitors (Baldick et al., 2010).
Since HCV only cause disease condition in human and chimpanzee, small animal
model for HCV investigation has been major obstacle in the past and for long time the
absence of an appropriate cell culture system have hampered the progress of HCV study.
The lack of virus particle production by full length replicon system was perhaps due to
the adaptive mutations required for the enhanced viral RNA production which on the
other hand were deleterious for the virus particle assembly and release. Kato et al.

Chapter 1
4
(2003) reported JFH1 replicon from genotype 2a that exhibited unusually high rate of
RNA replication in Huh-7 cells, 20 times higher than the Con1 (genotype 1a), without
requiring any adaptive mutations.
Several groups working on full length genome JFH1 (2a) or intragenomic or
intergenomic chimera including structural or non structural proteins between J6
(2a)/JFH1, S52(3a)/JFH1 or H77(1a)/ JFH1 observed that transfection in Huh-7-derived
cells, particularly the Huh-7.5 clone, results in HCV particles (HCVcc) which were
secreted in the extracellular medium (Lindenbach et al., 2005; Zhong et al., 2005; Yi et
al.,2006; Pietschmann et al., 2006; Rouille et al., 2006; Wakita et al.,2006; Gottwein et
al., 2007). Results from various chimeras revealed that structural proteins have a major
impact on kinetic and efficiency of virus assembly and release.
HCV exist as highly heterogenic virus and because of heterogeneity it has been
divided into six major genotypes and several sub types. It exists as quasi species even in
an infected individual (Xavier et al., 1999). Genotype 2a and 1a are more common in the
west (McOmish et al., 1994), most of the HCV research is based on HCV model system
(HCVcc or HCVpp) representing either 1a or 2a genotype. There is very little data
available on genotype 3a the most dominating genotype in Pakistan (Idrees et al., 2008).
Keeping in mind the geographical heterogeneity of the HCV, the current research was
aimed to generate a 3a genotype based viral entry model system for the local isolate,
especially for the screening of putative entry inhibitors and for the development of
vaccine based on structural proteins. Unless there is HCV model system available for the
local isolates any inhibitor or anti viral targets natural or synthetic, studied will present
false data. Thus there is an intense need to develop HCV model system from local isolate

Chapter 1
5
before stepping into the antiviral research for Pakistani population. Keeping the fact in
mind the present study was undertaken and the specific aims of the study are listed
below;
Specific Aims:
To clone and sequence HCV structural genes from HCV 3a local isolates.
Identify the important variation in structural region of the local isolates.
To develop and characterize HCV entry model (HCVpp).
To develop and characterize HCV chimera of Core- NS2 region (HCVcc) for the
Pakistani isolates.
Development of HCV entry model and practical HCV cell culture system is
mandatory for the study of anti viral targets against local viral population and for the
initial screening of any putative vaccine specifically for 3a genotype since 50 % of the
HCV infected population have genotype 3a in Pakistan. We cannot progress in HCV
research unless we have our own model system.

Chapter 2
6
LITERATURE REVIEW
Hepatitis C virus history dates back to 1975. The only known hepatitis viruses at
that time were HAV (hepatitis A virus) and HBV, and were diagnosed by serological
tests. Regardless of HAV and HBV screening, about 10% of blood transfusion was still
resulting in hepatitis (Feinstone et al., 1975) indicating the existence of a yet unknown
hepatitis virus (Alter et al.,, 1975; Feinstone et al., 1975). Filtration experiments and
sensitivity to organic solvents suggested that the so-called non-A non-B hepatitis
(NANBH) virus was small and enveloped (Bradley et al., 1983; Bradley et al., 1985;
Feinstone et al., 1983; He et al., 1987). Later was given name as NANBH by Michael
Houghton’s lab at Chiron. In 1978 a chimpanzee model was available for NANBH
hepatitis virus. As the molecular biology techniques were getting refined at that period,
attempts were made to isolate virus using molecular cloning techniques. Series of
successful experimentation led to the isolation of first HCV clone from infected
chimpanzee plasma using cDNA libraries, several hundred clones were screened before
Choo et al. (1989) could get his first clone (Fig. 2.1).

Chapter 2
7
Figure 2.1: HCV discovery, Choo et al (1989) isolated the first HCV clone by screening a cDNA library obtained from highly infectious chimpanzee plasma, and packaged into λ phages, with serum from a chronic NANBH patient, as a potential source of antibodies.

Chapter 2
8
2.1 CLASSIFICATION AND GENETIC VARIABILITY
HCV is the exclusive member of the hepacivirus genus belongs to the
Flaviviridae family (Choo et al., 1991). It differs in its biological and serological
properties from flaviviruses and pestiviruses, the other two genera of the Flaviviridae,
however at the same time shares with them a great deal of similarity in terms of virion
morphology, genome organization and in its replication strategy. It also show similarities
in mode of translation and the glycosylation pattern of its envelope glycoproteins and
seems to be a close relative of pestiviruses than flaviviruses.
HCV is an RNA virus and encodes its own RNA-dependent RNA polymerase
(RdRp) for genome replication. HCV RdRp is error-prone like all other RdRp with an
error rate up to 10-3 substitutions per nucleotide site in vitro (Bull et al., 2010)). Its lack
of proof-reading activity leads to generation of a multitude of variants, accompanied by
high levels of virus replication (around 1012 genome equivalents secreted per day
(Neumann et al., 1998)). All these facts led to much accelerated evolution of HCV
isolates consequential development into 7 major genotypes and several subtypes (Murphy
et al., 2007; Simmonds et al., 2005).
HCV genotypes show up to 70% similarity over the whole viral genome
(Simmonds et al., 1993) and posses a significant pattern of geographical distribution (Fig
2.2; WHO, 2009). These genotypes also differ in their response to treatment as well as
possible disease outcomes (Zein, 2000). This virus variability is so momentous that it
results in HCV circulating in its host as a constantly evolving quasi-species, giving the

Chapter 2
9
Figure2.2: Global distribution of HCV genome. Adapted from WHO (2009).
virus an inimitable opportunity to escape the adaptive immune responses and the current
antiviral treatments and thus pose a great challenge to vaccine development.
2.2 HCV VIRION ASSEMBLY AND RELEASE
HCV is a positive sense, single stranded RNA virus with a genome size of 9.5Kb
The RNA encodes for a polyprotein of approximately 3000 amino acid ,translated from
an internal ribosome entry site (IRES) which is the most conserved region of the genome
(Lindenbach et al., 2007). The polyprotein is cleaved off by the action of host and viral
protéases. Host protéases cleave off capsid, envelope glycoproteins E1and E2 and an ion
chennol protein P7,whereas the viral cystein protease (NS2/NS3) and serine protease,
comprised of NS3and NS4A, cleave off to release the non structural proteins NS2, NS3,
NS4A, NS4B, NS5A and NS5B which are essentially required for viral replication as
shown in figure 2.3 (Suzuki et al., 2000).

Chapter 2
10
Figure2.3: HCV polyprotein processing and topology.a. Structure of the viral genome, including the long open reading frame encoding structural and non-structural proteins, surrounded by 5' and 3' UTRs. The polyprotein processing scheme is shown below. Closed circles, signal peptidase cleavage sites; open circle, signal peptide peptidase cleavage site; open arrow, NS2-3 cleavage site; closed arrow, NS3-4A cleavage sites. b. Topology of HCV proteins with respect to the ER membrane after polyprotein cleavage. Membrane anchors represented with hatched pattern correspond to a post-translational membrane insertion while others are co-translationally inserted in the ER membrane. Scissors refer to the signal peptide peptidase cleavage of Core precursor. The topology shown for NS4B and NS2 is still matter of discussion. Note that the topology for E1 and E2 depicted here is only transient. Adapted from Lindenbach & Rice (2005).

Chapter 2
11
Structural proteins are the composite part of the virion, composed of complex
between genomic RNA and Core protein surrounded by an envelope of E1and E2,
heterodimerized and embedded in host derived membrane. Ectodomains of E1and E2 are
higly glycosylated and are responsible for viral attachment and entry into susceptible host
cells (Lavie et al., 2007). Core oligomerization is reported to be facilitated by 72-91
amino acids of the primary protein (Nakai et al., 2006).
Assembly of the virus is initiated by oligomerization of the capsid protein and its
interaction with genomic RNA. This interaction involves domain l and lll of the Core
protein with nucleotides 24-41 of the viral positive sense RNA (Tanaka et al., 2000).
Once nucleocapsid is formed it interact with E1and E2 heterodimer and is secreted by
budding through the plasma membrane so as some host membrane constituents are also
incorporated into the virus membrane (Ezelle et al., 2002; Murakami et al., 2006).
Current study by Masaki et al. (2008) has also shown the role of NS5A in regulation of
early phase of virus assembly and the C terminal serine clusters are reported to play key
role through interacting with the Core.
Recent advancements in the field of HCV assembly have revealed that virus
particle assemble in vesicles enriched in ApoB and MTT, a microsomal triglyceride
transfer protein, and the viral secretion is dependent on the expression of ApoB and
VLDL assembly in the ER (Masaki et al., 2008). These reports are in accordance with the
use of earlier reported lipoprotein receptors LDLR and SR-B1 as cellular entry sites.
Based on all these advancements in the research it seems likely that HCV uses lipoprotein
/cholesterol export system to escape from hepatocytes and it is further confirmed by a
report from Nahmias et al. (2008).

Chapter 2
12
Figure 2.4: The various steps of HCV entry in hepatocytes (Adopted from Gondeau et al., 2009)
The size of the virion is about 50 nm (Wakita et al., 2005) and is often isolated
with diverse densities ranging from < 1.06 to 1.30 g/ml. HCV virion form complex with
low and very low density lipoproteins including LDL, VLDL, apoB100, apoE, apoCII
and apoCIII, all of them are rich in triglycerides (Andre et al., 2005; Diaz et al., 2006).
These complexes are known as lipoviroparticles or LVPs and about 40% of plasma
derived HCV RNA is f coupled to these triglyceride loaded LVPs (Gondeau et al., 2009).
There are several reports detecting HCV RNA in hepatocytes from liver biopsies of
infected patients however it is well established that HCV also replicates in dendritic cells
and other cells of lymphoid origin (Shimizu et al., 1998; Pachiadakis et al., 2005).
2.3 HCV CORE IS A MULTIFUNCTIONAL PLEIOTROPIC
PROTEIN
The HCV Core protein is known for its pleotropic functions. The primary
function of the Core protein is to form the capsid shell that abode and protect the HCV

Chapter 2
13
Figure 2.5: Schematic representation of the assembly and release of HCV. Red blocks mark potential drug targets in the process. (Adapted from Tews et al., 2010)
genomic RNA as the virus enter from one cell to another or from one person to another.
In addition to this basic function HCV Core protein also modulates diverse host pathways
by interacting with an array of cellular factors.
Core plays a vital role in recruitment of HCV replicase proteins and lipid droplet
mobilization (Duvignaud et al., 2008, Barba et al., 1997). In addition Core protein is the
key player for nucleocapsid formation, assembly and release of viral particles from
infected cells (Shavinskay et al., 2007, Penin et al., 2004). The progression of events
leading to Core-orchestrated HCV particle assemblage is depicted in figure 5.
After translation the poly protein is directed to ER by the signal sequence located
at the C terminus of the Core protein adjacent to E1 structural protein. Two succeeding
cleavages, first by a cellular signal peptidase (Santolini et al., 1994) and the other by a
cellular signal peptide peptidase (Bartosch et al., 2003) result in release of mature,

Chapter 2
14
probably dimerized or oligomerized Core, to the surface of lipid droplets (LD) ( Ai et al.,
2009). Attached to the LD Core recruits other components of non-structural HCV
proteins from the ER viz; NS3 (Mancini et al., 2009), NS5A (Zeisel et al., 2007;
Bankwitz et al., 2010), NS5B and possibly p7 and NS2 (Jones et al., 2007, Yi et al.,
2007), which together constitute the replicas complex, responsible for RNA replication
(Fig 2.5). Core also interacts with number of cellular factors especially those involved in
Very Low Density Lipoproteins (VLDL) biogenesis particularly ApoB, ApoE and
Microsomal Transfer Protein (Popescu and Dubuisson, 2009; Targett-Adams, 2010).
Newly synthesized HCV RNA is finally encapcidated by Core oligomers and the
nucleocapsid formed is associated into lipid-encapsulated particles, together with E1 and
E2 glycoproteins (Fig 2.5) protruding from the surface. The exact sequence of events is
still not finalized.
Replication occurs at dedicated ER-derived membranes. Core and NS5A interact
with lipid droplets and recruit the other non-structural proteins, and with them assemble
the replication complex. Core and genomic RNA interact and oligomerize, and must
interact with the envelope glycoproteins to form the viral particle. The viral particle
interacts with the VLDL. ApoB interacts with triglycerides in an MTP-dependent manner
to form a pre-VLDL, which will accumulate more lipids and ApoE in an MTP dependent
process. Both the VLDL and the virus continue to undergo maturation during their
passage through the secretion pathway (Popescu, and Dubuisson, 2009).
Number of amino-acid residues in Core is implicated to play an essential role in
assembly and release of the viral particle (Kim et al., 2006). Various parts of the Core
are depicted to play different roles and are divided into 3 essential domains (Fig .2.6). At

Chapter 2
15
Figure 2.6: Structural and Functional domains of the HCV Core protein.(Adapted from Arthur et al., 2010).
the N-terminal the domain 1called “D1” is up to 117 residue is rich in basic amino acids
binds to and promotes dimerization of the viral RNA (Cristofari et al., 2004) resulting in
the formation of viral nucleocapsid (Ivanyi-Nagy et al., 2006; Boulant et al., 2006).
Recent studies based on mutations in the D1 revealed that D1 is also involved in Core
envelopment by endosomal membranes (Ai et al., 2009). The rest of the two domains are
generally more hydrophobic and are reported to interact with the E1 and E2
glycoproteins, with the lipid droplets, and with several host factors involved in lipid
transport (Meunier et al., 2008; Cocquerel et al., 2006). The mature Core of 177 residue
is translocated from the endoplasmic reticulum to the of lipid droplet (Okamoto et al.,
2008).
The transmembrane C-terminal Core peptide remains in the ER, where it serves as
the E1 glycoprotein signal sequence (Hüssy et al., 1996; Yasui et al., 1998). Core plays
an essential role in organization of particle assembly and as a mediator of host-pathogen
interactions. Core also acts as an influential chaperone, and as an inducer of dimerization

Chapter 2
16
of newly-made HCV RNA (Selby et al., 1994). The first 68 residues of Core are critical
for capsid formation in a cell-free system (Klein et al., 2004) and Core 1-82 aa is
designated as the minimal domain for nucleocapsid assembly (Fromentin et al., 2007;
Majeau et al., 2004). Alanine scanning revealed numerous Core residues essential for
infectious virus production, including a significant number in the first 120 positions of
the protein (Slater-Handshy et al., 2004). The C-terminal region comprised between
residues 118 and 169 is required for proper folding of the whole Core protein and
preservation of interactions not observed with D1 (Cocquerel et al., 2006). For instance,
mutations of residues at position 130, 138 or 143 all abrogate association with LD’s and
virus production (Duvignaud et al., 2008; Majeau et al., 2009). Core’s D2 C-terminal half
thus harbours several sites that are essential for the protein’s roles in HCV’s life cycle,
e.g. for binding and attracting other HCV proteins to LD’s. In addition, Core’s C-terminal
is involved in mechanisms of viral persistence, oncogenesis (Moriya et al., 1998) and
steatosis (Moriya et al., 1997).
2.4 HCV GLYCOPROTEINS, HETRODIMERIZATION, ENTRY,
INFECTIVITY, AND FUSION
Envelop glycoproteins E1and E2 play an essential role in HCV life cycle by
mediating viral binding and entry to susceptible cells. Their cleavage from the HCV
encoded poly protein is mediated by host signal peptidases residing in the endoplasmic
reticulum (Lindenbach et al., 2001; Jusoh et al., 2010). Upon cleavage these proteins
assemble to form non covalent heterodimer and are retained in the ER in cell culture
system (Cocquerel et al., 1998 and 1999). This glycoprotein complex is reported to be the
viral component seen at the surface of viral particles, and are expected to be the ligand

Chapter 2
17
for a cellular receptor (Cocquerel et al., 2006). These two membrane proteins are
classified as class I transmembrane proteins and possess a large N-terminal ectodomain
facing the ER lumen and a C-terminal hydrophobic anchor domain containing
membrane-spanning segments for both E1 and E2 also called the transmembrane (TM)
domains.
The TM domains of HCV glycoproteins display some atypical features and have
shown to play important role in glycoprotein assembly and localization. These domains
are reported to be less than 30 amino acid residues long and have two hydrophobic
stretches which are interrupted by a short segment with one or two conserved charged
residues (Cocquerel et al., 2000). The N terminal amino acids of these domains are
proposed to be the 353 and 718 residue for E1 and E2, respectively (Cocquerel et al.,
1999, 1998 and 2000).
Although E1and E2 do not posses classical KDEL or KKXXER signal for ER
retention, it has been proved that TMDs of these protein act as ER retention signal and
their sub cellular localization is not the result of cis-Golgi retrieval pathway. When CD4
or CD8 chimera proteins containing the TM domain of E1 was expressed both of these
surface proteins were retained in ER. Likewise switching of the TMD of E2 with the
anchor sequence of CD4 was shown to be adequate for its export on the cell surface, and
a chimera protein with an ectodomain of CD4 fused t the TMD of E2 was localized in the
ER (Cocquerel et al., 1998; Chambers et al., 1993).
Transmembranal domain of glycoprotein of HCV plays multiple roles in
biogenesis of E1E2 and in their hetrodimerization (Yann et al., 2006). Op De Beeck et
al. (2000) have reported by using Alanine insertions that the central and N-terminal part

Chapter 2
18
of the TM domain of E1 is involved in hetrodimerization. Further investigations by
Ciczora, et al, (2007) identified the residues involved in E1E2 interaction during
hetrodimerization. They used tryptophan replacement scan and reported four residues
involved in heterodimerization viz Gly 354, Gly 358, Lys 370, and Asp 728. They
identified Gly 354 and Gly 358 as a part of oligomerization motif GXXXG. They also
analyzed their tryptophan mutants for HCVpp infectivity and found that despite of proper
hetrodimerization and incorporation into pseudotype particles some mutants had very low
infectivity. Invitro fusion assay showed that the reduce infectivity was due to decrease in
fusion function. Thus they proposed that TM domains of HCV glycoprotein may
contribute in fusion properties of these proteins. These results are further supported by
Jusoh et al., (2010) who performed molecular dynamics simulations for the
transmembrane segments of the E1–E2 involve in hetrodimerization. They reported a
second GXXXG motif in E1 at position 350 to 354 and were very highly conserved
among all genotypes. In the structural model of the E1–E2 heterodimer developed in their
study, however, the Gly350, Gly354, and Gly358 residues were not located at the helix
interfacial region. Therefore, they did not observe any possible interaction between the
GXXXG motif of E1 and the residues from the TM domain of E2. However, this does not
exclude the probability of GXXXG segments to heterodimerize at the ectodomain region
of the E2 glycoprotein. The E1 helix conformation agrees nicely with an experimental
structure of E1 solvated in TFE. Whereas the NMR analysis revealed an unwinding of the
N-terminal end of the E1 helix between Gly354 and Gly358, this region stayed intact in a
α-helical conformation during the heterodimer simulations. The emerging structural
model of the helix dimer shows the importance of the Lys370–Asp728 ion pair at the

Chapter 2
19
centre of the lipid bilayer for the formation of the E1–E2 heterodimer. The proposed
structural model of the helix dimer revealed the significance of the Lys370–Asp728 ion
pair at the centre of the lipid bilayer during formation of the E1–E2 heterodimer (Jusoh et
al., 2010).
Envelope protein 2 interact with the host cell surface receptor leading to receptor
mediated endocytosis, taking up the virus into the target cells. The next step, viral
envelops fusion with the endosomal membrane triggered by the low pH of the endosome.
Acidic environment of the endosome is proposed to induce conformational changes in the
HCV glycoprotein as reported in other members of the flaviviruses such as tick borne
encephalitis virus (Bressanelli et al., 2004) dengue virus (Hsu et al., 2003), and West
Nile virus (Mukhopadhyay et al., 2003). Though exact fusion peptide of HCV is not
determined HCV glycoproteins are classified in to class II fusion proteins on the basis of
presence of type II fusion protein in other members of the flaviviruses Voisset and
Dubuisson, 2004).
The E1 protein of HCV has two distinct hydrophobic regions, a central domain
proposed to act as fusion peptide (FP), and a C terminal domain (CT) consisting of two
segments, a pre-anchor and a TM region. Bruni et al. (2009) used two different
computational approaches, the first one based on the co-evolution paradigm of interacting
peptides which depend on the correlation between the distance matrices obtained by the
sequence alignment for FC and CT region. Second approach was based on a non-linear
signal analysis of a protein called Recurrence Quantification Analysis (RQA) which
allows a direct relationship of domains for the presence of common hydrophobicity
patterns, on which the physical interaction is based.

Chapter 2
20
Hsiao et al. (2009) investigated a fusion peptide like domain of E1 positioned at
264 to 290 aa in HCV. This domain has some conserved residue among all genotypes and
among other members of the Flaviviruses. It also showed similarity with
paramyxoviruses (Fig. 2.7). They used site directed mutagenesis and substituted Ala and
Asn for the conserved residues among the two groups. HCVpp based infectivity assay
showed that the reduction in infectivity was not due to cell surface expression or
hetrodimirization of the envelop protein or E2 interaction with CD81. They proposed this
reduced infectivity was due to alteration in cell fusion and further suggested that specific
residues are involved in cell fusion and entry process and perhaps the whole domain’s
structure is not the major determinant.
Alignment of the E1 fusion peptide-like motif of HCV with the predicted fusion
peptide sequences of the E protein of flaviviruses and fusion protein of paramyxoviruses.
The E1 putative fusion peptide sequence derived from various HCV genotypes contains
similarities to the predicted fusion peptide of flavivirus E glycoproteins which are boxed
(Fig 2.7). The spacing of Gly residues, as indicated by shading in the HCV putative
fusion peptide, is also similar to those within the fusion peptides of paramyxovirus F
proteins.
E1 is believed to mediate fusion while E2 has been shown to bind cellular
receptors including CD81. The role of CD81 in HCV entry is well established (Heo et al.,
2006). CD 81 is non glycosylated membrane bound protein, present on all nucleated
cells. It is characteristically tetraspanine with a large (LEL) and a small (SEL) extra
cellular loop. The LEL is established site of E2 interaction (Hemler, 2005) while the
critical residues involved in this binding are not well determined. There are three discrete

Chapter 2
21
Figure 2.7: Analysis of the HCV E1 putative fusion domain. Adapted from Li et al.
segments that interact with CD81 LEL domain. The first segment is from amino acid
474–492 and spans the second hyper variable domain (Yagnik et al., 2000; Roccasecca et
al., 2003).
.
Second segment is attributed to position 522-551and the last one at 612-619 3536.
Even though the first presumed CD81 binding segment (residues 474–482) falls in hyper
variable region II (HVR II), residues Y474 and D481 are very highly conserved among
various genotypes (Rothwangl et al., 2008) have targeted highly conserved residues
across several strains of HCV. They generated individual Alanine substitutions for the
conserved amino acids and challenged a susceptible cell line with HCV E1E2 HCVpp

Chapter 2
22
Figure 2.8: Conserved residues within the putative CD81 binding domains of E2. HCV strains from the Los Alamos HCV sequence database were aligned. Three regions previously implicated in CD81 binding were analyzed. Amino acids are numbered relative to the AUG start codon of the H77 strain. The hyper conserved (black rectangles) targeted (asterisk) residues for alanine substitution are indicated (Adopted from Rothwangl et al., 2008).
containing the individual mutations. They mainly focused on charged, hydrophobic
residues conserved in these regions. Most Alanine substitutions within the putative CD81
binding region 1 (aa 474–492) resulted in reduced HCVpp infectivity but retained soluble
CD81 binding competence, with proper E2 conformation, E1E2 interaction and
incorporation into HCVpp. Thus they recommended that proposed region 1 of E2 does
not mediate binding to CD81. On the other hand conformationally correct E2 mutants
(Y527 and W529) within the second putative CD81 binding region (aa 522–551) lose
binding of E2 to CD81-GST, signifying that region 2 is vital to CD81 binding. They
further reported that all conformationally correct mutants within the third putative CD81
binding region (aa 612–619), except L615A, were imperative for E2 binding to CD81-
GST. The third region is highly conserved across genotypes, emphasizing its importance
in mediating viral entry.

Chapter 2
23
Hepatitis C virus glycoproteins E2 is highly variable not among different
genotypes but within same genotype and even in the same patient infected with one
isolate. Hyper variable region 1 (HVR1) is 27-amino-acid-long segment located at the N
terminus of E2, has also been suggested to play a role in cell attachment (Penin et al.,
2001). HVR1 has been anticipated to modulate accessibility to CD81 or SR-BI. Deletion
of HVR1 increased binding to CD81 (Roccasecca et al., 2003) but abrogated binding to
SR-BI (Scarselli et al., 2002). HVR1 is employed as a target for anti-HCV neutralizing
antibodies (Farci, et al., 1996). The envelope glycoprotein E2 of HCV exhibits a great
deal of heterogeneity, highest being found in HVR1 and its heterogeneity in infected
individuals suggest strong immune pressure (Mondelli et al., 2003). HCV clone lacking
HVR1 was infectious in chimpanzees; this mutant virus was attenuated, signifying that
HVR1 plays a facilitating role in HCV infectivity (Forns et al., 2000). Although it shows
strong amino acid sequence variability, the chemicophysical properties and conformation
of HVR1 are highly conserved with a conserved basic stretch, with basic residues
occupying the specific sequence positions (Penine et al., 2000).
Hepatitis C virus pseudotype paticles infectivity generally increased with an
increase of basic residues in HVR1 although interaction with CD81 was not affected.
However, a shift in position of some charged residues results in altered HCVpp
infectivity and in the absence of basic residues, infectivity reduced to the same level as
for a mutant lacking HVR1. Deletion of HVR1 abridged the recognition of E2 by CD81-
LEL, signifying that some residues within HVR1 might be involved in binding to CD81
(Callens et al., 2005). Quasispecies dynamics based on HVR1 are indicative of outcomes
such as spontaneous viral clearance (Farci et al., 2000; Chen and Wang, 2005), response

Chapter 2
24
to interferon treatment (Farci et al., 2002; Gaudy et al., 2003; Abbate et al., 2004), and
HCV-associated liver histopathology (Honda et al., 1994; Gonzalez-Peralta et al., 1996).
Comprised of 9 amino acids positioned at 474-482 downstream to HVR1 is the second
hyper variable region of E2, HVR2 (Kato et al., 1992). Structure predictions of HVR2 are
indicative of its potential participation in cell surface receptor binding (Yagnik et al.,
2000). HVR3 extending from amino acid 431–466 is further sub divided into two parts
HVR3a (aa position 431–449) and HVR3b (aa position 450–466) (Troesch et al., 2006).
A region corresponding to HVR3a is suspected to contain a fusion peptide-like
domain (amino acid positions 436–443) GWLAGLFY, homology found in TBEV,
dengue virus, and other flaviviruses are expected to have a bipartite functions in binding
CD81 and mediating fusion of the viral and host cell membranes (Drummer et al., 2005).
Troesch et al., (2006) studied antigenicity, secondary structure, accessibility predictions
and three-dimensional molecular modelling of HVR3. They proposed its sub domain
organization and suggested that a significant part of HVR3, including the C-terminal of
HVR3a, are exposed at the surface of E2. They further anticipated HVR3 had a role in
the process of binding with host cell receptors and viral entry.
Detailed examination of the amino acid sequence composition of HVR3a revealed
a domain comprised of relatively conserved hydrophobic residues surrounded by
relatively hydrophilic or neutral amino acids. Thus, in spite of the relative amino acid
sequence variability observed within this region, its overall hydropathic character appears
highly conserved, both in inter host and intra host comparisons (Fig. 2.9). In particular,
amino acid positions 433, 437, 438, 439, 441, 442 443, and 447 were almost exclusively
held by hydrophobic residues. Positions 432, 435, and 436 were occupied by neutral

Chapter 2
25
Figure 2.9: Amino acid sequence diversity in HCV E1–E2 envelope glycoproteins. Variability at each amino acid position was computed using a data set comprised of 391 E1–E2 sequences derived from third trimester samples from all study subjects and the Entropy-ONE Web tool. Amino acid positions correspond to the M62321 reference sequence (Choo et al., 1989). Boundaries of E1, E2, HVR1, and HVR2 were set in accordance with previous reports (Weiner et al., 1991; Kato et al., 1992). Adapted from Troesch et al. (2006).
residues, with a fully invariant glycine at position 436. The remaining positions were held
by hydrophilic (431, 434, 445, 446, and 448) or variable amino acids (440, 444, and 449)
the conservation of hydrophobic amino acids and charged residues suggests that the
overall conformation of HVR3a is likewise conserved among HCV variants comprised
within the quasispecies (Troesch et al., 2006).

