Embed Size (px)
Citation preview

RESEARCH ARTICLE SUMMARY◥
RNA IMAGING
Spatially resolved, highly multiplexedRNA profiling in single cellsKok Hao Chen,1* Alistair N. Boettiger,1* Jeffrey R. Moffitt,1*Siyuan Wang,1 Xiaowei Zhuang1,2†
INTRODUCTION: The copy number and in-tracellular localization of RNA are importantregulators of gene expression. Measurementof these properties at the transcriptome scalein single cells will give answers to many ques-tions related to gene expression and regulation.Single-molecule RNA imaging approaches, suchas single-molecule fluorescence in situ hybrid-ization (smFISH), are powerful tools for count-ing andmapping RNA; however, the numberof RNA species that can be simultaneously im-aged in individual cells has been limited. Thismakes it challenging to perform transcriptomicanalysis of single cells in a spatially resolvedmanner. Here, we report multiplexed error-robust FISH (MERFISH), a single-molecule im-aging method that allows thousands of RNA
species to be imaged in single cells by usingcombinatorial FISH labeling with encodingschemes capable of detecting and/or correct-ing errors.
RATIONALE: We labeled each cellular RNAwith a set of encoding probes, which containtargeting sequences that bind the RNA andreadout sequences that bind fluorescently la-beled readout probes. Each RNA species isencodedwith a particular combination of read-out sequences. We used successive rounds ofhybridization and imaging, each with a differ-ent readout probe, to identify the readout se-quences bound to each RNA and to decode theRNA. In principle, combinatorial labeling al-lows the number of detectable RNA species to
growexponentiallywith thenumber of imagingrounds, but the detection errors also increaseexponentially. To combat such accumulatingerrors, we exploited error-robust encodingschemes used in digital electronics, such asthe extended Hamming code, in the design of
our encoding probes butmodified these schemesin order to account for theerror properties in FISHmeasurements.Weassignedeach RNA a binary wordin ourmodifiedHamming
code and encoded the RNA with a combina-tion of readout sequences according to thisbinary word.
RESULTS:We first imaged 140 RNA speciesin human fibroblast cells using MERFISHwith 16 rounds of hybridization and a mod-ified Hamming code capable of both errordetection and correction. We obtained ~80%detection efficiency and observed excellentcorrelation of RNA copy numbers determinedwith MERFISH with both bulk RNA sequenc-ing data and conventional smFISH measure-ments of individual genes.Next, we used an alternative MERFISH en-
coding scheme, which is capable of detectingbut not correcting errors, to image 1001 RNAspecies in individual cells using only 14 roundsof hybridization. The observed RNA copy num-bers again correlate well with bulk sequencingdata. However, the detection efficiency is onlyone-third that of the error-correcting encod-ing scheme.We performed correlation analysis of the 104
to 106 pairs of measured genes and identifiedmany covarying gene groups that share com-mon regulatory elements. Such grouping allowedus to hypothesize potential functions of ~100unannotated or partially annotated genes ofunknown functions. We further analyzed cor-relations in the spatial distributions of differentRNA species and identified groups of RNAswith different distribution patterns in the cell.
DISCUSSION: This highly multiplexed imag-ing approach enables analyses based on thevariation and correlation of copy numbers andspatial distributions of a large number of RNAspecieswithin single cells. Such analyses shouldfacilitate the delineation of regulatory networksand in situ identification of cell types. We en-vision that this approach will allow spatiallyresolved transcriptomes to be determined forsingle cells.▪RELATED ITEMS IN SCIENCEJ. H. Lee et al., Science 343, 1360–1363 (2014).
RESEARCH
412 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
The list of author affiliations is available in the full article online.†Corresponding author. E-mail: [email protected] this article as K. H. Chen et al., Science 348, aaa6090(2015). DOI:10.1126/science.aaa6090
MERFISH for transcriptome imaging. Numerous RNA species can be identified, counted, andlocalized in a single cell by using MERFISH, a single-molecule imaging approach that uses combi-natorial labeling and sequential imaging with encoding schemes capable of detection and/orcorrection of errors. This highly multiplexed measurement of individual RNAs can be used tocompute the gene expression profile and noise, covariation in expression among different genes,and spatial distribution of RNAs within single cells.
ON OUR WEB SITE◥
Read the full articleat http://dx.doi.org/10.1126/science.aaa6090..................................................
on Septem
ber 10, 2020
http://science.sciencemag.org/
Dow
nloaded from

RESEARCH ARTICLE◥
RNA IMAGING
Spatially resolved, highly multiplexedRNA profiling in single cellsKok Hao Chen,1* Alistair N. Boettiger,1* Jeffrey R. Moffitt,1*Siyuan Wang,1 Xiaowei Zhuang1,2†
Knowledge of the expression profile and spatial landscape of the transcriptome inindividual cells is essential for understanding the rich repertoire of cellular behaviors.Here, we report multiplexed error-robust fluorescence in situ hybridization (MERFISH), asingle-molecule imaging approach that allows the copy numbers and spatial localizationsof thousands of RNA species to be determined in single cells. Using error-robust encodingschemes to combat single-molecule labeling and detection errors, we demonstrated theimaging of 100 to 1000 distinct RNA species in hundreds of individual cells. Correlationanalysis of the ~104 to 106 pairs of genes allowed us to constrain gene regulatorynetworks, predict novel functions for many unannotated genes, and identify distinct spatialdistribution patterns of RNAs that correlate with properties of the encoded proteins.
System-wide analyses of the abundance andspatial organization of RNAs in single cellspromise to transform our understandingin many areas of cell and developmentalbiology, such as the mechanisms of gene
regulation, the heterogeneous behavior of cells,and the development and maintenance of cellfate (1). Single-molecule fluorescence in situ hy-bridization (smFISH) has emerged as a powerfultool for studying the copy number and spatialorganization of RNAs in single cells either inisolation or in their native tissue context (2, 3).Taking advantage of its ability to map the spatialdistributions of specific RNAs with high resolu-tion, smFISH has revealed the importance of sub-cellular RNA localization in diverse processes suchas cell migration, development, and polarization(4–8). In parallel, the ability of smFISH to pre-ciselymeasure the copy numbers of specific RNAswithout amplification bias has allowed quantita-tive measurement of the natural fluctuations ingene expression, which has in turn elucidated theregulatory mechanisms that shape such fluctua-tions and their role in a variety of biological pro-cesses (9–13).Recent advances in imaging and analysismeth-
ods have allowed hundreds of smFISH measure-ments to be performed in an automatedmanner,substantially expanding our knowledge of theRNA expression profile and spatial organizationin different organisms (14, 15). However, applica-tion of the smFISH approach to many systems-level questions remains limited by the number ofRNA species that can be simultaneously mea-sured in single cells. State-of-the-art efforts by
using combinatorial labeling with either color-based barcodes or sequential hybridization haveenabled simultaneous measurements of 10 to 30different RNA species in individual cells (16–19),yet many interesting biological questions wouldbenefit from the measurement of hundreds tothousands of RNAs within a single cell. For ex-ample, analysis of how the expression profile ofsuch a large number of RNAs vary from cell tocell and how these variations correlate amongdifferent genes could be used to systematicallyidentify coregulated genes and map regulatorynetworks, knowledge of the subcellular organi-zations of numerous RNAs and their correlationscouldhelp elucidatemolecularmechanismsunder-lying the establishment andmaintenance ofmanylocal cellular structures, and RNA profiling of in-dividual cells in native tissues could allow in situidentification of cell type.Here, we report multiplexed error-robust FISH
(MERFISH), a highly multiplexed smFISH imag-ingmethod that substantially increases the num-ber of RNA species that can be simultaneouslyimaged in single cells by using combinatorial la-beling and sequential imaging with error-robustencoding schemes. We demonstrated this tran-scriptome imaging approach by simultaneouslymeasuring 140 RNA species with an encodingscheme that can both detect and correct errorsand 1001 RNA species with an encoding schemethat can detect but not correct errors. Correlationanalyses of the copy number variations and spa-tial distributions of these genes allowed us toidentify groups of genes that are coregulated andgroups of genes that share similar spatial distribu-tion patterns inside the cell.
Combinatorial labeling witherror-robust encoding schemes
Combinatorial labeling that identifies each RNAspecies by multiple (N) distinct signals offers a
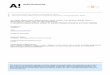
route to rapidly increase the number of RNA spe-cies that can be probed simultaneously in indi-vidual cells (Fig. 1A). However, this approach toscaling up the throughput of smFISH to the sys-tems scale faces a substantial challenge becausenot only does the number of addressable RNAspecies increases exponentially with N, but thedetection error rates also grow exponentially withN (Fig. 1, B to D). Imagine a conceptually simplescheme to implement combinatorial labeling, inwhich each RNA species is encoded with a N-bitbinary word, and the sample is probed with Ncorresponding rounds of hybridization, each roundtargeting only the subset of RNAs that shouldread “1” in the corresponding bit (fig. S1). Nrounds of hybridization would allow 2N – 1 RNAspecies to be probed. With just 16 hybridizations,more than 64,000 RNA species—which shouldcover the entire human transcriptome, includingboth messenger RNAs (mRNAs) and noncodingRNAs (20)—could be identified (Fig. 1B, black sym-bols). However, asN increases, the fraction of RNAsproperly detected (the calling rate) would rapidlydecrease and, more troublingly, the fraction ofRNAs that are identified as incorrect species (themisidentification rate) would rapidly increase (Fig.1, C and D, black symbols). With realistic errorrates per hybridization (measured below), the ma-jority of RNA molecules would be misidentifiedafter 16 rounds of hybridizations.To address this challenge, we designed error-
robust encoding schemes in which only a sub-set of the 2N – 1 words separated by a certainHammingdistance (21)wereused to encodeRNAs.In a codebook in which the minimumHammingdistance is 4 (HD4 code), at least four bits mustbe read incorrectly to change one valid code wordinto another (fig. S2A). As a result, every single-biterror produces a word that is exclusively close toa single RNA-encoding word, allowing such er-rors to be detected and corrected (fig. S2B).Double-bit errors produce words with an equalHamming distance of 2 frommultiple valid codewords and, thus, can be detected but not corrected(fig. S2C). Such a code should substantially increasethe calling rate and reduce the misidentificationrate (Fig. 1, C and D, blue symbols). To furtheraccount for the fact that it is more likely to miss ahybridization event (an 1→0 error) than to mis-identify a background spot as an RNA (an 0→1error) in smFISH measurements, we designed amodified HD4 (MHD4) code, in which the num-ber of 1 bits were kept both constant and rela-tively low—only four per word in this work—soas to reduce error and avoid biased detection. ThisMHD4 code should further increase the callingrate and reduce the misidentification rate (Fig. 1,C and D, purple symbols).In addition to the error considerations, several
practical challenges have also made it difficult toprobe a large number of RNA species, such as thehigh cost of themassive number of distinct FISHprobes needed and the long time required to com-plete many rounds of hybridization. An oligopaintapproachhas beenpreviously developed to generatea large number of oligonucleotide probes to labelchromosomeDNA and to introduce nontargeting
RESEARCH
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-1
1Howard Hughes Medical Institute, Department of Chemistryand Chemical Biology, Harvard University, Cambridge, MA02138, USA. 2Department of Physics, Harvard University,Cambridge, MA 02138, USA.*These authors contributed equally to this work. †Correspondingauthor. E-mail: [email protected]
on Septem
ber 10, 2020
http://science.sciencemag.org/
Dow
nloaded from

sites for secondary activities (22). Inspired bythis approach, we designed a two-step labelingscheme to encode and read out cellular RNAs(Fig. 1E). First, we label cellular RNAs with a setof encoding probes, each probe comprising a
RNA targeting sequence and two flanking readoutsequences. Four of the N distinct readout se-quences were assigned to each RNA species basedon the N-bit MHD4 code word of the RNA.Second, we identified these N readout sequences
with complementary FISH probes (the readoutprobes) via N rounds of hybridization and imag-ing, each round using a different readout probe.To increase the signal-to-background ratio, welabeled every cellular RNA with ~192 encodingprobes. Because each encoding probe containedtwo of the four readout sequences associated withthat RNA (Fig. 1E), a maximum of ~96 readoutprobes can bind to each cellular RNA per hybrid-ization round. To generate the massive number ofencoding probes required, we amplified themfromarray-derivedoligonucleotide pools contain-ing tens of thousands of custom sequences usinga modified form of the oligopaint protocol com-prising in vitro transcription followed by reversetranscription (fig. S3 and supplementary mate-rials, materials and methods, “Probe Synthesis”)(22, 23). This two-step labeling approach dra-matically diminished the total hybridization timefor an experiment; we found that efficient hybrid-ization to the readout sequences took only 15 min,whereas efficient direct hybridization to cellularRNA required more than 10 hours.
