Embed Size (px)
Citation preview

Using Saccharomyces cerevisiae for the Biosynthesis of Tetracycline Antibiotics
Ehud Herbst
Submitted in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy
in the Graduate School of Arts and Sciences
COLUMBIA UNIVERSITY
2019

© 2019
Ehud Herbst
All rights reserved

ABSTRACT
Using Saccharomyces cerevisiae for the biosynthesis of tetracycline antibiotics
Ehud Herbst
Developing treatments for antibiotic resistant bacterial infections is among the most urgent public
health challenges worldwide. Tetracyclines are one of the most important classes of antibiotics,
but like other antibiotics classes, have fallen prey to antibiotic resistance. Key small changes in
the tetracycline structure can lead to major and distinct pharmaceutically essential improvements.
Thus, the development of new synthetic capabilities has repeatedly been the enabling tool for
powerful new tetracyclines that combatted tetracycline-resistance. Traditionally, tetracycline
antibiotics were accessed through bacterial natural products or semisynthetic analogs derived from
these products or their intermediates. More recently, total synthesis provided an additional route
as well. Importantly however, key promising antibiotic candidates remained inaccessible through
existing synthetic approaches.
Heterologous biosynthesis is tackling the production of medicinally important and structurally
intriguing natural products and their unnatural analogs in tractable hosts such as Saccharomyces
cerevisiae. Recently, the heterologous biosynthesis of several tetracyclines was achieved in
Streptomyces lividans through the expression of their respective biosynthetic pathways. In
addition, the heterologous biosynthesis of fungal anhydrotetracyclines was shown in S. cerevisiae.
This dissertation describes the use of Saccharomyces cerevisiae towards the biosynthesis of target
tetracyclines that have promising prospects as antibiotics based on the established structure-
activity relationship of tetracyclines but have been previously synthetically inaccessible.
Chapter 1 provides an introduction to the pursuit of tetracycline antibiotics using S. cerevisiae.
Following an overview of tetracycline drugs, the chapter describes the methods for making

tetracyclines and their limitations in accessing the tetracycline analogs targeted in this study. The
desirability of making these target analogs as well as key desired properties are then exemplified
by natural products, totally synthetic and semisynthetic derivatives. The target tetracycline analogs
pursued in this study are then outlined and the considerations in choosing their desired properties
are discussed, as well as the reasons for employing S. cerevisiae in their synthesis.
Chapter 2 describes the use of Saccharomyces cerevisiae for the final steps of tetracycline
biosynthesis, setting the stage for total biosynthesis of tetracyclines in Saccharomyces cerevisiae.
Chapter 3 describes the work towards biosynthesis of the target tetracycline analogs using
Saccharomyces cerevisiae, utilizing successful expression optimization and gene biomining
approaches. Chapter 4 describes the work towards the target tetracycline analogs from fungal
anhydrotetracyclines in Saccharomyces cerevisiae.
The challenge of enzyme evolution towards unnatural substrates and the complex environment of
cells require metabolic engineering efforts to be performed in libraries, as it is currently impossible
to predetermine which modifications will prove beneficial. Traditional methods in DNA
mutagenesis and increasingly, advances in DNA synthesis, DNA assembly and genome
engineering are enabling high throughput strain construction. Thus, there is a need for a general,
high-throughput, versatile and readily implemented assay for the detection of target molecule
biosynthesis. The development of such an assay is described in Chapter 5. The assay is
demonstrated to detect tetracycline derivatives, and differentiate a producer and a nonproducer
strain of the fungal anhydrotetracycline TAN-1612. The yeast three hybrid assay for metabolic
engineering of tetracycline derivatives described in this chapter could be used in the next steps
towards the heterologous biosynthesis of the target tetracycline analogs in S. cerevisiae and
beyond.

i
Table of Contents
List of Figures……………………………………………………………………………………vii
List of Schemes……………………………………………………………………………………x
List of Tables……………………………………………………………………………………...xi
List of Abbreviations……………………………………………………………………………xiii
1 An introduction to tetracycline biosynthesis using Saccharomyces cerevisiae ...................... 1
1.1 Chapter outline ................................................................................................................. 2
1.2 An antibiotic crisis ........................................................................................................... 2
1.3 Tetracycline antibiotics .................................................................................................... 3
1.3.1 General historical context for tetracycline antibiotics and tetracycline-resistance ....... 3
1.3.2 Tetracycline analogs – small structural changes can lead to major pharmaceutical
improvements .......................................................................................................................... 3
1.3.3 A structural overview of the FDA-approved tetracyclines ........................................... 4
1.4 Making tetracycline antibiotics ........................................................................................ 8
1.4.1 Biosynthesis and semisynthesis of tetracyclines using the original microbial hosts .... 9
1.4.2 Total synthesis of tetracycline antibiotics ................................................................... 12
1.4.3 Heterologous biosynthesis and semisynthesis of tetracyclines ................................... 14
1.5 Tetracycline analogs in the 6-position ........................................................................... 17
1.5.1 Dactylocyclines – natural product 6-epiglycotetracyclines ........................................ 17
1.5.2 6-demethyl-6-deoxy-6α-aryltetracyclines – totally synthetic tetracyclines ................ 18

ii
1.5.3 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines – semisynthetic
tetracyclines ........................................................................................................................... 19
1.6 Design of the biosynthesis of 6-demethyl-6-epiglycotetracyclines using S. cerevisiae 21
1.6.1 Targeting 6-demethyl derivatives of 6-epiglycotetracyclines for acid stability ......... 21
1.6.2 Targeting glycoside libraries on 6-demethyl-6-epiglycotetracyclines for broad
spectrum activity.................................................................................................................... 23
1.6.3 Targeting the 6-epi forms of 6-demethylglycotetracyclines for antibiotic efficacy ... 25
1.6.4 S. cerevisiae as the heterologous host of choice for 6-demethyl-6-epiglycotetracycline
biosynthesis ........................................................................................................................... 26
1.7 Research progress and future goals in the biosynthesis of 6-demethyl-6-
epiglycotetracyclines using S. cerevisiae .................................................................................. 29
1.7.1 Chapter 2 – Using Saccharomyces cerevisiae for the final steps of tetracycline
biosynthesis ........................................................................................................................... 29
1.7.2 Chapter 3 – Towards the biosynthesis of 6-demethyl-6-epitetracyclines using S.
cerevisiae ............................................................................................................................... 30
1.7.3 Chapter 4 – Towards the biosynthesis of 6-demethyl-6-epitracyclines from TAN-1612
in Saccharomyces cerevisiae ................................................................................................. 30
1.7.4 Chapter 5 – a yeast three hybrid system for the metabolic engineering of tetracycline
derivatives .............................................................................................................................. 31
1.8 References ...................................................................................................................... 32
2 Using Saccharomyces cerevisiae for the final steps of tetracycline biosynthesis ................. 41

iii
2.1 Chapter outline ............................................................................................................... 42
2.2 Introduction .................................................................................................................... 42
2.3 Materials and methods ................................................................................................... 43
2.3.1 General protocol for hydroxylation/reduction assay in cell lysate ............................. 43
2.3.2 Protocol for hydroxylation/reduction assay in whole cells ......................................... 44
2.4 Results ............................................................................................................................ 45
2.4.1 6β-hydroxylation of anhydrotetracycline in S. cerevisiae .......................................... 45
2.4.2 5a(11a)-dehydrotetracycline reduction in S. cerevisiae .............................................. 50
2.5 Discussion ...................................................................................................................... 53
2.5.1 6β-hydroxylation of anhydrotetracycline in S. cerevisiae .......................................... 53
2.5.2 5a(11a)-dehydrotetracycline reduction in S. cerevisiae .............................................. 55
2.6 Conclusion ...................................................................................................................... 56
2.7 Appendix ........................................................................................................................ 58
2.8 References ...................................................................................................................... 64
3 Towards the biosynthesis of 6-demethyl-6-epitetracyclines using S. cerevisiae ................. 70
3.1 Chapter outline ............................................................................................................... 71
3.2 Introduction .................................................................................................................... 72
3.3 Materials and methods ................................................................................................... 72
3.3.1 General protocol for high throughput assay of hydroxylation/reduction in whole cells
in microtiter plates ................................................................................................................. 72

iv
3.3.2 General protocol for western blots .............................................................................. 73
3.4 Results ............................................................................................................................ 74
3.4.1 Optimizing DacO1 expression in S. cerevisiae – rational design ............................... 74
3.4.2 Searching the bacterial hydroxylase space for 6α-hydroxylation of
anhydrotetracyclines .............................................................................................................. 77
3.4.3 Developing a microtiter plate assay for anhydrotetracycline hydroxylation .............. 79
3.4.4 Evolving a 6α-hydroxylase of anhydrotetracyclines – DacO1 random mutagenesis . 80
3.4.5 Testing DacO1 expression optimization constructs and other bacterial hydroxylases 82
3.5 Discussion ...................................................................................................................... 97
3.5.1 Optimizing DacO1 expression in S. cerevisiae – rational design ............................... 97
3.5.2 Searching the bacterial hydroxylase space for 6α-hydroxylation of
anhydrotetracyclines .............................................................................................................. 98
3.5.3 Developing a microtiter plate assay for anhydrotetracycline hydroxylation .............. 98
3.5.4 Evolving a 6α-hydroxylase of anhydrotetracyclines – DacO1 random mutagenesis . 99
3.5.5 Testing DacO1 expression optimization constructs and other hydroxylases............ 100
3.6 Conclusion .................................................................................................................... 102
3.7 Appendix ...................................................................................................................... 105
3.8 References .................................................................................................................... 127
4 Towards the biosynthesis of 6-demethyl-6-epitracyclines from TAN-1612 in Saccharomyces
cerevisiae .................................................................................................................................... 131

v
4.1 Chapter outline ............................................................................................................. 132
4.2 Introduction .................................................................................................................. 132
4.3 Materials and methods ................................................................................................. 133
4.3.1 General protocol for high throughput assay of TAN-1612 hydroxylation in whole cells
in microtiter plates ............................................................................................................... 133
4.3.2 Protocol for selecting amino acids for mutagenesis in hypothesized binding pockets of
monooxygenases .................................................................................................................. 134
4.4 Results .......................................................................................................................... 135
4.4.1 Testing PgaE and other bacterial hydroxylases for TAN-1612 hydroxylation......... 135
4.4.2 Searching the fungal hydroxylase space for 6α-hydroxylation of TAN-1612 .......... 145
4.4.3 Evolving monooxygenases for 6α-hydroxylation of anhydrotetracyclines – OxyS
example ................................................................................................................................ 149
4.4.4 Library screening for TAN-1612 hydroxylation in S. cerevisiae ............................. 152
4.5 Discussion .................................................................................................................... 154
4.5.1 Testing PgaE and other bacterial hydroxylases for TAN-1612 hydroxylation......... 154
4.5.2 Searching the fungal hydroxylase space for 6α-hydroxylation of TAN-1612 .......... 159
4.5.3 Evolving monooxygenases for 6α-hydroxylation of anhydrotetracyclines .............. 159
4.5.4 Library screening for TAN-1612 hydroxylation in S. cerevisiae ............................. 160
4.6 Conclusion .................................................................................................................... 161
4.7 Appendix ...................................................................................................................... 163

vi
4.8 References .................................................................................................................... 179
5 A yeast three hybrid assay for metabolic engineering of tetracycline derivatives .............. 181
5.1 Chapter outline ............................................................................................................. 182
5.2 Introduction .................................................................................................................. 183
5.3 Materials and methods ................................................................................................. 185
5.4 Results .......................................................................................................................... 192
5.4.1 Y3H system design and CID design and synthesis ................................................... 192
5.4.2 Initial characterization of the Y3H assay for tetracycline derivatives ...................... 194
5.4.3 Applying the Y3H assay to the detection of target molecule biosynthesis ............... 198
5.5 Discussion .................................................................................................................... 200
5.6 Conclusion .................................................................................................................... 203
5.7 Appendix ...................................................................................................................... 204
5.7.1 Supplementary figures .............................................................................................. 204
5.7.2 Supplementary methods ............................................................................................ 205
5.7.3 Supplementary tables ................................................................................................ 206
5.8 References .................................................................................................................... 211

vii
List of Figures
Figure 1-1 Structural comparison of FDA-approved tetracyclines................................................. 5
Figure 1-2 Comparison of the tetracycline lipophilic and zwitterionic forms ................................ 7
Figure 1-3 Key steps in the Myers convergent total synthesis of tetracyclines ............................ 14
Figure 1-4 Structures of the dactylocycline family of natural products ....................................... 18
Figure 1-5 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines and their antibiotic activities
....................................................................................................................................................... 20
Figure 1-6 Two possible C-O bond cleavage pathways in glycoside hydrolysis ......................... 22
Figure 1-7 Retrosynthetic analysis of 6-demethyl-6-epiglycotetracyclines ................................. 27
Figure 1-8 Structural Comparison between anhydrotetracycline, chloranhydrotetracycline, TAN-
1612 and anhydrotdactylocyclinone ............................................................................................. 28
Figure 2-1 Mass spectrometry analysis of anhydrotetracycline hydroxylation in cell lysate
expressing OxyS ........................................................................................................................... 47
Figure 2-2 UV/Vis spectroscopy analysis of the reaction of anhydrotetracycline in whole cells
expressing OxyS ........................................................................................................................... 49
Figure 2-3 Mass spectrometry of anhydrotetracycline hydroxylation and reduction in cell lysate
expressing OxyS in the presence of G6P ...................................................................................... 52
Figure 3-1 Western Blot of DacO1 and OxyS .............................................................................. 75
Figure 3-2 A simplified outline of the microtiter plate assay for anhydrotetracycline hydroxylation.
....................................................................................................................................................... 79
Figure 3-3 DacO1 error-prone mutagenesis screen excerpt ......................................................... 81
Figure 3-4 ΔExcitation spectrum for DacO1 and DacO1 error-prone PCR mutant ..................... 82

viii
Figure 3-5 Western blot analysis of DacO1 fusion proteins and other bacterial hydroxylases in
FY251 ........................................................................................................................................... 86
Figure 3-6 Western blot analysis of bacterial hydroxylases in BJ-5464-NpgA ........................... 87
Figure 3-7 Western blot analysis of DacO1-DacO4 and DacO4-DacO1 fusion proteins ............ 88
Figure 3-8 Western blot analysis of DacO1 and its fusion proteins in BJ5464-NpgA ................. 90
Figure 3-9 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction by cell
lysates of strains expressing DacO1, its fusion proteins and other bacterial hydroxylases .......... 93
Figure 3-10 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction in cell
lysates of strains expressing DacO1, its fusion proteins and other bacterial hydroxylases .......... 96
Figure 3-11 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction in cell
lysates of strains expressing OxyS and PgaE ............................................................................... 97
Figure 4-1 UV/Vis analysis of hydroxylation attempt of TAN-1612 by bacterial hydroxylases 137
Figure 4-2 UV/Vis chromatogram of the HPLC separation of extracts from +/- PgaE strains
encoding the TAN-1612 pathway. .............................................................................................. 139
Figure 4-3 Mass spectrometry analysis of isolate from +/-PgaE strain encoding the TAN-1612
pathway. ...................................................................................................................................... 142
Figure 4-4 MSMS analysis of isolate from +PgaE strain encoding the TAN-1612 pathway..... 142
Figure 4-5 1H-NMR spectra of isolates from +/-PgaE strains encoding the TAN-1612 pathway.
..................................................................................................................................................... 143
Figure 4-6 COSY NMR spectrum of isolate from +PgaE strain encoding the TAN-1612 pathway.
..................................................................................................................................................... 144
Figure 4-7 Western blot analysis of fungal hydroxylase expression in BJ5464-NpgA .............. 148

ix
Figure 4-8 PyMOL illustrations of amino acids in proximity to the tetracycline substrate in
aklavinone-11-Hydroxylase and in OxyS ................................................................................... 151
Figure 4-9 Library screening for TAN-1612 hydroxylation in S. cerevisiae ............................. 153
Figure 4-10 Possible moieties in the product isolated from +PgaE strain coexpressing the TAN-
1612 bioysnthetic pathway ......................................................................................................... 155
Figure 4-11 Possible intermediates in the biosynthesis of TAN-1612 ....................................... 156
Figure 4-12 xanthurenic acid and its derivative that could correspond to experimental mass and
NMR spectra ............................................................................................................................... 158
Figure 5-1 Detection of a target molecule biosynthesis by the yeast three hybrid system. ........ 184
Figure 5-2 Characterization of the dynamic range of the Y3H assay for tetracyclines .............. 195
Figure 5-3 The modularity of the yeast three hybrid assay enables both screening and selecting for
biosynthesis of target tetracyclines ............................................................................................. 197
Figure 5-4 Differentiation of a producer and a non-producer strain of a target molecule by the Y3H
assay for tetracyclines, demonstrating the applicability of the Y3H assay to metabolic engineering.
..................................................................................................................................................... 199
Figure 5-5 LacZ readout of strain harboring plasmid encoding for LexA-TetR (PBA-8) and strain
without the plasmid (PBA-5). ..................................................................................................... 204
Figure 5-6 standard curve for TAN-1612 quantification in cultures. ......................................... 205

x
List of Schemes
Scheme 2-1 Two-step enzymatic conversion of anhydrotetracycline to tetracycline ................... 46
Scheme 3-1 Bacterial monooxygenases and their hypothesized substrates and products ............ 78
Scheme 3-2 Two-step enzymatic conversion of (6-demethyl-)anhydrotetracycline to (6-demethyl-
)6-epitetracycline .......................................................................................................................... 91
Scheme 4-1 Biosynthetic plan for TAN-1612 6α-hydroxylation ............................................... 135
Scheme 4-2 Bacterial monooxygenases and their hypothesized substrates and products .......... 136
Scheme 5-1 Synthesis of the chemical inducer of dimerization (CID) Min-Mtxa ...................... 193

xi
List of Tables
Table 1-1 FDA approved tetracycline derivatives and their differentiating functional groups ...... 6
Table 1-2 Rates of hydrolysis of β-D-glycopyranosides in 0.5 M aqueous sulfuric acid ............. 23
Table 2-1 Strains used in this study .............................................................................................. 58
Table 2-2 Plasmids used in this study ........................................................................................... 58
Table 2-3 Sequences used in this study ........................................................................................ 59
Table 3-1 Potential hits in the screen of DacO1 expression optimization constructs and other
bacterial hydroxylases ................................................................................................................... 85
Table 3-2 UV/Vis spectroscopy results for hydroxylase strains cell lysates incubated with
anhydrotetracycline and glucose-6-phosphate .............................................................................. 94
Table 3-3 Assay conditions for anhydrotetracycline hydroxylation and reduction in cell lysates of
strains expressing DacO1, its fusion proteins and other bacterial hydroxylases .......................... 95
Table 3-4 Strains used in this study ............................................................................................ 105
Table 3-5 Plasmids used in this study ......................................................................................... 106
Table 3-6 Sequences used in this study ...................................................................................... 107
Table 4-1 Fungal monooxygenases biomined for TAN-1612 6α-hydroxylation in S. cerevisiae
..................................................................................................................................................... 146
Table 4-2 Residues chosen for mutagenesis in OxyS ................................................................. 149
Table 4-3 Rank of PgaE positive control in screen for TAN-1612 hydroxylase ........................ 152
Table 4-4 Strains used in this study ............................................................................................ 163
Table 4-5 Plasmids used in this study ......................................................................................... 164
Table 4-6 Plasmid Library A – Strain EH-5-217-1 .................................................................... 165
Table 4-7 Plasmid Library B – Strain EH-5-217-3 ..................................................................... 165

xii
Table 4-8 Plasmid Library C – Strain EH-5-217-1 and EH-5-217-3.......................................... 165
Table 4-9 Plasmid Library D – Strains EH-5-217-2 and EH-217-4 ........................................... 166
Table 4-10 Sequences used in this study .................................................................................... 166
Table 4-11 Primers used for OxyS saturation mutagenesis in this study ................................... 177
Table 5-1 Strains used in this study ............................................................................................ 206
Table 5-2 Plasmids used in this study ......................................................................................... 207
Table 5-3 Sequences of the various TetR classes in the within the pMW103 backbone. .......... 207

xiii
List of Abbreviations
5-FOA 5-fluoroorotic acid
AD activation domain
DBD DNA binding domain
DCM dichloromethane
DHFR dihydrofolate reductase
DMF dimethylformamide
EtOAc ethyl acetate
FAD flavin adenine dinucleotide
G6P glucose-6-phosphate
HPLC high-pressure liquid chromatography
HRMS high resolution mass spectroscopy
kb kilobase pair
LCMS liquid chromatography mass spectrometry
MeCN Acetonitrile
MeOH methanol
MIC minimal inhibitory concentration
MS-MS tandem mass spectrometry
NADPH dihydronicotinamide adenine dinucleotide phosphate
NMR nuclear magnetic resonance
ONPG o-nitrophenyl β-d-galactopyranoside
PAGE polyacrylamide gel electrophoresis

xiv
PR protein receptor
PKS polyketide synthase
r.t. room temperature
SDS sodium dodecyl sulfate
TetR tetracycline repressor
TIC total ion count
TFA trifluoroacetic acid
Tris Tris(hydroxymethyl)aminomethane
w.t. wild type
X-Gal 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside

xv
Acknowledgments
I would like to thank my advisor Prof. Virginia Cornish for a wide cast mentorship, for providing
a very fertile environment for sciencemaking and for the pushes to dare scientifically.
I would like to thank my thesis committee members, Prof. Scott Snyder, Dr. George Ellestad, Prof.
Ruben Gonzalez and Prof. Anne-Catrin Uhlemann as well as my advisory committee member
Prof. Tristan Lambert. The valuable discussions with you throughout the years have been
tremendously helpful to shape this research direction. I consider myself extremely lucky to have
benefited from your highly relevant expertise to this research and your will to mentor.
I have profited immensely in various ways from interacting with the highly talented and committed
members, past and present, of the Cornish and neighboring labs. Specifically, I would like to thank
Arden Lee, Pedro Baldera-Aguayo and Hyunwook Lee for their large contribution to this research
and for being, each in their distinctive style, a pleasure to work with. I would like to thank Dr.
Andrew Anzalone, Dr. Nili Ostrov, Dr. Casey Brown, Dr. Sonja Billerbeck, Dr. Zhixing Chen, Dr.
Caroline Patenode, Dr. Marie Harton, Dr. Bertrand Adanve, Dr. Miguel Jimenez, Dr. Andy (Yao
Zong) Ng, Dr. Mia Shandell, Sean Bannier, Dr. Anna Kaplan and Dr. Andreas Hartel for
collaborations and/or mentorship and/or technical assistance.
I would like to thank Prof. Yi Tang for sharing his time and wide-ranging knowledge, as well as
plasmids pYR291 and pYR342 and yeast strain BJ5464-NpgA, Prof. Alan R. Davidson for sharing
pET21d plasmid encoding TetR(B), Prof. Frank Foss for sharing synthetic FO and Dr. Bertrand T.
Adanve for the synthesis of Methotrexate-α-OtBu.
I would like to thank my family for making the immense effort it takes to be close while an ocean
apart. I would like to thank my friends in Israel and the US for their support and company. Finally,
I would like to thank Jess for the love and support throughout the years. It’s been quite a journey.

xvi
To my Savtas. For your perseverance.

1
1 An introduction to tetracycline biosynthesis using
Saccharomyces cerevisiae

2
1.1 Chapter outline
This chapter provides an introduction to the pursuit of tetracycline antibiotics using S. cerevisiae.
First, the significance of the production of new antibiotic analogs from a human health perspective
is mentioned (Section 1.2). Then, the general historical context for developing tetracycline
antibiotics is given (Section 1.3.1), the antibiotic promise in making tetracycline analogs is
discussed (Section 1.3.2) and a structural overview of FDA-approved tetracyclines follows
(Section 1.3.3).
Three modes of synthesis of tetracycline antibiotic are then introduced in the historical order in
which they appeared, biosynthesis and semisynthesis using the original microbial hosts (Section
1.4.1), total synthesis (Section 1.4.2) and heterologous biosynthesis and semisynthesis (Section
1.4.3). The desirability of making 6-position analogs of tetracyclines as well as key desired
properties of such analogs is then exemplified by natural products (Section 1.5.1), totally synthetic
(Section 1.5.2) and semisynthetic derivatives (Section 1.5.3).
The target tetracycline analogs pursued in this study are then outlined and the considerations in
choosing their desired properties are then discussed (Sections 1.6.1, 1.6.2 and 1.6.3) as well as the
reasons for employing S. cerevisiae in their synthesis (Section 1.6.4). Finally, an overview of the
research plan and its execution throughout this dissertation is given (Section 1.7).
1.2 An antibiotic crisis
Developing treatments for antibiotic resistant bacterial infections is among the most urgent public
health challenges world-wide.(1) The US Center for Disease Control reports over 2 million
illnesses and 20,000 deaths annually in the United States as a result of antibiotic resistance.(1)
Novel antibiotic drug candidates have traditionally been accessed through natural product

3
screening, total synthesis, semisynthesis and combinatorial chemistry. However, the number of
new antibiotics approved by the FDA has dropped significantly from previous decades, from 29
approvals in the 1980s to 9 in the 2000s.(2, 3) To reverse this trend, transformative new
technologies for discovery of new antibiotics are needed.
1.3 Tetracycline antibiotics
1.3.1 General historical context for tetracycline antibiotics and tetracycline-
resistance
Tetracyclines are one of the most important classes of antibiotics,(4) but like other classes of
antibiotics, have fallen prey to resistance.(5) From their discovery in the 1940’s, tetracyclines were
used as broad spectrum antibiotics, as well as for other types of disease, such as periodontitis.(6)
The tetracyclines inhibit bacterial protein synthesis by binding reversibly to the 30S ribosomal
subunit and sterically hindering aminoacyl-tRNA binding to the ribosomal A-site.(7, 8) However,
bacteria have evolved a growing number of resistance mechanisms – including efflux pumps,(9)
ribosomal protection proteins,(10) rRNA mutations,(11) and enzymatic degradation.(12)
1.3.2 Tetracycline analogs – small structural changes can lead to major
pharmaceutical improvements
Small changes in the tetracycline structure can lead to major and distinct pharmaceutically
essential improvements. Examples include tetracycline analogs differing from tetracycline in only
up to three positions and showing improved pharmacokinetic properties, binding affinity to the
ribosome, activity against resistant strains and non-antimicrobial properties.(13-16)
Thus, for example, a difference in the position of one hydroxy group between tetracycline and
doxycycline improves the half-life, tissue penetration and matrix metalloproteinase (MMP)

4
inhibition properties of the latter (Figure 1-1 and Table 1-1).(13) Due to such advantages,
doxycycline is not only one of the most commonly clinically indicated tetracycline antibiotics, it
is the only clinically available MMP inhibitor as of 2010 and is indicated for the treatment of
periodontitis.(14)
Minocycline differs from tetracycline in only three functional groups, a hydroxy, a methyl and an
N,N-dimethylamino (Figure 1-1 and Table 1-1). Impressively, these differences are enough to
improve minocycline relative to tetracycline in its pharmacokinetic profile, radical oxygen species
scavenging and other anti-inflammatory properties.(13, 14) While tetracycline is unable to cross
the blood-brain barrier (BBB), minocycline is known for its neuroprotective properties that have
been demonstrated pre-clinically and clinically in the treatment of various neurological disease.
Examples include ischemia, traumatic brain injury and neuropathic pain, Parkinson's disease,
Huntington's disease, amyotrophic lateral sclerosis, Alzheimer's disease, multiple sclerosis and
spinal cord injury.(15)
Lastly, tigecycline, being identical to minocycline except for the 9-t-butyl-glycylamido group has
a better binding affinity to the A-site of the ribosome and an improved antimicrobial activity
against tetracycline-resistant strains (Figure 1-1 and Table 1-1).(16) On the other hand, these
changes also led to tigecycline being orally unavailable and furthermore, tigecycline use is
associated with an increased risk of death.(17)
1.3.3 A structural overview of the FDA-approved tetracyclines
As of 2018, all nine FDA approved tetracycline antibiotics were either bacterial natural products
or semisynthetic analogs derived from bacterial natural products or their intermediates, and all
nine are produced commercially partly or wholly by fermentation (Figure 1-1).(6, 18-20) Three of
these nine tetracycline antibiotics, chlortetracycline, meclocycline and metacycline, were

5
discontinued.(20) In 2018, three additional tetracycline antibiotics have received their FDA
approval, omadacycline, sarecycline and eravacycline.(20-22) Importantly, FDA approved
tetracycline derivatives are identical in all positions but for the 5α-, 6α-, 6β-, 7- and 9-positions
(Table 1-1).
Figure 1-1 Structural comparison of FDA-approved tetracyclines
The common name, a full name outlining the structural relationship between the different
derivatives and the year of FDA approval, are mentioned below the structures.(6, 20, 23)

6
Table 1-1 FDA approved tetracycline derivatives and their differentiating functional groups
Namea 5α 6α 6β 7 9
Chlortetracycline Me OH Cl
Oxytetracycline OH Me OH
Tetracycline Me OH
Demeclocycline OH Cl
Doxycycline OH Me
Minocycline dimethylamino
Meclocycline OH CH2 CH2 Cl
Metacycline OH CH2 CH2
Tigecycline dimethylamino tBu-glycylamido
Sarecycline (methoxy(methyl)amino)
methyl
Omadacycline dimethylamino neopentylaminomethyl
Eravacycline F pyrrolidinoacetamido aFDA approved tetracycline derivatives are identical in all positions but the 5α-, 6α-, 6β-, 7- and
9-positions. Empty cells indicate implicit Hs.
The identity of the FDA approved tetracyclines at all positions but for 5α, 6α, 7, 8 and 9 could be
explained as stemming from two major reasons. One is the antibacterial ineffectiveness of
tetracycline analogs generated so far in positions 10, 11, 11a, 12, 12a, 1, 2, 3 and 4. The other is
limited semisynthetic and total synthetic access to modifications in positions such as 4a and 6α
that could potentially yield successful tetracycline antibiotics but are awaiting efficient synthetic
methods to access them. Both of these topics will be expanded on below.
Given that positions 10, 12a, 1, 2 and 3, as well as the β-diketone of 11,11a and 12 of tetracyclines
(Figure 1-1) are considered to interact with the ribosome and thus to be directly responsible for
tetracyclines antibiotic activity,(8, 24) it is not surprising that modifications in these positions do
not yield effective antibiotics. The 2-position is somewhat of an exception as modification at this
position can yield tetracycline antibiotic prodrugs that hydrolyze to the unmodified 2-carboxamido
functional group (Section 1.4.1). Modifications at the 4-position have also not yielded
antibiotically active tetracyclines even though this position is not considered to be important for

7
ribosome binding. The prevalent hypothesis to explain the requirement for the 4α-dimethylamino
group in tetracycline antibiotics is the importance of maintaining an equilibrium between a
zwitterionic and a nonionic (lipophilic) structures (Figure 1-2, Section 1.4.1).(25) This equilibrium
is considered crucial for tetracycline antibiotic activity as the zwitterionic form is the form of
tetracycline that binds to the ribosome,(8) and the nonionic form is the form that can cross the
cytoplasmic membrane.(26) It is therefore clear that the promising positions for modifications that
could yield tetracycline antibiotics are likely to be limited to positions 4a, 5, 5a, 6, 7, 8 and 9.
Figure 1-2 Comparison of the tetracycline lipophilic and zwitterionic forms
Adapted from (26).
In order to explain which modifications in which positions are of promise to obtain tetracycline
analogs with desired antibiotic activity but have not yet been accessed, the following two
subsections of the introduction are given. The first (Section 1.4) introduces three different fronts
of tetracycline synthesis to access tetracycline antibiotic analogs, biosynthesis and semisynthesis
using the original microbial hosts (Section 1.4.1), total synthesis (Section 1.4.2) and heterologous
biosynthesis and semisynthesis (Section 1.4.3). Specifically, Section 1.4 discusses which
modifications were so far inaccessible by all existing methods. The following section introduces
desirable tetracyclines that are inaccessible by biosynthesis and semisynthesis using the original
microbical hosts and by total synthesis and could be conceived to made by heterologous
biosynthesis (Section 1.5). Finally, the last two sections of the introduction describe the plan to

8
access these target antibiotics using S. cerevisiae (Section 1.6) and the overview of the
experimental progress and future plans towards these analogs (Section 1.7).
1.4 Making tetracycline antibiotics
In this section an overview of the three main methods for the production of tetracycline antibiotics
is given, biosynthesis and semisynthesis using the original microbial hosts (Section 1.4.1), total
synthesis (Section 1.4.2) and heterologous biosynthesis and semisynthesis (Section 1.4.3). This
overview emphasizes two main points. For one, it is shown here that synthetic advancements in
the tetracycline field have time and again led to the development of FDA approved tetracyclines.
The second point emphasized in this overview is the inaccessibility of key tetracycline antibiotic
analogs through both of the established methods of tetracycline analog production, biosynthesis
and semisynthesis using the original microbial hosts (Section 1.4.1) and total synthesis (Section
1.4.2).
Some example for the first main point include the FDA approval of minocycline on 1971 following
the mutgenesis studies that enabled the fermentation of 6-demethyltetracyclines (e.g.
demeclocycline) and the followup synthetic studies that enabled aromatic substitutions in the 7-
position. This synthetic achievement was in turn followed up with studies on aromatic substitions
of minocycline in the 9-position that gave birth to the tigecycline that received its FDA approval
on 2005 (Section 1.4.1). Similarly, a breakthrough in the younger field of tetracycline total
synthesis, enabling unprecedented modifications in the tetracycline D-ring such as 7-
fluorotetracyclines, gave birth to eravacycline that was FDA approved on 2018 (Section 1.4.2).
Some examples for the second main point include promising tetracycline analogs at the 6- and 4a-
positions that are further discussed in Section 1.5. It remains to be seen whether unique

9
transformations enabled by the much younger field of heterologous biosynthesis and semisynthesis
of tetracyclines would yield FDA approved antibiotics (Section 1.4.3).
1.4.1 Biosynthesis and semisynthesis of tetracyclines using the original
microbial hosts
Tetracycline biosynthesis and semisynthesis using the original microbial hosts products can yield
modifications in positions 1, 2, 3, 4, 5, 5a, 6, 7, 9, 11, 11a, 12 and 12a.(25) The original microbial
hosts referred to here include Streptomyces aureofaciens and Streptomyces rimosus, the original
producers of chlortetracycline and oxytetracycline, as well as the S. aureofaciens mutants that
produce tetracycline and demeclocycline.(27) Despite the limitations on the positions that can be
modified and more importantly despite the limitations on the functional group that could be
introduced in these positions, this strategy yielded all FDA-approved tetracyclines that are not
natural products prior to 2018.(19)
Considering that positions 12a, 1, 2 and 3 and the β-diketone of 11,11a and 12 are known to be
important for the interaction of tetracyclines with the ribosome,(8, 24, 25) it is not surprising that
modifications in these positions have never led to a new tetracycline antibiotic drug. I will therefore
provide a short overview for biosynthetic and semisynthetic modifications using the original host
that led to tetracyclines modified in positions 4, 5, 5a, 6, 7, and 9. For a broader review of the
semisynthesis of tetracyclines using the original microbial hosts the reader is directed to the
excellent book chapter by Rogalski.(25) For more recent reviews in the field, covering tigecycline
and related derivatives as well, the reader is referred to references (6, 22, 28-31).
Modifications at the 4-position have so far not presented tetracycline analogs with improved
antibiotic activity and most commonly have worsened antibiotic outcomes in comparison to the
unmodified counterparts. For example, 4-epitetracycline, 4-dedimethylaminotetracycline and 4-

10
hydroxy-4-dedimethylaminotetracycline all present much higher MIC values relative to
tetracycline.(25) Increasing bulk on the 4-position in 6-demethyltetracycline derivatives leads to
reduced antibiotic activity as well. 6-demethyltetracycline derivatives containing
methylethylamino, methylpropylamino, diethylamino and methyl(2-hydroxyethyl)-amino at the
4α-position instead of the 4α-dimethylamino group of 6-demethyltetracycline show 75%, 50%,
25% and 12%, respectively, of the antibiotic activity of tetracycline against Staphylococcus aureus
while 6-demethyltetracycline maintains 96% of the activity.(32)
The study the effect of reducing bulk on the 4-amino group was attempted as well by synthesizing
4-monoalkylamines, but only the synthesis of 4β-methylamines has succeeded. Considering that
4-epitetracycline has much reduced antibiotic activity it is not surprising that these derivatives
were much less effective antibiotics than tetracycline as well. The one monoalkylamino
successfully generated, 6-demethyl-4-dedimethylamino-4-methylaminotetracycline has shown
only 7% of the antibiotic activity of tetracycline.(32)
While 6-methylene tetracycline has effective antibiotic activity (e.g., MIC value of 0.2 against
tetracycline-sensitive S. aureus), its 4-trimethylammonium derivative 6-methylenetetracycline
methiodide does not (e.g., MIC value of 12 against tetracycline-sensitive S. aureus).(23) While
some of the difference could stem from the partial epimerization at the 4-position for the
methiodide, the partial epimerization alone cannot explain the difference. The lack of the proton
on the 4-dimethylammonium required to form the characteristic H-bond of the zwitterionic form
(Figure 1-2) is hypothesized to explain why 4-quaternary ammonium derivatives cannot form the
zwitterionic tetracycline form,(33) and could be a reason for their higher MIC values. 4-
epitetracyclines would be incapable of forming the zwitterionic form (Figure 1-2) as well and
similar problems could arise with increasing bulk on the 4-amino group.

11
The 4a proton was unmodified by biosynthesis and semisynthesis of tetracycline derivatives using
the original microbial hosts, as is to be expected given that C-H activation of the tertiary 4a-proton
is challenging in a highly functionalized molecule such as tetracycline and its natural and
semisynthetic analogs.(25, 34) A 4a(12a)-anhydrotetracycline was made and found to have less
than 1% the activity of tetracycline.(25)
Modifications at the 5-position enabled by biosynthesis and semisynthesis using the original
microbial hosts were focused on modifications of oxytetracycline and doxycycline (Figure 1-1),
such as esterification, and oxidation. In general, these modifications have either yielded
tetracycline derivatives with similar or worse antibiotic activity.(25) Modifications at the 5a-
position were mostly limited to dehydration, isomerization and 5a-methyl installation but have not
produced improvements on biological activity.
Modifications at the 6-position have yielded many of the currently FDA-approved tetracyclines
(Figure 1-1, Table 1-1). Some of the 6-position analogs made had the following functional groups
in the α and β positions, respectively: Me,OH; Me,H; H,Me; Me,-; H,OH; H,H; CH2,-; RSCH2,H;
OH,-; OH, HOCH2; PhCH2,H; RSOCH2,H; CH3COCH2,H; HOCH2CH2CH2,H; PhCH2CH2,H;
F,H; H,F; F,-; F,Me; H, OAc; with R indicating and alkyl, phenyl or a derivatized alkyl or phenyl
and a hyphen indicating an SP2 carbon at the 6-position.(25) Thus, it is clear that alkyl
derivatizations of the 6α-position were explored, as well as some heteroatoms such as fluorine.
However, the scope was still limited and has not included 6-epi-6-demethytetracyclines, a point
that will be revisited in section 1.6. This is to be expected considering that natural tetracyclines
such as tetracycline, oxytetracycline and chlortetracycline give ready access to a 6β-
hydroxytetracyclines. Notably, 6-demethyltetracyclines such as demeclocycline (Figure 1-1) were
accessed by strain mutagenesis.(25)

12
Experimental success with aromatic substitutions enabled initially the semisynthetic derivatization
of the 7-position generating minocycline that later received FDA approval (Figure 1-1).(6) 9-
position substation followed and thus the road was paved for the newer generation of semisynthetic
tetracyclines, tigecycline, sarecycline and omadacycline (Figure 1-1).(21, 22, 25, 28, 35, 36)
However, the scope of possible substitutions in the D-ring has been significantly expanded with
the Myers convergent approach to tetracycline total synthesis, covered in Section 1.4.2.
Modifications at the 2-position have generated tetracycline antibiotics that were approved outside
the United States. Rolitetracycline, lymecycline (N-lysinomethyltetracycline) and clomocycline
are such examples. However, the modifications in general made for tetracycline prodrugs with
improved water solubility that hydrolyze to tetracyclines unmodified in the 2-position, which is
their active form.(25)
1.4.2 Total synthesis of tetracycline antibiotics
Early efforts at the total synthesis of tetracyclines include the first total synthesis of a biologically
active tetracycline, 6-demethyl-6-deoxytetracycline by the Woodward laboratory in the 1960s,
albeit as a racemic mixture.(37) Later synthetic efforts of interest to antibiotically active analog
generation included generating heteroatoms at the 6-position in derivatives such as 6-
thiatetracycline.(25) However, the latter was found to be acting as an antibiotic by an alternative
mechanism that does not involve the ribosome and was labeled, along with anhydrotetracycline,
anhydrochlortetracycline, chelocardin and 4-epianhydrochlortetracycline, as an atypical
tetracycline. Importantly, adverse side effects precluded the latter group from being perused as
antibiotic agents.(38)
While other total syntheses of tetracyclines appeared over the years, (39, 40) the Myers’ group
convergent enantioselective total synthesis of tetracyclines,(41) was the first one to produce a

13
totally synthetic FDA approved tetracycline.(20, 42) This convergent approach to tetracycline total
synthesis permitted tetracycline analog modifications previously impossible, particularly in the 7-
, 8- and 9-positions. These modifications enabled the synthesis of TP-271, a clinical candidate and
the FDA approved eravacycline, both 7-dedimethylamino-7-fluoro-9-amidominocyclines (Figure
1-1).(18, 43, 44) Additional tetracycline analogs that became accessible by later developments in
this total synthesis approach are 5-oxo analogs with an oxygen instead of the C5 carbon and 5a-
quaternary carbon analogs.(45, 46) Importantly, the Myers convergent synthesis of tetracyclines
does involve a fermentation process in its first step, the enantioselective dihydroxylation of
benzoic acid.(41, 47) However, this synthesis is clearly distinct from the semisynthetic efforts
towards tetracyclines that modify fully formed tetracycline fermentation products (Section 1.4.1).
On the other hand, the synthetic strategy for forming the AB-ring precursor and then coupling it
with the D-ring precursor across the C-ring limits modifications to the AB-ring junction and C-
ring.(41) Specifically, the Michael-Claisen cyclization that forms the ABCD tetracycline core from
the AB- and D-ring precursors demands a D-ring precursor that can undergo anionization at the
carbon precursor to the C6 carbon and then serve as a nucleophile for 1,4-addition reaction to an
α,β-unsaturated carbonyl (Figure 1-3-a). This demand limits the functional diversity that can be
convergently generated at the 6α-position (Figure 1-1). 6α-substituents that were thus published in
the Myers group include alkyl, aryl, heteroaryl, and proton substituents.(41, 42) Thus, some 6-
deoxytetracyclines such as doxycycline and some 6-deoxy-6-demethyl tetracyclines such as
eravacycline can be convergently synthesized using this total synthesis strategy.(41, 42, 44)
However, other simple tetracyclines, such as tetracycline itself, require an alternative cyclization
strategy and further oxygenation following the cyclization, and thus while the overall yield for
doxycycline was 8.3%, the overall yield for tetracycline was 1.1%.(41, 48)

14
Substitutions at the 4a-positions also appear limited by the convergent approach to tetracycline
total synthesis, as epoxide formation and opening on this position are critical to the formation of
the AB-ring intermediate (Figure 1-3-b).(41) Finally, it is also unclear at this point whether this
method of tetracycline total synthesis can compete economically with fermentation, or with
semisynthesis from a fermentation intermediate, for commercial process chemistry of a wide range
of tetracyclines indicated for a wide range of infectious diseases.
Figure 1-3 Key steps in the Myers convergent total synthesis of tetracyclines
The key step to form the (a) ABCD precursor and (b) AB-ring precursor in the total synthesis of
doxycycline. Adapted from (41)
1.4.3 Heterologous biosynthesis and semisynthesis of tetracyclines
Heterologous biosynthesis of natural products and their analogs is a rapidly developing field and
is importing important biosynthetic pathways into tractable hosts, such as Escherichia coli and
Saccharomyces cerevisiae. This field relies heavily on recent breakthrough in the fields of DNA
synthesis, DNA sequencing, methods in genetic manipulation of model organisms and information

15
regarding natural product pathways in a variety of organisms.(49) Heterologous biosynthesis is
tackling the production of medicinally important and structurally complex natural products such
as taxol,(50) erythromycin,(51) and lovastatin.(52) Notably, heterologous biosynthesis offers a
greener or more economically feasible approach to the production of known compounds.(53, 54)
While much of the effort in heterologous biosynthesis is still focused on the production of natural
products themselves, an ultimate goal is to obtain their unnatural derivatives as well.(55)
Heterologous biosynthesis in the tetracycline field was spearheaded by the Tang laboratory who
produced in Streptomyces lividans K4 by herelogous expression oxytetracycline (Figure 1-1),
dactylocyclinone (Figure 1-4) and SF2575 through the expression of their respective biosynthetic
pathways.(56) Analog production of the antitumor agent SF2575 was also shown in this study by
the elimination of biosynthetic enzymes from the SF2575 pathway. As is the common case for the
heterologous production of other glycosylated natural products,(57) the aglycone,
dactylocyclinone and not the glycosylated natural products dactylocyclines has been
produced.(56)
The power of genetic modifications to the biosynthetic pathway in yielding tetracycline derivatives
was historically used in the original hosts. For example, in the production of tetracycline and
demeclocycline by mutants of Streptomyces aureofaciens that naturally produces
chlortetracycline.(58) Demeclocycline is an intermediate in the semisynthesis of the important
drug minocycline and a tetracycline drug on its own right (Figure 1-1).(59) While impressive for
having taken place before full deciphering of the tetracycline biosynthetic pathway and prior to
the development of efficient tools for genomic engineering, these two analogs were obtained by
elimination of function rather than by introduction of function, which is more challenging. The

16
genetic revolution that intervened and the use of genetically tractable heterologous organisms such
as S. cerevisiae are a potential avenue towards such challenging transformations.(49)
Importantly the Tang group has also heterologously expressed in S. cerevisiae the biosynthetic
pathway of the fungal anhydrotetracycline derivative, TAN-1612.(60) Fungal
anhydrotetracyclines such as TAN-1612 and viridicatumtoxin serve as excellent scaffolds for
generating tetracycline analogs in S. cerevisiae (Section 1.6.4 and Chapter 4). While not pursued
thus far in tetracyclines, heterologous semisynthesis combining the power of heterologous
biosynthesis with the often complimentary advantages of synthetic chemistry is exciting. Such
combination has led to the production of artemisinin from artemisinic acid fermented in yeast,(61)
and could prove especially beneficial for generating tetracycline analogs (Section 1.6.4).
In conclusion, for each of the fronts of tetracycline synthesis, biosynthesis and semisynthesis using
the original microbial hosts, total synthesis and heterologous biosynthesis and semisynthesis, the
emergence of new capabilities is the enabling tool for powerful new antibiotics. Thus, the
development of semisynthetic capabilities in the 6-, 7- and 9-positions enabled the synthesis of
novel more powerful tetracycline antibiotics such as doxycycline, minocycline and tigecycline
(Section 1.4.1). The expansion of the range of possible modifications at the 7- and 9-positions by
total synthesis enabled the synthesis of 7-fluoro-9-amidotetracycline antibiotics such as
eravacycline (Section 1.4.2). Importantly however, key tetracycline analogs such as 6-demethyl-
6-epitetracyclines and 4a-hydroxytetracyclines, as well as their derivatives remained inaccessible
through the existing synthetic approaches (Sections 1.4.1 and 1.4.2) It is now to be tested whether
the new efforts enabling the heterologous expression of tetracycline biosynthetic pathways will
give rise to new tetracycline antibiotic drugs (Section 1.4.3). Section 1.5 describes exciting
potential structures for such tetracyclines.

17
1.5 Tetracycline analogs in the 6-position
The 6-position of tetracycline has been particularly useful for generating tetracycline analogs.
Indeed, no less than five different functional group combinations in the 6-position exist for FDA-
approved tetracyclines (Figure 1-1 and Table 1-1). As noted by Rogalski in 1985, “Modifications
at the C6 atom have produced by far the greatest success in evolving highly active
tetracyclines.”(25) Three different classes of 6-position tetracycline analogs will be outlined here
as inspiration for future tetracycline antibiotics analogs in the 6-position, the natural product
dactylocyclines (Section 1.5.1) the totally synthetic 6α-aryltetracyclines (Section 1.5.2) and the
semisynthetic 6α-methylenemercaptantetracyclines (Section 1.5.3).
1.5.1 Dactylocyclines – natural product 6-epiglycotetracyclines
Dactylocyclines are tetracycline antibiotics isolated in the early 1990’s from Dactylosporangium
sp. (ATCC 53693) in a screen by Bristol-Myers Squibb for antibiotics active against tetracycline-
resistant strains.(62) Dactylocyclines are 4a-hydroxy-6-epi-6-glyco-7-chloro-8-
methoxytetracyclines (Figure 1-4). While the 7-chloro substitution is known for existing
tetracycline antibiotics on the market, the 4a, 6, and 8 substitutions are unique (Figure 1-1).
Dactylocyclines were found to be antibiotically active, although not of an exceptional potency,
with MIC values of 3.1-6.3 μg/mL against gram-positive tetracycline-resistant strains of
Straphylococcus aureus, Staphylococcus epidermis and Streptococcus faecalis. However,
dactylocyclines exhibited high MIC values (>100 μg/mL) against gram-negative tetracycline-
resistant strains.(63) Importantly, the antibiotic activity of dactylocyclines against tetracycline-
resistant strains is dependent on the glycoside group, with dactylocyclinone, the aglycone of
dactylocycline, not presenting similar antibiotic properties.(63)

18
Despite their promising antibacterial activity outlook, two main disadvantages prevented
dactylocyclines’ further development as antibiotics. For one, dactylocyclines are extremely acid
labile. Specifically, the glycoside installed on the tertiary hydroxyl group of the 6-epitetracycline
aglycone dactylocyclinone is acid labile in mildly acidic conditions.(56, 62) Secondly, the fields
of synthetic biology and metabolic engineering were not yet ripe to generate dactylocycline
analogs at the early 1990’s when dactylocyclines were discovered. Twenty years later, with higher
maturity of these fields, the pathway towards the dactylocycline aglycone, dactylocyclinone, was
successfully heterologously expressed by the Tang group.(56) Thus, today there is an outlook for
generating dacylocycline analogs that build on the advantages of the natural dactylocyclines and
overcome their disadvantages (Section 1.6).
Figure 1-4 Structures of the dactylocycline family of natural products
Adapted from (62).
1.5.2 6-demethyl-6-deoxy-6α-aryltetracyclines – totally synthetic tetracyclines
6-demethyl-6-deoxy-6α-aryltetracyclines and 6-demethyl-6-deoxy-6α-heteroaryltetracyclines
have been accessed through the Myers convergent total synthesis of tetracyclines and described in
2008.(42) Among the 6α-aryls synthesized are included, phenyl, 4-toluyl and 3,5-eifluorophenyl.
While these derivatives showed good MIC values of <1 μg/mL against gram-positive tetracycline-

19
resistant strains, they showed values of ≥8 μg/mL against gram-negative tetracycline-resistant
strains.(42) However, despite promising MIC values, when one of these derivatives, 6-demethyl-
6-deoxy-6α-phenyltetracycline was tested in vivo in mice infected with tetracycline-sensitive
gram-positive S. aureus, it was found to be much less effective than tetracycline, requiring a
concentration at least 10 times higher to achieve the same rescuing ability.(42) A possible reason
for the ineffectiveness of 6α-aryltetracyclines against gram-negative tetracycline-resistant bacteria
in vitro and against tetracycline-sensitive infections in vivo could be their high expected
lipophilicity, a topic explored in Section 1.5.3.
1.5.3 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines –
semisynthetic tetracyclines
6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines are an example for the potential
importance of the polarity of 6α-substituents on the antibacterial activity of 6α-substituted 6-
demethyltetracycline analogs. 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines were
synthesized from the semisynthetic 6-methylenetetracycline and 6-methyleneoxytetracycline
(metacycline, Figure 1-1).(23)
Mercaptan derivatives of 6-methylenetetracycline with lipophilic substituents such as Ph and Bn
showed a much reduced activity relative to tetracycline against the gram-negative Klebsiella
pneumoniae (Figure 1-5-a). However, in the case of the phenyl and benzyl derivatives of 6-
methyleneoxytetracycline the reduction in activity was lower, presumably due to the mediating
hydrophilicity of the 5α-hydroxy (Figure 1-5-b). Similarly, in the case of the more polar benzyl
and phenyl sulfoxide derivatives of 6-methylenetetracycline the reduction in activity relative to
tetracycline was lower as well (Figure 1-5-c). The smaller and more polar acetyl derivatives

20
showed better activity in both mercaptan derivatives of 6-methylenetetracycline and 6-
methyleneoxytetracycline (Figure 1-5-a and Figure 1-5-b).(23)
Figure 1-5 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines and their antibiotic
activities
(a) 6-demethyl-6-deoxy-6α-methylenemercaptantetracyclines derived from 6-
methylenetetracycline, (b) 6-demethyl-6-deoxy-6α-methylenemercaptanoxytetracyclines derived
from 6-methyleneoxytetracycline (metacycline) and (c) 6-demethyl-6-deoxy-6α-
methylenemercaptantetracycline oxides derived from 6-methylenetetracycline. The numeric value
stated for each of the derivative is its normalized turbidimetric bioassay value (relative to
tetracycline) against the gram-negative Klebsiella pneumoniae.(23, 64)
Based on these results it was hypothesized that bulky and lipophilic substituents on the 6-position
reduce antibacterial activity, especially against gram-negative organisms. Importantly, however,
MIC values for the derivatives of Figure 1-5 did not follow the above mentioned trend and were
generally high, with the acetyl derivatives of 6-methyleneoxytetracycline performing better than
the others.(23) It is of interest to see whether this strategy of improving gram-negative efficacy by
reducing lipophilicity of the 6α-substituent, namely the glycoside, could be effective in the case of
6-demethyl-6-epiglycotetracyclines (Section 1.6).

21
1.6 Design of the biosynthesis of 6-demethyl-6-
epiglycotetracyclines using S. cerevisiae
Given the promise in tetracycline analogs in the 6-position to make effective antibiotics (Section
1.5) and given the limitations in existing synthetic methods to access key analogs in this position
(Section 1.4), I decided to focus my efforts on 6-position analogs. Specifically, inspired by the
natural products dactylocyclines (Section 1.5.1) I focused my efforts towards making acid stable
broad spectrum antibiotic derivatives of dactylocyclines effective against tetracycline-resistant
strains. The sections below outline the specific structural elements of the target tetracycline
analogs. Namely, making 6-demethyl derivatives for acid stability (Section 1.6.1), aiming for
broad spectrum activity using glycoside libraries (Section 1.6.2) and ensuring antibiotic efficacy
using 6-demethyl-6-epitetracyclines as opposed to 6-demethyltetracyclines (Section 1.6.3).
Finally, I describe why S. cerevisiae was chosen as the heterologous host for making these target
tetracycline analogs (Section 1.6.4).
1.6.1 Targeting 6-demethyl derivatives of 6-epiglycotetracyclines for acid
stability
To improve on the extreme acid lability of dactylocyclines, I propose making 6-demethyl-6-
epiglycotetracyclines, instead of 6-epiglycotetracyclines such as dactylocyclines. Dactylocyclines
show promising antibacterial activity against tetracycline-resistant strains. However,
dactylocyclines’ extreme acid lability prevents their development as antibiotics (Section 1.5.1). In
weak acidic conditions dactylocyclines are known to convert to their aglycone, dactylocyclinone
and thereby lose their antibiotic activity against tetracycline-resistant strains.(62, 63)

22
The extreme acid lability of dactylocyclines is likely originating in the tertiary 6α-hydroxy to
which the glycoside is bound in dactylocyclines (Figure 1-4). As Figure 1-6 shows, the acidic
hydrolysis bond splitting mechanism of the glycosidic bond is hypothesized to shift based on the
aglycone ability to form a stable carbonium ion. Thus, in aglycones that cannot form stable
carbonium ion, the oxonium ion would form on the glycoside and in aglycones that can form a
stable carbonium ion the carbonium ion would form on the aglycone. Examples for aglycones that
can form a stable carbonium ion are aglycones that are bound to the glycoside through a tertiary
or benzylic hydroxyl.(65)
As expected according to this hypothesis, the acid hydrolysis reaction rate is increased in the case
of a tert-butyl aglycone that can form a stable tertiary carbocation relative to the case of a methyl
aglycone (Table 1-2). However, the adamantyl carbocation is not similarly stabilized and
nevertheless when adamantyl is the aglycone the reaction rate is higher than in the case of other
substituents such as Me, Et and Pr. This incongruence led to the hypothesis that F-strain produced
by bulky aglycones enhances the hydrolysis rate as well.(65) Thus, in the case of the aglycone
dactylocyclinone, where the aglycone is both bulky tertiary and benzylic, it is indeed expected to
undergo facile hydrolysis in low acid concentrations.
Figure 1-6 Two possible C-O bond cleavage pathways in glycoside hydrolysis
Adapted from (65).