Chapter 2
26
Humphreys et al., (2009) have identified two smaller variable regions that specifically
found in genotype 3a only, HVR495 extending from position 495 to 502 and HVR 575
extending from position 575 to 584 in E2.
N-linked glycosylation of viral proteins are essential for a variety of biological
and immunological functions (Helenius, 1994). Glycosylation of proteins assist in the
formation and maintenance of native protein conformation masks the neutralization
epitopes and protect the protein from proteolytic degradation (Ellgaard et al., 1999;
Means and Desrosiers, 2000).
Hepatitis C virus envelope proteins are highly glycosylated. The ectodomain of
E1 is subjected to 4 or 5 potential glycosylation sites and E2 have up to 11 sites subjected
to N-linked glycosylation (Beyene et al., 2004; Goffard et al., 2005). Some of these
glycans have been known to play a critical role in protein folding and in HCV entry
(Choukhi et al., 1998). Extensive glycosylation suggests that these glycans may also be
involved in modulating the immunogenicity of HCV envelope glycoproteins and hamper
the binding of certain antibodies to their epitops on the virion surface, as is seen in case
of human immunodeficiency virus (HIV) (Pikora et al., 2004; Wei et al., 2003; Wyatt et
al., 1998).
Characterization of various mutants for glycosylation sites revealed their
significant role in HCV infectivity and protein folding. Goffard et al. (2005) have
grouped such mutants in three phenotypes. The first cluster (E1N3, E2N3, E2N5, E2N6,
E2N7, and E2N9), showed infectivities of the mutants similar to that of the wild type.
The second group (E1N1, E1N2, E1N4, E2N1, and E2N11) cluster harbor mutants with
infectivities reduced to <50% than the wild type and third cluster (E2N2, E2N4, E2N8,

Chapter 2
27
and E2N10) was with mutants that had almost lost infectivity. Immunoprecipitation with
conformation-sensitive antibodies lead to the conclusion that E2N8 and E2N10 lose
infectivity due incorporation failure of the E1E2 heterodimer into HCVpp and their
mutation resulted in misfolding of the heterodimer. Helle et al., (2007) used similar
approach for individual glycosylation sites to evaluate neutralization sensitivity of the
mutants to patient sera derived antibodies and for anti-E2 monoclonal antibodies. There
data showed that N-linked glycans of E1 are not involved in masking of neutralizing
epitops, whereas at least three glycans on E2 viz., E2N1, E2N6, and E2N11 not only
reduced the sensitivity of HCVpp to neutralizing antibody it also significantly hampered
access of CD81 to its E2 binding site. They advocate that glycans E2N1, E2N6, and
E2N11 are close to the binding site of CD81 and play a major role in evasion of HCV
from the humoral immune response (Fig 2.10).
DC-SIGN (dendritic cell-specific intercellular adhesion molecule CD209) and L-SIGN
(DC-SIGNR; liver and lymph node-specific; CD209L), are involved in arresting several
viruses, including HIV type 1 (HIV-1) (Geijtenbeek et al., 2000), Ebola virus (Alvarez et
al., 2002), cytomegalovirus (Halary et al., 2002), and dengue virus (Tassaneetrithep et
al., 2003). Both L-SIGN and DC-SIGN have an extracellular C-terminal region with
a calcium-dependent carbohydrate recognition domain (CRD) and a membrane-proximal
heptad-repeat domain essential for oligomerization (38–41). Viral particle attachment is
mediated by CRD and promotes infection of target cells (Engering et al., 2002; Gardner
et al., 2003).

Chapter 2
28
Figure 2.10: Conservation of sequences close to glycosylation sites E2N1, E2N6, and E2N11. The amino acid repertoires of E2 sequence segments including glycosylation sites N1 (417), N6 (532), and N11 (645), deduced from the multiple alignment sequences from the various patients infected with genotype 1a. Sequences were extracted from the euHCVdb database. Adapted from Helle at al. (2007).
Hepatic sinusoidal endothelial cells can distinctively internalize and process a
varied range of antigens. DC-SIGNR, a type 2 C-type lectin expressed on liver sinusoids,
has been shown to bind with high affinity to hepatitis C virus (HCV) E2 glycoprotein and
may assist infection of hepatocytes. DC-SIGN , a closely related homologue reported to
be expressed only on dendritic cells and a subset of macrophages and has similar binding
affinity to HCV E2 glycoprotein, is expected to transmit HCV to hepatocytes as well as
to a subset of B and or T lymphocytes (Cormier et al., 2004). Both receptors function as
adhesion and antigen presentation molecules. Lai et al., (2006) have reported a distinct

Chapter 2
29
pattern of DC-SIGNR and DC-SIGN expression in human liver tissue and showed that
both C-type lectins are expressed on sinusoidal endothelial cells. They showed HCV E2
binding is supported by these lectins on primary sinusoidal cells but they were unable to
support HCV entry. They strongly advocate for a model where DC-SIGN and DC-
SIGNR on sinusoidal endothelium provide a high affinity binding for capturing of
circulating HCV within the liver sinusoids and a allowing successive transfer of the
captured virus to underlying hepatocytes, an analogous approach used by dendritic cells
DC-SIGN presentation for human immunodeficiency virus.
Hepatitis C virus RNA have been shown to replicate in murine cells although
these cells are non infectious to HCVpp and cell culture derived virus particles. Ploss et
al., (2009) have demonstrated human occludin (OCLN) is a crucial HCV cell entry factor
that renders murine cells infectable with HCVpp. They further investigated that OCLN is
required for the HCV-susceptibility of human cells as well, because it’s over expression
in non permissive cells distinctively increase HCVpp uptake, while its silencing in
infectable cells impaired both HCVpp and HCVcc infection. They also demonstrated that
OCLN and CD81 must be of human origin for efficient infection. OCLN is another
tetraspanine present in the tight junction complex of polarized epithelial cells, where it
perhaps regulates paracellular permeability and cell adhesion (Chiba et al., 2008).
Expression of human SR-BI, CD81, CLDN1 and OCLN in non permissive NIH3T3 cells
improved HCVpp infectivity by approximately 120-fold (Rothwangl et al., 2008).
SR-B1 is a glycoprotein 509 amino acid long, anchored to plasma membrane at
both termini with a large extra cellular loop and a short extension into the cytoplasm
(Krieger, 2001). SR-B1 plays a crucial role in bidirectional cholesterol transport and can

Chapter 2
30
bind to high-density lipoprotein HDL and LDL. It also binds to modified lipoproteins
such as oxidized LDL. SR-B1 is highly expressed on steroidogenic tissue and in liver
(Pietschman et al., 2006).
SR-B1 as a putative HCV entry receptor has been investigated using HCVcc
system, results of the investigation demonstrate that antibodies against extra cellular loop
of the SR-B1 inhibit viral infectivity in dose dependent manner and down regulation of
SR-B1 by si-RNA significantly reduce hepatoma cell infectivity to cell culture derived
HCV. It has been further proposed that SR-B1 is involve at post binding perhaps at entry
level somewhere parallel to CD81 interaction. Although addition of HDL enhances HCV
infectivity however inhibition by SR-B1 antibodies and si RNA was shown be
independent of the HDL (Zeisel et al., 2007).
In Patient serum HCV particles have been shown to be associated with
lipoproteins, suggesting an indirect involvement of virus with lipoprotein receptors
(Andre et al., 2002; Burlone et al., 2009). There are recent reports showing HCVcc
association with VLDL containing ApoB and ApoE. Various factors influencing
lipoprotein trafficking are reported to have pleiotropic effects on HCV infectivity.
Antibodies to ApoB have antiviral activity against HCV (Dreux et al., 2007) where as
Lipoprotein lipase (LPL) not only increase trafficking of triglyceride-rich lipoproteins
into the liver it also enhances neutralized HCV infection (Andreo et al., 2007). These
observations leads to a suggestion that HCV uptake by the target cell is perhaps linked
with the normal hepatocellular processes of lipid transport and factors that interfere with
this regular process may influence HCV infection as well. Recent studies on cholesterol
lowering drugs of the statin family have shown a synergistic effect with current

Chapter 2
31
interferon/ribavirin combination therapies (Delang et al., 2010). Statins are reported to
inhibit HCV replication in vitro at high doses (Ikeda et al., 2006), they may also
influence cholesterol metabolism and lipoprotein trafficking via SR-BI.
CD81 is the most established and thoroughly characterized HCV entry receptor
(Heo et al., 2006). CD81 is a non-glycosylated, membrane bound, tetraspanine
possessing a small (SEL) and large (LEL) extracellular loop (Hemler, 2005). It is present
on almost all nucleated cells. A definitive role for CD81 in HCV infection has been
established using the retroviralbased HCVpp and in vitro HCV infectious clone systems
(Lindenbach et al., 2006). Critical role of CD81 in HCV entry was confirmed using
HepG2 and HH29 CD81-negative hepatoma cells, they were able to support HCVpp
entry after exogenous CD81 expression while silencing of CD81 expression in hepatoma
cells with siRNAs inhibited HCV infection (McKeating et al., 2004).
The LEL of CD81 has been recognized as the binding target of HCV E2 and vital
amino acids for maintaining this interaction are well established (Higginbottom et al.,
2000; Kery et al., 2010 see Fig.2.11). On the other hand critical residues of E2 involve in
this interaction are still under investigation and several putative CD81 binding sites of
HCV E2 have been recognized (Rothwangl et al., 2008). Anti-CD81 or soluble CD81
LEL antibodies are known to neutralize HCVpp and HCVcc infection (Zhang et al.,
2004). This neutralization seems to occur after viral attachment to the host cell,
signifying that CD81actually functions as a co-receptor for internalization and is not a
primary receptor (Flint et al., 2006).

Chapter 2
32
Figure 2.11: Tertiary structure of HCVE2 showing location of various domains, HVR are shown in brown color. CD81 interacting residues are encircled in dark blue. Whereas red encircled residue in domain 2 (yellow) are the putative fusion peptide in E2. Disulfide bridges are indicated with black bars and glycosylated asparagines are in green. Gray color at the C terminus marks the stem loop region. Adapted from Kerry et al. (2010).
Claudin-1 (CLDN1) a tight junction protein, was introduced to list of HCV
receptors in 2007 by Evanset et al. Although silencing of CLDN1 expression or
mutagenesis of the first extracellular loop of CLDN1 reduces HCV infection in hepatoma
cell yet to date, there is no experimental evidence of interaction between CLDN1 and the
HCV gps, which seems to be an essential requirement for the virus to bind its receptors in
a defined manner (Timpe and McKeating, 2008) have recently demonstrated that a

Chapter 2
33
Figure 2.12: Cellular receptors for hepatitis C virus. The transmembrane domains of CD81, scavenger receptor BI (SRBI) and claudin-1 (CLDN1) are depicted as cylinders. The cell membrane is shown in grey, above which are the extracellular domains. The CCG motif common to tetraspanins is shown as yellow and green circles. Adapted from Timpe and McKeating, (2008).
proportion of CLDN1 exist localized outside apical domains in hepatocytes and polarized
Caco-2 cells and was co-stained with CD81. Their results support independent existence
of co-receptor complexes other than TJs. Investigation involving non-polarized 293T
HEK and hepatoma cells revealed that anti-CD81 antibodies precipitate CLDN1 100 and
that fluorescence resonant energy transfer (FRET) was demonstrated between fluorescent
N-terminal tagged CLDN1 and CD81, representing homotypic CD81–CD81and
CLDN1–CLDN1 as well heterotypic (CLDN1–CD81) interactions 101 at non-junctional
areas of the plasma membrane (Fig. 2.12 and 2.13). Other members of the CLDN family,
CLDN6 and CLDN9 also support HCV entry (Meertens et al., 2008). However,
physiological relevance and expression levels within the liver are not clear and this area
is still vacant for exploration

Chapter 2
34
Figure 2.13: HCV first interact with HS and LDLR on the basolateral membrane surface of hepatocytes to allow concentration of the virion. Subsequently, interaction with other host factors such as SR-BI, CD81, CLDN1, and OCLN ultimately leads to viral internalization via clathrin mediated endocytosis. Fusion between viral and endosomal membranes is followed by release of the viral genome into the cytosol where translation and replication take place. HCV particles are then assembled and released from the host cell. Alternative route of viral entry is direct cell–cell transmission which is resistant to neutralizing antibodies. (Adapted from Zeisel et al., 2010)
2.5 P7 AN ION CHANNEL PROTEIN
P7 is the most small but intriguing intrinsic membrane protein of HCV. The p7
protein has a double membrane-spanning topology, with a short cytoplasmic loop and
both extremities facing the ER lumen (Carrere-Kremer et al.,, 2002). P7 belongs to the
viroporin family of proteins; it is able to oligomerise in vitro and form an hexameric 42-
kDa cation-selective ion channel (Luik et al.,, 2009), which constitutes a potential

Chapter 2
35
antiviral target .Many studies have revealed that p7 is indeed, critical for the release of
infectious virions in cell culture system (Jones et al., 2007; Steinmann et al., 2007) and in
the chimpanzee model (Sakai et al., 2003).Although not involved in RNA replication but
is required for late steps of virus assembly (Steinmann et al., 2007). A likely role in entry
has been evoked (Griffin et al., 2008) but demonstration of p7 incorporation in virions is
missing.
2.6 NS2 GENE AND PROTEIN
The non structural proteins ensure RNA replication and orchestrate viral
assembly.NS2 is transmembranal protein. The gene starts at 2769 nt of the genome and
extends till 3420 nt making the gene length of 651 bp (coordinates based on NCBI entry
H77 (accession No. NC_004102)) resulting in a protein product of 217 amino acids and
makes a 23 kDa protein, positioned between amino acid 810 to 1027of the poly protein.
The amino terminus of the protein is highly hydrophobic and is responsible for the
integration of the end product into the membrane of endoplasmic reticulum (ER). NS2 is
a non glycosylated transmembrane protein that is anchored to the ER (Yamaga et al.,
2002; Santolini et al., 1995).
The amino terminus of NS2 is dissociated from p7 by a host cell signal peptidase
in a membrane dependent manner. Research carried out to study p7/NS2 cleavage in cell
free environment showed that this cleavage was dependent upon addition of microsomal
membranes (Lin et al., 1994). Studies on the fusion of p7 with other genes have shown
that cleavage fails at the carboxy terminus of p7 if NS2 is not directly linked to it
(Carrere-Kremer et al., 2004) revealing that the signal for dissociation of p7 and NS2 lies
on both the genes. The c terminus of the protein is split from NS3 by the action of NS2/3

Chapter 2
36
autoprotease (Schregel et al., 2009). The vital players of this event includes the HCV
encoded protease, located between amino acid 827 and 1207 of the polyprotein, the
NS2/3 cleavage site, and the serine protease domain of NS3 (Grakoui et al., 1993). NS2,
soon after its dissociation from NS3, is inserted into the ER membrane through its amino
terminus hydrophobic domain (Santolini et al., 1995; Yamaga and Ou, 2002; Pallaoro et
al., 2001).
Most cellular proteins are incorporated into the plasma membrane or membrane
of any other organelle through the action of signal recognition particle (SRP) and signal
recognition particle receptor targeting. NS2 may employ the same machinery for its own
integration into the ER membrane. It has been clarified that the carboxy terminus of NS2
lies in the lumen of ER while the amino terminus is exposed in the cytosol (Santolini et
al., 1995). P7 also has a membrane integration signal and it was initially hypothesized
that NS2 incorporation into the membrane is P7 dependent but further analysis by various
groups has shown that this is not the case and that NS2 incorporation into the membrane
is P7 independent (Yamaga and Ou, 2002; Santolini et al., 1995). The mechanism that
ensures the incorporation of NS2 into the ER membrane and the topology acquired by the
protein after the insertion has been controversial. In the last decade research led to facts
that the amino terminal region of NS2 is highly hydrophobic (Yamaga and Ou, 2002;
Pallaoro et al., 2001) and might contain one to three TM segments followed by a globular
cytosolic protease domain (Jirasko et al., 2008; Lorenz et al., 2006). Most recent report
suggested that NS2 spans the membrane through three transmembrane alpha helices
(Lemon et al., 2010).

Chapter 2
37
Most sub genomic replicon systems that have been developed to date, to
investigate the replication process of HCV do not necessarily contain NS2and it seems to
be an indispensible protein as far as HCV replication is concern. NS2 has been found to
interact with all other HCV NS proteins in vitro, as studied by cell based co-localization
and co-immunoprecipitation assays (Dimitrova et al., 2003; Hijikata et al., 1993).
Therefore, although not required for RNA replication, the possible presence of NS2 in
this complex as an accessory protein is plausible and warrants further investigation.
Recent investigations have shown that HCV NS2 protein up regulated HCV internal
ribosomal entry site (IRES) dependent translation in a specific and dose-dependent
manner in Huh7 cells but not in HeLa and HepG2 cells (She et al., 2008). In the same
study, it was also suggested that NS2 protein inhibited NS5B RNA dependent RNA
polymerase activity in a dose-independent manner in all three cell lines. These findings
may suggest a novel mechanism by which HCV adjusts its replication and IRES
dependent translation to facilitate virus persistence.
For prediction of protein structure and function, one of the crucial steps is to
identify its partners. One of them is the viral protein NS5A. NS2 is involved in the
phosphorylation of this protein (Koch et al., 1999; Neddermann et al., 1999). The other is
NS3, after its cleavage from the precursor polyprotein; NS2 forms a biologically
important complex with NS3 which plays a significant role in the assembly of the virus
(Kiiver et al., 2006).
Among the cellular interacting partners of NS2, the first and foremost is human
chaperon protein Heat shock protein 90 (Hsp90), which aids the functioning of NS2/3
protease (Waxman et al., 2001). It is also known to interact with cellular pro-apoptotic

Chapter 2
38
protein Cell death-inducing DFFA-like effecter b, CIDE-B and block its functioning. As
the structure and function of NS2 has not yet been completely identified, the knowledge
regarding its interacting partners is still incomplete. Recent data has shed some light on
the interactions of NS2 with cellular protein cyclophilin A (Ciesek et al., 2009). Studies
have revealed that this protein, unlike all other known cellular interacting partners of
NS2, functions in the folding of NS2 and in turn is a crucial player in the functioning of
this protein.
2.7 HCV NEUTRALIZATION
A complete understanding of viral and host factors responsible for HCV clearance
or persistence during the various stages of infection is still incomplete. Research focused
on innate and cellular immune responses have revealed that a significant HCV inoculum
is competent enough to escape, subvert or outwit the host's defence system (Fournier et
al., 2007).
HCV-specific T-cell immunity play an important role in the control of HCV
infection (Rollier et al., 2004; Lechmann and Liang, 2000). Humoral immunity is shown
to play equally significant role in clearance of HCV during acute infection this aspect
however remains inadequately characterized. The E1 and E2 glycoproteins are
established to be the viral attachment proteins and thus are the focal targets for HCV-
neutralizing antibodies. A lot of effort is going on to identify protective epitops conserved
across different genotypes of HCV and pose foremost challenge in vaccine design. To
date several antibodies proficient in blocking E2 binding to cells or cell receptors have
been evaluated (Owsianka et al., 2001; Allander et al., 2000) and some of them have

Chapter 2
39
been found neutralizing for HCV entry in animal as well as in cellular models (Farci et
al., 1994).
Detection of neutralizing antibodies in human blood had been complicated until
recent development of reliable cell culture system. In vitro neutralization assay system
based on infectious retroviral pseudoparticles (HCVpp), harbouring HCV envelope
glycoproteins, and have inveterate that HCV-infected patient sera contain neutralizing
antibodies that can block HCVpp entry into susceptible cell lines (Shimizu et al., 1994;
Lavillette et al., 2005; Bartosch et al., 2003).
Various reports on neutralizing antibodies established the fact that, patients
capable of clearing HCV infection in acute phase has high titters of nAbs (Pestka et al.,
2007). Contrary to that chronic HCV infection is marked by complete or partial collapse
of nAbs either to neutralize transmitted virus at early stage or delayed induction of nAbs
in the late phase of infection (Meunier et al., 2005; Logvinoff et al., 2004). Cross-reactive
nAb responses is also reported to occur long after acute infection, resulting in an increase
in titter and extent to recognize various HCV genotypes (Von et al., 2007).
Anti-HCV-positive plasma contains neutralizing antibodies directed against
hepatitis C virus (HCV). These neutralizing antibodies dominated by IgG isotype. An
interesting report by Zhang et al. (2007) revealed a new aspect of hemmoral response
against HCV. They reported that a significant amount of HCV-specific Igs circulating in
patients’ plasma are ineffective in vivo. The mechanism for the poor effectiveness is
currently unknown; however they suggested that the presence of no neutralizing
antibodies in HCV specific Igs interferes with the function of neutralizing antibodies
leading to decline or blockage of their effect. In their study they identified two epitops

Chapter 2
40
downstream of the hyper variable region I within the glycoprotein E2 protein one at
amino acid residues 412–419 (epitope I) and the other at 434–446 (epitope II).Epitope I
was responsible for HCV neutralization awhile interaction of a non neutralizing antibody
to epitope II completely disrupted virus neutralization by epitope I specific antibodies.
This led to the suggestion that Huh-7 cells are unable to support HCV particle
assembly or release. Cell culture-adaptive changes were also found toxic or highly
deleterious for replication in chimpanzees (Pietschmann at al., 2oo6). They further
reported full-length HCV genomes replicated poorly but released HCV RNA and Core
protein into the cell culture medium without adaptive mutations (Holmes, 2008).

Chapter 3
41
MATERIAL AND METHODS
3.1 ENROLLMENT OF PATIENTS
For this study blood samples were collected from HCV infected patients. Patients
of age 24 and older who were HCV positive and negative for HBV and HIV were
enrolled in this study. HCV infection was confirmed by RT PCR and at the same time the
blood samples were tested for HBV surface antigen (HBsAg; DRG Germany) and (p24
core) using BKH ELISA kit (China).
3.2 SAMPLE COLLECTION AND STORAGE
Initially 22 patients were enrolled for the study. Blood sample of the patients were
collected in BD vacutainer collection tubes (Becton Dickenson, USA) and were shifted to
4°C. Serum was isolated from the blood after centrifugation at 12000 rpm for 2 minutes,
divided into 140 µl aliquots and was stored at -80°C for further use.
3.3 RNA ISOLATION
Serum was brought to room temperature prior to RNA extraction. Viral RNA was
isolated from 140 μl of serum using a Viral RNA Isolation Kit (Qiagen, USA), following
protocol as described in the manual. Briefly, 140 µl of serum was taken in a 1.5 ml
appendorf and was brought to room temperature. AVL buffer 560 µl along with 5.6 µl of
carrier RNA was added and vortexed for 15 seconds. The mixture was incubated at room
temperature for 10 minutes and centrifuged briefly. Chilled ethanol, 560 µl, was added
and was mixed for 15 seconds. Eventually 630 µl of the mixture was applied to the
column and centrifuged at 8000 rpm for one minute. Flow through was discarded and the

Chapter 3
42
rest of the mixture was applied to the column and centrifuged. The column was washed
with 500 µl of AW1 buffer followed by 500 µl of AW2 buffer and the column was
centrifuged at 14000 rpm for 3 minutes. Flow through was discarded.
Finally column was placed in new labelled microfuge tube. To elute RNA, 60 µl
of AVE (elusion buffer) was added and centrifuged at 8000 rpm for one minute. RNA
obtained was quantified using nanodrop spectrophotometer (Nanodrop, USA) and was
stored at -80 °C for further use.
3.4 GENOTYPING
NCVI diagnostic laboratory protocol was used for genotyping using Ohno et al.
(1997) method. 14 samples out of 22 were positive for HCV genotype 3a and were
selected for further research (Table 3.1).
3.5 PRIMER DESIGNING
For PCR amplification of the HCV Core-NS2 region, primers were designed by
retrieving sequences of the specific viral genes of genotype 3a from NCBI Nucleotide
database, especially NZL1 strain and HCV-K3A strains accession numbers NC-009824
and D28917K respectively. The sequence of the sense primers (F) contained restriction
endonuclease site EcoRI or NheI while reverse/antisense primers (R) were flanked by
restriction site of Not I or Hind III at 5ˈ end (Table 3.2).

Chapter 3
43
Table 3.1: HCV patient information enrolled in the current study
Sr no
Gender
Age
genotype
ALT
IU/ml
Viral Titer
Copies/ml
1- Male 39Y 56 4789163
2- Female 26Y 3a 210 671418
3- Female 40Y 3a 72 399155
4- Male 42Y 78 729081
5- Male 27Y 3a 78 729081
6- Female 28Y 3a 72 4653579
7- Female 45Y 3a 19 5492
8- Male 24Y 3a 20 64142
9- Male 37Y 97 991553
10- Male 45Y 3a 90 429681
6- Female 27Y 3a 14 3411
9- Male 47Y 3a 79 107593
13- Male 40Y 3a 76 1545678
14- Male 32Y 3a 99 1098730

Chapter 3
44
Table 3.2: Primers used for amplification and sequencing of core-NS2 region
Primer Sequence
Core-
F-
EcoR1
AAAGAATTCGCCACCATGCTAGAGTGGCGGAATACGTCTGGCC
Core-
R-Not1
CCCGCGGCCGCTTAACTGGCTGCTGGATGAATTAAG
E1- F-
EcoR1
AAAGAATTCGCCACCATGCTAGAGTGGCGGAATACGTCTGGCC
E1- R-
Not1
AAAGCGGCCGCCTATATGATGATTGCGACCTTGGCCCAGTGG
P7-R-
Not1
AAAGCGGCCGCTCACGGCGTTCAGCGTGACAAGGTTCTC
NS2-R-
Not1
AAAGCGGCCGCTCACCCCAGGTGATGACCTTGATTTCC
NS2 R-
IN-
HindIII
AGGAAGCTTCTGCGGCCCAATGTTGCATCGGC
COR
F-IN-
Nhe1
GGGGCTAGCACACTTCCTAAACCTCGAAGAAAAACCAAAAGAAACACC
P7 R -
HindIII
GGAAAGCTTTCACGGCGTTCAGCGTGACAAGGTTCTC

Chapter 3
45
3.6 cDNA SYNTHESIS
To generate an appropriate template for PCR amplification of Core to NS2 genes,
cDNA was generated using specific primer designed as complementary to conserved
region of the NS2 gene (NS2-R-Not I, table 3.2). An appropriate volume of RNA,
typically 8-10 μl was used for this purpose. RNA template was denatured in the presence
of 0.2-0.5 pmol of primer and 1 mM of dNTPs (Invitrogen, USA), in a final volume of 12
μl. The mixture was heated at 65°C for 5 minutes and then was rapidly cooled on ice. 4 μl
of 5 x reaction buffers, 10 mM DTT (In vitrogen, USA), 0.5ul of RNase inhibitor (In
vitrogen, USA), 1 unit of Thermoscript reverse transcriptase (In vitrogen, USA) was
added in a final volume of 20 µl. Reaction mix was incubated at 50°C for 1 hour and then
the polymerase was inactivated by heating at 85°C for 5 minutes. Finally 2 units of
RNase H (Invitrogen, USA) was added and incubated at 37°C for 20 minutes to remove
the template RNA and leaving single-stranded cDNA product. The cDNA produced was
stored at -20°C for further use.
3.7 CORE - NS2 GENE AMPLIFICATION
The cDNA generated using NS2 outer antisense primer was used as template for
amplification. The Core and NS2 specific outer primers were used to pick a 3kb amplicon
including Core, E1, E2, P7 and NS2 part of the genome. The region was successfully
amplified from four different samples. The amplification product was visualized on 0.8%
agarose gel using Ethidium Bromide staining. Inner primers (Table 3.2) were designed at
a distance of about 100 base pair from outer antisense primer to amplify a second round
of PCR using first round’s product as template.

Chapter 3
46
For amplification of Core-NS2 region Long High Fidelity Polymerase Mix
(Roche, Germany) was used. The reaction mixture for a single reaction consisted 1X
PCR Buffer, 1mMl DNTPs, 1 pmol outer antisense primer, 2 units Long High Fidelity
polymerase (Roche, Germany), about 100 ng RT-PCR product. Final volume was
adjusted (50 µl) with nuclease free water.
Amplification cycle parameters were optimized for each template and briefly
consisted of two denaturation steps at 95°C for 5 minutes (one cycle) followed by
denaturation at 94°C for 45 seconds, primer annealing at 56°C for 45 seconds, extension
at 72°C for 3 minutes. Synthesis reaction consisted of 35 cycles of denaturation,
annealing and extension followed by one longer cycle of extension for 10 minutes.
For the second round of PCR inner NS2R (reverse) primer was used with Core F
(forward) primer and first round PCR product was used as template instead of cDNA.
The second round amplification improved the yield of amplification. The PCR product
was visualized on 0.8% agarose gel dyed with Ethidium Bromide using gel
documentation system.
3.8 CLONING OF HCV CORE-NS2 REGION IN pCRII-TOPO
VECTOR
The 3kb PCR product was directly cloned in pCRII-TOPO VECTOR (Invitrogen,
Germany). For this purpose the amplification product was run on 0.8% agarose gel and
the desired bands were excised from a gel and was purified using the Gel Purification Kit
(Invitrogen, USA) according to the manufacturer protocol. Once pure products were
obtained, these products were accurately quantified using a spectrophotometer. To obtain

Chapter 3
47
an accurate reading, a negative PCR reaction was used as a blank sample. From the
absorbance recorded, the correct amount of product to be used in a cloning reaction was
calculated.
A
B
Figure 3.1: A Schematic presentation of pCRII-TOPO vector. A; linerized plasmid.
B; Circular plasmid after Core-NS2 (orange fragment) cloning between P lac and
lac Z sites.