Measuring 140 genes with MERFISHby use of a 16-bit MHD4 code
To test the feasibility of this error-robust, multi-plexed imaging approach,we performed a 140-genemeasurement on human fibroblast cells (IMR90)using a 16-bit MHD4 code to encode 130 RNAspecies while leaving 10 code words as misiden-tification controls (table S1). After each round ofhybridization with the fluorescent readout probes,cells were imaged bymeans of conventional wide-field imaging with an oblique-incidence illumina-tion geometry. Fluorescent spots correspondingto individual RNAswere clearly detected andwerethen efficiently extinguished via a brief photo-bleaching step (Fig. 2A). The sample was stablethroughout the 16 rounds of iterative labelingand imaging: The change in the number of fluo-rescent spots from round to round matched theexpected change predicted on the basis of the rel-ative abundances of RNA species targeted in eachround derived from bulk sequencing, and we didnot observe a systematic decreasing trend withincreasing number of hybridization rounds (fig.S4A). The average brightness of the spots variedfrom round to roundwith a standard deviation of40%, which is likely due to different binding ef-ficiencies of the readout probes to the differentreadout sequences on the encoding probes (fig.S4B).We observed only a small, systematic decreas-ing trend in the spot brightness with increasinghybridization rounds, which was on average 4%per round (fig. S4B).We then constructed binary words from the
observed fluorescent spots based on their on-offpatterns across the 16 hybridization rounds (Fig.2, B to D). If the word exactly matched one of the140MHD4 codewords (exactmatches) or differedby only one bit (error-correctable matches), weassigned it to the corresponding RNA species(Fig. 2D).Within the single cell depicted in Fig. 2,A and B, more than 1500 RNA molecules corre-sponding to 87% of the 130 encoded RNA spe-cies were detected after error correction (Fig. 2E).
aaa6090-2 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
0 8 16 24 3210
0
103
106
109
Number of bits (N ) Number of bits (N ) Number of bits (N )Num
ber
of R
NA
spe
cies
0 8 16 24 320
0.5
1
Cal
ling
rate
Mis
iden
tific
atio
n ra
te
0 8 16 24 320
0.5
1
Readouthyb 1
Readouthyb N
1 1 0 1 1
0 0 0 0 1
0 1 1 0 0
1 0 0 0 0
0 0 1 0 1
RNA Species
1
2
3
4Encoding
hyb
M
Targeting sequence
Readout sequence 2
Readout sequence 1
Distance 4 code
Decode
Readouthyb 2
Readouthyb 3
Targeting sequence
Readout sequence N
Readout sequence 4
Encoding probes
Image 1 Image 2 Image 3 Image N
1
0
1
0
01
1
11
01
10
01
0110
10
110
011
101
011
011101
101
110...1
011...1
101...0
011...0
011...1101...0
B
C
A
D
CA
Decoded Image
G101...1
Readouthyb 4
x96 x96
Encoding probes
Fig. 1. MERFISH: A highly multiplexed smFISH approach enabled by combinatorial labeling anderror-robust encoding. (A) Schematic depiction of the identification of multiple RNA species inN roundsof imaging. Each RNA species is encoded with aN-bit binary word, and during each round of imaging, onlythe subset of RNAs that should read 1 in the corresponding bit emit signal. (B to D) The number ofaddressable RNA species (B); the rate at which these RNAs are properly identified—the “calling rate” (C);and the rate at which RNAs are incorrectly identified as a different RNA species—the “misidentificationrate” (D); plotted as a function of the number of bits (N) in the binarywords encoding RNA.Black indicatesa simple binary code that includes all 2N-1 possible binary words. Blue indicates the HD4 code in which theHamming distance separating words is 4. Purple indicates a modified HD4 (MHD4) code where thenumber of 1 bits are kept at four. The calling and misidentification rates are calculated with per-bit errorrates of 10% for the 1→0 error and 4% for the 0→1 error. (E) Schematic diagramof the implementation of aMHD4 code for RNA identification. Each RNA species is first labeled with ~192 encoding probes thatconvert the RNA into a specific combination of readout sequences (Encoding hyb).These encoding probeseach contain a central RNA-targeting region flanked by two readout sequences, drawn from a pool of Ndifferent sequences, each associated with a specific hybridization round. Encoding probes for a specificRNA species contain a particular combination of four of theN readout sequences,which correspond to thefour hybridization rounds in which this RNA should read 1. N subsequent rounds of hybridization with thefluorescent readout probes are used to probe the readout sequences (hyb 1, hyb 2,…, hyb N).The boundprobes are inactivated by photobleaching between successive rounds of hybridization. For clarity, only onepossible pairing of the readout sequences is depicted for the encoding probes; however, all possible pairsof the four readout sequences are used at the same frequency and distributed randomly along eachcellular RNA in the actual experiments.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

Similar observations were made in ~400 cellsfrom seven independent experiments. On aver-age, ~4 times as many RNA molecules and ~2times as many RNA species were detected percell after error correction as compared with thevalues obtained before error correction (fig. S5).Two types of errors can occur in the copy
number measurement of each RNA species: (i)Some molecules of this RNA species are not de-tected, leading to a drop in calling rate, and (ii)somemolecules from other RNA species aremis-identified as this RNA species. To assess the extentof misidentification, we used the 10 misidentifi-cation control words—code words that were notassociatedwithany cellularRNA.Althoughmatchesto these controlwordswere observed, they occurredfar less frequently thandid the real RNA-encodingwords: 95% of the 130 RNA-encoding words werecounted more frequently than the median countfor these control words. Moreover, we typicallyfound the ratio of the number of exact matchesto the number of matches with one-bit errors fora real RNA-encoding word to be substantiallyhigher than the same ratios observed for the mis-identification controls, as expected (fig. S6, A andB). Using this ratio as ameasure of the confidencein RNA identification, we found that 91% of the130 RNA species had a confidence ratio greaterthan themaximum confidence ratio observed forthe misidentification controls (Fig. 2F), demon-strating a high accuracy of RNA identification.Subsequent analyses were conducted only onthese 91% of genes.To estimate the calling rate, we used the error-
correction ability of theMHD4 code to determinethe 1→0 error rates (10% on average) and 0→1error rates (4% on average) for each hybridiza-tion round (fig. S6, C and D). Using these errorrates, we estimated an ~80% calling rate for in-dividual RNA species after error correction—~80% of the fluorescent spots corresponding toa RNA species were decoded correctly (fig. S6E).Although the remaining 20% of spots contrib-uted to a loss in detection efficiency, most of themdid not cause species misidentification becausethey were decoded as double-bit error words anddiscarded.To test for potential technical bias in our mea-
surements, we probed the same 130 RNAs spe-cies with a differentMHD4 codebook by shufflingthe code words among different RNA species(table S1) and changing the encoding probe se-quences.Measurementswith this alternative codegave similar misidentification and calling rates(fig. S7). The copy numbers of individual RNAspecies per cell measured with these two code-books showed excellent agreementwith aPearsoncorrelation coefficient of 0.94 (Fig. 2G), indicatingthat the choice of encoding scheme did not biasthe measured counts.In order to validate the copy numbers derived
from our MERFISH experiments, we performedconventional smFISHmeasurements on 15 of the130 genes, spanning the full measured abun-dance range of three orders of magnitude. Foreach of these genes, both the average copy num-ber and the copy number distribution across
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-3
hyb 1 bleach 1 hyb 2 hyb 16
11
1
111
1
11
1
1
111
111
11
11
1
100
0
00
0
0
00
0
00
0
0
00
0
00
0
0
0
0
00
0
0
0
0
0
00
0
0
0
0
0
0
0
0
00
0
00
0
0
00
0
00
0
0
0
0
00
0
0
00
0
0
00
0
0
00
0
00
0
0
00
0
0
0
00
0
0
0
0
00
0
x
x
1
1
Hybridization round
Gene
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
DYNC1H1
EGFRFLNA
TLN1
TLN1LRP1
1
23
4
56
7
Spo
t num
ber
0 20 40 60 80 100 120 14001
10
100
Cop
y nu
mbe
r
Gene ID
10µm
1
2
3
4
5 6
710µm
0 20 40 60 80 100120140
1
4
7
10
13
Gene ID Copy number / cell
Cop
y nu
mbe
r / c
ell
codebook 2
code
book
1
1µm
hyb 1 hyb 2 hyb 3 hyb 4
hyb 5 hyb 6 hyb 7 hyb 8
hyb 9 hyb 10 hyb 11 hyb 12
hyb 13 hyb 14 hyb 15 hyb 16
1µm
100
101
102
103
100
101
102
103
r = 0.89
FPKM
Cop
y nu
mbe
r/ c
ell
100
101
102
103
100
101
102
103
r = 0.94
Fig. 2. Simultaneous measurement of 140 RNA species in single cells by use of MERFISH with a16-bit MHD4 code. (A) Images of RNA molecules in an IMR90 cell after each hybridization round (hyb1 to hyb 16). The images after photobleaching (for example, bleach 1) demonstrate efficient removal offluorescent signals between hybridizations. (B) The localizations of all detected singlemolecules in this cellcolored according to their measured binary words. (Inset) The composite, false-colored fluorescent imageof the 16 hybridization rounds for the boxed subregion with numbered circles indicating potential RNAmolecules. A red circle indicates an unidentifiable molecule, the binary word of which does not match anyof the 16-bit MHD4 code words even after error correction. (C) Fluorescent images from each round ofhybridization for the boxed subregion in (B), with circles indicating potential RNA molecules. (D)Correspondingwords for the spots identified in (C). Red crosses represent the corrected bits. (E) The RNAcopy number for each gene observed without (green) or with (blue) error correction in this cell. (F) Theconfidence ratiomeasured for the 130 RNA species (blue) and the 10misidentification control words (red)normalized to the maximum value observed from the misidentification controls (dashed line). (G) Scatterplot of the average copy number of each RNA species per cell measured with two shuffled codebooks ofthe MHD4 code. The Pearson correlation coefficient is 0.94 with a P value of 1 × 10−53. The dashed linecorresponds to the y = x line. (H) Scatter plot of the average copy number of each RNA species per cellversus the abundance determined by bulk sequencing in FPKM. The Pearson correlation coefficientbetween the logarithmic abundances of the two measurements was 0.89 with a P value of 3 × 10−39.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

many cells agreed quantitatively between ourMERFISH and conventional smFISH measure-ments (fig. S8, A and B). The ratio of the copynumbers determined by these two approacheswas 0.82 T 0.06 (mean T SEM across the 15 mea-sured RNA species) (fig. S8B), which agrees withthe estimated 80%calling rate for ourmultiplexedimaging approach. The quantitative match be-tween this ratio and our estimated calling rateover the full measured abundance range addi-tionally supports our assessment that themisiden-tification error was low. Given that the agreementbetween theMERFISHand conventional smFISHresults extended to the genes at the lowest mea-sured abundance (<1 copy per cell) (fig. S8B), weestimate that our measurement sensitivity wasbetter than 1 copy per cell.As a final validation, we compared the abun-
dance of each RNA species averaged over hun-dreds of cells to those obtained from a bulk RNAsequencing measurement that we performed onthe same cell line. Our imaging results correlatedremarkably well with bulk sequencing results,with a Pearson correlation coefficient of 0.89(Fig. 2H).