23
Table 1-2 Rates of hydrolysis of β-D-glycopyranosides in 0.5 M aqueous sulfuric acid
Aglycone Methyl Ethyl isopropyl tButyl Benzyl Adamantyl
Hydrolysis ratea 1 1.1 1.9 555.8 1.1 48.6
ahydroylsis rate is normalized to a methyl aglycone. The rate was calculated at 60 oC at 0.5 M
aqueous sulfuric acid or extrapolated or interpolated from data at different temperatures.(66, 67)
To produce 6-epiglycotetracycline analogs inspired by dactylocyclines but with a much increased
acid stability, I therefore propose to produce 6-demethyl-6-epiglycotetracyclines rather than 6-
epiglycotetracyclines. An example for such tetracycline derivatives would be 6-
demethyldactylocyclines. However, additional modifications are likely to be required to lead to
improved activity of 6-demethyl-6-epiglycotetracyclines against tetracycline-resistant gram-
positive and especially gram-negative strains. Such modifications are the subject of Section 1.6.2.
1.6.2 Targeting glycoside libraries on 6-demethyl-6-epiglycotetracyclines for
broad spectrum activity
For improving broad spectrum antibiotic activity against tetracycline-resistant strains I propose
installing a library of glycoside moieties on the 6-demethyl-6-epitetracycline aglycone. The
extremely limited tools in synthetic biology around the time of discovery of dactylocyclines
restricted exploring unnatural derivatives in this manner. However, throughout the intervening
years much progress has been achieved in producing diversified glycosylated natural products by
employing glycoside libraries for natural product aglycones.(68-70) Progress in this field is
fostered by the natural glycoside promiscuity of many glycosyltransferase.(68)
It was recently shown that installing sterically accessible amines on antibiotics that are only
effective against gram-positive strains can assist in transforming them into broad spectrum
antibiotics.(71) Thus, installing a library of glycosamines on 6-demethyl-6-epitetracyclines could

24
be a promising avenue to test whether tetracyclines active against gram-positive strains alone could
be transformed into broad-spectrum tetracyclines in this manner as well. Many glycosamines with
a primary amine have been previously reported in the literature and could be employed in this
glycodiversification effort.(69)
More specifically for tetracyclines, it was shown that reducing lipophilicity of 6α-substituents or
overall lipophilicity of 6-demethyl-6-deoxy-6α-methylmarcaptantetracyclines can increase their
gram-negative activity (Section 1.5.3). Importantly, dactylocyclines were noted for being
unusually more lipophilic than commercially available tetracyclines.(62) Their lipophilicity could
theoretically be stemming from any of the differentiating elements between dactylocyclines and
commercially available tetracyclines (Figure 1-1). These include a 4a-hydroxy, a 6-epi
configuration, a 7-chloro and an 8-methoxy (Section 1.5.1, Figure 1-4). Given that the 4a-hydroxy
and the 8-methoxy are unlikely to make dactylocyclines more lipophilic and given the presence of
the 7-chloro in commercially available tetracyclines (Table 1-1) it is likely the uniqueness in the
6-position that makes dactylocyclines more lipophilic. The 6-epi configuration of dactylocyclines
is unlikely to contribute to their unusually high lipophilicity relative to commercially available
tetracyclines because 6α-methyltetracyclines are generally known to be more lipophilic than 6β-
methyltetracyclines (Section 1.6.3). Thus, the 6-glycoside is the likely main contributor to
dactylocyclines lipophilicity, especially considering that the glycoside of dactylocyclines is rather
lipophilic with a 2,6-dideoxy and a 4-methoxy, leaving it with only one or zero hydroxyls (Figure
1-4). It would therefore be of interest to test whether 6-demethyl-6-epiglycotetracyclines with a
variety of less lipophilic glycosides have broad-spectrum activity against tetracycline-resistant
strains.

25
1.6.3 Targeting the 6-epi forms of 6-demethylglycotetracyclines for antibiotic
efficacy
C6 monosubstituted tetracyclines are generally more antibiotically active when substituted at the
α-position than when substituted at the β-position (Figure 1-1).(25) For example, doxycycline (6-
deoxyoxytetracycline) had lower MIC values than 6-epidoxycycline (6-epi-6-
deoxyoxytetracycline) in 7 / 7 gram-positive and gram-negative strains tested in one set of
experiments and its in vivo protective dose was lower as well.(23) Similar results, although less
pronounced, were reported for 6-deoxytetracycline versus 6-epi-6-deoxytetracycline,(23) and 6α-
fluoro-6-demethyl-6-deoxytetracycline was also shown to be more effective than its 6-epimer, 6β-
fluoro-6-demethyl-6-deoxytetracycline.
A possible explanation to the advantage 6α-tetracycline analogs have over their 6β counterparts is
that 6α-analogs more efficiently maintain the important balance between the zwitterionic and the
lipophilic forms of tetracyclines (Section 1.3.3 and Section 1.4.1). This explanation is supported
by the following known trend in lipophilicity, with lipophilicity levels ranking oxytetraycline < 6-
epi-6-deoxyoxytetracycline < 6-deoxyoxytetracycline, and with 6-epi-6-deoxyoxytetracycline
showing only a modest increase in lipophilicity relative to oxytetracycline. While the increase in
lipophilcity between oxytetracycline and its two 6-deoxy derivatives is expected based on a loss
of a hydrophilic hydroxy group, the increase in lipophilicity between the two deoxyoxytetracycline
epimers is less immediately reasoned. The commonly proposed reasoning is that a 6β-functional
group leads to steric clashes with the 4α-dimethylamino and thus destabilizes the lipophilic
conformation of the tetracycline, making the tetracycline derivative less lipophilic as a whole. In
contrast, an 6α-substiuent does not disrupt the important balance between the lipophilic and
zwitterionic forms of tetracycline derivatives.(72)

26
While these difference in lipophilicity and antibiotic activity are noted for 6α- and 6β-methyl
groups, they are all the more relevant when a much bulkier glycoside is installed on the 6-hydroxyl.
It is thus perhaps not surprising that dactylocyclines, the only known 6-glycotetrayclines natural
products, are 6-epitetracyclines while other tetracycline natural products such as oxytetracycline
and chlortetracyclines are not. Thus, 6-demethyl-6-epiglycotetracyclines are proposed in this study
and not 6-demethyl-6-glycotetracyclines. This point is of special importance given the higher
accessibility of 6β-hydroxytetracyclines over their 6α-hydroxytetracycline counterparts in all
methods of tetracycline preparation (Section 1.4.1 and Section 1.4.2 and Chapter 3).
1.6.4 S. cerevisiae as the heterologous host of choice for 6-demethyl-6-
epiglycotetracycline biosynthesis
Given that 6-demethyl-6-epiglycotetracyclines were marked here as highly desirable tetracycline
targets to synthesize and test as antibiotics (Sections 1.6.1, 1.6.2 and 1.6.3), the question arises
what would be the appropriate synthetic tool. Section 1.4.1 mentions the limitations in accessing
6-demethyl-6-epiglycotetracyclines by semisynthesis from the mainstream tetracycline
fermentation products, such as tetracycline, oxytetracycline, chlortetracycline and demeclocycline,
that are all 6β-hydroxy. Similarly, Section 1.4.2 mentions the difficulty in accessing 6-demethyl-
6-epitetracyclines by total synthesis due to the inherent limitations in the synthetic approach that
requires constructing the C-ring from AB- and D-ring precursors. Recent advances in metabolic
engineering and synthetic biology render it especially exciting to test heterologous biosynthesis
for generating 6-demethyl-6-epitetracyclines, the precursors to the desired 6-demethyl-6-
epiglycotetracyclines. Importantly, the following glycodiversification step can take place either in
vivo or by isolating 6-demethyl-6-epitetracyclines and appending activated glycosides on them
using glycosyltransferases in vitro or through chemical synthesis (Figure 1-7).

27
Figure 1-7 Retrosynthetic analysis of 6-demethyl-6-epiglycotetracyclines
Saccharomyces cerevisiae is uniquely positioned to the synthesis of 6-demethyl-6-
epiglycotetracyclines. First, as a highly genetically tractable model organism that is easy to culture
and quick to reproduce, S. cerevisiae has been demonstrated a useful heterologous host for the
production of a variety of natural products of importance to human health.(61, 73-75) The
advantages, sometimes unexpected, of performing chemistry in S. cerevisiae are also exemplified
in this study (e.g., Section 2.4.2).
Nevertheless, 6-demethyl-6-epiglycotetracyclines or 6-demethyl-6-epitetracyclines could also
theoretically be accessed from the native host Dactylosporangium sp. (ATCC 53693) or from an
alternative heterologous host, such as S. lividans K4 previously used for dactylocyclinone
biosynthesis.(56, 63) However, the native host Dactylosporangium sp. (ATCC 53693) is reported
to grow at an exceedingly slow rate and to be completely inaccessible for genetic
modifications.(56) It is yet to be determined whether an alternative heterologous host such as S.
lividans K4 has better or worse outcomes than S. cerevisiae for this biosynthesis goal.
Importantly, the Tang laboratory has reported the heterologous biosynthesis of the fungal
anhydrotetracycline TAN-1612 in S. cerevisiae (Section 1.4.3) and TAN-1612 is well positioned
to serve as a scaffold for the production of 6-demethyl-6-epiglycotetracyclines in S. cerevisiae.
TAN-1612 holds important structural similarities to anhydrodactylocyclinone, such as the 4a-
hydroxy and the 8-methoxy functional groups, that can render it a good starting point for further
derivatization. These substitutions are not observed in the anhydrotetracycline precursors of the

28
commonly fermented tetracyclines, such as chloranhydrotetracycline and anhydrotetracycline.
Also favorably to the target molecules outlined (Figure 1-7), TAN-1612 lacks a 6-methyl group
(Figure 1-8).
Figure 1-8 Structural Comparison between anhydrotetracycline, chloranhydrotetracycline,
TAN-1612 and anhydrotdactylocyclinone
In grey are marked potentially desirable functional groups TAN-1612 has relative to the
commercially available anhydrotetracycline as a scaffold for 6-demethyl-6-epiglycotetracycline
biosynthesis. The stereochemistry of TAN-1612 at positions 4a and 12a was not yet confirmed by
X-ray crystallography. Empty cells indicate implicit Hs.
However, TAN-1612 is also lacking structural features that are important for antibiotic activity in
tetracyclines in the A-ring such as the 4α-dimethylamino and the 2-carboxamido (Figure 1-8). The
furnishing of a 2-carboxamido derivative of TAN-1612 or a related molecule in S. cerevisiae could
be envisioned by heterologously expressing the relevant genes from the oxytetracycline
biosynthetic pathway of S. rimosus. An alternative pathway to harvest the relevant genes is the
biosynthetic pathway towards the fungal polyketide viridicatumtoxin, which is homologous to that
of TAN-1612. This effort would likely require evolving the TAN-1612 biosynthetic enzymes to
the unnatural substrate.(76, 77)
The Yeast three hybrid (Y3H) system for metabolic engineering of tetracycline analogs could be
crucial to assay mutants of the TAN-1612 pathway enzymes for relaxed substrate specificity
towards the new 2-carboxamido intermediates (Section 1.7.4 and Chapter 5). Likewise,
hydroxylation and oxidation of the 4-position of TAN-1612 can be envisioned by OxyE and OxyL

29
from the oxytetracycline pathway (78) evolved for TAN-1612 specificity by employing the Y3H
as well. These steps could be followed by reductive amination to furnish the 4α-dimethylamino or
a related functional group on TAN-1612 or a related derivative. Finally, fermentation of 6-
demethyl-6-epiglycotetracyclines or 6-demethyl-6-epitetracyclines in S. cerevisiae could readily
open up possibilities in the longer term for commercial production of new tetracycline
therapeutics.
1.7 Research progress and future goals in the biosynthesis of
6-demethyl-6-epiglycotetracyclines using S. cerevisiae
1.7.1 Chapter 2 – Using Saccharomyces cerevisiae for the final steps of
tetracycline biosynthesis
This chapter describes the use of Saccharomyces cerevisiae for the final steps of tetracycline
biosynthesis, setting the stage for total biosynthesis of tetracycline in S. cerevisiae. Specifically,
data presented in this chapter supports the conversion of anydrotetracycline to tetracycline or an
epimer in S. cerevisiae by heterologous expression of the 6β-hydroxylase enzyme form the
oxytetracycline pathway, OxyS. This result is surprising as it was expected that the expression of
a reductase enzyme that uses a cofactor not native to yeast would be required as well. It was also
expected that OxyS would lead to an oxytetracycline intermediate and not to tetracycline by
performing two hydroxylation steps as reported previously and not one as shown in this study. The
ability to perform the required steps for tetracycline biosynthesis from anhydrotetracycline
supports the ongoing efforts to produce 6-demethyl-6-epitetracyclines from anhydrotetracyclines
(Figure 1-7, Chapter 3 and Chapter 4).

30
1.7.2 Chapter 3 – Towards the biosynthesis of 6-demethyl-6-epitetracyclines
using S. cerevisiae
This chapter describes the work towards 6-demethyl-6-epitetracyclines biosynthesis in S.
cerevisiae. 6α-hydroxylation of a 6-demethylanhydrotetracycline by an enzyme such as DacO1 is
required for the synthesis of 6-demethyl-6-epitetracyclines as opposed to 6β-hydroxylation by
enzymes such as OxyS leading to 6-demethyltetracyclines. However, previous attempts using
multiple heterologous hosts to heterologously express DacO1 without the rest of the
dactylocyclinone pathway have not yielded stably expressed protein. The chapter describes the
successful development of a stably expressing form of DacO1 in S. cerevisiae by employing
rational design approaches for optimizing DacO1 expression. This chapter also describes the
successful hydroxylation of anhydrotetracycline by another 6α-hydroxylase candidate, PgaE. The
next step towards 6-demethyl-6-epitetracyclines is to test the stably expressed DacO1 and PgaE in
larger scale for 6α-hydroxylation of anhydrotetracycline and/or anhydrodactylocyclinone and/or
6-demethylanhydrodactylocyclinone.
1.7.3 Chapter 4 – Towards the biosynthesis of 6-demethyl-6-epitracyclines
from TAN-1612 in Saccharomyces cerevisiae
This chapter describes the work towards 6-demethyl-6-epitracycline derivatives of the fungal
anhydrotetracycline TAN-1612 in Saccharomyces cerevisiae. TAN-1612 is indicated as a unique
anhydrotetracycline scaffold for the production of 6-demethyl-6-epitetracyclines (Section 1.6.4),
the precursors to the target molecules 6-demethyl-6-epiglycotetracyclines (Figure 1-7). The
progress and future steps towards the directed evolution and biomining of a TAN-1612 6α-
hydroxylase are outlined and the isolation and characterization of a new major product in a strain
coexpressing PgaE and the TAN-1612 pathway is described.

31
1.7.4 Chapter 5 – a yeast three hybrid system for the metabolic engineering of
tetracycline derivatives
The transformation of anhydrotetracyline to tetracycline could be readily detected by a UV/Vis
based assay due to the blue-shift in the excitation and emission of the molecule (Chapters 2-4).
However, most transformations of tetracyclines and other molecules that do not involve such a
major UV/Vis shift, such as the proposed glycosylation and A-ring modifications (Section 1.6.4)
require the development of a general assay for their detection. This chapter describes the
development of a yeast three hybrid (Y3H) assay as such a general, high-throughput, versatile and
readily implemented approach for the detection of target molecule biosynthesis. The Y3H assay is
presented here as a tool for the reactions in the next steps towards the heterologous biosynthesis
of 6-demethyl-6-epiglycotetracyclines in S. cerevisiae and beyond.

32
1.8 References
1. Antibiotic Resistance Threats in the United States. US Department of Health and Human
Services, Center for Disease Control and Prevention, 2013.
2. Mullard A. Momentum builds around new antibiotic business models. Nature Reviews
Drug Discovery. 2014;13(10):711-713. doi: 10.1038/nrd4455.
3. Henssler EM, Scholz O, Lochner S, Gmeiner P, Hillen W. Structure-based design of Tet
repressor to optimize a new inducer specificity. Biochemistry. 2004;43(29):9512-9518. doi:
10.1021/bi049682j.
4. Tatsuta K. Total synthesis of the big four antibiotics and related antibiotics. The Journal of
Antibiotics. 2013;66(3):107-129. doi: 10.1038/ja.2012.126.
5. Eisen DP. Doxycycline. In: M. Lindsay Grayson SEC, Suzanne Crowe, William Hope,
James S. McCarthy, John Mills, Johan W. Mouton, David L. Paterson, editor. Kucers' The Use of
Antibiotics: A Clinical Review of Antibacterial, Antifungal, Antiparasitic, and Antiviral Drugs. 6
ed. Boca Raton, FL: CRC Press; 2010. p. 851-869.
6. Nelson ML, Levy SB. The history of the tetracyclines. Ann NY Acad Sci. 2011;1241:17-
32. doi: 10.1111/j.1749-6632.2011.06354.x.
7. Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular
biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. 2001;65(2):232-260.
doi: 10.1128/MMBR.65.2.232-260.2001.
8. Brodersen DE, Clemons WM, Jr., Carter AP, Morgan-Warren RJ, Wimberly BT,
Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin,
and hygromycin B on the 30S ribosomal subunit. Cell. 2000;103(7):1143-1154. doi:
10.1016/S0092-8674(00)00216-6.
9. Speer BS, Shoemaker NB, Salyers AA. Bacterial resistance to tetracycline: mechanisms,
transfer, and clinical significance. Clin Microbiol Rev. 1992;5(4):387-399. doi:
10.1128/CMR.5.4.387.

33
10. Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal protection proteins and their
mechanism of tetracycline resistance. Antimicrob Agents Chemother. 2003;47(12):3675-3681.
doi: 10.1128/AAC.47.12.3675-3681.2003.
11. Gerrits MM, de Zoete MR, Arents NL, Kuipers EJ, Kusters JG. 16S rRNA mutation-
mediated tetracycline resistance in Helicobacter pylori. Antimicrob Agents Chemother.
2002;46(9):2996-3000. doi: 10.1128/AAC.46.9.2996-3000.2002.
12. Speer BS, Salyers AA. Novel aerobic tetracycline resistance gene that chemically modifies
tetracycline. J Bacteriol. 1989;171(1):148-153.
13. Toon S, Rowland M. Quantitative Structure Pharmacokinetic Activity Relationships with
Some Tetracyclines. J Pharm Pharmacol. 1979;31(S1):43P-43P. doi: 10.1111/j.2042-
7158.1979.tb11591.x.
14. Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Tetracyclines: a pleitropic family of
compounds with promising therapeutic properties. Review of the literature. AJP: Cell Physiology.
2010;299(3):C539-C548. doi: 10.1152/ajpcell.00047.2010.
15. Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J
Pharmacol. 2013;169(2):337-352. doi: 10.1111/bph.12139.
16. Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC,
Wilson DN, Yusupova G. Structural basis for potent inhibitory activity of the antibiotic tigecycline
during protein synthesis. Proceedings of the National Academy of Sciences. 2013;110(10):3812-
3816. doi: 10.1073/pnas.1216691110.
17. Prasad P, Sun J, Danner RL, Natanson C. Excess Deaths Associated With Tigecycline
After Approval Based on Noninferiority Trials. Clin Infect Dis. 2012;54(12):1699-1709. doi:
10.1093/cid/cis270.
18. Liu F, Myers AG. Development of a platform for the discovery and practical synthesis of
new tetracycline antibiotics. Curr Opin Chem Biol. 2016;32:48-57. doi:
10.1016/j.cbpa.2016.03.011.
19. Nelson ML, Levy SB. 3.25 - Tetracyclines and Tetracycline Derivatives. In: Moo-Young
M, editor. Comprehensive Biotechnology (Second Edition). Burlington: Academic Press; 2011. p.
269-283.

34
20. Approved drug products with therapeutic equivalence evaluations (Orange book). U.S.
Department of Health and Human Services, 2019.
21. Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J
Antibiot. 2013;66(10):571-591. doi: 10.1038/ja.2013.86.
22. Honeyman L, Ismail M, Nelson ML, Bhatia B, Bowser TE, Chen J, Mechiche R, Ohemeng
K, Verma AK, Cannon EP, Macone A, Tanaka SK, Levy S. Structure-Activity Relationship of the
Aminomethylcyclines and the Discovery of Omadacycline. Antimicrob Agents Chemother.
2015;59(11):7044-7053. doi: 10.1128/aac.01536-15.
23. Blackwood RK, English AR. Structure–Activity Relationships in the Tetracycline Series.
In: Perlman D, editor. Adv Appl Microbiol: Academic Press; 1970. p. 237-266.
24. Khosla C, Tang Y. A new route to designer antibiotics. Science. 2005;308(5720):367-368.
doi: 10.1126/science.1111415.
25. Rogalski W. Chemical Modification of the Tetracyclines. In: Hlavka JJ, Boothe JH, editors.
The Tetracyclines. Berlin, Heidelberg: Springer Berlin Heidelberg; 1985. p. 179-316.
26. Rasmussen B, Noller HF, Daubresse G, Oliva B, Misulovin Z, Rothstein DM, Ellestad GA,
Gluzman Y, Tally FP, Chopra I. Molecular basis of tetracycline action: identification of analogs
whose primary target is not the bacterial ribosome. Antimicrob Agents Chemother.
1991;35(11):2306-2311. doi: 10.1128/AAC.35.11.2306.
27. Hošťálek Z, Vaněk Z. Biosynthesis of the Tetracyclines. In: Hlavka JJ, Boothe JH, editors.
The Tetracyclines. Berlin, Heidelberg: Springer Berlin Heidelberg; 1985. p. 137-178.
28. Sum PE, Petersen P. Synthesis and structure-activity relationship of novel glycylcycline
derivatives leading to the discovery of GAR-936. Bioorg Med Chem Lett. 1999;9(10):1459-1462.
doi: 10.1016/S0960-894X(99)00216-4.
29. Chopra I. New developments in tetracycline antibiotics: glycylcyclines and tetracycline
efflux pump inhibitors. Drug Resistance Updates. 2002;5(3-4):119-125. doi: 10.1016/s1368-
7646(02)00051-1.
30. Zhanel GG, Homenuik K, Nichol K, Noreddin A, Vercaigne L, Embil J, Gin A, Karlowsky
JA, Hoban DJ. The Glycylcyclines. Drugs. 2004;64(1):63-88. doi: 10.2165/00003495-200464010-
00005.

35
31. Wright PM, Seiple IB, Myers AG. The Evolving Role of Chemical Synthesis in
Antibacterial Drug Discovery. Angew Chem Int Ed. 2014;53(34):8840-8869. doi:
10.1002/anie.201310843.
32. Esse RC, Lowery JA, Tamorria CR, Sieger GM. Tetracycloxides. II. Transformations at
the C-4 Position. J Am Chem Soc. 1964;86(18):3875-3877. doi: 10.1021/ja01072a057.
33. Prewo R, Stezowski JJ. Chemical-structural properties of tetracycline derivatives. 3. The
integrity of the conformation of the nonionized free base. J Am Chem Soc. 1977;99(4):1117-1121.
doi: 10.1021/ja00446a024.
34. Mitscher LA. Degradation and Structure Proofs. The Chemistry of the Tetracycline
Antiobitcs. New York: Marcel Dekker, Inc.; 1978. p. 122-164.
35. Sum PE, Lee VJ, Testa RT, Hlavka JJ, Ellestad GA, Bloom JD, Gluzman Y, Tally FP.
Glycylcyclines .1. A New-Generation of Potent Antibacterial Agents through Modification of 9-
Aminotetracyclines. J Med Chem. 1994;37(1):184-188. doi: 10.1021/jm00027a023.
36. Tanaka SK, Steenbergen J, Villano S. Discovery, pharmacology, and clinical profile of
omadacycline, a novel aminomethylcycline antibiotic. Biorg Med Chem. 2016;24(24):6409-6419.
doi: 10.1016/j.bmc.2016.07.029.
37. Conover LH, Butler K, Johnston JD, Korst JJ, Woodward RB. The Total Synthesis of 6-
Demethyl-6-Deoxytetracycline. J Am Chem Soc. 1962;84(16):3222-3224. doi:
10.1021/ja00875a063.
38. Chopra I. Tetracycline analogs whose primary target is not the bacterial ribosome.
Antimicrob Agents Chemother. 1994;38(4):637-640. doi: 10.1128/aac.38.4.637.
39. Wasserman HH, Lu TJ, Scott IA. The total synthesis of tetracycline. J Am Chem Soc.
1986;108(14):4237-4238. doi: 10.1021/ja00274a072.
40. Stork G, La Clair JJ, Spargo P, Nargund RP, Totah N. Stereocontrolled Synthesis of (±)-
12a-Deoxytetracycline. J Am Chem Soc. 1996;118(22):5304-5305. doi: 10.1021/ja960434n.
41. Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. A convergent
enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science.
2005;308(5720):395-398. doi: 10.1126/science.1109755.

36
42. Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Charest M, Siegel DR,
Wang Y-M, Myers AG. A Robust Platform for the Synthesis of New Tetracycline Antibiotics. J
Am Chem Soc. 2008;130(52):17913-17927. doi: 10.1021/ja806629e.
43. Clark RB, Hunt DK, He M, Achorn C, Chen C-L, Deng Y, Fyfe C, Grossman TH, Hogan
PC, O’Brien WJ, Plamondon L, Rönn M, Sutcliffe JA, Zhu Z, Xiao X-Y. Fluorocyclines. 2.
Optimization of the C-9 Side-Chain for Antibacterial Activity and Oral Efficacy. J Med Chem.
2012;55(2):606-622. doi: 10.1021/jm201467r.
44. Xiao X-Y, Hunt DK, Zhou J, Clark RB, Dunwoody N, Fyfe C, Grossman TH, O’Brien WJ,
Plamondon L, Rönn M, Sun C, Zhang W-Y, Sutcliffe JA. Fluorocyclines. 1. 7-Fluoro-9-
pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial
Agent. J Med Chem. 2012;55(2):597-605. doi: 10.1021/jm201465w.
45. Liu F, Wright PM, Myers AG. Diastereoselective Michael–Claisen Cyclizations of γ-Oxa-
α,β-unsaturated Ketones en Route to 5-Oxatetracyclines. Org Lett. 2016;19(1):206-209. doi:
10.1021/acs.orglett.6b03491.
46. Wright PM, Myers AG. Methodological advances permit the stereocontrolled construction
of diverse fully synthetic tetracyclines containing an all-carbon quaternary center at position C5a.
Tetrahedron. 2011;67(51):9853-9869. doi: 10.1016/j.tet.2011.09.143.
47. Myers AG, Siegel DR, Buzard DJ, Charest MG. Synthesis of a Broad Array of Highly
Functionalized, Enantiomerically Pure Cyclohexanecarboxylic Acid Derivatives by Microbial
Dihydroxylation of Benzoic Acid and Subsequent Oxidative and Rearrangement Reactions. Org
Lett. 2001;3(18):2923-2926. doi: 10.1021/ol010151m.
48. Charest MG, Siegel DR, Myers AG. Synthesis of (−)-Tetracycline. J Am Chem Soc.
2005;127(23):8292-8293. doi: 10.1021/ja052151d.
49. Woolston BM, Edgar S, Stephanopoulos G. Metabolic engineering: past and future. Annu
Rev Chem Biomol Eng. 2013;4:259-288. doi: 10.1146/annurev-chembioeng-061312-103312.
50. Jiang M, Stephanopoulos G, Pfeifer BA. Downstream reactions and engineering in the
microbially reconstituted pathway for Taxol. Appl Microbiol Biotechnol. 2012;94(4):841-849.
doi: 10.1007/s00253-012-4016-1.
51. Pfeifer BA. Biosynthesis of Complex Polyketides in a Metabolically Engineered Strain of
E. coli. Science. 2001;291(5509):1790-1792. doi: 10.1126/science.1058092.

37
52. Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva
NA, Vederas JC, Tang Y. Complete Reconstitution of a Highly Reducing Iterative Polyketide
Synthase. Science. 2009;326(5952):589-592. doi: 10.1126/science.1175602.
53. Fasciotti M. Perspectives for the use of biotechnology in green chemistry applied to
biopolymers, fuels and organic synthesis: from concepts to a critical point of view. Sustainable
Chem Pharm. 2017;6:82-89. doi: 10.1016/j.scp.2017.09.002.
54. Keasling JD. Manufacturing Molecules Through Metabolic Engineering. Science.
2010;330(6009):1355-1358. doi: 10.1126/science.1193990.
55. Tsoi CJ, Khosla C. Combinatorial biosynthesis of 'unnatural' natural products: the
polyketide example. Chem Biol. 1995;2(6):355-362. doi: 10.1016/1074-5521(95)90214-7.
56. Wang P, Kim W, Pickens LB, Gao X, Tang Y. Heterologous Expression and Manipulation
of Three Tetracycline Biosynthetic Pathways. Angewandte Chemie-International Edition.
2012;51(44):11136-11140. doi: 10.1002/anie.201205426.
57. Feng Z, Kallifidas D, Brady SF. Functional analysis of environmental DNA-derived type
II polyketide synthases reveals structurally diverse secondary metabolites. Proc Natl Acad Sci U
S A. 2011;108(31):12629-12634. doi: 10.1073/pnas.1103921108.
58. Goodman JJ. Fermentation and Mutational Development of the Tetracyclines. In: Hlavka
JJ, Boothe JH, editors. The Tetracyclines. Berlin, Heidelberg: Springer Berlin Heidelberg; 1985.
p. 5-57.
59. Mccormick JRD, Sjolander NO, Hirsch U, Jensen ER, Doerschuk AP. A New Family of
Antibiotics - the Demethyl-Tetracyclines. J Am Chem Soc. 1957;79(16):4561-4563. doi:
10.1021/ja01573a089.
60. Li YR, Chooi YH, Sheng YW, Valentine JS, Tang Y. Comparative Characterization of
Fungal Anthracenone and Naphthacenedione Biosynthetic Pathways Reveals an alpha-
Hydroxylation-Dependent Claisen-like Cyclization Catalyzed by a Dimanganese Thioesterase. J
Am Chem Soc. 2011;133(39):15773-15785. doi: 10.1021/ja206906d.
61. Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus
RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Production of
the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940-
943. doi: 10.1038/nature04640.

38
62. Tymiak AA, Aklonis C, Bolgar MS, Kahle AD, Kirsch DR, O'Sullivan J, Porubcan MA,
Principe P, Trejo WH. Dactylocyclines: novel tetracycline glycosides active against tetracycline-
resistant bacteria. J Org Chem. 1993;58(3):535-537. doi: 10.1021/jo00055a002.
63. Wells JS, O'Sullivan J, Aklonis C, Ax HA, Tymiak AA, Kirsch DR, Trejo WH, Principe
P. Dactylocyclines, novel tetracycline derivatives produced by a Dactylosporangium sp. I.
Taxonomy, production, isolation and biological activity. J Antibiot. 1992;45(12):1892-1898. doi:
10.7164/antibiotics.45.1892.
64. Kersey RC. A Turbidimetric Assay for Terramycin. Journal of the American
Pharmaceutical Association (Scientific ed). 1950;39(5):252-253. doi: 10.1002/jps.3030390503.
65. Bochkov AF, Zaikov GE. Cleavage of O-glycosidic Bonds. Chemistry of the O-glycosidic
bond: Pergamon Press; 1979. p. 177-201.
66. Timell TE. The Acid Hydrolysis of Glycosides: I. General Conditions and the Effect of the
Nature of the Aglycone. Can J Chem. 1964;42(6):1456-1472. doi: 10.1139/v64-221.
67. Cocker D, Jukes LE, Sinnott ML. Alkyl–oxygen versus glycosyl–oxygen fission in the
acid-catalysed hydrolyses of some alkyl β-D-glycopyranosides. J Chem Soc, Perkin Trans 2.
1973(2):190-194. doi: 10.1039/p29730000190.
68. Thibodeaux CJ, Melançon CE, Liu H-w. Natural-Product Sugar Biosynthesis and
Enzymatic Glycodiversification. Angew Chem Int Ed. 2008;47(51):9814-9859. doi:
10.1002/anie.200801204.
69. Elshahawi SI, Shaaban KA, Kharel MK, Thorson JS. A comprehensive review of
glycosylated bacterial natural products. Chem Soc Rev. 2015;44(21):7591-7697. doi:
10.1039/c4cs00426d.
70. Jiang M, Zhang H, Park S-H, Li Y, Pfeifer BA. Deoxysugar pathway interchange for
erythromycin analogues heterologously produced through Escherichia coli. Metab Eng.
2013;20:92-100. doi: 10.1016/j.ymben.2013.09.005.
71. Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ.
Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature. 2017. doi:
10.1038/nature22308.

39
72. Stezowski JJ. Chemical-structural properties of tetracycline derivatives. 1. Molecular
structure and conformation of the free base derivatives. J Am Chem Soc. 1976;98(19):6012-6018.
doi: 10.1021/ja00435a039.
73. Galanie S, Thodey K, Trenchard IJ, Filsinger Interrante M, Smolke CD. Complete
biosynthesis of opioids in yeast. Science. 2015;349(6252):1095-1100. doi:
10.1126/science.aac9373.
74. Awan AR, Blount BA, Bell DJ, Shaw WM, Ho JCH, McKiernan RM, Ellis T. Biosynthesis
of the antibiotic nonribosomal peptide penicillin in baker’s yeast. Nature Communications. 2017;8.
doi: 10.1038/ncomms15202.
75. Luo X, Reiter MA, d’Espaux L, Wong J, Denby CM, Lechner A, Zhang Y, Grzybowski
AT, Harth S, Lin W, Lee H, Yu C, Shin J, Deng K, Benites VT, Wang G, Baidoo EEK, Chen Y,
Dev I, Petzold CJ, Keasling JD. Complete biosynthesis of cannabinoids and their unnatural
analogues in yeast. Nature. 2019;567(7746):123-126. doi: 10.1038/s41586-019-0978-9.
76. Lesnik U, Lukezic T, Podgorsek A, Horvat J, Polak T, Sala M, Jenko B, Harmrolfs K,
Ocampo-Sosa A, Martinez-Martinez L, Herron PR, Fujs S, Kosec G, Hunter IS, Muller R, Petkovic
H. Construction of a New Class of Tetracycline Lead Structures with Potent Antibacterial Activity
through Biosynthetic Engineering. Angewandte Chemie-International Edition. 2015;54(13):3937-
3940. doi: 10.1002/anie.201411028.
77. Chooi YH, Cacho R, Tang Y. Identification of the viridicatumtoxin and griseofulvin gene
clusters from Penicillium aethiopicum. Chem Biol. 2010;17(5):483-494. doi:
10.1016/j.chembiol.2010.03.015.
78. Pickens LB, Tang Y. Oxytetracycline biosynthesis. J Biol Chem. 2010;285(36):27509-
27515. doi: 10.1074/jbc.R110.130419.

40

41
2 Using Saccharomyces cerevisiae for the final steps of
tetracycline biosynthesis
*The contents of this chapter will be published in:
E. Herbst, V. W. Cornish, et al “Using Saccharomyces cerevisiae for the final steps of tetracycline
biosynthesis”, in preparation.

42
2.1 Chapter outline
This chapter describes the use of Saccharomyces cerevisiae for the final steps of tetracycline
biosynthesis, setting the stage for total biosynthesis of tetracycline in S. cerevisiae. Key results
described in this chapter are that OxyS performs in S. cerevisiae just one hydroxylation step as
opposed to two performed in vitro, thus enabling the biosynthesis of tetracycline instead of
oxytetracycline (Section 2.4.1 and 2.5.1). In addition, this chapter describes the reduction of the
hydroxylation product of OxyS in yeast cell lysate, without the need for heterologous expression
of a dedicated reduction enzyme, such as OxyR (Section 2.4.2). This result is important as OxyR
uses a cofactor not native to yeast. Thus, circumventing the need to express OxyR also circumvents
the need to heterologously express the biosynthetic pathway of the cofactor or to extragenously
supply it or its equivalent (Sections 2.4.2 and 2.5.2). Finally, this chapter describes mass
spectrometry and UV/Vis results supporting the production of 5a(11a)-dehydrotetracycline using
S. cerevsiae even though its further hydroxylation and/or degradation hampered previous analysis
when produced by OxyS in vitro and in vivo (Sections 2.4.2 and 2.5.2).
2.2 Introduction
S. cerevisiae has proven itself a useful heterologous host for the production of a variety of natural
products of importance to human health. A key example is the production of artemisinic acid, a
key intermediate in the production of artemisinin,(1) with more recent examples being the
production of hydrocodone, Δ9-tetrahydrocannabinolic acid (THCA) and penicillin.(2-4) One of
the promises in natural product biosynthesis using S. cerevisiae is the access to unnatural analogs
of natural products.(3) Tetracyclines are one of the most important classes of antibiotics and
tetracycline analogs are needed to combat the resistance to current tetracycline antibiotics on the

43
market given the current and impending antibiotic crisis (Chapter 1). Thus, from an applied science
perspective, research on heterologous biosynthesis of tetracyclines is important for its potential to
lead to new and effective tetracycline antibiotics. From a pure science perspective, heterologous
biosynthesis of is often highly instructive for the study of the biosynthetic steps that take place in
the natural hosts, as is exemplified throughout this chapter.
Granted, Heterologous biosynthesis of oxytetracycline was already shown in Streptomyces
lividans K4‐11. This is a more readily achievable feat given the existence in that host of a
biosynthetic pathway to cofactor F420, a required cofactor in the biosynthesis of tetracyclines that
is not native to S. cerevisiae, and the capacity of bacterial heterologous hosts to express whole
gene clusters from other bacteria.(5) However, its eukaryotic family lineage is also a key advantage
for S. cerevisiae as a heterologous host to bacterial natural products such as tetracycline.
Expression in S. cerevisiae allows orthogonal and complementary chemistry to be performed,
giving access to analogs that are otherwise inaccessible in heterologous expression in bacterial
hosts.(6) Examples for the rich, useful, and sometimes unexpected chemical possibilities of S.
cerevisiae in the heterologous biosynthesis of bacterial natural products such as tetracycline are
demonstrated in this chapter (2.4.2 and 2.5.2).
2.3 Materials and methods
See Chapter 5 for general methods.
2.3.1 General protocol for hydroxylation/reduction assay in cell lysate
Fresh patches of strains harboring the plasmid for the hydroxylation and/or reduction enzyme and
control strains were inoculated in 5 mL selective media (U- or HU-) in 15 mL culture tubes
(Corning 352059) and placed in shaker overnight to OD600 2-3. Overnight cultures were used to
inoculate 100 mL selective media (U- or HU-) cultures in 500 mL conical flasks with a starting OD

44
of 0.01-0.05. Cells were grown to final OD of 0.6-0.8 before pelleting in 2 50 mL tubes (Corning
352098) at 4 °C, 4,000 rpm for 20 min. Each pellet was redissolved in 0.5 mL H2O and the
suspension was distributed into two presterilized 1.5 mL Eppendorf tubes and pelleted at 14,000
rpm for 10 min at 4 °C. Pellets were stored at -20 °C prior to further use.
Pellets were weighed and thawn on ice. A 99:1 mixture of Y-PER yeast protein extraction reagent
(ThermoFisher Scientific 78991) and HALT protease inhibitor cocktail (ThermoFisher Scientific
PI87786) was added in a ratio of 3 μL mixture per mg pellet and placed on orbital shaker for 20
min at r.t., followed by 10 min centrifugation at 14,000 rpm at 4 °C and the cell lysate was
transferred to a new 1.5 mL Eppendorf tube, kept on ice and used within 1 h.
The cell lysate (80 μL) was added as the last component to a 4 mL vial (Chemglass CG-4900-01)
containing 280 μL of 143 mM Tris (pH 7.45), 7.7 mM anhydrotetracycline HCl (AdipoGen CDX-
A0197-M500), 4.3 mM NADPH tetrasodium hydrate (Sigma-Aldrich N7505), 26.4 mM
mercaptoehtanol and 14.285 mM glucose-6-phosphate (in experiments labeled +G6P, 0 mM
glucose-6-phophsate in all other experiments) and 40 μL glucose (278 mM) for final
concentrations of 100 mM Tris, 5.4 mM anhydrotetracycline HCl, 3 mM NADPH, 18.5 mM
mercaptoethanol, 0 or 10 mM G6P as indicated and 27.8 mM glucose. A septum was placed on
top of the vial through which a needle was inserted to allow air exchange and the reaction was left
at r.t. overnight. After night, 1 mL of MeOH was added, the contents were mixed and the reaction
was filtered through a PTFE 0.2 μm filter (Acrodisc 4423) prior to analysis by mass and UV/VIS
spectrometry.
2.3.2 Protocol for hydroxylation/reduction assay in whole cells
Fresh patches of strains harboring the plasmid for the hydroxylation and/or reduction enzyme and
control strains were inoculated in 5 mL selective media (U- or HU-) in 15 mL culture tubes

45
(Corning 352059) and placed in shaker overnight to OD600 2-3. Overnight cultures were used to
inoculate 100 mL selective media (U- or HU-) cultures in 500 mL conical flasks with a starting OD
of 0.08. Cells were grown to an OD of 1.3 before placing at 15 °C for an additional 10 h until a
final OD of 1.65. Cells were then pelleted in 2 50 mL tubes (Corning 352098) at 10 °C, 3,500 rpm
for 5 min. Each pellet was redissolved in 0.5 mL H2O and the suspension was distributed into a
presterilized 1.5 mL Eppendorf tube and pelleted at 11,000 rpm for 3 min at 10 °C. Pellets were
placed on ice and used within 1 h.
Pellets from 50 mL culture were redissolved in H2O (1,025 μL) and added as the last component
to 15 mL culture tubes (Corning 352059) containing 1,100 μL of 8 mg/mL anhydrotetracycline
HCl, 125 μL glucose solution in H2O (40%) and 250 μL 1 M Tris buffer pH 7.45 and were placed
in shaker at 350 rpm at 21 °C for 27 h. Cultures were then pelleted and the supernatant was diluted
10X into H2O before being used for UV/VIS spectral measurements.
2.4 Results
2.4.1 6β-hydroxylation of anhydrotetracycline in S. cerevisiae
The first step required to convert anhydrotetracyclines to tetracyclines is the 6-hydroxylation of
the anhydrotetracyclines. This step was studied in the oxytetracycline biosynthetic pathway and it
was shown that anhydrotetracycline 6-hydroxylation is catalyzed by OxyS. A similar step is
hypothesized to be catalyzed on 7-chloroanhydrotetracycline by the homologous enzyme Cts8 of
the chlortetracycline pathway.(7) As tetracycline is biosynthesized in 7-chlorotetracycline
producers with reduced chlorination ability,(8) anhydrotetracycline hydroxylation is likely to be
the first in the final sequence of steps in tetracycline biosynthesis.
To test the capacity of Saccharomyces cerevisiae to hydroxylate an anhydrotetracycline, the model
hydroxylating enzyme OxyS was used together with its native substrate, anhydrotetracycline

46
(Scheme 2-1). This enzyme-substrate pair was chosen as OxyS was shown to functionally express
in Escherichia coli by the Tang laboratory and anhydrotetracycline is commercially available.(7)
The expression plasmid pSP-G1 was chosen in order to facilitate the coexpression of a reductase
enzyme, to append antibody tags to both hydroxylase and reductase and to constitutively express
both enzymes.(9) OxyS was cloned into pSP-G1 under the transcriptional control of the strong
constitutive promoter TEF1 with a FLAG antibody tag at its C-terminus. The resulting plasmid
(AL-1-101) was transformed into FY251 and BJ5464-NpgA to generate strains EH-3-98-6 and
EH-3-248-1, respectively.
Scheme 2-1 Two-step enzymatic conversion of anhydrotetracycline to tetracycline
The OxyS-catalyzed reaction to convert anhydrotetracycline to 5a(11a)-dehydrotetracycline in S.
cerevisiae is supported by mass spectrometry. When a cell lysate expressing OxyS is added to
anhydrotetracycline the molecular ion corresponding to 5a(11a)-dehydrotetracycline ([M+H]+) has
much higher ion counts compared to the molecular ion corresponding to anhydrotetracycline
([M+H]+). However, the opposite is the case when a cell lysate not expressing OxyS is added to
anhydrotetracycline (Figure 2-1).

47
Figure 2-1 Mass spectrometry analysis of anhydrotetracycline hydroxylation in cell lysate
expressing OxyS
Cell lysate of OxyS expressing strain EH-3-248-1 (+OxyS) or a no-hydroxylase control EH-3-
248-8 (-OxyS) was placed overnight in Tris buffer (100 mM, pH 7.45) containing

48
anhydrotetracycline (5.4 mM), glucose (27.8 mM), NADPH (3 mM) and mercaptoethanol (18.5
mM). After night, MeOH was added, the contents were mixed and the reaction was filtered prior
to MS analysis.
Following this encouraging result with a cell lysate, the ability of OxyS to hydroxylate
anhydrotetracycline was tested in whole cells as well. As for the cell lysate, the molecular ion
corresponding to 5a(11a)-dehydrotetracycline had much higher ion counts in the +OxyS sample
relative to the -OxyS sample (data not shown). Given that anhydrotetracycline absorbs and
fluoresces in the visible range, it was logical to test whether the reaction could be monitored using
a simple UV/Vis assay using a spectrophotometer. Indeed, reducing the -OxyS spectrum from the
+OxyS spectrum shows a reduction in the 440 nm absorption and 570 nm emission peaks
corresponding to anhydrotetracycline and a formation of 380 nm absorption and 500 nm emission
peaks (Figure 2-2).
The blue shift in absorption and emission is supportive for the formation of 5a(11a)-
dehydrotetracycline from anhydrotetracycline as anhydrotetracycline’s conjugation in the CD-
rings is expected to be significantly reduced by 6-hydroxylation (Scheme 2-1). Indeed, the
corresponding red shift in the absorption spectrum for the opposite transformation, where
chlortetracycline is converted to anhydrochlortetracycline was previously reported (λmax = 373
nm to 438 nm, respectively).(10)

49
Figure 2-2 UV/Vis spectroscopy analysis of the reaction of anhydrotetracycline in whole cells
expressing OxyS
The spectra shown are Δ spectra, that is, the absorption/emission values for the OxyS expressing
cells EH-98-6 minus the absorption/emission values of the no hydroxylase control EH-3-80-3 are
shown.

50
2.4.2 5a(11a)-dehydrotetracycline reduction in S. cerevisiae
The second step in the synthesis of tetracycline from anhydrotetracycline is to reduce the 5a(11a)
α,β-unsaturated double bond (Scheme 2-1). The reduction at the 5a(11a) bond is known to be key
to tetracyclines’ antibiotic activity, with 7-chlorotetracycline being over 20X more potent than 7-
chloro-5a(11a)-dehydrotetracycline against Staphylococcus aureus.(11) In order to execute the
reduction step OxyR, the reductase of 5a(11a)-dehydrooxytetracycline,(7) was placed under the
control of a PGK1 promoter in the pSP-G1-OxyS plasmid AL-1-101 to generate AL-1-119-A-C7.
To function properly, OxyR requires cofactor F420, a cofactor that is not native to S. cerevisiae.
Cofactor Fo is an intermediate in cofactor F420 biosynthesis and is known to successfully replace
cofactor F420 in several F420 dependent enzymes with similar Km and Kcat values.(12-15) Given that
Fo synthases, employing known yeast metabolites to biosynthesize Fo were previously
characterized, an attempt was made to heterologousy produce Fo in S. cerevisiae. Fo synthase from
Thermobifida fusca and Chlamydomonas reinhardtii, was placed under the control of promoter
pADH1 to generate plasmids AL-1-183-C and AL-1-183-D, respectively.(16, 17) However,
following transformations of the plasmids to S. cerevisiae no Fo biosynthesis was confirmed by
MS (data not shown).
In order to test whether OxyR could be functional in S. cerevisiae with Fo as a cofactor, synthetic
Fo,(18) a generous gift from Frank Foss, was exogenously added to a yeast strain expressing OxyS,
OxyR and an Fo reductase. Fo reductase was placed under the control of pGPD (pTDH3) on PRS413 to
generate AL-215-D-C1, AL-255-C1 and AL-235-C5, encoding F420 reductase from Mycobacterium
tuberculosis, Archeoglobus fulgidus and Streptomyces griseus, respectively.(19-22) The reaction
mixture in Tris buffer (pH 7.45) also contained anhydrotetracycline, glucose, NADPH, and in the

51
case of Fo reductase from M. tuberculosis, glucose-6-phosphate for Fo reduction. It was found that
the levels of the molecular ion peak corresponding to tetracycline, the reduction product of
5a(11a)-dehydrotetracycline, are increased when glucose-6-phosphate is added (Figure 2-3).
Contrary to the expectation that glucose-6-phosphate serves as a substrate for the Fo reductase, it
was found that neither Fo, the Fo reductase or OxyR increased the levels of the molecular ion peak
corresponding to tetracycline either in the presence or absence of glucose-6-phosphate (data not
shown).
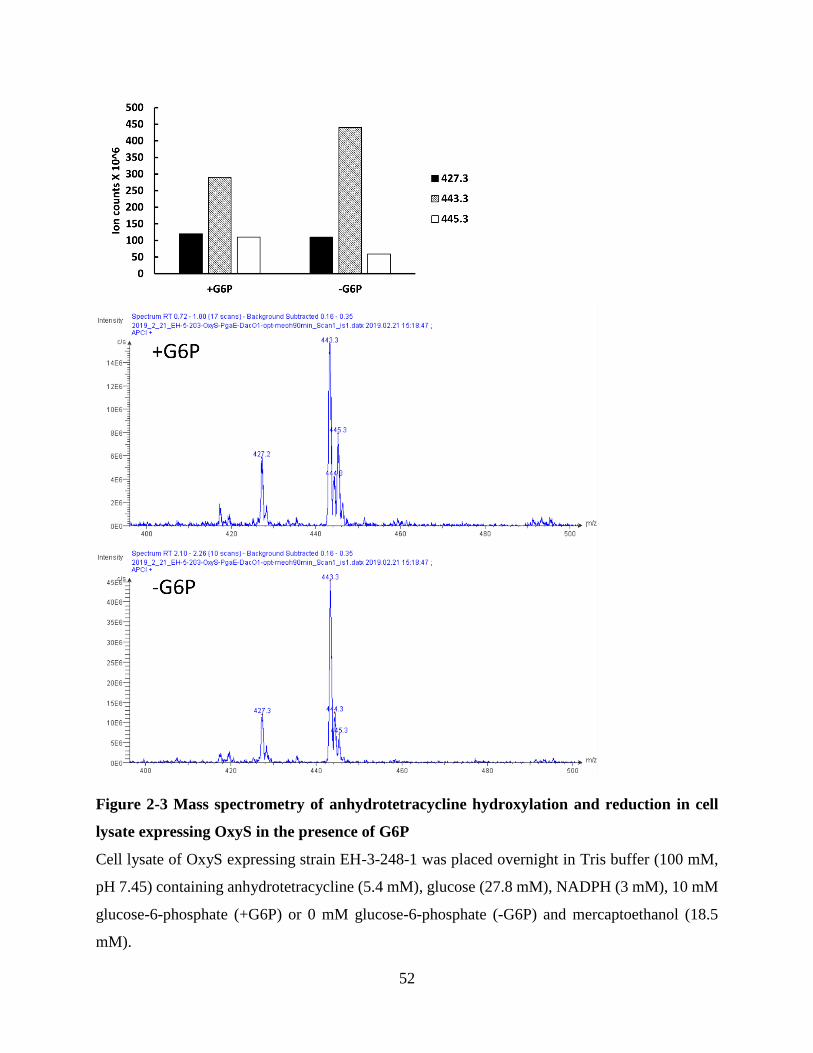
52
Figure 2-3 Mass spectrometry of anhydrotetracycline hydroxylation and reduction in cell
lysate expressing OxyS in the presence of G6P
Cell lysate of OxyS expressing strain EH-3-248-1 was placed overnight in Tris buffer (100 mM,
pH 7.45) containing anhydrotetracycline (5.4 mM), glucose (27.8 mM), NADPH (3 mM), 10 mM
glucose-6-phosphate (+G6P) or 0 mM glucose-6-phosphate (-G6P) and mercaptoethanol (18.5
mM).

53
To test whether a native reductase to S. cerevisiae is reducing 5a(11a)-dehydrotetracycline, the
native old yellow enzyme (OYE) reductases, OYE2 and OYE3 from S. cerevisiae were expressed
under the strong promoter pPGK1, along with their fungal homolog OYE1 from Saccharomyces
carlsbergensis.(23-26) OYE proteins are known to reduce the olefinic bond in α,β-unsaturated
carbonyls, such as the reduction of 2-cyclohexenone to cyclohexanone and are known to have a
flexible substrate specificity.(23) However, coexpression of the OYE proteins together with OxyS
did not enhance the reduction compared to the expression of OxyS alone (data not shown).
2.5 Discussion
2.5.1 6β-hydroxylation of anhydrotetracycline in S. cerevisiae
When OxyS and OxyR were used in vitro, oxytetracycline (5-hydroxytetracycline) and not
tetracycline was formed as the major product, indicating that OxyS is catalyzing both
hydroxylation steps at C5 and C6 prior to OxyR reduction of the 5a(11a) double bond (Scheme
2-1).(7) Interestingly however, when expressed in S. cerevisiae, both in cell lysate and whole cells,
OxyS is catalyzing just one hydroxylation step (Figure 2-1 and data not shown). It could be that
the absence of the 5-hydroxyl is the reason for the lack of contribution of OxyR to reduction as
OxyR’s native substrate is the doubly hydroxylated 5a(11a)-dehydrooxytetracycline and not the
singly hydroxylated 5a(11a)-dehydrotetracycline. Given that DacO4 and CtcR are known to
hydroxylate 5a(11a)-dehydrotetracycline,(7) an attempt of the hydroxylase-reductase pairs OxyS-
DacO4 and OxyS-CtcR is warranted in S. cerevisiae where OxyS performs only a single
hydroxylation step.
Furthermore, previously in the absence of OxyR the reaction product of OxyS rapidly degraded
both in vitro and in vivo in ΔoxyR Streptomyces lividans.(7) However, in the S. cerevisiae

54
expression system both in whole cells and in cell lysate the hydroxylation product of OxyS was
analyzable (Figure 2-1, Figure 2-2). This could be explained by the additional degradation
pathways possible due to the presence of the 5-hydroxy. For example, anhydrooxytetracycline (5-
hydroxyanhydrotetracycline) is known to undergo B-ring scission degradation that is not possible
in anhydrotetracycline.(27)
The ability to observe the hydroxylated product of anhydrotetracycline could also be because in
this study prior to mass spectrometry analysis MeOH added and the samples were filtered, while
in the in vitro study of OxyS acidic organic extraction using 1% AcOH in EtOAc was employed.(7)
Anhydrooxytetracycline is known to undergo B-ring scission specifically in the presence of dilute
acid.(27).
Importantly, it could be that some double hydroxylation product is formed in the reactions of the
+OxyS S. cerevisiae strain with anhydrotetracycline (Figures 2-1, 2-2 and 2-3) but degraded prior
to analysis. However, even if that is the case, it is still notable that a significant proportion of a
singly hydroxylated product is formed as well (Figure 2-1), in contrast to the previously reported
OxyS double hydroxylation in vitro and in S. lividans.(7)
The literature reported ability to isolate and analyze 7-chloro-5a(11a)-dehydrotetracycline could
provide further support for the potential role of the second hydroxylation step in destabilizing the
5a(11a)-dehydro product. 7-chloro-5a(11a)-dehydrotetracycline, lacking the 5α-hydroxy present
in 5a(11a)-dehydrooxytetracycline, is produced by mutant strain S-1308 of Streptomyces lividans
and can be isolated and analyzed.(11) The corresponding S. lividans wild type strain normally
produces 7-chlorotetracycline, which, like tetracycline is also lacking the additional 5-hydroxy
group. However, the 7-chloro group could also be responsible for the stability of 7-chloro-5a(11a)-
dehydrotetracycline, previously reported to be the only stable 5a(11a)-dehydrotetetracycline of the

55
ones that have been prepared.(28) Purification of the hydroxylation product of anhydrotetracycline
and NMR characterization will enable verifying whether 5a(11a)-dehydrotetracycline is indeed
produced.
2.5.2 5a(11a)-dehydrotetracycline reduction in S. cerevisiae
Recently, the biosynthetic pathway of cofactor F420 was fully characterized and F420 was
successfully heterologously produced in E. coli by recombinant expression of four genes.(29) The
substrates for the F420 pathway, lactate, tyrosine, and 5-amino-6-ribitylamino-2,4[1H,3H]-
pyrimidinedione, are native to yeast.(30, 31) Thus, a future attempt to natively produce F420 in
yeast to allow functional expression of cofactor F420-dependent enzymes such as OxyR is now
more feasible. Alternatively, cofactor F420 or its precursors might become more readily available
to be exogenously supplied to S. cerevisiae through heterologous production in another organism
such as E. coli. Importantly however, a reduction that takes place in S. cerevisiae in the absence
of OxyR and F420 (Figure 2-3) potentially circumvents the need to heterologously express F420 in
S. cerevisiae, a formidable challenge on its own right.
Another alternative route to tetracycline from 5a(11a)-dehydrotetracycline is chemical reduction.
However, the process is not necessarily stereoselective and can yield an additional stereoisomer to
tetracycline, 5a-epitetracycline, of significantly lesser antibiotic activity (e.g. against S. aureus)
than tetracycline.(11) Stereoselectivity was achieved however in the chemical reduction of the
5a(11a)-bond of 7-chloro-5a(11a)-dehydrotetracycline and of the 6-peroxidated derivative of
5a(11a)-dehydrotetracycline.(32-34) Larger scale culturing, isolation and NMR analysis of the
reduction product of OxyS hydroxylation (Figure 2-3) will be needed to confirm whether the
reduction takes place in the 5a(11a)-bond and if it does, whether it is stereoselective.

56
While the conversion percent for the hydroxylation of anhydrotetracycline by OxyS is high (Figure
2-1), the conversion percent of the reduction reaction is much lower (Figure 2-3), assuming equal
ionization capacity of anhydrotetracycline, its hydroxylated intermediate and its hydroxylated and
reduced product (Scheme 2-1). Thus, optimization of the reduction reaction is likely to be needed.
An obvious method for optimization of the reduction reaction would be to identify the native
reductase enzyme in S. cerevisiae and overexpress it. In order to identify the endogenous reductase
enzyme in S. cerevisiae a screen could be done using a plasmid collection of yeast genes and a
screening method such as described in the next chapter.(35) An alternative assay would be an
overlay of the yeast on bacteria given the much higher antibiotic activity of tetracycline over
5a(11a)-dehydrotetracycline.(11) Instead of a library of yeast genes, a library of yeast deletion
mutants could be screened, using similar assays, to identify yeast mutants that cannot reduce the
hydroxylation products of OxyS.(36, 37)
Importantly however, for these assays to function, the reduction of OxyS’s hydroxylation product
needs to happen in whole cells and not only in cell lysates and whether and in which conditions
this is the case is still unanswered. Thus, a cell lysate assay employing a more specific library of
yeast reductases or ene-reductases or even ene-reductases of α,β-unsat carbonyls is preferable.(38-
40) An alternative method will be to screen additional OYE proteins for the reduction step and
potentially perform directed evolution for improved substrate recognition and stereoselectivity in
similar methods to those described in the next chapters.(41-50) Finally, OxyR homologs with F420
or an intermediate in its biosynthesis are a potential alternative as well (Section 2.5.1).
2.6 Conclusion
The data presented in this chapter supports that the use of OxyS as the sole heterologously
expressed enzyme in S. cerevisiae allows both a single hydroxylation and reduction steps of

57
anhydrotetracycline to take place using S. cerevisiae in the presence of G6P (Figure 2-3). This
result is unexpected for two reasons. One, OxyS is known to perform two hydroxylation steps on
anhydrotetracycline in vitro and in vivo. Two, it was expected that the coexpression of the
dedicated reductase enzyme and the supply / heterologous biosynthesis of its nonnative cofactor
would be required for the reduction step. Work on isolation and NMR analysis of the hydroxylation
and hydroxylation + reduction products of anhydrotetracycline and 6-
demethylanhydrotetracycline to complement the results presented in this chapter is ongoing.
The work presented in this chapter paves the road for total biosynthesis of tetracyclines using S.
cerevisiae by expressing the additional bacterial biosynthetic enzymes from the oxytetracycline or
the chlortetracycline pathways.(51-54) In addition, the work presented in this chapter paves the
road for producing key tetracycline analogs from bacterial and fungal anhydrotetracyclines such
as anhydrotetracycline, anhydrodactylocyclinone and TAN-1612, the subject of the next chapters.