Chapter 3
48
3.8.1 DNA Ligation
The 3 kb purified product was cloned into pCRII-TOPO vector (Invitrogen, USA)
according to the manufacturer’s protocol. Approximately 150 ng of the PCR product was
added to 1 μl of plasmid (50 ng), 5 units of T4 DNA ligase, 2 µl of ligase buffer and
nuclease free water was added to a final volume of 20 μl. The mixture was incubated at
room temperature for one hour.
3.8.2 Bacterial Transformation
The entire ligation reaction was added to a 50 μl aliquot of TOP10 competent
cells. The mix was incubated on ice for 20 minutes. The cells were heat-shocked at 42°C
for 40s and returned to ice. 500 μl of SOC medium was added to the cells and incubated
at 37°C for one hour.
After one hour the culture was spread onto a Luria-Bertani (LB) agar plate
containing 100 μg/ml ampicillin. For blue and white colony selection 40 µl of IPTG
(complete) and X-gal (complete) was added to the plate prior to cell spreading. After one
night incubation white colonies were selected and cultured in liquid LB media for
plasmid amplification.
3.8.3 Plasmid DNA Isolation and Restriction Endonuclease Analysis
Plasmid mini prep kit (Invitrogen, France) protocol was followed according to
manufacturer’s instructions. Briefly, 5ml culture was spun at 8000 rpm to pellet down the
cells, media was removed. Cells were resuspended in buffer A containing RNase A,
buffer B (lysis buffer) was added and incubated at room temperature not more than 5

Chapter 3
49
minutes. Neutralization buffer (C) was added, mixed well and centrifuged at 12000 rpm
for 10 minutes and the supernatant was applied to a column and centrifuged for one
minute. Column was washed once with wash buffer and the plasmid DNA was finally
eluted in 50 µl TE buffer. Positive colonies were checked through restriction digestion
with EcoR1 digestion. The PCRII TOPO vector has two EcoR1sites at both sides of the
ligation site and provides an efficient system for clone confirmation.
For digestion 1 µg of the mini prep product was mixed with one unit of enzyme
with 1x EcoR1 buffer in a final volume of 30 µl and the digestion mix was incubated at
37°C for 3 hours. The digestion product was viewed on 1% agarose gel and positive
clones were selected. Clones were confirmed by sequencing.
3.8.4 Sequencing and Phylogenetic Analysis
To determine the nucleotide sequence of individual clones Big Dye chemistry
(Backman, England) system was used. To obtain complete sequence of the 3 kb product
Core F, E1F, E2F, P7F and NS2R primer were used (Table 3.2) in addition to T7
promoter specific primers, specific for the vector. Approximately 500 ng of plasmid was
used in a sequencing reaction containing 0.2 pmol of primer, 2 μl of big dye and 2 μl of
dilution buffer (Applied Bio systems, USA) in a total volume of 10 μl. Sequencing
reaction was performed in a thermal cycler with the parameters 94°C for 20 s, 50°C for
20 s 60°C for 4 min, repeated 25 times. The reaction mix was transferred to a 0.5 ml
microcentrifuge tube and 2 μl of 3 M sodium acetate (pH5.2) was added and 50 μl of
100% molecular grade ethanol was added to precipitate the labelled DNA. The DNA was
incubated at room temperature for at least 20 min and then centrifuged at 14,000 g for 45
min. The supernatant was discarded and pellet was washed twice with 250 μl of 70%

Chapter 3
50
ethanol, centrifuging at 14,000 g for 15 min each time. Pellet was air dried at room
temperature and analyzes the DNA using an ABI PRISM 3100 Genetic Analyzer
(Applied Biosystems, USA).
Sequence assembly and editing was carried out using appropriate DNA analysis
software, such as Lasergene (DNAStar Inc), or the freely available Bioedit software
(http://www.mbio.ncsu.edu/BioEdit/BioEdit.zip). Phylogenetic and molecular
evolutionary analysis of samples was performed with the MEGA3 software (10)
(www.megasoftware.net), using an implementation of the Clustal W algorithm.
3.9 IN SILICO MODELING AND PROTEIN-PROTEIN
INTERACTION FOR HCV CORE AND STAT1
The HCV Core sequence was submitted to I-TASSER (Wu et al., 2007) online
web server and the model of highest c value was chosen for further analysis. HCV Core
sequence, HQ 108092, was used for molecular modelling. The model was refined with
energy minimization by subjecting it to ionized water box and physiological
concentrations. Amber 99 force field was used to minimize its energy after protonation of
the system by fixing its charges and lone pairs. The minimized model was extracted from
the solvent system and was docked with the STAT 1 protein (pdb id 1yvl).
The protein interaction among Core and STAT was studied by using had dock
web server [17]. Residues 1-23 of the Core were selected as active site and residues 577-
684 of STST1 were chosen as passive residues for this interaction. Passive residues of
both proteins were automatically selected. Five clusters were generated by restraining 1-
23 amino acids of Core and residues 577-684 of STAT 1. Amongst the had dock
generated clusters best nr structure had been chosen on basis of had dock sCore, rmsd

Chapter 3
51
overall lowest energy level structure, vander waal energy, electrostatic energy,
desolvation energy, restraint violation energy and buried surface energy.
Different contact types including Ionic cut off 4.5, Hydrophobic cut off 4.5,
hydrogen bonds and Disulfide cut off 2.5 were evaluated between Core and STAT1 using
the default bond angles used by Molecular operating environment (MOE). The sequence
separation used was 4 residues apart. His was selected as a basic in character whereas the
Met was characterized with hydrophobic nature. The Formation of hydrogen bonds
within main chain and side chain were also studied in contact mapping.
3.10 CLONING OF HCV ENVELOPE PROTEINS E1E2 IN
MAMMALIAN EXPRESSION VECTOR pcDNA 3.1
The 3 kb TA clones were used as template to amplify full length E1E2 including
the signal sequence from 3' end of core. Core signal F (forward) and E2R (reverse)
primers were used for this amplification. Start codon was artificially introduced at 5'end
and stop codon at 3'end. About 100 ng cloned DNA was used as template using the same
reaction mix and PCR program as described earlier. 2 minutes extension was given
instead of three minutes for the1700 base pair product of E1 and E2. The specific primers
were designed for sub cloning into pcDNA 3.1 containing Nhe1 at 5' and HindIII
restriction sites at 3'end of the product (Table 3.2).
The amplified product was visualized on 1% agarose gel; specific band was
excised with sterile razor and was purified using gel purification kit according to the
recommended protocol (Invitrogen, Germany). The purified product was quantified on
spectrophotometer.

Chapter 3
52
One microgram of the purified PCR product was digested with Nhe1and HindIII,
parallel to this digestion reaction, one µg of pcDNA was also digested with the same
restriction enzymes for about 3 hours and the digested products were then column
purified using invitrogen PCR purification kit (Germany).
Ligation mix was prepared using 1:3 ratios of the purified digested vector and the
PCR product (quantified on agarose gel). Invitrogen conventional ligase was used for this
purpose and ligation was performed at room temperature for 2-3 hours. Entire ligation
was transformed into 60 µl aliquot of TOP10 competent cells, incubated for one hour and
the culture was spread on ampicillin agar plates for selection.
After overnight incubation at 37°C, 6 colonies were picked for each sample.
Culture was amplified in LB ampicillin media for overnight and plasmid was isolated
using plasmid prep kit (In vitrogen, USA) as described earlier. Plasmid DNA was
quantified on spectrophotometer. 1 µg of DNA was digested with Nhe1 and HindIII as
described earlier. Digestion products were visualized using 1% agarose gel and positive
clones were selected. Clones were also confirmed by sequencing and full length E1E2
sequencing was done using CorSig F, E1F, E2F and E2R primer (Table 3.2).

Chapter 3
53
Figure 3.2: Recombinant pcDNA3.1 showing cloning position (orange
fragment) of E1E2 genes of HCV along with signal sequence (Sc) from 3'end
of Core.
3.11 CHARACTERIZATION OF HCV GLYCOPROTEINS BY
USING HCV PSEUDOTYPE PARTICLES (HCVpp) SYSTEM.
For HCVpp production, E1E2 pcDNA clones and two other plasmids PTG
luciferase 126 containing luciferase marker and phCMV Gag-Pol (kindly provided by
Jean Dubuossion) were amplified using bacterial culture and the plasmid DNA was
isolated using Nucleospin midi prep kit as described earlier. For each plasmid 100 ng/µl
DNA was prepared by diluting in nuclease free water.
3.11.1 Transfection
Human epithelial kidney (HEK) 293T cells (ATCC CRL-1573) were grown in
P100 tissue culture plate using Dulbecco’s modified Eagle’s medium (DMEM, GIBCO
BRL) supplemented with 10 % fetal calf serum (FCS, heat inactivated at 56°C for 30

Chapter 3
54
min) and 5% non-essential amino acids). Cells were split when reached up to 80%
confluency. Following removal of the medium, 293T cells were washed once with sterile
PBS and then trypsinized using trypsin solution (Life Technologies). Fresh DMEM
media was used to resuspend cells and were quantified using grid slide system under
microscope.
Approximately 24 h prior to transfection, 2 x 106 cells were seeded in 100-mm
tissue culture dishes each containing 10 ml of DMEM. Next day, DNA transfection was
done (in 50 % confluent cells). Physiological sterile water (9 g/l NaCl in H2O), 50 µl ,
was mixed with 4 µl of EXGENE 500 and incubated at room temperature for five
minutes. 100 ng of each plasmid, pcDNA E1E2, pluciferase and phCMV gag pol, was
added in 50 µl of physiological water. To this mix 50 µl of EXGENE 500 mix was added,
mixed well and incubated for 20 minutes. The transfection DNA mix was then added drop
wise directly to cell medium such that it was distributed evenly over the cell monolayer,
DMEM was replaced with Opti MEM prior to transfection. Mixed gently and incubated at
37 °C for six hours. Medium was gently replaced with 10 ml of fresh DMEM and the
cells were incubated at 37C for 48 hours for HCVpp production.
3.11.2 Glycoprotein Analysis
The E1 and E2 proteins expressed in transfected HEK 293T cells were visualized
by Western blot and immunoprecipitation. Genotype 3a has conserved epitope for AP33
and 311 antibodies used against E2 protein however no antibody is available for E1.

Chapter 3
55
3.11.3 Western Blot
Transfected HEK 293 cells were lysed after 48 hours of transfection by adding 500 µl
of PBS containing 1% Triton X100 C630 for 30 minutes on ice. The lysate was spun at
13,000 rpm for 5 min and the clarified supernatant was collected. The clarified lysate
may be stored at -20 °C for later use. 60 μl volume of transfected cell lysate was mixed
with a 20 µl of 6X protein gel loading buffer (200 mM Tris-HCl, pH6.7; 0.5% SDS, 10%
glycerol, 20 mM DTT), and fractionated proteins was loaded on a SDS-polyacrlyamide
gel with a 9 % resolving gel. A molecular weight marker (Invitrogen, protein ruler) was
loaded alongside to assess the apparent molecular weight of proteins expressed.
The proteins were transferred from SDS-PAGE gel to nitrocellulose membranes
using a semi-dry blotting apparatus (Bio-Rad) at 25V for 60 minutes. The membrane was
blocked for 1 hour with a 5% milk solution in Phosphate Buffered Saline, 0.05% Tween-
20 (PBS-T) and washed three times with 50 ml of PBS-T. The membrane was incubated
with a mixture of primary mouse monoclonal antibodies (MAbs) AP33 or 311 to
visualize HCV E2, each at a concentration of 1μg/ml in 5 ml of blocking buffer for 1
hour at room temperature. After one hour the membrane was washed 3 times, each for 10
min with PBS-T. Subsequently membrane was incubated with secondary antibody, goat
anti-mouse IgG, conjugated to horse radish peroxidase (Sigma, France), at a dilution of
1/5000, in blocking buffer, at room temperature for 1 hour. Finally, the membrane was
washed 3 times, each for 10 min with PBS-T. Proteins were visualized using enhanced
chemiluminescence (ECL Plus, GE healthcare). The luminescence was detected using
Kodak Light-1 film, typically exposing the film for 1 minute and developing with Kodak
GBX developer.

Chapter 3
56
3.11.4 Immuno Fluorescent Labeling
Huh7 cells were grown on cover slips using same protocol as described for HEK
cells. About 50% confluent cells were transfected next day using XGENE500 method.
After 48 hours cells were washed twice with 1XPBS and were fixed using 3% PFA
(Paraformaldehyde in PBS) was added for 20 minutes. PFA was removed by washing
cells twice with 1 XPBS. 10% goat serum was added and cells were blocked for 10
minutes and then cells were washed twice with PBS. Primary antibody “AP33” diluted in
goat serum was added and cells were incubated at room temperature for 30 minutes (30-
50 µl per cover slip). Cells were washed 3 times in PBS and were incubated for 5 minutes
between each wash.
Secondary antibody fluorescent IGg1-A488 was diluted in goat serum (1:800) and
the cover slips were incubated with the antibody for 30 minutes. Cells were washed three
times; five minutes each, with PBS. DAPI (4', 6-diamidino-2-phenylindole) was added in
first wash. Cover slips were mounted using 7 µl of mounting media on glass slides and
the slides were left over night at room temperature for drying. Slides were observed under
fluorescent microscope and the photographs were saved.
3.11.5 Immunoprecipitation (CD81 Pull down Assay)
HEK 293 cells were transfected for the production of pseudotype particles as
described earlier. For analyzing HCVpps, medium was collected (after 48 hour of
transfection), spined at 4000 rpm for 1 minute to remove cell debris and the cell free
medium was collected.
Glutathion beads (glutathione sephrose 4B, Amersham Bioscience) were washed
twice with cold PBS to remove storage buffer. 50 µl of glutathione beads were incubated

Chapter 3
57
with 10 µg of LEL-GST/mCD81-GST in one ml cold PBS for at least 8 hours or
overnight at 4 °C. After incubation G-seahorse beads were washed with cold PBS. G-
sephrose beads incubated with CD81 were then re-incubated with HCVpp containing
media (800 µl) overnight at 4 °C. The beads were washed five times with PBS and 0.1%
triton X. Finally the beads were re-suspended in 30 µl of reducing Lamemmli buffer or
protein loading buffer (200 mM Tris-HCl, pH6.7; 0.5% SDS, 10% glycerol) containing
20 mM DTT (reducing buffer). Samples were boiled and loaded on to 10%
polyacrelamide gel electrophoresis (SDS-PAGE) and was immune labeled as described
for western blot.
3.11.6 HCVpp Infectivity Assay
The retrovirus-based HCVpp assay as described by Bartosch et al. (2003). HEK-
293T cells were co-transfected with plasmids expressing the HCV glycoproteins, the
murine leukemia virus (MLV) Gag-Pol, and the MLV transfer vector carrying the fire fly
fluorescent protein as reporter. Upon expression, the MLV gag-pol particles encapsidate
the replication-defective genome carrying the GFP sequence and acquire the HCV
glycoprotein-containing envelope before being released into the medium. The HCVpp
released in the medium are then used to infect the human hepatoma cells (Huh-7), and the
infection measured by detection of GFP following incubation at 37 °C.
For infectivity assessment, HEK-293T cells were co-transfected in 6-well dishes at
1x105 cells per well with 3 plasmids, carrying sequences encoding MLV gag-pol, MLV
GFP and HCV glycoproteins as previously described. 24 hours before the
HCVpp were ready for harvest, target cells Huh7 were prepared. For this purpose
Huh-7 cells were trypsinized as described for HEK-293Tcells. Cells were seeded in 24-

Chapter 3
58
well dishes at 1x105 cells per well in 600 µl of DMEM (Alternatively, use 12-well dishes,
increasing cell number and volumes accordingly). Following incubation at 37°C for 48
hours, medium containing HCVpp was collected from HEK-293T cells and was filtered
through 0.45μm pore-size Minisart singe- syringe filter. The HEK-293T cells should be
no more than 90% confluent at this stage. Medium from Huh-7 target cells was removed
and 0.6 ml of media containing HCVpp was added per well. Cells were incubated at 37°C
for 4 to 6 hours, then the inoculum was removed, cells were then re-fed with 0.6 ml of
fresh DMEM. After 48 hours of incubation at 37°C, infected cells were harvested for FLP
(fire fly luciferase protein) expression. For this purpose the media was removed, cells
were lysed with the firefly lucierase lysis buffer (promega France) and were incubated at
4 °C shaker at least for two hour before taking readings. The level of FLP in HCVpp
infected cells were analyzed using luminometer (Bio-Rad) and the data was recorded.
3.12 PCR BASED MUTAGENESIS IN E2 GENE
On the bases of sequence obtained a unique glycosylation site was observed in E2 not
reported before in any genotype. To see the impact of this unique site PCR based
mutations were introduced at that site using specific primers. For this purpose the E1E2
region along with signal sequence from cores 3'end was amplified from the pcDNA clone
using two separate sets of primers harbouring artificially introduced mutations (Table 3.3).
The same protocol and conditions were used for PCR as described earlier.

Chapter 3
59
Table3.3: Primers Used for Mutagenesis
Primer Sequence
M48CNhe1 AAAGCTAGCATGCCAGGGAACTTGCCCGGTTGCTC
E2RM HindIII 5'GGGAAGCTTTCATGCTTCTGCTTGTGATACCATTAGC
E1EPITOPE F 5'GAG TGG CGG AAT TCG TCT GGT CTC TAT CAT GTC ACC AAC GAC
E1EPITOPE R 5'GCTATTGGAACAGTCGTTGGTGACATGATAGAGACCAGACGAATTC
CG
E2 del.F 5'CCCCCTTGTGACATTTATGGGGGCAGTAACCAAGCCCTTTTCTGTCC
CACC
E2 del.R 5'GCAGTCGGTGGGACAGAAAAGGGCTTGGTTACTGCCCCCATAAATG
TCACAAGGGGG
HVR495 MUT F 5'GCGCCTAGACCTTGTGGCATCGTTCCTGCAACAAGCGTCTGCGGCCC
TGTGTACTGC
HVR495 MUT R 5'GCAGTACACAGGGCCGCAGACGCTTGTTGCAGGAACGATGCCACAA
GGTCTAGGCGC
M1PP DEL F 5'CCCCTTGTAACATTTATGGGGGCATTAACCGAACCCTCTTCTGTCCC
ACC
M1PP DEL R 5'GGTGGGACAGAAGAGGGTTCGGTTAATGCCCCCATAAATGTTACAA
GGGG
3AW DEL F 5'CCCCTTGTAACATCTATGGGGGTAATGAGTCAGACCTCTTCTGCCCC
ACC
3AW DEL R 5'GGTGGGGCAGAAGAGGTCTGACTCATTACCCCCATAGATGTTACAA
GGGG
M1PP HVR F 5'GCACCTAGAACTTGCGGCGTTGTCAATGCATCAACTGTCTGCGGCCC
M1PP HVR R 5'GGGCCGCAGACAGTTGATGCATTGACAACGCCGCAAGTTCTAGGTC
M1PP HVRG R 5'GGTGGGACAGAAAAGGTCGCTCTCGTTCTTCGAGTTCCCACCGCCCC
CATAAATGT
M1PP HVRG F 5'ACATTTATGGGGGCGGTGGGAACTCGAAGAACGAGAGCGACCTTTT
CTGTCCCACC
NRU1F 5'GCCAGCAACTTCGCGACCACGGATCTCGATGCTGACCG
3.12.1 Fusion PCR
In order to introduced mutations inside E2 fragments, the PCR products amplified
in first step were gel eluted and purified and used as template in the second round or

Chapter 3
60
fusion PCR. The eluted products were run on 1% agarose gel for approximate
quantification. Roughly equal numbers of molecules were used as template in the fusion
PCR. Same cyclic conditions were used for fusion PCR except addition of 5 cycles for
fusion. After one longer denaturation at 95°C for 5 minutes, denaturation at 94°C for 45
seconds, annealing at 65°C for 3 minutes and extension at 72 °C for 5 minutes was
repeated five times for fusion of the template fragments. The fusion was followed by 30
cycles of routine PCR as described earlier. Fusion product was visualized on agarose gel,
required band was than excised and purified as described earlier.
The purified product was digested with Nhe1and HindIII and was ligated into
pcDNA3.1 using the same protocol as described before. The ligation was than
transformed into TOP10 cells, Positive clones were confirmed first by restriction
digestion and then finally presences of mutations were confirmed by sequencing.
3.12.2 Characterization of HCVpp Mutants
Positive clones with mutations were transfected in 293T cells for the production
of HCVpp. The media containing HCVpp was removed after 48 hours and was used to
infect Huh7 cells. The infected target cells were quantified after 48 hours of infection
using firefly luciferase assay system.
3.13 NUTRALIZATION OF HCVpp
For neutralization assay antibodies were isolated from patient sera. Serum from
20 different patients infected with HCV 3a genotype, tested for absence of HBV and HIV
were pooled and antibodies were purified using NAB TM purification kit according to
manufacturer’s protocol.

Chapter 3
61
3.13.1 Isolation of Antibodies
Column containing IgG coated beads was washed twice to remove storage buffer,
for this purpose 1 ml wash buffer was added and centrifuged at 8000 rpm for one minute.
Serum sample (1 ml) was loaded on the column and was mixed well with the beads,
followed by centrifugation for one minute. Column was washed with wash buffer.
Elusion buffer (1 ml) was added mixed well and centrifuged. The flow through was
saved. This step was repeated three times and each time the flow through was saved and
labeled as fraction A, B, and C.
Column was washed once with wash buffer. Storage buffer was added mixed
well. The column was then saved for another use. The saved fractions of eluted
antibodies were quantified using BIO-RAD Assay. Various known concentrations of
BSA were used as control.
3.13.2 GNA Capture ELISA for HCV E2
Reactivity of the specific antibodies present in the patient sera was checked by
ELISA using HCV E2 protein expressed in HEK293T cells. Immulon2, 96 well plate was
coated with 0.25 µg/well Galanthus nivialis lectin (GNA), dissolved in PBS. Plates were
incubated at room temperature overnight. Each well was washed with PBST (0.05%
Tween 20) to remove excess GNA and then were blocked with 2% skimmed milk,
200µl/well. Plates were washed 3 times with PBST and were stored at 4 °C or -20 °C for
later use.
Test fraction of E2 (20 µl cell lysate containing E2) was added in 100 µl of PBST
for each well and incubated at room temperature for 2 hours. Plate was then washed three
times with PBST. Serum was diluted 1:15 times in PBST (stock) and than 8 serial

Chapter 3
62
dilutions were prepared in PBST using 1:3 of the stock prepared. AP33 was used as
positive control and the negative control had no antibody. The plate was incubated at
room temperature for one hour and then washed 3 times with PBST. Secondary antibody
anti mouse HRP conjugate was added for AP33 while anti human HRP was used for
serum treated wells.
The plates were incubated for one hour with secondary antibodies. The plates
were washed six times with PBST to remove unbound antibodies. 100 µl of TMB was
added per well and the plates were incubated in dark for 30 minutes, inspected every 10
minutes and the reaction was stopped by adding 50 µl of 0.05 M H2SO4 as soon the
colour become dark. ELISA plates were then analyzed using BIOTEK, XL80 reader.
3.13.3 Neutralization Assay
For Neutralization assay various concentrations of antibody were incubated with
HCVpp and infectivity assay was performed as described earlier. To determine EC 50
(effective dose) various concentrations of antibodies were incubated with HCVpp,
harvested from HEK 293T cells, prior to infection of Huh7 cells. Infectivity assay was
performed and EC 50 was determined. Neutralization assay was performed for each
HCVpp mutant using antibodies equallent to EC 50 and data was record3.14
CONSTRUCTION AND CHARACTERIZATION OF INTER‐ GENOTYPIC CHIMERA FOR STRUCTURAL
PROTEINS 3.14.1 Cloning of Core‐NS2 Region in pJFH1
pJFH1 used in the current study was provided by Jean Dubussion after a generous
MOI with T. Wakita (Wakita et al., 2005). The construct contain a monosistronic HCVcc
(cell culture adaptive) ubiquitine, Renalla luciferase expression system.

Chapter 3
63
Clone M1 and M2 were selected for cloning into pJFH1 because compactable
sites for the cloning were available in their sequences. The cloning was completed in
three sub cloning steps involving pJFH1del, pJFH1VPand pB24-fl-Gluc, ubi2A
JFH1.XDNA as.
3.14.2 Cloning of Core-NS2 region in pJFH1del
The pC2a-1a NS2.2a del a JFH1 chimera for short referred as pJFH1 del is a
JFH1 constructs without structural proteins and is used for study of replication process.
This particular construct had competent sites for cloning 3a sequence in it. pJFH1del and
the M1 and M2 TOPO clones were digested with Bst1107 (site in the 5' of core) and
BglII ( site in the second loop of NS2 region). The digested products were run on agarose
gel and the required bands were excised and purified by using columns for gel elusion
and purification. The purified products (pJFH1del and Core-N22 PCR fragment) were
run on 1% agarose gel for estimation and measured quantities were used for ligation
reaction. Ligation was carried out at 16°C for overnight.
DH5α cells were used for transformation of the ligation product and transformed
bacterial cells were spread on an ampicillin plate for positive selection. The plates were
incubated overnight and the next day 8 to 10 colonies were tested by using colony PCR
method. For this purpose a forward primer from 5ˈUTR in the JFH1del (NRU F, table
3.3) and reverse primer specific for 3a core region was used to pick a clone with a
successful chimera region. JFH1del-3a chimera was further confirmed by digesting the
DNA with Kpn1.There was only one site of Kpn1 in JFH1del and the JFH1del chimera
had two sites, one acquired from 3a sequence and released a 2kb fragment upon

Chapter 3
64
digestion. The positive clone was selected and plasmid DNA was amplified using mini
prep protocol.
3.14.3 Cloning of Core-NS2 region in pJFH1VP
PJFH1del M1 and M2 chimera were digested with EcoR1, a site in the upstream
of the plasmid, and BglII. EcoR1 site was unique and common both in JFH1del and
JFH1VP, a full length JFH1. The digestion product from JFH1del was used as insert, and
of pJFH1VP was used as back bone vector with non structural genes from 2a genotype.
The restriction products were purified and ligated according to the protocol described
earlier. The recombinant plasmid was transformed in DH5α. Positive clones were
confirmed by colony PCR and also by digestion with KpnI.
3.14.4 Cloning of Gossia luciferase repoter gene in 3a-JFH1VP chimera
pB24-fl-Gluc-ubi2A JFH1.XDNA (pB24JFH1-Gluc) with a gossia luciferase
reporter gene was used to clone the luciferase reporter in 3a-JFH1VP chimera. Both
plasmids had two Bst11071 sites. 3a JFH1VP chimera at position 463 and at 9093 while
pB24JFH1-Gluc at position 1223 and 9081. The purified digested fragment of 8630 base
pair from 3a-JFH1VP chimera was ligated into a 4578 backbone of pB24JFH1-Gluc
containing IRES–Gluc and Ubiquitien. Ampicillin resistant colonies were amplified and
positive clones were tested by digestion with EcoR1 and BglII. Positive clone have three
fragments of approximately 10kb, 3kb and 500 base pair size visible on 1% agarose gel.
The Final Chimera M2JFHVP+Gluc was named as 3aPAK-JFH1 Chimera.

Chapter 3
65
Figure 3.3: 3aPAK-JFH1 chimera. pB24-pFul-Gluc-Ubi2A-JFH1-XDNA back bone (shown
in italic) was used for final construct. Green fragment represent 3a sequence from Pakistani
isolate encoding Core, E1, E2, P7 and a part of NS2. The orange part of HCV genome
encodes partial NS2, NS3, NS4A, 4B, NS5A, NS5B from JFH1 (genotype 1a).
3.15 CHARACTERIZATION OF 3APAK-JFH1 (CHIMERA)
For Characterization 3aPAK-JFH1 chimera was transcribed using cell culture
system (Huh7) and full length virus was produced using protocol as described by Kato et
al. (2006).
3.15.1 Preparation of DNA for In vitro Transcription
For in vitro transcription the plasmid was amplified. About 300 ml transformed
bacterial culture was harvested for the plasmid DNA preparation using nucleobond midi
prep kit (Nucleobond, France) following manufacturers’ protocol. Briefly, 100 ml of

Chapter 3
66
bacterial culture containing plasmid was grown over night and processed for plasmid
isolation according to the manufactures’ instructions. Briefly 2 ml of equilibration buffer
was applied to the column and was allowed to drain by gravity flow. Cells were pellet
down using centrifugation at 8000 rpm for 10 minutes. Pellet was suspended in 5ml
suspension buffer (containing RNase A) and was vortexed until homogeneous. 5 ml of
cell lyses solution was added and mixed gently by inverting the capped tube five times.
The mix was incubated at room temperature for 5 minutes. 5ml of neutralization buffer
was added and mix immediately by inverting the tube until the solution was
homogeneous. The mixture was centrifuged at 12,000 rpm at room temperature.
Supernatant from above step was loaded onto the equilibrated column and the
solution in the column was allowed to drain by gravity. Flow-through was discarded. The
column was washed two times with 5 ml of Wash Buffer. The solution was allowed to
drain by gravity flow after each wash. Flow-through was discarded. Plasmid DNA was
eluted by adding 5 ml of elution buffer. The flow-through containing DNA was collected
in a clean 15 ml falcon tube.
For plasmid precipitation 2 ml of isopropanol was added to the elute, mixed and
was allowed to incubate at room temperature for 5 minutes. The mixture was centrifuged
at 12,000 rpm at 4°C for 30 minutes. Supernatant was discarded and DNA pellet was
washed with the 1 ml of 70% ethanol and centrifuge at 12,000 rpm at 4°C for 15 min.
Ethanol was carefully removed by pipetting and the pellet was air dried. DNA pellet was
dissolved in 800 µl of nuclease free water and the purified DNA was quantified by
spectrophotometer. The DNA obtained was diluted to 1µg/µl for further use.