High-throughput analysis ofcell-to-cell variation in gene expression
The MERFISH approach allows parallelizationof measurements of many individual RNA spe-cies and covariation analysis between differentRNA species. We first illustrated the paralleliza-tion aspect by examining the cell-to-cell variationin the expression level of each of the measuredgenes (Fig. 3A). To quantify the measured varia-tion, we computed the Fano factors, defined asthe ratio of the variance to the mean RNA copynumber, for all measured RNA species. The Fanofactors substantially deviated from 1, the valueexpected for a simple Poisson process, for manygenes and exhibited an increasing trend with themeanRNA abundance (Fig. 3B), which is consist-ent with a previous observation for other cell types(24). A simplemodel for promoter regulation—thepromoter stochastically switches between on andoff states with global constraints on the kineticrates—has been previously suggested to ratio-nalize such a trend (24, 25). According to thismod-el, this trend of increasing Fano factors with meanRNA abundance can be explained by changesin the transcription rate and/or promoter off-switching rates but not by changes in the pro-moter on-switching rate.Moreover, we identified several RNA species
with substantially larger Fano factors than thisaverage trend. For example,we found that SLC5A3,CENPF, MKI67, TNC, and KIAA1199 displayedFano factor values substantially higher than thoseof the other genes expressed at similar abundancelevels. The high variability of some of these genescan be explained by their association with thecell cycle. For example, two of these particularly“noisy” genes, MKI67 and CENPF, are both an-notated as cell-cycle related genes (26), and basedon their bimodal expression (Fig. 3C), we proposethat their transcription is strongly regulated bythe cell cycle. Other high-variability genes did not
show the same bimodal expression patterns andare not known to be associatedwith the cell cycle.Understanding the origin and implications ofnoisy gene expression is an active topic of currentresearch (24).
Analysis of expression covariationamong different genes
Analysis of covariations in the expression levelsof different genes can reveal which genes arecoregulated and elucidate gene regulatory path-ways. At the population level, such analysis oftenrequires the application of external stimuli to drivegene expression variation; hence, correlated ex-pression changes can be observed among genesthat share common regulatory elements influ-enced by the stimuli (27). At the single-cell level,one can take advantage of the natural stochasticfluctuations in gene expression for such analysisand can thus studymultiple regulatory networkswithout having to stimulate each of them indi-vidually. Such covariation analysis can constrainregulatory networks, suggest new regulatory path-ways, and predict function for unannotated genesbased on associationswith covarying genes (11, 28).We applied this approach to the 140-genemea-
surements and examined the ~10,000 pairwisecorrelation coefficients that describe how theexpression levels of each pair of genes covariedfrom cell to cell. Many of the highly variable genesshowed tightly correlated or anticorrelated varia-tions (Fig. 3C). To better understand the correla-tions for all gene pairs, we adopted a hierarchicalclustering approach, commonly used in the analy-sis of both bulk and single-cell expression data(29, 30), to organize these genes on the basis oftheir correlation coefficients (Fig. 3D). From thecluster tree structure, we identified seven groupsof genes with substantially correlated expressionpatterns (Fig. 3D and table S2). Within each ofthe seven groups, every gene showed significant-ly stronger average correlation with other mem-bers of the group than with genes outside thegroup (table S2). To further validate and under-stand these groups, we identified gene ontology(GO) terms (31) enriched in each of these sevengroups. The enrichedGO termswithin each groupshared similar functions and were largely specificto each group (Fig. 3E and table S2), validatingthe notion that the observed covariation in ex-pression reflects some commonalities in the regu-lation of these genes.Here, we describe two of these groups as il-
lustrative examples. The predominant GO termsassociated with group 1 were terms associatedwith the extracellular matrix (ECM) (Fig. 3, D andE, and table S2). Notable members of this groupincluded ECM components—such as FBN1, FBN2,COL5A, COL7A, and TNC—and glycoproteins link-ing the ECM and cell membranes, such as VCANand THBS1. The group also included an unanno-tated gene,KIAA1199, which we would predict toplay a role in ECMmetabolism on the basis of itsassociation with this cluster. Indeed, this genehas recently been identified as an enzyme in-volved in the regulation of hyaluronan, which isa major sugar component of the ECM (32).
Group 6 contained many genes that encodevesicle transport proteins and proteins associ-atedwith cell motility (Fig. 3, D and E, and tableS2). The vesicle transport genes included micro-tubulemotors and related genesDYNC1H,CKAP1,and factors associated with vesicle formation andtrafficking, such as DNAJC13 and RAB3B. Again,we found an unannotated gene, KIAA1462, with-in this cluster. On the basis of its strong cor-relation with DYNC1H1 and DNAJC13, we predictthat this gene may be involved in vesicle trans-port. The cell motility genes in this group includedgenes encoding actin-binding proteins such asAFAP1, SPTAN1, SPTBN1, andMYH10, and genesinvolved in the formation of adhesion complexes,such as FLNA and FLNC. Several guanosinetriphosphatase (GTPase)–associated factors in-volved in the regulation of cell motility, attach-ment, and contraction also fell into this group,including DOCK7, ROCK2, IQGAP1, PRKCA, andAMOTL1. The observation that some cellmotilitygenes correlated with vesicle transport genes isconsistent with the role of vesicle transport incell migration (33). An additional feature of group6 is that a subset of these genes—in particular,those related to cell motility—were anticorrelatedwith members of the ECM group discussed above(Fig. 3D). This anticorrelation may reflect regu-latory interactions that mediate the switching ofcells between adherent and migratory states.
Mapping spatial distributions of RNAs
As an imaging-based approach, MERFISH alsoallowed us to investigate the spatial distributionsof many RNA species simultaneously. Several pat-terns emerged from the visual inspection of indi-vidual genes, with someRNA transcripts enrichedin the perinuclear region, some enriched in thecell periphery, and some scattered throughoutthe cell (Fig. 4A). To identify genes with similarspatial distributions, we determined the correla-tion coefficients for the spatial density profilesof all pairs of RNA species and organized theseRNAs according to the pairwise correlationsagain using the hierarchical clustering approach.The correlation coefficient matrix showed groupsof genes with correlated spatial organizations, andthe two most notable groups with the strongestcorrelations are indicated in Fig. 4B. Group IRNAs appeared enriched in the perinuclear re-gion, whereas group II RNAs appeared enrichednear the cell periphery (Fig. 4C). Quantitativeanalysis of the distances between each RNAmol-ecule and the cell nucleus or the cell peripheryindeed confirmed this visual impression (Fig. 4D).Group I contained genes encoding extracel-
lular proteins such as FBN1, FBN2, and THSB1;secreted proteins such as PAPPA; and integralmembrane proteins such as LRP1 and GPR107.These proteins have no obvious commonalitiesin function. Rather, a GO analysis showed signif-icant enrichment for location terms, such as extra-cellular region, basementmembrane, or perivitellinespace (Fig. 4E). To reach these locations, proteinsmust pass through the secretion pathway, whichoften requires translation of mRNA at the endo-plasmic reticulum (ER) (34, 35). Thus, we propose
aaa6090-4 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-5
AFF4SIPA1L3
NF1PRKDC
GTF3C1RAB3B
SPTBN1ZC3H13SPTAN1
DYNC1H1DOCK7
SMARCA5FLNA
DIP2CSRRM2PRPF8
IQGAP1PRKCA
USP9XPTPN14ASCC3AFAP1CREBBPCHD8HERC2TPRKIAA1462ROCK2DNAJC13AMOTL1TLN1MYH10PDS5ACKAP5TEAD1FLNC
KIAA1199
SVEP1
LRP1
COL5A1
H6PD
VCAN
THBS1
FBN2
FBN1
IGF2R
COL7A1
NUMA1
TNC
Group 1
Group 6
12
34
5
6
7-0.5 0.5
0 2
cysteine−type peptidase activitycellular response to nutrient levels
Rho GTPase bindingregulation of GTPase activity
dynein complexcell projection assemblyArp2/3 protein complex
myosin complexcell leading edge
centromeric heterochromatinDNA replication
negative regulation of transcription during mitosis
peripheral nervous system developmentensheathment of neurons
notochord formationsynaptic transmission, dopaminergic
ciliummicrovillus
macropinocytic cupembryo implantationion channel complex
regulation of integrin activationresponse to gonadotropin
extracellular matrixproteinaceous extracellular matrix
basal laminaanchoring collagen
basement membrane organization
Group 1
0 5
Group 2
0 2
Group 3
0 5Enrichment (log2)
Group 4
0 5
Group 5
0 1 2
Group 6
0 5
Group 7
0 1 2
Ungrouped
Copy number, cell 1
Cop
y nu
mbe
r, c
ell
2
1 25 50 75 100
−0.50
0.51
Cell
Z−
scor
e
−0.5
00.5
1
−0.5
00.5
1
0
0.51
THBS1 CKAP5
MYH10 IGF2R
LRP1 FBN1
MKI67 CENPF
0 1 10 1000
1
10
100
100 102
101
102
MKI67
CENPF
KIAA1199
TNC
SLC5A3
Copy number / cellF
ano
fact
or
101
ubiquitin–specific protease activity
centromere–specific nucleosome
Fig. 3. Cell-to-cell variations and pairwise correlations for the RNA spe-cies determined from the 140-gene measurements. (A) Comparison ofgene expression levels in two individual cells. (B) Fano factors for individualgenes. Error bars represent standard error of themean determined fromsevenindependent data sets. (C) Z-scores of the expression variations of four ex-ample pairs of genes showing correlated (top two) or anticorrelated (bottomtwo) variation for 100 randomly selected cells. Z-score is defined as thedifference from the mean normalized by the standard deviation. (D) Matrix ofthe pairwise correlation coefficients of the cell-to-cell variation in expressionfor the measured genes, shown together with the hierarchical clustering tree.The seven groups identified by a specific threshold on the cluster tree (dashed
line) are indicated by the black boxes in the matrix and colored lines on thetree,with gray lines on the tree indicating ungrouped genes. Different thresholdchoices on the cluster tree could be made to select either smaller subgroupswith tighter correlations or larger super-groups containing more weakly cou-pled subgroups.Two of the seven groups are enlarged on the right. (E) Enrich-ment of 30 selected, statistically significantly enriched GO terms in the sevengroups. Enrichment refers to the ratio of the fraction of genes within a groupthat have the specific GO term to the fraction of all measured genes havingthat term. Top 10 statistically significantly enriched GO terms for each of theseven groups are shown in table S2.Not all of theGO terms presented here arein the top 10 list.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

that the spatial pattern that we observed forthese mRNAs reflects their cotranslational en-richment at the ER. The enrichment of thesemRNAs in the perinuclear region (Fig. 4, C and D,light blue), where the rough ER resides, supportsthis conclusion.Group II contained genes encoding the actin-
binding proteins, including filamins FLNA andFLNC, talin TLN1, and spectrins SPTAN1 andSPTBN1; themicrotubule-binding protein CKAP5;and the motor proteins MYH10 and DYNC1H1.This group was enriched with GO terms such ascortical actin cytoskeleton, actin filament bind-ing, and cell-cell adherens junction (Fig. 4E). Ithas been shown previously that b-actin mRNA isenriched near the cell periphery in fibroblasts, asare mRNAs that encode members of the actin-binding Arp2/3 complex (36, 37). The enrichmentof group II mRNAs in the peripheral region ofthe cells (Fig. 4, C and D) suggests that the spa-tial distribution of the group II genes mightbe related to the distribution of actin cytoskele-ton mRNAs.