58
2.7 Appendix
Table 2-1 Strains used in this study
Genotype Source/Reference
FY251 MATa leu2-Δ1 trpΔ63 ura3-52 his3-Δ200 ATCC 96098
BJ5464-
NpgA
MATα ura3-52 his3-Δ200 leu2-Δ1
trp1 pep4::HIS3 δ:: pADH2-npgA-tADH2 prb1Δ1.6R can1
GAL
a / (55)
EH-3-80-3 FY251 pSP-G1 This Study
EH-3-98-6 FY-251 AL-1-101-C10 This Study
EH-3-142-A AL-1-141-A-C6 This Study
EH-3-142-B AL-1-151-A-C4 This Study
EH-3-142-C AL-1-151-B-C2 This Study
ΕΗ-5-153-8 FY-251 AL-1-119-A-C7 This Study
EH-3-204-5 FY-251 AL-1-119-A-C7, AL-215-D-C1 This Study
EH-3-204-6 FY-251 AL-1-119-A-C7, AL-255-C1 This Study
EH-3-204-7 FY-251 AL-1-119-A-C7, AL-235-C5 This Study
EH-3-204-8 FY-251 AL-1-119-A-C7, pRS413-pGAL1 This Study
EH-3-204-9 FY-251 AL-1-101-C10, pRS413-pGAL1 This Study
EH-3-248-1 BJ5464-NpgA AL-1-101-C10 This Study
EH-3-248-8 BJ5464-NpgA pSP-G1 This Study a Y. Tang, University of California, Los Angeles, Department of Chemical and Biomolecular
Engineering & Department of Chemistry and Biochemistry, Department of Bioengineering
Table 2-2 Plasmids used in this study
Description Source/Reference
pSP-G1 pTEF1-FLAG-tADH1, pPGK1-MYC-tCYC1, 2 μ, Ura Addgene 64736
pRS413-
pGAL1
Shuttle vector (empty), His marker, CEN ATCC 87326
AL-1-101-
C10
pTEF1-oxyS-tADH1 in pSP-G1 This Study
AL-1-119-A-
C7
pTEF1-oxyS-tADH1, pPGK1-oxyR-tCYC1 in pSP-G1 This Study
AL-1-141-A-
C6
pTEF1-oxyS-tADH1, pPGK1-OYE1-tCYC1 in pSP-G1 This Study
AL-1-151-A-
C4
pTEF1-oxyS-tADH1, pPGK1-OYE2-tCYC1 in pSP-G1 This Study
AL-1-151-B-
C2
pTEF1-oxyS-tADH1, pPGK1-OYE3-tCYC1 in pSP-G1 This Study
AL-1-183-C-
C8
Fo synthase Thermobifida fusca This Study

59
AL-1-183-D-
C7
Fo synthase Chlamydomonas reinhardtii This Study
AL-1-215-D-
C1
FGD1 from Mycobacterium tuberculosis This Study
AL-1-255-C1 F420-dependent NADP+ oxidoreductase from
Archeoglobus fulgidus
This Study
AL-1-235-C5 NADPH-dependent F420 reductase from Streptomyces
griseus
This Study
Table 2-3 Sequences used in this study
The sequence for the hydroxylase OxyS from Streptomyces rimosus (GenBank AAZ78342.1) is
shown capitalized within the context of the pSP-G1 backbone containing pTEF1, the FLAG tag
and tADH1 (partial, decapitalized).
The sequence for the reductase OxyR from Streptomyces rimosus (GenBank: DQ143963.2) is
shown capitalized within the context of the pSP-G1 backbone containing pPGK1, the myc tag and
tCYC1 (partial, decapitalized).
Sequences for the three F420 reductases from M. tuberculosis, A. fulgidus and S. griseus are shown
along with the pGPD (pTDH3) promoter capitalized within the context of the pRS413 backbone
containing the tCYC1 terminator (partial, decapitalized). The NCBI/Genbank reference sequences
used for the Fo reductases from M. tuberculosis, A. fulgidus and S. griseus are CP023708.1,
NC_000917.1 and NC_010572.1 (6172267..6172977). Prior to codon optimization, leucine codon
in M. tuberculosis F420 reductases in subsequence LTGAACAACACCCGGTTT was changed to
methionine so that the total sequence matches the protein sequence used for M. tuberculosis F420
crystallization.(19)
pTEF1-
oxyS-
tADH1-in-
pSP-G1-
AL-1-101-
C10 and
AL-1-119-
A-C7
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGAGGTACGATGTTGTTATAGCTGGTGCAGGAC
CCACCGGTTTGATGTTAGCATGTGAACTTCGGCTGGCGGGTGCCAGA
ACTTTGGTTTTAGAAAGATTAGCCGAGCCTGTTGACTTCTCGAAAGCT
CTAGGAGTCCACGCTCGCACTGTTGAACTATTAGATATGAGAGGCCT
CGGTGAAGGATTCCAGGCTGAAGCACCAAAGTTAAGAGGTGGTAATT
TTGCCTCATTAGGCGTCCCCCTGGATTTCTCATCATTTGATACTAGAC
ACCCATATGCATTGTTTGTTCCACAAGTACGAACTGAAGAACTGCTA
ACAGGTAGAGCTTTGGAGCTAGGGGCGGAGCTGCGTCGTGGTCATGC
CGTGACCGCCTTGGAACAAGATGCTGATGGTGTTACTGTGAGCGTGA
CAGGCCCTGAAGGCCCATACGAAGTAGAATGTGCTTATTTGGTGGGC
TGCGACGGTGGAGGCAGTACGGTGAGGAAACTATTGGGCATAGATTT
TCCAGGTCAAGACCCACATATGTTTGCTGTCATCGCAGACGCAAGAT
TCAGAGAAGAGCTTCCTCACGGAGAAGGTATGGGTCCTATGCGTCCT

60
TATGGTGTCATGAGACATGACCTTCGTGCATGGTTCGCAGCATTTCCG
CTAGAACCAGACGTCTACAGAGCAACAGTCGCCTTTTTCGATAGACC
GTATGCTGACAGGAGGGCGCCGGTAACGGAGGAGGATGTCAGAGCC
GCGTTAACAGAAGTTGCTGGATCTGACTTTGGAATGCATGATGTAAG
ATGGTTATCTCGTTTGACCGATACAAGTAGACAAGCAGAAAGGTATA
GAGATGGGAGAGTTCTTTTGGCTGGTGATGCATGCCATATTCATTTGC
CCGCTGGTGGCCAGGGACTGAACTTAGGATTTCAAGATGCCGTTAAC
TTGGGTTGGAAGCTTGGTGCTACCATTGCCGGGACCGCTCCACCAGA
GTTATTGGATACGTATGAAGCTGAGAGAAGGCCGATAGCCGCCGGTG
TTTTGAGAAATACAAGGGCTCAAGCTGTTCTAATTGATCCAGATCCTC
GCTACGAGGGTTTAAGGGAATTAATGATTGAATTGTTGCACGTTCCT
GAGACTAATAGATATTTAGCGGGGCTAATCTCCGCATTAGACGTTAG
ATACCCAATGGCTGGGGAACATCCATTGCTGGGAAGAAGAGTACCCG
ACTTACCTCTTGTCACCGAAGATGGAACAAGACAGTTGTCCACTTATT
TCCATGCTGCACGTGGCGTATTATTAACGTTAGGTTGTGATCAACCAC
TAGCAGATGAAGCCGCTGCTTGGAAAGATAGGGTTGATTTAGTTGCA
GCTGAAGGTGTGGCGGACCCTGGTTCTGCAGTAGATGGCTTAACTGC
TTTACTTGTAAGGCCTGATGGTTACATTTGTTGGACAGCTGCCCCAGA
AACTGGTACTGATGGATTGACAGACGCGCTGCGTACTTGGTTCGGCC
CACCTGCAATGgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcg
ccagaggtttggtcaagtctccaatcaaggttgtcggct
FGD1-in-
pRS413-
pGPD-
tCYC1-AL-
1-215-D-
C1
ccctcactaaagggaacaaaagctggagctcAGTTTATCATTATCAATACTGCCATTTC
AAAGAATACGTAAATAATTAATAGTAGTGATTTTCCTAACTTTATTTA
GTCAAAAAATTAGCCTTTTAATTCTGCTGTAACCCGTACATGCCCAAA
ATAGGGGGCGGGTTACACAGAATATATAACATCGTAGGTGTCTGGGT
GAACAGTTTATTCCTGGCATCCACTAAATATAATGGAGCCCGCTTTTT
AAGCTGGCATCCAGAAAAAAAAAGAATCCCAGCACCAAAATATTGT
TTTCTTCACCAACCATCAGTTCATAGGTCCATTCTCTTAGCGCAACTA
CAGAGAACAGGGGCACAAACAGGCAAAAAACGGGCACAACCTCAAT
GGAGTGATGCAACCTGCCTGGAGTAAATGATGACACAAGGCAATTG
ACCCACGCATGTATCTATCTCATTTTCTTACACCTTCTATTACCTTCTG
CTCTCTCTGATTTGGAAAAAGCTGAAAAAAAAGGTTGAAACCAGTTC
CCTGAAATTATTCCCCTACTTGACTAATAAGTATATAAAGACGGTAG
GTATTGATTGTAATTCTGTAAATCTATTTCTTAAACTTCTTAAATTCTA
CTTTTATAGTTAGTCTTTTTTTTAGTTTTAAAACACCAAGAACTTAGTT
TCGACGGATACTAGTAAAATGGCTGAGTTGAAACTTGGCTACAAGGC
ATCAGCTGAACAGTTTGCCCCAAGGGAGTTAGTCGAACTAGCAGTCG
CTGCCGAAGCTCACGGTATGGATTCCGCTACTGTTTCCGACCATTTCC
AACCATGGAGACACCAAGGTGGCCATGCACCATTCTCACTCAGTTGG
ATGACAGCTGTTGGAGAAAGAACTAATAGATTGTTATTGGGGACGTC
GGTACTCACCCCGACGTTTAGATACAACCCTGCGGTAATAGCACAGG
CCTTTGCTACAATGGGATGTCTATATCCAAACCGGGTGTTTTTAGGTG
TTGGTACTGGAGAGGCCTTGAATGAAATCGCCACTGGTTATGAAGGT
GCTTGGCCTGAGTTCAAAGAAAGGTTTGCGCGTCTGCGCGAAAGCGT
GGGGCTGATGAGACAATTATGGTCTGGTGATAGGGTAGATTTTGATG
GAGATTATTATAGATTAAAAGGCGCGTCTATATATGACGTTCCGGAT

61
GGTGGTGTCCCTGTATATATTGCCGCCGGAGGACCAGCAGTTGCTAA
ATATGCTGGCCGAGCTGGTGACGGTTTCATATGCACTTCAGGCAAGG
GTGAAGAACTTTACACAGAAAAGTTGATGCCCGCCGTCAGAGAAGG
GGCGGCAGCCGCTGACAGGTCTGTTGATGGCATAGACAAAATGATTG
AGATCAAGATTTCATACGACCCAGATCCCGAATTAGCAATGAATAAC
ACAAGATTTTGGGCACCTCTTAGTTTGACCGCAGAACAAAAGCATAG
CATCGATGATCCAATTGAAATGGAAAAAGCTGCTGATGCTTTACCCA
TCGAGCAAATTGCAAAAAGATGGATTGTTGCAAGTGATCCTGATGAA
GCAGTGGAGAAAGTAGGACAGTACGTGACCTGGGGTTTAAATCATCT
AGTTTTCCACGCTCCTGGGCATGATCAAAGAAGATTCTTGGAATTGTT
TCAATCTGACCTAGCGCCAAGACTTCGTCGTCTGGGTTAACTCGAGtca
tgtaattagttatgtcacgcttacattcacgccctccccccacatccgctctaaccgaaaaggaaggagttagacaac
ctgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtac
agacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaaggctttaatttgcg
gccggtacccaat
FNO-A-
fulgidus-in-
pRS413-
pGPD-
tCYC1-
AL-1-255-
C1
ccctcactaaagggaacaaaagctggagctcAGTTTATCATTATCAATACTGCCATTTC
AAAGAATACGTAAATAATTAATAGTAGTGATTTTCCTAACTTTATTTA
GTCAAAAAATTAGCCTTTTAATTCTGCTGTAACCCGTACATGCCCAAA
ATAGGGGGCGGGTTACACAGAATATATAACATCGTAGGTGTCTGGGT
GAACAGTTTATTCCTGGCATCCACTAAATATAATGGAGCCCGCTTTTT
AAGCTGGCATCCAGAAAAAAAAAGAATCCCAGCACCAAAATATTGT
TTTCTTCACCAACCATCAGTTCATAGGTCCATTCTCTTAGCGCAACTA
CAGAGAACAGGGGCACAAACAGGCAAAAAACGGGCACAACCTCAAT
GGAGTGATGCAACCTGCCTGGAGTAAATGATGACACAAGGCAATTG
ACCCACGCATGTATCTATCTCATTTTCTTACACCTTCTATTACCTTCTG
CTCTCTCTGATTTGGAAAAAGCTGAAAAAAAAGGTTGAAACCAGTTC
CCTGAAATTATTCCCCTACTTGACTAATAAGTATATAAAGACGGTAG
GTATTGATTGTAATTCTGTAAATCTATTTCTTAAACTTCTTAAATTCTA
CTTTTATAGTTAGTCTTTTTTTTAGTTTTAAAACACCAAGAACTTAGTT
TCGACGGATACTAGTAAAATGAGGGTAGCTTTACTTGGTGGTACAGG
AAATCTTGGAAAAGGCCTTGCCCTACGACTGGCCACGCTGGGCCATG
AAATCGTAGTCGGAAGCAGAAGAGAAGAAAAGGCAGAGGCTAAAGC
AGCAGAATATAGGCGCATTGCAGGTGACGCTTCCATCACTGGTATGA
AAAATGAAGATGCCGCTGAGGCATGCGACATTGCTGTCTTGACCATT
CCTTGGGAACATGCTATTGACACTGCCAGAGATTTGAAGAATATTTT
ACGTGAGAAAATTGTTGTATCACCATTAGTTCCAGTGTCAAGAGGTG
CAAAGGGCTTCACCTACTCGTCCGAAAGATCAGCGGCCGAGATTGTG
GCTGAAGTTCTGGAATCTGAAAAAGTTGTTTCTGCGTTGCACACAAT
ACCTGCTGCAAGATTTGCCAACTTGGATGAAAAATTTGATTGGGATG
TTCCTGTTTGTGGTGATGATGACGAAAGTAAGAAAGTCGTCATGTCTT
TAATAAGTGAAATAGATGGGTTGAGGCCGTTAGATGCTGGTCCCCTC
TCTAACTCCCGTTTAGTGGAGAGCCTAACTCCATTGATATTGAACATC
ATGAGATTCAATGGAATGGGTGAACTAGGGATCAAGTTTTTATAACT
CGAGtcatgtaattagttatgtcacgcttacattcacgccctccccccacatccgctctaaccgaaaaggaagga
gttagacaacctgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttctt

62
ttttttctgtacagacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagg
ctttaatttgcggccggtacccaat
FNO-S-
Griseus-in-
pRS413-
pGPD-
tCYC1-
AL-1-235-
C5
ccctcactaaagggaacaaaagctggagctcAGTTTATCATTATCAATACTGCCATTTC
AAAGAATACGTAAATAATTAATAGTAGTGATTTTCCTAACTTTATTTA
GTCAAAAAATTAGCCTTTTAATTCTGCTGTAACCCGTACATGCCCAAA
ATAGGGGGCGGGTTACACAGAATATATAACATCGTAGGTGTCTGGGT
GAACAGTTTATTCCTGGCATCCACTAAATATAATGGAGCCCGCTTTTT
AAGCTGGCATCCAGAAAAAAAAAGAATCCCAGCACCAAAATATTGT
TTTCTTCACCAACCATCAGTTCATAGGTCCATTCTCTTAGCGCAACTA
CAGAGAACAGGGGCACAAACAGGCAAAAAACGGGCACAACCTCAAT
GGAGTGATGCAACCTGCCTGGAGTAAATGATGACACAAGGCAATTG
ACCCACGCATGTATCTATCTCATTTTCTTACACCTTCTATTACCTTCTG
CTCTCTCTGATTTGGAAAAAGCTGAAAAAAAAGGTTGAAACCAGTTC
CCTGAAATTATTCCCCTACTTGACTAATAAGTATATAAAGACGGTAG
GTATTGATTGTAATTCTGTAAATCTATTTCTTAAACTTCTTAAATTCTA
CTTTTATAGTTAGTCTTTTTTTTAGTTTTAAAACACCAAGAACTTAGTT
TCGACGGATACTAGTAAAATGACTACTCAAGACAGTGGGTCTGCACC
GAAGCCTCCCGCCAAAGATCCATGGGATTTGCCAGATGTCAGCGCAC
TGTCTGTTGGTGTCCTAGGTGGTACAGGACCACAGGGCAGGGGATTA
GCCTACAGATTGGCGCGTGCTGGTCAAAAAGTGACCTTGGGCTCAAG
AGATGCTGGACGCGCTGCTGATGCAGCGGCAGAGCTGGGCCATGGTG
TGGAAGGTACTGATAATGCAGAATGCGCAAGAAGATCCGACATCGTT
ATTGTTGCTGTACCTTGGGACGGTCATGCTAAAACTTTGGAAAGTTTA
AGAGAAGAGTTGGCTGGCAAGCTGGTTATCGACTGTGTTAACCCCTT
GGGGTTTGATAAGAAAGGTGCTTATGCTTTAAAACCAGAGGAGGGCT
CGGCCGCTGAACAAGCTGCGGCGCTTCTCCCTGATTCACGAGTAACA
GCAGCTTTCCACCATTTATCTGCTGTGCTTTTACAAGATGAATCCATT
GAAGAAATTGATACCGATGTTTTGGTCTTAGGTGAAGCAAGGGCAGA
CACGGATATTGTCCAGGCACTAGCAGGGCGTATACCAGGTATGAGAG
GAATATTTGCCGGTCGTCTAAGAGGTGCCCACCAAGTTGAATCACTT
GTAGCCAATTTAATATCTGTAAACAGAAGGTATAAGGCTCATGCCGG
ACTAAGGGCCACAGACGTGTAACTCGAGtcatgtaattagttatgtcacgcttacattcac
gccctccccccacatccgctctaaccgaaaaggaaggagttagacaacctgaagtctaggtccctatttatttttttat
agttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtacagacgcgtgtacgcatgtaacattatac
tgaaaaccttgcttgagaaggttttgggacgctcgaaggctttaatttgcggccggtacccaat
pPGK1-
oxyR-
tCYC1 in
pSP-G1
AL-1-119-
A-C7 and
AL-1-26-1-
C1
tggaatggcgggaaagggtttagtaccacatgctatgatgcccactgtgatctccagagcaaagttcgttcgatcgt
actgttactctctctctttcaaacagaattgtccgaatcgtgtgacaacaacagcctgttctcacacactcttttcttctaa
ccaagggggtggtttagtttagtagaacctcgtgaaacttacatttacatatatataaacttgcataaattggtcaatgc
aagaaatacatatttggtcttttctaattcgtagtttttcaagttcttagatgctttctttttctcttttttacagatcatcaagga
agtaattatctactttttacaacaaatataaaacaaggatccATGCCATTCACTCAAAAGGAGAT
CACGTATTTAAGGGCTCAAGGCTACGGCCGACTAGCCACCGTCGGTG
CTCACGGTGAACCTCACAACGTGCCGGTATCTTTTGAAATTGATGAA
GAAAGAGGTACCATTGAAATAACAGGAAGGGATATGGGACGTTCCA
GAAAATTCAGAAATGTTGCCAAAAGTGACAGAGTGGCATTTGTTGTT
GATGATGTTCCATGCAGAGACCCAGAAGTTGTCAGAGCTGTTGTAAT
ACATGGTACTGCACAGGCGCTTCCCACAGGCGGAAGAGAAAGGCGT

63
CCTCATTGTGCTGACGAAATGATTAGAATTCATCCAAGAAGAATAGT
AACATGGGGTATCGAAGGTGATTTGTCAACTGGTGTTCATGCAAGAG
ATATTACTGCTGAAGATGGTGGTAGAAGGgtcgacatggaacagaagttgatttccga
agaagacctcgagtaagcttggtaccgcggctagctaagatccgctctaaccgaaaaggaaggagttagacaacc
tgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtaca
gacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagatccagctgcatt
aatgaatcggccaacgcgcggggagaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgactcgct
gcgctcgg

64
2.8 References
1. Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus
RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Production of
the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940-
943. doi: 10.1038/nature04640.
2. Galanie S, Thodey K, Trenchard IJ, Filsinger Interrante M, Smolke CD. Complete
biosynthesis of opioids in yeast. Science. 2015;349(6252):1095-1100. doi:
10.1126/science.aac9373.
3. Luo X, Reiter MA, d’Espaux L, Wong J, Denby CM, Lechner A, Zhang Y, Grzybowski
AT, Harth S, Lin W, Lee H, Yu C, Shin J, Deng K, Benites VT, Wang G, Baidoo EEK, Chen Y,
Dev I, Petzold CJ, Keasling JD. Complete biosynthesis of cannabinoids and their unnatural
analogues in yeast. Nature. 2019;567(7746):123-126. doi: 10.1038/s41586-019-0978-9.
4. Awan AR, Blount BA, Bell DJ, Shaw WM, Ho JCH, McKiernan RM, Ellis T. Biosynthesis
of the antibiotic nonribosomal peptide penicillin in baker’s yeast. Nature Communications. 2017;8.
doi: 10.1038/ncomms15202.
5. Wang P, Kim W, Pickens LB, Gao X, Tang Y. Heterologous Expression and Manipulation
of Three Tetracycline Biosynthetic Pathways. Angewandte Chemie-International Edition.
2012;51(44):11136-11140. doi: 10.1002/anie.201205426.
6. Zhou K, Qiao K, Edgar S, Stephanopoulos G. Distributing a metabolic pathway among a
microbial consortium enhances production of natural products. Nat Biotechnol. 2015;33(4):377-
383. doi: 10.1038/nbt.3095.
7. Wang P, Bashiri G, Gao X, Sawaya MR, Tang Y. Uncovering the Enzymes that Catalyze
the Final Steps in Oxytetracycline Biosynthesis. J Am Chem Soc. 2013;135(19):7138-7141. doi:
10.1021/ja403516u.
8. Hošťálek Z, Vaněk Z. Biosynthesis of the Tetracyclines. In: Hlavka JJ, Boothe JH, editors.
The Tetracyclines. Berlin, Heidelberg: Springer Berlin Heidelberg; 1985. p. 137-178.
9. Partow S, Siewers V, Bjorn S, Nielsen J, Maury J. Characterization of different promoters
for designing a new expression vector in Saccharomyces cerevisiae. Yeast. 2010;27(11):955-964.
doi: 10.1002/yea.1806.

65
10. Tymiak AA, Aklonis C, Bolgar MS, Kahle AD, Kirsch DR, O'Sullivan J, Porubcan MA,
Principe P, Trejo WH. Dactylocyclines: novel tetracycline glycosides active against tetracycline-
resistant bacteria. J Org Chem. 1993;58(3):535-537. doi: 10.1021/jo00055a002.
11. McCormick JRD, Miller PA, Growich JA, Sjolander NO, Doerschuk AP. Two new
tetracycline-related compounds: 7-chloro-5a(11a)-dehydrotetracycline and 5a-epi-tetracycline. a
new route to tetracycline. J Am Chem Soc. 1958;80(20):5572-5573. doi: 10.1021/ja01553a066.
12. Greening C, Ahmed FH, Mohamed AE, Lee BM, Pandey G, Warden AC, Scott C,
Oakeshott JG, Taylor MC, Jackson CJ. Physiology, Biochemistry, and Applications of F420- and
Fo-Dependent Redox Reactions. Microbiol Mol Biol Rev. 2016;80(2):451-493. doi:
10.1128/mmbr.00070-15.
13. Jacobson FS, Daniels L, Fox JA, Walsh CT, Orme-Johnson WH. Purification and
properties of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from Methanobacterium
thermoautotrophicum. J Biol Chem. 1982;257(7):3385-3388.
14. Yamazaki S, Tsai L, Stadtman TC. Analogs of 8-hydroxy-5-deazaflavin cofactor: relative
activity as substrates for 8-hydroxy-5-deazaflavin-dependent NADP+ reductase from
Methanococcus vannielii. Biochemistry. 1982;21(5):934-939. doi: 10.1021/bi00534a019.
15. Eirich LD, Vogels GD, Wolfe RS. Distribution of coenzyme F420 and properties of its
hydrolytic fragments. J Bacteriol. 1979;140(1):20-27.
16. Decamps L, Philmus B, Benjdia A, White R, Begley TP, Berteau O. Biosynthesis of F0,
Precursor of the F420 Cofactor, Requires a Unique Two Radical-SAM Domain Enzyme and
Tyrosine as Substrate. J Am Chem Soc. 2012;134(44):18173-18176. doi: 10.1021/ja307762b.
17. Petersen JL, Ronan PJ. Critical role of 7,8-didemethyl-8-hydroxy-5-deazariboflavin for
photoreactivation in Chlamydomonas reinhardtii. J Biol Chem. 2010;285(42):32467-32475. doi:
10.1074/jbc.M110.146050.
18. Hossain MS, Le CQ, Joseph E, Nguyen TQ, Johnson-Winters K, Foss FW. Convenient
synthesis of deazaflavin cofactor FO and its activity in F420-dependent NADP reductase. Org
Biomol Chem. 2015;13(18):5082-5085. doi: 10.1039/c5ob00365b.
19. Bashiri G, Squire CJ, Moreland NJ, Baker EN. Crystal Structures of F420-dependent
Glucose-6-phosphate Dehydrogenase FGD1 Involved in the Activation of the Anti-tuberculosis
Drug Candidate PA-824 Reveal the Basis of Coenzyme and Substrate Binding. J Biol Chem.
2008;283(25):17531-17541. doi: 10.1074/jbc.M801854200.

66
20. Le CQ, Joseph E, Nguyen T, Johnson-Winters K. Optimization of Expression and
Purification of Recombinant Archeoglobus fulgidus F420H2:NADP+ Oxidoreductase, an F420
Cofactor Dependent Enzyme. The Protein Journal. 2015;34(6):391-397. doi: 10.1007/s10930-015-
9633-y.
21. Warkentin E. Structures of F420H2:NADP+ oxidoreductase with and without its substrates
bound. The EMBO Journal. 2001;20(23):6561-6569. doi: 10.1093/emboj/20.23.6561.
22. Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M,
Horinouchi S. Genome Sequence of the Streptomycin-Producing Microorganism Streptomyces
griseus IFO 13350. J Bacteriol. 2008;190(11):4050-4060. doi: 10.1128/jb.00204-08.
23. Karplus PA, Fox KM, Massey V. Flavoprotein structure and mechanism. 8. Structure-
function relations for old yellow enzyme. The FASEB Journal. 1995;9(15):1518-1526. doi:
10.1096/fasebj.9.15.8529830.
24. Saito K, Thiele DJ, Davio M, Lockridge O, Massey V. The cloning and expression of a
gene encoding Old Yellow Enzyme from Saccharomyces carlsbergensis. J Biol Chem.
1991;266(31):20720-20724.
25. Swiderska MA, Stewart JD. Stereoselective enone reductions by Saccharomyces
carlsbergensis old yellow enzyme. J Mol Catal B: Enzym. 2006;42(1-2):52-54. doi:
10.1016/j.molcatb.2006.06.023.
26. Hall M, Stueckler C, Hauer B, Stuermer R, Friedrich T, Breuer M, Kroutil W, Faber K.
Asymmetric Bioreduction of Activated C=C Bonds Using Zymomonas mobilis NCR Enoate
Reductase and Old Yellow Enzymes OYE 1–3 from Yeasts. Eur J Org Chem. 2008;2008(9):1511-
1516. doi: 10.1002/ejoc.200701208.
27. Mitscher LA. Degradation and Structure Proofs. The Chemistry of the Tetracycline
Antiobitcs. New York: Marcel Dekker, Inc.; 1978. p. 122-164.
28. Miller PA, Hash JH. [47] NADP:Tetracycline 5a(11a)dehydrogenase1975;43:606-607.
doi: 10.1016/0076-6879(75)43123-8.
29. Bashiri G, Antoney J, Jirgis ENM, Shah MV, Ney B, Copp J, Stuteley SM, Sreebhavan S,
Palmer B, Middleditch M, Tokuriki N, Greening C, Scott C, Baker EN, Jackson CJ. A revised
biosynthetic pathway for the cofactor F420 in prokaryotes. Nature Communications. 2019;10(1).
doi: 10.1038/s41467-019-09534-x.

67
30. Fischer M, Bacher A. Biosynthesis of flavocoenzymes. Nat Prod Rep. 2005;22(3). doi:
10.1039/b210142b.
31. Abbas CA, Sibirny AA. Genetic Control of Biosynthesis and Transport of Riboflavin and
Flavin Nucleotides and Construction of Robust Biotechnological Producers. Microbiol Mol Biol
Rev. 2011;75(2):321-360. doi: 10.1128/mmbr.00030-10.
32. Tatsuta K, Yoshimoto T, Gunji H, Okado Y, Takahashi M. The First Total Synthesis of
Natural (−)-Tetracycline. Chem Lett. 2000;29(6):646-647. doi: 10.1246/cl.2000.646.
33. Scott AI, Bedford CT. Simulation of the Biosynthesis of Tetracyclines. A Partial Synthesis
of Tetracycline from Anhydroaureomycin. J Am Chem Soc. 1962;84(11):2271-2272. doi:
10.1021/ja00870a061.
34. Charest MG, Siegel DR, Myers AG. Synthesis of (−)-Tetracycline. J Am Chem Soc.
2005;127(23):8292-8293. doi: 10.1021/ja052151d.
35. Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiae
genomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60(2-3):237-
243. doi: 10.1016/0378-1119(87)90232-0.
36. Giaever G, Nislow C. The Yeast Deletion Collection: A Decade of Functional Genomics.
Genetics. 2014;197(2):451-465. doi: 10.1534/genetics.114.161620.
37. Scherens B, Goffeau A. The uses of genome-wide yeast mutant collections. Genome Biol.
2004;5(7). doi: 10.1186/gb-2004-5-7-229.
38. Knaus T, Toogood HS, Scrutton NS. Ene-reductases and their Applications. Green
Biocatalysis2016. p. 473-488.
39. Winkler CK, Tasnádi G, Clay D, Hall M, Faber K. Asymmetric bioreduction of activated
alkenes to industrially relevant optically active compounds. J Biotechnol. 2012;162(4):381-389.
doi: 10.1016/j.jbiotec.2012.03.023.
40. Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR,
Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ,
Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M, Weng S, Wong ED. Saccharomyces
Genome Database: the genomics resource of budding yeast. Nucleic Acids Res.
2011;40(D1):D700-D705. doi: 10.1093/nar/gkr1029.

68
41. Hollmann F, Arends IWCE, Holtmann D. Enzymatic reductions for the chemist. Green
Chem. 2011;13(9). doi: 10.1039/c1gc15424a.
42. Hollmann F, Arends IWCE, Buehler K. Biocatalytic Redox Reactions for Organic
Synthesis: Nonconventional Regeneration Methods. ChemCatChem. 2010;2(7):762-782. doi:
10.1002/cctc.201000069.
43. Fryszkowska A, Toogood H, Sakuma M, Gardiner JM, Stephens GM, Scrutton NS.
Asymmetric Reduction of Activated Alkenes by Pentaerythritol Tetranitrate Reductase:
Specificity and Control of Stereochemical Outcome by Reaction Optimisation. Adv Synth Catal.
2009;351(17):2976-2990. doi: 10.1002/adsc.200900574.
44. Cheng JZ, Coyle CM, Panaccione DG, O’Connor SE. A Role for Old Yellow Enzyme in
Ergot Alkaloid Biosynthesis. J Am Chem Soc. 2010;132(6):1776-1777. doi: 10.1021/ja910193p.
45. Hall M, Stueckler C, Kroutil W, Macheroux P, Faber K. Asymmetric Bioreduction of
Activated Alkenes Using Cloned 12-Oxophytodienoate Reductase Isoenzymes OPR-1 and OPR-
3 fromLycopersicon esculentum (Tomato): A Striking Change of Stereoselectivity. Angew Chem
Int Ed. 2007;46(21):3934-3937. doi: 10.1002/anie.200605168.
46. Hall M, Stueckler C, Ehammer H, Pointner E, Oberdorfer G, Gruber K, Hauer B, Stuermer
R, Kroutil W, Macheroux P, Faber K. Asymmetric Bioreduction of C C Bonds using Enoate
Reductases OPR1, OPR3 and YqjM: Enzyme-Based Stereocontrol. Adv Synth Catal.
2008;350(3):411-418. doi: 10.1002/adsc.200700458.
47. Messiha HL, Munro AW, Bruce NC, Barsukov I, Scrutton NS. Reaction of Morphinone
Reductase with 2-Cyclohexen-1-one and 1-Nitrocyclohexene. J Biol Chem. 2005;280(11):10695-
10709. doi: 10.1074/jbc.M410595200.
48. Shimoda K, Kubota N, Hirata T, Kondo Y, Hamada H. Stereoselective reduction of 2-
butenolides to chiral butanolides by reductases from cultured cells of Glycine max. Tetrahedron
Lett. 2007;48(8):1345-1347. doi: 10.1016/j.tetlet.2006.12.126.
49. Shimoda K, Kubota N. Asymmetric reduction of 2-substituted 2-butenolides with
reductase from Marchantia polymorpha. Tetrahedron: Asymmetry. 2004;15(24):3827-3829. doi:
10.1016/j.tetasy.2004.11.006.
50. Wallwey C, Matuschek M, Xie X-L, Li S-M. Ergot alkaloid biosynthesis in Aspergillus
fumigatus: Conversion of chanoclavine-I aldehyde to festuclavine by the festuclavine synthase

69
FgaFS in the presence of the old yellow enzyme FgaOx3. Org Biomol Chem. 2010;8(15). doi:
10.1039/c003823g.
51. Pickens LB, Tang Y. Oxytetracycline biosynthesis. J Biol Chem. 2010;285(36):27509-
27515. doi: 10.1074/jbc.R110.130419.
52. Zhang W, Watanabe K, Cai X, Jung ME, Tang Y, Zhan J. Identifying the minimal enzymes
required for anhydrotetracycline biosynthesis. J Am Chem Soc. 2008;130(19):6068-6069. doi:
10.1021/ja800951e.
53. Zhu T, Cheng X, Liu Y, Deng Z, You D. Deciphering and engineering of the final step
halogenase for improved chlortetracycline biosynthesis in industrial Streptomyces aureofaciens.
Metab Eng. 2013;19:69-78. doi: 10.1016/j.ymben.2013.06.003.
54. Dairi T, Nakano T, Aisaka K, Katsumata R, Hasegawa M. Cloning and Nucleotide
Sequence of the Gene Responsible for Chlorination of Tetracycline. Biosci, Biotechnol, Biochem.
2014;59(6):1099-1106. doi: 10.1271/bbb.59.1099.
55. Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva
NA, Vederas JC, Tang Y. Complete Reconstitution of a Highly Reducing Iterative Polyketide
Synthase. Science. 2009;326(5952):589-592. doi: 10.1126/science.1175602.

70
3 Towards the biosynthesis of 6-demethyl-6-
epitetracyclines using S. cerevisiae
*The contents of this chapter will be published in:
E. Herbst, V. W. Cornish, et al “Biosynthesis of 6-demethyl-6-epitetracyclines using
Saccharomyces cerevisiae”, in preparation.

71
3.1 Chapter outline
This chapter describes the work towards 6-demethyl-6-epitetracyclines biosynthesis using S.
cerevisiae. 6α-hydroxylation by an enzyme such as DacO1 is required for the synthesis of 6-
demethyl-6-epitetracyclines as opposed to 6β-hydroxylation by enzymes such as OxyS leading to
6-demethyltetracycline (Scheme 3-1 and Scheme 3-2). However, previous attempts in multiple
heterologous hosts to heterologously express DacO1 without the rest of the dactylocyclinone
pathway have not yielded stably expressed protein. Thus, the chapter first describes rational design
approaches for optimizing DacO1 expression in S. cerevisiae (Section 3.4.1). The rationale behind
the choices for bacterial hydroxylases screened for 6α-hydroxylase activity on anhydrotetracycline
is explained (Section 3.4.2). Screening for 6-hydroxylase activity towards the biosynthesis of 6-
demethyl-6-epitetracyclines required the development of a multititer plate assay for
anhydrotetracycline biosynthesis (Section 3.4.3). This assay was put to the use in screening for
error-prone mutants of DacO1 in an attempt to identify 6α-hydroxylases of anhydrotetracycline
(Section 3.4.4). Finally, the results of testing the rational design DacO1 expression optimization
library as well as the other bacterial candidates for 6α-hydroxylation in S. cerevisiae are described
(Section 3.4.5). Key results in this chapter are the the development of a stably expressing form of
DacO1 in S. cerevisiae (Figure 3-8), the identification of PgaE as an anhydrotetracycline
hydroxylase (Figure 3-9 and Figure 3-10), and the discovery that a larger proportion of PgaE
hydroxylated intermediates undergo reduction in the presence of glucose-6-phosphate than is the
case in OxyS (Figure 3-11). Finally, the next required steps towards 6-demethyl-6-epitetracyclines
biosynthesis in S. cerevsiae are discussed (Section 3.5).

72
3.2 Introduction
As mentioned in Chapter 1, the synthesis of 6-demethyl-6-epiglycotetracyclines for testing is
highly desirable due to their highly promising outlook as antibiotics. The first two steps in the
synthesis of 6-demethyl-6-epiglycotetracyclines are the synthesis of 6-demethyl-6-epitetracyclines
from anhydrotetracyclines. This chapter shows the progress towards developing an S. cerevisiae
platform for 6-demethyl-6-epitetracyclines biosynthesis using bacterial hydroxylase enzymes as a
starting point for directed evolution and rational design as well as anhydrotetracycline as a model
substrate. DacO1, a bacterial flavin-dependent monooxygenase homologous to OxyS was
hypothesized to perform a 6α-hydroxylation on its native substrate that leads to 6-
epiglycotetracyclines in its native host, Dactylosporangium sp. SC 14051 (ATCC 53693).(1, 2)
Thus, DacO1, along with other bacterial hydroxylases, is used here as a template to evolve a 6α-
hydroxylase in S. cerevisiae towards the biosynthesis of 6-demethyl-6-epiglycotetracyclines.
3.3 Materials and methods
See Chapter 5 for general methods. See Chapter 2 for general protocol for hydroxylation/reduction
assay in cell lysate.
3.3.1 General protocol for high throughput assay of hydroxylation/reduction
in whole cells in microtiter plates
Fresh colonies of strains harboring a plasmid encoding a hydroxylase and/or reductase and control
strains (EH-3-98-6 encoding OxyS as positive control and EH-3-80-3 with no hydroxylase as
negative control) were inoculated in 200 μL selective media (U- or HU-) in 96-well plates (Corning
353072) and placed in shaker overnight. Overnight cultures were used to inoculate 1 mL selective
media (U- or HU-) cultures in deep well plates (Corning P-2ML-SQ-C-S) with an average starting

73
OD of 0.01. The plate was covered with two layers of SealMate film (Excel Scientific, SM-KIT-
BS) and placed in shaker overnight. Cells were then pelleted at 4 °C at 4,000 rpm. Each pellet was
redissolved in 0.3 mL containing a 150 μL solution of 5 mg / mL anhydrotetracycline HCl, 30 μL
1 M Tris buffer pH 7.45, 15 μL 40% glucose solution in H2O and 105 μL H2O for final
concentrations of 2.5 mg/mL anhydrotetracycline HCl, 100 mM Tris pH 7.45 and 2% glucose. The
plates were then covered with two layers of SealMate film (Excel Scientific, SM-KIT-BS) and
placed in shaker overnight at 800 rpm. 2 μL of Overnight suspensions were diluted into 198 μL of
H2O in Corning 3603 plates before UV/VIS spectroscopic measurements. UV/VIS spectroscopic
measurements taken were as follows: (i) absorption spectrum 350 nm – 500 nm, (ii) emission
spectrum 450 nm – 550 nm (λexcitation = 400 nm), (iii) excitation spectrum 305 nm – 455 nm (λemission
= 500 nm). Spectral steps in the spectra were 25 nm.
3.3.2 General protocol for western blots
Fresh patches or colonies of strains harboring the plasmid for the hydroxylation and/or reduction
enzyme and control strains were inoculated in 3 mL selective media (U-) in 15 mL culture tubes
(Corning 352059) and placed in shaker overnight. Overnight cultures were used to reinoculate 3
mL selective media (U-) in 15 mL culture tubes (Corning 352059) and placed in shaker overnight
with a starting OD of 0.01-0.05 (all western Blots shown in this study except for western blots
shown in Figure 3-1 which were pelleted and lysed immediately). Cells were grown to final OD
of 0.6-0.8 before pelleting in 2 separate microcentrifuge tubes (1 mL culture each) at 4 °C at 14,000
rpm, removing the supernatant and freezing at -20 °C prior to further use.
100 μL of a 99:1 mixture of Y-PER yeast protein extraction reagent (ThermoFisher Scientific
78991) and HALT protease inhibitor cocktail (ThermoFisher Scientific PI87786) were added to
each microcentrifuge tube. The tubes were placed on orbital shaker for 20 min at r.t., followed by

74
10 min centrifugation at 14,000 rpm at 4 °C and the cell lysate was transferred to a new 1.5 mL
microcentrifuge tube and kept on ice. Following 5 min at 95 °C with SDS loading buffer,(3) an
SDS-page was run with a Kaleidoscope Prestained Standard (Bio-Rad 161-0324, Figure 3-1) or a
Precision Plus Protein Kaleidoscope (Bio-Rad 161-0375, all other western blots shown),(4) the
gels were transferred to PVDF blots (Thermo Fisher Scientific IB24001) and western blots were
performed according to manufacturer guidelines (Thermo Fisher R951-25 and Sigma-Aldrich
A8592 for myc-and FLAG-tag, respectively) with BSA used instead of milk and analyzed with
Thremo Scientific 34000 substrate kit for horseradish peroxidase.
3.4 Results
3.4.1 Optimizing DacO1 expression in S. cerevisiae – rational design
Following the successful hydroxylation of anhydrotetracycline in S. cerevisiae by OxyS described
in Chapter 2, the 6α-hydroxylation of anhydrotetracycline in S. cerevisiae by DacO1 was
attempted. DacO1 is 66% homologous to OxyS and is assumed to perform the 6α-hydroxylation
on its native substrate, anhydrodactylocyclinone, as opposed to the 6β-hydroxylation that OxyS
performs on anhydrotetracycline (Scheme 3-1).(1)
DacO1 heterologous expression, along with the rest of the Dactylocyclinone pathway was
previously attempted in Streptomyces lividans K4‐114 and has led to the successful biosynthesis
of dactylocyclinone. Assuming correct assignment for the role of DacO1 in the dactylocycline
pathway, given its homology to OxyS, this result supports that DacO1 was successfully
heterologously expressed in S. lividans.(2) However, previous attempts to obtain soluble forms of
heterologously expressed DacO1 from Streptomyces and E. coli, when heterologously expressed
without the rest of its pathway, were not successful.(1)

75
My initial attempts to hydroxylate anhydrotetracycline with DacO1 yielded either little or no
hydroxylation when assayed by mass spectrometry (data not shown) but little hydroxylation was
supported by UV/Vis spectroscopy (Figure 3-4). Therefore, the expression levels of DacO1 were
examined by western blotting with FLAG-tag to determine whether a problem in expression leads
to the low/no hydroxylation levels with DacO1. Another likely explanation for low/no
hydroxylation is difference between anhydrotetracycline, the model substrate I used, and
anhydrodactylocyclinone, the native substrate of DacO1 (Scheme 3-1). Indeed, DacO1 expression
levels were found to be significantly lower than its counterpart OxyS (Figure 3-1-a).
Figure 3-1 Western Blot of DacO1 and OxyS
Cultures of DacO1, DacO4 and OxyS, OxyR encoding strains EH-3-153-7 and EH-3-153-8,
respectively were lysed with Y-PER and labeled with Monoclonal ANTI-FLAG HRP antibody.
Two biological replicates were used for each strain indicated as colony 1 and 2 (C1 and C2),
respectively. The expected sizes are 55.1, 55.9, 16.3 and 17.5 for DacO1, OxyS, DacO4 and OxyR,
respectively.
I therefore attempted to optimize the functional expression of DacO1 by a number of approaches.
Namely, fusion to a stable protein, switching to a transcriptional control of an inducible promoter,
expression in a strain background that has protease deletions, lower temperature culturing and

76
coexpression of pathway proteins. The testing of these expression optimization approaches is
described in Section 3.4.5.
Specifically, I attempted N-terminal fusions proteins previously shown to improve expression of
heterologous proteins in yeast, ubiquitin,(5-8) superoxide dismutase,(9-11) Gal1(8) and γ-
IFN.(12) I also attempted a C- and N-terminal fusion to DacO4, the 5a(11a) reductase enzyme of
the dactylocycline pathway that I verified to be stably expressed in yeast (Figure 3-1-b).
Given the homology between OxyS and DacO1, the successful heterologous expression of OxyS
and the potential importance of the N-terminus of the protein to its in vivo stability,(13) I attempted
three fusion proteins composed of an N-terminal portion of OxyS and a C-terminal portion of
DacO1. These fusion proteins include the first 37, 87 and 191 amino acids of OxyS, followed by
the last 461, 411 and 307 amino acids of DacO1, respectively. In order to determine where to fuse
the proteins I considered the structure of OxyS as determined previously by X-ray
crystallography,(1) and made fusions proteins in disordered regions of the structure around
positions 37, 87 and 191. The homology between DacO1 and OxyS made it simple to determine
what are the corresponding amino acids at the DacO1 protein, although there is no guarantee the
disordered regions in OxyS are also disordered in DacO1.
For inducible promoters I tested pGAL1, induced by galactose, and pADH2, a late stage promoter
induced in low glucose / high EtOH.(14) The alternative strain background I chose is BJ5464-
NpgA, as the BJ-5464 strain background was previously shown useful for heterologous protein
production in yeast.(15, 16) For a lower temperature culturing I attempted 25 °C.(17, 18)
The activity of polyketide biosynthetic enzymes is known to be dependent on an oligomeric
complexation of biosynthetic enzymes. This is claimed for instance in the case of JadF, JadG and
JadH of the Jadomycin biosynthetic pathway.(19) Similarly, in the case of an oxytetracycline

77
producing strain, deletion of the gene encoding for OtcC, which is 99% identical to OxyS used in
this study, led to production of a polyketide with a shorter chain length. Expression of a triply
mutated, catalytically disfunctional OtcC, restored production of a polyketide with the same chain
length. Thus, a post-polyketdie synthase (PKS) polyketide biosynthetic enzyme could have a
structural role in the oligomeric protein complex responsible for polyketide biosynthesis.(20)
Potential further support for the potential that coexpression of additional proteins from the
dactylocycline pathway has for improving DacO1 expression could be found in the case of DacO1
itself. As mentioned above, expression of DacO1 together with the rest of the dactylocyclinone
pathway in S. lividans led to the production of dactylocyclinone.(2) However, expression of
DacO1 in the absence of the rest of the dactylocycline pathway in E. coli, Streptomyces,(1) or S.
cerevisiae (Figure 3-1) yielded no/low levels of soluble protein. In order to provide DacO1 with
enzymes potentially required to form a functional oligomeric complex I attempted the
coexpression of DacJ and DacM2. DacJ and DacM2 were chosen since they are the closest
enzymes to DacO1 in the dactylocycline gene cluster and they are proposed to share with DacO1
the functional role of aglycone tailoring.(2)
3.4.2 Searching the bacterial hydroxylase space for 6α-hydroxylation of
anhydrotetracyclines
Given the goal to perform 6α-hydroxylation on anhydrotetracyclines it was clear that there is a
need to go beyond the 6β-hydroxylase OxyS. Expected challenges with the expression of Daco1
(Section 3.4.1) as well as incongruencies between the DacO1 native substrate
anhydrodactylocyclinone and anhydrotetracycline affirmed the need to test other hydroxylases as
well (Scheme 3-1). I therefore looked for homologous enzymes catalyzing hydroxylation in
polyketides that are structurally related to anhydrotetracycline.

78
Scheme 3-1 Bacterial monooxygenases and their hypothesized substrates and products
Bacterial flavin-dependent monooxygenases OxyS, DacO1, SsfO1, CtcN and PgaE and their
native substrates. Marked in red are differences between the native substrate and
anhydrotetracycline.
SsfO1, the hypothesized flavin-dependent monooxygenases of the 6α-position in SF2575 pathway,
with 54% homology to OxyS, was an obvious candidate (Scheme 3-1).(2) Flavin-dependent
monooxygenases of angucyclines, seemed like another promising source for relevant
monooxygenases, given the structural similarity between angucyclines and tetracyclines.
Specifically, enzymes that catalyze monooxygenation of position 12 of the angucyclines,

79
analogous to position 6 of the tetracyclines seemed of particular interest.(21) I chose to test PgaE
(56% homology to OxyS), catalyzing the hydroxylation of UWM6 (Scheme 3-1), in the
biosynthesis of gaudimycin C since it was previously both analyzed by X-ray crystallography and
used in chemoenzymatic synthesis.(22, 23) Finally, CtcN, with 69% homology to OxyS was
chosen since it was shown to be necessary for the 6-hydroxylation step in the chlortetracycline
pathway.(24) The testing of these bacterial hydroxylases is described in Section 3.4.5.
3.4.3 Developing a microtiter plate assay for anhydrotetracycline
hydroxylation
Given the need to screen for a 6α-hydroxylase of anhydrotetracyclines I developed a high
throughput screen for hydroxylation of anhydrotetracyclines based on the ready detection of
anhydrotetracycline hydroxylation by UV/Vis spectroscopy (Figure 2-2). Following colony
picking onto microtiter plates and overnight culturing to even out the OD across the different wells
the cells were reinoculated for a 2nd overnight culture, pelleted and resuspended in a Tris reaction
buffer containing anhydrotetracycline and glucose. The suspension was then diluted in H2O for
UV/VIS measurements (Figure 3-2).
Figure 3-2 A simplified outline of the microtiter plate assay for anhydrotetracycline
hydroxylation.

80
3.4.4 Evolving a 6α-hydroxylase of anhydrotetracyclines – DacO1 random
mutagenesis
Given the low/no hydroxylation activity of DacO1 on anhydrotetracycline, DacO1 optimization
was attempted by directed evolution. An error-prone mutagenesis library from DacO1 encoding
plasmid AL-1-31-C7 was made with an average of 3 mutations per 1 kb and transformed into
FY251. The library was then assayed for anhydrotetracycline hydroxylation by the microtiter plate
assay for anhydrotetracycline hydroxylation (Figure 3-2) with strains expressing OxyS, DacO1
(w.t.) and empty plasmid (EH-3-98-6, EH-3-80-2 and EH-3-80-3, respectively) as controls. Over
500 colonies were screened in the assay using 96-well plates with two potential hits identified. A
typical plate with no hits, showing only the OxyS expressing strain positive control above
background fluorescence as well as a plate displaying a hit are shown in Figure 3-3.