Chapter 3
67
For transcription 500 µg of DNA was over night incubated with 200 mg of CCl2
(Cesium Chloride). Following day the mix was extracted with isoamyl alcohol and
Chloroform (1:24) and the step was repeated twice. The extracted DNA was ethanol
precipitated using 1.5 volume of ethanol and 1/10 volume of acetate. The DNA pellet was
washed with 70% ethanol and was finally dissolved in Tris EDTA buffer. The amount
was quantified by spectrophotometer and 1 µg/ul stock was prepared.
40 µg of DNA was digested with Xba1using one unit of enzyme per µg of DNA
for 3-4 hours and then was heat inactivated at 65 °C for 10 minutes. To obtain blunt end
and or single stranded DNA the digested DNA was treated with Mung bean nuclease
(Invitrogen, USA) 1 unit per µg of DNA. The reaction mix was incubated for 30 minutes
at 37°C. The mung bean treated DNA was extracted with isoamyl alcohol and
chloroform mix, the process was repeated. The DNA was ethanol precipitated, washed
with 70% ethanol and the DNA pellet was dissolved in nuclease free water. Finally the
digested DNA was quantified by spectrophotometer.
3.15.2 In vitro Transcription of 3a-JFH1 Chimera
For in vitro transcription 1µg of linearized DNA (digested with Xba1) was used
as template and was transcribed in vitro using Mega script high yield transcription kit
(Applied Biosystems, USA). In brief 0.5mM dCTP, dGTP, dATP and dUTP were added
to 1µg of template and 1x transcriptase buffer. 1unit of transcriptase was added and the
reaction volume was maintained with nuclease free water. The Mix was incubated at
37°C for 5 to 6 hours. The reaction was stopped by adding equal volume of Lithium
Chloride and nuclease free water, the mix was chilled at -20°C for one hour and then
centrifuged at 4°C for 15 minutes. Supernatant was removed carefully and RNA pellet

Chapter 3
68
was first washed with 90% and then with 70% ethanol. The pellet was air dried for 10
minutes and RNA was gently resuspended in RNase free water. The RNA obtained was
visualized on 1% agarose gel for quality and the quantity was measured by using
spectrophotometer.
3.15.3 Electroprotaion of Huh7 cells with 3aPAK-JFH1 RNA
A day before electroporation confluent plate of Huh7 cells was split in 1:2 so that
next day nearly confluent plate was available with all cells in active growth phase. Cells
were trypsinized and 8ml DMEM was added. Resuspended cells were centrifuged at 800
rpm for 7 minutes at 4°C. Cells were resuspended in Opti MEM and were quantified by
using Grid system and approximately 106 cells were used for electroporation.
Cells were transferred in sterilized 0.4 cm gap Gene Pulser cuvette (Bio-Rad,
France) and 4 µg RNA was added just prior to electroporation. Cells were electroporated
at 100 Ohms, 280 volts and 975 UF (1puls) using a BioRad Genepulse Xcell
electroporator (Bio-Rad, France).
The electroporated cells were transferred to 12 ml cold DMEM and were
incubated for 10 minutes. 4 ml of the above media was transferred to P75 already
containing 6 ml of DMEM and the flask was shifted in 37°C incubator. At the same time
600 µl of the media containing electroporated cells was transferred to P24 and cells were
kept in incubator for luciferase assay and in another P24 containing cover slips for
immunoflorescent labeling.

Chapter 3
69
3.16 REPLICATION AND INFECTIVITY ASSESSMENT FOR M2-
JFH1VP CHIMERA
3.16.1 Replication Assay
To quantify the replication, cells were lysed after 48 hours and 72 hours interval
after electroporation with 100 µl of Renella Luciferase lyses buffer (Promega, UK). Cells
were incubated at room temperature for 3-4 hours and 50 µl of the lysate was then used to
measure luciferase activity using bright-GloTM luciferase Assay System (Promega, UK).
3.16.2 Infectivity Assay
For measuring infectivity of the 3aPAK-JFH1 progeny virus was harvested from
the cultured flask by filtering the media trough 0.5 µ filter (Millipore) at 72 hours post
electroporation and 500 µl of the media containing virus was used to infect naïve Huh7
cells grown in 24 well culture plates with and without cover slips. The one with cover
slips were used for immune fluorescent labelling. The infected cells were lysed at 24, 48
and 72 hours after infection and Luciferase assay was performed as described in earlier
section.
3.16.3 Immuno Fluorescent Labelling
For visualizing replication positive and infection positive cells, the cells grown on
cover slips were fixed and labelled. For this labelling Huh7 cells electroporated
(replication) or infected (infectivity) were grown on cover slips. After 48/72 hours cells
were washed twice with 1XPBS and were fixed using 3% PFA (Paraformaldehyde in
PBS) for 20 minutes. PFA was removed by washing cells twice with 1XPBS.

Chapter 3
70
Cells were blocked with 10% goat serum for 10 minutes. After blocking cells were
washed twice with PBS. Primary antibody ACAP 27, NS5a and AP33 was diluted in goat
serum and cover slips were incubated with antibodies at room temperature for 30 minutes
(30-50 µl per cover slip). Cells were washed 3 times in PBS and were incubated for 5
minutes between each wash.
Secondary antibody, fluorescent specific for each primary antibody, was diluted in
goat serum (1:800) and cover slips were incubated with the conjugate HRP antibody
(horse reddish peroxidase) for 30 minutes. Cells were washed three times; five minutes
each, with PBS. DAPI (4', 6-diamidino-2-phenylindole) was added in first wash. Cover
slips were mounted using 7 µl of mounting media on glass slides and the slides were left
over night at room temperature for drying. Slides were observed under fluorescent
microscope and the photographs were saved. ACAP 27, NS5a and AP33 were used
against core, NS5a and E2, respectively.

Chapter 4
71
A B
Figure 4.1: 1% agarose gel showing PCR amplification of Core-NS2 region. A;
First round PCR amplification. B; Second round PCR amplification.
RESULTS
4.1 AMPLIFICATION AND CLONING OF HCV CORE-NS2
REGION
4.1.1 Core to NS2 Amplification
HCV patient serum negative for HBV and HIV surface antigen and positive for
anti-hepatitis C virus antibody (HCV IgG) was used to extract viral RNA. Genotyping
was performed and 3a genotype positive RNA was used to generate cDNA using NS2
outer antisense primer. Specific primers were used to pick a 3kb amplicon including
Core, E1, E2, P7 and NS2 in the first round of the PCR (Fig. 1A). In second round of
PCR, the first round product was used as template (Fig. 1B).
4.1.2 Core-NS2 Cloning
Purified PCR product was cloned in PCRII-TOPO vector (Invitrogen) from four
different patients. Recombinant PCRII-TOPO vector designated as M, S, K and R

Chapter 4
72
Figure 4.3: 1% agrose gel showing restriction digestion of recombinant pCRII TOPO vector from patient S. Lane 1, 2,3,5,6 showing recombinant vector digested with EcoR1 and are the positive clones for Core-NS2 except lane 4. Lane 7 shows 1kb DNA marker. Core–NS2 region from patient S has an internal EcoR1 site showing two bands of insert on the gel.
(4kb+3kb) were digested with EcoRI and the digestion product were visualized on 1%
agarose gel. The positive clones were selected on the basis of insert released after EcoR1
digestion (3kb) as shown in figure 4.2, 4.3, 4.4 and 4.5. Positive clones from patient S
and R had an internal EcoRI site and the insert was digested into two fragments of 1300
and 1700 base pair size (Fig 4.3 and 4.5). Positive clones were amplified in bacterial
culture and further confirmed by sequencing.
Figure 4.2: 1% agrose gel showing restriction digestion of recombinent pCRII TOPO vector from patient M. Lane 1-5 are recombinant vectors digested with EcoR1. Lane 6 shows 1kb DNA marker.

Chapter 4
73
Figure 4.5: 1% agarose gel showing restiction analysis of recombinant pCRII TOPO vector from patient R. lane 1-7 are digestion products of EcoR1.lane 1, 2, 5 and 7 are positive clones. Core–NS2 region from patient R has an internal EcoR1 site showing two bands of insert on the gel.
Figure 4.4: 1% agarose gel showing restriction digestion of recombinant pCRII TOPO vector from patient k. Lane 1 to 6 are recombinant vectors digested with EcoR1 and are the positive clones for Core-NS2except lane 6. Lane 7 shows 1kb DNA marker.

Chapter 4
74
4.2. CORE-NS2 DNA SEQUENCING:
Nucleotide sequences of individual clones were obtained by using Big Dye
chemistry (Beckman Coulter system, CEQ8000). Three inner primers were used along
with T7F and T7R primer specific for the TOPO vector and complete nucleotide
sequence for an entire core-NS2 region was obtained. Sequences were aligned and
complete sequences were submitted to NCBI sequence data bank. Accession numbers are
tabulated in Table 4.1
Table 4.1: Core-NS2 Sequence Accession Number (NCBI). Table shows accession numbers
of sequences from patients K, R, S & M.
Serial number
Accession number
Patient code
Clone Code
01 HQ108092 K C1
02 HQ108093 K C2
03 HQ108094 K C3
04 HQ108095 K C4
05 HQ108096 R C5
06 HQ108097 R C6
07 HQ108098 R C7
08 HQ108099 R C8
09 HQ1080100 S C9
10 HQ1080101 S C10
11 HQ1080102 S C11
12 HQ1080103 S C12
13 HQ1080104 M C13
14 HQ1080105 M C14
15 HQ1080106 M C15
16 HQ1080107 M C16

Chapter 4
75
4.2.1 Amino Acid Sequence Analysis
Focus of this study was structural proteins; detail analysis of Core, E1 and E2 was
performed. Amino acid sequence of all the 16 clones were aligned and numbered using
HCV H77 (genotype 1a) as reference strain (Fig. 4.6). The most prominent amino acid
changes between 1a and 3a are complied in table 4.2.
Domain ICOREA.
Domain II DomainIII E1B.
1

Chapter 4
76
C. Fusion peptide
2 3
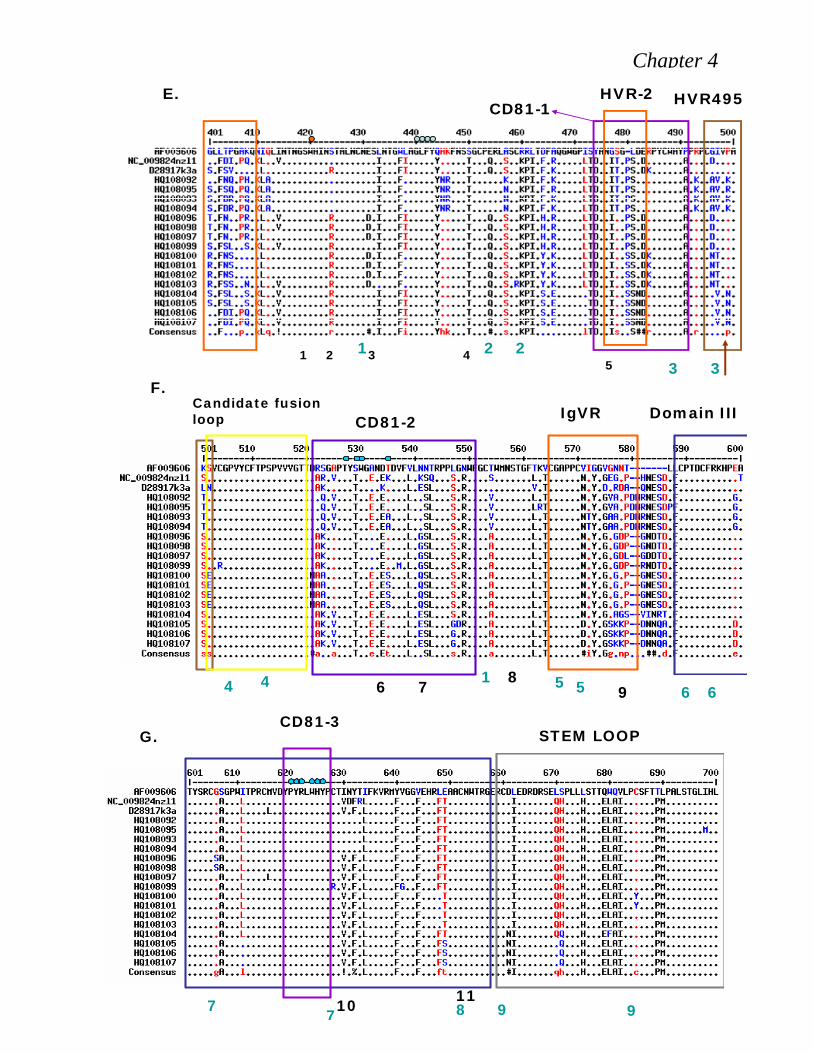
Chapter 4
77
G.CD81-3
STEM LOOP
7 710 811
9 9
E.CD81-1
HVR-2 HVR495
11 2 3 4 2 25 3 3
NNF.Candidate fusion loop CD81-2 IgVR Domain III
4 4 6 7 1 8 5 5 9 6 6

Chapter 4
78
H.
Figure 4.6: A & B; Amino acid sequence alignment; Amino acids are numbered with reference to H77 strain (1a). HCV core, position 1-191, is divided into 3 major domains domain I (aa 117), Domain II(118-177), domain III ( 178-19). HCV glycoprotein E1 extends from position 192 to 383.
C, D E and F; represents part of glycoprotein E1 and E2. Yellow rectangle (258 to 308) is the potential fusion peptide of E1. Gray rectangle is the trans membranel domain of E1, green rectangle shows the segment designated as pre anchor domain. Brown rectangle marks the 5'end of glycoprotein E2 and is designated as HVR1 ( 384-410) .Blue rectangles are showing CD81 interacting region and is distributed in three separate segments, segment I, 474- 492, segment II 527 -553 and segment III is 612-620.Critical residues for CD81 interaction are marked with blue circles at the top of the rectangle. Red rectangle are the hypervariable regions within E2.The second variable region, HVR2 lies within the CD81 interacting segment II sited at 475-485 and the third variable region designated as intergenotypic variable region (IgHVR) lies at 570-580. Within this IgHVR lies a small variable pocket ( HVR577) with 5 to 7 extra amino acid restricted to genotype 3a only. Yellow rectangle in E2 is the potential fusion peptide assigned to residue 501-520.
G and H shows terminal part of glycoprotein E2. Blue rectangle showing CD81 binding segment III, within are critical residue marked as blue circle at the top of rectangle. The C terminal of E2 is the stem loop region marked as gray rectangle and mark the end of E2 at position

Chapter 4
79
Table 4.2: Amino acid sequence variations of HCV (3a) structural proteins (core, E1, E2)
Sr no
AA position
Mutation
properties
1 2 S-R R: non polar
2 3 T-L L: hydrophobic
3 7 P-L L: hydrophobic
4 16 I-S/N S: polar, soluble
N: polar
5 47 A-C C: non polar, hydrophobic, thiol
6 190 D-A A: non polar,soluble
7 192 Y to L/F F: non polar
L: hydrophobic
8 193 Q-E in all 3a E: polar, acidic, hydrophilic
9 194 V-W in all 3a
10 202 H-V/I I: Hydrophobic, V: branched chain, hydrophobic
11 203 V-L L: hydrophobic
12 208 P-S/P S: polar, soluble P: secondary amino group, non polar
13 214 Y-C C: non polar, hydrophobic, thiol
14 217 A-D/G D: polar, acidic, less soluble
G:non polar
Table continued

Chapter 4
80
15 218 D-E E: polar, acidic, hydrophilic
16 232 E-D/A/S S: polar, soluble A: non polar,solubleD: polar, acidic, less soluble
17 237 R-T/M/K T: polar, less soluble M: non polar
K: polar, positive, soluble
18 240 V-T/I T: polar, less soluble
19 241 A-P/S S: polar, soluble
20 250 D-Y Y: polar side group
21 251 G-A/V V: branched chain, hydrophobic
22 254 L-A A: non polar,soluble
23 257 Q-S S: polar, soluble
24 258 L-I I: Hydrophobic
25 260 R-S S: polar, soluble
26 262 I-V V: branched chain, hydrophobic
27 285 F-S/L S: polar, soluble
L: hydrophobic
28 290 L-A A: non polar,soluble
29 294 S-R R: non polar
30 330 V-Q Q: polar,
31 349 L-A/V V: branched chain, hydrophobic
A: non polar,soluble
32 356 E-H/Q/S H: positively charged
S: polar, soluble
Q: polar,
Table continued

Chapter 4
81
33 372 H-S/Q S: polar, soluble
Q: polar,
34 384 V-Q Q: polar,
35 388 T-S/I I: Hydrophobic
S: polar, soluble
36 411 Q-A A: non polar,soluble
37 425 S-R R: non polar
38 444 Q-Y Y: polar, soluble
39 457 A-K/N K: polar, soluble
N: polar
40 459 C-R only in one R: non polar
41 461 R-P in 3a P: secondary amino group, non polar
42 464 D-F/H/Y/S H: positively charged
S: polar, soluble
Y: polar side group
F: non polar
43 465 A-K/R/E K: polar, soluble
R: non polar
G: polar, acidic, soluble
44 474 Y in 3a and D in 2a Y: polar side group
D: polar, acidic, less soluble
45 477 G in 2a and I in 3a G: polar, acidic, soluble
I: Hydrophobic
Table continued

Chapter 4
82
46 479 + P/S in all other genotype then 1a
P: secondary amino group, non polar
S: polar, soluble
47 490/491 P-A A: non polar,soluble
48 503/504 C-R only in one R: non polar
49 521/522 R-A in all except ‘92-‘95
A: non polar,soluble
50 522/523 S-R/Q/K/A A: non polar,soluble
R: non polar, K: polar, soluble, Q: polar,
51 528/529 S-T T: polar, less soluble
52 531/532 A-E E: polar, acidic, hydrophilic
53 534/535 D-E E: polar, acidic, hydrophilic
54 535/536 T-A/S A: non polar,soluble, S: polar, soluble
55 540/541 N-G/Q/E G: polar, acidic, soluble
Q: polar, E: polar, acidic, hydrophilic
56 542/543 T-L L: hydrophobic
57 546/547 L-S/G S: polar, soluble,G: polar, acidic, soluble
58 553/554 T-V/A A: non polar,soluble
V: branched chain, hydrophobic
59 561/562 T-L L: hydrophobic
60 570/571 V-N N: polar
61 522 E-G/D in HQ’92-95 G: polar, acidic, soluble
D: polar, acidic, less soluble
62 621 C-R in HQ’99 R: non polar
63 634 V-G in HQ’99 G: polar, acidic, soluble
Table continued

Chapter 4
83
4.3 IN SILICO CHARACTERIZATION OF STAT1 INTERACTING
DOMAIN OF HCV CORE:
Sequence analysis of the studied clones revealed that most of the mutations in
HCV Core protein were located in first 50 amino acids that are known to interact with
STAT1. In order to see the impact of these mutations on Core STAT1 interaction
molecular modelling and docking techniques were used. Results are shown in table 4.3,
4.4, 4.5 and in figure 4.7.
64 663 L-Q Q: polar
65 664 S-H H: positively charged
66 673 W-L/F L: hydrophobic
F: non polar
67 674 Q-A in all 3a A: non polar,soluble
68 678 C-Y Y: polar side group
69 682 T-P in all 3a P: secondary amino group, non polar
70 683 L-M in all 3a M: non polar
71 709 S-G G: polar, acidic, soluble
72 710 I-M M: non polar
73 711 A-V V: branched chain, hydrophobic
74 712 S-G G: polar, acidic, soluble

Chapter 4
84
Table 4.4: Results of four different models for Core, STAT1 interaction, showing interacting residues of Core and STAT1.
Table 4.3: In vivo Mutations observed in HCV Core and their Effect on STAT1 Interaction

Chapter 4
85
Table 4.5: Results of cluster model 1 showing the contact points between HCV Core and STAT1 along with bond type and net charge. Details of Core 23rd amino acid are taken from cluster model 3.

Chapter 4
86
Figure 4.7: HCV Core and STAT1 Interacting domains. (A) STAT1 has been shown in turquoise blue and Core in purple color. (B) Interaction points are marked in red color and numbers showing the positions repeatedly mutated in different strains of HCV Core and are involved in the interaction.
4.4 HCVpp PRODUCTION AND CHARACTERIZATION
Envelop proteins E1 and E2 along with signal sequence from C terminus of Core
were amplified using specific primers from TA clones and were sub cloned in expression
vector pCDNA3.1. pCDNA clones from four different patients (M, S, K, R, four clones
from each patient) were first tested for E2 expression by western blots (Fig. 4.8, 4.9)
using 311 anti E2 antibody. HCVpp were produced by cotransfection of pCDNA

Chapter 4
87
Figure 4.8: Western blot analysis showing E2 expression in HEK293T cells. Cells were lysed 48 hour post transfection and E2 expression was visualized using 311anti E2 antibody. Lane 2-5 represent 4 different clones from patient M and lane 6-9 represent 4 clones from patient S. 3awt was used as positive control (lane 1) for 3a E2 glycoprotein. βactin was used as loading control.
Figure 4.9: Western blot analysis showing E2 expression in HEK293T cells. Cells were lysed 48 hour post transfection and E2 expression was visualized using 311anti E2 antibody. Lane 2-5 represent 4 different clones from patient K and lane 6-9 represent 4 clones from patient R. 3awt was used as positive control (lane 1) for 3a E2 glycoprotein. βactin was used as loading control.
harbouring glycoprotein gag pol and luciferase expressing vectors. HCVpp produced in
HEK293T cells were collected from the media (secreted HCVpp) and E2 incorporated in
HCVpp was visualized by western blots using 311antibody (4.10, 4.11).

Chapter 4
88
Figure 4.11: Western blot analysis showing CD81 pull down of E2 incorporated in HCVpp. Secreted HCVpp were concentrated from HEK 293T media and were pull down using GST-LEL CD81. E2 was visualized using 311 antibody. Lane 2-5 represents 4 different clones from patient M and lane 6-9 represent 4 clones from patient S. 3awt was used as positive control for 3a E2 glycoprotein. βactin was used as loading control.
Figure 4.10: Western blot analysis showing CD81 pull down of E2 incorporated in HCVpp. Secreted HCVpp were concentrated from HEK 293T media and were pull down using GST-LEL CD81. E2 was visualized using 311 antibody. Lane 2-5 represents 4 different clones from patient M and lane 6-9 represent 4 clones from patient S. 3awt was used as positive control for 3a E2 glycoprotein. βactin was used as loading control.

Chapter 4
89
Figure 4.12: Bar graph showing HCV pp infectivity based on luciferase activity. pCDNA was used as negative control, 2A with E1E2 from 2a genotype, was used as positive control. M1-M4 represents HCVpp of four different clones from Patient M, S1-S4 from Patient S, K1-K4 from patient K and R1-R4 from patient R. Data is plotted on logarithmic scale.
HCVpp produced in HEK 293T cells were harvested from media and were used to infect
Huh7 cell. Infectivity of the HCVpp was measured 48 hours after infection using
luciferase reporter assay. Data indicate that all patient derived E1E2 clones had very low
infectivity in HCVpp system (Fig. 4.12) as compared to 2a positive controls (Accession
numbFer AB237837). The experiment was repeated using 1a, 2a, and 3a positive control
and with those clones that had at least two times infectivity than base line (pCDNA) (Fig.
4.13)

Chapter 4
90
Figure 4.13: Bar graph showing HCV pp infectivity based on luciferase activity. pCDNA was used as negative control 1A, 2A and 3A with E1E2 from genotype 1a,2a,and 3a respectively were used as positive control. M1-M3M represents HCVpp of four different clones from Patient M, S2, S3, S4 from Patient S, K3and K4 from patient K and R3and R4 from patient R. Data is plotted on logarithmic scale.
4.5 HCVpp MUTATAGENESIS AND CHARACTERIZATION
Significant variations observed in 3a E2 sequence included a novel glycosylation
site at position 499 in all clones from patient M and five additional amino acids found in
3a genotype at position 575. These two positions were subjected to PCR based
mutagenesis in order to investigate their role in HCV infectivity.

Chapter 4
91
Figure 4.14: 1% agarose gel showing first round PCR for the introduction of mutations in M2pp clone. E2 was amplified in two fragments a and b. Fragment a and b had an overlapping part of about 30-40 nucleotides incorporating the desired mutation in the overlapping part. E2M2-499 (lane 5, 6) is for removal of glycosylation site at position 499, E2M2∆575( lane 3,4) for deletion of 5 extra amino acids present in intergenotypic variable region at position 575, while E2M2-575wt(lane 1,2) is for switching intergenotypic region between M2 and 3awt positive control. Lane 7 represents 1kb DNA ladder.
4.5.1 Removal or addition of glycosylation site at position 499
Novel glycosylation site found in patient M, at position 499, was characterized
by removing or adding this particular glycosylation site in different clones. The site was
subjected to PCR based mutagenesis and the glycosylation site was removed from clone
M2 and from M3 (Fig. 4.14, 4.15, 4.16) and was added to the clone 3awt (Fig.4.17, 4.18,
4.19).
4.5.2 Deletion of 5 extra amino acids in intergenotypic variable region
In 3a genotype there are 5extra amino acids present in intergenotypic region. In
order to check its effect on infectivity these five extra amino acids were removed from
clone M2 and 3awt (Fig. 4.14, 4.15, 4.16, 4.17, 4.18, 4.19).

Chapter 4
92
Figure 4.16: 1% agarose gel showing cloning of fusion PCR products in PcDNA3.1 aVector. Lane 3-6 are positive clones digested with Nhe1 and HindiIII are showing E1E2 and core signal sequence fragment of about 1700 base pairs. Lane 1 showing pCDNA digested with same enzymes and lane 2 is showing positive control for E1E2. Lane 7 showing 1kb DNA marker.
Figure 4.15: 1% agarose gel showing second round PCR (fusion PCR) for the introduction of mutations in M2 clone (lane 1-3).E2M2HVR499 for mutation of glycosylation site position 499, E2M2∆575 for deletion of 5 extra aminoacids present in intergenotypic variable region “IgHVR” and E2M2-575wt is for switching of IgHVR with 3awt used as positive control.

Chapter 4
93
Figure 4.17: 1% agarose gel showing first round PCR for the introduction of mutation in 3awtpp clone E2 was amplified in two fragments a and b. Fragment a and b had an overlapping part of about 30-40 nucleotides incorporating the desired mutation in the overlapping part. 3aE2wt499 (lane 1, 2) is for addition of glycosylation site, 3aE2wt∆575 (lane 3,4 ) for deletion of 5 extra amino acids present in intergenotypic variable region at position 575, while 3aE2wt575M2 (lane 6,7) is for switching intergenotypic region between M2 and 3awt positive control. Lane 7 shows 1kb DNA ladder.
4.5.3 Switching of intergenotypic region
The wild type 3awt clone was much more infectious than M2. Their
intergenotypic regions were switched by PCR based mutations and their infectivity was
recorded to see if the switching had any effect on infectivity (Fig. 4.17, 4.18, 4.19).

Chapter 4
94
Figure 4.19: Cloning of fusion PCR products in pCDNA3.1 vector. Positive clones digested with Nhe1 and HindIII are showing E1E2 and core signal sequence fragment of about 1700 base pairs, lane 1-4 and 6-9. Lane 5 and 12 shows 1kb DNA marker. Lane 10 is pCDNA digested with the same enzymes. Lane 11 shows positive control for E1E2.
Figure 4.18: 1% agarose gel showing results of second round PCR (fusion PCR) for the introduction of mutations in 3awt clone (alane 1, 2, 4, 5, and 6). 3aE2wt499 for mutation of glycosylation site, 3aE2wtM2∆575 for deletion of 5 extra amino acids present in intergenotypic hyper variable region “IgHVR” and 3aE2wtM2-575wt is for switching of IgHVR with E2M2. Lane 3 represents 1kb DNA ladder.

Chapter 4
95
Figure 4.21: CD81 pull down from HCVpp (3a). HEK293T cells were transfected and pseudotype particles secreted in the media were concentrated. E2 incorporated in HCVpp was pull down usiang GSTLEL-CD81, E2 interaction with CD81 was visualized using anti E2 311. βacatin was used as loading control.
Figure 4.20: Western blot showing CD81 pull down from cell lysate. HCVpp produced in HEK293T cells were pull down using GSTLEL-CD81, E2 interaction with CD81 was visualized using antiE2 311. βactin was used as loading control.
4.5.4 Characterization of E2 Mutants for CD81 binding and Infectivity
Infectivity of the E2 mutants was checked by using HCVpp system. Mutation
bearing pseudotype particles were produced and a pull down assay was performed to
analyze their interaction with GSTLEl CD81 (Fig. 4.20 and 4.21). Results indicate that
removal of extra glycosylation site slightly increase CD81 binding.

Chapter 4
96
Figure 4.22: Bar graph showing HCVpp infection in Huh7 cells measured as luciferase activity. 3aE2wt499M2 bearing extra glycosylation site at 499 position as compared to 3aE2wt. Glycosylation site 499 was removed from E2M2-499 and from E2M3-499 while E2M2 and E2M3 are clones with novel glycosylation site at position 499. The data is presented in logarithmic scale.
4.5.5 Characterization of E2 Mutants for infectivity
HCVpp produced with mutated glycoprotein E2 were used to infect target Huh7
cells and their infectivity was assessed after 48 hours using Firefly luciferase reporter
gene activity (Fig. 4.22).
HVRIG in genotype 3a distinctively had 5 to7 extra amino acid lying at position 575. In
order to see its effect on HCV infectivity, HCVpp were generated bearing deletions and
switched extra amino acids from a clone with low infectivity to a clone with a high
infectivity and vice versa (Fig. 4.21). HCVpp infection of Huh7 cells was recorded as

Chapter 4
97
Figure 4.23: Bar graph showing HCVpp infection in Huh7 cells, measured as luciferase activity. In 3aE2wt575 M2 and E2M2-575wt 5 extra amino acid in IgHVR have been switched as compared to 3aE2wt and E2M2. In 3aE2∆575 and E2M2∆575 these extra amino acids have been deleted. The data is presented in logarithmic scale.
luciferase reporter gene activity and data was plotted on graphs using Microsoft Excel
Worksheet (Fig. 4.23).
4.5.6 Incorporation of A4 Epitope In Glycoprotein E1
Antibody for glycoprotein E1 from 3a genotype was not available. For genotype
1a and 2a antibody A4 is well characterized. A4 epitope was cloned in E1 (3a) by fusion
PCR and the recombinant protein was used to generate HCVpp. Infectivity analysis of
HCVpp bearing recombinant protein is shown in figure 4.24.