Measuring 1001 genes with MERFISHby use of a 14-bit MHD2 code
Last, we sought to further increase the through-put of our MERFISH measurement by simulta-neously imaging ~1000RNA species. This increasecould be achieved with our MHD4 code by in-creasing the number of bits per code word to 32while maintaining the number of 1 bits per word
at four (Fig. 1B). This could be implemented byeither increasing the number of hybridizationround to 32 or maintaining 16 rounds of hybrid-ization, but using two-color imaging in each round.We pursued an alternative approach that didnot require an increase in the number of hybrid-izations or color channels by relaxing the errorcorrection requirement but keeping the error-detection capability. For example, by reducing theHamming distance from 4 to 2, we could use all14-bit words that contain four 1 bits to encode1001 genes and probe these RNAs with only 14rounds of hybridization. However, because a sin-gle error can produce a word equally close to twodifferent code words, error correction is no longerpossible for this modified Hamming-distance-2(MHD2) code. Hence, we expect the calling rateto be lower and the misidentification rate to behigher with this encoding scheme.To evaluate the performance of this 14-bit
MHD2 code, we set aside 16 of the 1001 possiblecode words as misidentification controls andused the remaining 985 words to encode cellularRNAs (table S3). Among these 985 RNAs, we in-cluded 107 RNA species probed in the 140-geneexperiments as an additional control. We per-formed the 1001-gene experiments in IMR90 cellsby using a similar procedure as described above.To allow all encoding probes to be synthesizedfrom a single 100,000-member oligopool, we re-duced the number of encoding probes per RNAspecies to ~94. Fluorescent spots corresponding
to individual RNA molecules were again clearlydetected in each round of hybridization with thereadout probes, andbased on their on-off patterns,these spots were decoded into RNA (Fig. 5A andfig. S9, A and B). In the cell shown in Fig. 5A, 430RNA species were detected, and similar resultswere obtained in ~200 imaged cells in three in-dependent experiments.As expected, the misidentification rate of this
scheme was higher than that of the MHD4 code.Of all real RNA words, 77% were detected morefrequently than the median count for the mis-identification controls, instead of the 95% valueobserved in theMHD4measurements. Using thesame confidence ratio analysis as described above,we found that 73% (instead of 91% for the MHD4measurements) of the 985 RNA species weremeasured with a confidence ratio larger thanthe maximum value observed for the misidenti-fication controls (fig. S9C). RNA copy numbersmeasured from these 73% RNA species showedexcellent correlation with our bulk RNA sequenc-ing results (Pearson correlation coefficient r =0.76) (Fig. 5B, black). The remaining 27% of thegenes still exhibit good, albeit lower, correlationwith the bulk RNA sequencing data (r = 0.65)(Fig. 5B, red), but we took the conservative mea-sure of excluding them from further analysis.The lack of an error correction capability also
decreased the calling rate of each RNA species:When comparing the 107 RNA species commoninboth the 1001-gene and 140-genemeasurements,
aaa6090-6 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
THBS1 FBN2 FLNA TLN1
10 µm
10 µm
-0.2 0.2
0.8 0.9 1 1.1 1.2
0.85 0.9 0.95 1 1.05 1.1 1.15
Group II
Group I
Averagegene
Relative distance to nucleus
Relative distance to cell edge
Group II
Group I
Averagegene
II
I0 2
cell−cell adherens junction
cortical actin cytoskeleton
perivitelline space
extracellular region
basement membrane
Enrichment (log2)
Group I
0 2
Group II
Group IGroup II
Fig. 4. Distinct spatial distributions of RNAs observed in the 140-genemeasurements. (A) Examples of the spatial distributions observed for fourdifferent RNA species in a cell. (B) Matrix of the pairwise correlation co-efficients describing the degree with which the spatial distributions of eachgene pair is correlated, shown together with the hierarchical clustering tree.Two strongly correlating groups are indicated by the black boxes on thematrix
and color on the tree. (C) The spatial distributions of all RNAs in the two groupsin two example cells. Light blue symbols, group I genes; red symbols, group IIgenes. (D) Average distances for genes in group I and genes in group II to thecell edge or the nucleus normalized to the average distances for all genes. Errorbars represent SEM across seven data sets. (E) Enrichment of GO terms ineach of the two groups.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

we found that the copy numbers per cell of theseRNA species were lower in the 1001-gene mea-surements (Fig. 5C and fig. S9D). The total countof these RNAs per cell was ~1/3 of that observedin the 140-gene measurements. Thus, the lackof error correction in the MHD2 code reducedthe calling rate to ~30% of that of the MHD4code, which is consistent with the decrease incalling rate observed for the MHD4 code whenerror correction was not applied. As expectedfrom the quantitative agreement between 140-genemeasurements and conventional smFISH results,comparison of the 1001-gene measurements withconventional smFISH results for 10 RNA speciesalso indicated a calling rate that is ~1/3 of thatobserved for the MHD4 code (fig. S8C). Despitethe expected reduction in calling rate, the goodcorrelations found between the copy numbersobserved in the 1001-gene measurements andthose observed in the 140-gene measurements, aswell as in conventional smFISH and bulk RNAsequencing measurements, indicates that the rel-ative abundance of these RNAs can be quantifiedwith the MHD2 encoding scheme.Simultaneously imaging ~1000 genes in indi-
vidual cells substantially expanded our ability todetect coregulated genes. The matrix of pairwisecorrelation coefficients determined from the cell-to-cell variations in the expression levels of thesegenes is shown in Fig. 6A. Using the same hier-archical clustering analysis as described above,
we identified ~100 groups of genes with corre-lated expression (table S4). Nearly all of these~100 groups showed statistically significant en-richment of functionally related GO terms (Fig.6B and table S4). These included some of thegroups identified in the 140-genemeasurements,such as the group associated with cell-replicationgenes and the group associated with cell-motilitygenes (Fig. 6, A and B, groups 7 and 102), as wellas many new groups. The groups identified hereincluded 46 RNA species lacking any previousGO annotations, for which we can now hypoth-esize function on the basis of their group asso-ciation (table S4). For example, KIAA1462 is partof the cell motility group, as also shown in the140-gene experiments, suggesting a potential roleof this gene in cell motility (Fig. 6A, group 102).Likewise, KIAA0355 is part of a new group en-riched in genes associated with heart develop-ment (Fig. 6A, group 79), and C17orf70 is part of agroup associatedwith ribosomal RNAprocessing(Fig. 6A, group 22). Using these groupings, wecan also hypothesize cellular functions for 61transcription factors and other partially annotatedproteins of unknown functions (table S4). Forexample, the transcription factors Z3CH13 andCHD8 are bothmembers of the cell-motility group,suggesting their potential role in the transcrip-tional regulation of cell-motility genes. Althoughthesepredicted functionsbasedongene-associationanalysis require further validation, our covaria-
tion data provide a resource for generating hy-potheses on gene function and regulation.
Discussion
We have developed a highly multiplexed detec-tion scheme for transcriptomic-scale RNA imag-ing in single cells. Using combinatorial labeling,sequential hybridization and imaging, and twodifferent error-robust encoding schemes,we simulta-neously imaged either 140 or 1001 genes in hun-dreds of individual human fibroblast cells. Of thetwo encoding schemes presented here, theMHD4code is capable of both error detection and errorcorrection and hence can provide a higher callingrate and a lower misidentification rate than cantheMHD2 code, which instead can only detect butcannot correct errors. MHD2, on the other hand,provides a faster scaling of the degree of multi-plexing with the number of bits than canMHD4.Other error-robust encoding schemes can also beused for such multiplexed imaging, and experi-menters can set the balance between detectionaccuracy and ease of multiplexing according tothe specific requirements of the experiments.By increasing the number of bits in the code
words, it should be possible to further increasethe number of detectable RNA species by usingMERFISH with either MHD4 or MHD2 codes.Because of their much slower increase in errorrates with the number of bits, we expect the error-correcting encoding schemes, such as MHD4, tobemore favorable for scalingup themeasurements.For example, using the MHD4 code with 32 totalbits and four or six 1 bits would increase the num-ber of addressable RNA species to 1240 or 27,776,respectively; the latter is the approximate scale ofthe human transcriptome. The predicted misiden-tification and calling rates are still reasonable forthe 32-bit MHD4 code (shown in Fig. 1, C and D,purple for the MHD4 code with four 1 bits, andsimilar rates were calculated for the MHD4 codewith six 1 bits). If more accuratemeasurements aredesired, an additional increase in the number ofbits would allow the use of encoding schemeswith a Hamming distance greater than 4, furtherenhancing the error detection and correction ca-pability. Although an increase in the number ofbits by adding more hybridization rounds wouldincrease the data collection time and potentiallylead to sample degradation, these problems couldbe mitigated by using multiple colors to readoutmultiple bits in each round of hybridization.As the degree of multiplexing is increased, it is
important to consider the potential increase inthe density of RNAs that need to be resolved ineach roundof imaging.On the basis of our imagingand sequencing results, we estimate that includingthe whole transcriptome of the IMR90 cells wouldlead to a totalRNAdensity of ~200molecules/mm3.Using our current imaging and analysis methods,we could resolve 2 to 3 molecules/mm3 per hy-bridization round (38), which would reach atotal RNA density of ~20 molecules/mm3 after32 rounds of hybridization. This density shouldallow all but the top 10%most expressed genes tobe imaged simultaneously or a subset of geneswith even higher expression levels to be included.