81
Figure 3-3 DacO1 error-prone mutagenesis screen excerpt
DacO1 error-prone mutagenesis screen using the microtiter plate assay for anhydrotetracycline
hydroxylation. (a) a plate with only the OxyS positive control significantly above background
fluorescence. (b) a plate with both the OxyS positive control and a DacO1 mutant hit with
fluorescence significantly above background. Wells 60 and 72 contain the positive control OxyS
encoding strain EH-3-98-6. λex = 400 nm.
A followup on the two potential DacO1 mutant hits in the screen showed that one of them has a
slightly increased fluorescence in the Δexcitation spectrum relative to DacO1 (Figure 3-4). The

82
new excitation peak at 375 nm, in 25 nm resolution, is similar to that found for OxyS hydroxylation
of anhydrotetractycline at 380 nm, in 10 nm resolution, but of a much lower intensity (Figure 2-
2). Considering that the increase in fluorescence is rather minor I decided to focus in the interim
on alternatives such as rational design approaches to optimize hydroxylation by DacO1 and
exploring DacO1 homologs (Sections 3.4.1 and 3.4.2, respectively). The testing of these
candidates is described in Section 3.4.5.
Figure 3-4 ΔExcitation spectrum for DacO1 and DacO1 error-prone PCR mutant
The spectrum shown is a Δ spectrum, that is, the values shown are the emission values for the
hydroxylase expressing cells minus the emission values of the no hydroxylase control EH-3-80-3.
λemission = 500 nm. Each value is the average of six biological replicates and the error bars represent
standard error.
3.4.5 Testing DacO1 expression optimization constructs and other bacterial
hydroxylases
I screened the strains generated for the DacO1 optimization attempts as well as their controls, EH-
5-98-1 through EH-5-98-19, EH-3-248-1 through EH-3-248-8, EH-3-80-2, EH-3-80-3, EH-3-98-
6 and EH-5-115-1 through EH-5-115-5 (Sections 3.4.1 and 3.4.2) using the microtiter plate

83
UV/Vis assay (Section 3.4.3). Each of the 35 strains was assayed in four biological replicates, as
well as in two culturing temperatures, 30 °C and 25 °C, starting from the 2nd inoculation stage
(Figure 3-2). The protocol for the microtiter plate UV/Vis assay (Section 3.3.1) was slightly
modified to accommodate the strains encoding DacO1 controlled by inducible promoters. Thus,
EH-5-98-7 and EH-5-98-8 encoding pADH2-dacO1 were inoculated in YPD in the 2nd inoculation
and EH-5-98-4 and EH-5-98-5, encoding pGAL1-dacO1 were inoculated in U- Raffinose and
supplemented in the next morning with 66.7 μL of 30% galactose in H2O for a total concentration
of 2%. UV/Vis measurements were taken following one night and after three nights of incubation
with anhydrotetracycline for the 30 °C plates and after one night only for the 25 °C plates.
Strains that showed a blue shift in their peak in the Δexcitation spectrum from that of negative
control strain EH-3-80-2 harboring empty pSP-G1 were indicated as potential hits in the assay and
are shown in Table 3-1. Specifically, strains listed in the table have shown in the Δexcitation
spectrum a peak of 405 nm, 380 nm or 355 nm as opposed to the 430 nm peak of negative control
EH-3-80-3, associated with anhydrotetracycline. Results from three measurements are shown, two
measurements after overnight incubation of the cells suspensions in the buffer containing
anhydrotetracycline, one for each of the culturing temperatures (25 °C and 30 °C) and another
final measurement for the 30 °C culturing condition after 3 nights of suspension in the buffer
containing anhydrotetracycline. The rank was calculated per plate measured for one of the
measurements according to the emission at the peak maximum, and should be interpreted only as
a rough indicator as peak emissions at three different wavelengths are ranked on the same scale.
The number 1 or 2 after the dot indicates the plate number from which the measurement was taken.
Reassuringly, the positive control OxyS, whose native substrate is anhydrotetracycline is leading
the rank in highest emission resulting from a blue-shifted excitation. It is followed by the various

84
DacO1 fusion proteins as well as by hydroxylase alternatives to OxyS and DacO1. Surprisingly,
empty pSP-G1 and OxyR in the alternative background strain, BJ-5464-NpgA have also displayed
a blue-shifted peak in the excitation spectrum, although they are the lowest ranking (Table 3-1).
Also surprisingly, EH-3-80-2 encoding unoptimized DacO1 has not displayed a blue shifted
excitation peak in the reduction spectrum in contrast to previous experiments (Figure 3-4). Strains
that exhibited a blue-shifted excitation peak at 25 °C and not in 30 °C are: DacO1-DacO4,
OxySDacO1-191, no-AB-tags and DacO1-JCAT-BJ. I proceeded to analyze by western blot the
hydroxylase expression of the strains that displayed a blue-shifted excitation peak in all three
measurements (Figure 3-5, Figure 3-6 and Figure 3-7).
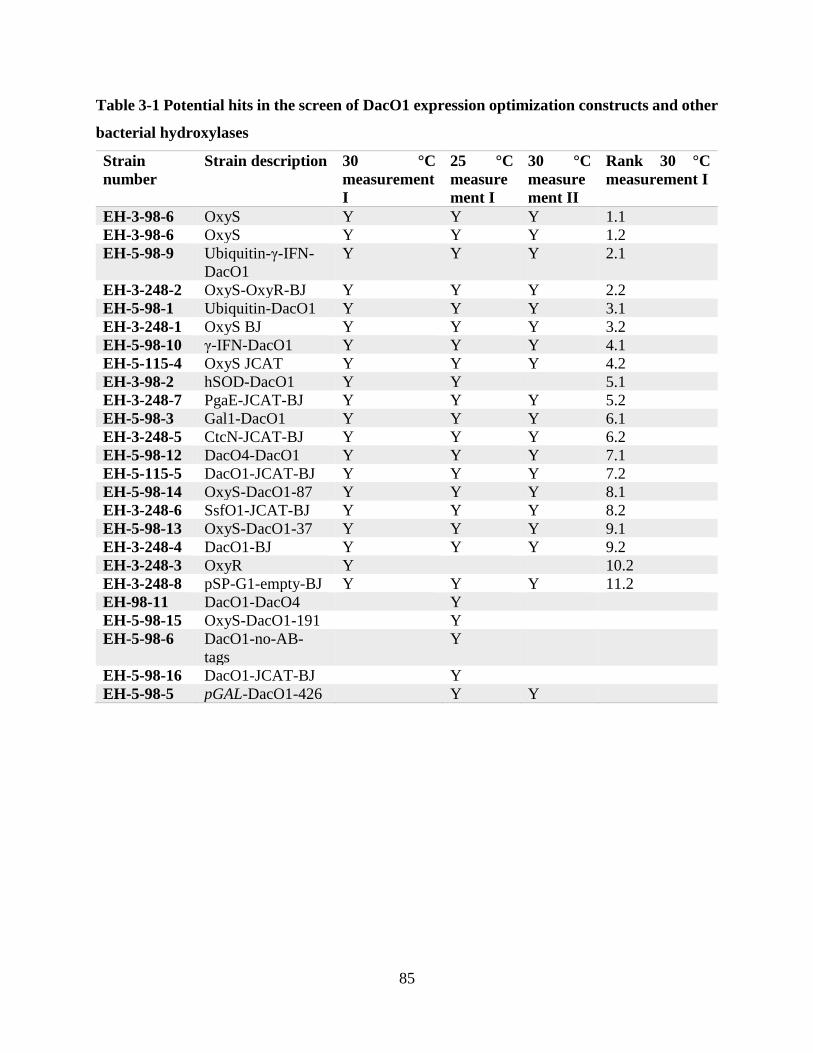
85
Table 3-1 Potential hits in the screen of DacO1 expression optimization constructs and other
bacterial hydroxylases
Strain
number
Strain description 30 °C
measurement
I
25 °C
measure
ment I
30 °C
measure
ment II
Rank 30 °C
measurement I
EH-3-98-6 OxyS Y Y Y 1.1
EH-3-98-6 OxyS Y Y Y 1.2
EH-5-98-9 Ubiquitin-γ-IFN-
DacO1
Y Y Y 2.1
EH-3-248-2 OxyS-OxyR-BJ Y Y Y 2.2
EH-5-98-1 Ubiquitin-DacO1 Y Y Y 3.1
EH-3-248-1 OxyS BJ Y Y Y 3.2
EH-5-98-10 γ-IFN-DacO1 Y Y Y 4.1
EH-5-115-4 OxyS JCAT Y Y Y 4.2
EH-3-98-2 hSOD-DacO1 Y Y 5.1
EH-3-248-7 PgaE-JCAT-BJ Y Y Y 5.2
EH-5-98-3 Gal1-DacO1 Y Y Y 6.1
EH-3-248-5 CtcN-JCAT-BJ Y Y Y 6.2
EH-5-98-12 DacO4-DacO1 Y Y Y 7.1
EH-5-115-5 DacO1-JCAT-BJ Y Y Y 7.2
EH-5-98-14 OxyS-DacO1-87 Y Y Y 8.1
EH-3-248-6 SsfO1-JCAT-BJ Y Y Y 8.2
EH-5-98-13 OxyS-DacO1-37 Y Y Y 9.1
EH-3-248-4 DacO1-BJ Y Y Y 9.2
EH-3-248-3 OxyR Y 10.2
EH-3-248-8 pSP-G1-empty-BJ Y Y Y 11.2
EH-98-11 DacO1-DacO4 Y
EH-5-98-15 OxyS-DacO1-191 Y
EH-5-98-6 DacO1-no-AB-
tags
Y
EH-5-98-16 DacO1-JCAT-BJ Y
EH-5-98-5 pGAL-DacO1-426 Y Y

86
Figure 3-5 Western blot analysis of DacO1 fusion proteins and other bacterial hydroxylases
in FY251
Strain cultures were lysed with Y-PER and labeled with monoclonal ANTI-FLAG HRP antibody.
Two biological replicates were used for each strain indicated as colony 1 and 2 (C1 and C2),
respectively. Strain EH-5-98-1, EH-5-98-3, EH-5-98-9, EH-5-98-10 and EH-5-98-13 encode
DacO1 fusions with ubiquitin, Gal1, Ubiquitin-γ-IFN, γ-IFN, and OxyS (first 37 amino acids)
respectively. In the latter only the last 461 amino acids of DacO1 are encoded, while in the other
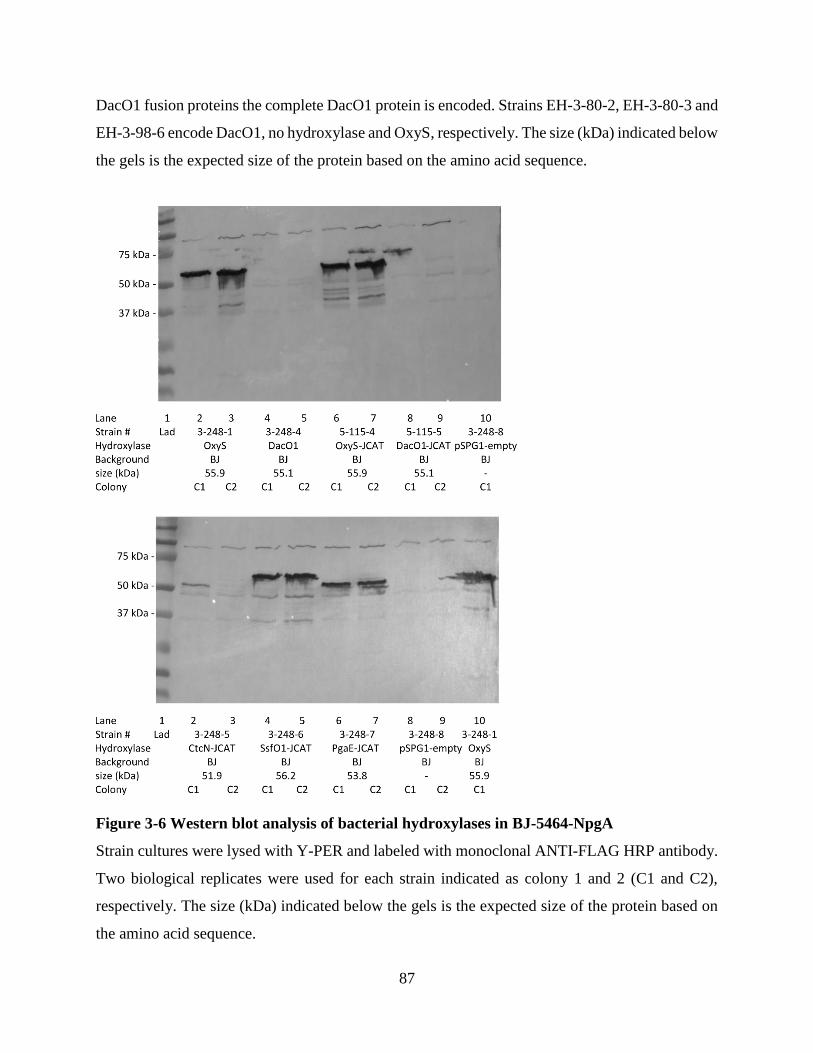
87
DacO1 fusion proteins the complete DacO1 protein is encoded. Strains EH-3-80-2, EH-3-80-3 and
EH-3-98-6 encode DacO1, no hydroxylase and OxyS, respectively. The size (kDa) indicated below
the gels is the expected size of the protein based on the amino acid sequence.
Figure 3-6 Western blot analysis of bacterial hydroxylases in BJ-5464-NpgA
Strain cultures were lysed with Y-PER and labeled with monoclonal ANTI-FLAG HRP antibody.
Two biological replicates were used for each strain indicated as colony 1 and 2 (C1 and C2),
respectively. The size (kDa) indicated below the gels is the expected size of the protein based on
the amino acid sequence.

88
Figure 3-7 Western blot analysis of DacO1-DacO4 and DacO4-DacO1 fusion proteins
Strain cultures were lysed with Y-PER and labeled with monoclonal ANTI-Myc HRP antibody.
Two biological replicates were used for each strain indicated as colony 1 and 2 (C1 and C2),
respectively. The size (kDa) indicated below the gels is the expected size of the protein based on
the amino acid sequence.
For the fusion protein constructs in FY251, ubiquitin-DacO1-C1, Gal1-DacO1-C1 and Ubiquitin-
IFN-DacO1-C1 slight bands are observed in the expected sizes that are not observed in the DacO1-
C1 control (55.0, 86.5 and 72.0 kDa, respectively, Figure 3-5). Bands at the expected sizes were
also observed for OxyS-DacO1-37, SsfO1, OxyS and DacO1-C2, with SsfO1 and OxyS bands
looking more prominent (55.5, 56.2, 55.9 and 55.1 kDa, respectively, Figure 3-5). OxyS-DacO1-
37 and DacO1-C2 show a 50 < band < 75 kDa that is not observed in the empty pSP-G1 negative

89
control. In all bands, proteins of lower mass are noticeable, potentially corresponding to
proteolysis products of the hydroxylases retaining the C-terminal FLAG-tag.
For the protein constructs in BJ-5464-NpgA, clearly less degradation products are observed
(Figure 3-6). OxyS, SsfO1 and PgaE show very prominent bands at the expected sizes (55.9, 56.2
and 53.8 kDa) for both colonies; CtcN-C1 shows a slighter band in the expected size (51.9 kDa)
and DacO1-JCAT-C2 shows an even slighter band of the expected size (55.1 kDa). In addition,
the codon optimization method (COOL vs JCAT) does not seem to significantly alter expression
levels in the case of OxyS, or significantly remediate expression levels in the case of DacO1
(Compare strains EH-3-248-1 and EH-3-248-4 with strains EH-5-115-4 and EH-5-115-5,
respectively, Figure 3-6). Finally, even though BJ-5464-NpgA expression apparently reduced
protein degradation for other hydroxylases (e.g. OxyS and SsfO1) it does not seem to have
promoted significantly expression levels for DacO1 (Figure 3-5 and Figure 3-6).
The DacO4-DacO1 fusion did not yield a stably expressed protein. While DacO4 under pPGK1
and labeled with myc-tag is clearly expressed when unfused to DacO1, the DacO4-DacO1 fusion
is not observed in the gel when under pPGK1 and labeled with myc-tag, in either FY251 or
BJ5464-NpgA background strain (Figure 3-7).
Since less protein degradation was observed for OxyS and SsfO1 in BJ5464-NpgA relative to
FY251 (Compare strain EH-3-248-1 and EH-3-248-6 to strains EH-3-98-6 and EH-5-98-17,
Figure 3-6 and Figure 3-5, respectively) it was logical to test whether the expression of the DacO1
N-terminal fusion proteins would be enhanced in BJ5464-NpgA as well. Strains EH-5-163-1
through EH-5-163-7 were therefore cloned and their DacO1 fusion expression was tested by
western blot (Figure 3-8).
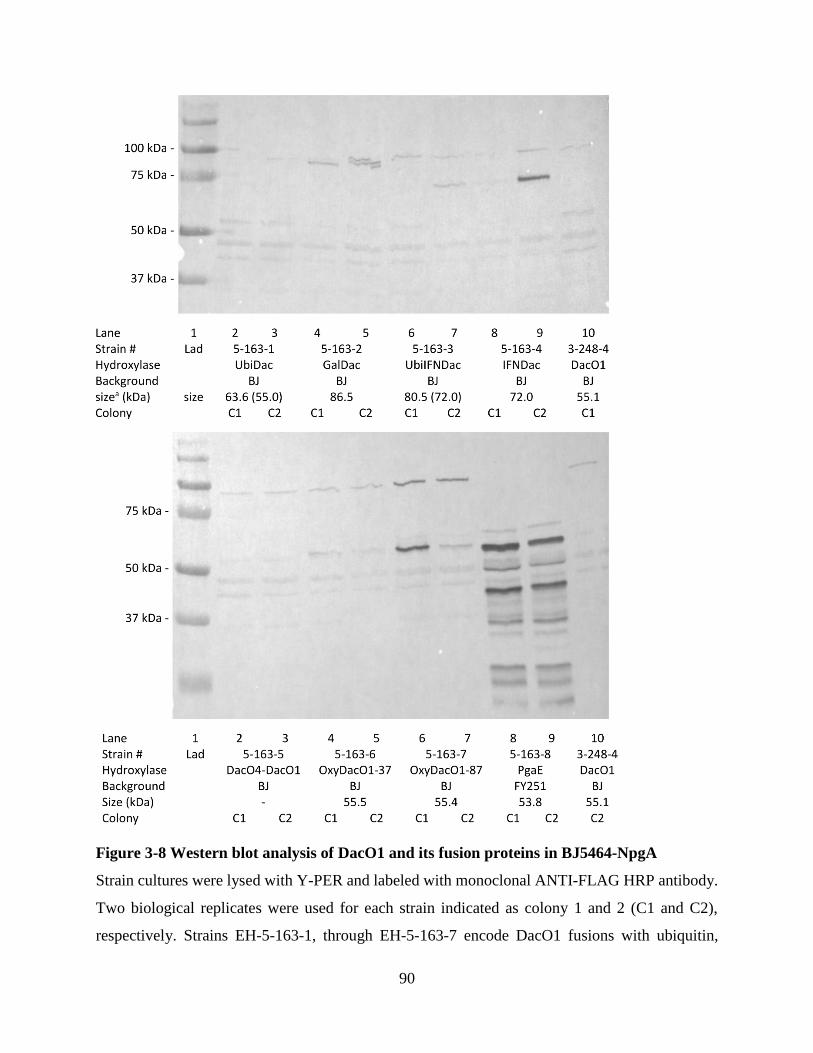
90
Figure 3-8 Western blot analysis of DacO1 and its fusion proteins in BJ5464-NpgA
Strain cultures were lysed with Y-PER and labeled with monoclonal ANTI-FLAG HRP antibody.
Two biological replicates were used for each strain indicated as colony 1 and 2 (C1 and C2),
respectively. Strains EH-5-163-1, through EH-5-163-7 encode DacO1 fusions with ubiquitin,

91
Gal1, Ubiquitin-γ-IFN, γ-IFN, DacO4, OxyS (first 37 amino acids) and OxyS (first 87 amino
acids), respectively. In the last two strains only the last 461 and 411 amino acids of DacO1 are
encoded, respectively, while the other DacO1 fusion proteins the complete DacO1 protein is
encoded. In contrast to DacO1 and its fusion proteins, PgaE expressing strain in this assay has an
FY251 background. The size (kDa) indicated below the gels is the expected size of the protein
based on the amino acid sequence.
In BJ5464-NpgA background, Ubiquitin-DacO1 fusion expression levels seem similar to that of
unfused DacO1; Gal1-DacO1, Ubiquitin-IFN-DacO1-C2 and IFN-DacO1-C2 fusions gave bands
at the expected sizes of 86.5 and 72.0, respectively, with IFN-DacO1-C2 showing the most
prominent band; The OxyS N-terminal fusions to C-terminal DacO1, OxyS-DacO1-37 and OxyS-
DacO1-87 both gave bands at the expected size with OxyS-DacO1-87-C1 showing the more
prominent band (Figure 3-8). Based on these results, the colonies showing better DacO1
expression levels were chosen for analysis of anhydrotetracycline hydroxylation by mass
spectrometry (Scheme 3-2).
Scheme 3-2 Two-step enzymatic conversion of (6-demethyl-)anhydrotetracycline to (6-
demethyl-)6-epitetracycline
Cell lysates of strains expressing hydroxylases that showed stronger bands of the expected sizes
in the Western Blots were incubated in a Tris buffer with anhydrotetracycline and glucose-6-
phosphate. The overnight incubations were then assayed by mass and UV/Vis spectrometry
(Figure 3-9 and Table 3-2). The strains that show hydroxylation levels potentially above
background, as indicated by 443.4 ion counts that are more than double those for the no

92
hydroxylase negative control, are those encoding OxyS, PgaE and Ubiquitin-γ-IFN-DacO1
(labeled as 1, 4 and 9, respectively in Figure 3-9). These three strains, along with three additional
ones encoding γ-IFN-DacO1, OxySDac37 and OxySDac87, also show a maximal emission and/or
excitation in the Δspectra that differs from that of the background strain EH-3-248-8 encoding
empty pSP-G1 (Table 3-2). Therefore, these six strains along with EH-3-248-4 and EH-3-248-8
as controls were assayed again by mass spectrometry in a wider set of conditions prior to larger
scale reactions, isolation and analysis by NMR.

93
Figure 3-9 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction
by cell lysates of strains expressing DacO1, its fusion proteins and other bacterial
hydroxylases
The analyzed strains shown are EH-3-248-1, EH-3-248-4, EH-3-248-6, EH-3-248-7, EH-3-248-8,
EH-5-163-1-C1, EH-5-163-2-C1, EH-5-163-2-C2, EH-5-163-3-C2, EH-5-163-4-C2, EH-5-163-
6-C1, EH-5-163-7-C1 encoding hydroxylases OxyS, DacO1, SsfO1, PgaE, pSPG1 (no
hydroxylase negative control), Ubiquitin-DacO1, Gal1-DacO1, Gal1-DacO1, Ubiquitin-γ-IFN-
DacO1, γ-IFN-DacO1, OxyS-DacO1-37 and OxyS-DacO1-87, respectively. Cell lysates were
placed overnight in Tris buffer (100 mM, pH 7.45) containing anhydrotetracycline (5.4 mM),

94
glucose (27.8 mM), NADPH (3 mM), glucose-6-phosphate (10 mM) and mercaptoethanol (18.5
mM).
Table 3-2 UV/Vis spectroscopy results for hydroxylase strains cell lysates incubated with
anhydrotetracycline and glucose-6-phosphate
Strain no. Hydroxylase Max emission
(λexcitation = 280 nm)
Max excitation
(λemission = 500 nm)
1 EH-3-248-1 OxySb 490 390
2 EH-3-248-4 DacO1 590 350
3 EH-3-248-6 SsfO1 580 350
4 EH-3-248-7 PgaEb 580 370
5 EH-3-248-8 pSPG1a 580 350
6 EH-5-163-1-C1 UbiDac 570 350
7 EH-5-163-2-C1 GalDac 570 350
8 EH-5-163-2-C2 GalDac 570 350
9 EH-5-163-3-C2 UbiIFNDacb 690 350
10 EH-5-163-4-C2 IFNDacb 460 350
11 EH-5-163-6-C1 OxySDac37b 420 370
12 EH-5-163-7-C1 OxySDac87b 430 470 a The maximum emission and maximum absorption spectra of EH-3-248-8 encoding no
hydroxylase (empty pSP-G1) are shown in italics. For all other strains, the maximum value shown
are from the Δspectrum of the corresponding strain minus the spectrum of the EH-3-248-8 strain
encoding no hydroxylase (empty pSP-G1).
b Maximum emission and excitation values for the Δspectra that are more than 10 nm different
than the corresponding values obtained for strain EH-3-248-8 encoding no hydroxylase (empty
pSP-G1) are shown in bold.
In order to select strains and conditions for larger scale reactions, strains encoding hydroxylases
that performed favorably according to the Western Blot, mass spectrometry and UV/Vis
spectroscopy results were analyzed in a larger set of conditions using mass spectrometry.
Following cell lysis, lysates were incubated overnight in four different conditions (Table 3-3) and
analyzed by mass spectrometry. The rationale behind trying different conditions was that given
that the conversion rates for all non-OxyS hydroxylases were significantly lower than for OxyS
(Figure 3-9), the reaction should be optimized to allow enough product to be isolated for NMR

95
analysis. The strains that show hydroxylation levels potentially above background, as indicated by
443.4 ion counts that are more than double those for the no hydroxylase negative control encoding
empty pSP-G1, are OxyS and PgaE, with OxyS-DacO1-37 as the closest runner up (Figure 3-10).
These four strains will be used for a larger scale lysate experiment followed by product isolation
and analysis by NMR spectroscopy. Condition D, maximizing the m/z peak associated with
tetracycline ([M + H]+) per amount of cell lysate used will be used to attempt the isolation of
tetracycline or its 6-epimer and condition B maximizing the m/z peak associated with 5a(11a)-
dehydrotetracycline ([M + H]+) will be used to attempt the isolation of the latter or its 6-epimer.
Table 3-3 Assay conditions for anhydrotetracycline hydroxylation and reduction in cell
lysates of strains expressing DacO1, its fusion proteins and other bacterial hydroxylases
Condition no.a Atc (mM) G6P (10 mM) Cell lysate dilution factor into reaction mix
A 5.4 + 5X
B 5.4 - 5X
C 21.6 + 5X
D 21.6 + 10X aCell lysates were placed overnight in Tris buffer (100 mM, pH 7.45) containing in addition to
anhydrotetracycline and glucose-6-phosphate at the concentrations mentioned above, glucose
(27.8 mM), NADPH (3 mM) and mercaptoethanol (18.5 mM).

96
Figure 3-10 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction
in cell lysates of strains expressing DacO1, its fusion proteins and other bacterial
hydroxylases
The analyzed strains shown EH-3-248-1, EH-3-248-4, EH-3-248-7, EH-3-248-8, EH-5-163-3-C2,
EH-5-163-4-C2, EH-5-163-6-C1, EH-5-163-7-C1 are encoding hydroxylases OxyS, DacO1,
PgaE, pSPG1 (no hydroxylase negative control), Ubiquitin-γ-IFN-DacO1, γ-IFN-DacO1, OxyS-
DacO1-37 and OxyS-DacO1-87, respectively.
While for both OxyS and PgaE, in the presence of G6P there is an increase in 445 ion counts
associated with the product of both hydroxylation and reduction reactions (Figure 2-3 and Figure
3-10), the PgaE samples are consistently associated with higher 445/443 ion count ratios relative

97
to the OxyS samples in the presence of G6P (Figure 3-11). This supports that in the case of PgaE
hydroxylation a larger fraction of the hydroxylation product ([M-H]+ = 443) gets further reduced
([M-H]+ = 445).
Figure 3-11 Mass spectrometry analysis of anhydrotetracycline hydroxylation and reduction
in cell lysates of strains expressing OxyS and PgaE
The analyzed strains shown are EH-3-248-1 and EH-3-248-7 encoding hydroxylases OxyS and
PgaE, respectively in condition A (Table 3-3). TIC is total ion count. 443 and 445 refer to ion
counts of these m/z values.
3.5 Discussion
3.5.1 Optimizing DacO1 expression in S. cerevisiae – rational design
DacO1 is hypothesized to be the 6-hydroxylase enzyme in the dactylocycline pathway based on
its homology to OxyS, the 6-hydroxylase of the oxytetracycline pathway. Based on this
assignment, DacO1 is performing its 6-hydroxylase function when heterologously expressed
together with the rest of the dactylocyclinone pathway in S. lividans.(2) Heterologous expression
of DacO1 exclusively does not yield a stably expressed protein in either E. coli or Stryptomyces,(1)
and as shown in this study neither does it stably heterolougsly expressed in S. cerevisiae (Figure
3-1). These results taken together with the importance of coexpression of other pathway proteins
to the function of other polyketide enzymes,(19, 20) led to the hypothesis that coexpression of

98
pathway proteins might be beneficial for DacO1 stable expression in S. cerevisiae. For the
particular proteins whose coexpression with DacO1 in S. cerevisiae was attempted, DacJ and
DacM2, this hypothesis was not confirmed (Table 3-1). However, it could be that other or
additional proteins from the dactylocycline pathway are needed for stable heterologous expression
of DacO1. In contrast, the strategy to improve DacO1 expression by N-terminal fusions was
successful (Sections 3.4.5 and 3.5.5).
3.5.2 Searching the bacterial hydroxylase space for 6α-hydroxylation of
anhydrotetracyclines
The successful identification of PgaE as a hydroxylase of anhydrotetracycline is shown in Section
3.4.5 and discussed in Section 3.5.5. PgaE heterologous expression in S. cerevisiae thus
exemplifies the power of natural enzyme diversity harvesting and testing in S. cerevisiae as a
heterologous host.
3.5.3 Developing a microtiter plate assay for anhydrotetracycline
hydroxylation
The microtiter plate assay for hydroxylation of anhydrotetracyclines is functional, as confirmed
by differentiation between the OxyS encoding strain and the negative control strain (Figure 3-3)
and its throughput could be further increased. One approach to increase the assay throughput would
be to employ an automatic liquid dispenser / colony picker as well as to reduce the volume of each
well in the assay.(25, 26) In the current assay format only 2 μL of the 300 μL suspension are
needed for the final UV/Vis analysis. An alternative approach to increase the assay throughput
would be to grow colonies on agar plates supplemented with anhydrotetracycline and monitor the
change in fluorescence by a fluorescence agar plate imager and thus circumventing the need for

99
microtiter plate handling. However, given that anhydrotetracycline is toxic to S. cerevisiae at
elevated concentrations it remains to be determined whether an agar plate UV/Vis assay is
practical.(27, 28) On the other hand, that very toxicity could prove useful to develop a growth-
based assay, as tetracyclines eukaryotic toxicity is significantly lower than that of
anhydrotetracyclines. Thus, conversion of anhydrotetracycline to tetracycline could lead to an
increased growth rate in media containing anhydrotetracycline.(27, 28)
The stereochemical difference between the expected products of anhydrotetracycline
hydroxylation by a 6β-hydroxylase such as OxyS and a 6α-hydroxylase such as DacO1, 5a(11a)-
dehydrotetracycline and 5a(11a)-6-epidehydrotetracycline, respectively, might lead to a difference
in the absorption/emission of the respective products. Thus, an optimized spectrophotometer
measurement protocol for the microtiter plate assay for anhydrotetracycline hydroxylation might
be desired. However, in both cases both absorption and emission are expected to be blue-shifted,
a difference that the current protocol is capable of detecting. DacO1 hydroxylation is supported by
UV/Vis evidence of the current protocol (Figure 3-4) although it is still to be determined whether
5a(11a)-6-epidehydrotetracycline is indeed synthesized from the reaction of DacO1 with
anhydrotetracycline (Section 3.5.5).
3.5.4 Evolving a 6α-hydroxylase of anhydrotetracyclines – DacO1 random
mutagenesis
Importantly, the library-based (Section 3.4.4 and Section 4.4.3) and rational design-based (Section
3.4.1) approaches for optimizing DacO1 for 6α-hydroxylation of anhydrotetracycline in S.
cerevisiae are not mutually exclusive. The winner of the DacO1 error-prone mutagenesis screen
and additional winners of future screens could be sequenced to identify the amino acids at which
mutations occurred and these sites could be chosen for saturation mutagenesis. The resulting

100
library of size ~100 could be then sequenced by the microtiter plate assay for anhydrotetracycline
hydroxylation with a followup on screen hits as shown (Figure 3-4). The sequence of the confirmed
hits can then be fused in DacO1-fusions that were confirmed to be effective in enhancing DacO1
expression levels such as UbiIFN-DacO1, IFN-DacO1, OxyS-DacO1-37 and OxyS-DacO1-87
(Figure 3-8). Alternatively the expression optimized DacO1 fusion porteins could be used for EP-
PCR or saturation mutagenesis as per Section 3.4.4 and Section 4.4.3.
3.5.5 Testing DacO1 expression optimization constructs and other
hydroxylases
The attempt to fuse a stable protein or protein fragment at the N-terminus of DacO1, or a DacO1
fragment in the case of the OxyS fusions, to improve DacO1 expression levels was successful
(Figure 3-8). However, the more stably expressed DacO1 fusions are still poor hydroxylases of
anhydrotetracycline at best compared to OxyS (Figure 3-9 and Figure 3-10). A very likely
hypothesis to explain the poor hydroxylation activity is that the natural substrate of DacO1,
anhydrodactylocyclinone, is different enough from anhydrotetracycline to impede efficient
hydroxylation of the latter. In fact, the hydroxylase SsfO1, whose heterologous expression was
confirmed to be effective in S. cerevisiae (Figure 3-5 and Figure 3-6) did not prove an effective
hydroxylase of anhydrotetracycline at all (Figure 3-9). The native substrate of DacO1 differs from
anhydrotetracycline by having 8-methoxy, 7-chloro and 4a-hydroxy instead of protons in
anhydrotetracycline and it could be that these functional groups serve as recognition elements in
the substrate (Scheme 3-1). The 8-methoxy and 7-chloro substituents can could further have an
electronic contribution to the activation energy of the hydroxylation reaction.
With stably expressed DacO1 fusion proteins at hand such as ubiquitin-γ-IFN-DacO1, γ-IFN-
DacO1, OxyS-DacO1-37 and OxyS-DacO1-87 (Figure 3-8), a few avenues could now be

101
explored: (i) upscale current hydroxylation setup (Table 3-3 and Section 3.4.5) and attempt
isolation of product and analysis by NMR to verify whether the desired 6α-hydroxylation occurred.
(ii) mutagenize stably expressed DacO1 fusion proteins by error-prone PCR (Section 3.4.4) or
saturation mutagenesis (Section 4.4.3) or a combination thereof and then test for
anhydrotetracycline or 6-demethylanhydrotetracycline(29) hydroxylation by the mutants using the
microplate UV/Vis assay (Section 3.4.3); (iii) test DacO1 fusion proteins hydroxylation of the
DacO1 native substrate, anhydrodactylocyclinone (Scheme 3-1) or 6-
demethyanhydrodactylocyclinone; and (iv) test TAN-1612 hydroxylation by coexpressing the
DacO1 fusion proteins and the TAN-1612 pathway (Chapter 4). Given the high biological
variation between the different colonies expressing DacO1-fusion proteins (Figure 3-8), testing
several biological replicates for each strain for these approaches might be crucial.
Specifically for (iii), Anhydrodactylocyclinone and 6-demethylanhydrodactylocyclinone could be
obtained either by deleting the dacO1 and dacO4 or the dacO1, dacO4 and dacM2 genes from
plasmid pDac, respectively, and transforming the resulting plasmid into a K4-114 strain harboring
pDacT1O3E.(2) Importantly, isolating anhydrodactylocyclinone and/or 6-
demethylanhydrodactylocyclinone using such plasmids would support the functional assignment
of DacO1, DacO4 and DacM2. Alternatively the DacO1 native substrate could be pursued by
acidic dehydration of dactylocyclinone,(29) the aglycone of dactylocycline A, which can be
purified either from its native producer Dactylosporangium sp. SC 14051 (ATCC 53693) or from
the heterologous producer K4/pDac-T1O3E.(2, 30, 31) A coculture of a (6-demethyl-
)anhydrodactylocyclinone producing strain and the strain encoding the DacO1 fusion protein is
another potential alternative. Finally, it is also potentially possible to feed the strain encoding the

102
DacO1 fusion protein with crude (6-demethyl-)anhydrodactylocyclinone without prior
purification.
PgaE, a bacterial monooxygenase tested as a potential 6α-hydroxylase candidate (Section 3.4.2)
was found to hydroxylate anhydrotetracycline (Sections 3.4.5 and 3.5.2). Furthermore, in the
presence of PgaE more reduced product was obtained relative to the hydroxylated product, when
compared to OxyS (Figure 3-11). Possible reasons for increased reduction proportion of
hydroxylated anhydrotetracycline in the PgaE sample (Figure 3-11) could be (i) a lower
hydroxylation rate in the case of PgaE results in less overload on reduction capacities (ii) a different
hydroxylation product between OxyS and PgaE, such as different stereochemistry that leads to a
more ready reduction of the PgaE hydroxylation product, (iii) an involvement of OxyS and/or
PgaE in the reduction process either directly by catalyzing it or indirectly by facilitating the process
through product binding. These reasons could be tested by isolating the hydroxylation product
from two different cell lysates expressing OxyS or PgaE and not containing G6P and analyzing
the hydroxylated product by NMR. Studying and comparing the spectra would support or reject
reason (ii). The isolated hydroxylated product can then be added to one of three cell lysates in the
presence of G6P, expressing OxyS or PgaE or no hydroxylase. This will eliminate hydroxylation
levels influence as per reason (i) and will enable testing whether OxyS and/or PgaE are involved
in the reduction as per reason (iii), although it is still perhaps possible that involvement of OxyS
and/or PgaE in the reduction happens only immediately after the hydroxylation before product
release.
3.6 Conclusion
This chapter describes the progress towards 6-demethyl-6-epitetracyclines biosynthesis in S.
cerevisiae (Scheme 3-2). The biosynthesis of 6-demethyl-6-epitetracyclines as opposed to 6-

103
demethyltetracyclines requires a 6α-hydroxylase, such as DacO1, as opposed to a 6β-hydroxylase
such as OxyS (Scheme 3-1). However, past attempts to stably heterologoulsy express DacO1
without the rest of its pathway have been unsuccessful.(1) With the rational design attempt to
stabilize DacO1 expression by N-terminal fusion proteins proving successful (Figure 3-8), the next
step is to test the stably expressed DacO1 fusion proteins in larger scale for 6α-hydroxylation of
anhydrotetracycline and/or anhydrodactylocyclinone and/or 6-demethylanhydrodactylocyclinone
(Section 3.5.5). This will allow product isolation followed by NMR analysis. In addition, given
the differences between DacO1’s native substrate and anhydrotetracycline (Scheme 3-1) and given
that DacO1 fusion proteins are poor hydroxylases of anhydrotetracycline at best (Figure 3-9 and
Figure 3-10), the stably expressed DacO1 fusion proteins could be subjected to mutagenesis
(Section 3.4.4 and Section 4.4.3) to identify mutants that readily hydroxylate the desired
anhydrotetracycline substrate.
In addition to the use of rational design to improve DacO1 expression, this chapter also describes
harvesting natural diversity in bacterial hydroxylases towards the search for a 6α-hydroxylase
(Section 3.4.2). With PgaE, one of these bacterial hydroxylases, proving as a hydroxylase of
anhydrotetracycline (Figure 3-9 and Figure 3-10) the next step is to analyze its product after large
scale preparation and isolation and to verify whether it is performing 6α-hydroxylation or 6-
hydroxylation at all on anhydrotetracycline. It also remains to be determined why a larger
proportion of the hydroxylation product of anhydrotetracycline undergoes reduction when PgaE is
used as the hydroxylase in comparison to when OxyS is used (Figure 3-11) and whether either of
these hydroxylases participates in reduction as well. Participation of OxyS or PgaE in the reduction
reaction could theoretically take place by direct catalysis of the reduction or by facilitating catalysis
through product binding. These open questions could be better understood by adding the isolated

104
hydroxylation product of OxyS and of PgaE, each to three separate cell lysates of strains encoding
OxyS, PgaE and no hydroxylases.
Finally, using a variant of the microtiter plate assay for anhydrotetracycline hydroxylation (Figure
3-2 and Figure 3-3), the promising 6α-hydroxylation candidates identified in this chapter (Table
3-2) were screened for hydroxylation of the fungal anhydrotetracycline TAN-1612 in the next
chapter.

105
3.7 Appendix
Table 3-4 Strains used in this study
Genotype Source/Reference
FY251 MATa leu2Δ1 trpΔ63 ura3-52 his3-200 ATCC 96098
BJ5464-
NpgA
MATα ura3-52 his3-Δ200 leu2-Δ1
trp1 pep4::HIS3 δ:: pADH2-npgA-tADH2 prb1Δ1.6R can1
GAL
a / (15)
EH-3-80-2 FY251 AL-1-31-C7 This Study
EH-3-80-3 FY251 pSP-G1 Chapter 2
EH-3-98-6 FY-251 AL-1-101-C10 Chapter 2
ΕΗ-5-153-7 FY-251 AL-1-173-B-C4 This Study
ΕΗ-5-153-8 FY-251 AL-1-119-A-C7 Chapter 2
EH-3-248-1 BJ5464-NpgA AL-1-101-C10 Chapter 2
EH-3-248-2 BJ5464-NpgA AL-1-119-A-C7 This Study
EH-3-248-3 BJ5464-NpgA AL-1-26-1-C1 This Study
EH-3-248-4 BJ5464-NpgA AL-1-31-C7 This Study
EH-3-248-5 BJ5464-NpgA AL-1-234-B-C2 This Study
EH-3-248-6 BJ5464-NpgA AL-1-239-B-C3 This Study
EH-3-248-7 BJ5464-NpgA AL-1-234-C-C1 This Study
EH-3-248-8 BJ5464-NpgA pSP-G1 Chapter 2
EH-3-270-9 FY-251 AL-2-8-D This Study
EH-5-19-2 FY-251 AL-2-8-D This Study
EH-5-98-1 FY251 EH-5-49-A This Study
EH-5-98-2 FY251 EH-5-49-B This Study
EH-5-98-3 FY251 EH-5-49-C This Study
EH-5-98-4 FY251 EH-5-80-1 This Study
EH-5-98-5 FY251 EH-5-80-2 This Study
EH-5-98-6 FY251 EH-5-80-3 This Study
EH-5-98-7 FY251 EH-5-80-4 This Study
EH-5-98-8 FY251 EH-5-80-5 This Study
EH-5-98-9 FY251 EH-5-80-6 This Study
EH-5-98-10 FY251 EH-5-80-7 This Study
EH-5-98-11 FY251 EH-5-80-8 This Study
EH-5-98-12 FY251 EH-5-80-9 This Study
EH-5-98-13 FY251 EH-5-80-10 This Study
EH-5-98-14 FY251 EH-5-80-11 This Study
EH-5-98-15 FY251 EH-5-80-12 This Study
EH-5-98-16 FY251 AL-1-239-A This Study
EH-5-98-17 FY251 AL-1-239-B This Study
EH-5-98-18 FY251 EH-5-90-A This Study
EH-5-98-19 FY251 EH-5-90-A This Study
EH-5-115-1 FY251 EH-5-90-A EH-5-105-Α This Study
EH-5-115-2 FY251 AL-1-31-C7 EH-5-105-A This Study

106
EH-5-115-3 FY251 pSP-G1 EH-5-105-A This Study
EH-5-115-4 BJ5464-NpgA AL-1-226-A This Study
EH-5-115-5 BJ5464-NpgA AL-1-239-A This Study
EH-5-163-1 BJ5464-NpgA EH-5-49-A This Study
EH-5-163-2 BJ5464-NpgA EH-5-49-C This Study
EH-5-163-3 BJ5464-NpgA EH-5-80-6 This Study
EH-5-163-4 BJ5464-NpgA EH-5-80-7 This Study
EH-5-163-5 BJ5464-NpgA EH-5-80-9 This Study
EH-5-163-6 BJ5464-NpgA EH-5-80-10 This Study
EH-5-163-7 BJ5464-NpgA EH-5-80-11 This Study
EH-5-163-8 FY251 AL-1-234-C-C1 This Study a Y. Tang, University of California, Los Angeles, Department of Chemical and Biomolecular
Engineering & Department of Chemistry and Biochemistry, Department of Bioengineering
Table 3-5 Plasmids used in this study
Description Source/Reference
pSP-G1 pTEF1-FLAG-tADH1, pPGK1-MYC-tCYC1, 2 μ, Ura Addgene 64736
pRS413-pGAL1 Shuttle vector (empty), His marker, CEN ATCC 87326
pRS416- pGAL1 Shuttle vector (empty), Ura marker, CEN ATCC 87332
pRS426- pGAL1 Shuttle vector (empty), Ura marker, 2 μ ATCC 87361
AL-1-26-1-C1 pPGK1-oxyR-tCYC1 in pSP-G1 This Studya
AL-1-31-C7 pTEF1-dacO1-tADH1 in pSP-G1 This Study
AL-1-101-C10 pTEF1-oxyS-tADH1 in pSP-G1 Chapter 2
AL-1-119-A-C7 pTEF1-oxyS-tADH1, pPGK1-oxyR-tCYC1 in pSP-G1 Chapter 2
AL-1-173-B-C4 pTEF1-dacO1-tADH1, pPGK1-dacO4-tCYC1 in pSP-
G1
This Study
AL-1-226-A-C2 pTEF1-oxyS-tADH1 in pSP-G1 JCAT codon opt. This Study
AL-1-234-B-C2 pTEF1-ctcN-tADH1 in pSP-G1 JCAT codon opt. This Study
AL-1-234-C-C1 pTEF1-pgaE-tADH1 in pSP-G1 JCAT codon opt. This Study
AL-1-239-A-C2 pTEF1-dacO1-tADH1 in pSP-G1 JCAT codon opt. This Study
AL-1-239-B-C3 pTEF1-ssfO1-tADH1 in pSP-G1 JCAT codon opt. This Study
AL-2-8-D Error-prone mutagenesis library of AL-1-31-C7 This Study
EH-5-49-A pTEF1-UBI4-dacO1-tADH1 in pSP-G1 This Study
EH-5-49-B pTEF1-SOD1-dacO1-tADH1 in pSP-G1 This Study
EH-5-49-C pTEF1-GAL1-dacO1-tADH1 in pSP-G1 This Study
EH-5-80-1 pGAL1-dacO1-tCYC1 in pRS416 This Study
EH-5-80-2 pGAL1-dacO1-tCYC1 in pRS426 This Study
EH-5-80-3 pTEF1-dacO1-tADH1 in pSP-G1 (no antibody tag) This Study
EH-5-80-4 pADH2-dacO1-tCYC1 in pRS416 This Study
EH-5-80-5 pADH2-dacO1-tCYC1 in pRS426 This Study
EH-5-80-6 pTEF1-UBI4-IFNG-dacO1-tADH1 in pSP-G1b This Study
EH-5-80-7 pTEF1-IFNG-dacO1-tADH1 in pSP-G1b This Study
EH-5-80-8 pPGK1-dacO4-dacO1-tCYC1 in pSP-G1 This Study

107
EH-5-80-9 pPGK1-dacO1-dacO4-tCYC1 in pSP-G1 This Study
EH-5-80-10 pTEF1-oxyS-dacO1-37-tADH1 in pSP-G1 This Study
EH-5-80-11 pTEF1-oxyS-dacO1-87-tADH1 in pSP-G1 This Study
EH-5-80-12 pTEF1-oxyS-dacO1-191-tADH1 in pSP-G1 This Study
EH-5-90-A pTEF1-dacO1-tADH1, pPGK1-dacJ-tCYC1 in pSP-G1 This Study
EH-5-105-Α pGPD-dacM2-tCYC1 in pRS413 This Study a See Chapter 2 for sequence.
b the sequence of UBI4 and of IFNG used in the gene contrasts here is partial. See Table 3-6
Table 3-6 Sequences used in this study
Sequences for the hydroxylases DacO1 (GenBank: AFU65892.1), PgaE (PDB: 2QA1_A), SsfO1
(GenBank: GQ409537.1), CtcN (GenBank: D38214.1) and the DacO1 fusion proteins with UBI4
(GenBank: AF134781.1), SOD1 (GenBank: KJ892183.1), GAL1 (NM_001178368.1), IFNG
(GenBank: X62468.1) and OxyS (AAZ78342.1) are shown capitalized within the context of the
pSP-G1 backbone containing pTEF1, the FLAG tag and tADH1 (partial, decapitalized).
Sequences for DacO4 (GenBank: JX262387.1), the DacO1-DacO4 and DacO4-DacO1 fusions and
DacJ (GenBank: JX262387.1) are shown capitalized within the context of the pSP-G1 backbone
containing pPGK1, the myc tag and tCYC1 (partial, decapitalized).
The sequence for DacM2 (GenBank: JX262387.1) is shown along with the pGPD (pTDH3)
promoter capitalized within the context of the pRS413 backbone containing the tCYC1 terminator
(partial, decapitalized).
The sequences for DacO1 in pRS backbone under the control of pGAL1 or pADH2 is shown
capitalized along with the promoter (decapitalized in the case of pGAL1 and capitalized in the case
of pADH2) within the context of the pRS backbone containing the tCYC1 terminator (partial,
decapitalized).
pTEF1-
dacO1-
tADH1-in-
pSP-G1-
AL-1-31-
C7 and AL-
1-173-B-
C4
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGTTAGTAGCCGGTGGTGGACCAACAGGGTTAT
GGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTA
GAGCGTAGGACAGAACCCTTTCCACATAGCAAAGCACTGGGTATCCA
TCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGAT
TTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTC
CTGTGCCCTTAGACTATGGTGCAATGGATACTCGACATCCGTACGGT
GTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGC
AACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCC
TAAGACAGCACGATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAA
GGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGG

108
AGGTTCTACCACAAGACGCCTGGCTGGGATAGATTTCCCTGGACAAG
ATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCCGGGACCCAT
TACCCTCTGATCCAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGA
GACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAA
CAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAA
GATAGACGTGCTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGT
TGATCTAGCGGGAACTGACTTTGGTATGTACGATGTTCAATGGTTGA
CCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGA
AGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGT
GGCCCAGGTTTGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTG
GAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATT
CATATCATGATGAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGAC
ACAAGAACACAATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCA
TTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAACCCGTAAACA
GATATTTCACCGGACACAACACGGCTCTAGCAGTTAGATACGATTTG
GGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGT
CGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCG
GGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTG
TTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAGGTACCGTGG
CAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCTGATGGTTATG
TTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGGTTTAGAAACT
GCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgatggattacaaggatgac
gacgataaAatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pPGK1-
dacO4-
tCYC1-AL-
1-173-B-
C4
tggaatggcgggaaagggtttagtaccacatgctatgatgcccactgtgatctccagagcaaagttcgttcgatcgt
actgttactctctctctttcaaacagaattgtccgaatcgtgtgacaacaacagcctgttctcacacactcttttcttctaa
ccaagggggtggtttagtttagtagaacctcgtgaaacttacatttacatatatataaacttgcataaattggtcaatgc
aagaaatacatatttggtcttttctaattcgtagtttttcaagttcttagatgctttctttttctcttttttacagatcatcaagga
agtaattatctactttttacaacaaatataaaacaaggatccATGTCATTCACTGCCAAGGAAGT
TCAATATTTGCGTTCTCAGCAATTAGGAAGATTAGCCACCGTTGGTGC
TGATGGTACACCACATAATGTTCCAGTAGGCTACAGATACAACGCTG
ACTTGGGGACCATTGACATCACAGGAAGGGACTTAAGAAGAAGTAG
GAAATATAGAGATGTTCAGGCTGGCTCGCGGGTGGCATTTATTGTTG
ATGATCTACCGAGCACAGCACCTATAGTCGCCCGCGGGCTGGAGGTG
AGGGGAACAGCTGAGGCGCTTTCTGGTGAAGAAGATTTGATACGTGT
ACGACCTGCAAGAATCGTCACTTGGGGTATTGAAGCAGATTGGCAAG
CTGGTCCTACTGGTAGAACGGTAAGGCCCACCACTCCAAGATCCGCG
ACGgtcgacatggaacagaagttgatttccgaagaagacctcgagtaagcttggtaccgcggctagctaagatc
cgctctaaccgaaaaggaaggagttagacaacctgaagtctaggtccctatttatttttttatagttatgttagtattaag
aacgttatttatatttcaaatttttcttttttttctgtacagacgcgtgtacgcatgtaacattatactgaaaaccttgcttgag
aaggttttgggacgctcgaagatccagctgcattaatgaatcggccaacgcgcggggagaggcggtttgcgtattg
ggcgctcttccgcttcctcgctcactgactcgctgcgctcgg
pTEF1-
oxyS-
tADH1 in
pSP-G1
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGAGATACGACGTTGTTATCGCTGGTGCTGGTCC
AACTGGTTTGATGTTGGCTTGTGAATTGAGATTGGCTGGTGCTAGAA
CTTTGGTTTTGGAAAGATTGGCTGAACCAGTTGACTTCTCTAAGGCTT
TGGGTGTTCACGCTAGAACTGTTGAATTGTTGGACATGAGAGGTTTG

109
JCAT
codon opt.
(AL-1-226-
A)
GGTGAAGGTTTCCAAGCTGAAGCTCCAAAGTTGAGAGGTGGTAACTT
CGCTTCTTTGGGTGTTCCATTGGACTTCTCTTCTTTCGACACTAGACA
CCCATACGCTTTGTTCGTTCCACAAGTTAGAACTGAAGAATTGTTGAC
TGGTAGAGCTTTGGAATTGGGTGCTGAATTGAGAAGAGGTCACGCTG
TTACTGCTTTGGAACAAGACGCTGACGGTGTTACTGTTTCTGTTACTG
GTCCAGAAGGTCCATACGAAGTTGAATGTGCTTACTTGGTTGGTTGT
GACGGTGGTGGTTCTACTGTTAGAAAGTTGTTGGGTATCGACTTCCCA
GGTCAAGACCCACACATGTTCGCTGTTATCGCTGACGCTAGATTCAG
AGAAGAATTGCCACACGGTGAAGGTATGGGTCCAATGAGGCCATAC
GGTGTTATGAGACACGACTTGAGAGCTTGGTTCGCTGCTTTCCCATTG
GAACCAGACGTTTACAGAGCTACTGTTGCTTTCTTCGACAGACCATA
CGCTGACAGAAGAGCGCCAGTTACTGAAGAAGATGTTAGAGCTGCTT
TGACTGAAGTTGCTGGTTCTGACTTCGGTATGCACGACGTTAGATGGT
TGTCTCGTTTGACTGACACTTCTCGTCAAGCTGAAAGATACAGAGAT
GGTAGAGTTTTGTTGGCTGGTGACGCTTGTCACATCCACTTGCCAGCT
GGTGGTCAAGGTTTGAACTTGGGTTTCCAAGACGCTGTTAACTTGGG
TTGGAAGTTGGGTGCTACTATCGCTGGTACTGCTCCACCAGAATTGTT
GGACACTTACGAAGCTGAAAGAAGGCCAATCGCTGCTGGTGTTTTGA
GAAACACTAGAGCGCAAGCTGTTTTGATCGACCCAGACCCAAGATAC
GAAGGTTTGAGAGAATTGATGATCGAATTGTTGCACGTTCCAGAAAC
TAACAGATACTTGGCTGGTTTGATCTCTGCTTTGGACGTTAGATACCC
AATGGCTGGTGAACACCCATTGTTGGGTAGAAGAGTTCCAGACTTGC
CATTGGTTACTGAAGATGGTACTAGACAATTGTCTACTTACTTCCACG
CTGCTAGAGGTGTTTTGTTGACTTTGGGTTGTGACCAACCATTGGCTG
ACGAAGCTGCTGCTTGGAAGGACAGAGTTGACTTGGTTGCTGCTGAA
GGTGTTGCTGACCCAGGTTCTGCTGTTGACGGATTAACTGCTTTGTTG
GTTAGACCAGACGGTTACATCTGTTGGACTGCTGCTCCAGAAACTGG
TACTGACGGTTTGACTGACGCTTTGAGAACTTGGTTCGGTCCACCTGC
TATGgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttg
gtcaagtctccaatcaaggttgtcggct
pTEF1-
dacO1-
tADH1 in
pSP-G1
JCAT
codon opt. (
AL-1-239-
A)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGTTGGTTGCTGGTGGTGGTCCAACTGGTTTGTG
GTTGGCTGGTGAATTGAGATTGGCTGGTGTTAGACCATTGGTTTTGGA
AAGAAGAACTGAACCATTCCCACACTCTAAGGCTTTGGGTATCCACC
CAAGAACTATCGAAATGTTGGAATTGAGAGGTTTGGCTGGTAGATTC
TTGGACGGTGCTCCAAAGTTGCCAAAGGGTCACTTCGCTTCTTTGCCA
GTTCCATTGGACTACGGTGCTATGGACACTAGACACCCATACGGTGT
TTACAGAAAGCAAGTTGAAACTGAAGCTTTGTTGGCTGGTCACGCTA
CTAGATTGGGTGTTCCAGTTAGAAGAGGTCACGAATTGGTTGGTTTG
AGACAACACGACGACGCTGTTGTTGGTACTGTTCAAGGTCCACAAGG
TAGATACGAAGTTAGAGCTGCTTACTTGGTTGGTTGTGACGGTGGTG
GTTCTACTACTAGAAGATTGGCTGGTATCGACTTCCCAGGTCAAGAC
CCAGGTGTTGCTGTTTTGATGGCTGACGCTAGATTCAGAGATCCATTG
CCATCTGACCCAGCTATGGGTCCATTCAGAAGATACGGTGTTTTAAG
ACCAGACTTGAGAGCTTGGTTCTCTGCTATCCCATTGCCAGAAGAAC
AAGGTTTGTGTAGAGTTATGGTTTTCTGGTACGGTAGACAATTCGAA

110
GATAGAAGAGCGCCAGTTACTGAAGATGAAATGAGAGCTGCTTTGGT
TGACTTGGCTGGTACTGACTTCGGTATGTACGACGTTCAATGGTTGAC
TAGATTCACTGACGCTTCTCGTCAAGCTGCTAGATACAGATCGGGTA
GAGTTTTCTTGGCTGGTGACGCTGCTCACACTGTTTTCCCAACTGGTG
GTCCAGGTTTGAACGCTGGTTTGCAAGACGCTATGAACTTGGGTTGG
AAGTTGGCTGGTGCTGTTCACGGTTGGGGTGGTGCTTTGTTGGACTCT
TACCACGACGAAAGATACCCAGTTGCTGAACAAGTTTTGAGAGACAC
TAGAACTCAATGTTTGTTGTTGGACCCAGACCCAAGATTCGTTCCATT
GAGAGAAACTTTGGCTGAATTGTTGAGATTGGAACCAGTTAACAGAT
ACTTCACTGGTCACAACACTGCTTTGGCTGTTAGATACGACTTGGGTG
GTGCTCACCCAGCTACTGGTGGTAGAATGCCAGACGTTTTGGTTGCT
ACTGGTGCTGGTCAAAGAAGATTCTCTGACTGTTGTGTTACTGGTAG
AGGTGTTTTGTTGGTTACTTCTGGTGCTGACAGAGGTGAATTGTTGAG
AGGTTGGAAGGACAGAGTTGACGTTGTTACTGGTACTGTTGCTGGTC
CATTGGACGCTGCTGACCACTTGGTTAGACCAGACGGTTACGTTGCTT
GGGCTGGTGGTGCTGGTCACGACGAACCAGGTTTGGAAACTGCTTTG
AGATTGTGGTTCGGTGAACCAGTTTCTgtatcgatggattacaaggatgacgacgataag
atctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
ctcN-
tADH1 in
pSP-G1
JCAT
codon opt.
(AL-1-234-
B-C2)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGTTGTTGCTGGTGCTGGTCCAACTGGTTTGAT
GTTGGCTTGTGAATTGGCTTTGGGTGGTGCTAGAGCTGTTGTTGTTGA
AAGAAGAAGAGAACCAGAAAAGCACTCTAAGGCTATGGGTATGCAA
GGTAGAACTGTTGAATTGTTGGAATTGAGAGGTTTGTTGGACAGATT
CAAGGAAGGTGCTGGTGTTTTGCAAGGTGGTAACTTCGCTTCTTTGG
GTGTTCCAATGAGATTCGAAGAATTTGACACTCAACCAACTCAATAC
GTTTTGTTGGTTCCACAATTGAGAACTGAAGAATTGTTGGCTGAAAG
AGCTAGAGAATTGGGTGTTAGAATCGTTAGAGGTTCTGGTGTTACTG
GTTTCGCTCAAGACGCTGACGGTGTTACTGTTGAAACTGACACTGGTT
TGTTGAGAGCTAGATACTTGGTTGGTTGTGACGGTGGTTCTACTGTTA
GAAAGGCTGCTGGTATCGGTTTCACTGGTCAAGACCCACACATGTAC
GCTTTGATCGGTGACATGAGATTCTCTGGTGACTTGCCAAGAGGTGA
AGGTTTGGGTCCAATGAGGCCAGTTGGTTTGGTTTACAGAGCTACTG
TTGCTTGGTTCGACAGACCATTCGCTGACAGAAGAGCGCCAGTTACT
GAAGAAGAAATGAGAGCTGCTTTGGTTGAACACACTGGTTCTGACCA
CGGTATGCACGACGTTACTTGGTTGTCTCGTTTGACTGACGTTTCTCG
TTTGGCTGACTCTTACAGATTGGGTAGAGTTTTGTTGGCTGGTGACGC
TGCTCACATCCACTTGCCAGCTGGTGGTCAAGGTTTGAACTTGGGTTT
CCAAGACGCTGTTAACTTGGGTTGGAAGTTGGCTGCTGTTGTTAGAG
GTCACGGTACTGAAGAATTGTTGGACTCTTACGGTAGAGAAAGAAGG
CCAATCGCTGACGGTGTTGTTAGAAACACTAGAACTCAAGCTGTTTT
GATCGACCCAGACCCAAGATACGAAGCTCTCAGACAAACTTTGCCAC
ACTTGATGGCTTTGCCAGACACTAACAGACACATGGCTGGTATGTTG
TCTGGTTTCGACGTTGCTTACGGTGGTGGTGACCACCCATTGGTTGGT
AGAAGAATGCCAGACGCTGAATTGATCACTGCTGACGGTCCAAGAA
GAATCTCTGACTGTTTCGCTGGTGCTAGAGGTTTGTTGTTGTTGCCAG
AACAAGGTCCAACTGCTTCTCCATTGGCTGCTTGGGCTGACAGAGTT