Chapter 4
98
Figure 4.24: Bar graph showing HCVpp infection in Huh7 cells. Infectivity of HCVpp was measured as luciferase activity and is plotted on logarithmic scale. M14A4, M2A4, S4A4, and K4A4 are with recombinant E1 bearing A4 epitope from1a genotype. Infectivity of recombinant M1A4 has shown a significant increase when compared to non recombinant M1.
4.6 HCVpp NEUTRALIZATION BY SERUM DERIVED IgG
Serum from HCV positive patients was isolated and serum IgG was purified.
Various concentrations of IgG were used to determine Effective Dose 50 (EC50). HCVpp
bearing E1E2 from 3awt and M1 clone were used to determine EC50. HCVpp treated
with various concentrations of IgG (Fig. 4.25 and 4.26) showed approximately 50%
neutralization at concentration 33 ug/ml.
HCVpp produced from 3awt, M2 and M3 harbouring mutations for glycosylation
site at position 499 and for extra 5 amino acid in IgHVR were treated with 33ug/ml
serum derived IgG prior to infecting Huh7 cells. Results clearly showed that
Glycosylation at position 499 help HCVpp to escape neutralization. Deletion of 5 extra

Chapter 4
99
Figure 4.25: Graph showing HCVpp neutralization measured as luciferase activity by HCVpp infected cells. HCVpp were incubated with various concentrations of serum derived IgG for 4 hours prior to infection of Huh7 cells. 50% neutralization was observed at 33ug/ml of IgG used.
amino acids in IgHVR or difference in amino acids in this region does not have any effect
on HCVpp neutralization (Fig. 4.27, 4.28).

Chapter 4
100
Figure 4.26: Graph showing HCVpp neutralization measured as luciferase activity by HCVpp infected cells. HCVpp were incubated with various concentrations of serum derived IgG for 4 hours prior to prior to infection of Huh7 cells. 50% neutralization was observed at 33ug/ml of IgG used.

Chapter 4
101
Neutralization
Figure 4.27: Graph showing serum IgG derived neutralization of HCVpp bearing mutations in E2 glycoprotein. Neutralization was measured as luciferase activity by HCVpp infected Huh7 cells. Data is plotted on logarithmic scale. 3aE2wt499M2 harbor extra glycosylation site at 499 position as compared to 3aE2wt .Glycosylation site 499 has been removed from E2M2-499 and from E2M3-499 while E2M2 and E2M3 are clones with novel glycosylation site at position 499.

Chapter 4
102
Figure 4.28: Graph showing serum IgG derived neutralization of HCVpp bearing mutations in E2 IgHVR. Neutralization was measured as luciferase activity by HCVpp infected Huh7 cells. Data is plotted on logarithmic scale. In 3aE2wt575 M2 and E2M2-575wt 5 extra amino acid in IgHVR have been switched as compared to 3aE2wt and E2M2. In 3aE2∆575 and E2M2∆575 these extra amino acids have been deleted.

Chapter 4
103
4.7 PHYLOGENETIC ANALYSIS OF HCV STRUCTURAL
PROTEINS CORE, E1 AND E2
Nucleotide sequences of 3a genotype reported from other parts of the world were
retrieved from NCBI data base and were used to establish phylogenetic link of the HCV
3a genotype from Pakistan (Fig. 4.29 and 4.30).
The data retrieved from NCBI Gene Bank for this study indicate an independent
origin of several clades all over the world. HCV core protein from the Pakistani isolates
appeared in seven different clades (Fig. 4.29).
Limited data for 3a genotype is available from Pakistan as well from the rest of
the world. In this analysis 16 clones of HCV 3a genotype from present study and two
clones from previous report were integrated to construct phylogenetic relation of 3a
glycoprotein genes to the rest of the 3a genotype reported from all over the world.
Phylogenetic analysis of HCV glycoprotein E1 and E2 revealed a parallel origin of 3a
genotype across the world. From the data set used for this study 13 major clades appeared
while all Pakistani isolates appeared in two main clades (Fig. 4.30).

Chapter 4
104
Figure 4.29: Phylogenetic relation of HCV core protein, genotype 3a

Chapter 4
105
Figure 4.29 A & B

Chapter 4
106
Figure 4.30: Phylogenetic relation of HCV glycoprotein E1E2 for 3a genotype.

Chapter 4
107
Figure 4.31: 1%agarose gel showing purified products digested with Bst11071
and BglII. Lane 1, 2 and 4 represents M1, M2 (Core-NS2) PCR products and
pJFH1del respectively. Lane 3 shows 1kb DNA marker.
4.8 CONSTRUCTION AND CHARACTERIZATION OF
INTERGENOTYPIC CHIMERA FOR STRUCTURAL PROTEINS
4.8.1 Cloning of Core-NS2 region in pJFH1del
Clone M1 and M2 were selected for recombination into JFH1del as competent
sites for the cloning were available in their sequences. M1and M2 TOPO clones were
digested with Bst11071 (site in the 5'of core) and Bgl II (in the second loop of NS2
region), (Fig. 4.31). After ligation, positive clone were screened by colony PCR using
forward primer from 5’UTR in the JFH1del and reverse primer specific for 3a core region
(Fig. 4.32).

Chapter 4
108
Figure 4.32: 1% agarose gel showing results of colony PCR. Lane 1, 2, 6 and 8 are potential recombinant clones, clone 8 was selected for further processing.
Figure 4.33: Purified digestion fragments loaded on 0.8% agarose gel showing pJFH1VP, M1JFH1del, M2JFH1del in lane 1, 3 and 4 respectively. Plasmids were digested with EcoR1and BglII.
4.8.2 Cloning of Core-NS2 region in pJFH1VP.
pJFH1del M1 and M2 chimera were digested with EcoR1, a site at very upstream
of the plasmid and with BglII. pJFH1VP was digested with the same enzymes. The
fragments were gel purified and were visualized on 0.8% gel (Fig. 4.33). Appropriate
quantities were ligated and positive clones were selected by colony PCR (Fig. 4.34) and
were further confirmed by sequencing. M2JFH1VP (Fig. 4.35 & 4.36) was used to
produced viral progeny in Huh 7 cells.

Chapter 4
109
Figure 4.34: 1% agarose gel showing results of colony PCR (lane 1-4). 5'UTR forward primer from 2a and E1R (reverse) from 3a glycoprotein E1 was used to screen 3aJFH1VP recombinant. Clone with best amplification (lane 2) was selected for further use. Lane 5 shows 1kb
Figure 4.35: Schematic representation of JFH1VP chimera, green box represents 3a genotype sequence and red box represent part of genome from genotype 2a (JFH1).

Chapter 4
110
Figure 4.37: 1% agarose gel showing results of In vitro transcription, Lane 1
JFH1C6S4, Lane2 M1JFH1VP, lane3 M2JFH1VP, Lane 4 1kb ladder.
4.8.3 Characterization of M2JFH1VP Chimera
Recombinant M2JFHVP (chimera) was transcribed in vitro and the RNA was
used to transfect Huh7 cells (Fig 4.37).
Transfected cell were allowed to produce viral progeny. Viral replication was visualized
by Immunolabelling of various viral proteins, in particular Core and E2 from 3a part and
Figure 4.36: Schematic representation of NS2 topology. The star and arrow represents
site within NS2 employed to join 3a and 2a fragments of the chimera.

Chapter 4
111
Figure 4.39: Huh7cells showing expression of core 72 hours post electroporation with M2J-JFH1VP RNA. The cells are labeled with Acap 27 an anti Core antibody
Figure 4.38: Huh7 cells showing expression of Core 48 hours post electroporation with M2FH1VP RNA. The cells are labeled with Acap27 an anti Core antibody.
NS5a from 2a genome of the chimera (Fig. 4.37-4.41). Susceptible Huh7 cells were
infected with cell culture derived chimera virus and infection was confirmed by
Immunolabelling with E2 antibody AP33 (Fig. 4.42).
.

Chapter 4
112
Figure 4.41: Huh7cells showing expression of NS5a 72hr post electroporation with M2JFH1VP RNA. The cells are labeled with NS5a
Figure 4.40: Huh7cells showing expression of Core 72 hours post electroporation with
M2J-JFH1VP RNA, The nuclei are stained with DAPI and Core is labeled with Acap27.
.

Chapter 4
113
Figure 4.42: Huh7cells showing expression of E2 72hr post infection with M2 JFH1VP virus produced in cell culture. Nuclei of the cells are labeled with DAPI and the E2 with AP33.
4.9 CLONING OF GAUSSIA LUCIFERASE REPORTER GENE IN
M2JFH1VP CHIMERA.
pBJFH1CS-N6A4 with a gaussia luciferase reporter gene was used as backbone
vector to clone luciferase reporter in 3a-JFH1VP chimera (Fig. 4.43). Positive clone was
also confirmed by nested PCR using Ubiquitin forward primer and E1A4 reverse primer
from 3a genotype (Fig. 4.44) and by sequencing. Finally the 3a-JFH1VP chimera with
gaussia luciferase was named as 3aPAK -JFH1 chimera.

Chapter 4
114
Figure 4.43: 0.8% agarose gel of Bst11071 digested and purified fragments of M2J-
JFH1VP and JFH1CS-N6A4-Gluc.2a (lane 2 and 3), 1ul of each loaded on
0.8%agarose gel.
Figure 4.44: 1% agarose gel showing 3aPAK-JFH1 recombinant positive PCR.
Lane 1. Amplification with Ubiquitine forward primer (for back bone vector) and
E1A4 reverse primer specific for 3a.

Chapter 4
115
Figure 4.45: o.8% gel showing 3aPAK-JFH1 and JFH1C6S4 digested with Xba1, lane 1and 2 respectively.1 ul product was loaded on gel.
4.10 CHARACTERIZATION OF 3aPAK -JFH1 (CHIMERA)
3aPAK -JFH1 chimera was characterized for its replication and its infectivity.
4.10.1: In vitro Transcription and Electroporation of 3aPAK -JFH1
p3aPAK-JFH1 was in vitro transcribed, the RNA was electroporated in Huh7 cell
and the chimera virus produced was used to infect naïve Huh7 cells.
.

Chapter 4
116
Figure 4.46: 1% agarose gel showing in vitro transcribed RNA (lane
1,lane 2) from 3aPAK-JFH1and JFH1C6S4 respectively. Lane 3
shows 1Kb marker.
4.10.2 Immuno fluorescent labeling.
For visualizing replication and infection positive cells, cells grown on cover slips
were fixed and labelled as described in section 3. For this labelling ACAP 27, NS5a
and AP33 was used as anti Core, NS5a and E2 antibodies respectively and the images
were taken using LSM710 Confocal microscope (Zeiss) images were assembled by
using Adobe Photoshop software (Fig. 4.47-4.50).

Chapter 4
117
Figure 4.48: Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour
after transformation with 3a PAK-JFH1 RNA.
Figure 4.47: Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour
after transformation with JFH1C6S4 RNA.

Chapter 4
118
Figure 4.49: Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour
after infection with cell cultured derived JFHI virus.
Figure 4.50: Huh 7 cells fixed and stained with E2 antibody AP33, 48 hour
after infection with cell culture derived 3a PAK-JFH1 recombinant virus.

Chapter 4
119
Replication
10000
100000
1000000
10000000
100000000
24 hr 28 hr 72 hr 96 hr
Gluc‐R
LU
3aP AK ‐J F H1J F H1C S A4
Figure 4.51: 3aPAK-JFH1 replication vs JFH1CSA4. Replication is shown as
Gluc/Rluc based luminescence. Data is represented in logarithmic scale.
4.10.3 Luciferase Assay for Replication and Infectivity of 3aPAK-JFH1 Chimera
In order to quantify chimeric virus replication, electroporated cells were lysed at
48 hours and 72 hours interval. Luciferase activity was measured using bright-GloTM
luciferase Assay System (Fig. 4.51). For measuring infectivity of the 3aPAK-JFH1
chimera the infected Huh 7 cells were harvested at 24, 48 and 72 hours post infection and
luciferase activity was measured (Fig 4.52).

Chapter 4
120
1000
10000
100000
1000000
10000000
100000000
48 hr 72 hr 92 hr
Gluc‐RLU
Infection 3aPAK‐JFH1
JFH1CSA4
Figure 4.52: 3aPAK-JFH1 infection vs JFH1CSA4. Infection is shown as Gluc/Rluc base luminescence. Data is represented in logarithmic scale.

121
DISSCUSSION
The present study characterizes structural proteins of HCV 3a genotype isolated
from HCV infected Pakistani patients. HCV Core-NS2 region were amplified and cloned
from patients diagnosed with 3a genotype. The clones were sequenced and multiple
sequence alignment was performed. Since the focus of the study was structural proteins
and development of invitro models HCVpp and HCVcc, a separate mutational and
phylogenetic analysis was performed for HCV Core and envelope proteins E1E2.
Viral capsid proteins have been proposed previously as targets for anti-viral drugs
because they are highly conserved, are essential for viral assembly, self-assemble in well-
controlled conditions and easy to assay in vitro (Prevelige, 1998). HCV Core is the most
conserved of all HCV proteins, across the 6 major genotypes (Meurs and Breiman, 2007).
In addition to its putative role as a potential vaccine candidate it is an important factor for
viral morphogenesis, interaction with various cellular proteins, involved in multiple
cellular and/or pathogenic processes, such as apoptosis, gene transcription, lipid retention
and cell signalling (Giannini and Brechot, 2003). There is limited data available on
sequence analysis of HCV 3a genotype (Waheed et al., 2010) from Pakistan, therefore
the recent study was undertaken to characterize HCV from this region. In the current
study 16 clones of Core-NS2 regions are aligned and separate mutational (as seen in
genomic sequence from patients) and phylogenetic analyses are performed for Core and
envelop proteins of HCV (Table 4.1).

122
Various studies on molecular characterization of HCV Core protein have identified three
major domains and key amino acids within these domains involved in viral assembly,
interaction with cellular factors leading to cell transformation and viral survival.
Amino acid sequence analysis of present study (Fig. 4.6) illustrate the major
changes in Pakistani isolates are in the amino terminus of Core. N-terminus of HCV Core
plays a significant role in capsid assembly. The most important residue for capsid
assembly are basic amino acids present with in two separate clusters (8-25 amino acid
and 39-64 amino acid) (Klein et al., 2005). Two of the analyzed clones i.e. HQ108093
and HQ108094 (C2, C3; table 4.2) showed an amino acid shift S2R and T3L. P7L
transition in clone HQ108097 (C6) and HQ108098 (C7) is noticed. The most consistent
mutation is Q8R and is present in 12 out of 16 clones included in this study. These amino
acid changes as observed in patients may modulate virus assembly and interaction with
various host factors.
Core-STAT-1 interaction is reported to results in down regulation of interferon
signalling, leading to immuno suppression (Lin et al., 2005). N terminus (1-23) residues
of HCV Core are known to block interferon signalling of infected cells by interaction
with STAT-1 (SH2 domain) and helps virus to resist the interferon therapy (Lin et al.,
2006). Interferon resistance in HCV is on the rise which is an alarming situation,
resulting in therapy failure, leading to chronicity. Interestingly all the four patients
included in this study were under Interferon Ribavirin treatment. HCV isolates included
in the present study have shown significant changes in N terminus that fall in Core-
STAT-1 interaction domain. The patient serum was taken in July/August 2009 and when
followed after the completion of the study in April 2011; three out of four patients

123
cleared HCV. We were interested to see whether these mutations have any role in viral
clearance. In slico interaction showed that all of HCV isolates from these patients
exhibited mutations that results in interrupted interaction between Core and STAT-1 at
position 2, 3, 8 and 23. There is a possibility that these contact losses resulted in
improved interferon response and helped in viral clearance. Interestingly in C6 and C7
HCV has managed to counteract the loss of STAT1 interaction at position 8 by P>L shift
at position 7 that resulted in establishment of new interaction with V 642/ Ile 647 of
STAT-1(Table 4.3). This new contact may modulate STAT-1 signalling. Core is
relatively conserved however this study suggests that Interferon Ribavirin therapy may
confer mutational pressure on HCV that results in loss of Core STAT1 interaction. Data
from previous studies support introduction of mutations in capsid and NS3as one of the
antiviral mechanisms of Ribavirin against HCV infection (Vuillermoz et al., 2004; Mihm
et al., 2010; Hofmann et al., 2008). In the present study some other adaptive mutations
are observed that lead to evolution of HCV variants capable of escaping neutralizing
antibodies. Particularly, in C13-C16 where two STAT1 interacting residues at position 8
and 23 are mutated. These HCV isolates perhaps have adapted to some extent by
producing escape variants, utilizing glycosylation site shift in E2 that help virus to escape
neutralizing antibodies, a very critical observation of this study. However, Insilco results
of this study need to be further corroborate utilizing the experimental model in the wet
lab. These results may contribute towards characterization of the STAT1 interaction with
Core and help in understanding of interferon responsiveness.
An interferon induced double stranded RNA activated PKR is a key component of
antiviral and anti proliferative effect of interferon. Yan et al. (2007) reported that 1-58

124
amino acids at N-terminus of Core protein are responsible for direct interaction with PKR
and results in transformation of the cells. Major mutations as described in the current
studies are in the PKR interacting domain thus suggesting that PKR interaction
modulations may contribute to interferon response.
Additional cellular factors that may contribute to interferon responsiveness
include the Dead-box RNA helicase protein (DDX3X) which is an essential cellular
protein for the HCV life cycle. The HCV Core amino acid 16-36 has been shown to
interact with DDX3X and helps virus survival in host (Sun et al., 2010). This domain is
necessary for viral survival in the host and is almost conserved in our local isolates.
In Core protein two domains, amino acids 1-48 and 178-187 show the molecular
mimicry with cellular proteins. First domain is homologous to GOR (Vitozzi et al., 2002)
and second to p450. These regions may involve in autoimmunity due to mimicry with
cellular proteins (Barban et al., 2000). There were certain mutations in GOR homology
domain in the studied Pakistani HCV isolates which may no longer show the molecular
mimicry with GOR but the p450 homologous domain is very stable and is almost
conserved in all isolates except HQ108092 which has H187R mutation.
The patients infected with 3a genotype show higher prevalence and severity of
liver steatosis. The Y164F mutation in 3a genotype results in severe steatosis because
phenylalanine has high affinity for lipids than tyrosine (Hourioux et al., 2007). The
sequencing results of this study are consistent with previous findings about 3a genotype
and all the Pakistani viral isolates harbour the phenylalanine at 164 hence showing
functional significance of this residue in HCV induced steatosis.

125
The amino acids 88-106 in Core protein are reported to be essential for HCV-LP
assembly and morphogenesis (Hourioux et al., 2007). This domain is quite stable, not
even a single amino acid change was visible thus this domain may be a potential
candidate for vaccine development.
Further experimental studies are recommended for the N terminal domain of core
involve in capsid assembly, PKR and STAT-1 interaction. Viral C-PKR and STAT-1
interactions are critical determinant for the outcome of liver disease. The patients
included in this study were with active HCV infection so the investigation of the changes
in these areas may help to some extent in understanding the underlying mechanism of
disease progression.
Envelop glycoproteins of HCV are the subject of intensive investigation due to its
interaction with cellular receptors and immune system, therefore, this study was further
extended to define the structural functional motifs involve in these pathways. Marked
differences exist within E1/E2 of various HCV genotypes. Tryptophan 192 marks the
initiation of E1 preceded by a small stretch of hydrophobic residues 172-191 (Walewski
et al., 2002; Fig. 4.6) characterized as signal sequence for E1 (Okamoto et al., 2004).
Envelop protein E1 of 3a GT differ from 1a genotype at 5'end. Most significant change is
at position 193 where glutamine is changed to Glutamic acid followed by an introduction
of bulky hydrophobic amino acid Tryptophan replacing much smaller Valine. Since this
amino acid lie very close to proteolytic signal sequence an introduction of negatively
charged amino acid may influence the enzymatic activity of host signal peptidase (Liu et
al., 2007).

126
Cysteine is involved in formation of disulphide bridges in protein structure. In
two of the studied clones HQ108093 and HQ108094 Tyrosine 214 is being replaced with
Cysteine and this position is followed by presence of charged residues like Aspartic acid
or Glysine at position 217 and Glutamic acid at position 218. Presence of this Cysteine
might have some significance in protein folding as indicated by surrounding charged
residues. In contrast to E2, tertiary structure of E1 is not well characterized and therefore
one can only speculate role of Cysteine in E1 protein folding at certain position.
A hot debate is going on about the central hydrophobic domain of E1
glycoprotein being a fusion peptide, though the exact location of the fusion peptide is not
yet confirmed. Recent studies proposed amino acids 260 to 290 as a putative fusion
peptide (Hasio et al., 2009). All critical residues G/M at 267, C 272, G278, D 279, C 281,
G 282, and G/A/S 288 of the proposed fusion peptide like domain of E1 are well
conserved in all clones of 3a included in this study. An interesting shift at position 260
Argenine in 1a to Serine in 3a and a contrary shift at 294 Serine in 1a to Argenine in 3a
seem important. These position shifts of Serine might have some implication in fusion
process or in post fusion signalling as most Serine or Threonine residues are often
involve in signal transduction.
Knowledge about the topology of the HCV E1/E2 is still emerging (Albeca et al.,
2011) and particularly the role of amino acids from 300 to 330 is not well characterized.
Key alterations were observed with in this segment of E1 including Threonine 301 to
Valine in HCV 3a, whereas Aspartic acid 303 to Threonine in 1a. Maintaining of a
Threonine in this segment of the protein with a slight shift in position may indicate its
putative functional role.

127
In glycoprotein E2, 18 highly conserved Cysteine residues are reported to
maintain 9 disulfide bonds that help in maintaining the secondary structure of the protein
(Fenouillet et al., 2008). Cysteine at position 459 is involved in E2 folding and makes a
disulfide bridge with another Cysteine at position at 452. In one of the studied clones HQ
108103 Cysteine 459 is mutated to Arginine. Another disulfide bridge reported at
position 503 and 508 (Kery et al., 2010) is mutated in clone HQ10809, Cysteine to
Argenine. Despite of the fact that both of these Cysteine (Cysteine bridge 2 and 4) are
very important, their mutation to Argenine do not have detrimental effects on protein
since in both clones pseudotype particles were produced and secreted although had less
infectivity (S4 and R4 respectively Fig. 4.36). Based on the results of present study, it
remains to be investigated to what extant various Cysteine play a role in E2 folding. In
two other clones “Hq108100 and HQ108101” (S1 and S2) Cysteine at position 685 have
been mutated to Tyrosine, at this position the chemical nature of the replaced amino
group is maintained in being the hydrophilic group and the Cysteine at this position is not
involve in any Cysteine bridges and the residue lies in stem loop of the E2 (Kery et al.,
2010).
HCV envelope glycoproteins have highly glycosylated N-terminal ectodomain
(Helle et al., 2007) and a C-terminal TM domain (Cocquerel et al., 2002) referred as type
I TM proteins (Albecka et al., 2011). The glycoprotein E2 heterodimerizes with E1
(Deleersnyder et al., 1997) and interact with several cellular receptors. The most well
characterize CD81 interact with E2 via its large extra cellular loop (Petracca et al., 2000)
and the interacting amino acids are assigned to three different segments in E2. The first
segment (aa 474-492) spans the hyper variable region 1. The CD81 binding motif “Trp-

128
437, Leu-438, Leu-441, Phe-442” that directly involve with the LEL interaction
(Drummer et al., 2006) is well conserved in all of our clones except Trp-437 which is
occupied by Phenyl alanine (F) in genotype 3a and hence may differ in . An interesting
addition of proline is illustrious at position 480 of 3a genotype when compared to
genotype 1a H77 strain. There are two other Proline residues conserved in surrounding,
one at position 484 and other at 494 and seems to involve in maintaining a narrow bend
at this position (Fig. 2.11; Kery et al., 2010). Proline added at 480 and one mutated at
491 (to Alanine) in genotype 3a might be involve in maintaining the narrow bend at this
position and thus maintain the structure of the protein that might be slightly different in
3a.
The second CD81 binding segment is attributed to residue 522-551, Alanine
substitutions of Y527 and W529 results in loss of CD81 binding to E2 (Rothwangl et al.,
2008). Current study is in line with this report and both residues are well conserved in all
the studied clones of 3a genotype.
In CD 81 binding segment II there are 4 Serine/Threonine sites. Threonine 526
and Serine 528 are well conserved across all genotypes. Position 529 is either occupied
by Threonine or Serine in various genotypes and exclusively serine in the present
sequences of genotype 3a. Presence of Serine or Threonine at this location seem
significant and maintenance of Serine /Threonine at this position points towards its
functional involvement or downstream signalling and characterization of these particular
residues seems essential
Residues 530G and 535D are also considered vital for CD81 binding in region II
and are well conserved in all studied clones. In most of the genotypes position 531 is

129
occupied by Glutamic acid except in genotype 1a and 3a (Kery et al., 2010). Most of our
isolates 12 out of 16 contain Glutamic acid at this place while the rest of them had
Alanine. Position 533 is also occupied by acidic residues either Glutamic acid or Aspartic
acid. Presence of negatively charged residues at these positions seems to be of
considerable importance since they lie very close to imperative residues for CD81
binding region II as distribution of charges play critical role in protein protein interaction.
Lying in the CD81binding segment II, position 541 or 542 is occupied by serine or
Threonine. Threonine at 542 in genotype 1a, 1b and genotype 4 while Serine at 541 in 2a,
3a, 2b, genotype 5 and genotype 6. Maintenance of Serine or Threonine at one of the
position points towards its implication in signalling related to post CD81 binding event
and a wet lab study is needed to evaluate this proposition.
Recently a new function is attributed to the stem region of the E2 protein, a small
segment comprising of amino acid 705-715 in the stem region of E2 is found essential for
HCVcc entry. CD and NMR structural analyses of the synthetic peptide containing this
segment revealed the presence of a central amphipathic helix, which is expected to folds
upon membrane binding. Due to its location in the stem region, segment 705-715 is most
likely involved in the re organization of the glycoprotein complexes in the fusion process
(Albecka et al., 2011). Most of the stem region of E2, 660 to 722 is conserved and amino
acid 705 -715 are very well conserved in all the studied clones of 3a genotype and are
expected to have significant functional implications as suggested by Albecka et al.
(2011).
In the present study HCV glycoproteins were cloned and sequenced from
Pakistani patients infected with 3a genotype. The major aim of this study was to develop

130
a surrogate entry model for HCV Pakistani isolate. HCV show geographical tropism and
circulate as highly diverse viral population among various areas of the world is one of the
reasons to develop entry model for particular isolate from this area. HCV glycoproteins
were successfully cloned and expressed in 293T cells and in Huh7 cells. Retrovirus based
pseudotype particles were produced in 293T cells, were collected from the media with
sucrose gradient based ultra centrifugation. These particles were then used to infect naïve
Huh7 cells and their infectivity was measured as luciferase activity of the reporter gene
incorporated into the HCVpp system. Results plotted in figure 4.12 and 4.13 clearly
shows that HCVpp generated from various clones vary in their infectivity. Data clearly
shows that even the clones from same patient are variable in their infectivity. This might
is a display of intra patient quasispecies diversity as HCV circulates in vivo as a complex
population of different but closely related variants (Cristina, 2005). Mutations in HCV
are not homogeneously distributed rather are over-represented in confined hypervariable
regions of E2 and may contribute to structural and functional limitations of various
isolates (Simmonds , 2004; Kuntzen et al., 2007; Ray et al., 2005). Envelop protein differ
in their amino acid composition and shows about 70% homology when intra patient and
90-85% homology when interpatient clones are aligned. This difference in their
composition is responsible for variable infectivity when expressed on the surface of
pseudotype particles.
HCV envelope proteins E1 and E2 are heavily glycosylated and N-linked
glycosylation of these envelop proteins have been implicated in immunogenicity (Liu et
al., 2007). E1 and E2 proteins contain multiple N-glycosylation sites that are well
conserved across various genotypes (Goffard et al., 2005). In the current study all

131
glycosylation sites of E1 are well conserved, however some variations were observed in
glycosylation sites of E2. In E2 glycosylation site E2N7 found in 1a and 2a is absent in
genotype 3a. Thus genotype 3a glycoprotein E2 harbour 10 glycosylation sites instead of
11. In one of the patients a novel glycosylation site was observed at position 499 in all
most all the clones from this patient (patient M, HQ108104-HQ108107; Fig. 4.6),
interestingly most of these clones have lost glycosylation E2N9 except HQ108106/M3.
Using retroviral particles pseudotyped with genotype 1a envelop proteins, recent
studies have depicted potential role of these glycans in protein folding, in HCV entry and
in protection against neutralization (Falkowska et al., 2007; Goffard et al., 2005; Helle et
al., 2007). Certain glycans are essential for correct protein folding, deficient in glycan
E1N1, E1N4, E2N8 or E2N10 strongly affects the incorporation of HCV glycoproteins
into HCVpp (Goffard et al., 2005). In addition mutation of glycosylation sites E2N2 or
E2N4 modify HCVpp infectivity. All of these glycans are well conserved in clones
included in this study.
E2N7 glycan has Genotype-specific role. Recent reports revealed that mutation of
E2N7 glycosylation site showed a robust reduction in HCVcc infectivity (5% compared
to that of the WT).It also caused a slight decrease in secretion of viral particles in
genotype 2a, indicative of a defect in virus entry. Same mutation although decreased the
infectivity of genotype 2a HCVpp but contrary to that resulted in the production of
genotype 1a HCVpp as infectious as wild-type particles (Helle et al., 2010). This
glycosylation site is among the least conserved and is even missing in sequences from
genotypes 3 and 6 (Helle et al., 2007) including all clones of the present study.