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-7
1
2
3 4
5 6 7
8
910
11
12
500 nm
100 101
100
101r = 0.89
Copy number / cell MHD4
Cop
y nu
mbe
r / c
ell
MH
D2
100 101
10−1
100
101
Cop
y nu
mbe
r / c
ell
FPKM
r = 0.76r = 0.65
10µm
Fig. 5. Simultaneous measurements of 1001 RNA species in single cells by using MERFISH with a14-bit MHD2 code. (A) The localizations of all detected single molecules in a cell colored based on theirmeasured binary words. (Inset) The composite, false-colored fluorescent image of the 14 hybridizationrounds for the boxed subregion with numbered circles indicating potential RNA molecules. Red circlesindicate unidentifiable molecules, the binary words of which do not match any of the 14-bit MHD2 codewords. Images of individual hybridization round are shown in fig. S9A. (B) Scatter plot of the average copynumber per cell measured in the 1001-gene experiments versus the abundance measured via bulksequencing.The black symbols are for the 73% of genes detected with confidence ratios higher than themaximum ratio observed for themisidentification controls.The Pearson correlation coefficient is 0.76 witha P value of 3 x 10−133. The red symbols are for the remaining 27% of genes. The Pearson correlationcoefficient is 0.65with aP value of 3 x 10−33. (C) Scatter plot of the average copy number for the 107 genesshared in both the 1001-genemeasurementwith theMHD2 code and the 140-genemeasurement with theMHD4 code. The Pearson correlation coefficient is 0.89 with a P value of 9 × 10−30. The dashed linecorresponds to the y = x line.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

By using more advanced image analysis algo-rithms to better resolve overlapping images ofindividual molecules, such as compressed sensing(39, 40), it would be possible to extend the re-solvable density by approximately fourfold andthus allow nearly the entire transcriptome, ex-cept for the top 2% most expressed genes, to beimaged all together. Last, theoretical predictions(17) indicate that the use of superresolutionimaging (41, 42) could increase the resolvabledensity to ~105 molecules/mm3, which should beample to address the entire transcriptome, evenin cell types with RNA densities substantiallyhigher than that of IMR90. However, RNAs indensely packed structures, such as p-bodies andstress granules, may still elude measurement.We have illustrated the utility of the data de-
rived from highly multiplexed RNA imaging byusing covariation and correlation analysis to re-veal distinct subcellular distribution patterns ofRNAs, to constrain gene regulatory networks, andto predict functions formany previously unanno-tated or partially annotated geneswith unknownfunctions. We anticipate that many more quan-titative analyses could be applied to such datasets that include the spatial localization and copynumber information of many RNA species in
individual cells. Given its ability to quantify RNAsacross a wide range of abundances withoutamplification bias while preserving native con-text, we envision that MERFISH will enablemany applications of in situ transcriptomic anal-yses of individual cells in culture or complextissues.
Materials and MethodsProbe design
Each RNA species in our target set was randomlyassigned a binary code word either from all 140possible code words of the 16-bit MHD4 code orfrom all 1001 possible code words of the 14-bitMHD2 code, as we describe in the main text.The encoding schemes are provided in tablesS1 and S3.We used array-synthesized oligopools as tem-
plates to make the encoding probes (22, 23). Thetemplate molecule for each encoding probe con-tains three components: (i) a central targetingsequence for in situ hybridization to the targetRNA, (ii) two flanking readout sequences de-signed to hybridize each of two distinct readoutprobes, and (iii) two flanking primer sequencesto allow enzymatic amplification of the probes
(fig. S3). The readout sequences were taken fromthe 16 possible readout sequences, each corre-sponding to one hybridization round. The read-out sequences were assigned to the encodingprobes so that for any RNA species, each of thefour readout sequences were distributed uni-formly along the length of the target RNA andappeared at the same frequency. Template mol-ecules for the 140-gene library also included acommon 20-nucleotide (nt) priming region be-tween the first polymerase chain reaction (PCR)primer and the first readout sequence. This prim-ing sequence was used for the reverse transcrip-tion step described below. All template sequencesare provided in table S5.We embedded multiple experiments in a sin-
gle array-synthesized oligopool and used PCR toselectively amplify only the oligos required for aspecific experiment. Primer sequences for thisindexed PCR reaction were generated from a setof orthogonal 25-nt sequences (43). These sequenceswere trimmed to 20 nt and selected for (i) anarrow melting temperature range (70 to 80°C),(ii) the absence of consecutive repeats of 3 ormore identical nucleotides, and (iii) the presenceof a GC clamp—one of the two 3′ terminal basesmust be G or C. To further improve specificity,
aaa6090-8 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
Group 7
Group 22
Group 79
Group 102
-0.4 0.5
7
22
79
102
2 4
cell proliferationG2/M transition of mitotic cell cycle
DNA replication
mitotic G2/M transition checkpoint
cleavage involved in rRNA processingmaturation of 5S rRNAreceptor internalization
pericardium morphogenesisheart valve morphogenesis
cardiac cell fate determinationpulmonary valve developmentpost−embryonic hemopoiesisestablishment of cell polarity
cell projection assemblyArp2/3 protein complex
myosin complexmicrotubule
Mon1−Ccz1 complex
Group 7
2 4
Enrichment(log2)
Group 22
2 4 6
Group 79
2 4
Group 102
centromere–specific nucleosome
rRNA modification
Fig. 6. Covariation analysis of the RNA species measured in the 1001-gene measurements. (A) Matrix of all pairwise correlation coefficients of the cell-to-cell variation in expression for the measured genes shown with the hierarchical clustering tree. The ~100 identified groups of correlated genes are indicated bycolor on the tree. Zoom-in of four of the groups described in the text are shown on the right. (B) Enrichment of 20 selected, statistically significantly enriched GOterms in the four groups.The statistically most significantly enriched GO terms (maximum 10) for each of the ~100 groups are shown in table S4.
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

these sequences were then screened against thehuman transcriptome by using Basic Local Align-ment Search Tool+ (BLAST+) (44), and primerswith 14 or more contiguous bases of homologywere eliminated. Last, BLAST+ was again used toidentify and exclude primers that had an 11-nthomology region at the 3′ end of any otherprimer or a 5-nt homology region at the 3′ endof the T7 promoter. The forward primer sequences(primer 1) were determined as described above,whereas the reverse primers each contain a 20-ntsequence as described above plus a 20-nt T7promoter sequence to facilitate amplification viain vitro transcription (primer 2). The primer se-quences used in the 140-gene and 1001-gene ex-periments are listed in Table 1.Thirty-nt-long readout sequenceswere created
by concatenating fragments of the same orthog-onal primer set generated above by combiningone 20-nt primer with a 10-nt fragment of an-other. These readout sequenceswere then screened,by using BLAST+, for orthogonality with the in-dex primer sequences andother readout sequences(no more than 11 nt of homology) and for poten-tial off-target binding sites in the human genome(no more than 14 nt of homology). Fluorescentlylabeled readout probes with sequences comple-mentary to the readout sequences were used toprobe these readout sequences, one in each hy-bridization round. All used readout probes se-quences are listed in Table 2.The readout probes used for the 140-gene
libraries were probes 1 through 16. The readoutprobes used for the 1001-gene experiment wereprobes 1 through 14. “/3Cy5Sp/” indicates a 3′ Cy5modification.To design the central targeting sequences of
the encoding probes, we first compiled the abun-dance of different transcripts in IMR90 cellsusing Cufflinks v2.1 (45), total RNA data from theEncyclopedia of DNA Elements (ENCODE)project (46), and human genome annotationsfrom Gencode v18 (20). Probes were designedfrom gene models corresponding to the mostabundant isoform by using OligoArray2.1 (47)with the following constraints: The target se-quence region is 30-nt long; the melting tem-peratures of the hybridized region of the probeand cellular RNA target is greater than 70°C;there is no cross hybridization targets with melt-ing temperatures greater than 72°C; there is nopredicted internal secondary structures withmelting temperatures greater than 76°C; andthere is no contiguous repeats of six or moreidentical nucleotides. Melting temperatures wereadjusted to optimize the specificity of theseprobes and minimize secondary structure while
still producing sufficient numbers of probes forour libraries. To decrease computational cost,isoforms were divided into 1-kb regions for probedesign. Using BLAST+, all potential probes thatmapped to more than one cellular RNA specieswere rejected. Probes with multiple targets onthe same RNA were kept.For each gene in the 140-gene experiments, we
generated 198 putative encoding probe sequencesby concatenating the appropriate index primers,readout sequences, and targeting regions as shownin fig S3. To address the possibility that con-catenation of these sequences introduced newregions of homology to off-target RNAs, we usedBLAST+ to screen these putative sequences againstall human ribosomal RNA (rRNA) and transferRNA (tRNA) sequences aswell as highly expressedgenes [genes with fragments per kilobase per mil-lion reads (FPKM) > 10,000]. Probes with greaterthan 14 nt of homology to rRNAs or tRNAs orgreater than 17 nt of homology to highly expressedgenes were removed. After these cuts, we had~192 (with a standard deviation of 2) probes pergene for both MHD4 codebooks used in the 140-gene experiments. We followed the same proto-col for the 1001-gene experiments: Starting with96 putative targeting sequences per gene, weobtained ~94 (with a standard deviation of 6)encoding probes per gene after these additionalhomology cuts. We decreased the number of en-coding probes per RNA for the 1001-gene experi-ments so that these probes could be synthesizedfrom a single 100,000-member oligopool as op-posed to two separate pools. We designed eachencoding probe to contain two of the four read-
out sequences associated with each code word;hence, only half of the bound encoding probescan bind readout probe during any given hybrid-ization round. We used ~192 or ~94 encodingprobes perRNA to obtainhigh signal-to-backgroundratios for individual RNA molecules. The num-ber of encoding probes per RNA could be sub-stantially reduced but still allow single RNAmolecules to be identified (17, 48, 49). In addi-tion, increasing the number of readout sequencesper encoding probe or using optical sectioningmethods to reduce the fluorescence backgroundmay allow further reduction in the number ofthe encoding probes per RNA.We designed two types of misidentification
controls. The first control—blank words—werenot represented with encoding probes. The sec-ond type of control—no-targetwords—had encod-ing probes that were not targeting any cellularRNA. The targeting regions of these probes werecomposed of random nucleotide sequences sub-ject to the same constraints used to design theRNA targeting sequences described above.More-over, these random sequences were screenedagainst the human transcriptome to ensure thatthey contain no substantial homology (>14-nt) toany human RNA. The 140-gene measurementscontained five blank words and five no-targetwords. The 1001-gene measurements contained11 blank words and five no-target words.
Probe synthesis
The encoding probes were synthesized by usingthe following four steps, and this synthesis pro-tocol is illustrated in fig. S3.
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-9
Table 2. All used readout probes sequences.