111
GACACTTTGACTGTTAAGTCTGGTGGTCCAGACCCAGACACTGCTCA
CTTGGTTAGACCAGACGGTTACGTTGCTTGGGCTGGTGAACCAGCTA
GAACTGAAGAATTGCACCACGCTGCTACTACTTGGTTCGGTGCTGCT
GCTgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggt
caagtctccaatcaaggttgtcggct
pTEF1-
pgaE-
tADH1 in
pSP-G1
JCAT
codon opt.
(AL-1-234-
C-C1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGACGCTGCTGTTATCGTTGTTGGTGCTGGTCC
AGCTGGTATGATGTTGGCTGGTGAATTGAGATTGGCTGGTGTTGAAG
TTGTTGTTTTGGAAAGATTGGTTGAAAGAACTGGTGAATCTCGTGGTT
TGGGTTTCACTGCTAGAACTATGGAAGTTTTCGACCAAAGAGGTATC
TTGCCAAGATTCGGTGAAGTTGAAACTTCTACTCAAGGTCACTTCGGT
GGTTTGCCAATCGACTTCGGTGTTTTGGAAGGTGCTTGGCAAGCTGCT
AAGACTGTTCCACAATCTGTTACTGAAACTCACTTGGAACAATGGGC
TACTGGTTTGGGTGCTGACATCAGAAGAGGTCACGAAGTTTTGTCTTT
GACTGACGACGGTGCTGGTGTTACTGTTGAAGTTAGAGGTCCAGAAG
GTAAGCACACTTTGAGAGCTGCTTACTTGGTTGGTTGTGACGGTGGT
AGATCGTCTGTTAGAAAGGCTGCTGGTTTCGACTTCCCAGGTACTGCT
GCTACTATGGAAATGTACTTGGCTGACATCAAGGGTGTTGAATTGCA
ACCAAGAATGATCGGTGAAACTTTGCCAGGTGGTATGGTTATGGTTG
GTCCATTGCCAGGTGGTATCACTAGAATCATCGTTTGTGAAAGAGGT
ACTCCACCACAAAGAAGAGAAACTCCACCATCTTGGCACGAAGTTGC
TGACGCTTGGAAGAGATTGACTGGTGACGACATCGCTCACGCTGAAC
CAGTTTGGGTTTCTGCTTTCGGTAACGCTACTAGACAAGTTACTGAAT
ACAGAAGAGGTAGAGTTATCTTGGCTGGTGACTCTGCTCACATCCAC
TTGCCAGCTGGTGGTCAAGGTATGAACACTTCTATCCAAGACGCTGT
TAACTTGGGTTGGAAGTTGGGTGCTGTTGTTAACGGTACTGCTACTGA
AGAATTGTTGGACTCTTACCACTCTGAAAGACACGCTGTTGGTAAGA
GATTGTTGATGAACACTCAAGCTCAAGGTTTGTTGTTCTTGTCTGGTC
CAGAAGTTCAACCATTGAGAGATGTTTTGACTGAATTGATCCAATAC
GGTGAAGTTGCTAGACACTTGGCTGGTATGGTTTCTGGTTTGGAAATC
ACTTACGACGTTGGTACTGGTTCTCACCCATTGTTGGGTAAGAGAAT
GCCAGCTTTGGAATTGACTACTGCTACTAGAGAAACATCTTCTACTG
AATTATTACACACTGCTAGAGGTGTTTTGTTGGACTTGGCTGACAACC
CAAGATTGAGAGCTAGAGCTGCTGCTTGGTCTGACAGAGTTGACATC
GTTACTGCTGTTCCAGGTGAAGTTTCTGCTACTTCTGGTTTGAGAGAC
ACTACTGCTGTTTTGATAAGACCAGACGGTCACGTTGCTTGGGCTGCT
CCAGGTTCTCACCACGACTTGCCAATGGCTTTGGAAAGATGGTTCGG
TGCTCCATTGACTGGTgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaac
aattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
ssfO1-
tADH1 in
pSP-G1
JCAT
codon opt.
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGAACCAGAAGTTGTTGTTGCTGGTGCTGGTCC
AGTTGGTTTGATGTTGGCTTGTGAATTGAGAAGGCAAGGTGTTGGTG
TTGTTTTGGTTGAAAGAACTACTGAACCAGCTAACGACAACAGAGCG
CAAGCTCTCCACGGTAGAACTATCCCAGTTTTGGACAGAAGAGGTTT
GTTGCCAAGATTCAGAGCTGCTGAAAGAGAATTGAACGGTGGTGGTT
CTGGTGGTACTGAATCTCACAGAAAAAGACCATTGCCAAGAGGTCAC

112
(AL-1-239-
B-C3)
TTCGCTGGTATCACTGGTTTATTACCATCTGCTCCAGACCCAGACCCA
GACGTTCCACCAGTTGTTTTCGTTCCACAATGGGTTACTACTAGATTG
TTGACTGAACACGCTGCTGAATTGGGTGTTGTTTTGCACAGAGGTTCT
GAAATCACTGCTGTTGAACAAGACGCTGACGGTGTTACTGTTGCTGT
TTCTGGTGGTACTGCTCCAGCTAGATTGAGAGCTAGATACTTGATAG
GTTGTGACGGTGCTAGATCGGTTGTTAGAAGATCGGCTGGTATCGAC
TTCGCTGGTACTGGTGCTACTTTGTCTACTTTGGCTGCTGAAGTTCAC
TTGGACGACCCAGCTGACTTGCCAGTTGGTTGGCACAGAACTCAACA
CGGTTGTACTGTTATCTCTCCACACCCAGAAGGTGGTCACTCTCGTGT
TGTTGTTATCGACTTCTCTGGTCCAGACGCTGACAGAGAAGCTCCAGT
TACTTTGACTGAATTGGAAGAAAGAATCGCTGGTGTTTTGGGTAGAA
AGGTTGCTATGTCTGACTTGAAGAACGCTCAAAGATACGGTGACGCT
GCTAGATTGGCTGACAGATACAGAGTTGGTAGAGTTTTCTTGGCTGG
TGACGCTGCTCACATCCACTACCCAGTTGGTGGTCAAGGTTTGAACA
TCGGTTTGCAAGACGCTGTTAACTTGGCTTGGAAGTTGGCTGCTGACT
TGAGAGGTTGGGCTCCAGACGGTTTGTTGGACACTTACCACACTGAA
AGATACCCAGTTGCTGACTCTGTTTTGACTCACACTAGAGCGCAAGTT
GCTTTGTTGGCTCCAGGTCCAAGAATCGACGCTTTGAGAACTTTGTTC
ACTGACTTGATCGGTTTGGCTGACGTTTCTCGTTACTTGTCTGAAAAG
ATGTCTGCTGCTGACGTTAGATACGACATGGGTGAAGCTGAACCACA
CCCATTGACTGGTAGATTCGCTCCATCTTTGAGATTGGCTACTGAACA
AGGTGAAAGAAGATTGGCTGACTTGTTGGCTGGTGGTAGACCATTGT
TGTTGGACTTGTCTGGTAGAGCTGCTACTGCTGCTGTTGCTGCTAGAT
GGGCTGACAGAGTTGACGTTCACGCTGCTACTTGTCCAGCTGAACCA
GCTTTGGGTTCTTTGTTGATCAGACCAGACTCTTACGTTGCTTGGGCT
TGTGCTCCAGGTGACGCTCCAGACCAAGTTGAAAGAGGTTTGGAAGC
TGCTTTGAGAAGATGGTTCGGTGCTCCAGACCACCAAgtatcgatggattacaa
ggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtc
ggct
pTEF1-
UBI4-
dacO1-
tADH1 in
pSP-G1
(EH-5-49-
A)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgccattATGCAGATCTTCGTCAAGACGTTAACCGGTAA
AACCATAACTCTAGAAGTTGAATCTTCCGATACCATCGACAACGTTA
AGTCGAAAATTCAAGACAAGGAAGGCATTCCACCTGATCAACAAAG
ATTGATCTTTGCCGGTAAGCAGCTCGAGGACGGTAGAACGCTGTCTG
ATTACAACATTCAGAAGGAGTCGACCTTACATCTTGTCTTAAGACTA
AGAGGTGGTATGTTAGTAGCCGGTGGTGGACCAACAGGGTTATGGTT
GGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTAGAGC
GTAGGACAGAACCCTTTCCACATAGCAAAGCACTGGGTATCCATCCA
AGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGATTTTT
AGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTCCTG
TGCCCTTAGACTATGGTGCAATGGATACTCGACATCCGTACGGTGTCT
ACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGCAACC
AGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCCTAAG
ACAGCACGATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAAGGTA
GATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGGAGGT
TCTACCACAAGACGCCTGGCTGGGATAGATTTCCCTGGACAAGATCC

113
TGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCCGGGACCCATTACC
CTCTGATCCAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGAGAC
CTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAACAG
GGTTTATGCAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAAGAT
AGACGTGCTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGTTGA
TCTAGCGGGAACTGACTTTGGTATGTACGATGTTCAATGGTTGACCA
GATTCACAGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGAAGG
GTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGTGGC
CCAGGTTTGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTGGAA
GTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATTCATA
TCATGATGAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGACACAA
GAACACAATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCATTAA
GAGAGACGTTAGCAGAACTATTGAGACTAGAACCCGTAAACAGATA
TTTCACCGGACACAACACGGCTCTAGCAGTTAGATACGATTTGGGTG
GTGCCCATCCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGTCGCA
ACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCGGGAG
AGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTGTTAC
GAGGATGGAAAGATAGAGTGGATGTGGTAACAGGTACCGTGGCAGG
ACCTCTTGATGCAGCTGATCATTTAGTTAGACCTGATGGTTATGTTGC
ATGGGCCGGTGGAGCTGGACACGATGAACCTGGTTTAGAAACTGCTC
TTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgatggattacaaggatgacgacgat
aagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
SOD1-
dacO1-
tADH1 in
pSP-G1
(EH-5-49-
B)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgccattATGGCGACGAAGGCCGTGTGCGTGCTGAAGGG
CGACGGCCCAGTGCAGGGCATCATCAATTTCGAGCAGAAGGAAAGT
AATGGACCAGTGAAGGTGTGGGGAAGCATTAAAGGACTGACTGAAG
GCCTGCATGGATTCCATGTTCATGAGTTTGGAGATAATACAGCAGGC
TGTACCAGTGCAGGTCCTCACTTTAATCCTCTATCCAGAAAACACGGT
GGGCCAAAGGATGAAGAGAGGCATGTTGGAGACTTGGGCAATGTGA
CTGCTGACAAAGATGGTGTGGCCGATGTGTCTATTGAAGATTCTGTG
ATCTCACTCTCAGGAGACCATTGCATCATTGGCCGCACACTGGTGGT
CCATGAAAAAGCAGATGACTTGGGCAAAGGTGGAAATGAAGAAAGT
ACAAAGACAGGAAACGCTGGAAGTCGTTTGGCTTGTGGTGTAATTGG
GATGTTAGTAGCCGGTGGTGGACCAACAGGGTTATGGTTGGCTGGTG
AACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTAGAGCGTAGGACA
GAACCCTTTCCACATAGCAAAGCACTGGGTATCCATCCAAGAACTAT
TGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGATTTTTAGATGGCG
CCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTCCTGTGCCCTTAG
ACTATGGTGCAATGGATACTCGACATCCGTACGGTGTCTACCGTAAA
CAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGCAACCAGATTAGG
AGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCCTAAGACAGCACG
ATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAAGGTAGATACGAA
GTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGGAGGTTCTACCAC
AAGACGCCTGGCTGGGATAGATTTCCCTGGACAAGATCCTGGTGTTG
CCGTTTTGATGGCTGACGCAAGATTCCGGGACCCATTACCCTCTGATC
CAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGAGACCTGATTTA

114
AGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAACAGGGTTTATG
CAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAAGATAGACGTG
CTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGTTGATCTAGCG
GGAACTGACTTTGGTATGTACGATGTTCAATGGTTGACCAGATTCAC
AGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGAAGGGTTTTCT
TAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGTGGCCCAGGTT
TGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTGGAAGTTAGCT
GGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATTCATATCATGAT
GAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGACACAAGAACACA
ATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCATTAAGAGAGAC
GTTAGCAGAACTATTGAGACTAGAACCCGTAAACAGATATTTCACCG
GACACAACACGGCTCTAGCAGTTAGATACGATTTGGGTGGTGCCCAT
CCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGTCGCAACTGGTGC
TGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCGGGAGAGGGGTTC
TTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTGTTACGAGGATGG
AAAGATAGAGTGGATGTGGTAACAGGTACCGTGGCAGGACCTCTTGA
TGCAGCTGATCATTTAGTTAGACCTGATGGTTATGTTGCATGGGCCGG
TGGAGCTGGACACGATGAACCTGGTTTAGAAACTGCTCTTAGATTAT
GGTTTGGTGAACCAGTTAGTgtatcgatggattacaaggatgacgacgataagatctgagctctt
aattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
GAL1-
dacO1-
tADH1 in
pSP-G1
(EH-5-49-
C)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACTAAATCTCATTCAGAAGAAGTGATTGTACC
TGAGTTCAATTCTAGCGCAAAGGAATTACCAAGACCATTGGCCGAAA
AGTGCCCGAGCATAATTAAGAAATTTATAAGCGCTTATGATGCTAAA
CCGGATTTTGTTGCTAGATCGCCTGGTAGAGTCAATCTAATTGGTGAA
CATATTGATTATTGTGACTTCTCGGTTTTACCTTTAGCTATTGATTTTG
ATATGCTTTGCGCCGTCAAAGTTTTGAACGAGAAAAATCCATCCATT
ACCTTAATAAATGCTGATCCCAAATTTGCTCAAAGGAAGTTCGATTT
GCCGTTGGACGGTTCTTATGTCACAATTGATCCTTCTGTGTCGGACTG
GTCTAATTACTTTAAATGTGGTCTCCATGTTGCTCACTCTTTTCTAAA
GAAACTTGCACCGGAAAGGTTTGCCAGTGCTCCTCTGGCCGGGCTGC
AAGTCTTCTGTGAGGGTGATGTACCAACTGGCAGTGGATTGTCTTCTT
CGGCCGCATTCATTTGTGCCGTTGCTTTAGCTGTTGTTAAAGCGAATA
TGGGCCCTGGTTATCATATGTCCAAGCAAAATTTAATGCGTATTACG
GTCGTTGCAGAACATTATGTTGGTGTTAACAATGGCGGTATGGATCA
GGCTGCCTCTGTTTGCGGTGAGGAAGATCATGCTCTATACGTTGAGTT
CAAACCGCAGTTGAAGGCTACTCCGTTTAAATTTCCGCAATTAAAAA
ACCATGAAATTAGCTTTGTTATTGCGAACACCCTTGTTGTATCTAACA
AGTTTGAAACCGCCCCAACCAACTATAATTTAAGAGTGGTAGAAGTC
ACTACAGaattccggATGTTAGTAGCCGGTGGTGGACCAACAGGGTTATG
GTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTAG
AGCGTAGGACAGAACCCTTTCCACATAGCAAAGCACTGGGTATCCAT
CCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGATT
TTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTC
CTGTGCCCTTAGACTATGGTGCAATGGATACTCGACATCCGTACGGT
GTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGC

115
AACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCC
TAAGACAGCACGATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAA
GGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGG
AGGTTCTACCACAAGACGCCTGGCTGGGATAGATTTCCCTGGACAAG
ATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCCGGGACCCAT
TACCCTCTGATCCAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGA
GACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAA
CAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAA
GATAGACGTGCTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGT
TGATCTAGCGGGAACTGACTTTGGTATGTACGATGTTCAATGGTTGA
CCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGA
AGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGT
GGCCCAGGTTTGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTG
GAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATT
CATATCATGATGAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGAC
ACAAGAACACAATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCA
TTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAACCCGTAAACA
GATATTTCACCGGACACAACACGGCTCTAGCAGTTAGATACGATTTG
GGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGT
CGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCG
GGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTG
TTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAGGTACCGTGG
CAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCTGATGGTTATG
TTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGGTTTAGAAACT
GCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgatggattacaaggatgac
gacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pGAL1-
dacO1-
tCYC1 in
pRS416
and
pGAL1-
dacO1-
tCYC1 in
pRS426
(EH-5-80-1
and EH-5-
80-2)
ccctcactaaagggaacaaaagctggagctctagtacggattagaagccgccgagcgggtgacagccctccgaa
ggaagactctcctccgtgcgtcctcgtcttcaccggtcgcgttcctgaaacgcagatgtgcctcgcgccgcactgct
ccgaacaataaagattctacaatactagcttttatggttatgaagaggaaaaattggcagtaacctggccccacaaac
cttcaaatgaacgaatcaaattaacaaccataggatgataatgcgattagttttttagccttatttctggggtaattaatc
agcgaagcgatgatttttgatctattaacagatatataaatgcaaaaactgcataaccactttaactaatactttcaacat
tttcggtttgtattacttcttattcaaatgtaataaaagtatcaacaaaaaattgttaatatacctctatactttaacgtcaag
gagaaaaaaccccggattctagaactagtATGTTAGTAGCCGGTGGTGGACCAACAGG
GTTATGGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTCTTG
TTTTAGAGCGTAGGACAGAACCCTTTCCACATAGCAAAGCACTGGGT
ATCCATCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGCTGG
TAGATTTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTGCAT
CGCTTCCTGTGCCCTTAGACTATGGTGCAATGGATACTCGACATCCGT
ACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCCGGC
CACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACTTGT
AGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGTTCAAGGTC
CTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGTGAT
GGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGATTTCCCTGG
ACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCCGGG
ACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACGCTATGGA
GTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTACCA

116
GAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGACGACA
ATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGAGAGCA
GCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGATGTTCAA
TGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGGTATAG
ATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTTCCC
CACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCATGAATT
TAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCTTTA
CTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAAGTTTTG
CGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCCTCGCTT
TGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAACCCG
TAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTTAGATAC
GATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAGATGT
CTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTGTGT
TACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGGGGG
AACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAGGTAC
CGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCTGATG
GTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGGTTTA
GAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgatggattac
aaggatgacgacgataagatctgaaagcttatcgataccgtcgacctcgagtcatgtaattagttatgtcacgcttac
attcacgccctccccccacatccgctctaaccgaaaaggaaggagttagacaacctgaagtctaggtccctatttatt
tttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtacagacgcgtgtacgcatgtaaca
ttatactgaaaaccttgcttgagaaggttttgggacgctcgaaggctttaatttgcggccggtacccaat
pTEF1-
dacO1-
tADH1 in
pSP-G1 (no
antibody
tag) (EH-5-
80-3)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgCATGTTAGTAGCCGGTGGTGGACCAACAGGGTTAT
GGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTA
GAGCGTAGGACAGAACCCTTTCCACATAGCAAAGCACTGGGTATCCA
TCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGAT
TTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTC
CTGTGCCCTTAGACTATGGTGCAATGGATACTCGACATCCGTACGGT
GTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGC
AACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCC
TAAGACAGCACGATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAA
GGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGG
AGGTTCTACCACAAGACGCCTGGCTGGGATAGATTTCCCTGGACAAG
ATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCCGGGACCCAT
TACCCTCTGATCCAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGA
GACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAA
CAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAA
GATAGACGTGCTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGT
TGATCTAGCGGGAACTGACTTTGGTATGTACGATGTTCAATGGTTGA
CCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGA
AGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGT
GGCCCAGGTTTGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTG
GAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATT
CATATCATGATGAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGAC
ACAAGAACACAATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCA

117
TTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAACCCGTAAACA
GATATTTCACCGGACACAACACGGCTCTAGCAGTTAGATACGATTTG
GGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGT
CGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCG
GGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTG
TTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAGGTACCGTGG
CAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCTGATGGTTATG
TTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGGTTTAGAAACT
GCTCTTAGATTATGGTTTGGTGAACCAGTTAGTTGAgagctcttaattaacaattct
tcgccagaggtttggtcaagtctccaatcaaggttgtcggcttgtctaccttgccagaaatttacgaaaagatggaaa
agggtcaaatcgttggtagatacgttgttgacacttctaaataagcgaatttcttatgatttatgatttttattattaaataa
gttataaaaaaaataagtgtatacaaattttaaagtgactcttaggttttaaaacgaaaattcttattcttgagtaactcttt
cctgtaggtcaggttgctttctcaggtatagcatgaggtcgctccaattcagctggcgtaatagcgaaga
pADH2-
dacO1-
tCYC1 in
pRS416
and
pADH2-
dacO1-
tCYC1 in
pRS426
(EH-5-80-4
and EH-5-
80-4)
ccctcactaaagggaacaaaagctggagctcGCAAAACGTAGGGGCAAACAAACGGA
AAAATCGTTTCTCAAATTTTCTGATGCCAAGAACTCTAACCAGTCTTA
TCTAAAAATTGCCTTATGATCCGTCTCTCCGGTTACAGCCTGTGTAAC
TGATTAATCCTGCCTTTCTAATCACCATTCTAATGTTTTAATTAAGGG
ATTTTGTCTTCATTAACGGCTTTCGCTCATAAAAATGTTATGACGTTT
TGCCCGCAGGCGGGAAACCATCCACTTCACGAGACTGATCTCCTCTG
CCGGAACACCGGGCATCTCCAACTTATAAGTTGGAGAAATAAGAGA
ATTTCAGATTGAGAGAATGAAAAAAAAAAAAAAAAAAAAGGCAGAG
GAGAGCATAGAAATGGGGTTCACTTTTTGGTAAAGCTATAGCATGCC
TATCACATATAAATAGAGTGCCAGTAGCGACTTTTTTCACACTCGAA
ATACTCTTACTACTGCTCTCTTGTTGTTTTTATCACTTCTTGTTTCTTCT
TGGTAAATAGAATATCAAGCTACAAAAAGCATACAATCAACTATCAA
CTATTAACTATATCGTAATACCATATGGCTAtctagaactagtATGTTAGTAG
CCGGTGGTGGACCAACAGGGTTATGGTTGGCTGGTGAACTAAGGTTG
GCCGGGGTTAGACCTCTTGTTTTAGAGCGTAGGACAGAACCCTTTCC
ACATAGCAAAGCACTGGGTATCCATCCAAGAACTATTGAAATGTTGG
AGTTGAGAGGCCTTGCTGGTAGATTTTTAGATGGCGCCCCGAAGCTG
CCGAAAGGTCACTTTGCATCGCTTCCTGTGCCCTTAGACTATGGTGCA
ATGGATACTCGACATCCGTACGGTGTCTACCGTAAACAAGTGGAAAC
GGAAGCTTTATTAGCCGGCCACGCAACCAGATTAGGAGTTCCAGTCC
GCAGAGGTCATGAACTTGTAGGCCTAAGACAGCACGATGATGCTGTA
GTCGGTACTGTTCAAGGTCCTCAAGGTAGATACGAAGTCAGAGCCGC
TTATCTTGTTGGTTGTGATGGAGGAGGTTCTACCACAAGACGCCTGG
CTGGGATAGATTTCCCTGGACAAGATCCTGGTGTTGCCGTTTTGATGG
CTGACGCAAGATTCCGGGACCCATTACCCTCTGATCCAGCTATGGGT
CCATTCAGACGCTATGGAGTCCTGAGACCTGATTTAAGGGCATGGTT
TAGTGCTATTCCTTTACCAGAAGAACAGGGTTTATGCAGAGTTATGG
TTTTTTGGTATGGACGACAATTTGAAGATAGACGTGCTCCAGTAACT
GAGGATGAAATGAGAGCAGCACTTGTTGATCTAGCGGGAACTGACTT
TGGTATGTACGATGTTCAATGGTTGACCAGATTCACAGACGCTTCCA
GGCAAGCTGCAAGGTATAGATCTGGAAGGGTTTTCTTAGCTGGAGAT
GCTGCTCATACTGTCTTCCCCACTGGTGGCCCAGGTTTGAATGCAGGT
TTACAAGATGCCATGAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCA

118
TGGCTGGGGTGGTGCTTTACTAGATTCATATCATGATGAGCGTTACCC
TGTAGCTGAACAAGTTTTGCGTGACACAAGAACACAATGTCTATTGT
TAGATCCAGACCCTCGCTTTGTTCCATTAAGAGAGACGTTAGCAGAA
CTATTGAGACTAGAACCCGTAAACAGATATTTCACCGGACACAACAC
GGCTCTAGCAGTTAGATACGATTTGGGTGGTGCCCATCCTGCAACGG
GAGGCAGAATGCCAGATGTCTTGGTCGCAACTGGTGCTGGTCAAAGA
AGGTTTTCAGATTGTTGTGTTACCGGGAGAGGGGTTCTTTTAGTCACT
TCTGGTGCAGATAGGGGGGAACTGTTACGAGGATGGAAAGATAGAG
TGGATGTGGTAACAGGTACCGTGGCAGGACCTCTTGATGCAGCTGAT
CATTTAGTTAGACCTGATGGTTATGTTGCATGGGCCGGTGGAGCTGG
ACACGATGAACCTGGTTTAGAAACTGCTCTTAGATTATGGTTTGGTG
AACCAGTTAGTgtatcgatggattacaaggatgacgacgataagatctgaaagcttatcgataccgtcga
cctcgagtcatgtaattagttatgtcacgcttacattcacgccctccccccacatccgctctaaccgaaaaggaagga
gttagacaacctgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttctt
ttttttctgtacagacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagg
ctttaatttgcggccggtacccaat
pTEF1-
UBI4-
IFNG-
dacO1-
tADH1 in
pSP-G1
(EH-5-80-
6)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgccattATGCAGATCTTCGTCAAGACGTTAACCGGTAA
AACCATAACTCTAGAAGTTGAATCTTCCGATACCATCGACAACGTTA
AGTCGAAAATTCAAGACAAGGAAGGCATTCCACCTGATCAACAAAG
ATTGATCTTTGCCGGTAAGCAGCTCGAGGACGGTAGAACGCTGTCTG
ATTACAACATTCAGAAGGAGTCGACCTTACATCTTGTCTTAAGACTA
AGAGGTGGTATGCAAGACCCATATGTAAAAGAAGCAGAAAACCTTA
AGAAATATTTTAATGCAGGTCATTCAGATGTAGCGGATAATGGAACT
CTTTTCTTAGGCATTTTGAAGAATTGGAAAGAGGAGAGTGACAGAAA
AATAATGCAGAGCCAAATTGTCTCCTTTTACTTCAAACTTTTTAAAAA
CTTTAAAGATGACCAGAGCATCCAAAAGAGTGTGGAGACCATCAAG
GAAGACATGAATGTCAAGTTTTTCAATAGCAACAAAAAGAAACGAG
ATGACTTCGAAAAGCTGACTAATTATTCGGTAACTGACTTGAATGTC
CAACGCAAAGCAATACATGAACTCATCCAAGTGATGGCTGAACTGTC
GCCAGCAGCTAAAACAGGGAAGCGAAAAAGGAGTCAGATGCTGTTT
CGAGGTCGAAGAGCATCCCAGATGTTAGTAGCCGGTGGTGGACCAAC
AGGGTTATGGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGACCTC
TTGTTTTAGAGCGTAGGACAGAACCCTTTCCACATAGCAAAGCACTG
GGTATCCATCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCTTGC
TGGTAGATTTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACTTTG
CATCGCTTCCTGTGCCCTTAGACTATGGTGCAATGGATACTCGACATC
CGTACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTAGCC
GGCCACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGAACT
TGTAGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGTTCAAG
GTCCTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGTTGT
GATGGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGATTTCCC
TGGACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGATTCC
GGGACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACGCTAT
GGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTTTA
CCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGACG

119
ACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGAGA
GCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGATGTT
CAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGGTA
TAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTCTT
CCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCATGA
ATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTGCT
TTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAAGTT
TTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCCTCG
CTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAAC
CCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTTAGA
TACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAGA
TGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTTG
TGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGGG
GGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAGG
TACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCTG
ATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGGT
TTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgatgg
attacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaa
ggttgtcggct
pTEF1-
IFNG-
dacO1-
tADH1 in
pSP-G1
(EH-5-80-
7)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgccattATGCAAGACCCATATGTAAAAGAAGCAGAAAA
CCTTAAGAAATATTTTAATGCAGGTCATTCAGATGTAGCGGATAATG
GAACTCTTTTCTTAGGCATTTTGAAGAATTGGAAAGAGGAGAGTGAC
AGAAAAATAATGCAGAGCCAAATTGTCTCCTTTTACTTCAAACTTTTT
AAAAACTTTAAAGATGACCAGAGCATCCAAAAGAGTGTGGAGACCA
TCAAGGAAGACATGAATGTCAAGTTTTTCAATAGCAACAAAAAGAA
ACGAGATGACTTCGAAAAGCTGACTAATTATTCGGTAACTGACTTGA
ATGTCCAACGCAAAGCAATACATGAACTCATCCAAGTGATGGCTGAA
CTGTCGCCAGCAGCTAAAACAGGGAAGCGAAAAAGGAGTCAGATGC
TGTTTCGAGGTCGAAGAGCATCCCAGATGTTAGTAGCCGGTGGTGGA
CCAACAGGGTTATGGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAG
ACCTCTTGTTTTAGAGCGTAGGACAGAACCCTTTCCACATAGCAAAG
CACTGGGTATCCATCCAAGAACTATTGAAATGTTGGAGTTGAGAGGC
CTTGCTGGTAGATTTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCA
CTTTGCATCGCTTCCTGTGCCCTTAGACTATGGTGCAATGGATACTCG
ACATCCGTACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTAT
TAGCCGGCCACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCAT
GAACTTGTAGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGT
TCAAGGTCCTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTG
GTTGTGATGGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGAT
TTCCCTGGACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAG
ATTCCGGGACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACG
CTATGGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCC
TTTACCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGG
ACGACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGA
GAGCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGAT

120
GTTCAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAG
GTATAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGT
CTTCCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCA
TGAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGT
GCTTTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAA
GTTTTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCC
TCGCTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAG
AACCCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTT
AGATACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCC
AGATGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATT
GTTGTGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATA
GGGGGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAAC
AGGTACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGAC
CTGATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCT
GGTTTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatc
gatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctcca
atcaaggttgtcggct
pPGK1-
dacO4-
dacO1-
tCYC1 in
pSP-G1
(EH-5-80-
8)
tggaatggcgggaaagggtttagtaccacatgctatgatgcccactgtgatctccagagcaaagttcgttcgatcgt
actgttactctctctctttcaaacagaattgtccgaatcgtgtgacaacaacagcctgttctcacacactcttttcttctaa
ccaagggggtggtttagtttagtagaacctcgtgaaacttacatttacatatatataaacttgcataaattggtcaatgc
aagaaatacatatttggtcttttctaattcgtagtttttcaagttcttagatgctttctttttctcttttttacagatcatcaagga
agtaattatctactttttacaacaaatataaaacaaggatccATGTCATTCACTGCCAAGGAAGT
TCAATATTTGCGTTCTCAGCAATTAGGAAGATTAGCCACCGTTGGTGC
TGATGGTACACCACATAATGTTCCAGTAGGCTACAGATACAACGCTG
ACTTGGGGACCATTGACATCACAGGAAGGGACTTAAGAAGAAGTAG
GAAATATAGAGATGTTCAGGCTGGCTCGCGGGTGGCATTTATTGTTG
ATGATCTACCGAGCACAGCACCTATAGTCGCCCGCGGGCTGGAGGTG
AGGGGAACAGCTGAGGCGCTTTCTGGTGAAGAAGATTTGATACGTGT
ACGACCTGCAAGAATCGTCACTTGGGGTATTGAAGCAGATTGGCAAG
CTGGTCCTACTGGTAGAACGGTAAGGCCCACCACTCCAAGATCCGCG
ACGATGTTAGTAGCCGGTGGTGGACCAACAGGGTTATGGTTGGCTGG
TGAACTAAGGTTGGCCGGGGTTAGACCTCTTGTTTTAGAGCGTAGGA
CAGAACCCTTTCCACATAGCAAAGCACTGGGTATCCATCCAAGAACT
ATTGAAATGTTGGAGTTGAGAGGCCTTGCTGGTAGATTTTTAGATGG
CGCCCCGAAGCTGCCGAAAGGTCACTTTGCATCGCTTCCTGTGCCCTT
AGACTATGGTGCAATGGATACTCGACATCCGTACGGTGTCTACCGTA
AACAAGTGGAAACGGAAGCTTTATTAGCCGGCCACGCAACCAGATTA
GGAGTTCCAGTCCGCAGAGGTCATGAACTTGTAGGCCTAAGACAGCA
CGATGATGCTGTAGTCGGTACTGTTCAAGGTCCTCAAGGTAGATACG
AAGTCAGAGCCGCTTATCTTGTTGGTTGTGATGGAGGAGGTTCTACC
ACAAGACGCCTGGCTGGGATAGATTTCCCTGGACAAGATCCTGGTGT
TGCCGTTTTGATGGCTGACGCAAGATTCCGGGACCCATTACCCTCTGA
TCCAGCTATGGGTCCATTCAGACGCTATGGAGTCCTGAGACCTGATTT
AAGGGCATGGTTTAGTGCTATTCCTTTACCAGAAGAACAGGGTTTAT
GCAGAGTTATGGTTTTTTGGTATGGACGACAATTTGAAGATAGACGT
GCTCCAGTAACTGAGGATGAAATGAGAGCAGCACTTGTTGATCTAGC

121
GGGAACTGACTTTGGTATGTACGATGTTCAATGGTTGACCAGATTCA
CAGACGCTTCCAGGCAAGCTGCAAGGTATAGATCTGGAAGGGTTTTC
TTAGCTGGAGATGCTGCTCATACTGTCTTCCCCACTGGTGGCCCAGGT
TTGAATGCAGGTTTACAAGATGCCATGAATTTAGGTTGGAAGTTAGC
TGGAGCTGTTCATGGCTGGGGTGGTGCTTTACTAGATTCATATCATGA
TGAGCGTTACCCTGTAGCTGAACAAGTTTTGCGTGACACAAGAACAC
AATGTCTATTGTTAGATCCAGACCCTCGCTTTGTTCCATTAAGAGAGA
CGTTAGCAGAACTATTGAGACTAGAACCCGTAAACAGATATTTCACC
GGACACAACACGGCTCTAGCAGTTAGATACGATTTGGGTGGTGCCCA
TCCTGCAACGGGAGGCAGAATGCCAGATGTCTTGGTCGCAACTGGTG
CTGGTCAAAGAAGGTTTTCAGATTGTTGTGTTACCGGGAGAGGGGTT
CTTTTAGTCACTTCTGGTGCAGATAGGGGGGAACTGTTACGAGGATG
GAAAGATAGAGTGGATGTGGTAACAGGTACCGTGGCAGGACCTCTTG
ATGCAGCTGATCATTTAGTTAGACCTGATGGTTATGTTGCATGGGCCG
GTGGAGCTGGACACGATGAACCTGGTTTAGAAACTGCTCTTAGATTA
TGGTTTGGTGAACCAGTTAGTgtcgacatggaacagaagttgatttccgaagaagacctcgagt
aagcttggtaccgcggctagctaagatccgctctaaccgaaaaggaaggagttagacaacctgaagtctaggtcc
ctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtacagacgcgtgtacgc
atgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagatccagctgcattaatgaatcggcca
acgcgcggggagaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgactcgctgcgctcgg
pPGK1-
dacO1-
dacO4-
tCYC1 in
pSP-G1
(EH-5-80-
9)
tggaatggcgggaaagggtttagtaccacatgctatgatgcccactgtgatctccagagcaaagttcgttcgatcgt
actgttactctctctctttcaaacagaattgtccgaatcgtgtgacaacaacagcctgttctcacacactcttttcttctaa
ccaagggggtggtttagtttagtagaacctcgtgaaacttacatttacatatatataaacttgcataaattggtcaatgc
aagaaatacatatttggtcttttctaattcgtagtttttcaagttcttagatgctttctttttctcttttttacagatcatcaagga
agtaattatctactttttacaacaaatataaaacaaggatccATGTTAGTAGCCGGTGGTGGACC
AACAGGGTTATGGTTGGCTGGTGAACTAAGGTTGGCCGGGGTTAGAC
CTCTTGTTTTAGAGCGTAGGACAGAACCCTTTCCACATAGCAAAGCA
CTGGGTATCCATCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCCT
TGCTGGTAGATTTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCACT
TTGCATCGCTTCCTGTGCCCTTAGACTATGGTGCAATGGATACTCGAC
ATCCGTACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTA
GCCGGCCACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGA
ACTTGTAGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGTTC
AAGGTCCTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGT
TGTGATGGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGATTT
CCCTGGACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGAT
TCCGGGACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACGCT
ATGGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTT
TACCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGA
CGACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGAG
AGCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGATG
TTCAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGG
TATAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTC
TTCCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCAT
GAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTG
CTTTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAAG

122
TTTTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCCTC
GCTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAA
CCCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTTAG
ATACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAG
ATGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTT
GTGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGG
GGGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAG
GTACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCT
GATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGG
TTTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTATGTC
ATTCACTGCCAAGGAAGTTCAATATTTGCGTTCTCAGCAATTAGGAA
GATTAGCCACCGTTGGTGCTGATGGTACACCACATAATGTTCCAGTA
GGCTACAGATACAACGCTGACTTGGGGACCATTGACATCACAGGAAG
GGACTTAAGAAGAAGTAGGAAATATAGAGATGTTCAGGCTGGCTCG
CGGGTGGCATTTATTGTTGATGATCTACCGAGCACAGCACCTATAGT
CGCCCGCGGGCTGGAGGTGAGGGGAACAGCTGAGGCGCTTTCTGGTG
AAGAAGATTTGATACGTGTACGACCTGCAAGAATCGTCACTTGGGGT
ATTGAAGCAGATTGGCAAGCTGGTCCTACTGGTAGAACGGTAAGGCC
CACCACTCCAAGATCCGCGACGgtcgacatggaacagaagttgatttccgaagaagacctcg
agtaagcttggtaccgcggctagctaagatccgctctaaccgaaaaggaaggagttagacaacctgaagtctaggt
ccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtacagacgcgtgtac
gcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagatccagctgcattaatgaatcggc
caacgcgcggggagaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgactcgctgcgctcgg
pTEF1-
oxyS-
dacO1-37-
tADH1 in
pSP-G1
(EH-5-80-
10)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGAGGTACGATGTTGTTATAGCTGGTGCAGGAC
CCACCGGTTTGATGTTAGCATGTGAACTTCGGCTGGCGGGTGCCAGA
ACTTTGGTTTTAGAAAGATTAGCCGAGCCTTTTCCACATAGCAAAGC
ACTGGGTATCCATCCAAGAACTATTGAAATGTTGGAGTTGAGAGGCC
TTGCTGGTAGATTTTTAGATGGCGCCCCGAAGCTGCCGAAAGGTCAC
TTTGCATCGCTTCCTGTGCCCTTAGACTATGGTGCAATGGATACTCGA
CATCCGTACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTATT
AGCCGGCCACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATG
AACTTGTAGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGTT
CAAGGTCCTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGG
TTGTGATGGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGATT
TCCCTGGACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGA
TTCCGGGACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACGC
TATGGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCT
TTACCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGG
ACGACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGA
GAGCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGAT
GTTCAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAG
GTATAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGT
CTTCCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCA
TGAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGT
GCTTTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAA

123
GTTTTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCC
TCGCTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAG
AACCCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTT
AGATACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCC
AGATGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATT
GTTGTGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATA
GGGGGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAAC
AGGTACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGAC
CTGATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCT
GGTTTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatc
gatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctcca
atcaaggttgtcggct
pTEF1-
oxyS-
dacO1-87-
tADH1 in
pSP-G1
(EH-5-80-
11)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGAGGTACGATGTTGTTATAGCTGGTGCAGGAC
CCACCGGTTTGATGTTAGCATGTGAACTTCGGCTGGCGGGTGCCAGA
ACTTTGGTTTTAGAAAGATTAGCCGAGCCTGTTGACTTCTCGAAAGCT
CTAGGAGTCCACGCTCGCACTGTTGAACTATTAGATATGAGAGGCCT
CGGTGAAGGATTCCAGGCTGAAGCACCAAAGTTAAGAGGTGGTAATT
TTGCCTCATTAGGCGTCCCCCTGGATTTCTCATCATTTGATACTCGAC
ATCCGTACGGTGTCTACCGTAAACAAGTGGAAACGGAAGCTTTATTA
GCCGGCCACGCAACCAGATTAGGAGTTCCAGTCCGCAGAGGTCATGA
ACTTGTAGGCCTAAGACAGCACGATGATGCTGTAGTCGGTACTGTTC
AAGGTCCTCAAGGTAGATACGAAGTCAGAGCCGCTTATCTTGTTGGT
TGTGATGGAGGAGGTTCTACCACAAGACGCCTGGCTGGGATAGATTT
CCCTGGACAAGATCCTGGTGTTGCCGTTTTGATGGCTGACGCAAGAT
TCCGGGACCCATTACCCTCTGATCCAGCTATGGGTCCATTCAGACGCT
ATGGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCTT
TACCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGGA
CGACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGAG
AGCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGATG
TTCAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAGG
TATAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGTC
TTCCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCAT
GAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGTG
CTTTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAAG
TTTTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCCTC
GCTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAGAA
CCCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTTAG
ATACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCCAG
ATGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATTGTT
GTGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATAGG
GGGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAACAG
GTACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGACCT
GATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCTGG
TTTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatcgat

124
ggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatc
aaggttgtcggct
pTEF1-
oxyS-
dacO1-
191-tADH1
in pSP-G1
(EH-5-80-
12)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGAGGTACGATGTTGTTATAGCTGGTGCAGGAC
CCACCGGTTTGATGTTAGCATGTGAACTTCGGCTGGCGGGTGCCAGA
ACTTTGGTTTTAGAAAGATTAGCCGAGCCTGTTGACTTCTCGAAAGCT
CTAGGAGTCCACGCTCGCACTGTTGAACTATTAGATATGAGAGGCCT
CGGTGAAGGATTCCAGGCTGAAGCACCAAAGTTAAGAGGTGGTAATT
TTGCCTCATTAGGCGTCCCCCTGGATTTCTCATCATTTGATACTAGAC
ACCCATATGCATTGTTTGTTCCACAAGTACGAACTGAAGAACTGCTA
ACAGGTAGAGCTTTGGAGCTAGGGGCGGAGCTGCGTCGTGGTCATGC
CGTGACCGCCTTGGAACAAGATGCTGATGGTGTTACTGTGAGCGTGA
CAGGCCCTGAAGGCCCATACGAAGTAGAATGTGCTTATTTGGTGGGC
TGCGACGGTGGAGGCAGTACGGTGAGGAAACTATTGGGCATAGATTT
TCCAGGTCAAGACCCACATATGTTTGCTGTCATCGCAGACGCAAGAT
TCAGAGAAGAGCTTCCTCACGATCCAGCTATGGGTCCATTCAGACGC
TATGGAGTCCTGAGACCTGATTTAAGGGCATGGTTTAGTGCTATTCCT
TTACCAGAAGAACAGGGTTTATGCAGAGTTATGGTTTTTTGGTATGG
ACGACAATTTGAAGATAGACGTGCTCCAGTAACTGAGGATGAAATGA
GAGCAGCACTTGTTGATCTAGCGGGAACTGACTTTGGTATGTACGAT
GTTCAATGGTTGACCAGATTCACAGACGCTTCCAGGCAAGCTGCAAG
GTATAGATCTGGAAGGGTTTTCTTAGCTGGAGATGCTGCTCATACTGT
CTTCCCCACTGGTGGCCCAGGTTTGAATGCAGGTTTACAAGATGCCA
TGAATTTAGGTTGGAAGTTAGCTGGAGCTGTTCATGGCTGGGGTGGT
GCTTTACTAGATTCATATCATGATGAGCGTTACCCTGTAGCTGAACAA
GTTTTGCGTGACACAAGAACACAATGTCTATTGTTAGATCCAGACCC
TCGCTTTGTTCCATTAAGAGAGACGTTAGCAGAACTATTGAGACTAG
AACCCGTAAACAGATATTTCACCGGACACAACACGGCTCTAGCAGTT
AGATACGATTTGGGTGGTGCCCATCCTGCAACGGGAGGCAGAATGCC
AGATGTCTTGGTCGCAACTGGTGCTGGTCAAAGAAGGTTTTCAGATT
GTTGTGTTACCGGGAGAGGGGTTCTTTTAGTCACTTCTGGTGCAGATA
GGGGGGAACTGTTACGAGGATGGAAAGATAGAGTGGATGTGGTAAC
AGGTACCGTGGCAGGACCTCTTGATGCAGCTGATCATTTAGTTAGAC
CTGATGGTTATGTTGCATGGGCCGGTGGAGCTGGACACGATGAACCT
GGTTTAGAAACTGCTCTTAGATTATGGTTTGGTGAACCAGTTAGTgtatc
gatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctcca
atcaaggttgtcggct
pPGK1-
dacJ-
tCYC1 in
pSP-G1
(EH-5-90-
A)
tggaatggcgggaaagggtttagtaccacatgctatgatgcccactgtgatctccagagcaaagttcgttcgatcgt
actgttactctctctctttcaaacagaattgtccgaatcgtgtgacaacaacagcctgttctcacacactcttttcttctaa
ccaagggggtggtttagtttagtagaacctcgtgaaacttacatttacatatatataaacttgcataaattggtcaatgc
aagaaatacatatttggtcttttctaattcgtagtttttcaagttcttagatgctttctttttctcttttttacagatcatcaagga
agtaattatctactttttacaacaaatataaaacaaggatccATGACCGCAGGACAGCTGCATCT
TGACGTTCCGGAACCGCCTGGTTCATGGGTAGATGAAGCCACATTTA
GGGCTGTTATGGGTGCTTTGCCATCCGGTGTTACAGTCGTGACGACCT
TGGACCCAGATGGCAGACCAGTAGGCTTAACTTGTTCTGCGGCTTGT
TCAGTTTCTCAAAGGCCTCCCTTATTACTTGTCTGCTTAAACGATAGA

125
AGCAGGGTACTAGATGCAATACTCAGAAGAGGTCAGTTCATAGTAAA
TGTCCTACGCGACCGTAGAGAAGCAACATCGGCAGTCTTTGCCAGTA
GAGCCGAAGATAGATTCGCAGCTGTGCCTTGGCAACCTGGGAGGCGA
ACTGGTGTCCCTTGGTTGTTAGCAGACACGGTTGCTCATGCAGAATGT
GAGGTGGCAGGTACTTTGCATGCTGGCGATCACGTTATCGTTGTTGG
AGGTATTGTTGCGGGTGATGCGCACCCCGACCGTGTGGGGCCATTAA
TGTATTGGAGACAAAGATACGGTGGATGGCCAGTAACAGATGATGCT
AGAGAAATTGCATTGACTCTAGCTGCCGAGGGAgtcgacatggaacagaagttga
tttccgaagaagacctcgagtaagcttggtaccgcggctagctaagatccgctctaaccgaaaaggaaggagttag
acaacctgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttc
tgtacagacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaagatccagc
tgcattaatgaatcggccaacgcgcggggagaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgac
tcgctgcgctcgg
pGPD-
dacM2-
tCYC1 in
pRS413
(EH-5-105-
Α)
ccctcactaaagggaacaaaagctggagctcAGTTTATCATTATCAATACTGCCATTTC
AAAGAATACGTAAATAATTAATAGTAGTGATTTTCCTAACTTTATTTA
GTCAAAAAATTAGCCTTTTAATTCTGCTGTAACCCGTACATGCCCAAA
ATAGGGGGCGGGTTACACAGAATATATAACATCGTAGGTGTCTGGGT
GAACAGTTTATTCCTGGCATCCACTAAATATAATGGAGCCCGCTTTTT
AAGCTGGCATCCAGAAAAAAAAAGAATCCCAGCACCAAAATATTGT
TTTCTTCACCAACCATCAGTTCATAGGTCCATTCTCTTAGCGCAACTA
CAGAGAACAGGGGCACAAACAGGCAAAAAACGGGCACAACCTCAAT
GGAGTGATGCAACCTGCCTGGAGTAAATGATGACACAAGGCAATTG
ACCCACGCATGTATCTATCTCATTTTCTTACACCTTCTATTACCTTCTG
CTCTCTCTGATTTGGAAAAAGCTGAAAAAAAAGGTTGAAACCAGTTC
CCTGAAATTATTCCCCTACTTGACTAATAAGTATATAAAGACGGTAG
GTATTGATTGTAATTCTGTAAATCTATTTCTTAAACTTCTTAAATTCTA
CTTTTATAGTTAGTCTTTTTTTTAGTTTTAAAACACCAAGAACTTAGTT
TCGACGGATACTAGTAAAATGACTGATGTATCCGCCCATACTACAAC
CGATGGGGCGGTTGCAAGGGCTCGCGCTGTGGTCAGACTAAATACTG
CATATTTTCGGGCTAAGGTACTTCAAAGTGCTGTCGAACTAGGTGTTT
TTGATCTTTTAGATGGAGGCCCACAACCTGCCGCTGACATTTGCGGA
AAGTTAGGTATTAGACATAGACTCGCAGTGGACTATTTGGACGCATT
GACGGGTTTAGGGTTGCTTGAAAGGGATGGTGAACGTTACAGAAACT
CGCCAACAGCGGCTGAGTTCTTGGTCCAAGATCGACCCGCCTATTTG
GGTGGAACCGCGAGGCAACATGCCAAATTACATTATCACGCTTGGGG
TAAACTGGCCGATGCAATGAGAGAAGGCCGTGCTCAGAGCGCCGTG
GCAGCTCAAGGTGCAAAAGCCTATGTAAATTACTATGAAGATTTGGG
CCAGGCCAGGAGGGTCATGACACATATGGATGCTCATAACGGTTTCA
CAGCGGATGAAATCGCTAGACAATTGGACTGGAGAGGATACGCCAC
AGTAACAGATGTGGGCGGCGCTCGTGGGAATGTTGCAGCTCGAATTG
TTCAAGCAGTGCCTCATTTGAGAGCAAATGTAGTTGACCTACCCGCA
TTGCGACCTTTATTCGATGAGCTGATGGCCCAACTAGGAACTGCTGG
TCAGGTTGAGTTTCACGGTGGTGACTTCTTTGCGGACCCGATACCAG
AGTCTGACGTTATCGTTGTGGGCCACGTCCTGGCGGATTGGCCACAG
CCAAGACGTGTGGAATTACTTAGAAGATGTCACGCAGCATTAAGACC
TGGTGGAACGGTAGTTGTTTACGATGTTATGGTTGATGATGATAGAG

126
ATGACGTCGAAGCACTCCTACAAAGATTAAACTCTGCAATGATAAGG
GACGATATGGGTGCTTACGCTGTTACTGAATGTGCTGGATATTTAAG
AGAAGCTGGATTTACCGTAGAAAGGGCCCTGCGTACGGATACCATAA
CTCGCGATCATTTTGTTATTGGTAGAAAAGCTGCATCATAACTCGAGt
catgtaattagttatgtcacgcttacattcacgccctccccccacatccgctctaaccgaaaaggaaggagttagaca
acctgaagtctaggtccctatttatttttttatagttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgt
acagacgcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcgaaggctttaatttg
cggccggtacccaat

127
3.8 References
1. Wang P, Bashiri G, Gao X, Sawaya MR, Tang Y. Uncovering the Enzymes that Catalyze
the Final Steps in Oxytetracycline Biosynthesis. J Am Chem Soc. 2013;135(19):7138-7141. doi:
10.1021/ja403516u.
2. Wang P, Kim W, Pickens LB, Gao X, Tang Y. Heterologous Expression and Manipulation
of Three Tetracycline Biosynthetic Pathways. Angewandte Chemie-International Edition.
2012;51(44):11136-11140. doi: 10.1002/anie.201205426.
3. SDS gel-loading buffer (2X). Cold Spring Harbor Protocols. 2006;2006(1). doi:
10.1101/pdb.rec407.
4. Gallagher SR. One-Dimensional SDS Gel Electrophoresis of Proteins. Curr Protoc Mol
Biol. 2006;75(1):10.12.11-10.12A.37. doi: 10.1002/0471142727.mb1002as75.
5. Ecker DJ, Stadel JM, Butt TR, Marsh JA, Monia BP, Powers DA, Gorman JA, Clark PE,
Warren F, Shatzman A, et al. Increasing gene expression in yeast by fusion to ubiquitin. J Biol
Chem. 1989;264(13):7715-7719.
6. Ecker DJ, Khan MI, Marsh J, Butt TR, Crooke ST. Chemical synthesis and expression of
a cassette adapted ubiquitin gene. J Biol Chem. 1987;262(8):3524-3527.
7. Schuster M, Einhauer A, Wasserbauer E, Süßenbacher F, Ortner C, Paumann M, Werner
G, Jungbauer A. Protein expression in yeast; comparison of two expression strategies regarding
protein maturation. J Biotechnol. 2000;84(3):237-248. doi: 10.1016/s0168-1656(00)00355-2.
8. Stȩpień PP, Brousseau R, Wu R, Narang S, Thomas DY. Synthesis of a human insulin gene
VI. expression of the synthetic proinsulin gene in yeast. Gene. 1983;24(2-3):289-297. doi:
10.1016/0378-1119(83)90089-6.
9. Cousens LS, Shuster JR, Gallegos C, Ku L, Stempien MM, Urdea MS, Sanchez-Pescador
R, Taylor A, Tekamp-Olson P. High level expression of proinsulin in the yeast, Saccharomyces
cerevisiae. Gene. 1987;61(3):265-275. doi: 10.1016/0378-1119(87)90190-9.
10. Steimer KS, Higgins KW, Powers MA, Stephans JC, Gyenes A, George-Nascimento C,
Luciw PA, Barr PJ, Hallewell RA, Sanchez-Pescador R. Recombinant polypeptide from the

128
endonuclease region of the acquired immune deficiency syndrome retrovirus polymerase (pol)
gene detects serum antibodies in most infected individuals. J Virol. 1986;58(1):9-16.
11. Barr P, Steimer K, Sabin E, Parkes D, Georgenascimento C, Stephens J, Powers M, Gyenes
A, Vannest G, Miller E. Antigenicity and immunogenicity of domains of the human
immunodeficiency virus (HIV) envelope polypeptide expressed in the yeast Saccharomyces
cerevisiae. Vaccine. 1987;5(2):90-101. doi: 10.1016/0264-410x(87)90053-3.
12. Barr PJ, Inselburg J, Green KM, Kansopon J, Hahm BK, Gibson HL, Lee-Ng CT, Bzik DJ,
Li W-b, Bathurst IC. Immunogenicity of recombinant Plasmodium falciparum SERA proteins in
rodents. Mol Biochem Parasitol. 1991;45(1):159-170. doi: 10.1016/0166-6851(91)90038-8.
13. Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its
amino-terminal residue. Science. 1986;234(4773):179-186. doi: 10.1126/science.3018930.
14. Peng B, Williams TC, Henry M, Nielsen LK, Vickers CE. Controlling heterologous gene
expression in yeast cell factories on different carbon substrates and across the diauxic shift: a
comparison of yeast promoter activities. Microbial Cell Factories. 2015;14(1). doi:
10.1186/s12934-015-0278-5.
15. Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva
NA, Vederas JC, Tang Y. Complete Reconstitution of a Highly Reducing Iterative Polyketide
Synthase. Science. 2009;326(5952):589-592. doi: 10.1126/science.1175602.
16. Jones EW. Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol.
1991;194:428-453. doi: 10.1016/0076-6879(91)94034-A.
17. Romanos MA, Scorer CA, Clare JJ. Foreign gene expression in yeast: a review. Yeast.
1992;8(6):423-488. doi: 10.1002/yea.320080602.
18. Tøttrup HV, Carlsen S. A process for the production of human proinsulin inSaccharomyces
cerevisiae. Biotechnol Bioeng. 1990;35(4):339-348. doi: 10.1002/bit.260350403.
19. Rix U, Wang C, Chen Y, Lipata FM, Remsing Rix LL, Greenwell LM, Vining LC, Yang
K, Rohr J. The Oxidative Ring Cleavage in Jadomycin Biosynthesis: A Multistep Oxygenation
Cascade in a Biosynthetic Black Box. ChemBioChem. 2005;6(5):838-845. doi:
10.1002/cbic.200400395.

129
20. Perić-Concha N, Borovička B, Long PF, Hranueli D, Waterman PG, Hunter IS. Ablation
of theotcC Gene Encoding a Post-polyketide Hydroxylase from the Oxytetracyline Biosynthetic
Pathway inStreptomyces rimosusResults in Novel Polyketides with Altered Chain Length. J Biol
Chem. 2005;280(45):37455-37460. doi: 10.1074/jbc.M503191200.
21. Kharel MK, Pahari P, Shepherd MD, Tibrewal N, Nybo SE, Shaaban KA, Rohr J.
Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat Prod Rep.
2012;29(2):264-325. doi: 10.1039/c1np00068c.
22. Koskiniemi H, Metsä-Ketelä M, Dobritzsch D, Kallio P, Korhonen H, Mäntsälä P,
Schneider G, Niemi J. Crystal Structures of Two Aromatic Hydroxylases Involved in the Early
Tailoring Steps of Angucycline Biosynthesis. J Mol Biol. 2007;372(3):633-648. doi:
10.1016/j.jmb.2007.06.087.
23. Kallio P, Liu Z, Mäntsälä P, Niemi J, Metsä-Ketelä M. Sequential Action of Two
Flavoenzymes, PgaE and PgaM, in Angucycline Biosynthesis: Chemoenzymatic Synthesis of
Gaudimycin C. Chem Biol. 2008;15(2):157-166. doi: 10.1016/j.chembiol.2007.12.011.
24. Dairi T, Nakano T, Aisaka K, Katsumata R, Hasegawa M. Cloning and Nucleotide
Sequence of the Gene Responsible for Chlorination of Tetracycline. Biosci, Biotechnol, Biochem.
2014;59(6):1099-1106. doi: 10.1271/bbb.59.1099.
25. Kong F, Yuan L, Zheng YF, Chen W. Automatic Liquid Handling for Life Science. Journal
of Laboratory Automation. 2012;17(3):169-185. doi: 10.1177/2211068211435302.
26. Dörr M, Fibinger MPC, Last D, Schmidt S, Santos-Aberturas J, Böttcher D, Hummel A,
Vickers C, Voss M, Bornscheuer UT. Fully automatized high-throughput enzyme library screening
using a robotic platform. Biotechnol Bioeng. 2016;113(7):1421-1432. doi: 10.1002/bit.25925.
27. Oliva B, Gordon G, McNicholas P, Ellestad G, Chopra I. Evidence that tetracycline analogs
whose primary target is not the bacterial ribosome cause lysis of Escherichia coli. Antimicrob
Agents Chemother. 1992;36(5):913-919. doi: 10.1128/aac.36.5.913.
28. Gossen M, Bujard H. Anhydrotetracycline, a novel effector for tetracycline controlled gene
expression systems in eukaryotic cells. Nucleic Acids Res. 1993;21(18):4411-4412. doi:
10.1093/nar/21.18.4411.
29. Asai M. Studies on Pillaromycin A. II. The Structure of Pillaromycinone. Chem Pharm
Bull (Tokyo). 1970;18(9):1706-1712. doi: 10.1248/cpb.18.1706.

130
30. Wells JS, O'Sullivan J, Aklonis C, Ax HA, Tymiak AA, Kirsch DR, Trejo WH, Principe
P. Dactylocyclines, novel tetracycline derivatives produced by a Dactylosporangium sp. I.
Taxonomy, production, isolation and biological activity. J Antibiot. 1992;45(12):1892-1898. doi:
10.7164/antibiotics.45.1892.
31. Tymiak AA, Aklonis C, Bolgar MS, Kahle AD, Kirsch DR, O'Sullivan J, Porubcan MA,
Principe P, Trejo WH. Dactylocyclines: novel tetracycline glycosides active against tetracycline-
resistant bacteria. J Org Chem. 1993;58(3):535-537. doi: 10.1021/jo00055a002.