132
Glycans associated with viral envelope proteins can alter the entry functions of
these proteins by modifying their affinities with receptors or by affecting their fusion
activities (Sterjovski et al., 2007; Willett et al., 2008). In the present study glycosylation
site E2N9 was not well conserved and was absent in all clones from patient M
(Q1080104-HQ108107). Interestingly these clones had an extra glycosylation site at
position 499 between E2N5 and E2N6. At this site the glycosylation has not been
reported before in any genotype. Since this glycosylation site was present in almost all
clones from Patient M, this mutation seems quite stable and characteristic of the main
isolate from Patient “M. It appears to be an adaptive mutation of this isolate.
The most important finding of the current study reveals the significance of
glycosylation at residue 499. In order to characterize this novel glycosylation, the site
was subjected to PCR based mutagenesis and the Asparagine in Asn-X-Ser/Thr motifs
was replaced with Glutamine, structurally the closest amino acid. HCVpp were produced
with E2 bearing mutation at 499. The Glycosylation site was removed from two clones
M2 and M3and was added to the 3a positive control 3aE2wt. The HCVpp harvested from
293T cell media were used to infect naïve Huh7 cells. Results of the present study (Fig.
4.22) clearly revealed that this novel glycosylation site reduced HCVpp infectivity to
permissive cells. CD81 pull down assay shows an increase in CD81 LEL interaction after
mutation of this glycosylation (Fig. 4.21) from clone M2 and M3; however a clear
increase in CD81interaction is visible when the glycosylation site added to 3aE2wt
contrary to that HCVpp infectivity was significantly reduced. This increase in CD81
interaction suggest that though addition of glycosylation site at position 499 limits CD81
interaction in clone M2 and M3, it’s not only dependent on this particular glycosylation

133
of the residue 499, but is also modulated by other amino acid present in close proximity
as sequence variations exist in 3aE2wt and M2 and M3 clone in this region. This fact is
further supported by the increased CD81 interaction of E2M-499wt when the HVR495 of
3awt was replaced with HVR495 of M2 as compared to the clone (E2M499) from which
only glycosylation site was removed. These results are in line with some recent reports
that point towards some amino acid mutations positioned outside CD81 binding site
modulate neutralizing antibody response and CD81 interaction (Keck et al., 2009). Some
other reports indicate glycosylation sites significance in HCV infectivity such as removal
of a glycan at positions E2N1 and E2N6 increases HCVcc infectivity of JFH1 (GT 2a)
viral progenies, although the increase was not statistically significant these mutations also
render viral particles more sensitive to CD81-LEL inhibition (Helle et al., 2010). In one
of their previous report based on HCVpp system Helle et al. (2007) have demonstrated
that glycans E2N1, E2N6, and E2N11 lie close to the CD81 binding site and modulate
both CD81 and neutralizing antibody binding to E2.
These results are further supported by Owsianka et al. (2006) who identified
conserved residues involved in CD81 interaction close to these glycosylation sites and by
Falkowska et al. (2007) who showed that a soluble form of E2 lacking a glycan at
position E2N1 or E2N6 exhibited improved binding to CD81. All these results indicate
an improved fitness of the E2N1 and E2N6 mutants may be due to a better interaction of
E2 with CD81. Our results indicate the involvement of other amino acid though augment
CD81 interaction, presence of glycosylation at 499 modulate both CD81 interaction and
HCVpp infectivity.

134
Modulation of the humoral immune response by glycans have been reported for
HIV gp120, another highly glycosylated envelope protein. It has been shown that
manifestation and repositioning of glycans on HIV gp120 modify recognition by
neutralizing antibodies and leads to generation of escape variants (Wei et al., 2003).
Individual mutations of glycosylation sites on the HIV envelope glycoprotein could
modify the EC50 of neutralizing antibodies by 1.2- to 2.6-fold, surprisingly simultaneous
deletions of several glycans could change the EC50 of neutralizing antibodies by more
than 100-fold (Wei et al., 2003). For HCV mutants lacking a glycan at position E2N1,
E2N6, or E2N11 show similar results (Helle et al., 2007). In contrast to HIV, removal of
several glycans robustly affected the infectivity of HCVpp. According to the previous
reports glycosylation sites on HCV envelope glycoproteins are much more conserved and
shifting sites are rarely observed as compared to HIV (Zhang et al., 2004). However
recent reports from Helle et al. (2010) and others (Cerino et al., 2001; Cocquerel et al.,
2006 ) documented emergence of adaptive mutants presenting a shifting site at position
E2N1 or lacking a glycan at position E2N6 after serially passaging JFH-1 or J6/JFH-1 in
cell culture. In the human host, however, these glycans must play a central role in
recognition of the CD81 binding site by neutralizing antibodies and in avoiding too-
rapid elimination of HCV by the immune system. This may explain the high level of
conservation of these glycosylation sites in HCV genomic sequences (Ploss et al., 2009).
Although various previous reports advocate conservations of Glycosylation sites in HCV
envelope proteins in genomic sequences, data of the present study revealed that as seen in
HIV, HCV also employ glycosylation shift In vivo to escape neutralization antibodies.
The particular patient in which this rare event was recorded, the Core protein had lost two

135
contact points with STAT1 at position 8 and 23, perhaps due to Ribavirin induced
mutations, and possibly the isolate was under sever interferon stress and used
glycosylation shift as a survival strategy.
Phylogenetic Analysis for HCV Core and Envelop Protein
To establish phylogenetic link of the HCV 3a genotype from Pakistan, nucleotide
sequences of 3a genotype reported from other parts of the world were retrieved from
NCBI data base and were used to set up a phylogenetic link. More data was available for
HCV Core protein as compared to envelop proteins, a separate phylogenetic tree was
generated for the two sets of genes using separate set of data (Fig. 4.29 and 4.30).
Phylogenetic analyses based on gene sequence of HCV Core protein as well as of
envelop proteins from various 3a isolates showed a parallel evolution of HCV in various
parts of the world including Pakistan. Pakistani isolates appeared in separate clades,
seven lines of evolution in case of Core and four in case of glycoproteins E1and E2. This
clearly indicate that 3a genotype in Pakistan is not from a single source, rather might
have various origins.
The rapid evolution of RNA viruses is well documented fact and is the
consequence of the high mutation frequency in RNA virus population. The genomic
RNA of HCV is translated into a single polyprotein in vivo mutation rate is estimated of
1.2 ± 0.3 × 10–4 for and is within the range generally accepted for RNA viruses (Duffy et
al., 2008; Cuevas et al., 2009).
The High rate of mutation had given origin to another theory of quasispecies that states
that an RNA virus population does not consist of a single ‘‘wild-type’’ but instead is an

136
ensemble of complex closely related viruses (Cristina, 2005). This quasispecies is
capable to accomplish very rapid evolution in new environments because it already
harbours large number of potentially favourable mutations within the population. Though
there is a limit to the maximum variability of viral genetic information before it loses
compatibility for its host (Crotty et al., 2001). Extent of quasispecies however may have
important implication in antiviral therapy response. In patients with high genetic
diversity, especially with HVR1 genetic distance more than 0.53 has a significant early
viral response to anti viral therapy (Fan et al., 2009).
The existence of quasispecies is clearly visible in the phylogenetic assemblage of
both Core and Glycoprotein E1E2. For Core protein Clade PK5 and PK7 are the clusters
of HCV clones from the same patient (PK5 patient R and PK7 patient K) and For E1E2
clade PK2 and PK3 are from the same patient while Clade PK1 had two sub clades each
from a different source. Diversity within these clades 1 to 0.92 is a clear evidence of
existence of quasispecies within the patients. The isolates included in PK1 and PK2
showed a considerable diversity having an evolutionary distance from 1 to 0.54. PK3 and
PK4 showing the evolutionary distance of 0.52 between them clearly showing emergence
of divergence. These results indicate that in Pakistan different isolates of 3a genotype in
circulation are evolving.. Isolates in clad PK3, Pk4, Pk5 and PK7 are from the same
patient so showed the strong linkage (1) within the clad. Isolates within PK5 and PK7
have the linkage of less than 1, which is an indicative that divergence has started even in
the isolates of the same patient which support the established fact of quasi specie
existence in HCV.

137
HCV Core protein is the most conserve among all the HCV proteins (Strosberg et
al., 2010) and the appearance of evolution in intra and inter PK clades is of significant
importance. An interesting observation is the evolutionary differences among various
clades emerged in this study particularly from the data of current investigation. In case of
Core gene PK3 and PK4 clades are 0.52 distance apart showing a significant variation
among these two clades, though they had some common origin in past but now have
shown significant divergence and in due course of time seems to have potential to
develop into two independent strain. Similar diversity is also visible within clade PK1
and PK2. Clones included in PK3are from same patient, likewise for PK4, however the
clones in PK1 and PK2 showing diversity ranging from 1 to 0.54 clearly indicate they are
from different source of infection. The divergent lines in both clades 1 and 2 have some
common origin point.
The data is further supported by the second cladogram (Fig 4.8) based on gene
sequence of HCV glycoprotein E1E2. Figure 4.8, clade PK1 is sub divided to two clades
being with 0.61 genetic diversity, like wise clade PK4 with accession numbers
GQ411068 and GQ411069 are with 0.54 genetic diversity as compared to closely linked
clade PK3.
HCV lineages in areas of endemic infection are highly divergent and in general
are isolated from emigrants or residents of restricted geographic regions. This diversity
suggests a long duration of constant infection in these areas. Endemic strains can be
identified for genotypes 1 and 2 in West Africa (Candotti et al., 2003; Jeannel et al.,
1998) and a similar patterns of genetic diversity have been found for genotype 3 in South
Asia, for genotype 6 in East Asia, for genotype 4 in Middle East and Central Africa

138
(Mellor et al., 1995; Ndjomou et al., 2003; Liu et al., 2007). The genetic diversity or
evolutionary divergence as seen among PK3, PK4, within PK1 and PK2 for HCV Core
clearly marks a long term existence of HCV in the population and clearly indicate that
viral population in circulation had undergone some evolutionary advancement and in
future may lead to emergence of new strain. Current diagnostic techniques are unable to
detect about <3% (Safi et al., 2010) to 37 % (Inamullah et al., 2011) HCV genotypes
from Pakistan, 1.7% from Italy (Furion et al., 1999), 5.3% from India (Chaudhuri et al.,
2005). There is a chance that in Pakistan the virus had already diverged enough to escape
current diagnostics and might be prevalent as new isolate at least in some part of the
country as 37% untypable HCV is reported from Swat region of Pakistan (Inamullah et
al., 2011).
Although Pakistani isolates appeared in separate independent clades and showed
parallel origin of genotype 3a in Pakistan with other parts of the world; however there are
some clues indicating a close relation or common origin of some Pakistani isolates with
Japanese and Indian isolates included in this study. Phylogram based on Core sequence
(Fig.4.7) indicate a common origin of PK2 cluster with a Japanese isolate NC 009824.
Another interesting observation in the phylogram is the common origin of Indian isolate
with another Japanese isolate D28917 (K3a). This link is further supported by the second
phylogram based on HCV envelop protein sequence (Fig 4.8). The largest Pakistani clade
PK1 in this phylogram is pointing toward some common line of origin with Japanese
isolate K3a while PK2 had an ancestral link to some Indian isolates (HQ738645,
GU172375, and GQ275355) and one of the isolate from UK (GQ356213). The
appearance of common origin of some of Pakistani isolates with Indian and Japanese and

139
that of Japanese and Indian isolate indicate presence of some common ancestor in the
area. That common ancestor might have diverged into separate but still closely related
viral progenies in these closely located geographical areas especially India and Pakistan
which used to be one geographical unit little more than half century back. Likewise the
link between Pakistani and UK isolate can be attributed to Pakistan being a colony of
England in past. Molecular clock analyses of HCV suggest that various strains have been
present in their particular geographical regions for at least several centuries (Okamoto et
al., 2004). Presence of several lines of origin of HCV 3a is in line with the fact that RNA
viruses have high rate of evolution and HCV had also evolved on several independent
lines in the world including Pakistan.
The results of the present study show an alarming situation as the 3a genotype is
evolving in Pakistan and all over the world. Pakistan is a developing country of 170
million people with low health and educational standards. According to the human
development index of the United Nations, it was ranked 134th out 174 countries (UNDP,
1996). Standard interferon therapy against HCV is effective against 50% of treated
patients (Hadziyannis et al., 2004), in Pakistan 10 million people are presumed to be
infected with HCV (Waheed et al., 2010) and with this evolving divergence it might
possible that local Pakistani isolates may result in more interferon resistance and poor
management of the disease and loss of money as well.
HCVcc Model system for Pakistani Isolate
An appropriate model system for HCV has been a great challenge. Full-length
genomes harbouring adaptive mutations for cell culture system replicated efficiently in
Huh-7 cells but failed to release infectious particles even though the structural proteins

140
were expressed (Hill, 1965; Gao et al., 2004; Khakoo et al., 2004). HCV isolate JFH-1,
genotype 2a obtained from a patient with fulminant hepatitis from Japan changed the
direction of HCV research. For reasons that are yet not understood, subgenomic replicon
derived from JFH-1 cDNA could efficiently replicate in cell culture without any adaptive
mutations (Wakita et al., 2005), later they found even full-length JFH-1 genome was able
to produces infectious particles in cell culture with a moderate titers.
Lindenbach et al. (2006) later made a chmeric genome with FH-1 replicase and
the Core–NS2 region from a related genotype 2a strain, J6. The chimera virus replicated
in cell culture and produced vigorous levels of infectious virus (HCVcc), nearly 105
infectious units ml–1 within 48 h of infection in Huh-7.5 cells. Pietschman et al., (2006)
constructed and characterized a panel of intragenotypic and intergenotypic HCV
chimeras that were as competent or even superior to the JFH1 WT genome with respect
to assembly and secretion of infectious virus particles. They maped alternative junctions
between the Con1-derived structural region and JFH1and identified a crossover site in
the N-terminal region in the loop connecting NS2TMD1and TMD2 that allowed much
higher virus production when compared with the original crossover site at the C terminus
of NS2 (13).
In the present study JFH1 chimera was constructed with structural proteins
encoded by 3a isolate from Pakistan using the optimal reported site between loop
connecting NS2 TMD1 and TMD2 (Fig 4.35). The infectivity and replication of the
chimeric virus 3aPAK-JFH1 was assessed using Gousia luciferase reporter assay.
Replication and infection of the recombinant virus was found 1 log less than the JFH1
counterpart (Fig. 4.51, 4.52). The results are in line with other reports for JFH Chimera.

141
In most cases chimera viruses are less infectious when Core-NS2 region is incorporated
from a distal genotype and may have equal or better when the same region is taken from
some other isolate of the same genotype such as J6 from 2a (Pietschman et al., 2006).
Another important factor responsible for low infectivity of the chimera could be
the presence of Glycosylation site at N6 of E2. N6 in E2 is located at position 533 in the
poly protein and lies within CD81 binding region (Fig.4.6). Removal of this
glycosylation site increase infectivity in HCVcc as well modulates infection of HCVpp
system. This particular glycosylation site has been removed in JFH1C6S4 used as
positive control. In addition to N6, at position 499 there is an extra glycosylation site
present between N6 and N5 in E2 of the particular clone used for construction of the
chimera. This novel glycosylation site reduces infectivity of HCVpp in Huh7 cells and
might also be responsible for reduced infectivity in HCVcc system.

142
SUMMARY
HCV show geographical tropism and circulate as highly diverse viral population
among various areas of the world. Continuous surveillance remains a high priority for
vaccine development as antigenic variability and immune escape of the virus is
widespread. To contribute in the efforts of vaccine development this study was initiated
to incorporate relevant genetic information and detail analysis of HCV structural proteins
(3a GT) from Pakistan.
Virological parameters were assessed for fourteen patients and Core-NS2 region
was cloned and characterized from four Ribavirin treated patients. Sequence analysis of
the clones revealed presence of significant mutations in STAT1 interacting domain of
Core (1-34 aa). Results of the in slico modelling suggest that these mutations have the
potential to modulate Core-STAT1 interaction resulting in better interferon response and
hence could be one possible explanation of viral clearance in ¾ patients.
To my knowledge, my data reveals for the first time that HCV also employ
glycosylation shift to escape neutralizing antibodies circulating in patient’s serum, a
phenomena previously known for HIV. A novel glycosylation shift in E2 was recorded in
the current study and was characterized by employing HCVpp system. This glycosylation
shift though reduce HCV infectivity in HCVpp system, at the same time provide
significant escape from neutralizing antibodies circulating in pooled serum from HCV
infected patients. Interestingly in this fastidious patient, the Core protein had lost two
contact points with STAT1 at position 8 and 23 and suggesting that perhaps the isolate
was under severe interferon stress and used glycosylation shift as a survival strategy.
Here I postulate that Interferon Ribavirin therapy induce mutations in selected regions of

143
HCV (N terminal domain of Core) that may favour a better response of the host immune
system; however at the same time these mutations may confer pressure on virus that
consequence in mutations, in other regions, favouring viral survival. An extensive
sequence comparison of treated vs untreated patients is recommended for more
conclusive statement in this regard.
Phylogenetic analyses of HCV Core and E1/E2 proteins from various 3a isolates
indicated a parallel evolution of HCV in different parts of the world including Pakistan.
This is consistent with several evolutionary lines indicating a long term existence of HCV
in this area. Phylogram based on Core and E1E2 sequence indicate a common
evolutionary origin for few Pakistani isolates with HCV 3a GT reported from Japan and
India. The results indicate a noteworthy situation as the 3a genotype is evolving in
Pakistan and all over the world. The dynamic flux within the HCV genome may
contribute for genesis of new genotypes as it has been reported 37% untypable cases
occur in some parts of Pakistan (Swat region).
Another major aim of the study was to develop HCVpp and HCVcc system from
local isolate. HCVpp system from the local isolate was well employed for
characterization of the novel glycosylation site. Replication and infection of the
recombinant virus, JFH1 chimera; HCVcc, was found 1 log less than the JFH1
counterpart (JFH1C6S4) used as positive control. Presence of E2N6 and the novel
glycosylation shift reduces infectivity of HCVpp in Huh7 cells and is expected to be
responsible for reduced infectivity in HCVcc system. In summary this study highlighted
the importance of both HCVcc and HCVpp system are functional for screening of
antiviral targets.

144
References
Abbate, I., Lo Iacono, O., Di Stefano, R., Cappiello, G., Girardi, E., Longo, R., Ferraro,
D., Antonucci, G., Di-Marco, V., Solmone, M., Craxì, A., Ippolito, G., and
Capobianchi, M.R. (2004). HVR-1 quasispecies modifications occur early and
are correlated to initial but not sustained response in HCV-infected patients
treated with pegylated- or standard-interferon and ribavirin. J. Hepatol.,
40(5):831-836.
Ai, L., Lee, Y.W., and Chen, S.S. (2009). Characterization of hepatitis C virus core
protein multimerization and membrane envelopment: revelation of a cascade of
core-membrane interactions. J. Virol., 83: 9923-9939.
Ait-Goughoulte, M., Hourioux, C., Patient, R., Trassard, S., Brand, D., and Roingeard, P.
(2006). Core protein cleavage by signal peptide peptidase is required for hepatitis
C virus-like particle assembly. J. Gen. Virol., 87:855-60.
Albecka, A., Montserret, R., Krey, T., Tarr, A.W., Diesis, E., Ball, J.K., Descamps, V.,
Duverlie, G., Rey, F., Penin, F., and Dubuisson, J. (2011). Identification of New
Functional Regions in Hepatitis C Virus Envelope Glycoprotein E2. J. Virol.,
85(4):1777-92.
Allander, T., Drakenberg, K., Beyene, A., Rosa, D., Abrignani, S., Houghton, M., Widell,
A., Grillner, L., and Persson, M.A. (2000). Recombinant human monoclonal
antibodies against different conformational epitopes of the E2 envelope

145
glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen.
Virol., 81(Pt 10):2451-9.
Alsaleh, K., Delavalle, P.Y., Pillez, A., Duverlie, G., Descamps, V., Rouillé, Y.,
Dubuisson, J., and Wychowski, C. (2010). Identification of basic amino acids at
the N-terminal end of the core protein that are crucial for hepatitis C virus
infectivity. J. Virol., 84(24):12515-28.
Alter, H.J., Holland, P.V., Morrow, A.G., Purcell, R.H., Feinstone, S.M., and Moritsugu,
Y. (1975). Clinical and serological analysis of transfusion-associated hepatitis.
Lancet, 2(7940):838-41.
Alvarez, C.P., Lasala, F., Carrillo, J., Muñiz, O., Corbí, A.L., and Delgado, R. (2002). C-
Type Lectins DC-SIGN and L-SIGN Mediate Cellular Entry by Ebola Virus in
cis and in trans. J. Virol., 76(13):6841-4.
André, P., Perlemuter, G., Budkowska, A., Bréchot, C., and Lotteau, V. (2005). Hepatitis
C virus particles and lipoprotein metabolism. Semin. Liver Dis., 25(1):93-104.
Andréo, U., Maillard, P., Kalinina, O., Walic, M., Meurs, E., Martinot, M., Marcellin, P.,
and Budkowska, A. (2007). Lipoprotein lipase mediates hepatitis C virus (HCV)
cell entry and inhibits HCV infection. Cell Microbiol., 9 (10):2445-56.
Baldick, C.J., Wichroski, M.J., Pendri, A., Walsh, A.W., Fang, J., Mazzucco, C.E.,
Pokornowski, K.A., Rose, R.E., Eggers, B.J., Hsu, M., Zhai, W., Zhai, G.,
Gerritz, S.W., Poss, M.A., Meanwell, N.A., Cockett, M.I., and Tenney, D.J.

146
(2010). A novel small molecule inhibitor of hepatitis C virus entry. PLoS
Pathog., 2:6(9).
Bankwitz, D., Steinmann, E., Bitzegeio, J., Ciesek, S., Friesland, M., Herrmann, E.,
Zeisel, M.B., Baumert, T.F., Keck, Z.Y., Foung, S.K., Pécheur, E.I., and
Pietschmann, T. (2010). Hepatitis C virus hypervariable region 1 modulates
receptor interactions, conceals the CD81 binding site, and protects conserved
neutralizing epitopes. J. Virol., 84(11):5751-63.
Barba, G., Harper, F., Harada, T., Kohara, M., Goulinet, S., Matsuura, Y., Eder, G.,
Schaff, Z., Chapman, M.J., Miyamura, T., and Brechot, C. (1997). Hepatitis C
virus core protein shows a cytoplasmic localization and associates to cellular
lipid storage droplets. Proc. Natl. Acad. Sci. USA, 94: 1200–1205.
Barban, V., Fraysse-Corgier, S., Paranhos-Baccala, G., Petit, M, Manin, C., Berard, Y.,
Prince, A.M., Mandrand, B., and Meulien, P. (2000). Identification of a human
epitope in hepatitis C virus (HCV) core protein using a molecularly cloned
antibody repertoire from a non-symptomatic, anti-HCV-positive patient. J. Gen.
Virol., 81( 2):461-9
Bartosch, B., Bukh, J., Meunier, J.C., Granier, C., Engle, R.E., Blackwelder, W.C.,
Emerson, S.U., Cosset, F.L., and Purcell, R.H. (2003). In vitro assay for
neutralizing antibody to hepatitis C virus: evidence for broadly conserved
neutralization epitopes. Proc. Natl. Acad. Sci. USA, 100(24):14199-204.

147
Bartosch, B., Thimme, R., Blum, H.E., and Zoulim, F. (2009). Hepatitis C virus-induced
hepatocarcinogenesis. J. Hepatol., 51(4):810-20.
Baumert, T.F., Ito, S., Wong, D.T., and Liang, T.J. (1998). Hepatitis C virus structural
proteins assemble into virus like particles in insect cells. J. Virol., 72(5):3827-36.
Beyene, A., Basu, A., Meyer, K., and Ray, R. (2004). Influence of N-linked glycans on
intracellular transport of hepatitis C virus E1 chimeric glycoprotein and its role
in pseudotype virus infectivity. Virology, 324(2):273-85.
Blanchard, E., Belouzard, S., Goueslain, L., Wakita, T., Dubuisson, J., Wychowski, C.,
and Rouillé, Y. (2006). Hepatitis C virus entry depends on clathrin-mediated
endocytosis. J. Virol., 80(14):6964-72.
Blanchard, E., Hourioux, C., Brand, D., Ait-Goughoulte, M., Moreau, A., Trassard, S.,
Sizaret, P.Y., Dubois, F., and Roingeard, P. (2003b). Hepatitis C virus-like
particle budding: role of the core protein and importance of itsAsp111. J. Virol.,
77:10131–10138.
Boulant, S., Montserret, R., Hope, R.G., Ratinier, M., Targett-Adams, P., Lavergne, J.P.,
Penin, F., and McLauchlan, J. (2006). Structural determinants that target the
hepatitis C virus core protein to lipid droplets. J. Biol. Chem., 28:22236-22247.
Bradley, D., McCaustland, K., Krawczynski, K., Spelbring, J., Humphrey, C., and Cook,
E.H. (1991). Hepatitis C virus: buoyant density of the factor VIII-derived isolate
in sucrose. J. Med. Virol., 34(3):206-8.

148
Bradley, D.W., Maynard, J.E., Popper, H., Cook, E.H., Ebert, J.W. McCaustland, K.A.,
Schable, C.A., and Fields, H.A. (1983). Posttransfusion non-A, non-B hepatitis:
physicochemical properties of two distinct agents. J. Infect. Dis., 148(2):254-65.
Bradley, D.W., McCaustland, K.A., Cook, E.H., Schable, C.A., Ebert, J.W. and Maynard,
J.E. (1985). Post-transfusion non-A, non-B hepatitis in chimpanzees.
Physicochemical evidence that a tubule-forming agent is a small, enveloped
virus. Gastroenterology, 85:773-779.
Bressanelli, S., Stiasny, K., Allison, S.L., Stura, E.A., Duquerroy, S., Lescar, J., Heinz,
F.X., and Rey, F.A. (2004). Structure of a flavivirus envelope glycoprotein in its
low-pH-induced membrane fusion conformation. EMBO J., 23(4):728-38.
Bruni, R., Costantino, A., Tritarelli, E., Marcantonio, C., Ciccozzi, M., Rapicetta, M., El
Sawaf, G., Giuliani, A., and Ciccaglione, A.R. (2009). A computational approach
identifies two regions of Hepatitis C Virus E1 protein as interacting domains
involved in viral fusion process. BMC Struct. Biol., 9:48.
Bull, R.A., Hyde, J., Mackenzie, J.M., Hansman, G.S., Oka, T., Takeda, N., and White,
P.A. (2011). Comparison of the replication properties of murine and human
calicivirus RNA-dependent RNA polymerases. Virus Genes, 42(1):16-27.
Burlone, M.E., and Budkowska, A. (2009). Hepatitis C virus cell entry: role of
lipoproteins and cellular receptors. J. Gen. Virol., 90(5):1055-70.
Callens, N., Ciczora, Y., Bartosch, B., Vu-Dac, N., Cosset, F.L., Pawlotsky, J.M., Penin,
F., and Dubuisson, J. (2005). Basic residues in hypervariable region 1 of

149
hepatitis C virus envelope glycoprotein e2 contribute to virus entry. J. Virol.,
79(24):15331-41.
Candotti, D., Temple, J., Sarkodie, F., and Allain, J.P. (2003). Frequent recovery and
broad genotype 2 diversity characterize hepatitis C virus infection in Ghana,
West Africa. J. Virol., 77(14): 7914-23.
Carrère-Kremer, S., Montpellier, C., Lorenzo, L., Brulin, B., Cocquerel, L., Belouzard,
S., Penin, F., and Dubuisson, J. (2004). Regulation of hepatitis C virus
polyprotein processing by signal peptidase involves structural determinants at the
p7 sequence junctions. J. Biol. Chem., 279(40):41384-92.
Carrère-Kremer, S., Montpellier-Pala, C., Cocquerel, L., Wychowski, C., Penin, F., and
Dubuisson, J. (2002). Subcellular localization and topology of the p7 polypeptide
of hepatitis C virus. J. Virol., 76(8):3720-30.
Cerino, A., Meola, A., Segagni, L., Furione, M., Marciano, S., Triyatni, M., Liang, T.J.,
Nicosia, A., and Mondelli, M.U. (2001). Monoclonal antibodies with broad
specificity for hepatitis C virus hypervariable region 1 variants can recognize
viral particles. J. Immunol., 167(7):3878-86.
Chambers, T.J., Nestorowicz, A., Amberg, S.M., and Rice, C.M. (1993). Mutagenesis of
the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-
NS3 complex formation, and viral replication. J. Virol., 67(11):6797-807.

150
Chaudhuri, S., Das, S., Chowdhury, A., Santra, A., Bhattacharya, S.K., and Naik, T.N.
(2005). Molecular epidemiology of HCV infection among acute and chronic
liver disease patients in Kolkata, India. J. Clin. Virol., 32 38–46.
Chen, S., and Wang, Y.M. (2005). Multigene tracking of quasispecies in viral persistence
and clearance of hepatitis C virus.World J. Gastroenterol., 11(19):2874-84.
Chiba, H., Osanai, M., Murata, M., Kojima, T., and Sawada, N. (2008). Transmembrane
proteins of tight junctions. Biochim. Biophys. Acta, 1778(3):588-600.
Choo, Q.L., Kuo, G., Weiner, A.J., Overby, L.R., Bradley, D.W. and Houghton, M.
(1989). Isolation of a cDNA clone derived from a blood-borne non-A, non-B
viral hepatitis genome. Science, 244: 359-362.
Choo, Q.L., Richman, K.H., Han, J.H., Berger, K., Lee, C., Dong, C., Gallegos, C., Coit,
D., Medina-Selby, A., Barr, P.J., Weiner, A.J., Bradley, D.W., Kuo, G.
Houghton, M. (1991). Genetic organization and diversity of the hepatitis C virus.
Proc. Natl. Acad. Sci. USA, 88:2451-2455.
Choukhi, A., Ung, S., Wychowski, C., and Dubuisson, J. (1998). Involvement of
endoplasmic reticulum chaperones in the folding of hepatitis C virus
glycoproteins. J. Virol., 72(5):3851-8.
Ciczora, Y., Callens, N., Penin F,,Pécheur, E.I., and Dubuisson, J. (2007).
Transmembrane domains of hepatitis C virus envelope glycoproteins: residues
involved in E1E2 heterodimerization and involvement of these domains in virus
entry. J. Virol., 81(5):2372-81.