Bit Readout probes
1 CGCAACGCTTGGGACGGTTCCAATCGGATC/3Cy5Sp/2 CGAATGCTCTGGCCTCGAACGAACGATAGC/3Cy5Sp/3 ACAAATCCGACCAGATCGGACGATCATGGG/3Cy5Sp/4 CAAGTATGCAGCGCGATTGACCGTCTCGTT/3Cy5Sp/5 GCGGGAAGCACGTGGATTAGGGCATCGACC/3Cy5Sp/6 AAGTCGTACGCCGATGCGCAGCAATTCACT/3Cy5Sp/7 CGAAACATCGGCCACGGTCCCGTTGAACTT/3Cy5Sp/8 ACGAATCCACCGTCCAGCGCGTCAAACAGA/3Cy5Sp/9 CGCGAAATCCCCGTAACGAGCGTCCCTTGC/3Cy5Sp/10 GCATGAGTTGCCTGGCGTTGCGACGACTAA/3Cy5Sp/11 CCGTCGTCTCCGGTCCACCGTTGCGCTTAC/3Cy5Sp/12 GGCCAATGGCCCAGGTCCGTCACGCAATTT/3Cy5Sp/13 TTGATCGAATCGGAGCGTAGCGGAATCTGC/3Cy5Sp/14 CGCGCGGATCCGCTTGTCGGGAACGGATAC/3Cy5Sp/15 GCCTCGATTACGACGGATGTAATTCGGCCG/3Cy5Sp/16 GCCCGTATTCCCGCTTGCGAGTAGGGCAAT/3Cy5Sp/
Table 1. Primer sequences used in the 140-gene and 1001-gene experiments.
Experiment namePrimer 1 sequence(index primer 1)
Primer 2 sequence(T7 promoter plus the reverse complement of index primer 2)
140-gene codebook 1 GTTGGTCGGCACTTGGGTGC TAATACGACTCACTATAGGGAAAGCCGGTTCATCCGGTGG140-gene codebook 2 CGATGCGCCAATTCCGGTTC TAATACGACTCACTATAGGGTGATCATCGCTCGCGGGTTG1001-gene CGCGGGCTATATGCGAACCG TAATACGACTCACTATAGGGCGTGGAGGGCATACAACGC
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

Step 1: The template oligopool (CustomArray)was amplified via limited-cycle PCR on a Bio-RadCFX96 by using primer sequences specific to thedesired probe set. To facilitate subsequent ampli-fication via in vitro transcription, the reverseprimer contained the T7 promoter. All primerswere synthesized by Integrated DNA Technol-ogies (IDT). This reaction was column purified(Zymo DNA Clean and Concentrator, D4003).Step 2: The purified PCR products were then
further amplified ~200-fold and converted intoRNA via a high yield in vitro transcription ac-cording to the manufacturer’s instructions [NewEngland Biolabs (NEB), E2040S]. Each 20 mL re-action contained ~1 mg of template DNA fromabove, 10 mM of each NTP, 1× reaction buffer,1× RNase inhibitor (Promega RNasin, N2611)and 2 mL of the T7 polymerase. This reaction wasincubated at 37°C for 4 hours to maximize yield.This reaction was not purified before the fol-lowing steps.Step 3: The RNA products from the above in
vitro transcription reaction were then con-verted back into DNA via a reverse transcrip-tion reaction. Each 50-mL reaction containedthe unpurified RNA produce from step 2 sup-plemented with 1.6 mM of each dNTP, 2 nmolof a reverse transcription primer, 300 units ofMaxima H- reverse transcriptase (Thermo Sci-entific, EP0751), 60 units of RNasin, and a final1× concentration of the Maxima RT buffer. Thisreaction was incubated at 50°C for 45 min, andthe reverse transcriptase was inactivated at 85°Cfor 5 min. The templates for the 140-gene librariescontain a common priming region for this re-verse transcription step; thus, a single primerwas used for this step when creating these probes.Its sequence was CGGGTTTAGCGCCGGAAATG.A common priming region was not included forthe 1001-gene library; thus, the reverse transcrip-tion was conducted with the forward primer:CGCGGGCTATATGCGAACCG.Step 4: To remove the template RNA, 20 mL of
0.25 M EDTA and 0.5 N NaOH was added to theabove reaction to selectively hydrolyze RNA, andthe sample was incubated at 95°C for 10 min.This reaction was then immediately purified bymeans of column purification using a 100-mg-capacity column (Zymo Research, D4030) andthe Zymo Oligo Clean and Concentrator proto-col. The final probes were eluted in 100 mL ofribonuclease (RNase)–free deionized water, evap-orated in a vacuum concentrator, and then re-suspended in 10 mL of encoding hybridizationbuffer (recipe below). Probeswere stored at –20°C.Denaturing polyacrylamide gel electrophoresisand absorption spectroscopy were used to con-firm the quality of the probes and revealed thatthis probe synthesis protocol converts 90 to 100%of the reverse-transcriptionprimer into full-lengthprobe and of the probe that is constructed, 70 to80% is recovered during the purification step.This protocol is similar to another recently pub-lished protocol (23) but provides a substantiallylarger yield.Fluorescently labeled readout probes have se-
quences complementary to the readout sequences
described above and a Cy5 dye attached at the 3′end. These probes were synthesized and purifiedby means of high-performance liquid chroma-tography (HPLC) by IDT.
Sample preparation and labelingwith encoding probes
Human primary fibroblasts (American Type Cul-ture Collection, IMR90), a commonly used cellline with a previously determined transcriptome(46), were used in this work. These cells are rel-atively large and flat, facilitating wide-field im-aging without the need for optical sectioning.Cells were cultured with Eagle’s Minimum Es-sential Medium. Cells were plated on 22-mm, #1.5coverslips (Bioptechs, 0420-0323-2) at 350,000cells per coverslip and incubated at 37°C with 5%CO2 for 48 to 96 hours within petri dishes. Cellswere fixed for 20 min in 4% paraformaldehyde(ElectronMicroscopy Sciences, 15714) in 1× phos-phate buffered saline (PBS; Ambion, AM9625) atroom temperature, reduced for 5 min with 0.1%w/v sodium borohydride (Sigma, 480886) in wa-ter to reduce background fluorescence, washedthree times with ice-cold 1× PBS, permeabilizedfor 2 min with 0.5% v/v Triton (Sigma, T8787) in1× PBS at room temperature, and washed threetimes with ice cold 1× PBS.Cells were incubated for 5 min in encoding
wash buffer comprising 2× saline-sodium citratebuffer (SSC) (Ambion, AM9763), 30% v/v form-amide (Ambion, AM9342), and 2 mM vanadylribonucleoside complex (NEB, S1402S). Ten mi-croliters of 100 mM (140-gene experiments) or200 mM (1001-gene experiments) encoding probesin encoding hybridization buffer was added tothe cell-containing coverslip and spread uniformlyby placing another coverslip on top of the sample.Sampleswere then incubated in a humid chamberinside a 37°C-hybridization oven for 18 to 36 hours.Encoding hybridization buffer is composed of en-coding wash buffer supplemented with 1 mg/mLyeast tRNA (Life technologies, 15401-011) and 10%w/v dextran sulfate (Sigma, D8906-50G).Cells were then washed with encoding wash
buffer, incubated at 47°C for 10min, and thiswashwas repeated for a total of three times. A 1:1000dilution of 0.2-mm-diameter carboxylate-modifiedorange fluorescent beads (Life Technologies,F-8809) in 2×SSC was sonicated for 3 min andthen incubated with the sample for 5 min. Thebeads were used as fiducial markers to alignimages obtained frommultiple successive roundsof hybridization, as described below. The samplewas washed once with 2×SSC, and then post-fixed with 4% v/v paraformaldehyde in 2×SSC atroom temperature for 30 min. The sample wasthenwashed three times with 2×SSC and eitherimaged immediately or stored for no longer than12 hours at 4°C before imaging. All solutionswere prepared as RNase-free.
MERFISH imaging
The sample coverslip was assembled into aBioptech’s FCS2 flow chamber, and the flowthrough this chamber was controlled via a home-built fluidics system composed of three computer-
controlled eight-way valves (Hamilton, MVP andHVXM 8-5) and a computer-controlled peristalticpump (Rainin, Dynamax RP-1). The sample wasimaged on a home-built microscope constructedaround an Olympus IX-71 body and a 1.45 NA,100× oil immersion objective and configured foroblique incidence excitation. The objective washeated to 37°C with a Bioptechs objective heater.Constant focus was maintained throughout theimaging process with a home-built, autofocusingsystem. Illumination was provided at 641, 561,and 405 nm by using solid-state lasers (MPBcommunications, VFL-P500-642; Coherent, 561-200CWCDRH; and Coherent, 1069413/AT) forexcitation of our Cy5-labeled readout probes,the fiducial beads, and nuclear counterstains, re-spectively. These lines were combined with acustom dichroic (Chroma, zy405/488/561/647/752RP-UF1) and the emission was filtered witha custom dichroic (Chroma, ZET405/488/561/647-656/752m). Fluorescence was separated witha QuadView (Photometrics) by using the dichroicsT560lpxr, T650lpxr, and 750dcxxr (Chroma) andthe emission filters ET525/50m, WT59550m-2f,ET700/75m, and HQ770lp (Chroma) and imagedwith an EMCCD camera (Andor, iXon-897). Thecamera was configured so that a pixel corre-sponds to 167 nm in the sample plane. The en-tire system was fully automated, so that imagingand fluid handling were performed for the entireexperiment without user intervention.Sequential hybridization, imaging, and bleach-
ing proceeded as follows. One milliliter of 10 nMof the appropriate fluorescently labeled readoutprobe in readout hybridization buffer (2×SSC;10% v/v formamide, 10% w/v dextran sulfate,and 2 mM vanadyl ribonucleoside complex) wasflown across the sample, flow was stopped, andthe sample was incubated for 15 min. Then 2 mLof readout wash buffer (2×SSC, 20% v/v form-amide, and 2 mM vanadyl ribonucleoside com-plex) was flown across the sample, flow wasstopped, and the samplewas incubated for 3min.Two milliliters of imaging buffer comprising2×SSC, 50 mM TrisHCl pH 8, 10% w/v glucose,2 mMTrolox (Sigma-Aldrich, 238813), 0.5 mg/mLglucose oxidase (Sigma-Aldrich, G2133), and40 mg/mL catalase (Sigma-Aldrich, C30) was flownacross the sample (50). Flow was then stopped,and then ~75 to 100 regions were exposed to~25mW 642-nm and 1 mW of 561-nm light andimaged. Each region was 40 by 40 mm. The laserpowers were measured at the microscope back-port. Because the imaging buffer is sensitive tooxygen (51), the ~50 mL of imaging buffer usedfor a single experiment was made fresh at thebeginning of the experiment and then storedunder a layer of mineral oil throughout the mea-surement. Buffer stored in this fashion was sta-ble for more than 24 hours.After imaging, the fluorescence of the readout
probes was extinguished via photobleaching.The sample was washed with 2 mL of photo-bleaching buffer (2×SSC and 2 mM vanadylribonucleoside complex), and each imaged re-gion of the sample was exposed to 200 mW of641-nm light for 3 s. To confirm the efficacy of
aaa6090-10 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

this photobleaching treatment, imaging bufferwas reintroduced, and the sample was imagedas described above.The above hybridization, imaging, and photo-
bleaching process was repeated either 16 timesfor the 140-genemeasurementsbyusing theMHD4code or 14 times for the 1001-genemeasurementsby using the MHD2 code. An entire experimentwas typically completed in ~20 hours.After completion of imaging, 2 mL of a 1:1000
dilution of Hoescht (ENZ-52401) in 2×SSC wasflown through the chamber to label the nucleiof the cells. The sample was then washed im-mediately with 2 mL of 2×SSC followed by 2 mLof imaging buffer. Each region of the samplewas then imaged once again with ~1 mW of405-nm light.Because we imaged cells using wide-field im-
agingwith oblique-incidence illumination, withoutoptical sectioning and z-scanning, we quantifiedthe fraction of individual RNA species that wasoutside the axial range of our imaging geometryfor six different RNA species using conventionalsmFISH. For this purpose, we optically sectionedthese cells by collecting stacks of images at differ-ent focal depths through the entire depth of thecells. We aligned the images in consecutive focalplanes and then computed for each cell the frac-tion of RNAs that were detected in the three-dimensional stack but not in the basal focalplane.We found that only a small fraction, 15 T 1%(mean T SEM across six different RNA species) ofRNA molecules were outside the imaging rangeof a fixed focal plane without z-scanning. Thesemeasurements also confirmed that our excita-tion geometry illuminated the full depth of ourcells. From an imaging perspective, any opticalsectioning technique could be used inMERFISHto allow the imaging of RNAs in thicker cells ortissues.