131
4 Towards the biosynthesis of 6-demethyl-6-
epitracyclines from TAN-1612 in Saccharomyces
cerevisiae
*The contents of this chapter will be published in:
E. Herbst, V. W. Cornish, et al “Biosynthesis of 6-demethyl-6-epitetracycline analogs of TAN-
1612 in Saccharomyces cerevisiae”, in preparation.

132
4.1 Chapter outline
This chapter describes the work towards 6-demethyl-6-epitracycline derivatives of the fungal
anhydrotetracycline TAN-1612 in Saccharomyces cerevisiae. Building on the previous chapters
describing anhydrotetracycline hydroxylations, this chapter begins by exploring the product
formed by coexpressing the TAN-1612 pathway and the bacterial hydroxylase PgaE (Section
4.4.1). The testing of fungal hydroxylase homologs to DacO1, the 6α-hydroxylase of
anhydrodactylocyclinone is then described (Section 4.4.2) followed by the description of a method
for bacterial monooxygenase saturation mutagenesis near the anhydrotetracycline binding site,
with OxyS as an example (Section 4.4.3). Finally, the screening process of this natural diversity
and generated diversity by mutagenesis is described (Section 4.4.4) with future directions for
followup discussed. A key result described in this chapter is the isolation and characterization of
a new major product in a strain coexpressing PgaE and the TAN-1612 pathway that differs from
the TAN-1612 major product in the strain expressing the TAN-1612 pathway without PgaE
(Figure 4-2, Figure 4-3 and Figure 4-5). The followup plan on this result and on the screen for
TAN-1612 hydroxylases is outlined in the Discussion.
4.2 Introduction
TAN-1612, a fungal anhydrotetracycline derivative originally produced by Aspergillus niger, has
been introduced to S. cerevisiae by the Tang laboratory and our laboratory has further improved
its titers in synthetic media, thus allowing the heterologous production of TAN-1612
derivatives.(1) TAN-1612 differs from anhydrotetracycline in five functional groups, an A-ring
methyl ketone instead of the A-ring amide, a 4α-proton instead of the 4α-dimethylamino, a 4a-
hydroxy instead of the 4a-proton, a 6-proton instead of the 6-methyl and an 8-methoxy instead of

133
the 8-proton (Scheme 4-2). The stereochemistry of TAN-1612 at the 4a,12a positions has not yet
been verified, although the stereochemistry of the product of the homologous fungal polyketide
pathway, viridicatumtoxin, has been determined to be the same as anhydrotetracycline in these
positions.(2, 3) As outlined in the introduction chapter, the synthesis of 6-demethyl-6-
epiglycotetracyclines is highly desirable (Section 1.6). TAN-1612 presents a unique scaffold for
the biosynthesizing these derivatives in S. cerevisiae due to its ready biosynthesis in this
heterologous host and its unique functional groups among anhydrotetracycline (Section 1.6.4).
Much of the knowledge generated in the process of anhydrotetracycline hydroxylation by OxyS
and PgaE, as well as DacO1 expression optimization, described in the previous chapters, is directly
relevant to the hydroxylation of TAN-1612 in S. cerevisiae as outlined in this chapter.
4.3 Materials and methods
See Chapter 5 for general methods. See Chapter 3 for general protocol for high throughput assay
of hydroxylation/reduction assay in whole cells in microtiter plates and a general protocol for
western blots.
4.3.1 General protocol for high throughput assay of TAN-1612 hydroxylation
in whole cells in microtiter plates
Fresh colonies of strains harboring a plasmid encoding the TAN-1612 pathway and a hydroxylase
and control strains (EH-5-212-2 encoding PgaE and the TAN-1612 pathway as positive control
and EH-5-212-4 encoding the TAN-1612 pathway and no hydroxylase as a negative control) were
inoculated in 200 μL selective media (UT -) in 96-well plates (Corning 353072) and placed in
shaker overnight. Overnight cultures were used to inoculate 300 μL selective media (UT-) cultures
in deep well plates (Corning P-2ML-SQ-C-S) with an average starting OD of 0.01. The plate was
covered with two layers of SealMate film (Excel Scientific, SM-KIT-BS) and placed in shaker at

134
800 rpm for three nights. 200 μL H2O were then added and after resuspension 20 μL were diluted
into 180 μL of H2O in Corning 3603 plates before UV/VIS spectroscopic measurements. UV/VIS
spectroscopic measurements taken were as follows: (i) absorption spectrum 350 nm – 600 nm, (ii)
emission spectrum 450 nm – 650 nm (λexcitation = 400 nm), (iii) excitation spectrum 300 nm – 450
nm (λemission = 500 nm), (iii) excitation spectrum 300 nm – 450 (λemission = 560 nm). Spectral steps
in the spectra were 50 nm.
4.3.2 Protocol for selecting amino acids for mutagenesis in hypothesized
binding pockets of monooxygenases
The structure of aklavinone-11 hydroxylase with FAD and aklavinone (PDB ID 3IHG) was loaded
on PyMOL. Residues that are within 5 Å from the substrate aklavinone were selected as follows:
PyMOL>hide everything, all
PyMOL>select contacts, (resn VAK and chain A) around 5
Selector: selection "contacts" defined with 88 atoms.
PyMOL>select contacts_res, byres contacts
Selector: selection "contacts_res" defined with 268 atoms.
PyMOL>show sticks, contacts_res
Setting: seq_view set to on.
PyMOL>show sticks, resn vak

135
4.4 Results
4.4.1 Testing PgaE and other bacterial hydroxylases for TAN-1612
hydroxylation
Considering the mass spectrometry support for hydroxylation of anhydrotetracycline by OxyS and
PgaE (Chapter 3, Section 3.4.5) it was logical to test them for TAN-1612 hydroxyaltion (Scheme
4-1). SsfO1 was tested as well because of its efficient expression in S. cerevisiae and 6α-
hydroxylation of its native substrate (Section 3.4.5 and Scheme 4-2).
Scheme 4-1 Biosynthetic plan for TAN-1612 6α-hydroxylation

136
Scheme 4-2 Bacterial monooxygenases and their hypothesized substrates and products
Bacterial flavin-dependent monooxygenases OxyS, DacO1, SsfO1, CtcN and PgaE and their
native substrates. Marked in blue are differences between the native substrate and TAN-1612.
The strain expressing the TAN-1612 pathway without an additional hydroxylase has an excitation
peak at 420 nm for 560 nm emission and an emission peak of 530 nm for 400 nm excitation (Figure
4-1). This strain was confirmed to produce TAN-1612 as the major compound of 400 nm
absorption (Figure 4-5-a and Figure 4-2). The strains expressing OxyS and SsfO1 have similar
excitation and emission peaks, although both are increased for the sample encoding OxyS.
Notably, the strain encoding PgaE has a significantly reduced emission in 560 nm when excited at
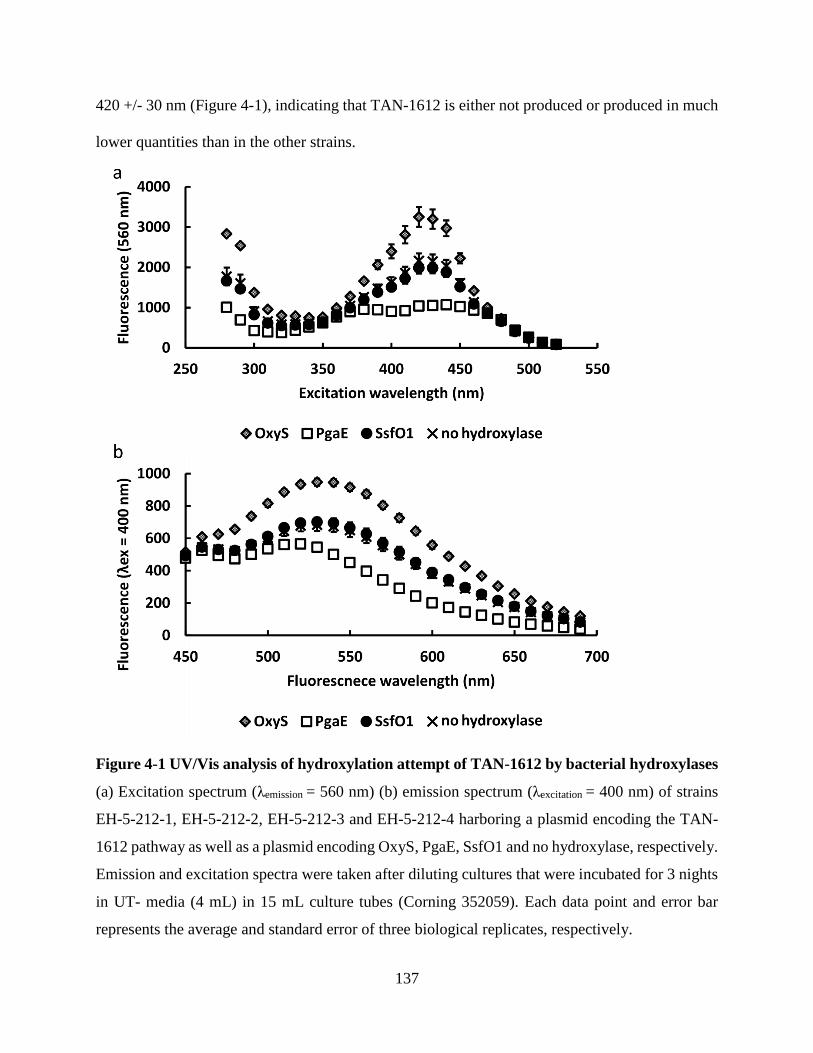
137
420 +/- 30 nm (Figure 4-1), indicating that TAN-1612 is either not produced or produced in much
lower quantities than in the other strains.
Figure 4-1 UV/Vis analysis of hydroxylation attempt of TAN-1612 by bacterial hydroxylases
(a) Excitation spectrum (λemission = 560 nm) (b) emission spectrum (λexcitation = 400 nm) of strains
EH-5-212-1, EH-5-212-2, EH-5-212-3 and EH-5-212-4 harboring a plasmid encoding the TAN-
1612 pathway as well as a plasmid encoding OxyS, PgaE, SsfO1 and no hydroxylase, respectively.
Emission and excitation spectra were taken after diluting cultures that were incubated for 3 nights
in UT- media (4 mL) in 15 mL culture tubes (Corning 352059). Each data point and error bar
represents the average and standard error of three biological replicates, respectively.

138
In order to test which compound or compounds are being produced instead of TAN-1612 in the
strain expressing the TAN-1612 pathway and PgaE, both that strain and the control strain
expressing no hydroxylase were cultured in 500 mL scale to allow purification and further analysis
by mass spectrometry and NMR. Following three nights of culturing each culture was extracted
twice with EtOAc and the combined organic extract was washed with H2O and dried with Na2SO4
with the organic solvent removed under reduced pressure. The extract was then purified by
semiprepative HPLC prior to NMR and mass spectrometry analysis.
As the absorption chromatogram of the semipreparative HPLC separation shows, the main product
with regards to both 400 nm absorption and 254 nm absorption is modified in the +PgaE sample
relative to the -PgaE control. While the main product in the -PgaE control elutes after 37 minutes,
the main product in the +PgaE sample elutes after 34 minutes (Figure 4-2). Interestingly the -PgaE
sample has a minor product eluting after 34 minutes, which could be the same product as the main
product of the +PgaE sample, or a different one.

139
Figure 4-2 UV/Vis chromatogram of the HPLC separation of extracts from +/- PgaE strains
encoding the TAN-1612 pathway.
Strains EH-5-212-2 and EH-5-212-4 harboring a plasmid encoding the TAN-1612 pathway as well
as a plasmid encoding PgaE and no hydroxylase, respectively are indicated as +PgaE and -PgaE,
respectively. Both strains were cultured in 500 mL scale for three nights. Prior to HPLC separation
the culture was extracted twice with EtOAc and the combined organic extract was washed with
H2O and dried with Na2SO4, with the organic solvent removed under reduced pressure. Absorption
chromatograms are shown for 254 and 400 nm for an HPLC separation method of 10:90 to 90:10
MeCN in 99.9% H2O/0.1% TFA over 1 h.

140
The main product of each HPLC separation was isolated and analyzed by NMR and mass
spectrometry. The mass spectrometry analysis of the -PgaE compound clearly supports that TAN-
1612 was isolated (MS (ES+): m/z calc’d for C21H19O9+, 415.1029; found 415.1026 [M + H]+
(Figure 4-3-a) and the NMR spectrum matches the published TAN-1612 spectrum (Figure 4-5-
a).(1) For the +PgaE sample, the mass spectrum shows four major peaks for an elution peak with
absorbance at both 254 nm and 400 nm: 593.1218, 298.0743, 279.0966 and 278.0483 (Figure 4-3-
b). The NMR spectrum of the sample does not show the methoxy and methyl ketone protons of
shifts 3.89 and 2.58 ppm as well as the two aromatic protons of chemical shifts 7.05 and 6.45 ppm
and does show additional 7 aromatic protons in their stead (Figure 4-5-b). The possible identity of
the newly isolated major compound of the +PgaE +TAN-1612 pathway sample is discussed in
Section 4.5.1.

141

142
Figure 4-3 Mass spectrometry analysis of isolate from +/-PgaE strain encoding the TAN-
1612 pathway.
Liquid chromatography mass spectrometry analyses for isolates from strains EH-5-212-2 and EH-
5-212-4 harboring a plasmid encoding the TAN-1612 pathway as well as a plasmid encoding PgaE
and no hydroxylase, respectively are shown in (b) and (a), respectively. Both strains were cultured
in 500 mL scale for three nights. Prior to HPLC separation the culture was extracted twice with
EtOAc and the combined organic extract was washed with H2O and dried with Na2SO4, with the
organic solvent removed under reduced pressure. The LCMS separation method is 5:95 to 95:5
MeCN in 99.9% H2O/0.1% formic acid over 2 min.
Figure 4-4 MSMS analysis of isolate from +PgaE strain encoding the TAN-1612 pathway.
Liquid chromatography MSMS analyses for isolate from strain EH-5-212-2 harboring a plasmid
encoding the TAN-1612 pathway as well as a plasmid encoding PgaE with ion selection at mass
593. The LCMSMS separation method is 5:95 to 95:5 MeCN in 99.9% H2O/0.1% formic acid over
2 min.

143
Figure 4-5 1H-NMR spectra of isolates from +/-PgaE strains encoding the TAN-1612
pathway.

144
1H-NMR spectra of isolates from strains EH-5-212-2 and EH-5-212-4 harboring a plasmid
encoding the TAN-1612 pathway as well as a plasmid encoding PgaE and no hydroxylase, are
shown in (b) and (a), respectively. Both strains were cultured in 500 mL scale for three nights.
Prior to HPLC separation the culture was extracted twice with EtOAc and the combined organic
extract was washed with H2O and dried with Na2SO4, with the organic solvent removed under
reduced pressure. NMR spectra shown are in MeOD-d4 for the major product of each strain
according to HPLC chromatogram (Figure 4-2). 1H NMR (for (b), δ>4 ppm, 500 MHz, MeOD-
d4): δ 7.61 (dd, J = 8.0, 1.0 ,1H), 7.47 (d, J = 8.6, 2H), 7.22 (dd, J = 8.0, 1.1, 1H), 6.88 (dd, J =
8.6, 2H), 6.85 (dd, J = 8.0, 1H), 6.72 (s, 1H).
Figure 4-6 COSY NMR spectrum of isolate from +PgaE strain encoding the TAN-1612
pathway.
Strain EH-5-212-2 harboring a plasmid encoding the TAN-1612 pathway as well as a plasmid
encoding PgaE. Both strains were cultured in 500 mL scale for three nights. Prior to HPLC
separation the culture was extracted twice with EtOAc and the combined organic extract was

145
washed with H2O and dried with Na2SO4, with the organic solvent was removed under reduced
pressure. NMR spectra shown is in MeOD-d4 for the major product according to the HPLC
chromatogram (Figure 4-2).
4.4.2 Searching the fungal hydroxylase space for 6α-hydroxylation of TAN-
1612
Given the expression challenges with the bacterial monooxygenase DacO1 (Section 3.4.1) it was
logical to test fungal monooxygenases as well. Fungal monooxygenases were chosen based on
homology to DacO1 and/or based on the presence of anhydrotetracycline monooxygenase in their
CDS product name. Table 4-1 shows the first twelve such enzymes attempted. Western blot
analysis revealed that expression challenges could be prevalent with fungal hydroxylases as well,
with only 25% of the fungal hydroxylases assayed displaying a band at the expected size (Figure
4-7). The strains that did not indicate any protein expression at the expected size encoded fungal
monooxygenases of entries 1, 2, 4, 5, 6, and 11 from Table 4-1. The two strains that did exhibit
protein expression at the expected size encoded fungal hydroxylases entries 8 and 12 from Table
4-1 (Figure 4-7). For the small sample tested, the two codon optimized fungal monooxygenases
did not present a band of the expected size in S. cerevisiae. Two of the six non-codon optimized
did presented such a band. The two strains that exhibited a protein of the expected size had a higher
identity %, positives % and query cover with both DacO1 and OxyS than the six strains that did
not exhibit a band of the expected size but not necessarily to a statistically significant degree
(Figure 4-7, Table 4-1 and data not shown).

146
Table 4-1 Fungal monooxygenases biomined for TAN-1612 6α-hydroxylation in S. cerevisiae
Entry Gene product
label
Source organism GenBank
entry
Codon
optimi
zed
Identity
(%) /
positives
(%) /
cover
with
DacO1a
Identity
(%) /
positives
(%) /
cover
with
OxySb
1 Anhydrotetracy
cline
monooxygenas
e
Valsa mali strain
03-8
CM00310
4.1
Y 30/44/266 25/39/388
2 Anhydrotetracy
cline
monooxygenas
e
Penicillium
subrubescens
strain CBS 132785
MNBE01
000128.1
N 32/43/533 35/46/541
3 Anhydrotetracy
cline
monooxygenas
e
Cercospora
beticola
NC_036772
NC_0367
72.1
N 30/41/510 34/45/537
4 related to
tetracenomycin
polyketide
synthesis
hydroxylase
tcmG
Phialocephala
subalpina strain
UAMH 11012
FJOG010
00008.1
N 31/46/370 30/43/547
5 Anhydrotetracy
cline
monooxygenas
e
Golovinomyces
cichoracearum
MCBR01
007648.1
N 28/43/398 30/40/564
6 Anhydrotetracy
cline
monooxygenas
e
Valsa mali var.
pyri strain
SXYL134
KN71467
1.1
N 23/37/345 24/38/405
7 tetracycline 6-
hydroxylase
protein
Aureobasidium
pullulans var.
pullulans
KL584983
.1
N 26/40/416 25/39/455
8 pentachlorophe
nol 4-
monooxygenas
e
Hypoxylon sp.
EC38
KZ111246
.1
N 34/45/520 38/49/531
9 probable 2-
polyprenyl-6-
methoxyphenol
Phialocephala
subalpina strain
UAMH 11012
FJOG010
00031.1
N 32/46/505 39/51/506
147
hydroxylase
and related
FAD-
dependent
oxidoreductase
s
10 2-polyprenyl-6-
methoxyphenol
hydroxylase
Aspergillus oryzae
100-8
AMCJ010
00140.1
N 32/47/501 34/50/503
11 pentachlorophe
nol 4-
monooxygenas
e
Colletotrichum
orbiculare MAFF
240422
KB72597
6.1
Y 34/44/531 34/47/531
12 unnamed
protein product;
2-polyprenyl-6-
methoxyphenol
hydroxylase
and related
FAD-
dependent
oxidoreductase
s"
Aspergillus oryzae
RIB40
AP007159
.1
N 32/45/521 34/47/529
aDacO1 [Dactylosporangium sp. SC14051] GenBank: AFU65892.1
b OxyS [Streptomyces rimosus] GenBank AAZ78342.1

148
Figure 4-7 Western blot analysis of fungal hydroxylase expression in BJ5464-NpgA
Strain cultures were lysed with Y-PER and labeled with Monoclonal ANTI-FLAG HRP antibody.
The size (kDa) indicated below the gels is the expected size of the protein based on the amino acid
sequence. Strains EH-5-227-1 through EH-5-227-9 encode Fungal monooxygenase entry 6, 5, 1,
4, 2, 2, 8, 11, 12, from Table 4-1, respectively.

149
4.4.3 Evolving monooxygenases for 6α-hydroxylation of anhydrotetracyclines
– OxyS example
Considering the effective hydroxylation of anhydrotetracycline in yeast by OxyS (Chapter 2), its
apparent lack of hydroxylation activity on TAN-1612 (Section 4.4.1) and an effective UV/Vis
assay for anhydrotetracyclines hydroxylation (Sections 3.4.3-3.4.5), directed evolution of OxyS to
accept TAN-1612 as a substrate seemed logical. In addition, OxyS structure was previously probed
by X-ray crystallography, as well as the structure of a homologous protein, Aklavinone-11-
Hydroxylase, along with its native substrate, aklavinone (42% homology, PDB ID 4K2X and
3IHG, respectively).(4, 5) Thus, the substrate binding pocket of OxyS could be identified with
some confidence and mutated accordingly.
In order to perform saturation mutagenesis on residues that are in proximity to the hypothesized
substrate binding pocket of OxyS, residues of Aklavinone-11-Hydroxylase that are within 5 Å of
aklavinone were listed (Section 4.3.2). Then the homologous residues in OxyS were noted as well
(Table 4-2). Some of these homologous residues in OxyS indeed sit in proximity to a cavity that
is a continuation of the cavity in which FAD is situated in the OxyS crystal structure (Figure 4-8).
Table 4-2 Residues chosen for mutagenesis in OxyS
Residue
number
in 3IHG
Residue context in
3IHG
Residue in
OxyS
Residue context
in OxyS
Distance
from VAK
(Å)
1 R45 PYPRAAG K42 FSKALGV 4-5
2 A47 PRAGQN A43 SKALGVH <4
3 G48 AAGQNPR G45 KALGVHAR 4-5
4 I72 ADDIRG - - <4
5 F79 QGDFVIR - - <4
6 I81 QGDFVIR - - 4-5
7 M101 DDMVAA - - 4-5
8 W113 PAGWAM L95 PYALFV 4-5
9 M115 PAGWAMLSQD F96 PYAL-FVPQV <4
10 M201 LTHMVGV M176 --HMFAV <4
11 W221 GTTGWYYL W211 DLRAWFAAFP <4

150
12 Y223 TGWYYLH F212 DLRAWFAAFPL <4
13 T232 PEFKGTFGPTDRP T225X ATVA <4
14 P235 TFGPTDRP F228 TVAFFDRP 4-5
15 F245 RHTLFVEYDPDEG V240X RRAPVTEED <4
16 W287 VDIQGWEMAARIA - - <4
17 M289 WEMAARIA - - 4-5
18 P314 VTPPTGGMSG P295X IHLPAGGQGL <4
19 T315 VTPPTGGMSG A296X IHLPAGGQGL <4
20 G316 VTPPTGGMSG G297X IHLPAGGQGL <4
21 G317 VTPPTGGMSG G298X IHLPAGGQGL <4
22 R373 AIYAQRMAP P357 LIDPDPRYE 4-5
23 M374 AIYAQRMAP R358 LIDPDPRYE 4-5
24 Y387 SVGYPET V372 LLHVPET 4-5

151
Figure 4-8 PyMOL illustrations of amino acids in proximity to the anhydrotetracycline
substrate in aklavinone-11-Hydroxylase and in OxyS
PyMOL illustration of (a) alkavinone and FAD (stick representation, carbons colored white)
surrounded by amino acids of aklavinone-11-Hydroxylase within 5 Å of alkavinone (stick
representation, carbons colored green, PDB ID 3IHG) and (b) FAD (carbons colored white)
surrounded by OxyS (surface representation, carbons colored green, PDB ID 4K2X) and its amino

152
acids that are homologous to those of aklavinone-11-Hydroxylase amino acids that are within 5 Å
of alkavinone in the structure shown in (a).
4.4.4 Library screening for TAN-1612 hydroxylation in S. cerevisiae
Following the assembly of the libraries of fungal monooxygenases (Section 4.4.2) and OxyS
saturation mutagenesis (Section 4.4.3) the libraries were screened for TAN-1612 hydroxylation
using a variant of the microtiter plate assay for anhydotetracycline hydroxylation (Section 3.4.3
and 4.3.1). As negative control a strain encoding the TAN-1612 pathway without an additional
hydroxylase was used. As positive control a strain encoding the TAN-1612 pathway and PgaE was
used, as that strain was already shown to produce an alternative major product instead of TAN-
1612 (Figure 4-2).
Given that the positive control strain expressing PgaE showed a significantly reduced emission at
560 nm upon 400 and 450 nm excitation, colonies were plotted according to their absorbance at
these wavelengths and normalized to OD6oo: (400 + 450 nm) / 600 nm. When colonies of each 96-
well plate was ranked according to this absorption criteria the PgaE positive control was ranked
on average 83.6 (out of 96) with a standard deviation of 10.0 (Table 4-3 and Figure 4-9). A low
rank of PgaE is expected in the likely scenario that most strains screened do not encode an efficient
TAN-1612 hydroxylase. Wells 84 and 96 containing no cells consistently ranked the lowest in the
measurement of (400 + 450)/600 nm (Figure 4-9).
Table 4-3 Rank of PgaE positive control in screen for TAN-1612 hydroxylase
Plate Strain Absorbance C1
(400 + 450)/600
Absorbance C2
(400 + 450)/600
a EH-5-217-2 89 59
b EH-5-217-2 91 92
c EH-5-217-2 94 68
d EH-5-217-4 88 79
e EH-5-217-4 78 85
f EH-5-217-4 93 87

153
g EH-5-217-1 and
EH-217-3
81 87
Average (all) 83.6
Standard
deviation (all)
10.0
Figure 4-9 Library screening for TAN-1612 hydroxylation in S. cerevisiae
Y axes shows the sum of absorption at 400 and 450 nm divided by absorption at 600 nm while the
X axes show the colony number. Plates a, b and c are screens of strain library EH-5-217-2. Plates
d, e and f are screens of strain library EH-5-217-4. Both strain libraries EH-5-217-2 and EH-5-
217-4 encode the same OxyS saturation mutagenesis library and differ in the background TAN-
1612 producing strain. Plate g contains strain libraries EH-5-217-1 and EH-5-217-3 encoding
bacterial and fungal hydroxylases, including selected DacO1 fusion proteins. Strains EH-5-212-1,

154
EH-5-212-2 and EH-5-212-4 encoding the TAN-1612 pathway along with OxyS, PgaE and no
hydroxylase, are labeled in blue, red and white, respectively.
4.5 Discussion
4.5.1 Testing PgaE and other bacterial hydroxylases for TAN-1612
hydroxylation
Despite the structural dissimilarities between UWM6, the native susbstrate of PgaE and TAN-
1612 (Scheme 4-2) it is PgaE and not OxyS or SsfO1 whose coexpression with the TAN-1612
pathway has led to the production of a different major product of 400 and 254 nm absorption
instead of TAN-1612. Both the chromatograms for the HPLC separation of the +PgaE and -PgaE
samples, as well as the excitation and emission spectra support that PgaE is converting TAN-1612
or an intermediate in its biosynthesis (Figure 4-2, Figure 4-1). It would therefore be of interest to
test other enzymes involved in the 12-position hydroxylation of UWM6, such as CabE, LanE,
UrdE and GilOI.(6, 7)
The protons of chemical shifts 7.47 and 6.88 ppm with coupling constant of 8.6 Hz are likely two
pairs of equivalent aromatic protons at ortho to each other and they are shown to interact in the
COSY spectrum as well (Figure 4-5 and Figure 4-6); the protons of chemical shifts 7.61, 7.22 and
6.85 ppm are likely three adjacent protons in one aromatic ring that has three other substituents,
with the protons of chemical shifts 7.22 and 7.61 ppm likely meta to each other. Interactions
between the protons of chemical shifts 7.61 and 6.85 as well as 7.22 and 6.85 ppm are also readily
observed in the COSY spectrum and the coupling constants, characteristic of ortho interactions,
are 8.0 Hz each (Figure 4-5 and Figure 4-6). The singlet at 6.72 ppm is likely a single proton in an
aromatic ring, possibly the only remaining proton in the D-ring of the substituted TAN-1612 or its

155
intermediate (Figure 4-5). The absence of the methoxy shift in the new major product at 3.89 ppm
(Figure 4-5) can indicate that PgaE derivatization occurs prior to AdaD methylation, which is
assumed to be the last step in TAN-1612 biosynthesis.(1) It could be that following hydroxylation
by PgaE and a potential further derivatization by a yeast endogenous molecule, substrate affinity
of AdaD to the modified product is too low to allow methylation. Examples for the moieties that
might be part of the newly formed major compound are shown in Figure 4-10. Importantly, not all
moieties are necessarily part of the same molecule, as can be verified by larger scale fermentation
followed by further purification and NMR analysis. The aromatic moiety of Figure 4-10-c could
theoretically represent a doubly hydroxylated TAN-1612 or any of the TAN-1612 intermediates
of Figure 4-11. However such an assignment is questionable, given the lack of the protons of
chemical shift 3.89 and 2.58 ppm corresponding to the methoxy and methyl ketone at the NMR
spectrum of the newly formed major compound (compare Figure 4-5-a and Figure 4-5-b).
Figure 4-10 Possible moieties in the product isolated from +PgaE strain coexpressing the
TAN-1612 bioysnthetic pathway
Either of the two aromatic moieties of (a) could be responsible for generating the protons of
chemical shifts 7.61, 7.22 and 6.85 ppm, the aromatic moiety of (b) could be responsible for
generating protons of chemical shifts 7.47 and 6.88 ppm and either of the aromatic moieties of (c)
could be responsible for generating a proton of chemical shifts 6.72 ppm in the 1H-NMR spectrum
of the product isolated from +PgaE strain coexpressing the TAN-1612 bioysnthetic pathway
(Figure 4-5). The squiggly lines represent any non-proton substituent.

156
Figure 4-11 Possible intermediates in the biosynthesis of TAN-1612
TAN-1612 (1) and its intermediates 2-10 that can occur in the case of exclusion or disfunction of
AdaB, AdaC or AdaD, the three post PKS biosynthetic enzymes of the TAN-1612 biosynthetic
pathway, or combinations thereof.(1)

157
The mass spectrum of the isolated compound from the +PgaE culture has major peaks with m/z
values of 593.1218, 298.0743, 279.0966 and 278.0483 (Figure 4-3). The 593.1218 ion has an
elution peak after 1.60 and after 1.47 min, but an absorption peak at 254 nm and 400 +/- 60 nm is
noted only slightly before the 1.60 min (Figure 4-3) and not slightly before the 1.47 peak (the ions
are first detected at the diode array before mass spectrometry scanning). Assuming that the two
593.1218 ions could be related stereoisomers of the same exact mass, it is perhaps doubtful
whether any of them corresponds to a derivative of the TAN-1612 intermediates shown in Figure
4-11. This is because at any stereoisomeric form such compounds would be expected to have
absorption at 400 and/or 254.
The 298.0743 and 278.0483 ions are four protons and one oxygen different in their m/z values.
Possibly a molecule of H2O + oxidation / reduction apart. In the MSMS spectrum for the ions of
m/z 593 both the ions 278.0452 and 296.0551 appear (Figure 4-4), the former being the mass of
H2O less than the latter and the latter being H2 away from 298.0743 in its mass. Also of note is
that the 593.1218 ion could be a result of a dimer of the 296.0551 ion plus the mass of a proton.
Thus, the 593.1218, 298.0743, 296.0551 and 278.0483 ions could correspond to chemical formulas
of C32H21N2O10+, C16H12NO5
+, C16H10NO5+, C16H8NO4
+ as they differ in 0.0022, 0.0028, 0.0008
and 0.0030 u, respectively, from the expected masses of these ions. These could be ions of some
xanthurenic acid derivatives that PgaE might or might not have a contribution to their biosynthesis
(Figure 4-12). While the derivatives of xanthurenic acid shown in Figure 4-12 are not known in
yeast, xanthurenic acid is a known yeast metabolite.(8) This proposal of structure or any other
structure that involves only yeast metabolites and their derivatives could be explaining the mass
and NMR spectra of the isolated major compound in the +PgaE sample (Figure 4-3, Figure 4-4,
Figure 4-5 and Figure 4-6). However, a major compound that does not involve a derivative of

158
TAN-1612 or its intermediate still does not directly explain how the expression of PgaE leads to
the reduction in the TAN-1612 absorbance and fluorescence (Figure 4-1 and Figure 4-2).
Figure 4-12 xanthurenic acid and its derivatives that could correspond to experimental mass
and NMR spectra
The xanthurenic acid derivatives presented in this figure have [M + H]+ values of 593.1196,
298.0715 and 296.0559 that are 0.0022, 0.0028 and 0.0008 amu from the experimental values
detected 593.1218, 298.0743 and 296.0551 (Figure 4-3 and Figure 4-4). In addition, the protons
of a molecule such as the one shown with chemical formula C16H11NO5 can correspond to the 6
proton types identified in the NMR spectrum (Figure 4-5 and Figure 4-6) and identified to
potentially belong to the moieties described in Figure 4-10.
In order to determine the structure of the newly formed major compound with regards to 254 nm
and 400 nm absorption in the +PgaE sample coexpressing the TAN-1612 pathway, the reaction
would be repeated in larger scale to isolate enough of the newly formed compound for HSBC and
HMBC proton coupled carbon NMR spectra. Furthermore, the minor compound eluting at 34 min
for the -PgaE sample will be isolated and analyzed as well to identify whether it is identical or at
all related to the product isolated from the +PgaE sample that elutes at 34 min (Figure 4-2). This

159
characterization can assist in understanding the role PgaE has on the biosynthesis of the newly
formed major compound with respect to 400 nm and 254 nm absorption in the +PgaE sample
(Figure 4-2). Another source of information would be the culturing and isolation of another control
strain encoding PgaE but not encoding the TAN-1612 pathway and examining whether such strain
displays a similar major peak in the HPLC chromatogram of 254 and 400 nm absorption as the
strain that encodes the TAN-1612 pathway.
4.5.2 Searching the fungal hydroxylase space for 6α-hydroxylation of TAN-
1612
Importantly, the library generation approaches used to generate 6α-hydroxylases of
anhydrotetracycline (Subsections 3.4.2 and 3.4.4) could be used for generating TAN-1612
hydroxylases (Subsections 4.4.2 and 4.4.3) and vice versa. The expression analysis of fungal
monooxygenases showed that while branching towards fungal monooxygenases might be a useful
strategy to find 6α-hydroxylases and specifically 6α-hydroxylases of TAN-1612, the fungal
monooxygenases do not necessarily present an advantage over their bacterial counterparts as far
as expression challenges in S. cerevisiae are concerned (Figure 4-7 and Chapter 3).
4.5.3 Evolving monooxygenases for 6α-hydroxylation of anhydrotetracyclines
Amino acids in OxyS were chosen for mutagenesis based on homology to amino acids in the
proximity of the substrate in the crystal structure of RdmE and this approach could be implemented
in other monooxygenases, such as PgaE or expression optimized DacO1 (Chapter 3). Importantly,
implementing this approach does not require an existing crystal structure of the monooxygenase
of interest. Also, as seen in the case of OxyS, at least some of the amino acids chosen for
mutagenesis according to this strategy actually sit in proximity to the anhydrotetracycline cavity
of OxyS (Figure 4-8). However, in the case of OxyS, an approach that uses the published crystal

160
structure of OxyS to choose additional amino acids for mutagenesis or to avoid mutating some of
the amino acids that are not in proximity to the cavity might have been preferred. Further
mutagenesis is being performed to explore the effect of additional mutations of OxyS in positions
that are homologous to amino acids in RdmE structure that are up to 8 Å from aklavinone in 3IHG.
Finally, similar followup mutagenesis studies on stably expressed DacO1 fussion proteins, as well
as PgaE will be directly relevant for testing 6α-hydroxylation of TAN-1612. Specifically for PgaE,
much structural information is available to guide the amino acid choice for saturation mutagenesis
from its crystal structure and from biochemical analyses.(6, 9-11)
4.5.4 Library screening for TAN-1612 hydroxylation in S. cerevisiae
In the screen for TAN-1612 hydroxylation, a variant of the microtiter plate assay for
anhydrotetracycline hydroxylation described in Chapter 3 was used. Chapter 3 also discusses non-
microtiter plate avenues for the anhydrotetracycline hydroxylation (Section 3.5.3) and these would
be especially interesting to examine in the context of TAN-1612 hydroxylation, as TAN-1612 does
not need to be exogenously supplied to the cells.
Similarly to the followup on the +PgaE +TAN-1612 encoding strain (Sections 4.4.1 and 4.5.1),
strains with a lower (400 + 450)/600 nm absorption than the PgaE positive control (Figure 4-9)
will be assayed for TAN-1612 hydroxylation by UV/Vis and mass spectrometry. If data supports
production of an alternative major product to TAN-1612 these strains will be cultured in larger
scale to allow purification and analysis of the product by NMR. If data does not support TAN-
1612 hydroxylation in any of the assayed strains, strain selection for further assay will be attempted
based on other criteria such as 550 + 600 – 450 nm emission upon 400 nm excitation or 400 + 450
– 350 nm excitation for 560 nm emission. These criteria are based on the reduced emission and
excitation peaks observed for the PgaE encoding positive control strain (Figure 4-1).

161
4.6 Conclusion
This chapter describes the progress towards TAN-1612 hydroxylation in S. cerevisiae, the first
step in the synthesis of 6-demethyl-6-epitetracycline from TAN-1612 in S. cerevisiae. This chapter
shows that a strain expressing both PgaE and the TAN-1612 pathway produces a major compound
with regards to 400 nm and 254 nm absorption that differs from TAN-1612 (Sections 4.4.1 and
4.5.1, Figure 4-1, Figure 4-2, Figure 4-3 and Figure 4-5). Future work towards TAN-1612
hydroxylation in S. cerevisiae should include a larger scale culturing of the +PgaE +TAN-1612
pathway strain to enable followup NMR experiments. In addition, the minor compound eluting at
34 min for the -PgaE sample (Figure 4-2) will be isolated and analyzed to verify whether it is
identical or at all related to the product isolated from the +PgaE sample that elutes at 34 min.
Another control strain encoding PgaE but not encoding the TAN-1612 pathway will be cultured
and examined to discover whether it displays a similar major peak in the HPLC chromatogram to
that of the +PgaE +TAN-1612 pathway strain.
This chapter also describes a hydroxylase harvesting and mutagenesis strategy as well as a UV/Vis
assay as a platform for identifying TAN-1612 hydroxylation. This platform was applied to the
screening for TAN-1612 hydroxylation (Section 4.4.4) by OxyS mutants (Section 4.4.3), DacO1
fusion proteins (Sections 3.4.1 and 3.4.5), and other hydroxylases from bacterial (Sections 3.4.2
and 3.4.5) and fungal sources (Section 4.4.2). Followup culturing and spectroscopic analysis will
be done on strains coexpressing the TAN-1612 pathway and one of the above mentioned
hydroyxlases, identified as potentially hydroxylating TAN-1612 in the UV/Vis assay (Figure 4-9).
In addition to the followup on PgaE and on the other hydroxylases described in this chapter, other
enzymes involved in the 12-position hydroxylation of UWM6, such as CabE, LanE, UrdE and
GilOI should also be tested for TAN-1612 hydroxylation.(6, 7) With a mutagenesis strategy that

162
does not require a crystal structure that could be used for DacO1 (Figure 4-8) and with the
additional benefit of the PgaE crystal structure,(9) followup mutagenesis studies such as shown on
OxyS (Section 4.4.3) and on DacO1 (Section 3.4.4) on stably expressed DacO1 fusion proteins
(Section 3.4.5), as well as on PgaE could prove beneficial for 6α-hydroxylation of TAN-1612.
Identification and overexpression of a reductase responsible for 5a(11a)-dehydrotetracycline
reduction in S. cerevisiae (Section 2.4.2 and 2.5.2) will be potentially useful for the reduction of
the hydroxylation product of TAN-1612 as would be other efforts in the reduction of 5a(11a)-
dehydrotetracycline in S. cerevisiae (Section 3.4.5 and 3.5.5). Finally, the anhydrotetracycline
hydroxylations described in this chapter and in Chapters 2 and 3 relies heavily on the UV/Vis assay
developed for the hydroxylation of anhydrotetracyclines (Section 3.4.3, 3.4.5, and 4.4.4). More
broadly, high-throughput assays are of very high importance to small molecule production in
microorganisms because of the inability to predict in advanced the successful strain modifications
needed in the complex environment of the cell.(12) TAN-1612 and anhydrotetracycline
hydroxylation belong to a special group of biochemical transformations that are easy to assay in
high throughput because of the inherent change in the chromophore of the CD ring (Scheme 4-2).
However, the next steps for 6-demtehyl-6-epiglycotetracyclines from TAN-1612 will require a
general, readily implemented, high-throughput assay for tetracycline biosynthesis in yeast. The
development and characterization of such an assay is described in Chapter 5.

163
4.7 Appendix
Table 4-4 Strains used in this study
Strain Genotype Source/Reference
BJ5464-
NpgA
MATα ura3-52 his3-Δ200 leu2-Δ1
trp1 pep4::HIS3 δ:: pADH2-npgA-tADH2 prb1Δ1.6R
can1 GAL
a / (13)
PBA-482 BJ5464 ΔPEP4::pTDH3-Sb-NpgA-tENO2
ΔPRB1::pTDH3-Sb-NpgA-tENO2
P. A. Baldera-
Aguayob
PBA-1277 BJ5464 ΔPEP4::pHSP26-Sb-NpgA-tENO2
ΔPRB1::pHSP26-Sb-NpgA-tENO2
P. A. Baldera-
Aguayob
PBA-1333 PBA482 pPBA-L2BWG9 P. A. Baldera-
Aguayob
PBA-1337 PBA1277 pPBA-L6BWG9 P. A. Baldera-
Aguayob
EH-5-212-1 PBA-1337 AL-1-101-C10 This Study
EH-5-212-2 PBA-1337 AL-234-C-C1 This Study
EH-5-212-3 PBA-1337 AL-239-B This Study
EH-5-212-4 PBA-1337 pSP-G1 (Addgene 64736) This Study
EH-5-217-1 PBA-1337 Plasmid Libraries A + C This Studyc
EH-5-217-2 PBA-1337 Plasmid Library D This Studyc
EH-5-217-3 PBA-1333 Plasmid Libraries B + C This Studyc
EH-5-217-4 PBA-1333 Plasmid Library D This Studyc
EH-5-227-11 BJ5464-NpgA AL-2-72-B-C6 This Study
EH-5-227-12 BJ5464-NpgA AL-2-72-D-C5 This Study
EH-5-227-13 BJ5464-NpgA AL-2-72-F-C4 This Study
EH-5-227-14 BJ5464-NpgA AL-2-79-J-C4 This Study
EH-5-227-15 BJ5464-NpgA AL-2-79-K-C5 This Study
EH-5-227-16 BJ5464-NpgA AL-2-79-L-C6 This Study
EH-5-227-17 BJ5464-NpgA AL-2-86-D-C5 This Study
EH-5-227-18 BJ5464-NpgA AL-2-86-I-C5 This Study
EH-5-227-19 BJ5464-NpgA AL-2-86-A-C1 This Study a Y. Tang, University of California, Los Angeles, Department of Chemical and Biomolecular
Engineering & Department of Chemistry and Biochemistry, Department of Bioengineering
b Integrated Program in Cellular, Molecular and Biomedical Studies, Cornish Laboratory,
Columbia University, New York, NY 10032, USA
c Plasmid libraries compositions are described below

164
Table 4-5 Plasmids used in this study
Description Source/Reference
pSP-G1 pTEF1-FLAG-tADH1, pPGK1-MYC-tCYC1, 2 μ, Ura Addgene 64736
AL-1-101-C10 pTEF1-oxyS-tADH1 in pSP-G1 Chapter 2
AL-1-234-C-C1 pTEF1-pgaE-tADH1 in pSP-G1 JCAT codon opt. Chapter 3
AL-1-239-B-C3 pTEF1-ssfO1-tADH1 in pSP-G1 JCAT codon opt. Chapter 3
AL-2-72-B-C6 pTEF1-Fungal-monooxygenase-6-tADH1 in pSP-G1c This Study
AL-2-72-D-C5 pTEF1-Fungal-monooxygenase-5-tADH1 in pSP-G1c This Study
AL-2-72-F-C4 pTEF1-Fungal-monooxygenase-1-tADH1 in pSP-G1c This Study
AL-2-79-J-C4 pTEF1-Fungal-monooxygenase-4-tADH1 in pSP-G1c This Study
AL-2-79-K-C5 pTEF1-Fungal-monooxygenase-2-tADH1 in pSP-G1c This Study
AL-2-79-L-C6 pTEF1-Fungal-monooxygenase-2-tADH1 in pSP-G1c This Study
AL-2-86-D-C5 pTEF1-Fungal-monooxygenase-8-tADH1 in pSP-G1c This Study
AL-2-86-I-C5 pTEF1-Fungal-monooxygenase-11-tADH1 in pSP-G1c This Study
AL-2-86-A-C1 pTEF1-Fungal-monooxygenase-12-tADH1 in pSP-G1c This Study
EH-5-49-C pTEF1-GAL1-dacO1-tADH1 in pSP-G1 Chapter 3
EH-5-80-6 pTEF1-UBI4-IFNG-dacO1-tADH1 in pSP-G1 Chapter 3
EH-5-80-7 pTEF1-IFNG-dacO1-tADH1 in pSP-G1 Chapter 3
EH-5-80-10 pTEF1-oxyS-dacO1-37-tADH1 in pSP-G1 Chapter 3
EH-5-80-11 pTEF1-oxyS-dacO1-87-tADH1 in pSP-G1 Chapter 3
pAV124 Trp1, 2 μ acceptor vector for yeast golden gate Addgene
63190/(14)
pPBA-AV124 Modified Trp1, 2 μ acceptor vector for yeast golden gate P. A. Baldera-
Aguayoa
pPBA-
L2BWG9
pHSP26-adaA-tPGK1_pPGK1-adaB-tADH1_pTEF1-
adaC_tENO1_pTDH3-adaD_-tRPL15A on pPBA-
AV124
P. A. Baldera-
Aguayoa
pPBA-
L6BWG9
pHSP26-adaA-tPGK1_pHSP26-adaB-tADH1_pTEF1-
adaC_-tENO1_pTDH3-adaD-tRPL15A on pPBA-
AV124
P. A. Baldera-
Aguayoa
a Integrated Program in Cellular, Molecular and Biomedical Studies, Cornish Laboratory,
Columbia University, New York, NY 10032, USA
b Fungal-monooxygenase-X refers to the fungal monooxygenase from Table 4-1 entry X. Thus,
for example Fungal-monooxygenase-2 refers to the fungal monooxgenase from Table 4-1 entry 2.

165
Plasmid libraries used for making strains EH-5-217-1 through EH-5-217-4
Table 4-6 Plasmid Library A – Strain EH-5-217-1
Library component
no.
Plasmid Plasmid common
name
Colony #
1 EH-5-49-C Gal1-DacO1 1
2 EH-5-80-6 9 - ubiIFN H
3 EH-5-80-7 9 - IFN H
4 EH-5-80-10 11-OxyS-DacO1-37 G
5 EH-5-80-11 11-OxyS-DacO1-87 H
Table 4-7 Plasmid Library B – Strain EH-5-217-3
Library component
no.
Plasmid Plasmid common
name
Colony #
1 EH-5-49-C Gal1-DacO1 1
2 EH-5-80-6 9 - ubiIFN H
3 EH-5-80-7 9 - IFN H
4 EH-5-80-10 11-OxyS-DacO1-37 G
5 EH-5-80-11 11-OxyS-DacO1-87 H
6 AL-234-C PgaE in pSPG1 1
7 AL-1-101 OxyS 10
8 AL-239-B SsfO1 3
Table 4-8 Plasmid Library C – Strain EH-5-217-1 and EH-5-217-3
* AL-2-79-K and AL-2-86-L are the same plasmid but one of them does and the other does not
encode a mutation in the sequence.
no. Plasmid Plasmid common name Colony #
1 AL-2-72-B Atc Moase Valsa Mali Var Pyr C6
2 AL-2-72-D Atc Moase G Cichoaracearumi C5
3 AL-2-72-F Atc Moase Valsa Mali CM C4
4 AL-2-79-J Related to tetra hydroxylase P subalpina C4
5 AL-2-79-K Atc Moase P subrubescens C5
6 AL-2-79-L* Atc Moase P subrubescens C6
7 AL-2-86-D Pentachlorophenol-4-Moase Hypoxylon C5
8 AL-2-86-I Pentachlorophenol-4-Moase Colletotrichum orbiculare C5
9 AL-2-86-A 2-ployprenyl-6-methoxyphenol-hydroxylase Aspergillus
Oryzae RIB40
C1

166
Table 4-9 Plasmid Library D – Strains EH-5-217-2 and EH-217-4
no. Plasmid Plasmid common name
1 AL-2-57-A F96X OxyS Library
2 AL-2-57-B M176X OxyS Library
3 AL-2-57-C W211X OxyS Library
4 AL-2-57-D F212X OxyS Library
5 AL-2-57-E T225X OxyS Library
6 AL-2-57-F V240X OxyS Library
7 AL-2-57-G P295X OxyS Library
8 AL-2-57-H A296X OxyS Library
9 AL-2-83-A A43X OxyS Library
10 AL-2-83-B G297X OxyS Library
11 AL-2-83-C G298X OxyS Library
Table 4-10 Sequences used in this study
Sequences for the hydroxylases fungal hydroxylases of Table 4-1 are shown capitalized within the
context of the pSP-G1 backbone containing pTEF1, the FLAG tag and tADH1 (partial,
decapitalized).
pTEF1-
CM003104
.1-tADH1-
in-pSP-G1
(Entry 1
Table 4-1,
AL-2-72-
F-C4)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGATGGGTGCAAAACACGTTATTAGAAGAGTGA
CCAATTCGGAAAATGATACAGGATCAAGTTCGAAATCCACTACTTCA
TCTAATTCCGACAGGGACCATAATCCCTCTTCCTCTAATAGAGACAG
ATATCACAATGAAACATCACCTGCCAATATGATATTGATAAACGAGC
CACCTCCTACCACCTCCACAGAATTGGAGAGCGACACTCATATTAAC
GTACGTAGCGTTTCTCCATGTTTGGAAACTGGTTCCCTAAAAGCAGA
GCAAGCGCCTCAGAATGTCCAGAACCCACCACAGACACAGGACCAA
GCTACTGAAGATACTGCCACCAACTCTCAACCGGCCTCAACGCAGTT
CAGAGCTATCATTGTGGGAGGTGGCCCAAACGGTTTGTGTCTGGCAC
ATGCGTTATACCTAGCTGGTATTGATTATGTTTTGCTAGAGCGCGGTG
GTGAAATCATCAACCAATCTGGAGCCGGACTTGCTTTATGGCCTCATT
CAGTTCGTATATTAGATCAATTGGGTCTGCTAGACGAAGCTAGGAAA
CATTATTTCCCGATTAAGACAAAGCATAACCACCGCCCCGACGGGTC
AGTCAGAGATGTTAACGATATGTTTGCCAAGGTAGAAGTTAATCACG
GTCACCCCTGGATGCTGTTCCATCGAGTAACATTGCTAGAACTCTTGT
GGGAAAATTTGCCTGATAAAGAGACTAGAGTAAAAGTCAACAAGAA
GGTGGAAAGTGTAGTCTCCAACCATGATGGCGTCGTGGTTACATGTT
CAGATGGAACATCTGAAGCAGGAAGTATAGTGATAGGTTGCGATGGT
GTACACTCGACTGTTAGACAAGTCATGCATGACTTGAGAAGCGAGAA
AAAGAAGAAAAGGCCAGGCCGAAAGCTCTCTCTGGGAAGGTCGCCG
TCACAAGACAAGCCAATGAGAGCCCACTACTATGGGCTTATTGGTTG
GATTCCGCTTCTTGATGGGTTACAACCTGCTGCTTGCTACGAAGTGAG
AAGTGAGCCTAAAGGTAAAACCTTTACCATTTTAACGGGCGAAGATA
CCGCTTACTTCATCGTGTATATTCATTTAGAAAAACCCACGAGAGAG

167
CGCAGCAGGCACACAGACGAGGATGCCGAAAGGTTGGCAGCCGCAT
TGGCCGAGAACAAGATAACGAGGGAAATTACTTTTGGTGACCTATGG
AGATCAAGGCGTTGGGGCAAAATGCTTGATTTTCAAGAAGGGTTCGT
AGATAAATGGTATCATGAAAGAATCGTTTTAGTTGGCGATGCTGTCC
ATAAGATGACTCCAAATGCGGGACTAGGCTTGAATGCTGGCTGGCAA
GGTATCGCTGAATTAACGAACAGATTGCGGAGATTAGTTGTTGCAGA
AGGACAAAGACCAGATGCAAGGTGCGTTGAAAAAGTTTTTCGAGGTT
ACCAAGATAGTAGAAAGGGTATGGCAAAGAAAACTATGAGGTTTTCT
TCTCTGTACACCCGTGTGGTAGCTAATCAGAGCTTACTGTATCGTTTT
TGTGATAGAATGACACCAGCGGTTGGTGGGGATGTCGCATTATTAAA
TACAATGGCTAGTCCTATTGTTAAAAAAGGTGTCACTTTCGACTTTGT
AGCGGAACGTGATCATAAAGAAGGTAGAGTAAGATGGGTTCATCCA
CAACATGTTCCAGCTGAAgtatcgatggattacaaggatgacgacgataagatctgagctcttaatt
aacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
MNBE010
00128.1-
tADH1-in-
pSP-G1
(Entry 2
Table 4-1,
AL-2-79-
K-C5 or
AL-2-79-
L-C6)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACCAAGTCCAAGTTATACGACGTGATAATCG
CGGGCGCCGGACCCGTCGGCCTCTTCCTCGCCCACGAACTCAGTCTC
CGCCAAATAGCAGTCCTAGTCCTAGAACGAACAACCCAACAAGACTC
GCCCTGGAAATCCGGACCTCTAGGCCTCCGAGGCCTAAACATCCAAT
CCATCGAAGCATTCTATCGCCGCGGTTTACTAGGCCAATTATTCAATC
TGGACGAAAGGCAAAAGTACCAACCGAGCAAGAAACCAGGTTTCCA
ATTCGGCGGCCACTTCGCCGGCATCATAATAAATGCCAATCAATTCG
ATTTGCAACGCTGGAAGTACCGCCTCTCAGGTCCGGCGCTTGGTCCG
GGTCCAACGACGATGGATCAAGTGGAGAAGGTCCTAACGGCGCGGG
TGGAGAGTCTAGGCGTGAAGATTCTCCGCGGTCGATCTGTTAGCCGT
ATCTCGCAGGATGAGGCAGGCGTTACAGTCGAAACAGACGATGACC
ATTCCGAGTCATTTCGGGGCAAATGGCTCGTTGGCTGTGACGGTGGG
CGCAGCGTTGTTCGAAAAGAAGCCGGCTTCGAATTCGTCGGTACAGA
TGCCCAATTCACCGGCTACGCAACGCATGTCGAGATAGAAGGTCGCG
AGAAGTTGAAGCCTGGTCTCAACGTTTCTGAAACGGGGATGTATATC
AACGTAGTGCAGAATGGCGCTTTTCATCTCCTTGAATTTGACGGGGC
GAACTTCGATCGCGCGGGTGATATCACGATCGATCACCTTCAGCGGG
TCCTTGATCGGGCTATTGGTCGGACAGATGTGAAGATTACAAATGTG
CATCTTGCTTCTTGTTATACCGATCGTTCAATGCAGGCGGCAAGTTAC
CGGAGGAACCGCGTTCTTCTTGCCGGTGATGCAGCGCATATTCATTCT
CCGCTCGGCGGGCAAGGACTGAATGTCGGACTAGGTGATGCGATGA
ATTTGGGGTGGAAGCTTGCGGCTATAGTTCAGCGGGACCAAGGCCAG
GGAGAGGATTCTGGGAATACGCATGATCTGACTCTTCTCGATACCTA
CGAGAGCGAGAGACACCCCATTGCTGCGTCGGTTCTTGATTGGACTC
GTGCGCAGGTCACGGCGCTGAGGCCGGATGCCTTTGGTCGGGCTACG
CGCGCCCTGACGGAAGATCTGATCAAGACGGATGATGGGGCTAATCT
GTTTATTGATCGGATCTGGGGACTTTCGCAGCGATATAAACTTGGCG
AAGAGTCGAGGCCTACACATCCGCTTCTTGGGTCTAGTGCGCCTGAT
TTCGAGTTTGGGAATGGGTCTAGGCTTGGCTCGAAGCTTGAGGAGGG
ACGGGGGTTACTTCTTGATTTTGGGAACAGTAGTGAACTGGAATCAG
TTGTTGATAGAAAATATTCTGGGAAGGTAGATTATCTTGCTCCAGAG