151
Ciesek , S., Steinmann, E., Wedemeyer, H., Manns, MP., Neyts, J., Tautz, N., Madan,
V., Bartenschlager, R., von Hahn, T., and Pietschmann, T. (2009). Cyclosporine
A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A.
Hepatology, 50(5):1638-45.
Cocquerel, L., Duvet, S., Meunier, J.C., Pillez, A., Cacan, R., Wychowski, C., and
Dubuisson, J. (1999). The transmembrane domain of hepatitis C virus
glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J.
Virol., 73(4):2641-9. (PMID: 10074109).
Cocquerel, L., Meunier, J.C., Pillez, A., Wychowski, C., and Dubuisson, J. (1998). A
retention signal necessary and sufficient for endoplasmic reticulum localization
maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J.
Virol., 72(3):2183-91.
Cocquerel, L., Op de Beeck, A., Lambot, M., Roussel, J., Delgrange, D., Pillez, A.,
Wychowski, C., Penin, F., and Dubuisson, J. (2002). Topological changes in the
transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J.,
21(12):2893-902.
Cocquerel, L., Voisset, C., and Dubuisson, J. (2006). Hepatitis C virus entry: potential
receptors and their biological functions. J. Gen. Virol., 87(Pt 5):1075-84.
Cocquerel, L., Wychowski, C., Minner, F., Penin, F., and Dubuisson, J. (2000). Charged
residues in the transmembrane domains of hepatitis C virus glycoproteins play a

152
major role in the processing, subcellular localization, and assembly of these
envelope proteins. J. Virol., 74(8):3623-33.
Cormier, E.G., Durso, R.J., Tsamis, F., Boussemart, L., Manix, C., Olson, W.C., Gardner,
J.P., and Dragic, T. (2004). L-SIGN (CD209L) and DC-SIGN (CD209) mediate
transinfection of liver cells by hepatitis C virus. Proc. Natl. Acad. Sci. USA,
101(39):14067-72.
Cribier, B., Schmitt, C., Bingen, A., Kirn, A., and Keller, F. (1995). In vitro infection of
peripheral blood mononuclear cells by hepatitis C virus. J. Gen. Virol.,
76( 10):2485-91.
Cristina, J. (2005). Genetic diversity and evolution of hepatitis C virus in the Latin
American region. J. Clin.Virol., 34 Suppl 2:S1-7.
Cristofari, G., Ivanyi-Nagy, R., Gabus, C., Boulant, S., Lavergne, J.P., Penin, F., and
Darlix, J.L. (2004). The hepatitis C virus Core protein is a potent nucleic acid
chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic
Acids Res., 32: 2623-2631.
Crotty, S., Cameron, C.E., and Andino, R. (2001). RNA virus error catastrophe: direct
molecular test by using ribavirin. Proc.Natl. Acad. Sci. USA, 98(12):6895-900.
Cuevas, J.M, González-Candelas, F., Moya, A., Sanjuán, R. (2009). Effect of ribavirin on
the mutation rate and spectrum of hepatitis C cirus In Vivo. J. Virol.,
83(11):5760-4.

153
Deforges, S., Evlashev, A., Perret, M., Sodoyer, M., Pouzol, S., Scoazec, J.Y., Bonnaud,
B., Diaz, O., Paranhos-Baccalà, G., Lotteau, V., and André, P. (2004).
Expression of hepatitis C virus proteins in epithelial intestinal cells in vivo. J.
Gen. Virol., 85:2515-23.
Delang, L., Coelmont, L., and Neyts J. ( 2010). Antiviral Therapy for Hepatitis C Virus:
Beyond the Standard of Care. Viruses, 2(4):826–866.
Delang, L., Coelmont, L., and Neyts, J. (2010). Antiviral Therapy for Hepatitis C Virus:
Beyond the Standard of Care. Viruses, 2(4):826–866.
Deleersnyder, V., Pillez, A., Wychowski, C., Blight, K., Xu, J., Hahn, YS., Rice, C.M.,
and Dubuisson, J. (1997). Formation of native hepatitis C virus glycoprotein
complexes. J. Virol., 71(1):697-704.
Diaz, O., Delers, F., Maynard, M., Demignot, S., Zoulim, F., Chambaz, J., Trépo, C.,
Lotteau, V., and André, P. (2006). Preferential association of Hepatitis C virus
with apolipoprotein B48-containing lipoproteins. J. Gen. Virol., 87(Pt 10):2983-
91.
Dimitrova, M., Imbert, I., Kieny, M.P., and Schuster, C. (2003). Protein-protein
interactions between hepatitis C virus nonstructural proteins. J. Virol.,
77(9):5401-14.
Dreux, M., Boson, B., Ricard-Blum, S., Molle, J., Lavillette, D., Bartosch, B., Pécheur,
E.I., and Cosset, F.L. (2007). The exchangeable apolipoprotein ApoC-I promotes
membrane fusion of hepatitis C virus. J. Biol. Chem., 282(44):32357-69.

154
Drummer, H.E., Boo, I., Maerz, A.L., and Poumbourios, P. (2006). A conserved Gly436-
Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a
determinant of CD81 binding and viral entry. J. Virol., 80(16):7844-53.
Drummer, H.E., Wilson, K.A., and Poumbourios, P. (2005). Determinants of CD81
dimerization and interaction with hepatitis C virus glycoprotein E2. Biochem.
Biophys. Res. Commun., 328(1):251-7.
Duffy, S., Shackelton, L.A., and Holmes, E.C. (2008). Rates of evolutionary change in
viruses: patterns and determinants. Nat. Rev. Genet., 9(4):267-76.
Duvignaud, J.B., Savard, C., Fromentin, R., Majeau, N., Leclerc, D., and Gagné, S.M.
(2009). Structure and dynamics of the N-terminal half of hepatitis C virus core
protein: an intrinsically unstructured protein. Biochem. Biophys. Res. Commun.,
378(1):27-31.
Ellgaard, L., Molinari, M. and Helenius, A. (1999). Setting the standards: quality control
in the secretory pathway. Science, 286(5446):1882-8.
Engering, A., Geijtenbeek, T.B., van Vliet, S.J., Wijers, M., van Liempt, E., Demaurex,
N., Lanzavecchia, A., Fransen, J., Figdor, C.G., Piguet, V., and Van-Kooyk, Y.
(2002). The dendritic cell-specific adhesion receptor DC-SIGN internalizes
antigen for presentation to T cells. J. Immunol, 168(5):2118-26.
Eyre, N.S., Drummer, H.E., and Beard, M.R. (2010). The SR-BI partner PDZK1
facilitates hepatitis C virus entry. PLoS Pathog., 6(10).

155
Ezelle , H.J., Markovic, D., and Barber , GN. (2002). Generation of hepatitis C virus-like
particles by use of a recombinant vesicular stomatitis virus vector. J. Virol.,
76(23):12325-34.
Falkowska, E., Kajumo, F., Garcia, E., Reinus, J., and Dragic, T. (2007). Hepatitis C
virus envelope glycoprotein E2 glycans modulate entry, CD81 binding and
neutralization. J. Virol., 81:8072-8079.
Fan, X., Mao, Q., Zhou, D., Lu, Y., Xing, J., Xu, Y., Ray, S.C., and Di Bisceglie, A.M.
(2009). High Diversity of Hepatitis C Viral Quasispecies is Associated with
Early Virological Response in Patients undergoing Antiviral Therapy.
Hepatology, 50(6):1765-1772.
Farci, P., Alter, H.J., Wong, D.C., Miller, R.H., Govindarajan, S., Engle, R., Shapiro, M.,
and Purcell, R.H. (1994). Prevention of hepatitis C virus infection in
chimpanzees after antibody-mediated in vitro neutralization. Proc. Natl. Acad.
Sci. USA, 91(16):7792-6.
Farci, P., and Purcell, R.H. (2000). Clinical significance of hepatitis C virus genotypes
and quasispecies. Semin. Liver Dis., 20(1):103-26.
Farci, P., Shimoda, A., Wong, D., Cabezon, T., De Gioannis, D., Strazzera, A., Shimizu,
Y., Shapiro, M., Alter, H.J., and Purcell, R.H. (1996). Prevention of hepatitis C
virus infection in chimpanzees by hyperimmune serum against the hypervariable
region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA, 93(26):15394-9.

156
Farci, P., Strazzera, R., Alter, H.J., Farci, S., Degioannis, D., Coiana, A., Peddis, G.,
Usai, F., Serra, G., Chessa, L., Diaz, G., Balestrieri, A., and Purcell, R.H. (2002).
Early changes in hepatitis C viral quasispecies during interferon therapy predict
the therapeutic outcome. Proc. Natl. Acad. Sci. USA, 99(5):3081-6.
Feinstone, S.M., Kapikian, A.Z., Purcell, R.H., Alter, H.J., and Holland, P.V. (1975).
Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J.
Med., 292(15):767-70.
Feinstone, S.M., Mihalik, K.B., Kamimura, T., Alter, H.J., London, W.T., and Purcell,
R.H. (1983). Inactivation of hepatitis B virus and non-A, non-B hepatitis by
chloroform. Infect. Immun., 41(2):816-21.
Fenouillet, E., Lavillette, D., Loureiro, S., Krashias, G., Maurin, G., Cosset, F.L., Jones,
I.M., and Barbouche, R. (2008). Contribution of Redox Status to Hepatitis C
Virus E2 Envelope Protein Function and Antigenicity. J. Biol. Chem.,
283(39):26340-26348.
Flint, M., Maidens, C., Loomis-Price, L.D., Shotton, C., Dubuisson, J., Monk, P.,
Higginbottom, A., Levy, S., and McKeating, J.A. (1999). Characterization of
hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor,
CD81. J. Virol., 73(8):6235-44.
Flint, M., von Hahn, T., Zhang, J., Farquhar, M., Jones, C.T., Balfe, P., Rice, C.M., and
McKeating, J.A. (2006). Diverse CD81 proteins support hepatitis C virus
infection. J. Virol., 80(22):11331-42.

157
Forns, X., Purcell, R.H., and Bukh, J. (1999). Quasispecies in viral persistence and
pathogenesis of hepatitis C virus. Trends Microbiol., 7(10):402-10.
Forns, X., Thimme, R., Govindarajan, S., Emerson, S.U., Purcell, H.R., Francis, V., and
Bukh, C.J. (2000). Hepatitis C virus lacking the hypervariable region 1 of the
second envelope protein is infectious and causes acute resolving or persistent
infectionin chimpanzees. . Proc. Natl. Acad. Sci. USA, 97(24):13318 –13323.
Fournier, C., Duverlie, G., François, C., Schnuriger, A., Dedeurwaerder S., Brochot E.,
Capron D., Wychowski C., Thibault V., and Castelain S. (2007). A focus
reduction neutralization assay for hepatitis C virus neutralizing antibodies. Virol.
J., 4:35.
Fournillier Jacob, A., Cahour, A., Escriou, N., Girad, M., and Wychowski, C. (1996).
Processing of the E1 glycoprotein of hepatitis C virus expressed in mammalian
cells. J. Gen. Virol., 77:1055-64.
Fournillier, A., Wychowski, C., Boucreux, D., Baumert, T.F., Meunier, J.C., Jacobs, D.,
Muguet, S., Depla, E., and Inchauspé, G. (2001 Dec). Induction of hepatitis C
virus E1 envelope protein-specific immune response can be enhanced by
mutation of N-glycosylation sites. J. Virol., 75(24):12088-97.
Fromentin, R., Majeau, N., Laliberté-Gagné, M.E., Boivin, A., Duvignaud, J.B., and
Leclerc, D. (2007). A method for in vitro assembly of hepatitis C virus core
protein and for screening of inhibitors. Anal. Biochem., 366(1):37-45.

158
Furione, M., Simoncini, L., Gatti, M., Baldanti, F., GraziaRevello, M., and Gerna, G.
(1999). HCV genotyping by three methods: Analysis of discordant results based
on sequencing. J. Clin. Virol., 13:121-130.
Gal-Tanamy, M., Keck, Z.Y., Yi, M., McKeating, J.A., Patel, A.H., Foung, S.K., and
Lemon, S.M. (2008). In vitro selection of a neutralization-resistant hepatitis C
virus escape mutant. Proc. Natl. Acad. Sci. USA, 105(49):19450-5.
Gao, B., Hong, F., and Radaeva, S. (2004). Host factors and failure of interferon-alpha
treatment in hepatitis C virus. Hepatology, 39: 880–890.
Gardner, J.P., Durso, R.J., Arrigale, R.R., Donovan, G.P., Maddon, P.J., Dragic, T., and
Olson, W.C. (2003). L-SIGN (CD 209L) is a liver-specific capture receptor for
hepatitis C virus. Proc. Natl. Acad. Sci. USA, 100(8):4498-503.
Garry, R.F., and Dash, S. (2003 Mar 15). Proteomics computational analyses suggest that
hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class
II fusion proteins. Virology, 307(2):255-65.
Gaudy, C., Moreau, A., Veillon, P., Temoin, S., Lunel, F., and Goudeau, A. (2003).
Significance of pretreatment analysis of hepatitis C virus genotype 1b
hypervariable region 1 sequences to predict antiviral outcome. J. Clin.
Microbiol., 41(8):3615-22.
Geijtenbeek, T.B., Kwon, D.S., Torensma, R., van Vliet, S.J., van Duijnhoven, G.C.,
Middel, J., Cornelissen, I.L., Nottet, H.S., KewalRamani, V.N., Littman, D.R.,
Figdor, C.G., and van Kooyk, Y. (2000). DC-SIGN, a dendritic cell-specific

159
HIV-1-binding protein that enhances trans-infection of T cells. Cell, 100(5):587-
97.
Giannini, C., and Brechot, C. (2003). Hepatitis C virus biology. Cell Death Differ., 10,
S27-S38.
Goffard, A., and Dubuisson, J. (2003). Glycosylation of hepatitis C virus envelope
proteins. Biochimie, 85(3-4):295-301.
Goffard, A., Callens, N., Bartosch, B., Wychowski, C., Cosset, F.L., Montpellier, C., and
Dubuisson, J. (2005). Role of N-linked glycans in the functions of hepatitis C
virus envelope glycoproteins. J. Virol., 79(13):8400-9.
Gondeau, C., Pichard-Garcia, L., and Maurel, P. (2009 Oct). Cellular models for the
screening and development of anti-hepatitis C virus agents. Pharmacol. Ther.,
124(1):1-22.
González-Peralta, R.P., Qian, K., She, J.Y., Davis, G.L., Ohno, T., Mizokami, M., and
Lau, J.Y. (1996). Clinical implications of viral quasispecies heterogeneity in
chronic hepatitis C. J. Med. Virol., 49(3):242-7.
Gottwein, J.M., Scheel, T.K., Jensen, T.B., Lademann, J.B., Prentoe, J.C., Knudsen,
M.L., Hoegh, A.M., and Bukh, J. (2009). Development and characterization of
hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger
receptor class B type I and effect of antiviral drugs. Hepatology, 49(2):364-77.
Grakoui, A., McCourt, D.W., Wychowski, C., Feinstone, S.M., and Rice, C.M. (1993).
Characterization of the hepatitis C virus-encoded serine proteinase:

160
determination of proteinase-dependent polyprotein cleavage sites. J.Virol.,
67(5):2832-43.
Griffin, S., Stgelais, C., Owsianka, A.M., Patel, A.H., Rowlands, D., and Harris M.
(2008). Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the
p7 ion channel. Hepatology, 48(6):1779-90.
Hadziyannis, S.J., Sette, H. Jr., Morgan, T.R., Balan, V., Diago, M., Marcellin, P.,
Ramadori, G.,Bodenheimer, H, Jr., Bernstein, D., Rizzetto, M., Zeuzem, S.,
Pockros, P.J., Lin, A., Ackrill, A.M.; and PEGASYS International Study Group.
(2004). Peginterferon-alpha2a and ribavirin combination therapy in chronic
hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann.
Intern. Med., 2;140(5):346-55.
Halary, F., Amara, A., Lortat-Jacob, H., Messerle, M., Delaunay, T., Houlès, C., Fieschi,
F., Arenzana-Seisdedos, F., Moreau, J.F., and Déchanet-Merville, J. (2002).
Human cytomegalovirus binding to DC-SIGN is required for dendritic cell
infection and target cell trans-infection. Immunity, 17(5):653-64.
He, L. F., Alling, D., Popkin, T., Shapiro, M., Alter, H. J. and Purcell, R. H. (1987).
Determining the size of non-A, non-B hepatitis virus by fltration. Journal of
Infectious Diseases, 156: 636-640.
Helenius, A. (1994). How N-linked oligosaccharides affect glycoprotein folding in the
endoplasmic reticulum. Mol. Biol. Cell, 5(3):253-65.

161
Helle, F., Goffard, A., Morel, V., Duverlie, G., McKeating, J., Keck, Z.Y., Foung, S.,
Penin, F., Dubuisson, J., and Voisset, C. (2007). The neutralizing activity of anti-
hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope
protein. J. Virol., 81(15):8101-11.
Helle, F., Vieyres, G., Elkrief, L., Popescu, C.I., Wychowski, C., Descamps, V.,
Castelain, S., Roingeard, P., Duverlie, G., and Dubuisson, J. (2010 Nov). Role of
N-linked glycans in the functions of hepatitis C virus envelope proteins
incorporated into infectious virions. J. Virol,. 84(22):11905-15.
Hemler, M.E. (2005). Tetraspanin functions and associated microdomains. Nat. Rev.
Mol. Cell Biol., 6(10):801-11.
Heo, T.H., Lee, S.M., Bartosch, B., Cosset, F.L., and Kang, C.Y. (2006). Hepatitis C
virus E2 links soluble human CD81 and SR-B1 protein. Virus Res., 121(1):58-
64.
Higginbottom, A., Quinn, E.R., Kuo, C.C., Flint, M., Wilson, L.H., Bianchi, E., Nicosia,
A., Monk, P.N., McKeating, J.A., and Levy, S. (2000). Identification of amino
acid residues in CD81 critical for interaction with hepatitis C virus envelope
glycoprotein E2. J. Virol., 74(8):3642-9.
Hijikata, M., Mizushima, H., Akagi, T., Mori, S., Kakiuchi, N., Kato, N., Tanaka, T.,
Kimura, K., and Shimotohno, K. (1993). Two distinct proteinase activities
required for the processing of a putative nonstructural precursor protein of
hepatitis C virus. J. Virol., 67(8):4665-75.

162
Hill, A.B. (1965).The environment and disease: association or causation? Proc. R. Soc.
Med., 58:295-300.
Hofmann, WP, Kronenberger, B., Bojunga, J., Stamm, B., Herrmann, E., Bücker, A., et
al., (2008). Prospective study of bone mineral density and metabolism in patients
with chronic hepatitis C during pegylated interferon alpha and ribavirin
therapy. J. Viral. Hepat., 15:790–796.
Holmes, E.C. (2008). Evolutionary history and phylogeography of human viruses. Annu.
Rev. Microbiol., 62:307-28.
Honda, A., and Matsuzaki, Y. (2011). Cholesterol and chronic hepatitis C virus infection.
Hepatol. Res., 41:697-710.
Hourioux, C., Ait-Goughoulte, M., Patient, R., Fouquenet, D., Arcanger-Doudet, F.,
Brand, D., Martin, A., and Roingeard, P. (2007). Core protein domains involved
in hepatitis C virus-like particle assembly and budding at the endoplasmic
reticulum membrane. Cell Microbio., 9(4):1014-27.
Hsu, M., Zhang, J., Flint, M., Logvinoff, C., Cheng-Mayer, C., Rice, C.M., and
McKeating, J.A. (2003). Hepatitis C virus glycoproteins mediate pH-dependent
cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA,
100(12):7271-6.
Hüssy , P., Langen , H., Mous , J., and Jacobsen, H. (1996). Hepatitis C virus core
protein: carboxy-terminal boundaries of two processed species suggest cleavage
by a signal peptide peptidase. Virology, 224(1):93-104.

163
Idrees, M., and Riazuddin, S. (2008). Frequency distribution of hepatitis C virus
genotypes in different geographical regions of Pakistan and their possible routes
of transmission. BMC Infect. Dis., 23; 8:69.
Ikeda, M., Abe, K., Yamada, M., Dansako, H., Naka, K., and Kato, N. (2006). Different
anti-HCV profiles of statins and their potential for combination therapy with
interferon. Hepatology, 44(1):117-25.
Inamullah, Idrees, M., Ahmed, H., Sajid-ul-ghafoor, Ali, M., Ali, L., and Ahmed, A.,
(2011). Hepatitis C virus genotypes circulating in district Swat of Khyber
Pakhtoonkhaw, Pakistan. Virol. J., 8:16.
Ivanyi-Nagy, R., Kanevsky, I., Gabus, C., Lavergne, J.P., Ficheux, D., Penin, F., Fosse,
P., and Darlix, J.L. (2006). Analysis of hepatitis C virus RNA dimerization and
core-RNA interactions. Nucleic Acids Res., 2006, 34: 2618-2633.
Jaeckel, E., Cornberg, M., Wedemeyer, H., Santantonio, T., Mayer, J., Zankel, M.,
Pastore, G., Dietrich, M., Trautwein, C., and Manns, M.P. (2001). German Acute
Hepatitis C Therapy Group. Treatment of acute hepatitis C with interferon alfa-
2b. N. Engl. J. Med., 345(20):1452-7.
Jeannel, D., Fretz, C., Traore, Y., Kohdjo, N., Bigot, A., Pê Gamy, E., Jourdan, G.,
Kourouma, K., Maertens, G., Fumoux, F., Fournel, J.J., and Stuyver, L. (1998).
Evidence for high genetic diversity and long-term endemicity of hepatitis C virus
genotypes 1 and 2 in West Africa. J. Med. Virol., 55:92–97.

164
Jirasko, V., Montserret , R., Appel, N., Janvier, A., Eustachi , L., Brohm, C., Steinmann,
E., Pietschmann, T., Penin, F., and Bartenschlager , R. S. (2008). Structural and
functional characterization of nonstructural protein 2 for its role in hepatitis C
virus assembly. J. Biol. Chem., 283(42):28546-62.
Jones, C.T., Murray, C., Eastman, D.K., Tassello, J., and Rice, C.M. (2007). Hepatitis C
virus p7 and NS2 proteins are essential for production of infectious virus. J.
Virol., 81: 8374-8383.
José, M. Cuevas, Pilar Domingo-Calap, Marianoel Pereira-Gómez and Rafael Sanjuán.
(2009). Experimental Evolution and Population Genetics of RNA Viruses. The
Open Evolution Journal, 3:9-16.
Jusoh, S.A., Welsch, C., Siu, S.W., Böckmann, R.A., and Helms, V. (2010 Oct).
Contribution of charged and polar residues for the formation of the E1-E2
heterodimer from Hepatitis C Virus. J. Mol. Model, 16(10):1625-37.
Kato, N., Ootsuyama, Y., Tanaka, T., Nakagawa, M., Nakazawa, T, et al. (1992). Marked
sequence diversity in the putative envelope proteins of hepatitis C viruses. Virus
Res., 22: 107–123.
Kato, T., Date, T., Miyamoto, M., Furusaka, A., Tokushige, K., Mizokami, M., and
Wakita, T. (2003). Efficient replication of the genotype 2a hepatitis C virus
subgenomic replicon. Gastroenterology, 125(6):1808-17.
Keck, Z.Y., Li, S.H., Xia, J., von Hahn, T., Balfe, P., McKeating, J.A., Witteveldt, J.,
Patel, A.H., Alter, H., Rice, C.M., and Foung, S.K. (2009). Mutations in hepatitis

165
C virus E2 located outside the CD81 binding sites lead to escape from broadly
neutralizing antibodies but compromise virus infectivity. J. Virol., 83(12):6149-
60.
Keck, Z.Y., Op De Beeck, A., Hadlock, K.G., Xia, J., Li, T.K., Dubuisson, J., and Foung,
SK. (2004). Hepatitis C virus E2 has three immunogenic domains containing
conformational epitopes with distinct properties and biological functions. J.
Virol., 78(17):9224-32.
Keck, Z.Y., Sung, V.M., Perkins, S., Rowe, J., Paul, S., Liang, T.J., Lai, M.M., and
Foung, S.K. (2004). Human monoclonal antibody to hepatitis C virus E1
glycoprotein that blocks virus attachment and viral infectivity. J. Virol.,
78(13):7257-63.
Khakoo, S.I., Thio, C.L., Martin, M.P., Brooks, C.R., Gao, X., Astemborski, J., Cheng, J.,
Goedert, J.J., Vlahov, D., Hilgartner, M., Cox, S., Little, A.M., Alexander, G.J.,
Cramp, M.E., O'Brien, S.J., Rosenberg, W.M., Thomas, D.L, and Carrington, M.
(2004). HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus
infection. Science, 305: 872–874.
Kiiver, K., Merits, A., Ustav, M., and Zusinaite, E. (2006).Complex formation between
hepatitis C virus NS2 and NS3 proteins. Virus Res., 117(2):264-72.
Kim, M., Ha, Y., and Park, H.J. (2006). Structural requirements for assembly and
homotypic interactions of the hepatitis C virus core protein. Virus Res., 122:137-
43.

166
Klein, K.C., Dellos, S.R., and Lingappa , J.R. (2005). Identification of residues in the
hepatitis C virus core protein that are critical for capsid assembly in a cell-free
system. J. Virol., (11):6814-26.
Klein , K.C., Polyak , S.J., and Lingappa , J.R. (2004). Unique features of hepatitis C
virus capsid formation revealed by de novo cell-free assembly. J. Virol.,
78(17):9257-69.
Koch, J.O., and Bartenschlager, R. (1999). Modulation of hepatitis C virus NS5A
hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J.
Virol., 73(9):7138-46. .
Krey, T., d'Alayer, J., Kikuti, C.M., Saulnier, A., Damier-Piolle, L., Petitpas, I.,
Johansson, D.X., Tawar, R.G., Baron, B., Robert, B., England, P., Persson, M.A.,
Martin, A., and Rey, F.A. (2010). The disulfide bonds in glycoprotein E2 of
hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog.,
6(2):e1000762.
Krieger, M. (2001). Scavenger receptor class B type I is a multiligand HDL receptor that
influences diverse physiologic systems. J. Clin. Invest., 108(6):793-7.
Kuntzen, T., Timm, J., Berical, A., Lewis-Ximenez, L.L., Jones, A., Nolan, B., Schulze
zur Wiesch, J., Li, B., Schneidewind, A., Kim, A.Y., Chung, R.T., Lauer, G.M.,
and Allen, T.M. (2007 ). Viral sequence evolution in acute hepatitis C virus
infection. J. Virol., 81(21):11658-68.

167
Kushima, Y., Wakita, T., and Hijikata, M. (2010). A disulfide-bonded dimer of the core
protein of hepatitis C virus is important for virus-like particle production. J.
Virol., 84(18):9118-27.
Kwon, D.S., Gregorio, G., Bitton, N., Hendrickson, W.A., and Littman, D.R. (2002).
DC-SIGN-mediated internalization of HIV is required for trans-enhancement of
T cell infection. Immunity, 16(1):135-44.
Lai, W.K., Sun, P.J., Zhang, J., Jennings, A., Lalor, P.F., Hubscher, S., McKeating, J.A.,
and Adams, D.H. (2006). Expression of DC-SIGN and DC-SIGNR on human
sinusoidal endothelium: a role for capturing hepatitis C virus particles. Am. J.
Pathol., 169(1):200-8. (PMID: 16816373).
Lavie, M., Goffard, A., and Dubuisson, J. (2007). Assembly of a functional HCV
glycoprotein heterodimer. Curr Issues Mol Biol. 9(2):71-86.
Lavillette, D., Morice, Y., Germanidis, G., Donot, P., Soulier, A., Pagkalos, E.,
Sakellariou, G., Intrator, L., Bartosch, B., Pawlotsky, J.M., and Cosset, F.L.
(2005). Human serum facilitates hepatitis C virus infection, and neutralizing
responses inversely correlate with viral replication kinetics at the acute phase of
hepatitis C virus infection. J. Virol., 79(10):6023–6034.
Lechmann, M., and Liang , T.J. (2000) Vaccine development for hepatitis C. Semin.
Liver Dis., 20(2):211-26.

168
Lemon, S.M., McKeating, J.A., Pietschmann, T., Frick, D.N., Glenn, J.S., Tellinghuisen,
T.L., Symons, J., and Furman, PA. (2010). Development of novel therapies for
hepatitis C. Antiviral Res., 86(1):79-92.
Li, H.F., Huang, C.H., Ai, L.S., Chuang, C.K., and Chen, S.S. (2009). Mutagenesis of the
fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in
cell fusion and virus entry. J. Biomed. Sci., 16(1): 89.
Lin, C., Lindenbach, B.D., Prágai, B.M., McCourt, D.W., and Rice, C.M. (1994).
Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two
distinct E2-specific products with different C termini. J. Virol., 68(8):5063-73.
Lin, W., Choe, W.H., Hiasa, Y., Kamegaya, Y., Blackard, J.T., Schmidt, E.V., and
Chung, R.T. (2005). Hepatitis C virus expression suppresses interferon signaling
by degrading STAT1. Gastroenterology, 128:1034-1041.
Lin, W., Kim, S.S., Yeung, E., Kamegaya, Y., Blackard, J.T., Kim, K.A., Holtzman,
M.J., and Chung, R.T. (2006). Hepatitis C Virus Core Protein Blocks Interferon
Signaling by Interaction with the STAT1 SH2 Domain. J. Virol., 80(18):9226-
35.
Lindenbach, B.D., and Rice, C.M. (2005). Unravelling hepatitis C virus replication from
genome to function. Nature, 436(7053):933-8.
Lindenbach, B.D., Evans, M.J., Syder, A.J., Wölk, B., Tellinghuisen, T.L., Liu, C.C.,
Maruyama, T., Hynes, R.O., Burton, D.R., McKeating, J.A., and Rice, C.M.