Construction of measured words
Fluorescent spots were identified and localizedin each image by using a multi-Gaussian-fittingalgorithm (38) assuming a Gaussian with a uni-form width of 167 nm. This algorithm was usedto allow partially overlapping spots to be distin-guished and individually fit. RNA spots weredistinguished from background signal—signalarising from probes bound nonspecifically, bysetting the intensity threshold required to fit aspot with this software. Because of variation inthe brightness of spots between rounds of hy-bridization, this threshold was adjusted appro-priately for each hybridization round in order tominimize the combined average of the 1→0 and0→1 error rates across all hybridization rounds(140-gene measurements) or to maximize theratio of the number ofmeasuredwords with four1 bits to those with three or five 1 bits (1001-genemeasurements). The location of the fiducial beadswas identified in each frame by using a fastersingle-Gaussian fitting algorithm.Images of the same sample region in different
rounds of hybridization were registered by ro-tating and translating the image to align the twofiducial beads within the same image that were
most similar in location after a coarse initial align-ment via image correlation.All imageswere alignedto a coordinate system established by the imagescollected in the first round of hybridization. Thequality of this alignment was determined fromthe residual distance between five additionalfiducial beads, and alignment error was typically~20 nm.Fluorescence spots in different hybridization
rounds were connected into a single string, cor-responding to a potential RNA molecule, if thedistance between spots was smaller than 1 pixel(167 nm). For each string of spots, the on-offsequence of fluorescent signals in all hybridiza-tion rounds were used to assign a binary word tothe potential RNA molecule, in which 1 was as-signed to the hybridization rounds that con-tained a fluorescent signal above threshold and 0was assigned to the other hybridization rounds.Measured words were then decoded into RNAspecies by using the 16-bit MHD4 code or the 14-bitMHD2 code discussed in themain text. In thecase of the 16-bit MHD4 code, if the measuredbinary word matched the code word of a specificRNA perfectly or differed from the code word byone single bit, it was assigned to that RNA. In thecase of the 14-bit MHD2 code, only if the mea-sured binary word matched the code word of aspecific RNA perfectly was it assigned to thatRNA. To determine the copy number per cell, thenumber of each RNA species was counted in in-dividual cells within each 40- by 40-mm imagingarea. This number accounts for the majority butnot all RNA molecules within a cell because afraction of the cell could be outside the imagingarea or focal depth. Tiling images of adjacentareas and adjacent focal planes could be used toimprove the counting accuracy.In the 140-gene experiments, some regions of
the cell nucleus occasionally contained toomuchfluorescence signal to properly identify individ-ual RNA spots. In the 1001-gene experiments, thecell nucleus generally contained too much fluo-rescent signal to allow identification of individ-ual RNA molecules. These bright regions wereexcluded from all subsequent analysis. This workfocuses on mRNAs, which are enriched in thecytoplasm. To estimate the fraction of mRNAsmissed by excluding the nucleus region, we usedconventional smFISH to quantify the fraction ofmolecules found inside the nucleus for six dif-ferentmRNAs species.We found that only 5 T 2%(mean T SEM across six RNA species) of theseRNA molecules are found in the nucleus. Useof super-resolution imaging and/or optical sec-tioning could potentially allow individual mole-cules in these dense nucleus regions to beidentified, which will be particularly useful forprobing those noncoding RNAs that are enrichedin the nucleus.
smFISH measurements of individual genes
Pools of 48 fluorescently labeled (Quasar 670)oligonucleotide probes per RNA were purchasedfrom Biosearch Technologies. Thirty-nucleotideprobe sequences were taken directly from a ran-dom subset of the targeting regions used for the
multiplexed measurements. Cells were fixedand permeabilized as described above. Ten mi-croliters of 250 nM oligonucleotide probes inencoding hybridization buffer (described above)was added to the cell-containing coverslip andspread uniformly by placing another coverslipon top of the sample. Samples were then in-cubated in a humid chamber inside a 37°C-hybridization oven for 18 hours. Cells were thenwashed with encoding wash buffer (describedabove) at 37°C for 10 min, and this wash wasrepeated for a total of three times. The samplewas then washed three times with 2×SSC andimaged in imaging buffer by using the same im-aging geometry as described above forMERFISHimaging.
Bulk RNA sequencing
Total RNA was extracted from IMR90 cells cul-tured as above using the Zymo Quick RNAMiniPrep kit (R1054) according to the manufac-turer’s instructions. Polyadenylated [poly(A)] RNAwas then selected (NEB, E7490), and a sequencinglibrary was constructed by using the NEBNextUltra RNA library preparation kit (NEB, E7530),amplified with custom oligonucleotides, and150–base pair (bp) reads were obtained on aMiSeq. These sequences were aligned to the hu-man genome (Gencode v18) and isoform abun-dance was computed with cufflinks (45).
Calculation of the predicted scalingand error properties of differentencoding schemes
Analytic expressions were derived for the depen-dence of the number of possible code words, thecalling rate, and the misidentification rate on N.The calling rate is defined as the fraction of RNAmolecules that are properly identified. The mis-identification rate is defined as the fraction ofRNAmolecules that aremisidentified as a wrongRNA species. For encoding schemeswith an error-detection capability, the calling rate and misiden-tification rate does not add up to 1 because afraction of the molecules not called properly canbe detected as errors and discarded and, hence,not misidentified as a wrong species. These cal-culations assume that the probability of misread-ing bits is constant for all hybridization roundsbut differs for the 1→0 and 0→1 errors. Exper-imentally measured average 1→0 and 0→1 errorrates (10 and 4%, respectively) were used for theestimates shown in Fig. 1, B to D. For simplicity,the word corresponding to all 0s was not re-moved from calculations.For the simple binary encoding scheme in
which all possibleN-bit binarywords are assignedto different RNA species, the number of possiblecode words is 2N. The number of words thatcould be used to encode RNA is actually 2N −1 because the code word “00…0” does not con-tain detectable fluorescence in any hybridizationround, but for simplicity theword correspondingto all 0s was not removed from subsequent cal-culations. The error introduced by this approxi-mation is negligible. For any given word withm1s and N − m 0s, the probability of measuring
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-11
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

that word without error—the fraction of RNAsthat is properly called—is
ð1 − p1Þmð1 − p0ÞN−m ð1Þwhere p1 is 1→0 error rate and p0 is 0→1 errorrate per bit. Because different words in this sim-ple binary encoding scheme can have differentnumbers of 1 bits, the calling rate for differentwords will differ if p1 ≠ p0. The average callingrate, reported in Fig. 1C, was determined fromthe weighted average of the value of Eq. 1 for allwords. This weighted average is
1
2N∑
m¼0
N�Nm
�ð1 − p1Þmð1 − p0ÞN−m ð2Þ
where
�Nm
�is the binomial coefficient and cor-
responds to the number of words withm 1 bits inthis encoding scheme. Because in this encodingscheme every error produces a binary word thatencodes a different RNA, the average misidenti-fication rate for this encoding scheme, reportedin Fig. 1D, follows directly from Eq. 2:
1 −1
2N∑
m¼0
N�Nm
�ð1 − p1Þmð1 − p0ÞN−m ð3Þ
To calculate the scaling and error properties ofthe extended Hamming distance 4 (HD4) code, wefirst created the generator matrix for the desirednumber of data bits using standardmethods (21).The generatormatrix determines the specific wordsthat are present in a given encoding scheme andwas used to directly determine the number of en-codedwords as a function of the number of bits. Inthis encoding scheme, the calling rate correspondsto the fraction of wordsmeasured without error aswell as the fraction of words measured with asingle-bit error. For codewordswithm 1 bits, thisfraction is determinedby the following expression:
ð1 − p1Þmð1 − p0ÞN−m þmp11ð1 − p1Þm−1ð1 − p0ÞN−m þðN −mÞp1
0ð1 − p1Þmð1 − p0ÞN−m−1 ð4Þwhere the first term is theprobability ofnotmakingany errors, the second termcorresponds to the totalprobability of making one 1→0 error at any of them 1 bits withoutmaking any other 0→1 errors, andthe final term corresponds to the total probabilityof making one 0→1 error at any of theN-m 0 bitswithoutmaking any 1→0 errors. Because the num-ber of 1 bits can differ between words in thisencoding scheme, the average calling rate reportedin Fig. 1C was computed from a weighted averageover Eq. 4 for different values ofm. The weight foreach term was determined from the number ofwords that contain m 1 bits as determined fromthe generator matrix described above.Because RNA-encoding words are separated
by a minimumHamming distance of 4, at least 4errors are required to switch one word into an-other. If error correction is applied, then 3 or 5errors could also convert one RNA into another.Thus, we estimate themisidentification rate fromall possible combinations of 3-bit, 4-bit, and 5-biterrors for code words with m 1 bits. Technically,>5-bit errors could also convert one RNA into
another, but the probability of making such er-rors is negligible because of the small per-bit er-ror rate. We approximate this expression with
∑i¼0
4�mi
��N−m4− i
�pi1p
4−i0 ð1−p1Þm−ið1−p0ÞN−m−ð4−iÞþ
∑i¼0
3�mi
��N −m3− i
�pi1p
3−i0 ð1−p1Þm−ið1−p0ÞN−m−ð3−iÞþ
∑i¼0
5�mi
��N −m5 − i
�pi1p
5−i0 ð1−p1Þm−ið1−p0ÞN−m−ð5−iÞ
ð5ÞThe first sum corresponds to all of the ways in
which exactly four mistakes can be made. Sim-ilarly, the second and third sums correspond toall of the ways in which exactly three or fivemistakes can be made. Equation 5 provides anupper bound for the misidentification rate be-cause not all 3-, 4-, or 5-bit errors produce a wordthat matches or would be corrected to anotherlegitimate word. Again because the number of1 bits can differ between words, the average mis-identification rate reported in Fig. 1D is calcu-lated as a weighted average of Eq. 5 over thenumber of words that have m 1 bits.To generate our MHD4 code in which the
number of 1 bits for each codeword is set to 4, wefirst generated the HD4 codes as described above,and then removed all code words that did notcontain four 1s. The calling rate of this code,reported in Fig. 1C, was directly calculated fromEq. 4, but with m = 4 because all code words inthis code have four 1 bits. The misidentificationrate of this code, reported in Fig. 1D, was cal-culated by modifying Eq. 5 with the followingconsiderations: (i) the number of 1 bits, m, wasset to 4 and (ii) errors that produce words thatdo not contain three, four, or five 1 bits wereexcluded. Thus, the expression in Eq. 5 wassimplified to
�42
��N − 42
�p21p
20ð1 − p1Þ2ð1 − p0ÞN−6 þ�
41
��N − 42
�p11p
20ð1 − p1Þ3ð1 − p0ÞN−6 þ
�42
��N − 41
�p21p
10ð1 − p1Þ2ð1 − p0ÞN−5
�42
��N − 43
�p21p
30ð1 − p1Þ2ð1 − p0ÞN−7 þ
�43
��N − 42
�p31p
20ð1 − p1Þ1ð1 − p0ÞN−6
ð6ÞAgain, this expression is an upper bound on theactualmisidentification rate because not allwordswith four 1s are valid code words.