168
GCAAAGGAGAACTGTGGGCTGAGTGCCTTGTTGATTCGACCTGATGG
AATCGTTGCATGGGTGGTGGAAGAAGATGCGCAGCACGATATTGATG
CTTTGAAAGCTGCATTAGGGGATTGGTTCACTTTGgtatcgatggattacaaggat
gacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
NC_03677
2.1-
tADH1-in-
pSP-G1
(Entry 3
Table 4-1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACAACTAATCCAGAAGTCATCATCGTGGGCG
GCGGAGCAGTCGGCCTGGCCGCTGCATGCGAATTGGCCATCAAGAAT
GTCTCTGTTACGATCTTGGAGCGGGAATCTGGACCTGCAGCGCCATG
GAAGAACGAGAAGCTGGGCTTTCGCGAGCTGTTTGTGCCTGCCATCG
AGCACTTGTACCGCCGTGGCCTGCTGAACGACATGTTCCGCAATGAA
GAACGACCACAGACTATGCCGGAGAAGAAGGGTGGCTTTGAGTTTGC
TGGTCATTTTGCTGGTATGATGATCAATGGAAACGATGTCAACTATTC
ACACTGGGACGATACACACCTTCCCGGGCCTGCTTGTGTACCTGGAT
CTTCGACACTGGGTCACATTGAGAATGTTCTGCGTGCACGTGCTGAG
CAACTCAAAGTGCCTATTCTTACTGGCAAGAATGTGACTGGCATCGA
AGACACAGACAGCGGCGTCAAGGTGTTCTGCGGCGAGGAAACATTC
GAGGCTCAATATCTTGTCGGATGCGATGGTGGTCGGAGCACCATTCG
CAAGAAAGCTGGGATTCCATTCATCGGCACAGAGCCCGAGTTCACTG
GCTATGCTGTCCATTGCAAGCTCGACAACCCCTTTTTGCTCAAGCCCG
GCTTCAATCGATGCCCAAATGGCGGACTCTACATCATGGCCGGCCCA
CAGCACATCTATGCTGTAGATCACGACATCAGCTTTGACCGAGACCA
AGAAGTAACAGCGGAACACTTTCAAAAGGTTCTGAGAAAGCTTTCCG
GTACCAACGTGGTTGTTGAGCAGGTCGTTCTCGCATCCGCTTTCACCG
ATCGCTGCAGACAAGCTGAGCAGTACCGCAAAGGCCGAGTCTTCCTT
GCTGGCGATGCTGCCCACATCTACCCTCCATTCAGCGCCCAAGGACT
CAACTGCGGTTACATCGATGCAGTGAATCTCGGTTGGAAGCTCGCTG
CTTTGGTCAAGGGCTATGCTACTCCGGGCTTGATCGATACATACCAG
CAAGAGCAACACCCAATGGCTGCGCGAGTACTTGATTGGGTGCGAGC
ACAAATTGTTACCCTTCGACCTGACGCTCATGGCCGGGCTGTGGGCA
ATGTTATGCGCGAGTTCCTCTACACCGAAGCTGGTGTGACATACTGC
ATCGGTCGACAATGGGGTCTTGATGTGCGGTATGAGATCGGAGAGGG
ACATAAGCTTGTCGGTCGCAGCGCTCTGGATTACGAGCTGGACGATG
GCTCTCGACTGGGAACTAAGCTCGGCGCCGCGAACTTCACACTCGTG
GCATTTGGTGACTGCAGTTCAGAGCTCACCAGCTCCGTCAACGAGCT
TAACCCAGTGCTCGGATTCTGCAGAGCGAAGGGCGAGGAGAAGTCA
GGCCTCCAGGGCGTCCTTGTACGTCCTGACGGGATCGTGTCGTGGGC
ATCTGCAGATGCCCTCAACATCGAGACGATCAAGGCCAGCCTCGAAC
GCTGGGTCGCGCTGCCAGCTGCTGTTGAGAAATTTGAATCTGTGTCTT
ACGAGGCCAACCAGTTGTCCAACATTCAGACGCCATTGACGACTACT
GTTCGATCGAATATCGGACGTGTATTTGGCAAGGATGGAGAGAAGTA
CACTGGCCTTCCGACAGTGGTGACGGAAGTCCCGGCAACGGCTGATG
CAgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtc
aagtctccaatcaaggttgtcggct
pTEF1-
FJOG0100
0008.1-
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGGCGAGACCGATGTTCTGATAGTTGGTGGAG
GGCCCACTGGATTGATGCTCGCCCTCGAGCTTACCAGCCAAGACATC

169
tADH1-in-
pSP-G1
(Entry 4
Table 4-1,
AL-2-79-J-
C4)
CCATTCCGCATCATCGACTCAAGTCCTATTCGAAGCGACAAAAGCCG
GGCCTTGGTTCTCCATTCACGGACTCTGGAACTTCTCAATCGCCATGG
AATTGCTCAGGAGTTCGTTGACCGTGGAAATTTCAATTTGGCCGTGC
GCATCTTCGCCAACCAGAAGTTCGTGTTTGAGAACGATTTCAACATC
ATCGCTTTCAATGACACTATGTACCAGAACCCCTTGATTATCAGTCAG
GCAGAGATTGAATCGATCCTGGACGAAGTCTTGATGAAACATCAAGT
GAAGGTGGAACGGCCCGTGACAGCGGAGAAGATATCACAAGATGAG
ACTGGAGTTACTGCCGTACTACGCCACGAGGATGGAAGCGAAGAGTC
ACTCCGTTGCAAATATGCTGTAGGCTGTGACGGCCCTCGCAGCATTG
TACGAAATTCGGCAGGTCTACAATTCGAAGGAGCGGCGTACCCGCAG
GATTTCATCCTCGCAGATGTGCACATGAAATGGGAGACTAGAGAATG
CCTTCATATCTATCTTGGCGCCTCTGGGTTCATGATTGTCTTTCCGATG
AAGGACAATGTCTGGCGTCTGATCTGTTCTCGCAGGGAAGCACTGGG
CGCTGACGCTGAGCCAACTCTTGAGAATTTCCAGGAGATGTTGGACA
AGCTGCTTCCTGAACCTGTTCAAATATTTGATCCAGTTTGGATCAGTC
GATTTAAATTACACCATCGAATTGCAGACAATTACCGCGCTGGTCGA
ATGCTCGTCGCGGGCGATGCAGCCCATGTCCATTCTCCTGCTGGTGG
GCAAGGGATGAACACCGGTATGCACGATGCAGTCAATCTTGGCTGGA
AGCTTGCAAGTGTCATTCGGGGCGAGAACGATGACTCTCTGTTGGAC
TCGTACAATATCGAACGGCGTAGAGTTGGCCAGACTCTCCTGCAAGG
CACAGATAGACTTTTCGAGTTTATGGCAACAACAAATCCTTTGTATCT
CTTCATAAGGAACTACGTCATGCCTTTCATTATGCCCTGGGCCATGGC
GATACCTGGCCGTCGAGCTCTTGCATATAGATTTGTATCCGAGTTGGG
GATTCGATATCGCAAAAGTCCCATCGTTGGACAGGCAACGACTTGGA
AAGGAACACTGAAAGGTGGAGATCGAGTACCCGACGGGAGACTCCT
GAAGGGGAATATTGAGATCACTGTTCACTCGTTGCTTGGAGCACGCA
AGCACAGTCTACTTCTGTTCTCGGGCGTGGACGGGGTCACCAGCACA
GACGATCTGGAGAAAGCTCATGTGGAGTTCATCGAGGCCAGTGGTCG
GTCGATCCCAGTATACACAATCACGAAATCATCCTCGGAGAACATAG
AGATCATGGATCCGGAGGGGCTAGTGCATAGGCTGTTTGGCTTTACA
GCATCCGGCTTTGTGTTAGTTCGACCCGATGGTCACATTGCGTTCATC
GGGCCTCTCACGTCAATGGACGAGCTCAAGACGTGGATGGAGAGAgta
tcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctc
caatcaaggttgtcggct
pTEF1-
MCBR010
07648.1-
tADH1-in-
pSP-G1
(Entry 5
Table 4-1,
AL-2-72-
D-C5)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGATTGTGAAGTATTAATTATCGGTGCTGGGCC
CACAGGACTTATGCTTGCCCTCGAACTTTCCCGCGAACAAATCCCCTT
CCGAATTCTCGATAGTCATTATTCTACTAAGACAGCCCATCAATCTCG
GTCTATAGTGGTTCACTCACGTTCCCTGGAGTTACTTGCCCGGCATGA
CTTAGCCGAAAGCTTCATCGCTAAAGGATTACTCATGGAAGAGATTC
GCTTCTTCTTGAAGCAAACCCTTAATGGGAAGAGTAGTTACCCTGGA
AGTCTCATGAATGATACAATTTATAAAAAACCACTTATCATTAGACA
ACCACTATCAGAGCAGCTTCTGGAAAAACGTCTTTGCGAGTATGGAG
GCATTATTGAAAGGGGGGCAAGCGCTAAAAAAATGGTGATAGATGA
AGATGGCGAGCGGGTGTCTGTTTTGGTACAGCATAGAATTCAGGAAG
AAGAAGAGAAAGTAAGAGAGGAATTAATCAATTGTAAGTATGTTGT

170
AGGTTGTGATGGAGCTAACAGTATGGTACGAAAATTTGCGGGTATCG
ACTTTCCGGGTACTTTATACCCTCAAGAGCTCATTCTCATTGATGCAC
AGGTAGACTGGAACAATAAAATATGTCCACATGTTTTTATTGATAAG
GAATTCGTGATCTTTATACCGCTAGACGAAACAGGTATCAACCGAAT
CATCTGCCGGCGTCCTCCAAATGACACCAGCACTATCTCATCCAGAT
CAAATCTGTTATCAGGATCATCGCCTTACCAAGAGCCTAGCCTATCC
GAGTTTCATGATACAGTTATTTCTTCCGTTCCTGGTACGACTCGTATT
CACAGCCCGATTTGGTCTTCCTCATTCCGTGTCTCACGTCGACTTGCT
AGTACATATAGTGTGGGGCGGATCTTCCTTGCTGGTGACGCCGCTCA
TGTCCATGCACCTATTGGCGGTCAAGGTATGAATACCGGTATTCAAG
ATGCCGTAAACCTGGGTTGGAAACTTGCGCGTGTTATAAAATCAACT
GCTCCGTCCTCTTTACTCAACTCATATAATGTAGAGCGATATAAAGTT
GGTAGTGACACTGTTGAAAATACAGATCGCATGTTCAATATTTTAAT
AACTACAAACCCTGTAAAAATCTGGTTACGAAACTTTCTTTTTCGCTG
GGTTTTGCCATATTTCCTCAAACCAGATTTTGCAGACAGAAAAATTCG
TTACATATCACAATTGTCAATTCGATATCGAAATAGTCCCGTTGTAGG
CACGGCTTCTGTGTGGAAAGGAATTCTAAGAGGCGGAGATCGGGCAC
CAGATGGATGGATGATGAATCCTGAAGGAGAAAATATCACTTTGCAC
AGGCTTTTTAGGACATCAAATGATCATCTTTTCTTATTCAGCGGCCTA
GACACTCTTGAATCGCGGATGGTCAATGATACTGTCCAGCAGCTAAA
ACTCTGCAGCTCAGTTCTTGTGGTACACAAAATATATGACGGGAATT
TTAAAGATAAAACAATTATCGATGGTTACATAGACCCAGGGGGTAAA
GTTCACACCTTGTATCAGTTTATTGAGCCTGGTTACGTTCTTGTTCGA
CCCGACGGATATATCTCATTTATAGGACCTATGACTTCACTAGATGA
ACTGAAGAATTGGATGAATTCATATATGCAGGGAgtatcgatggattacaaggat
gacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
KN714671.
1-tADH1-
in-pSP-G1
(Entry 6
Table 4-1,
AL-2-72-
B-C6)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGAGGCTAAGAAATCTTTCAAAGTTATCATTGC
TGGTGGCAGTATCGTCGGCCTAACGCTGGCCAACGCGCTCGAGAAGG
CGGGTATTGATTTTCTTGTCCTCGAGAGGGGCGACATTGCTCCACAGC
TCGGCGCATCAGTCTCAATCTACTGCAATGCGTCCAGAGTCCTCGAC
CAGCTGGGAGTTTGGAAAGGCATTCACCAGAGCACAATCCCGCTGAC
TGATCGGCTATACTTCGATGAACATGGCCGCTTGTTTGAGGACAACA
AGATGATGAGGCTCATAGACGACAAGACTAAGCGGCCTATCACGTTC
TTGAGAAGGGAGGCATATATCAGGGCTCTTTACGAAAACCTGAAGGA
CAAGTCGAAGGTGAAGGGTCACTCTGGGCTGGTGTCCTTTGCCGAAG
ATGATGAAGGCGTTTCAGTATTCACCAGTACCGGCGAGGAAATCAGG
GGCAGCATTCTTGTGGGAGCAGACGGCATCCATAGTACCGTCCGGAA
GCTCATGGCTGAGTCTGTCGCCAAGTCGGACCCTCAGAGGGCTAAGA
ATCTCATCGAGGGCTTCACGGCAAACTACCGGACGATCTTCGGAACG
TCCAAAAACCAGCGAAAAGACGACCCGAGCGTGCAGATGGTGGCAG
ATAGTCTTTCCCATACCTCCTATTACCGTGGCGTCACGGGGGTCTGCG
CCGTCGGCAACAGTGGGGTGATCTACTGGTTCCTCTTGGTGAAGGAG
GACACGCCTTCGACAATGCCGAACTGCCCGCGCTACTCGGAGGCGGA
TGCCGAAGAGACGCTTCGTAAATTCGGCCACCTTCATATGGGACCGG
GATATACTTTCAATGACCTCTGGGAACTGAGGGAGAAAGGAGTCATG

171
GTACCGTTGCAGGAAGGCATCGTCGAAGGAAGCTGGAATAGTAGCG
GGAGGGTTGTACTCATGGGCGACGCCGTGCACAAGTTCACTATAAGC
GCGGGGCTGGGCGCCAATTTAGGCGTAGAAGGCGCTTGCCACCTGGT
GAATGAGCTCCTCCCTGTCCTCAAAGAGACGGAAAACCCCGGAAAGC
AAGAGATAAAGGATGTGCTCGACAGATACGAGGAGAAGCATCGTCC
TCGAACTAAGATCTGTGCCGTATTGTCTAACTATTTGACCAAGTACGA
GGCGATGGAGACCTGGTGGCTCAGACTTCTGAGGTTCATTATTCCTC
GGATCCCGGACAGTTACAAGGCGAAGTCATTTGTGGACTTCATGTAT
GGTGCTCCGATTCTCGATTTCCTGCCTCACCCGGGCAAGAGCTCTGCG
gtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagt
ctccaatcaaggttgtcggct
pTEF1-
KL584983.
1-tADH1-
in-pSP-G1
(Entry 7
Table 4-1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGTCGGTCTTACAATTCAATCTGAACGGCTTCGT
GCCTGGCGACCCTCATGATCTCTACCGTGTCCACCAAGAAACCGCCG
AGATCGCAAACAATGTCCCGCAAGAAGTCGACGTCCTCATCATTGGC
TCTGGTCCAGCTGGTCTGGCTCTAGCAGCACAGCTATCTCTCTTTCCA
AGCATCACCACAAGAATCATTGAACAAAAGGCTGAACGACTAATCCT
AGGCCAAGCCGATGGCCTCCAAGTCCGAAGCATCGAAATGTTCGAAG
CATATGGCTTCAGCGAAAGAGTCCTCAAGGAAGCGTACTACATCAAC
GAAACCACCTTCTGGAAGCCTGACGAGCAGCAACCGGACCGCATCGT
GAGAAGTGGCAGGATTAAGGATACTGTAGACGATCTCTCCGAGTTCC
CGCACGTCATCCTCAATCAGGCGAGAGTTCATGACTTCTTCCTTGATG
TCATGCTCATGTCGCCGACAAGATTGCAACCGAGCTATTCACGCAAA
CTCAAGCAGCTCAGCCTTCAACCAGCACAGGGCAACTCCTCGCACGC
TACGTCCTCCGTGCAAGTTCAACTCGAGAGAACAGACTCCAGCCACG
AGGGCGAAGTCGAGGATGTACGTGCTCGCTACGTCGTAGGCTGTGAT
GGAGCCCGCAGTGCCGTCCGTCAAGCCCTCGGTCTCAAGCTGGAAGG
CGACTCCGCAAACCAAGCTTGGGGTGTCATGGACGTACTTGCTGTAA
CCGACTTCCCAGACTGGAGGACCAAGGTTGCAATCCACTCCGCGACA
CAAGGCAGCATCTTGATCATCCCCAGAGAAGGAGGTTACCTTGTGCG
CCTCTACATTGAACTCGACAAGCTCGACGCCAACGAACGTATCGCAA
ACAAGAAGATTACCGCCGACAAGCTCATCGAGACCGCACAGCGCAT
CTTCCATCCTTATGCTTTGGATGTCAAAGAAGTAGTCTGGCACTCTGT
CTACGAGATTGGCCAGCGCCTGTGTACCAGATTCGACAACCTCACTC
CGGAGACCGCTACAACAGAATCCCCAAACATCTTTATCGCAGGCGAC
GCATGCCACACCCACAGCCCTAAAGCAGGTCAAGGCATGAATGTCAG
CATGCAAGATGGCTACAATCTAGGCTGGAAGCTCGCCTCCGTGCTCA
ATGGAATCTCTAACCCGGCCATCCTCCACACCTACTCCGCCGAACGT
CAAAAAGTCGCTCAAGACCTCATCAACTTTGACCGAGAGATTGCTTC
AATGTTTAGCGCCAGACCCAAGAGCCATGCCAAAGACACAGAGGGA
GTCGACCCTGCTGAATTCCAAAAGTACTTTGTCAAGCAGGGCCTCTTC
ATGGCTGGACTGGGCACAGCTTACTCGACTTCTGCTATCACCGCTGG
ATCAGAGCATCAACACCTGGCCAAAGGTTTCCCCATCGGCATGAGAT
TGCACTCTTCTCCAGTCATAAGATTGGCAGATGCAAAGCCGATCCAA
CTAGGTCATGTGGTAAAAGCAGATGGAAGATGGAGAGTCTTCGTCTT
CGCATCAGCCGAGGATCCCGCTTCGACCTCGTCAAGCTTCCACTTGG

172
CCTGCGAATCCCTGACCGAGCTTGTCAACAAGGTCAACCCTACTGGT
GCTGATATCGACTCCGTCATCGATGTCAGAGGTATTCTGCAGCAGGG
CCATGCTTCACTGAACATCAACCACATGCCTGCTATCGTGTGGCCGC
AAAAGGGAAAGTATGGACTCAGAGATTACGAAAAGGTCTTCTGTGCT
GAAGATGTGGATGGACATAGGGATATCTTTAATGCTAGGGATATCGA
TCGCAGAGGCTGTATCGTGGTCGTACGACCTGACCAGTACGTCGCCA
ACGTTCTGCCGTTGTGCGATCATGATGGTTTGTCAAGGTTCTTCGGAG
GTTTCATGTTTGCAAGGGATgtatcgatggattacaaggatgacgacgataagatctgagctctt
aattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
KZ111246.
1-tADH1-
in-pSP-G1
(Entry 8
Table 4-1,
AL-2-86-
D-C5)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACCAAGACTTACGATGTTATCGTTGCCGGCGC
AGGCCCCATCGGCTTGTTTCTCGCCTGTGAGCTGAAGTTGGCGAACG
TTGACGTGCTAGTACTAGAGCGTGACACCAACCCCGAATCCCCCTGG
AAAAGCGAACCACTCGGCATACGAGGCTTGAATACGGTATCTGTCGA
GTCCTTCTATCGACGTGGGATGCTAGACAAGGTCACCTCCATGGAGC
GACCGAACTTCTTCAAGAAAGGCCCGGGGTTCCAATTCGGAGGCCAC
TTCGCCGGCATGCTACTGAATGCCAACAAGCTCGAAATGTCCCGTTA
TAAGTACCGCCTCCAAGGCCCAGCATTGGTTCCCAACGGATCAAGTA
TGGAACGTGTTGAGAAGGCCCTAACAGAGCGCGCTGAGAGCATTGG
AGTAGATATTCTCAGGGGGAAGGGAGTCACGGAAGTCTCGCAGGAT
GACAACGGCGTTACGGTGGAAGCAGGAGGCGAGACTTTCCATGCGA
AATGGCTTGTCGGATGTGATGGCGGGCGGAGCACAGTTCGCAAGGCC
GCTGGCTTCGACTTCGTCGGAACAAATCCTGAATGTACCGGTTACGC
TTTTTTGGCCGATTTAGACCATCCCGAAAAGCTGAAGCCGGGGTTCC
AACCCTCTGTAGGCGGCATGTACATCATAGGACATTGGGGTAACATG
TACTTGCTAGACTTCGATGGTGGCGCTTTCGATCGTACGCAGGAGAT
CACACCCGAGCACCTTAACGAAGTCATGGCCCGTGTGACAGGCACCG
ACGTCAAGGTCACCAAGATTCATCAGGCATCCTCGTTTACCGACCGA
TCTAAACAAGCGTCGACTTATCGAAAAGGCCGCGTTCTCTTAGCCGG
CGACGCGGCGCACATCCATTCGCCTCTGGGAGCACAAGGATTGAATC
TAGGGTTGGGAGATGCCGTGAACCTTGGCTGGAAATTGGCAGCGACA
GTTCAGGCTTCTAAATCTGGAGAGGAACCAAAGGACCTCAGCCTACT
CGATTCCTACGAGAAAGAGCGATATCCGATCGGGTCTTGGGTTCTTG
AATGGACACGCGCTCAGATCTCGACGCTAAAGCCCGACATGTTCGGT
ATCGCTCTTCGTAGGCTCATGAGCGATTTATTGGAGACGACTGATGG
AACGAATTTATTCATCGATCGCATCTGGGGTCTGTCGCTGCGGTACG
ATCTTGGCGACGAACACCCCGCCGTCGGCTGTAGCGCTCCCGATTTC
GAATTCCACGACGGAACGAGGCTAGGTCCCAAGCTAGCGACGGGGC
GAGGTCTATTGCTCGATTTCGGATCTAATGCAGAACTCAAGGATCTT
GTCGGGAAATACGAGAGCAAGGTTGATTATCTTAGCACAGAGGCAA
AGGACCAGCTGGGCCTGAAATCCTTGTTGATCCGCCCCGATGGCGTT
GTGGCTTGGGTGGCTGAAGAGAAGCCTGATATGGACTCGGCGAAGG
CCGCTCTGGAGAGATGGTTTGGGCTGGGTTCGAAAgtatcgatggattacaagga
tgacgacgataagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggc
t

173
pTEF1-
FJOG0100
0031.1-
tADH1-in-
pSP-G1
(Entry 9
Table 4-1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGAAAACCAGCTCTACGACGTCATCATTGTTGG
CCGCGGTCCTGTTGGGCTATTTTTAGCATGCGAACTTCGCCTCACAGG
GCTCTCTGTCTTGGTTGTTGAGCGCCGCTCGAGCTCAGAGGGAGTGG
CGGAGACTCGCGCTTTTGTTGTCCATTGCCGCACACTCGAGATCCTCG
CATTCCGTGGTCTTCTTGGCAAGTTCCTCCAGGAGGGCACGAAGTCTC
CTTGGTGGCACTATGGCGTCTTGGACACCCGTTTGGATTTTAGCAGAT
TTGGCCACGAAACGAAGCAGAATTTTGTCCTGTTAAGCCCACAGTTC
AGAACTGAGGAGATTTTTCTGGAGCGTGCGAAGGAATTAGGCGTCGA
CATTGTTCTGGGAATTCGCGTCGAGTCCGTGGAGCCTTCGACTACGTC
TGTTACGATATATGGAACATCAAGCAGTCCAGATGGCGGCGCTAGAA
GTTCGTTCAAGGCGAGAGGAAAGTATCTTGTCGGTGCGGATGGAGTG
CGCAGTACCATCCGGAAGGCTACGGGCTTTGAATTCCCAGGTACGCC
GGCAACACACACGGGTTTGACCGGCGAGGCAACATTGGGAGTGCCC
ATGCCCTATCCCTACATTGCCAAAAACGAAGCAGGGCTTGTGATCGC
CGTACAGCTGAATATTCCCAGCGGCCGGGCTCGCCTCAATGTTTTCAC
CCCTGCTCGAGCTAATGTACCCGATTCGACCCCGGTAACGCTGGAAG
ATTTCAACGAAGCATTACAAGAAGTCACTGGTGTTGACTACAAGCTC
TCCAACCCTTGCATGCTTGGACGCTTCAACAACGAAGCCCGCTGCGT
CGATACTTACCGGAAGGGACGCATCTTTCTCGCCGGTGATGCAGGCC
ACCAGCACCTTCCAGCTGGCGGCCAAGGGCTCAACGTCGGTATTCAG
GAAGCAACTAACCTTGGATGGAAACTTGGGGCCGTGATTCGCGGCTA
TGCTCCGGACTCGCTATTGGATACTTATGAGACCGAAAGACTTCCAA
TCGCACAAGGCGTCGTGCAGAGCACCACTGCTCAGTCTGTTCTCTTTT
TTGCGCACAGCGGGCCGGAGTTGGCAATACGAAGTGTGGTGAACAC
GTTGCTCAAAATACCAACAGCAAACCACGACATTGCTGTAGGAGTTA
GTGGTTTCGGCGTCTCATACCCAAAGCTTCTTGACATGATACTTCCAG
CTGGATGGGAGGCACTTCCAGAGGCGATCGCCGGGAAGCGAGCCCT
AGATGTGAAGTTACGCGTCGGGAATGGCGAGGAAAAGCAGCTAAGC
GACTATATGAGGGGAGGGAAGTGGGTTCAACTTCGGTTCCTTGACAA
ACTCGCAGGAAGGCCGCCACTGCCGGCGTTCGAGGGGGCGACAGAG
GTTGTGGATGTCGCTGAAGTTCTGGAGGGGAAAGGGAGCATGTACCG
GGGCGCCTTGAGCGAACTGCTAATTCGCCCGGACGGTTATCTAGGGT
TTGGAAAGCGCgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcg
ccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
AMCJ0100
0140.1-
tADH1-in-
pSP-G1
(Entry 10
Table 4-1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACCTACACTACTCATGACGTGGTGATTGTGGG
AGCTGGTCCAGTTGGACTCTTCCTCGCTTGCGAACTACGCCTTGCCGG
TCTCTCCGTTCTGGTGGTCGAGAAACGAACAAATTCGGACGGCATGG
CGGAAACGCGTGCCTTTGTGATGCATGGTCGATCTTTGGAAATCTTCG
CCTCTCGCGGTCTGCTCGACTCTTTCGTTGAAGCGGGTCAAAAGACC
GACTGGTGGCATTACGGTGTTCTCGACACTCGTCTTGATTACAGTGTT
TTCGGCCGTGAAACGGACCAGAACTACGTGTTACTCGTGCCTCAGTA
CAAAACCGAGATGATCTTATTTCAACGCGCCGTGGATCTGGGTGCAG
TGATTATCAAGGGTGTCCAGGTCGATTCAATACACGAATCCGGGCCT
TATGTCGTTGCGAGGGGTTTCTACTGCAATAACAAACCTTTCATTGCC

174
AGCGGGAAATACCTCGTCGGTGCGGACGGTGTTCGCAGCACGATCCG
CAAAATCGCCAACATCGAGTTCACTGGCAATCCACCTGTCAATACAG
TCATGAGTGGCGAGGCTACACTAGGCACGGCCATGCCGAATCCCTAC
ATTGTTCACAACGAGCACGGCCTAGTTATCGCCGCTGACCTGAGGGT
CCCCAGCGGAAGAACCCGCCTAAATGTGTTCGCGAGTGATCGCGGCA
CGGTTCCTGAGTCAGTTGAAGTGACTCTTGAGGAGATGAATCAGAGC
TTGCAGAAAATAACCGGCGTTGACTACAAGCTTTCTAACCCTTGCAT
GCTAAAGCGTTTCAGCAACGAACAACGGCTAGCGACAACGTATCGCC
AAAATCGGATTTTCATCGTCGGAGATGCATGCCATAAACACCTCCCA
GCCGGCGGCCAGGGCTTAAATGTCGGTCTCCAAGAAGCTCTGAATTT
AGGATGGAAACTTGCCGCAGTCATCTCCAAGTCCGCCCCTGCTTCGTT
ACTCGATACGTACGAAGAACGATGGCCAGTTGCCAAAGCTGTTGTAC
AAAACACAACTTCACAGTCACTCTTATTTTTTGCCTCGTCTGGCCCAG
AATGGGCGGTTAGGGAAGCGATCGACAAGCTTCTGCGTGTACCAGAG
GCCAACAAGCGTTTGGCTAGGGAGATCAGCGGATTTTCTGTTGCCTA
CCCCAAATCGCTGGATATGATACTTCCAGACGGATGGCGGGCCCTAC
CAGAGAACATCCAGGGAAAGCGTGCATTGAATGTAAAGATGAGATT
ACCAGATGGGATGGTAACCGAACTTCGTGATTACACCCAAGACGGAC
GATGGATCCAGCTGCACCTTCCTGGAAAGCATATCATTGAACTCCGG
CCTCCTCCGGCATTTGGTAACTGGACGACGGTAGTGGAAGTTGTCGA
CATGCCAGATGAAGAAGAGAAGACGAGCTTGTATATGTGCGGAGTG
AGAGAGATGCTGATCCGCCCGGATGGCTATTTGGCCTTTGGTCGAAT
GGACGACgtatcgatggattacaaggatgacgacgataagatctgagctcttaattaacaattcttcgccaga
ggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
KB725976.
1-tADH1-
in-pSP-G1
(Entry 11
Table 4-1,
AL-2-86-I-
C5)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGACTAATCAATATGATGTTGTAATTATTGGAGC
TGGGCCAGTTGGATTATTATTGGCTTGTGAATTAGCTTTGGGAAAAA
CAAGCGTCATAGTTCTGGAAAGAGAAGCGTCGCCCGTATCTCCGTGG
AAAGAAGAACCGACTGGTATGAGAGGTTTGCACCTGCCTTCTAGTGA
AATTTTGTGGCGTAGGGGTTTGTTAGAAAAAATCTGGACATTGGAGG
GTAGACCACATGGCCCGACTAAGACTCCTGGATTTCAATTTGCAGGC
CATTTCGCCGGGATACCTCTTAACCTATCTCAGCTAGACTTAGATAGG
TGGAAATATCGATTGAGGGGTCCATCATTAATGGGGGGTCCAATAAC
AATTGCATTAATTGAGAGAGTGTTAGCAGAACGCGCTGAATCATTGG
GAGTCGAAATTCATCGGGGTTGCGGGTTCGAGAGGATAGTCCAACAA
TCCGCTGACGGCGTAACAATAGAAGCGGGTGAAGAAACCCAGACAT
TTCATGCAAAATGGCTTATTGGTTGCGATGGCGGTCGTTCCCAAGTCC
GTAAGGCTGCCGAAATCGACTTTCCTGGCACTGACGCGACCTTAACT
GGCTACGTGATCCATTGTGATCTTGACCATCCTGAACGATTGTCACCT
GGTTTCCAACCTACCAAACAGGGAATGTATATTTTTAGAAAGCCCGG
CGCTCTCTACCTGATGGATTTCGACGGTGGTGCCGGCCAAACAAGAG
AACCAAGCCAGCAGAGACTACAAGAAGTGATGGAGAGGGTTGTAGG
GAGAACTGATATCAAGCTAGAGAAGGTTCACCTAGCAAGTGCGTTTA
CGGACAGGGGCAAACAAGCCACAACGTATAGGAAGGGAAGAGTCCT
CTTGGCTGGTGACGCAGCGCACATCCACCCACCACTTGGTGCACAAG
GTATGAACGCCGGTTTGGGTGATGCCATGAATCTAGGATGGAAGTTA

175
GCTGCAACCGTTCGTGGAGAAGAGAAAGGAGTCCGGGCTTTCACCGT
GTTGGATACCTATACTTCAGAGAGACATCCCGTTGGTCAATGGGTCTT
AGAATGGAATTACGCACAAGTTGCTGCTCTAAAACCCGATGTAACAG
GTTACGCCGTACAAAAATTAATGAGGGATTTGATTGCAACTGATGAC
GGTACGAATTACTTCATAGATAATGTGTGGGGATTGTCTCAGCGCTA
TGGTGACGGTGAAGGTGTTCATCCAGTAGTTGGAAGATCGGCCCCTG
ATTTCACATTTAAAGATGGTAGTAGACTTGGGCCAAAATTAGAGGGT
GGCAGAGGACTTTTTATAGATTTTGAAGATGTGGATCTGGAGAAAGC
AGTTAAGCGATTCGAAAGAGTTGATTATCTCGGACAAAACGTGGAAG
ATAGACGTGGAATCAGAGCTTTACTTATTAGACCAGATGGATTTGTT
GCTTGGGCTGTAGAAGAGGGAGATGAACCAGACGCAGAGGGTTTAG
AAACTGAATTAAAAAAGTGGTTTTCTTACgtatcgatggattacaaggatgacgacgat
aagatctgagctcttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct
pTEF1-
AP007159.
1-tADH1-
in-pSP-G1
(Entry 12
Table 4-1,
AL-2-86-
A-C1)
tcaagtttcagtttcatttttcttgttctattacaactttttttacttcttgctcattagaaagaaagcatagcaatctaatctaa
gttttaattacaagcggccgcATGGGCAAGACCTGGGACGTTATTATTGCTGGCG
CTGGGCCGGTCGGTCTTTTCCTCGCCTGTGAACTGGCCATAGCTGGCG
TGTCGGTACTTGTACTGGAGCGAGAGATGCAGCCAGAGTCGCCTTGG
AAAGAGGGATTGTTTGGGCGTCGCGGTCTCTATACCCCGGCGGTCGA
GGCATTTTACCGCCGTGGCATCTTGAAGAGAATTTTCGGTGATGATG
AGCGTCCGACGCATTTGAAGAAGACTGAAGGCTTTCAATATGGAGGA
CACTTTGCGGGCTTGGTGCTTAATGCAAACAACATCGAGTTTTCACGC
TGGCCATATCGCTTGCCAGGACCGTCCTTTCTTCCTGGTCCGACTTCT
CTTGGACGTCTTGAGGCTGTGTTGTCGGAACGTGCAGAGACGCTGGG
TGTCCAGATCCTTAGAGGTATGGAAGTCTCCCGGCTCGCTGATGAAG
GTGAATCAGTCAAGGTATGGGCGGGCGACCAGTGGTTTACGGCTCAA
TGGCTTGTCGGATGTGACGGTGGACGCAGCACAGTTCGGAAAACTGC
TGGATTCGAGTTTGTCGGGACCGAAGCAGAATTCACTGGTTATATTG
CGGTATGTGATCTCGACCGACCAGACTTATTGAAGTCAGGTTTCAAG
CACACAAACTCGGGGATGTACATAGTTAGCGGACCTGGACAGTTGCA
TGTTATCGATTTTGATAGGAGCTTTGATCGGTCACAAACTATCACGCG
GGAACACTTCCAGGAAGTGCTACGGCGAGTGTCGGGCACAAACATC
GTTGTCGAAGCGCTTCACCTAGCCTCCTCCTTCACCGATCGCTCGAAG
CAAGCAACAGAATATCGTAGAGGACGCATCCTTCTGGCTGGTGACAG
TGCCCACACACACTCTCCACTGGGCGCGCAAGGATTGAGTACAGGAA
TTGGCGATGCGATGAATCTCGGCTGGAAGCTTGCGGCTACCGTCAAG
GGGTTTGCATCTCCTGGTCTGCTAGACACCTACCACCAAGAGCGGCA
TCCAGAGGCAGCTCGAGTGCTTGAGTGGACTCGCGCTCAGGTGGCGG
CCTTACGCCCTGATCCATATGGCCAGGCTATTGCGAGTCTCATGCGG
GACATGATCAATACCCAAGATGGGGCGACTTATCTCGCCGATCGCAT
CTGGGGACTTTCAGTGCGCTATGGCCCGGGCGACGCGCATCCACTTG
TCGGTTCTAGCGCTCCGGATTTTGAATTCGATGATGGAATGCGACTTG
GAGCTAAGTTAGAGACAGGATCCTTCTTGGTGATTGACTTTGGGAGC
AACAACCAGGTGGCAGAGCATGTGCAGTCCTTGCAGTCTCTGCAGTT
CATGATTCAATACTGTGCATGTAGCGCAAAGGAGGAATTCGGGCTCA
AGGGGTTACTCTTGCGACCTGATGGTGTGGTCGCATGGGTGTCGACG
GAGGAGATAAATATAATACGACTGCATGTTGCATTGTCTCGTTGGAT

176
AAGTCTCCCTAGTTTCGAGGCTgtatcgatggattacaaggatgacgacgataagatctgagct
cttaattaacaattcttcgccagaggtttggtcaagtctccaatcaaggttgtcggct

177
Table 4-11 Primers used for OxyS saturation mutagenesis in this study
To create a saturation mutagenesis of the positions outlined in Table 4-2 the even numbered
primers were used for PCR amplification with EH414 and the odd numbered primers were used
for PCR amplification with EH745. The resulting amplicons were gel purified and used for Gibson
Assembly with pSP-G1 (empty) digested with SpeI and NotI.
EH414 GCTCATTAGAAAGAAAGCATAGC
EH745 CTGGCGAAGAATTGTTAATTAAGAGC
EH723 AGTGCGAGCGTGGACTCCTAGMNNTTTCGAGAAGTCAACAGGC
TC
A43X
EH724 CTAGGAGTCCACGCTCGC A43X
EH725 CAATGCATATGGGTGTCTAGTATCAA F96X
EH726 CATCATTTGATACTAGACACCCATATGCATTGNNKGTTCCACAA
GTACGAACTGAAGAAC
F96X
EH727 ATGTGGGTCTTGACCTGGAAAATC M176X
EH728 AACTATTGGGCATAGATTTTCCAGGTCAAGACCCACATNNKTTT
GCTGTCATCGCAGACG
M176X
EH729 TGCACGAAGGTCATGTCTCATG W211X
EH730 TCATGAGACATGACCTTCGTGCANNKTTCGCAGCATTTCCGCTA
G
W211X
EH731 CCATGCACGAAGGTCATGTCTC F212X
EH732 GAGACATGACCTTCGTGCATGGNNKGCAGCATTTCCGCTAGAA
CC
F212X
EH733 CAGCATACGGTCTATCGAAAAAGGCGACMNNTGCTCTGTAGAC
GTCTGGTTC
T225X
EH734 GTCGCCTTTTTCGATAGACCG T225X
EH735 CGGCGCCCTCCTGTCA V240X
EH736 GTATGCTGACAGGAGGGCGCCGNNKACGGAGGAGGATGTCAG
AGC
V240X
EH737 CTAAGTTCAGTCCCTGGCCACCAGCMNNCAAATGAATATGGCA
TGCATCACC
P295X
EH738 GCTGGTGGCCAGGGACTG P295X
EH739 ATCTTGAAATCCTAAGTTCAGTCCCTGGCCACCMNNGGGCAAA
TGAATATGGCATGCATC
A296X
EH740 GGTGGCCAGGGACTGAACTTAG A296X
EH741 TCCTAAGTTCAGTCCCTGGCCMNNAGCGGGCAAATGAATATGG
CA
G297X
EH742 GGCCAGGGACTGAACTTAGGA G297X
EH743 TTAACGGCATCTTGAAATCCTAAGTTCAGTCCCTGMNNACCAG
CGGGCAAATGAATATGG
G298X
EH744 CAGGGACTGAACTTAGGATTTCAAG G298X
EH812 GTGCGAGCGTGGACTCCTAGAGCMNNCGAGAAGTCAACAGGC
TCG
K42X
EH813 GCTCTAGGAGTCCACGCTCGCACTG K42X

178
EH814 CAACAGTGCGAGCGTGGACMNNTAGAGCTTTCGAGAAGTCAA
CAG
G45X
EH815 GTCCACGCTCGCACTGTTG G45X
EH816 TGGGTGTCTAGTATCAAATGATGAG L95X
EH817 TCTCATCATTTGATACTAGACACCCATATGCANNKTTTGTTCCA
CAAGTACGAACTGAAG
L95X
EH818 GGCGACTGTTGCTCTGTAGA F228X
EH819 TCTACAGAGCAACAGTCGCCNNKTTCGATAGACCGTATGCTGA
CA
F228X
EH820 CATTAATTCCCTTAAACCCTCGTAGCGMNNATCTGGATCAATT
AGAACAGCTTGAG
P357X
EH821 CGCTACGAGGGTTTAAGGGA P357X
EH822 AGGATCTGGATCAATTAGAACAGCT R358X
EH823 CTCAAGCTGTTCTAATTGATCCAGATCCTNNKTACGAGGGTTTA
AGGGAATTAATGATTG
R358X
EH824 TAGCCCCGCTAAATATCTATTAGTCTCAGGMNNGTGCAACAAT
TCAATCATTAATTCCCT
V372X
EH825 CCTGAGACTAATAGATATTTAGCGG V372X

179
4.8 References
1. Li YR, Chooi YH, Sheng YW, Valentine JS, Tang Y. Comparative Characterization of
Fungal Anthracenone and Naphthacenedione Biosynthetic Pathways Reveals an alpha-
Hydroxylation-Dependent Claisen-like Cyclization Catalyzed by a Dimanganese Thioesterase. J
Am Chem Soc. 2011;133(39):15773-15785. doi: 10.1021/ja206906d.
2. Chooi YH, Cacho R, Tang Y. Identification of the viridicatumtoxin and griseofulvin gene
clusters from Penicillium aethiopicum. Chem Biol. 2010;17(5):483-494. doi:
10.1016/j.chembiol.2010.03.015.
3. Nicolaou KC, Hale CR, Nilewski C, Ioannidou HA, ElMarrouni A, Nilewski LG, Beabout
K, Wang TT, Shamoo Y. Total synthesis of viridicatumtoxin B and analogues thereof: strategy
evolution, structural revision, and biological evaluation. J Am Chem Soc. 2014;136(34):12137-
12160. doi: 10.1021/ja506472u.
4. Lindqvist Y, Koskiniemi H, Jansson A, Sandalova T, Schnell R, Liu Z, Mäntsälä P, Niemi
J, Schneider G. Structural Basis for Substrate Recognition and Specificity in Aklavinone-11-
Hydroxylase from Rhodomycin Biosynthesis. J Mol Biol. 2009;393(4):966-977. doi:
10.1016/j.jmb.2009.09.003.
5. Wang P, Bashiri G, Gao X, Sawaya MR, Tang Y. Uncovering the Enzymes that Catalyze
the Final Steps in Oxytetracycline Biosynthesis. J Am Chem Soc. 2013;135(19):7138-7141. doi:
10.1021/ja403516u.
6. Patrikainen P, Kallio P, Fan K, Klika Karel D, Shaaban Khaled A, Mäntsälä P, Rohr J,
Yang K, Niemi J, Metsä-Ketelä M. Tailoring Enzymes Involved in the Biosynthesis of
Angucyclines Contain Latent Context-Dependent Catalytic Activities. Chem Biol.
2012;19(5):647-655. doi: 10.1016/j.chembiol.2012.04.010.
7. Kharel MK, Pahari P, Shepherd MD, Tibrewal N, Nybo SE, Shaaban KA, Rohr J.
Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat Prod Rep.
2012;29(2):264-325. doi: 10.1039/c1np00068c.
8. Kim J, Kim KH. Effects of minimal media vs. complex media on the metabolite profiles
of Escherichia coli and Saccharomyces cerevisiae. Process Biochem. 2017;57:64-71. doi:
10.1016/j.procbio.2017.04.003.

180
9. Koskiniemi H, Metsä-Ketelä M, Dobritzsch D, Kallio P, Korhonen H, Mäntsälä P,
Schneider G, Niemi J. Crystal Structures of Two Aromatic Hydroxylases Involved in the Early
Tailoring Steps of Angucycline Biosynthesis. J Mol Biol. 2007;372(3):633-648. doi:
10.1016/j.jmb.2007.06.087.
10. Kallio P, Liu Z, Mäntsälä P, Niemi J, Metsä-Ketelä M. Sequential Action of Two
Flavoenzymes, PgaE and PgaM, in Angucycline Biosynthesis: Chemoenzymatic Synthesis of
Gaudimycin C. Chem Biol. 2008;15(2):157-166. doi: 10.1016/j.chembiol.2007.12.011.
11. Kallio P, Patrikainen P, Belogurov GA, Mäntsälä P, Yang K, Niemi J, Metsä-Ketelä M.
Tracing the Evolution of Angucyclinone Monooxygenases: Structural Determinants for C-12b
Hydroxylation and Substrate Inhibition in PgaE. Biochemistry. 2013;52(26):4507-4516. doi:
10.1021/bi400381s.
12. Rogers JK, Taylor ND, Church GM. Biosensor-based engineering of biosynthetic
pathways. Curr Opin Biotechnol. 2016;42:84-91. doi: 10.1016/j.copbio.2016.03.005.
13. Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva
NA, Vederas JC, Tang Y. Complete Reconstitution of a Highly Reducing Iterative Polyketide
Synthase. Science. 2009;326(5952):589-592. doi: 10.1126/science.1175602.
14. Agmon N, Mitchell LA, Cai Y, Ikushima S, Chuang J, Zheng A, Choi W-J, Martin JA,
Caravelli K, Stracquadanio G, Boeke JD. Yeast Golden Gate (yGG) for the Efficient Assembly of
S. cerevisiae Transcription Units. ACS Synthetic Biology. 2015;4(7):853-859. doi:
10.1021/sb500372z.

181
5 A yeast three hybrid assay for metabolic engineering
of tetracycline derivatives
*The contents of this chapter were published in:
E. Herbst, P. A. Baldera-Aguayo, H. Lee and V. W. Cornish “A Yeast Three Hybrid Assay for
Metabolic Engineering of Tetracycline Derivatives” Biochemistry, 2018, 57, 4726-4734.
https://pubs.acs.org/doi/abs/10.1021/acs.biochem.8b00419

182
5.1 Chapter outline
Metabolic engineering stands to transform the discovery and production of a wide range of
chemicals, but metabolic engineering currently demands considerable resource investments that
restrict commercial application. To facilitate the applicability of metabolic engineering, general
high-throughput and readily-implemented technologies are needed to assay vast libraries of strains
producing desirable chemicals. Towards this end we describe here the development of a yeast three
hybrid (Y3H) assay as a general, high-throughput, versatile and readily-implemented approach for
the detection of target molecule biosynthesis. Our system detects target molecule biosynthesis
through a change in reporter gene transcription that results from the binding of the target molecule
to a modular protein receptor. We demonstrate the use of the Y3H assay for detecting the
biosynthesis of tetracyclines, a major class of antibiotics, based on the interaction between
tetracyclines and the tetracycline repressor protein (TetR). Various tetracycline derivatives can be
detected using our assay, whose versatility enables its use both as a screen and a selection to match
the needs and instrumentation of a wide range of end-users. We demonstrate the applicability of
the Y3H assay to metabolic engineering by differentiating between producer and non-producer
strains of the natural product anhydrotetracycline TAN-1612. The Y3H assay is superior to state-
of-the-art HPLC-MS methods in throughput and limit of detection of tetracycline derivatives.
Finally, our establishment of the Y3H assay for detecting the biosynthesis of a tetracycline
supports the generality of the Y3H assay for detecting the biosynthesis of many other target
molecules.

183
5.2 Introduction
Metabolic engineering is a potentially greener and more economical approach than traditional
organic synthesis for the production of a wide range of organic chemicals. As opposed to organic
synthesis, metabolic engineering is capable of producing valuable chemicals from readily available
and renewable carbon sources and significantly reducing the use of organic solvents and
reagents.(1, 2) Traditional methods in DNA mutagenesis and increasingly, advances in DNA
synthesis, DNA assembly and genome engineering are enabling high throughput strain
construction in metabolic engineering.(3-8) High-throughput strain construction is necessary
because not only the multi-gene biosynthetic pathway for the natural product, but also the
underlying metabolism of the host must be optimized.(9) Assuming a biosynthetic pathway of 5
genes and 10 interacting host genes, to test just 5 variants for each of these genes results already
in a library size exceeding 1010. However, the throughput of the most highly employed assaying
methods such as LC-MS is only ~102-103 samples per day and is thus heavily limiting in assaying
strains for successful metabolic engineering.(5, 9)
The currently employed methods for assaying metabolite production in metabolic engineering are
either low-throughput and general or high-throughput but not general. LC-MS methods are general
but low-throughput, enabling the screening of only about 102-103 samples per day.(9) Colorimetric,
fluorometric and growth assays for molecules that are colored, fluorescent, essential for producer
strain growth or could be easily converted to one of these, are high-throughput but not general.(10)
Attempts have been made to develop assays that are both high-throughput and general, but it is yet
to be determined whether or not these assays could be widely implemented.(10, 11) Versatile,
general, readily implemented high-throughput assays are required to transform metabolic

184
engineering from a field with a high potential to a field with an extensive real-world impact on the
way chemicals are made.
We set out to adapt the Y3H assay, previously employed for various basic science applications by
our laboratory and others,(12-18) to a general, readily implemented, high-throughput versatile
assay for metabolic engineering. We wished to develop an assay that can be readily adapted to
new target molecules, used as either a screen or a selection, and minimizes handling, to match
varying needs and instrumentation of end-users.
Here, we demonstrate the application of the Y3H assay to detect the biosynthesis of tetracyclines,
one of the major classes of antibiotics. We describe the engineering of the Y3H system to detect
tetracyclines by synthesizing a minocycline-methotrexate chemical inducer of dimerization (CID)
and cloning a LexA-TetR (tetracycline repressor) fusion protein.(14) Reporter gene expression
responds to tetracycline in our system by competitive binding of tetracycline and the CID to the
LexA-TetR protein fusion (Figure 5-1). Our assay can report the presence of tetracyclines using
various reporter gene outputs, enabling the use of the assay both as a screen and a selection to
match the needs and instrumentation of a variety of end-users. We show the applicability of our
assay to metabolic engineering through the differentiation between producer and non-producer
strains of the natural product anhydrotetracycline, TAN-1612.
Figure 5-1 Detection of a target molecule biosynthesis by the yeast three hybrid system.

185
Low/no production of a target molecule is detected by high reporter gene expression (left). High
production titers of the target molecule outcompete the chemical inducer of dimerization (CID)
from the fusion protein receptor and are detected by lower gene expression (right). DBD = DNA
binding domain; AD = activation domain; PR = protein receptor (e.g., TetR); DHFR =
dihydrofolate reductase.
5.3 Materials and methods
General methods. Absorption and fluorescence spectra were recorded on Infinite-M200
fluorescent spectrometer. DNA sequences were purchased from IDT. Polymerases, restriction
enzymes and Gibson Assembly mix were purchased from New England Biolabs. Sanger
sequencing was performed by Genewiz. Yeast strains were grown at 30 °C and shaker settings
were 200 rpm, unless otherwise indicated. Yeast transformations were done using the lithium
acetate method.(19) Plasmids were cloned and amplified using Gibson Assembly and cloning
strain C3040 (New England Biolabs).(20) Unless otherwise indicated, yeast strains were grown on
synthetic minimal media lacking histidine and/or uracil and/or tryptophan and/or leucine, as
indicated by the abbreviation HUTL-.(21) Yeast strain patches were obtained from glycerol stocks
by streaking on an agar plate of synthetic medium lacking the appropriate amino acid markers,
incubating at 30 °C for 3 days, patching single colonies onto a fresh agar plate and incubating at
30 °C overnight. Unless otherwise indicated, when 96 well plates were placed in the yeast shaker,
95% of the interface between the plate and its cover was wrapped with laboratory film (Parafilm,
Bemis PM999). All Y3H assays were performed with sterile components and three biological
replicates. Protein homology was calculated by BLAST (https://blast.ncbi.nlm.nih.gov) using the
standard sequence alignment parameters. DataExpress was used to analyze mass spectrometry

186
peaks. Protein visualization was performed using The PyMOL Molecular Graphics System,
Version 2.3 Schrödinger, LLC.
Codon optimization by COOL (http://cool.syncti.org/index.php) was used for all hydroxylases and
reductases unless noted explicitly that JCAT or no optimization was used (http://www.jcat.de/).
Optimization parameters chosen were individual codon use, codon context GC content of 39.3%
and S. cerevisiae organism. The following restriction sites were generally excluded: GAGACC,
GGTCTC, GGATCC, GAGCTC, CTCGAG, GAAGAC, GTCTTC, CGTCTC, GAGACG,
GAATTC, TTAATTAA, TCTAGA, ACTAGT, CCCGGG, CTGCAG, AAGCTT, GTCGAC,
ACGCGT, GGTACC, GCGGCCGC, AGATCT, GGCCGGCC, CCGCGG, GCTAGC,
CCATGG.
All chemical reactions were carried out under Argon atmosphere with anhydrous solvents unless
otherwise indicated. Analytical high-pressure liquid chromatography (HPLC) was performed with
a C-18 column, 2.6 μ, 250x4.6 mm, eluent given in parentheses. Unless stated otherwise, reactions
were monitored using analytical HPLC with a separation gradient of 10% to 90% MeCN in 0.1%
(v/v) TFA aqueous solution over 20 minutes. Preparative HPLC was carried out with a C-18 5 μ
column, 250x10 mm, eluent given in parentheses. NMR spectra were obtained using Bruker 400
MHz or 500 MHz instruments, as indicated. Unless stated otherwise, mass spectroscopy
measurements were performed on Advion CMS mass spectrometer equipped with atmospheric
pressure chemical ionization (APCI) and electrospray ionization (ESI) sources. TAN-1612
quantification was performed using a phenyl-hexyl 1.7 μ 100x3 mm column, on a Waters SQD2
quadrupole mass spectrometer equipped with a UPC2 SFC inlet, a photodiode array (PDA) UV-
Vis detector, and a dual ESI/APCI probe. HRMS spectra were taken on a Waters ACQUITY UPLC
BEH C18 column (2.1 x 50 mm) at 30 °C with a flow rate of 0.8 mL/min. Unless stated otherwise,

187
all reagents, salts and solvents were purchased from commercial sources and used without further
purification.
Synthesis of the Min-Mtx CID (1, Scheme 5-1). Step a. To 9-NH2-minocycline (100 mg, 0.2
mmol), Boc-11-aminoundecanoic acid (89 mg, 0.3 mmol), EDC (75 mg, 0.4 mmol) and HOBt (60
mg, 0.4) were added DMF (1 mL) and Et3N (139 μL, 1.0 mmol) and the reaction was mixed at r.t
for 6 h. The reaction progress was monitored by RP-HPLC. Aqueous NaHCO3 (0.05 M)(22) was
then added and the aqueous phase was extracted three times with CHCl3. The combined organic
fraction was extracted with brine, the combined brine extracts were extracted twice with CHCl3,
the combined organic fraction was dried with Na2SO4 and the solvent was removed under reduced
pressure. The crude product was purified in two batches by preparative RP-HPLC (30-35% MeCN
in 99.9%:0.1% H2O:TFA, 45 min) to afford compound 3 (53.4 mg, 36%) as a yellow solid. 1H-
NMR (400 MHz, MeOD d4) δ 8.60 (s, 1H), 4.11 (s, 1H), 3.24 (dd, J = 15.4, 4.2, 1H), 3.14 (s, 6H),
3.08 (m, 3H), 3.02 (s, 6H), 2.50 (t, J = 7.4, 2H), 2.44 (t, J = 14.3, 1H), 2.24 (m, 1H), 1.72 (m, 1H),
1.62 (m, 1H), 1.43 (s, 9H), 1.42 (m, 4H) 1.38 (m, 2H), 1.32 (m, 10H). MS (APCI+): m/z calc. for
C39H58N5O10+: 756.42; found: 756.4 [M + H]+; MS (APCI-): m/z calc. for C39H56N5O10
-: 754.40;
found: 754.3 [M – H]-.
Steps b-d. To compound 3 (27.3 mg, 0.036 mmol) were added DCM (0.5 mL) and TFA (0.5 mL)
and the reaction was mixed at r.t. for 12 min. The solvent was removed under reduced pressure.
PhMe was added and the solvent was again removed under reduced pressure. PhMe addition and
solvent removal was repeated once more. Methotrexate-α-OtBu (27.5 mg, 0.054 mmol)(23, 24)
and PyBOP (37.5 mg, 0.072 mmol) were added, followed by DIPEA (11.5 mg, 0.09 mmol) in
DMF (1 mL) and the reaction was mixed at r.t.. After 22 h EtOAc and aqueous NaHCO3 were
added and the aqueous phase was extracted twice with EtOAc. The combined organic fraction was

188
extracted with brine and the brine extract was filtered. The solid residue was redissolved in MeOH
and the solvent was removed under reduced pressure. The crude product was purified in two
batches by preparative RP-HPLC (20-30% MeCN in 99.9%:0.1% H2O:TFA, 50 min). Following
solvent removal under reduced pressure, TFA (2 mL) was added and the reaction was stirred at r.t.
for 90 min. The reaction progress was monitored by RP-HPLC. The solvent was removed under
reduced pressure to afford compound 1 (4.1 mg, 10%) as a yellow solid. 1H-NMR (500 MHz,
MeOD d4) δ 8.64 (s, 1H), 8.25 (s, 1H), 7.76 (d, J = 8.8, 2H), 6.86 (d, J = 8.8, 2H), 4.92 (s, 2H),
4.54 (dd, J = 8.8, 4.5, 1H), 4.08 (s, 1H), 3.27 (s, 3H), 3.11 (m, 2H), 3.00 (s, 6H), 2.83 (s, 3H), 2.80,
(s, 3H), 2.47 (m, 2H), 2.41-2.05 (m, 6H), 1.70-1.59 (m, 4H), 1.48-1.28 (m, 16H) MS (ESI+): m/z
calc. for C54H71N13O122+: 1093.54; found: 546.9 [M + 2H]2+.
Y3H strain construction
LexA-TetR plasmids. Plasmids PBA-Gib5 and HL-249-1 through HL-249-6 encoding LexA-
TetR protein fusions for the various TetR classes were cloned by Gibson Assembly of the
appropriate TetR sequence with pMW103 digested with restriction enzymes BamHI-HF and
EcoRI-HF (Table S3). All TetR sequences with the exception of TetR(B) were obtained as IDT
gBlocks. TetR(B) sequence was amplified from pET21d-TetR, courtesy of A. Davidson.(25, 26)
LacZ assay strain construction. V506 was grown overnight in UT- media.(27) The culture was
then diluted 104-105X with UT- and plated on UT- plates. Single colonies were streaked on HTU-
and UT- plates and a streak growing on UT- but not on HTU-, indicating the loss of the
pMW3(GSG)2rGR2 plasmid, was glycerol stocked as PBA5. This strain was further used for the
transformation of the LexA-TetR plasmids to generate LacZ assay strains PBA8 and HL-260-1
through HL-260-7.