169
(2005). Complete replication of hepatitis C virus in cell culture. Science,
309(5734):623-6.
Lindenbach, B.D., Meuleman, P., Ploss, A., Vanwolleghem, T., Syder, A.J., McKeating,
J.A., Lanford, R.E., Feinstone, S.M., Major, M.E., Leroux-Roels, G., and Rice,
C.M. (2006). Cell culture-grown hepatitis C virus is infectious in vivo and can be
recultured in vitro. Proc. Natl. Acad. Sci. USA, 103(10):3805-9.
Lindenbach, B.D., Prágai, B.M., Montserret, R., Beran, R.K., Pyle, A.M., Penin, F., and
Rice, C.M. (2007). The C terminus of hepatitis C virus NS4A encodes an
electrostatic switch that regulates NS5A hyperphosphorylation and viral
replication. J. Virol., 81(17):8905-18.
Liu, M., Chen, H., Luo, F., Li, P., Pan, Q., Xia, B., Qi, Z., Ho, W.Z., and Zhang, X.L.
(2007 Sep 4). Deletion of N-glycosylation sites of hepatitis C virus envelope
protein E1 enhances specific cellular and humoral immune responses. Vaccine,
25(36):6572-80.
Logvinoff, C., Major, M.E., Oldach, D., Heyward , S., Talal , A., Balfe, P., Feinstone,
S.M., Alter, H.J., Rice , C.M., and McKeating , J.A. (2004). Neutralizing
antibody response during acute and chronic hepatitis C virus infection. Proc.
Natl. Acad. Sci. USA. 101(27):10149-54.
Lorenz, I. C., Marcotrigiano, J., Dentzer, T.G., and Rice, C.M. (2006). Structure of the
catalytic domain of the hepatitis C virus NS2-3 protease. Nature,
442(7104):8315.

170
Loriot, M.A., Bronowicki, J.P., Lagorce, D., Lakehal, F., Persico, T., Barba, G., Mergey,
M., Vons, C., Franco, D., Belghiti, J., Giacca, M., Housset, C., and Bréchot, C.
(1999). Permissiveness of human biliary epithelial cells to infection by hepatitis
C virus. Hepatology, 29(5):1587-95.
Luik, P., Chew, C., Aittoniemi, J., Chang, J., Wentworth, P. Jr, Dwek, R.A., Biggin, P.C.,
Vénien-Bryan, C., and Zitzmann, N. (2009). The 3-dimensional structure of a
hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci.
USA, 106(31):12712-6.
Majeau, N., Fromentin, R., Savard, C., Duval, M., Tremblay, M.J., and Leclerc, D.
(2009). Palmitoylation of hepatitis C virus core protein is important for virion
production. J. Biol. Chem., 284(49):33915-25.
Majeau, N., Gagné, V., Boivin, A., Bolduc, M., Majeau, J.A., Ouellet, D., and Leclerc, D.
(2004). The N-terminal half of the core protein of hepatitis C virus is sufficient
for nucleocapsid formation. J. Gen. Virol., 85:971-81.
Mancini, N., Diotti, R.A., Perotti, M., Sautto, G., Clementi, N., Nitti, G., Patel, AH., Ball,
J.K., Clementi, M., and Burioni, R. (2009). Hepatitis C virus (HCV) infection
may elicit neutralizing antibodies targeting epitopes conserved in all viral
genotypes. PLoS One. 4(12):e8254.
Masaki, T., Suzuki, R., Murakami, K., Aizaki, H., Ishii, K., Murayama, A., Date, T.,
Matsuura, Y., Miyamura, T., Wakita, T.,and Suzuki, T. (2008). Interaction of
hepatitis C virus nonstructural protein 5A with core protein is critical for the
production of infectious virus particles. J. Virol., 82(16):7964-76.

171
McKeating, J.A., Zhang, L.Q., Logvinoff, C., Flint, M., Zhang, J., Yu, J., Butera, D., Ho,
D.D., Dustin, L.B., Rice, C.M., and Balfe, P. (2004). Diverse hepatitis C virus
glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol.,
78(16):8496-505.
McOmish, F., Yap, P.L., Dow, B. C., Follett, E.A., Seed, C., Keller, A.J., Cobain, T.J.,
Krusius, T., Kolho, E., Naukkarinen, R, and et al. (1994). Geographical
distribution of hepatitis C virus genotypes in blood donors: an international
collaborative survey. J. Clin. Microbiol., 32(4):884-92.
Means, R.E. and Desrosiers, R.C. (2000). Resistance of native, oligomeric envelope on
simian immunodeficiency virus to digestion by glycosidases. J. Virol.,
74(23):11181-90.
Meertens, L., Bertaux, C., Cukierman, L., Cormier, E., Lavillette, D., Cosset, F.L., and
Dragic, T. (2008). The tight junction proteins claudin-1, -6, and -9 are entry
cofactors for hepatitis C virus. J. Virol., 82(7):3555-60.
Mellor, J., Holmes, E.C., Jarvis, L.M., Yap, P.L., and Simmonds, P. (1995). Investigation
of the pattern of hepatitis C sequence diversity in different geographical regions:
implantations for virus classification. J. Gen. Virol., 76:2493–2507.
Meunier, J.C., Engle, R.E., Faulk, K., Zhao, M., Bartosch, B., Alter, H., Emerson, S.U.,
Cosset, Purcell, F.L., and R.H, Bukh, J. (2005). Evidence for cross-genotype
neutralization of hepatitis C virus pseudo-particles and enhancement of
infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. USA, 102(12):4560-5.

172
Meunier, J.C., Russell, R.S., Goossens, V., Priem, S., Walter, H., Depla, E., Union, A.,
Faulk, K.N., Bukh, J., Emerson, S.U., and Purcell, R.H. (2008). Isolation and
characterization of broadly neutralizing human monoclonal antibodies to the
glycoprotein of hepatitis C virus. J. Virol., 82(2):966-73.
Meurs, E.F., and Breiman, A. (2007). The interferon inducing pathways and the hepatitis
C virus. World J. Gastroenterol., 13(17):2446-54.Mihm, U., Hofmann, W.P.,
Welsch, C., Polta, A., Lengauer, T. (2010). Effect of ribavirin on the frequency
of RNase L cleavage sites within the hepatitis C viral genome. J. Viral. Hepat.,
17(3):217-21
Mondelli, M.U., Cerino, A., Meola, A., and Nicosia, A. (2003). Variability or
conservation of hepatitis C virus hypervariable region 1? Implications for
immune responses. J. Biosci., 28(3):305-10.
Montpellier, C., Tews, B.A., Poitrimole, J., Rocha-Perugini, V., D'Arienzo, V., Potel, J.,
Zhang, X.A., Rubinstein, E., Dubuisson, J., and Cocquerel, L. (2011). Interacting
Regions of CD81 and Two of Its Partners, EWI-2 and EWI-2wint, and Their
Effect on Hepatitis C Virus Infection. J. Biol. Chem., 286(16):13954-65.
Moradpour, D., and Blum, H.E. (2005). Pathogenesis of hepatocellular carcinoma. Eur. J.
Gastroenterol. Hepatol., 17(5):477-83.
Moradpour, D., Penin, F., and, Rice, C.M. (2007). Replication of hepatitis C virus. Nat.
Rev. Microbiol., 5(6):453-63.

173
Moriya, K., Fujie, H., Shintani, Y., Yotsuyanagi, H., Tsutsumi, T., Ishibashi, K.,
Matsuura, Y., Kimura, S., Miyamura, T., and Koike, K. (1998). The core protein
of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat.
Med., 4(9):1065-7.
Moriya, K., Yotsuyanagi, H., Shintani, Y., Fujie, H., Ishibashi, K., Matsuura, Y.,
Miyamura, T., and Koike, K. (1997). Hepatitis C virus core protein induces
hepatic steatosis in transgenic mice. J. Gen. Virol., 78:1527-31.
Mukhopadhyay, S., Kim, B.S., Chipman, P.R., Rossmann, M.G., and Kuhn, R.J. (2003).
Structure of West Nile virus. Science, 302(5643):248.
Murakami, K., Ishii, K., Ishihara, Y., Yoshizaki , S., Tanaka , K., Gotoh, Y., Aizaki, H.,
Kohara, M., Yoshioka , H., Mori , Y., Manabe, N., Shoji , I., Sata , T.,
Bartenschlager, R., Matsuura, Y., Miyamura , T., and Suzuki , T. (2006).
Production of infectious hepatitis C virus particles in three-dimensional cultures
of the cell line carrying the genome-length dicistronic viral RNA of genotype 1b.
Virology, 351(2):381-92.
Nahmias, Y., Goldwasser , J., Casali , M., van Poll ,D., Wakita, T., Chung , R.T., and
Yarmush , M.L. Hepatology. (2008). Apolipoprotein B-dependent hepatitis C
virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatalogy,
47(5):1437-45.
Nakai, K., Okamoto, T., Kimura-Someya, T., Ishii, K., Lim, C.K., Tani, H., Matsuo, E.,
Abe, T., Mori, Y., Suzuki, T., Miyamura, T., Nunberg, J.H., Moriishi, K., and
Matsuura, Y. (2006). Oligomerization of hepatitis C virus core protein is crucial

174
for interaction with the cytoplasmic domain of E1 envelope protein. J. Virol.,
80(22):11265-73.Ndjmou, J., Pybus, O.G., and Matz, B. (2003). Phylogenetic
analysis of hepatitis C virus isolates indicates a unique pattern of endemic
infection in Cameroon. J. Gen. Virol., 84: 2333–2341.
Neddermann, P., Clementi, A., and De Francesco, R. (1999). Hyperphosphorylation of
the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A,
NS4B, and NS5A encoded on the same polyprotein. J. Virol., 73(12):9984-91.
Neumann, A.U., Lam, N.P., Dahari, H., Gretch, D.R., Wiley, T.E., Layden, T.J., and
Perelson, A.S. (1998). Hepatitis C viral dynamics in vivo and the antiviral
efficacy of interferon-alpha therapy.Science, 282(5386):103-7.
Njouom, R., Pasquier, C, Ayouba, A., Gessain, A., Froment, A., Mfoupouendoun, J.,
Pouillot, R., Dubois, M., Sandres-Saune,´ K., Thonnon, J., Izopet, J., and
Nerrienet, E. (2003). High rate of Hepatitis C virus infection and predominance
of genotype A among elderly inhabitants of a remote village of the rain forest of
south Cameroon. J. Med. Virol., 71:219–225.
Okamoto, K., Mori, Y., Komoda, Y., Okamoto, T., Okochi, M., Takeda, M., Suzuki, T.,
Morishi, K., and Matsuura, Y. (2008). Intramembrane processing by signal
peptide peptidase regulates the membrane localization of hepatitis C virus core
protein and viral propagation. J. Virol., 82: 8349-8361.
Okamoto, K., Moriishi, K., Miyamura, T., and Matsuura, Y. (2004). Intramembrane
proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein.
J. Virol., 78(12):6370-80.

175
Op De Beeck, A., Montserret, R., Duvet, S., Cocquerel, L., Cacan, R., Barberot, B., Le
Maire, M., Penin, F., and Dubuisson, J. (2000). The transmembrane domains of
hepatitis C virus envelope glycoproteins E1 and E2 play a major role in
heterodimerization. J. Biol. Chem., 275(40):31428-37.
Owsianka, A., Clayton, R.F., Loomis-Price, L.D., McKeating, J.A., and Patel, A.H.
(2001). Functional analysis of hepatitis C virus E2 glycoproteins and virus-like
particles reveals structural dissimilarities between different forms of E2. J. Gen.
Virol., 82:1877-83.
Owsianka, A.M., Timms, J.M., Tarr, A.W., Brown, R.J., Hickling, T.P., Szwejk, A.,
Bienkowska-Szewczyk, K., Thomson, B.J., Patel, A.H., and Ball, J.K. (2006).
Identification of conserved residues in the E2 envelope glycoprotein of the
hepatitis C virus that are critical for CD81 binding. J. Virol., 80(17):8695-704.
Pachiadakis, I., Pollara, G., Chain, B.M., and Naoumov, N.V. (2005). Is hepatitis C virus
infection of dendritic cells a mechanism facilitating viral persistence? Lancet
Infect. Dis., 5(5):296-304.
Pallaoro, M., Lahm, A., Biasiol, G., Brunetti, M., Nardella, C., Orsatti, L., Bonelli, F.,
Orrù, S., Narjes, F., and Steinkühler, C. (2001). Characterization of the hepatitis
C virus NS2/3 processing reaction by using a purified precursor protein. J. Virol.,
75(20):9939-46.
Pang, P.S., Planet, P.J., and Glenn, J.S. (2009). The evolution of the major hepatitis C
genotypes correlates with clinical response to interferon therapy. PLoS One.
4(8):e6579.

176
Penin, F., Combet, C., Germanidis, G., Frainais, P.O., Deléage, G., and Pawlotsky, J. M.
(2001). Conservation of the conformation and positive charges of hepatitis C
virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell
attachment. J. Virol., 75(12):5703-10.
Penin, F., Dubuisson, J., Rey, F.A., Moradpour, D., and Pawlotsky, J.M. (2004).
Structural biology of hepatitis C virus. Hepatology, 39: 5-19.
Pestka, J.M., Zeisel, M.B., Bläser, E., Schürmann, P., Bartosch, B., Cosset, F.L., Patel,
A.H., Meisel, H., Baumert, J., Viazov, S., Rispeter, K., Blum, H.E., Roggendorf,
M., and Baumert, T.F. (2007). Rapid induction of virus-neutralizing antibodies
and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad.
Sci. USA. 104(14):6025-30.
Petracca, R., Falugi, F., Galli, G., Norais, N., Rosa, D., Campagnoli, S., Burgio, V., Di
Stasio, E., Giardina, B., Houghton, M., Abrignani, S.,and Grandi, G. (2000).
Structure-Function Analysis of Hepatitis C Virus Envelope-CD81 Binding. J.
Virol., 74(10):4824-4830.
Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis, S., Steinmann, E.,
Abid, K., Negro, F., Dreux, M., Cosset, F.L., and Bartenschlager R. (2006).
Construction and characterization of infectious intragenotypic and intergenotypic
hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA, 103(19):7408-13.
Pikora, C. A. (2004). Glycosylation of the ENV spike of primate immunodeficiency
viruses and antibody neutralization. Curr. HIV Res., 2:243–254.

177
Ploss, A., Evans, M.J., Gaysinskaya, V.A., Panis, M., You, H., de Jong, Y.P., and Rice,
C.M. (2009). Human occludin is a hepatitis C virus entry factor required for
infection of mouse cells. Nature, 457(7231):882-6.
Popescu, C.I., and Dubuisson, J. (2009). Role of lipid metabolism in hepatitis C virus
assembly and entry. Biol. Cell, 102: 63-74.
Prevelige, P.E. (1998). Inhibiting Virus Capsid Assembly by Altering the Polymerization
Pathway. Trends Biotechnol., 16(2):61-5.
Raja, N.S., and Janjua, K.A. (2008). Epidemiology of hepatitis C virus infection in
Pakistan. J. Microbiol. Immunol. Infect., 41(1):4-8.
Ray, S.C., Fanning, L., Wang, X.H., Netski, D.M., Kenny-Walsh, E., and Thomas, D.L.
(2005). Divergent and convergent evolution after a common-source outbreak of
hepatitis C virus. J. Exp. Med., 201(11):1753-9.
Roccasecca, R., Ansuini, H., Vitelli, A., Meola, A., Scarselli, E., Acali, S., Pezzanera, M.,
Ercole, B.B., McKeating, J., Yagnik, A., Lahm, A., Tramontano, A., Cortese, R.,
and Nicosia, A. (2003). Binding of the hepatitis C virus E2 glycoprotein to CD81
is strain specific and is modulated by a complex interplay between hypervariable
regions 1 and 2. J. Virol., 77(3):1856-67.
Rodríguez-Rodríguez, M., Tello, D., Yélamos, B., Gómez-Gutiérrez, J., Pacheco, B.,
Ortega, S., Serrano, A.G., Peterson, D.L., and Gavilanes, F. (2009). Structural
properties of the ectodomain of hepatitis C virus E2 envelope protein. Virus
Res., 139(1):91-9.

178
Rollier, C., Depla, E., Drexhage, J.A., Verschoor, E.J., Verstrepen, B. E., Fatmi, A.,
Brinster, C., Fournillier, A., Whelan, J.A., Whelan, M., Jacobs, D., Maertens, G.,
Inchauspé, G., Heeney, J. L. (2004). Control of heterologous hepatitis C virus
infection in chimpanzees is associated with the quality of vaccine-induced
peripheral T-helper immune response. J. Virol., 78(1):187-96.
Rothwangl, K.B., Manicassamy, B., Uprichard, S.L., and Rong L. (2008). Dissecting the
role of putative CD81 binding regions of E2 in mediating HCV entry: Putative
CD81 binding region 1 is not involved in CD81 binding. Virol. J., 5: 46.
Rothwangl, K.B., Manicassamy, B., Uprichard, S.L., and Rong, L. (2008). Dissecting the
role of putative CD81 binding regions of E2 in mediating HCV entry: putative
CD81 binding region 1 is not involved in CD81 binding. Virol. J., 5:46.
Russell, R.S., Kawaguchi, K., Meunier, J.C., Takikawa, S., Faulk, K., Bukh, J., Purcell,
R.H., and Emerson, S. U. (2009). Mutational analysis of the hepatitis C virus E1
glycoprotein in retroviral pseudoparticles and cell-culture-derived H77/JFH1
chimeric infectious virus particles. J. Viral. Hepat., 16(9):621-32.
Sakai, A., Claire, M.S., Faulk, K., Govindarajan, S., Emerson, S.U., Purcell, R.H., and
Bukh, J. (2003). The p7 polypeptide of hepatitis C virus is critical for infectivity
and contains functionally important genotype-specific sequences. Proc. Natl.
Acad. Sci. USA, 100(20):11646-51.
Santolini, E., Migliaccio, G., and LaMonica, N. (1994). Biosynthesis and biochemical
properties of the hepatitis C virus core protein. J. Virol., 68: 3631-3641.

179
Santolini, E., Pacini, L., Fipaldini, C., Migliaccio, G., and Monica, N. (1995). The NS2
protein of hepatitis C virus is a transmembranepolypeptide. J. Virol.,
69(12):7461-71.
Scarselli, E., Ansuini, H., Cerino, R., Roccasecca, R.M., Acali, S., Filocamo, G., Traboni,
C., Nicosia, A., Cortese, R., and Vitelli, A. (2002). The human scavenger
receptor class B type I is a novel candidate receptor for the hepatitis C virus.
EMBO J., 21(19):5017-25.
Schregel, V., Jacobi, S., Penin, F., and Tautz, N. (2009). Hepatitis C virus NS2 is a
protease stimulated by cofactor domains in NS3. Proc. Natl. Acad. Sci. USA,
106(13):5342-7.
Selby, M. J., Glazer, E., Masiarz, F., and Houghton, M. (1994). Complex processing and
protein: protein interactions in the E2:NS2 region of HCV. Virology,
204(1):114-22.
Shavinskay, A., Boulant, S., Penin, F., McLauchlan, J., and Bartenschlager, R. (2007).
The lipid droplet binding domain of hepatitis C virus core protein is a major
determinant for efficient virus assembly. J. Biol. Chem., 282:37158-37169.
Shimizu, Y.K., Hijikata , M., Iwamoto, A., Alter, H.J., Purcell, R. H., and Yoshikura, H.
(1994). Neutralizing antibodies against hepatitis C virus and the emergence of
neutralization escape mutant viruses. J. Virol., 68(3):1494-500.

180
Shimizu, Y.K., Igarashi, H., Kiyohara, T., Shapiro, M., Wong, D.C., Purcell, R.H., and
Yoshikura, H. (1998). Infection of a chimpanzee with hepatitis C virus grown in
cell culture. J. Gen. Virol., 79:1383-6.
Simmonds, P. (1995). Variability of hepatitis C virus. Hepatol., 21(2):570-83.
Simmonds, P. (2004). Genetic diversity and evolution of hepatitis C virus–15 years on. J.
Gen. Virol., 85:3173–3188.
Simmonds, P., McOmish, F., Yap, P.L., Chan, S.-W., Lin, C.K., Dusheiko, G., Saeed, A.
A. and Holmes, E. C. (1993). Sequence variability in the 5ˈ non-coding region of
hepatitis C virus: identification of a new virus type and restrictions on sequence
diversity. J. Gen. Virol., 74: 661-668.
Slater-Handshy, T., Droll, D.A., Fan, X., Di Bisceglie, A.M., and Chambers, T.J. (2004).
HCV E2 glycoprotein: mutagenesis of N-linked glycosylation sites and its effects
on E2 expression and processing. Virology, 319(1):36-48.
Steinmann, E., Penin, F., Kallis, S., Patel, A.H., Bartenschlager, R., and Pietschmann, T.
(2007). Hepatitis C virus p7 protein is crucial for assembly and release of
infectious virions. PLoS Pathog., 3(7):e103.
Sterjovski, J., Churchill, M.J., Ellett, A., Gray, L.R., Roche, M.J., Dunfee, R.L., Purcell,
D.F., Saksena, N., Wang, B., Sonza, S., Wesselingh, S.L., Karlsson, I., Fenyo,
E.M., Gabuzda, D., Cunningham, A.L., and Gorry, P.R. (2007). Asn 362 in
gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope
glycoprotein variants from patients with AIDS. Retrovirology, 4:89.

181
Strosberg, A.D., Kota, S., Takahashi, V., Snyder, J.K., and Mousseau, G. (2010). Core as
a Novel Viral Target for Hepatitis C Drugs. Viruses, 2: 1734-1751.
Sun, C., Pager, C.T., Luo, G., Sarnow, P., and Cate, J.H. (2010). Hepatitis C virus core-
derived peptides inhibit genotype 1b viral genome replication via interaction
with DDX3X.PLoS One. 5(9):e12826.
Tanaka, Y., Shimoike, T., Ishii, K., Suzuki, R., Suzuki, T., Ushijima, H., Matsuura, Y.,
and Miyamura, T. (2000). Selective binding of hepatitis C virus core protein to
synthetic oligonucleotides corresponding to the 5' untranslated region of the viral
genome. Virology, 270(1):229-36.
Targett-Adams, P., Boulant, S., Douglas, M.W., and McLauchlan, J. (2010). Lipid
Metabolism and HCV Infection. Viruses, 2:1195-1217.
Tassaneetrithep, B., Burgess, T.H., Granelli-Piperno, A., Trumpfheller, C., Finke, J., Sun,
W., Eller, M.A., Pattanapanyasat, K., Sarasombath, S., Birx, D. L., Steinman, R.
M., Schlesinger, S., and Marovich, M.A. (2003). DC-SIGN (CD209) mediates
dengue virus infection of human dendritic cells. J. Exp. Med., 197(7):823-9.
Tews, B.A., Popescu, C.-I., and Dubuisson, J. (2010). Last Stop Before Exit – Hepatitis C
Assembly and Release as Antiviral Drug Targets. Viruses, 2: 1782-1803.
Timpe, J.M., and McKeating, J.A. (2008). Hepatitis C virus entry: possible targets for
therapy. Gut, 57(12):1728-37.

182
Troesch, M., Meunier, I., Lapierre, P., Lapointe, N., Alvarez, F., Boucher, M., and
Soudeyns, H. (2006). Study of a novel hypervariable region in hepatitis C virus
(HCV) E2 envelope glycoprotein. Virology, 352(2):357-67.
Vieyres, G., Thomas, X., Descamps, V., Duverlie, G., Patel, A.H., and Dubuisson, J.
(2010). Characterization of the envelope glycoproteins associated with infectious
Hepatitis C Virus. J. Virol., 84(19): 10159–10168.
Vitozzi, S., Lapierre, P., Djilali-Saiah, I., and Alvarez, F. (2002). Autoantibody detection
in type 2 autoimmune hepatitis using a chimera recombinant protein. J. Immunol.
Methods, 262: 103–110.
Voisset, C., and Dubuisson, J. (2004). Functional hepatitis C virus envelope
glycoproteins. Biol. Cell, 96(6):413-20.
Von Hahn, T., Yoon, J.C., Alter, H.J., Rice, C.M., Rehermann, B., Balfe, P., and
McKeating J.A. (2007). Hepatitis C virus continuously escapes from neutralizing
antibody and T-cell responses during chronic infection in vivo.
Gastroenterology, 132(2):667-78.
Vuillermoz, I., Khattab, E., Sablon, E., Ottevaere, I., Durantel, D., Vieux, C., Trepo, C.,
and Zoulim, F. (2004). Genetic variability of hepatitis C virus in chronically
infected patients with viral breakthrough during interferon-ribavirin therapy. J.
Med. Virol., 74(1):41-53.

183
Waheed, Y., Tahir, S., Ahmad, T., and Qadri, I. (2010). Sequence comparison and
phylogenetic analysis of core gene of hepatitis C virus from Pakistani population.
Afr. J. Biotechnol., 9(29):4561-4567.
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., Murthy, K.,
Habermann, A., Kräusslich, H.G., Mizokami, M., Bartenschlager, R., and Liang,
T.J. (2005). Production of infectious hepatitis C virus in tissue culture from a
cloned viral genome. Nat. Med., 11(7):791-6.
Walewski, J. L., Gutierrez J.A., Branch-Elliman, W., Stump, D., Keller T.R., Rodriguez,
A., Benson, G., and Branch, A.D. (2002). Mutation Master: profiles of
substitutions in hepatitis C virus RNA of the core, alternate reading frame, and
NS2 coding regions. RNA, 8(5):557-71.
Wands, J. R. (2004). Prevention of hepatocellular carcinoma. N. Engl. J. Med.,
351(15):1567-70.
Waxman , L., Whitney, M., Pollok, B.A., Kuo , L.C., and Darke, P.L. (2001). Host cell
factor requirement for hepatitis C virus enzyme maturation. Proc. Natl. Acad.
Sci. USA, 98(24):13931-5.
Wei, X., Decker, J.M., Wang, S., Hui, H., Kappes, J.C., Wu, X., Salazar-Gonzalez, J. F.,
Salazar, M. G., Kilby, J. M., Saag, M. S., Komarova, N.L., Nowak, M.A., Hahn,
B.H., Kwong, P.D., and Shaw, G.M. (2003). Antibody neutralization and escape
by HIV-1. Nature, 422(6929):307-12.

184
Willett, B. J., McMonagle, E.L., Logan, N., Samman, A., and Hosie, M.J. (2008). A
single site for N-linked glycosylation in the envelope glycoprotein of feline
immunodeficiency virus modulates the virus-receptor interaction. Retrovirology,
5:77.
Wyatt, R., Kwong, P.D., Desjardins, E., Sweet, R.W., Robinson, J., Hendrickson, W.A.,
and Sodroski, J.G. (1998). The antigenic structure of the HIV gp120 envelope
glycoprotein. Nature, 393(6686):705-11.
Yagnik, A.T., Lahm, A., Meola, A., Roccasecca, R.M., Ercole, B.B., Nicosia, A., and
Tramontano, A. (2000).A model for the hepatitis C virus envelope glycoprotein
E2. Proteins, 40(3):355-66.
Yamaga, A.K., and Ou, J.H. (2002). Membrane topology of the hepatitis C virus NS2
protein. J Biol Chem., 277(36):33228-34.
Yan, X.B., Battaglia, S., Boucreux, D., Chen, Z., Brechot, C., and Pavio, N. (2007).
Mapping of the interacting domains of hepatitis C virus core protein and the
double-stranded RNA-activated protein kinase PKR. Virus Res., 125(1):79-87.
Yasui , K., Wakita, T., Tsukiyama-Kohara, K., Funahashi , S.I., Ichikawa, M., Kajita , T.,
Moradpour , D., Wands, JR., and Kohara, M. (1998). The native form and
maturation process of hepatitis C virus core protein. J Virol., 72(7):6048-55.
Yi, M., Ma, Y., Yates, J., and Lemon, S.M. (2007). Compensatory mutations in E1, p7,
NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric
hepatitis C virus. J. Virol., 81: 629-638.

185
Yi, M., Villanueva, R.A., Thomas, D.L., Wakita, T., and Lemon, S.M. (2006). Production
of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human
hepatoma cells. Proc. Natl. Acad. Sci. USA, 103(7):2310-5.
Zein, N.N. (2000). Clinical Significance of Hepatitis C Virus Genotypes. Clin. Microbiol.
Rev., 13(2): 223–235.
Zeisel, M.B., Barth, H., Schuster, C., and Baumert, T.F. (2009). Hepatitis C virus entry:
molecular mechanisms and targets for antiviral therapy. Front. Biosci., 14:3274-
85.
Zeisel, M.B., Fofana, I., Fafi-Kremer, S., and Baumert, T.F. (2011). Hepatitis C virus
entry into hepatocytes: molecular mechanisms and targets for antiviral therapies.
J. Hepatol., 54(3):566-76.
Zeisel, M.B., Koutsoudakis, G., Schnober, E.K., Haberstroh, A., Blum, H.E., Cosset,
F.L., Wakita, T., Jaeck, D., Doffoel, M., Royer, C., Soulier, E., Schvoerer, E.,
Schuster, C., Stoll-Keller, F., Bartenschlager, R., Pietschmann, T., Barth, H., and
Baumert, T.F. (2007). Scavenger receptor class B type I is a key host factor for
hepatitis C virus infection required for an entry step closely linked to CD81.
Hepatology, 46(6):1722-31.
Zhang, M., Gaschen, B., Blay, W., Foley, B., Haigwood, N., Kuiken, C., and Korber, B.
(2004). Tracking global patterns of N-linked glycosylation site variation in
highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza
hemagglutinin. Glycobiology, 14(12):1229-46.

186
Zhang, P., Wu, C.G., Mihalik, K., Virata-Theimer, M.L., Yu, M.Y., Alter, H.J., and
Feinstone, S.M. (2007). Hepatitis C virus epitope-specific neutralizing antibodies
in Igs prepared from human plasma. Proc. Natl. Acad. Sci. USA, 104(20):8449-
54.
Zhong, J., Gastaminza, P., Cheng, G., Kapadia, S., Kato, T., Burton, D.R., Wieland, S.F.,
Uprichard, S.L., Wakita, T., and Chisari, F.V. (2005). Robust hepatitis C virus
infection in vitro. Proc. Natl. Acad. Sci. USA, 102(26):9294-9.
Zoulim, F., Chevallier, M., Maynard, M., and Trepo, C. (2003). Clinical consequences of
hepatitis C virus infection. Rev. Med. Virol., 13(1):57-68.