189
Growth assay strain construction. Yeast strain LW2635(28) was transformed with plasmid
PBA-Gib5 to generate strain PBA6. PBA6 was transformed with pMW2eDHFR(27) to generate
the Y3H growth assay strain PBA14.
mCherry reporter plasmid. Reporter plasmid HL-174-2 encoding mCherry under the
transcriptional control of LexA operators was cloned by Gibson Assembly of an mCherry sequence
amplified from yEpGAP-Cherry(29) and an 8LexA-min-pGal1 sequence amplified from
pMW112(14) into pRS416 digested with SacI and XhoI.
Fluorescent protein assay strain construction. V506 was grown overnight in T- media. The
culture was then diluted 104-105X with T- and plated on T- plates. Single colonies were streaked
on TU-, HT- and T- plates and a streak growing on T- but not on UT- or on HT-, indicating the loss
of the pMW106 and pMW3(GSG)2rGR2 plasmids, was glycerol stocked as PBA3. This strain was
transformed with plasmid HL-174-2 to generate yeast strain HL-294-7. The latter was transformed
with PBA-Gib5 to generate the Y3H fluorescent reporter assay strain HL-2-4-1.
TAN-1612 producer/non-producer strain construction. TAN-1612 producer and non-producer
strains EH-3-54-4 and EH-3-54-2 were obtained by transforming plasmids pYR342 and pYR291
or pYR342 and pRS426, respectively, into yeast strain BJ5464-NpgA (Table 5-1 and Table
5-2).(30)
Protocol for fluorescent protein Y3H assay
Fresh patches of Y3H strain HL-2-4-1 were inoculated into HTU- media (1 mL) in 24-well plates
(flat, clear bottom, Corning #3526) and placed in shaker. After 24 hours the cells were pelleted by
centrifugation at 3,250 rpm and resuspended in H2O (1 mL). Pelleting and resuspension were
repeated once more. OD600 was measured by diluting 10X into H2O and the cells were diluted
accordingly into H2O for OD600 = 1 to be used as a 10X solution. 20 μL cells of OD600 = 1 were

190
added to the assay plate as the last component in the assay plate (flat, clear bottom, black 96-well
plate, Corning #3603). Each well of the assay plate contained 200 μL total liquid volume, 20 μL
cells, 100 μL of 2X HTU- media of 4% raffinose and no glucose, 20 μL of 10X 20% galactose
aqueous solution, 20 μL of 10X CID solution of 100 μM in 10% DMSO aqueous solution, 20 μL
supernatant and 20 μL purified TAN-1612 in a 10% DMSO aqueous solution,(30) or a 10% DMSO
aqueous solution. Starting conditions in the assay plate were OD600 = 0.1, 2% galactose, 2%
raffinose, 0% glucose, 1X HTU-, 10 μM CID, 2% DMSO. The assay plate was placed in the 30 °C
shaker overnight before measuring OD600 and 620 nm fluorescence (λex = 588 nm).(31)
TAN-1612 supernatants from flask cultures were obtained by inoculating TAN-1612 producing
and non-producing strains, EH-3-54-4 and EH-3-54-2 respectively, in UT- media (5 mL) in 15 mL
culture tubes (Corning 352059) and placing in shaker overnight; Overnight cultures were used to
inoculate 100 mL YPD in 500 mL conical flasks with a starting OD600 of 0.08. After 68 h, 1 mL
of culture was pelleted in 1.5 mL Eppendorf tubes at 14,000 rpm, sterile filtered and diluted 20X
into H2O to be used as a 10X stock solution.
TAN-1612 supernatants from 96-well plate cultures were obtained by inoculating 12 and 84
colonies of TAN-1612 producing and non-producing strains, EH-3-54-4 and EH-3-54-2
respectively, in UT- media (0.5 mL) in a 96-well plate (Corning P-2ML-SQ-C-S). The plate was
covered with two layers of SealMate film (Excel Scientific, SM-KIT-BS) and placed in a shaker
overnight. Overnight cultures were used to inoculate 300 μL YPD in an identical setup with a
starting OD600 of 0.1 and placed in 800 rpm shaker overnight. After 54 h, 150 μL of H2O were
added to the cultures to prevent drying of cultures due to evaporation and two new layers of
Sealmate film were replaced. After 78 h, the cultures were pelleted at 3,250 rpm and the
supernatant was diluted 20X into H2O to be used as a 10X stock solution.

191
Protocol for Y3H growth assay
Fresh patches of Y3H strain PBA14 were inoculated in HT- media (1 mL) and placed in shaker
overnight. Cultures were then centrifuged at 3,000 rpm for 3 min and the pellets were resuspended
with H2O (1 mL). Pelleting and resuspension was repeated twice more. OD600 was measured and
the cells were diluted accordingly into H2O to OD600 = 1 to be used as a 10X stock solution. 20 μL
cells of OD600 = 1 were added as the last component to the assay plate (flat, clear bottom, black
96-well plate, Corning #3603). Each well of the assay plate contained 200 μL total liquid volume,
20 μL cells, 100 μL of 2X HT- media with 4% raffinose, 4% galactose, 0% glucose, 0.4% 5-FOA,
20 μL of 0/250 μM 10X CID in 10% DMSO aqueous solution, 20 μL of doxycycline (0/50 μM in
10% EtOH aqueous solution) and 40 μL H2O. Starting conditions in the assay plate were OD600 =
0.1, 2% galactose, 2% raffinose, 0% glucose, 0.2% 5-FOA, 1X HT-, 25 μM CID, 1% DMSO, 1%
EtOH. The assay plate was placed in the 30 °C shaker for 5 days with daily measurements of
OD600.
Protocol for Y3H LacZ assay
Fresh patches of Y3H strains PBA-8 and HL-260-1 through HL-260-7 were inoculated into HTU-
media (1 mL) in 24-well plates (flat, clear bottom, Corning #3526) and place at 30 °C shaker. After
24 hours the cells were pelleted by centrifugation at 3000 rpm for 5 min and resuspended in H2O
(1 mL). Pelleting and resuspension were repeated once more. OD600 was measured and the cells
were diluted accordingly into H2O to OD600 = 1 to be used as a 10X solution. 20 μL cells of OD600
= 1 were added to the assay plate as the last component in the assay plate (clear v-bottom, clear
96-well plate, Corning #3894). Each well of the assay plate contained 200 μL total liquid volume,
20 μL cells, 100 μL of 2X HTU- media of 4% raffinose and no glucose, 20 μL of 10X 20%
galactose aqueous solution, 20 μL of 10X CID solution in 10% DMSO aqueous solution or a 10%

192
DMSO aqueous solution, 20 μL of the tetracycline ligand in 10% EtOH aqueous solution and 20
μL water. Starting conditions in the assay plate are OD600 = 0.1, 2% galactose, 2% raffinose, 0%
glucose, 1X HTU-, varying concentration CID, varying concentration of tetracycline ligand, 1%
DMSO and 1% EtOH. The assay plate was placed in the shaker overnight. OD measurements were
then taken after 24 h and the cells were pelleted and resuspended in 100 μL Z-buffer (60 mM
Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 2 mM MgSO4, pH adjusted to 7, 2.7 mL/L β-
mercaptoethanol added fresh the day of the assay). The cells were pelleted again, resuspended in
100 μL Y-PER buffer (Fisher Scientific, #78990) and lysed for 30 min at r.t. before the addition
of o-Nitrophenyl β-D-galactopyranoside (ONPG) in Z-buffer (8.5 μL, 10 mg/mL). After 90 min a
Na2CO3 solution (1 M, 110 μL) was added to quench the reaction. After pelleting at 3,000 rpm,
175 μL of the supernatant was transferred into flat clear-bottom 96-well plates (Corning #353072)
and OD420 was measured. The limit of detection was calculated as the concentration of molecule
at which the signal-to-noise ratio of the assay was > 3:1.(32, 33)
5.4 Results
5.4.1 Y3H system design and CID design and synthesis
For a Y3H system detecting tetracyclines, we cloned a Y3H strain encoding LexA-TetR and
DHFR-B42 fusion proteins and synthesized a minocycline-methotrexate (Min-Mtx) CID (Figure
5-1). We chose to base our Y3H system on well-studied components. Namely, the LexA DNA
binding domain (DBD) and B42 activation domain (AD),(34) respectively, introduced by Brent
and coworkers,(14, 27, 35, 36) as well as the DHFR- methotrexate and TetR-tetracycline
interactions. The latter two are highly characterized receptor-ligand interactions of picomolar
affinity,(37) and the methotrexate-DFHR interaction has been used numerous times by our
laboratory.(27, 35) We used LexA-TetR fusion proteins of TetR variants that were highly studied,

193
such as the variants from classes B and D,(37-39) as well TetR variants from the much less studied
TetR classes A,(40, 41) C,(42, 43) E,(44, 45) G,(46) and H.(47-50) TetR and LexA form stable
homodimers, with each of the TetR monomers binding tetracyclines and LexA binding DNA as a
dimer.(38, 51) Thus it is possible that the monomer ratio of TetR / LexA in the LexA-TetR fusion
protein is unequal to 1, affecting accordingly the local concentrations of the Min-Mtx CID and
DHFR-B42 as well. In any case, increased levels of tetracyclines are expected to outcompete the
CID from the LexA-TetR protein fusion and lower reporter protein expression (Figure 5-1).
Our design and synthesis of the Min-Mtx CID is based on the wealth of structure activity
relationship (SAR) data for the interaction of tetracyclines with TetR, as well as our laboratory’s
previously reported CIDs.(27, 35) It is known from inspection of the high-resolution structure of
TetR bound to tetracycline derivatives as well as biochemical characterization that tetracyclines
can be derivatized at the D ring without disrupting binding to TetR (Scheme 5-1).(37, 38, 52)
Moreover, the FDA-approved analog tigecycline and the clinical candidates eravacycline and TP-
271 are all 9-amidotetracyclines;(53, 54) thus, synthesis of a 9-amidotetracycline CID was logical.
Specifically, 9-aminominocycline was chosen as the starting material because of its high acid
stability relative to tetracycline as well as its commercial availability.(55) The design and synthesis
of the Mtx component of the Min-Mtx CID was based on our laboratory’s previously reported
methotrexate CIDs (Scheme 5-1).(27, 35)
Scheme 5-1 Synthesis of the chemical inducer of dimerization (CID) Min-Mtxa

194
aReagents and conditions: (a) Boc-11-aminoundecanoic acid (1.5 equiv), EDC (2 equiv), HOBt (2
equiv), Et3N (5 equiv), DMF, 22 oC, 6 h, 36%; (b) TFA:DCM (1:1), 22 oC, 12 min; (c)
Methotrexate-α-OtBu (1.5 equiv),(23, 24) PYBOP (2 equiv), DIPEA (2.5 equiv), DMF, 22 oC, 22
h; (d) TFA, 22 oC, 1.5 h, 10% over three steps. EDC = N-(3-Dimethylaminopropyl)-N′-
ethylcarbodiimide hydrochloride, HOBt = 1-hydroxybenzotriazole, TFA = trifluoroacetic acid,
PyBOP = (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate, DIPEA = N-
Ethyldiisopropylamine.
5.4.2 Initial characterization of the Y3H assay for tetracycline derivatives
With the Y3H strains and Min-Mtx CID at hand we verified the functionality of our Y3H system
and determined the optimal CID concentration and TetR variant to further use in the assay. First,
we confirmed that reporter gene output in our system is dependent upon the presence of the CID
and determined the desired range of CID concentration to use in the assay (Figure 5-2a). We then
tested candidates from each of the TetR classes as protein receptors and observed that the Y3H
system is responsive to the CID with all known TetR classes except for the TetR(C) variant we
used (Figure 5-2b). Given that TetR(B) and TetR(G) showed the best response to the CID and to
maintain consistency with the highly studied TetR(B),(37-39) we focused our further experiments
on TetR(B).

195
We show that our assay can differentiate sub-μM concentrations of various tetracyclines (Figure
5-2c), key for its applicability to differentiate between high and low producer strains. Our Y3H
system differentiates doxycycline, tetracycline and 9-NH2-minocycline concentrations in the range
of 2 – 200 nM, 0.02 – 2 μM and 0.2 – 20 μM, respectively. The limit of detection in our Y3H assay
is ≤ 0.2 μM for doxycycline and ≤ 2 μM for tetracycline and 9-NH2-minocycline.(32)
Figure 5-2 Characterization of the dynamic range of the Y3H assay for tetracyclines
Characterization of the dynamic range of the Y3H assay for tetracyclines in (a) varying
concentrations of the Min-Mtx CID, (b) varying TetR classes and (c) varying target molecule
structure and concentrations. LacZ was used as a reporter gene. Miller units were calculated as

196
(1000*OD420)/(well-volume(mL)*reaction-time(min)*OD600).(56) “+TetR” and “-TetR” are
strains HL-260-5 and HL-260-7 encoding LexA-TetR(G) and LexA, respectively; Error bars
represent the standard error of the mean (s.e.m) from three biological replicates.
After confirming the functionality of the Y3H system for tetracyclines we proceeded to show that
it can report the presence of tetracyclines with a modular output, key for enabling versatile use by
various end-users. Using a URA3 reporter gene and growing the yeast in the presence of 5-
fluorouracil (5-FOA) we show that yeast growth is doxycycline-dependent in the presence, but not
in the absence of the Min-Mtx CID (Figure 5-3a). We additionally show that the Y3H assay for
tetracyclines can be used as a colorimetric as well as a fluorometric assay, by employing a LacZ
or and mCherry reporter gene, respectively (Figure 5-3b, Figure 5-3c). As expected, while reporter
gene activity is dependent on CID and doxycycline in the Y3H strain, such dependence is not
detected in a control strain excluding the eDFHR-AD fusion protein (Figure 5-5).

197
Figure 5-3 The modularity of the yeast three hybrid assay enables both screening and
selecting for biosynthesis of target tetracyclines
The modularity of the yeast three hybrid assay enables both screening and selecting for
biosynthesis of target tetracyclines by enabling (a) a growth assay, (b) a colorimetric assay and a
(c) fluorometric assay. (a) Yeast growth, (b) LacZ transcription and (c) mCherry transcription are
dependent on doxycycline (Dox) in the presence, but not in the absence of the CID. CID, Dox
concentrations = (a) 25 μM, 5 μM (b) 25 μM, 0.2 μM (c) 10 μM, 1 μM. Error bars represent the
standard error of the mean (s.e.m) from three biological replicates. Miller units were calculated as
(1000*OD420)/(well-volume(mL)*reaction-time(min)*OD600).(56) mCherry (620 nm)/OD600
was calculated by dividing mCherry fluorescence (620 nm, λex = 588 nm) by OD600.

198
5.4.3 Applying the Y3H assay to the detection of target molecule biosynthesis
We demonstrate the applicability of the Y3H assay to metabolic engineering by differentiating
between producer and non-producer strains of TAN-1612 (Figure 5-4a).(30) For the TAN-1612
producer strain we used S. cerevisiae strain EH-3-54-4, encoding all four genes of the TAN-1612
biosynthetic pathway as previously reported by Tang and coworkers and producing ~60 μM TAN-
1612 (Supplementary methods).(30) The non-producer control we used, S. cerevisiae strain EH-
3-54-2, does not encode the polyketide synthase AdaA and has no measurable production of TAN-
1612 (data not shown).
The Y3H assay for tetracyclines can differentiate between supernatants of producer and non-
producer strains of TAN-1612 (Figure 5-4b). When purified TAN-1612 is spiked into non-
producer supernatants, the resulting Y3H signal resembles the signal obtained with producer
supernatant (Figure 5-4b). These results agree with our hypothesis that TAN-1612, present in the
producer strain supernatant but not in the non-producer strain supernatant, outcompetes the CID
from the LexA-TetR fusion protein.
The Y3H assay for tetracyclines also differentiates TAN-1612 producer colonies from a non-
producing population cultured in a 96-well plate (Figure 5-4c). The 12 producer colonies gave an
average mCherry fluorescence / OD600 signal of 132 with a standard deviation of 17 and the 84
non-producer colonies gave an average mCherry fluorescence / OD600 signal of 210 with a standard
deviation of 16. Thus, assuming normal distribution there is less than 2.5% overlap in the Y3H
signal from producer and non-producer colonies.

199
Figure 5-4 Differentiation of a producer and a non-producer strain of a target molecule by
the Y3H assay for tetracyclines, demonstrating the applicability of the Y3H assay to
metabolic engineering.
(a) Workflow for screening colonies for production of a target molecule using the Y3H assay. (b)
The Y3H assay for tetracyclines differentiates between the supernatants of TAN-1612 producer
and TAN-1612 non-producer cultured in shake flasks. (c) The Y3H assay for tetracyclines
differentiates the supernatants of TAN-1612 producer colonies from a non-producing population
cultured in a 96-well plate. The non-producer strain does not encode AdaA, the polyketide
synthase of the TAN-1612 biosynthetic pathway, while the producer strain encodes the complete

200
pathway (Table 5-1 and Table 5-2). The concentration of TAN-1612 in the Y3H assay well from
the supernatant and from the purified sample is ~0.3 μM and 0.5 μM, respectively (Supplementary
methods). mCherry (620 nm)/OD600 was calculated by dividing mCherry fluorescence (620 nm,
λex = 588 nm) by OD600. Error bars represent the standard error of the mean (s.e.m) from three
biological replicates of the Y3H strain and two biological replicates of the TAN-1612
producer/non-producer.
5.5 Discussion
The Y3H assay for tetracyclines is advantageous compared to the state-of-the-art methods for
tetracycline detection in terms of throughput, cost, instrumentation requirements and limit of
detection. Being readily performed in 96-well microtiter plates, the Y3H assay throughput is >1000
samples / hour when measured by a standard laboratory plate reader (Infinite-M200). By
comparison, the throughput of the previous state-of-the-art HPLC-based methods for tetracycline
detection is <10 samples/hour.(33, 57) The use of a plate reader to measure the Y3H assay output
is also advantageous in terms of cost to state-of-the-art chromatography-based methods such as
HPLC and even more so, LCMS. The price difference is both in the initial investment in
instrumentation, as spectrophotometers are cheaper than LC instruments, and in the reagent costs
of 96 well plates vs LC-grade solvents. In terms of limit of detection the Y3H assay has a limit of
detection of ≤ 200 nM for doxycycline (Figure 5-2c), which is better than the state-of-the-art HPLC
limit of detection of ≥ 4.5 μM for doxycycline.(33, 57) The low limit of detection our assay exhibits
enables it to differentiate producer from non-producer strains even in low titers of initial strain
development (Figure 5-4 and Section 5.7.1 5.7.2, ~60 μM). Differentiating between production
levels in higher titer ballparks in later stages of producer strain optimization would simply require
a higher dilution factor of supernatants into the Y3H strain media. Future generations of this Y3H

201
assay might include TetR mutants that are more specific and have a lower detection limit to
tetracycline derivatives of interest.
The Y3H assay for metabolic engineering also compares well with our laboratory’s recently
published FP assay,(58) because of two key differences in setup requirements and output. First,
since the Y3H assay does not require the purification of the protein receptor, it is used much more
readily to screen for the ultimate protein receptor for the assay. Thus, we show here the first
comparison of all TetR classes for binding of a small molecule in an attempt to identify the best
TetR variant for differentiating between + and – CID (Figure 5-2b). Further optimization of a
protein receptor for the assay of a specific target molecule is readily enabled in the Y3H assay
simply by cloning the protein variants into the same plasmid backbone and transforming into yeast.
The versatility in choice of output in the Y3H assay is a key enabling factor for the advantages the
Y3H assay has over the FP assay since it makes the assay accessible to a wider range of users. The
fluorescent output in the Y3H assay enables measurements with a standard laboratory plate reader,
eliminating the requirement for anisotropy capacities (Figure 5-3c). The LacZ output could also
be measured by a standard plate reader as well as by the naked eye (Figure 5-3b).
If the Y3H system and the metabolic pathway for the small molecule of interest are cloned into
the same strain, colonies can be assayed for production even more directly, increasing throughput
and minimizing handling. The Y3H assay can then enable screening colonies for production of the
target molecule directly on an agar plate without the need for further handling of the colonies in
microtiter plates. The agar plate screen could be performed with the naked eye or a simple camera
by using X-Gal plates and the LacZ reporter or fluorescent camera/microscope by using the
mCherry reporter. Finally, the Y3H assay throughput could be vastly increased by selecting for
high producing strains en masse in flask or fermenter culturing using the growth assay or

202
fluorescence assisted cell sorting (FACS). Such en masse screening methods not only greatly
enhance the throughput but also minimize the labor, instrumentation, cost and time, required to
obtain results. The Y3H assay for tetracyclines can report the presence of a target molecule with
any of the above mentioned outputs (Figure 5-3), enabling metabolic engineering applications for
a range of end users with varying needs and instrumentation availabilities.
While a TetR-tetO-based assay could conceivably be developed for the metabolic engineering of
tetracycline derivatives,(59) our TetR-based Y3H assay is advantageous for two main reasons. For
one, the Y3H assay can potentially be applied to a greater variety of tetracycline derivatives as
TetR-binding to the tetracycline suffices and there is no demand for a TetR conformational change.
By comparison, signal transduction in the TetR-tetO system is dependent on coupling between
tetracycline binding and a conformational change in TetR.(26, 38) It has been hypothesized that
high-affinity binding may be a necessary but not sufficient property for TetR induction in the TetR-
TetO system.(60) In practice, while it was possible to detect binding to a tetracycline analog, 4-
de(dimethylamino)-anhydrotetracycline, no induction was detected using a TetR-TetO-based
assay.(37) Thus, the use of TetR in a Y3H system can facilitate the evolvement of TetR mutants
with specific binding to tetracycline derivatives of interest in future generations of the assay.
Furthermore, the Y3H system for metabolic engineering can be readily modified to apply for many
metabolites of interest beyond tetracyclines, as detailed below. In contrast, the TetR-TetO system
is much more limited in the scope of molecules it could be applied to, namely transcriptional
regulators.(61)
A Y3H system as described here for the metabolic engineering of tetracycline derivatives can be
designed for any target molecule with a receptor. Given T, a desirable target, PR, a protein receptor
for T, and T’, a readily derivatizable analog of T, two new components need to be generated for a

203
Y3H assay to detect biosynthesis of T. The first is a Mtx-T’ CID and the second is a LexA-PR
fusion protein (Figure 5-1). This approach is especially amenable for the development of assays
for molecules of pharmacological interest. The reason is that such molecules often have a known
protein target as well as an established derivatization chemistry and SAR that greatly support the
design and synthesis of a CID. More broadly, building from the Cornish Laboratory’s early reports
of chemical complementation,(17, 62, 63) this work advances the enabling technology of the Y3H
system for the emerging field of synthetic biology for handling the diversity of engineering multi-
gene pathways and assaying for chemistry not natural to the cell.
5.6 Conclusion
This chapter describes the development of a yeast three hybrid (Y3H) assay as a general, high-
throughput, versatile and readily-implemented approach for the detection of target molecule
biosynthesis. We demonstrate the use of the Y3H assay for detecting the biosynthesis of
tetracyclines, a major class of antibiotics, based on the interaction between tetracyclines and the
tetracycline repressor protein (TetR). We demonstrate the applicability of the Y3H assay to
metabolic engineering by differentiating between producer and non-producer strains of the natural
product anhydrotetracycline TAN-1612. The Y3H assay is superior to state-of-the-art HPLC-MS
methods in throughput and limit of detection of tetracycline derivatives. Finally, our establishment
of the Y3H assay for detecting the biosynthesis of an anhydrotetracycline supports the generality
of the Y3H assay for detecting the biosynthesis of many other target molecules. Specifically, future
versions of this assay to screen or select for further modifications of TAN-1612 that could not be
assayed by the UV/Vis assay for hydroxylation of anhydrotetracycline used in the previous
chapters are envisioned.

204
5.7 Appendix
5.7.1 Supplementary figures
Figure 5-5 LacZ readout of strain harboring plasmid encoding for LexA-TetR (PBA-8) and
strain without the plasmid (PBA-5).
Only the strain with the LexA-TetR fusion protein responds to the CID. Addition of Dox lowers
reporter gene expression only in the strain with LexA-TetR in the presence of the CID.
Concentrations of CID and Dox are 25 μM and 200 nM, respectively. Miller units were calculated
as (1000*OD420)/(well-volume(mL)*reaction-time(min)*OD600).(56) Error bars represent the
standard error of the mean (s.e.m) from three biological replicates.
0
100
200
300
400
500
+ CID+Dox
+ CID-Dox
− CID +Dox
− CID -Dox
+ CID+Dox
+ CID-Dox
− CID +Dox
− CID -Dox
+LexA-TetR -LexA-TetR
Mill
er
Un
its

205
Figure 5-6 standard curve for TAN-1612 quantification in cultures.
A Purified TAN-1612 was analyzed at different concentrations on a separation gradient of 5% to
95% MeCN in supercritical CO2 over 5.5 minutes. Absorption between 390 nm to 410 nm was
integrated at retention times of 0.87 – 1.50 min, which corresponds to TAN-1612 (MS (AI-): m/z
calc. for C21H17O9-: 413.09; found: 413.4 [M - H]-; MS (EI-): m/z calc. for C21H17O9
-: 413.09;
found: 413.4 [M - H]-).
5.7.2 Supplementary methods
Quantification of TAN-1612 concentration in cultures.
TAN-1612 cultures for quantification were obtained by inoculating two biological replicates of
each of TAN-1612 producing and non-producing strains, EH-3-54-4 and EH-3-54-2 respectively,
in UT- media (5 mL) in 15 mL culture tubes (Corning 352059) and placing in shaker overnight.
Overnight cultures were used to inoculate 100 mL YPD in 500 mL conical flasks with a starting
OD600 of 0.08. After 68 h, the 100 mL from the two biological replicates were combined and
extracted with 200 mL of EtOAc in 8 x 50 mL falcon tubes (Corning 352098) by mixing at
maximal speed in Orbital Shaker (Bellco Glass) at r.t. for 1.5 h and centrifuging at 3,300 rpm at
r.t. for 15 min. The solvent of the combined organic phase was removed under reduced pressure
y = 0.016x + 8.1328R² = 0.9933
0
2
4
6
8
10
12
14
0 50 100 150 200 250 300
Ln
(ab
so
rpti
on
39
0-4
10
nm
)
TAN-1612 (μM)

206
and the sample was redissolved in MeOH (1.5 mL) and filtered (Acrodisc® Syringe Filters, 13mm,
0.2 μm, Pall Laboratory, PTFE Membrane, 4422). The filtered sample was diluted 50X in MeOH
and analyzed by Supercritical Fluid Chromatography / Mass spectrometry (SFC/MS) equipped
with a photodiode array (PDA) UV-vis detector. Absorption between 390 nm to 410 nm was
integrated at retention times of 0.87 – 1.50 min, which corresponds to TAN-1612 (MS (AI-): m/z
calc. for C21H17O9-: 413.09; found: 413.4 [M - H]-. The TAN-1612 concentration in the extract
was calculated to be 161 μM according to the following linear function:
concentration=[ln(absorption 390-410 nm)-8.1328]/0.016 (Figure 5-6). The concentration of
TAN-1612 in the culture is therefore estimated to be ~60 μM, assuming a quantitive yield for the
extraction process.
5.7.3 Supplementary tables
Table 5-1 Strains used in this study
Strain Genotype Source/Reference
FY250 MATα ura3-52 trp1 63 his3 200 leu2 1 gal L.P. Guarentea
LW2635 MATa trp1Δ63 his3Δ200 8lexAop-Spo13-URA3
leu2Δ1 Gal+
/(28)
BJ5464-
NpgA
MATα ura3-52 his3-Δ200 leu2-Δ1
trp1 pep4::HIS3 δ:: pADH2-npgA-tADH2 prb1Δ1.6R can1
GAL
Y. Tangb/(64)
V506 FY250 pMW106, pMW3(GSG)2rGR2, pMW2eDHFR /(27)
PBA-5 FY250 pMW106, pMW2eDHFR This study
HL-260-1 FY250 pMW106, pMW103TetR(A), pMW2eDHFR This study
PBA-8 FY250 pMW106, pMW103TetR(B), pMW2eDHFR This study
HL-260-2 FY250 pMW106, pMW103TetR(C), pMW2eDHFR This study
HL-260-3 FY250 pMW106, pMW103TetR(D), pMW2eDHFR This study
HL-260-4 FY250 pMW106, pMW103TetR(E), pMW2eDHFR This study
HL-260-5 FY250 pMW106, pMW103TetR(G), pMW2eDHFR This study
HL-260-6 FY250 pMW106, pMW103TetR(H), pMW2eDHFR This study
HL-260-7 FY250 pMW106, pMW103(empty), pMW2eDHFR This study
PBA-6 LW2635 PBA-Gib5 This study
PBA-14 LW2635 PBA-Gib5, pMW2eDHFR This study
PBA-3 FY250 pMW2eDHFR This study
HL-294-7 FY250 pMW2eDHFR HL-174-2 This study
HL-2-4-1 FY250 pMW2eDHFR pMW103TetR(B), HL-174-2 This study

207
EH-3-54-4 BJ5464-NpgA pYR291, pYR342 This study
EH-3-54-2 BJ5464-NpgA pRS426, pYR342 This study a L. P. Guarente, Massachusetts Institution of Technology, Biology Department
b Y. Tang, University of California, Los Angeles, Department of Chemical and Biomolecular
Engineering & Department of Chemistry and Biochemistry, Department of Bioengineering
Table 5-2 Plasmids used in this study
Plasmid Description Source/Reference
pMW2eDHFR pGal1-eDHFR-B42, trp marker, 2 μ /(27)
pMW106 LacZ under control of 8 tandem LexA operators, Ura
marker
/(27, 65)
pMW103 pADH1-LexA (empty), His marker, 2 μ R. Brenta/(65)
pMW112 LacZ under control of 8 tandem LexA operators, Ura
marker
R. Brent/(14, 65)
HL-249-1 pADH1-LexA-TetR(A)-tADH1 on pMW103 This study
PBA-Gib5 pADH1-LexA-TetR(B)-tADH1 on pMW103 This study
HL-249-2 pADH1-LexA-TetR(C)-tADH1 on pMW103 This study
HL-249-3 pADH1-LexA-TetR(D)-tADH1 on pMW103 This study
HL-249-4 pADH1-LexA-TetR(E)-tADH1 on pMW103 This study
HL-249-5 pADH1-LexA-TetR(G)-tADH1 on pMW103 This study
HL-249-6 pADH1-LexA-TetR(H)-tADH1 on pMW103 This study
pRS416 Shuttle vector (empty), uracil marker, CEN ATCC87521
HL-174-2 8LexAOp-minpGal1-mCherry-tCyc1, Ura marker, CEN This study
pYR291 AdaA expression in S. cerevisiae, uracil marker, 2 μ Y. Tangb/(30)
pYR342 AdaB-D expression in S. cerevisiae, Tryp marker, 2 μ Y. Tang/(30)
pRS426 Shuttle vector (empty), uracil marker, 2 μ ATCC77107
pET21d-TetR TetR(B) with C-term Histidine tag A.
Davidsonc/(25)
yEpGAP-
Cherry
mCherry, uracil marker, 2 μ Neta Deand/(29)
a R. Brent, Fred Hutchinson Cancer Research Center, Division of Basic Sciences
b Y. Tang, University of California, Los Angeles, Department of Chemical and Biomolecular
Engineering & Department of Chemistry and Biochemistry, Department of Bioengineering
c A. Davidson, Dept. of Molecular Genetics, Dept. of Biochemistry, Faculty of Medicine,
University of Toronto
d Neta Dean, Department of Biochemistry and Cell Biology, Stony Brook University
Table 5-3 Sequences of the various TetR classes in the within the pMW103 backbone.

208
Sequences for the TetR classes are shown decapitalized within the context of LexA-DBD (partial,
capitalized) in the 5’ end and the tADH1 terminator at the 3’ end (partial, capitalized).
The NCBI and GenBank Reference Sequences used to order/clone DNA for the different TetR
classes A, B, C, D, E, G and H are NC_006143.1, CP012140.1, NC_002056.1, CP000650.1,
LKJQ01000048.1, CP003225.1 and AJ514834.1, respectively.
TetR(A) –
HL-249-1
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatgacaa
agttgcagccgaatacagtgatccgtgccgccctggacctgttgaacgaggtcggcgtagacggtctgacgacac
gcaaactggcggaacggttgggggttcagcagccggcgctttactggcacttcaggaacaagcgggcgctgctc
gacgcactggccgaagccatgctggcggagaatcatacgcattcggtgccgagagccgacgacgactggcgct
catttctgatcgggaatgcccgcagcttcaggcaggcgctgctcgcctaccgcgatggcgcgcgcatccatgccg
gcacgcgaccgggcgcaccgcagatggaaacggccgacgcgcagcttcgcttcctctgcgaggcgggtttttcg
gccggggacgccgtcaatgcgctgatgacaatcagctacttcactgttggggccgtgcttgaggagcaggccgg
cgacagcgatgccggcgagcgcggcggcaccgttgaacaggctccgctctcgccgctgttgcgggccgcgata
gacgccttcgacgaagccggtccggacgcagcgttcgagcagggactcgcggtgattgtcgatggattggcgaa
aaggaggctcgttgtcaggaacgttgaaggaccgagaaagggtgacgattagGATCCGTCGACCAT
GGCGGCCGCTCGAGTCGACCTGCAGCCAAGCTAATTCCGGGCGAATT
TCTTATGATTTATGATTTTTATTATTAAATAAGTTATAAAAAAAATAA
GTGTATACAAATTTTAAAGTGACTCTTAGGTTTTAAAACGAAAATTCT
TATTCTTGAGTAACT
TetR(B) –
PBA-Gib5
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatgtcta
gattagataaaagtaaagtgattaacagcgcattagagctgcttaatgaggtcggaatcgaaggtttaacaacccgt
aaactcgcccagaagctaggtgtagagcagcctacattgtattggcatgtaaaaaataagcgggctttgctcgacg
ccttagccattgagatgttagataggcaccatactcacttttgccctttagaaggggaaagctggcaagattttttacgt
aataacgctaaaagttttagatgtgctttactaagtcatcgcgatggagcaaaagtacatttaggtacacggcctaca
gaaaaacagtatgaaactctcgaaaatcaattagcctttttatgccaacaaggtttttcactagagaatgcattatatgc
actcagcgctgtggggcattttactttaggttgcgtattggaagatcaagagcatcaagtcgctaaagaagaaaggg
aaacacctactactgatagtatgccgccattattacgacaagctatcgaattatttgatcaccaaggtgcagagccag
ccttcttattcggccttgaattgatcatatgcggattagaaaaacaacttaaatgtgaaagtgggtcttagGATCC
GTCGACCATGGCGGCCGCTCGAGTCGACCTGCAGCCAAGCTAATTCC
GGGCGAATTTCTTATGATTTATGATTTTTATTATTAAATAAGTTATAA
AAAAAATAAGTGTATACAAATTTTAAAGTGACTCTTAGGTTTTAAAA
CGAAAATTCTTATTCTTGAGTAACT
TetR(C) –
HL-249-2
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG

209
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatgaaca
agctccaacgcgaggccgtgatccgaaccgcgctcgaactgcttaacgacgtgggcatggaaggtctaacgacg
cgccgactggctgagcgcctcggggtgcaacagccagcgctctactggcatttcaagaacaagcgtgcgttgctc
gacgcacttgccgaagccatgctgacgataaatcacacgcattcgacgccaagggatgacgacgactggcgttc
gttcctgaagggcaatgcatgcagttttcgacgggcgttgctcgcttatcgcgatggcgcgcgtattcatgccggga
cgcggccagccgcgccgcagatggaaaaagccgacgcgcagcttcgcttcctttgcgatgctggcttttcggcag
gtgacgcgacctatgcgttgatggcaatcagctacttcaccgtcggcgctgttcttgagcagcaagctagcgaggc
agacgccgaggagcggggcgaagatcagttgaccacctcagcgtctacgatgccggcgcgcctacagagcgc
gatgaaaatcgtctacgaaggcggtccggacgcggcattcgagcgaggcctggctctcatcatcggcggtcttga
aaaaatgaggctcactacgaacgacattgaggtgctgaagaatgttgacgaatagGATCCGTCGACCA
TGGCGGCCGCTCGAGTCGACCTGCAGCCAAGCTAATTCCGGGCGAAT
TTCTTATGATTTATGATTTTTATTATTAAATAAGTTATAAAAAAAATA
AGTGTATACAAATTTTAAAGTGACTCTTAGGTTTTAAAACGAAAATT
CTTATTCTTGAGTAACT
TetR(D) –
HL-249-3
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatggcac
ggctgaacagagaatcggttattgatgcggcactggaactgctgaatgagacagggattgacgggctgacgaccc
gcaagctggcgcagaagctgggaatagaacagccgacactttactggcatgtgaaaaataaacgggcgttactgg
atgcgctggcggtggagatcctggcgcgtcatcatgattattcactgcctgcggcgggggaatcctggcagtcattt
ctgcgcaataatgcaatgagtttccgccgggcgctgctgcgttaccgtgacggggcaaaagtgcacctcggcacc
cgccctgatgaaaaacagtatgatacggtggaaacccagttacgctttatgacagaaaacggcttttcactgcgcga
cgggttatatgcgatttcagcggtcagtcattttacccttggtgccgtactggagcagcaggagcatactgccgccct
gaccgaccgccctgcagcaccggacgaaaacctgccgccgctattgcgggaagcgctgcagattatggacagt
gatgatggtgagcaggcctttctgcatggcctggagagcctgatccgggggtttgaggtgcagcttacggcactgt
tgcaaatagtcggtggtgataaacttatcatccccttttgctagGATCCGTCGACCATGGCGGCC
GCTCGAGTCGACCTGCAGCCAAGCTAATTCCGGGCGAATTTCTTATG
ATTTATGATTTTTATTATTAAATAAGTTATAAAAAAAATAAGTGTATA
CAAATTTTAAAGTGACTCTTAGGTTTTAAAACGAAAATTCTTATTCTT
GAGTAACT
TetR(E) –
HL-249-4
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatggcac
gactaagcttggacgacgtaatttcaatggcgctcaccctgctggacagcgaagggctagagggcttgactacgc
gtaagctggcgcagtccctaaaaattgagcaaccgactctgtattggcacgtgcgcaacaagcagactcttatgaa
catgctttcagaggcaatactggcgaagcatcacacccgttcagcaccgttaccgactgagagttggcagcagtttc
tccaggaaaatgctctgagtttccgtaaagcattactggtccatcgtgatggagcccgattgcatatagggacctctc

210
ctacgcccccccagtttgaacaagcagaggcgcaactacgctgtctatgcgatgcagggttttcggtcgaggagg
ctcttttcattctgcaatctatcagccattttacgttgggtgcagtattagaggagcaagcaacaaaccagatagaaaa
taatcatgtgatagacgctgcaccaccattattacaagaggcatttaatattcaggcgagaacctctgctgaaatggc
cttccatttcgggctgaaatcattaatatttggattttctgcacagttagatgaaaaaaagcatacacccattgaggatg
gtaataaatagGATCCGTCGACCATGGCGGCCGCTCGAGTCGACCTGCAGC
CAAGCTAATTCCGGGCGAATTTCTTATGATTTATGATTTTTATTATTA
AATAAGTTATAAAAAAAATAAGTGTATACAAATTTTAAAGTGACTCT
TAGGTTTTAAAACGAAAATTCTTATTCTTGAGTAACT
TetR(G) –
HL-249-5
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatgacca
aactggacaagggcaccgtgatcgcggcggcgctagagctgttgaacgaggttggcatggacagcctgacgac
gcggaagctcgctgaacgcctcaaggttcagcagcctgcgctttactggcatttccagaacaagcgagcgctgctt
gatgcgctcgccgaggcgatgctggcggaacgccatacccgctcgctacccgaagagaatgaggactggcggg
tgttcctgaaagagaatgccctgagcttcagaacggcgttgctctcttatcgggacggcgcgcgtatccatgccgg
cactcgaccgacagaaccgaattttggcaccgccgagacgcaaatacgctttctctgcgcggagggcttttgtccg
aagcgcgccgtttgggcgctccgggcggtcagtcactatgtggtcggttccgttctcgagcagcaggcatctgatg
ccgatgagagagttccggacaggccagatgtgtccgagcaagcaccgtcgtccttcctgcacgatctgtttcacga
gttggaaacagacggcatggatgctgcgttcaacttcggactcgacagcctcatcgctggtttcgagcggctgcgtt
catctacaacagattagGATCCGTCGACCATGGCGGCCGCTCGAGTCGACCTGC
AGCCAAGCTAATTCCGGGCGAATTTCTTATGATTTATGATTTTTATTA
TTAAATAAGTTATAAAAAAAATAAGTGTATACAAATTTTAAAGTGAC
TCTTAGGTTTTAAAACGAAAATTCTTATTCTTGAGTAACT
TetR(H) –
HL-249-6
GCTGCGCGTCAGCGGGATGTCGATGAAAGATATCGGCATTATGGATG
GTGACTTGCTGGCAGTGCATAAAACTCAGGATGTACGTAACGGTCAG
GTCGTTGTCGCACGTATTGATGACGAGGTTACCGTTAAGCGCCTGAA
AAAACAGGGCAATAAAGTCGAACTGTTGCCAGAAAATAGCGAGTTT
AAACCAATTGTCGTAGATCTTCGTCAGCAGAGCTTCACCATTGAAGG
GCTGGCGGTTGGGGTTATTCGCAACGGCGACTGGCTGGAATTCatggcaa
agctagataaagaacaagttattgatgatgcgttgattttacttaatgaagttggtattgaaggattaacaacgcgtaac
gtggcgcaaaaaataggtgtggaacaacccacattgtattggcatgtaaaaaataaacgcgctttgttagatgcatta
gcagaaactattttgcaaaagcaccatcatcatgttttgccattgccgaatgaaacatggcaggactttttgcgaaata
acgcgaaaagcttccgccaagccttattaatgtatcgtgatggtggcaaaattcatgcgggaacacgcccctctgaa
agtcaatttgagacatcagaacagcaactacagtttttgtgtgatgctgggtttagtctatctcaagccgtgtatgcatt
aagctctattgcgcattttacattaggctccgtactggaaactcaagagcatcaagaaagccaaaaagagcgtgaaa
aagtagagacggatactgttgcctatccgccattattaacccaagccgttgcaattatggatagtgataatggtgatg
ctgcatttttgtttgtccttgatgtgatgatctctggacttgaaacagtattaaagagcgctaaatagGATCCGTC
GACCATGGCGGCCGCTCGAGTCGACCTGCAGCCAAGCTAATTCCGGG
CGAATTTCTTATGATTTATGATTTTTATTATTAAATAAGTTATAAAAA
AAATAAGTGTATACAAATTTTAAAGTGACTCTTAGGTTTTAAAACGA
AAATTCTTATTCTTGAGTAACT

211
5.8 References
1. Fasciotti M. Perspectives for the use of biotechnology in green chemistry applied to
biopolymers, fuels and organic synthesis: from concepts to a critical point of view. Sustainable
Chem Pharm. 2017;6:82-89. doi: 10.1016/j.scp.2017.09.002.
2. Keasling JD. Manufacturing Molecules Through Metabolic Engineering. Science.
2010;330(6009):1355-1358. doi: 10.1126/science.1193990.
3. Chao R, Yuan Y, Zhao H. Recent advances in DNA assembly technologies. FEMS Yeast
Res. 2014;15(1):1-9. doi: 10.1111/1567-1364.12171.
4. Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications.
Nat Methods. 2014;11(5):499-507. doi: 10.1038/nmeth.2918.
5. Jullesson D, David F, Pfleger B, Nielsen J. Impact of synthetic biology and metabolic
engineering on industrial production of fine chemicals. Biotechnol Adv. 2015;33(7):1395-1402.
doi: 10.1016/j.biotechadv.2015.02.011.
6. Richardson SM, Mitchell LA, Stracquadanio G, Yang K, Dymond JS, DiCarlo JE, Lee D,
Huang CLV, Chandrasegaran S, Cai Y, Boeke JD, Bader JS. Design of a synthetic yeast genome.
Science. 2017;355(6329):1040-1044. doi: 10.1126/science.aaf4557.
7. Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells
by multiplex genome engineering and accelerated evolution. Nature. 2009;460(7257):894-898.
doi: 10.1038/nature08187.
8. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9.
Science. 2014;346(6213):1258096-1258096. doi: 10.1126/science.1258096.
9. Rogers JK, Taylor ND, Church GM. Biosensor-based engineering of biosynthetic
pathways. Curr Opin Biotechnol. 2016;42:84-91. doi: 10.1016/j.copbio.2016.03.005.
10. Schallmey M, Frunzke J, Eggeling L, Marienhagen J. Looking for the pick of the bunch:
high-throughput screening of producing microorganisms with biosensors. Curr Opin Biotechnol.
2014;26:148-154. doi: 10.1016/j.copbio.2014.01.005.

212
11. Feng J, Jester BW, Tinberg CE, Mandell DJ, Antunes MS, Chari R, Morey KJ, Rios X,
Medford JI, Church GM, Fields S, Baker D. A general strategy to construct small molecule
biosensors in eukaryotes. eLife. 2015;4. doi: 10.7554/eLife.10606.
12. Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor
interactions. Proc Natl Acad Sci U S A. 1996;93(23):12817-12821. doi:
10.1073/pnas.93.23.12817.
13. Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL. Controlling protein association and
subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc
Natl Acad Sci U S A. 1996;93(10):4604-4607. doi: 10.1073/pnas.93.10.4604.
14. Golemis EA, Serebriiskii I, Finley RL, Kolonin MG, Gyuris J, Brent R. Interaction
Trap/Two-Hybrid System to Identify Interacting Proteins. Curr Protoc Cell Biol.
2008;53(1):17.13.11-17.13.35. doi: 10.1002/0471142727.mb2001s82.
15. Bernstein DS, Buter N, Stumpf C, Wickens M. Analyzing mRNA-protein complexes using
a yeast three-hybrid system. Methods. 2002;26(2):123-141. doi: 10.1016/S1046-2023(02)00015-
4.
16. Henthorn DC, Jaxa-Chamiec AA, Meldrum E. A GAL4-based yeast three-hybrid system
for the identification of small molecule-target protein interactions. Biochem Pharmacol.
2002;63(9):1619-1628. doi: 10.1016/S0006-2952(02)00884-5.
17. Baker K, Bleczinski C, Lin H, Salazar-Jimenez G, Sengupta D, Krane S, Cornish VW.
Chemical complementation: a reaction-independent genetic assay for enzyme catalysis. Proc Natl
Acad Sci U S A. 2002;99(26):16537-16542. doi: 10.1073/pnas.262420099.
18. Fields S, Song OK. A Novel Genetic System to Detect Protein Protein Interactions. Nature.
1989;340(6230):245-246. doi: DOI 10.1038/340245a0.
19. Morita T, Takegawa K. A simple and efficient procedure for transformation of
Schizosaccharomyces pombe. Yeast. 2004;21(8):613-617. doi: 10.1002/yea.1104.
20. Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic
assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343-345.
doi: 10.1038/nmeth.1318.

213
21. Green SR, Moehle CM. Media and Culture of Yeast. Curr Protoc Cell Biol.
2001;4(1):1.6.1-1.6.12. doi: 10.1002/0471143030.cb0106s04.
22. Krishnan L, Sum P-e, Daigneault S, Bernatchez M, Pilcher AS, Horne JM, Tuper AJ,
Mccauley III JJ, Michaud AP, inventorsMethods of purifying tigecycline patent U.S. Patent
2007/0049561 A1. 2006 March 1, 2017.
23. Nagy A, Szoke B, Schally AV. Selective Coupling of Methotrexate to Peptide-Hormone
Carriers through a Gamma-Carboxamide Linkage of Its Glutamic-Acid Moiety - Benzotriazol-1-
Yloxytris(Dimethylamino)Phosphonium Hexafluorophosphate Activation in Salt Coupling. Proc
Natl Acad Sci U S A. 1993;90(13):6373-6376. doi: 10.1073/pnas.90.13.6373.
24. Castex U, Lalanne C, Mouchet P, Lemaire M, Lahana R. Regioselective synthesis of
peptidic derivatives and glycolamidic esters of methotrexate. Tetrahedron. 2005;61(4):803-812.
doi: 10.1016/j.tet.2004.11.049.
25. Reichheld SE, Davidson AR. Two-way interdomain signal transduction in tetracycline
repressor. J Mol Biol. 2006;361(2):382-389. doi: 10.1016/j.jmb.2006.06.035.
26. Reichheld SE, Yu Z, Davidson AR. The induction of folding cooperativity by ligand
binding drives the allosteric response of tetracycline repressor. Proc Natl Acad Sci U S A.
2009;106(52):22263-22268. doi: 10.1073/pnas.0911566106.
27. Abida WM, Carter BT, Althoff EA, Lin H, Cornish VW. Receptor-dependence of the
transcription read-out in a small-molecule three-hybrid system. ChemBioChem. 2002;3(9):887-
895. doi: 10.1002/1439-7633(20020902)3:9<887::AID-CBIC887>3.0.CO;2-F.
28. Harton MD, Wingler LM, Cornish VW. Transcriptional regulation improves the
throughput of three-hybrid counter selections in Saccharomyces cerevisiae. Biotechnol J.
2013;8(12):1485-1491. doi: 10.1002/biot.201300186.
29. Keppler-Ross S, Noffz C, Dean N. A New Purple Fluorescent Color Marker for Genetic
Studies in Saccharomyces cerevisiae and Candida albicans. Genetics. 2008;179(1):705-710. doi:
10.1534/genetics.108.087080.
30. Li YR, Chooi YH, Sheng YW, Valentine JS, Tang Y. Comparative Characterization of
Fungal Anthracenone and Naphthacenedione Biosynthetic Pathways Reveals an alpha-
Hydroxylation-Dependent Claisen-like Cyclization Catalyzed by a Dimanganese Thioesterase. J
Am Chem Soc. 2011;133(39):15773-15785. doi: 10.1021/ja206906d.

214
31. Ostrov N, Jimenez M, Billerbeck S, Brisbois J, Matragrano J, Ager A, Cornish VW. A
modular yeast biosensor for low-cost point-of-care pathogen detection. Science Advances.
2017;3(6):e1603221. doi: 10.1126/sciadv.1603221.
32. ICH Expert Working Group, editor. Validation of analytical procedures: text and
methodology Q2 (R1). International Conference on Harmonization, Geneva, Switzerland; 2005.
33. Monser L, Darghouth F. Rapid liquid chromatographic method for simultaneous
determination of tetracyclines antibiotics and 6-Epi-doxycycline in pharmaceutical products using
porous graphitic carbon column. J Pharm Biomed Anal. 2000;23(2-3):353-362. doi:
10.1016/s0731-7085(00)00329-0.
34. Ruden DM, Ma J, Li Y, Wood K, Ptashne M. Generating Yeast Transcriptional Activators
Containing No Yeast Protein Sequences. Nature. 1991;350(6315):250-252. doi:
10.1038/350250a0.
35. Lin H, Abida WM, Sauer RT, Cornish VW. Dexamethasone−Methotrexate: An Efficient
Chemical Inducer of Protein Dimerization In Vivo. J Am Chem Soc. 2000;122(17):4247-4248.
doi: 10.1021/ja9941532.
36. Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein
phosphatase that associates with Cdk2. Cell. 1993;75(4):791-803. doi: 10.1016/0092-
8674(93)90498-F.
37. Lederer T, Kintrup M, Takahashi M, Sum PE, Ellestad GA, Hillen W. Tetracycline analogs
affecting binding to Tn10-Encoded Tet repressor trigger the same mechanism of induction.
Biochemistry. 1996;35(23):7439-7446. doi: 10.1021/bi952683e.
38. Hinrichs W, Kisker C, Duvel M, Muller A, Tovar K, Hillen W, Saenger W. Structure of
the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science.
1994;264(5157):418-420. doi: 10.1126/science.8153629.
39. Saenger W, Hinrichs W, Orth P, Schnappinger D, Hillen W. Structural basis of gene
regulation by the tetracycline inducible Tet repressor–operator system. Nat Struct Biol.
2000;7(3):215-219. doi: 10.1038/73324.
40. Waters SH, Rogowsky P, Grinsted J, Altenbuchner J, Schmitt R. The tetracycline
resistance determinants of RP1 and Tnl721: nucleotide sequence analysis. Nucleic Acids Res.
1983;11(17):6089-6105. doi: 10.1093/nar/11.17.6089.

215
41. Rhodes G, Parkhill J, Bird C, Ambrose K, Jones MC, Huys G, Swings J, Pickup RW.
Complete Nucleotide Sequence of the Conjugative Tetracycline Resistance Plasmid pFBAOT6, a
Member of a Group of IncU Plasmids with Global Ubiquity. Appl Environ Microbiol.
2004;70(12):7497-7510. doi: 10.1128/aem.70.12.7497-7510.2004.
42. Brow MAD, Pesin R, Sutcliffe JG. The Tetracycline Repressor of Psc101. Mol Biol Evol.
1985;2(1):1-12. doi: 10.1093/oxfordjournals.molbev.a040334.
43. Bernardi A, Bernardi F. Complete sequence of pSC101. Nucleic Acids Res.
1984;12(24):9415-9426. doi: 10.1093/nar/12.24.9415.
44. Tovar K, Ernst A, Hillen W. Identification and Nucleotide-Sequence of the Class-E Tet
Regulatory Elements and Operator and Inducer Binding of the Encoded Purified Tet Repressor.
Mol Gen Genet. 1988;215(1):76-80. doi: 10.1007/Bf00331306.
45. Gao F, Webb HE, Bugarel M, den Bakker HC, Nightingale KK, Granier SA, Scott HM,
Loneragan GH. Carbapenem-Resistant Bacteria Recovered from Faeces of Dairy Cattle in the
High Plains Region of the USA. PLoS One. 2016;11(1):e0147363. doi:
10.1371/journal.pone.0147363.
46. Liu P, Li P, Jiang X, Bi D, Xie Y, Tai C, Deng Z, Rajakumar K, Ou HY. Complete Genome
Sequence of Klebsiella pneumoniae subsp. pneumoniae HS11286, a Multidrug-Resistant Strain
Isolated from Human Sputum. J Bacteriol. 2012;194(7):1841-1842. doi: 10.1128/jb.00043-12.
47. Hansen LM, Mcmurry LM, Levy SB, Hirsh DC. A New Tetracycline Resistance
Determinant, Tet H, from Pasteurella-Multocida Specifying Active Efflux of Tetracycline.
Antimicrob Agents Chemother. 1993;37(12):2699-2705. doi: 10.1128/Aac.37.12.2699.
48. Kehrenberg C, Tham NTT, Schwarz S. New Plasmid-Borne Antibiotic Resistance Gene
Cluster in Pasteurella multocida. Antimicrob Agents Chemother. 2003;47(9):2978-2980. doi:
10.1128/aac.47.9.2978-2980.2003.
49. Mendez B, Tachibana C, Levy SB. Heterogeneity of tetracycline resistance determinants.
Plasmid. 1980;3(2):99-108. doi: 10.1016/0147-619x(80)90101-8.
50. Hillen W, Berens C. Mechanisms Underlying Expression of TN10 Encoded Tetracycline
Resistance. Annu Rev Microbiol. 1994;48(1):345-369. doi:
10.1146/annurev.mi.48.100194.002021.

216
51. Mohana-Borges R, Pacheco ABF, Sousa FJR, Foguel D, Almeida DF, Silva JL. LexA
Repressor Forms Stable Dimers in Solution. J Biol Chem. 2000;275(7):4708-4712. doi:
10.1074/jbc.275.7.4708.
52. Kisker C, Hinrichs W, Tovar K, Hillen W, Saenger W. The Complex Formed between Tet
Repressor and Tetracycline-Mg2+ Reveals Mechanism of Antibiotic-Resistance. J Mol Biol.
1995;247(2):260-280. doi: 10.1006/jmbi.1994.0138.
53. Greer ND. Tigecycline (Tygacil): the first in the glycylcycline class of antibiotics. Proc
(Bayl Univ Med Cent). 2006;19(2):155-161. doi: 10.1080/08998280.2006.11928154.
54. Liu F, Myers AG. Development of a platform for the discovery and practical synthesis of
new tetracycline antibiotics. Curr Opin Chem Biol. 2016;32:48-57. doi:
10.1016/j.cbpa.2016.03.011.
55. Rogalski W. Chemical Modification of the Tetracyclines. In: Hlavka JJ, Boothe JH, editors.
The Tetracyclines. Berlin, Heidelberg: Springer Berlin Heidelberg; 1985. p. 179-316.
56. Zhang X, Bremer H. Control of the Escherichia coli rrnB P1 promoter strength by ppGpp.
J Biol Chem. 1995;270(19):11181-11189. doi: 10.1074/jbc.270.19.11181.
57. Kogawa AC, Salgado HRN. Quantification of Doxycycline Hyclate in Tablets by HPLC–
UV Method. J Chromatogr Sci. 2013;51(10):919-925. doi: 10.1093/chromsci/bms190.
58. Ng YZ, Baldera-Aguayo PA, Cornish VW. Fluorescence Polarization Assay for Small
Molecule Screening of FK506 Biosynthesized in 96-Well Microtiter Plates. Biochemistry.
2017;56(40):5260-5268. doi: 10.1021/acs.biochem.7b00602.
59. Gossen M, Bujard H. Tight Control of Gene-Expression in Mammalian-Cells by
Tetracycline-Responsive Promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547-5551. doi:
10.1073/pnas.89.12.5547.
60. Scholz O, Kostner M, Reich M, Gastiger S, Hillen W. Teaching TetR to recognize a new
inducer. J Mol Biol. 2003;329(2):217-227. doi: 10.1016/S0022-2836(03)00427-3.
61. Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X,
Gallegos MT, Brennan R, Tobes R. The TetR Family of Transcriptional Repressors. Microbiol
Mol Biol Rev. 2005;69(2):326-356. doi: 10.1128/mmbr.69.2.326-356.2005.

217
62. Lin H, Tao H, Cornish VW. Directed evolution of a glycosynthase via chemical
complementation. J Am Chem Soc. 2004;126(46):15051-15059. doi: 10.1021/ja046238v.
63. Peralta-Yahya P, Carter BT, Lin H, Tao H, Cornish VW. High-throughput selection for
cellulase catalysts using chemical complementation. J Am Chem Soc. 2008;130(51):17446-17452.
doi: 10.1021/ja8055744.
64. Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva
NA, Vederas JC, Tang Y. Complete Reconstitution of a Highly Reducing Iterative Polyketide
Synthase. Science. 2009;326(5952):589-592. doi: 10.1126/science.1175602.
65. Watson MA, Buckholz R, Weiner MP. Vectors encoding alternative antibiotic resistance
for use in the yeast two-hybrid system. BioTechniques. 1996;21(2):255-259. doi:
10.2144/96212st02.





