Estimates of the 1→0 and 0→1 error ratesfor each hybridization round
To compute the probability ofmisreading a bit ata given hybridization round, we used the error-correcting properties of the MHD4 code. Briefly,the probabilities of 1→0 or 0→1 errors were de-rived in the following way. Let the probability of
making an error at the ith bit,—ith hybridizationround—be pi and the actual number of RNAmol-ecules of the given species be A, then the num-ber of exact matches for this RNA will be
WE ¼ A∏i¼1
16ð1 − piÞ, and the number of one-bit
error corrected matches for this RNA corre-sponding to errors at the ith bit will be Wi ¼A pi
ð1 − piÞ ∏j¼1
16ð1 − pjÞ. The pi can be directly derived
from the ratio: Wi=WE ¼ pið1 − piÞ. This ratio as-
sumes that the 1-bit error-corrected counts wereonly generated from single-bit errors from thecorrect word and thatmulti-error contaminationfrom other RNA words is negligible. Given thatour error rate per hybridization round is smalland that it takes at least three errors to convertone RNA-encoding word into a word that wouldbe misidentified as another RNA, the above ap-proximation should be a good one.To compute the average 1→0 or 0→1 error
probabilities for each of the 16 hybridizationrounds, we use the above approach to calculatethe per-bit error rates for each bit of every gene,sort these errors on the basis of whether theycorrespond to a 1→0 or a 0→1 error, and thentake the average of these errors for each bitweighted by the number of counts observed forthe corresponding gene.
Estimates of the calling rate for individualRNA species from actual imaging data
With the estimates of the 1→0 or 0→1 errorprobabilities for each round of hybridization asdetermined above, it is possible to estimate thecalling rate for each RNA according to the spe-cific word used to encoded it. Specifically, thefraction of an RNA species that is called correctlyis determined by
∏i¼1
Nð1 − piÞ þ ∑
j¼1
N pj
ð1 − pjÞ∏i¼1
Nð1 − piÞ ð7Þ
where the first term represents the probability ofobserving an exact match of the code word andthe second term represents the probability of ob-serving an error-corrected match (with 1-bit er-ror). The values of the per-bit error rate pi foreach RNA species are determined by the specificcode word for that RNA and the measured 1→0or 0→1 error rates for each round of hybridization.If the code word of the RNA contains a 1 in the ithbit, then pi is determined from the 1→0 error ratefor the ith hybridization round; if the word con-tains a 0 in the ith bit, pi is determined from the0→1 error rate for the ith hybridization round.
Hierarchical clustering analysis of theco-variation in RNA abundance
Hierarchical clustering of the covariation in geneexpression for both the 140- and 1001-gene ex-periments was conducted as follows. First, thedistance between every pair of genes was deter-mined as 1 minus the Pearson correlation coef-ficient of the cell-to-cell variation of the measuredcopy numbers of these two RNA species, bothnormalized by the total RNA counted in the cell.Thus, highly correlated genes are “closer” to
aaa6090-12 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

one another, and highly anticorrelated genes are“further” apart. An agglomerative hierarchicalcluster tree was then constructed from these dis-tances using the unweighted pair group methodwith arithmeticmean (UPGMA). Specifically, start-ing with individual genes, we constructed hier-archical clusters by identifying the two clusters(or individual genes) that are closest to one an-other according to the arithmetic mean of thedistances between all intercluster gene pairs. Thepairs of clusters (or individual genes) with thesmallest distance are then grouped together, andthe process is repeated. The matrix of pairwisecorrelations was then sorted according to theorder of the genes within these trees.Groups of genes with substantial covariations
were identified by selecting a threshold on thehierarchical cluster tree (indicated by the dashedlines in Figs. 3D and 6A) that produced ~10groups of genes, each of which contains at leastfour members for the 140-gene experiments or~100 groups each of which contains at least threemembers for the 1001-gene experiments. Onecan change the threshold in order to identify ei-thermore tightly coupled smaller groups or largergroups with relatively loose coupling.A probability value for the confidence that a
gene belongs to a specific group was determinedby computing the difference between the averagecorrelation coefficient between that gene and allother members of that group and the averagecorrelation coefficient between that gene and allother measured genes outside that group. Thesignificance (P value) of this difference was de-terminedwith the student’s t test and is providedin tables S2 and S4.Because hierarchical clustering is inherently a
one-dimensional analysis—any given genes canonly be amember of a single group—this analysisdoes not allow all correlated gene groups to beidentified. Higher-dimension analysis, such asprincipal component analysis or k-means clus-tering, could be used to identify more covaryinggene clusters (30).
Analysis of RNA spatial distributions
To identify genes that have similar spatial dis-tributions, we subdivided each of the measuredcells into 2 by 2 regions and calculated the frac-tion of each RNA species present in each of thesebins. To control for the fact that some regions ofthe cell naturally contain more RNA than others,we calculate the enrichment for each gene—theratio of the observed fraction in a given regionfor a given RNA species to the average fractionobserved for all genes in that same region. Foreach pair of RNA species, we then determinedthe Pearson correlation coefficient of the region-to-region variation in enrichment of these twoRNA species for each cell and averaged the cor-relation coefficients over ~400 cells imaged inseven independent data sets. We then clusteredRNA species on the basis of these average cor-relation coefficients using the same hierarchicalclustering algorithm described above. Because ofthe large number of cells used for the analysis,we found that the coarse spatial binning (2 by 2
regions per cell) was sufficient to capture the spa-tial correlation between genes, and finer binningdid not produce more significantly correlatedgroups.To measure the distances of genes from the
nuclei and from the cell edge, we first used bright-ness thresholds on our cell images to segmentthe nuclei and identify the cell edge. We thenmeasured the distance from every RNAmoleculeto the nearest part of the nucleus and nearestpart of the cell edge. For each data set, we com-puted the average distance for each RNA speciesaveraged over all the cells measured. We thenaveraged these distances for the group I genes,group II genes, or all genes. Only those RNAspecies with at least 10 counts per cell were usedin this analysis to minimize statistical error onthe distance values.
GO analysis
Groups of genes were selected from the hierar-chical trees as discussed above. A collection ofGO terms (31) was determined for all measuredRNA species as well as the RNA species asso-ciated with each group from the most recent hu-man GO annotations (http://geneontology.org/page/download-annotations) by using both theannotated GO terms and terms immediately up-stream or downstream of the found annotations.The enrichment of these annotations was calcu-lated from the ratio of the fraction of geneswithin each group that have this term to thefraction of all measured genes that have thisterm and the P value for this enrichment wascalculated via the hypergeometric function. Onlystatistically significantly enriched GO terms witha P value less than 0.05 were considered.
REFERENCES AND NOTES
1. N. Crosetto, M. Bienko, A. van Oudenaarden, Spatially resolvedtranscriptomics and beyond. Nat. Rev. Genet. 16, 57–66(2015). doi: 10.1038/nrg3832; pmid: 25446315
2. A. M. Femino, F. S. Fay, K. Fogarty, R. H. Singer, Visualization ofsingle RNA transcripts in situ. Science 280, 585–590 (1998).doi: 10.1126/science.280.5363.585; pmid: 9554849
3. A. Raj, P. van den Bogaard, S. A. Rifkin, A. van Oudenaarden,S. Tyagi, Imaging individual mRNA molecules using multiplesingly labeled probes. Nat. Methods 5, 877–879 (2008).doi: 10.1038/nmeth.1253; pmid: 18806792
4. A. J. Rodriguez, K. Czaplinski, J. S. Condeelis, R. H. Singer,Mechanisms and cellular roles of local protein synthesis inmammalian cells. Curr. Opin. Cell Biol. 20, 144–149 (2008).doi: 10.1016/j.ceb.2008.02.004; pmid: 18378131
5. V. Balagopal, R. Parker, Polysomes, P bodies and stressgranules: States and fates of eukaryotic mRNAs. Curr. Opin.Cell Biol. 21, 403–408 (2009). doi: 10.1016/j.ceb.2009.03.005;pmid: 19394210
6. H. Jung, C. G. Gkogkas, N. Sonenberg, C. E. Holt, Remotecontrol of gene function by local translation. Cell 157, 26–40(2014). doi: 10.1016/j.cell.2014.03.005; pmid: 24679524
7. T. Gregor, H. G. Garcia, S. C. Little, The embryo as a laboratory:Quantifying transcription in Drosophila. Trends Genet. 30,364–375 (2014). doi: 10.1016/j.tig.2014.06.002; pmid: 25005921
8. A. R. Buxbaum, G. Haimovich, R. H. Singer, In the right place atthe right time: Visualizing and understanding mRNAlocalization. Nat. Rev. Mol. Cell Biol. 16, 95–109 (2015).doi: 10.1038/nrm3918; pmid: 25549890
9. D. R. Larson, R. H. Singer, D. Zenklusen, A single moleculeview of gene expression. Trends Cell Biol. 19, 630–637 (2009).doi: 10.1016/j.tcb.2009.08.008; pmid: 19819144
10. A. Raj, A. van Oudenaarden, Single-molecule approaches tostochastic gene expression.Annu. Rev. Biophys.38, 255–270 (2009).doi: 10.1146/annurev.biophys.37.032807.125928; pmid: 19416069
11. B. Munsky, G. Neuert, A. van Oudenaarden, Using geneexpression noise to understand gene regulation. Science 336,183–187 (2012). doi: 10.1126/science.1216379; pmid: 22499939
12. M. Lagha, J. P. Bothma, M. Levine, Mechanisms of transcriptionalprecision in animal development. Trends Genet. 28, 409–416(2012). doi: 10.1016/j.tig.2012.03.006; pmid: 22513408
13. T. Ha, Single-molecule methods leap ahead. Nat. Methods 11,1015–1018 (2014). doi: 10.1038/nmeth.3107; pmid: 25264779
14. Y. Taniguchi et al., Quantifying E. coli proteome and transcriptomewith single-molecule sensitivity in single cells. Science 329,533–538 (2010). doi: 10.1126/science.1188308; pmid: 20671182
15. N. Battich, T. Stoeger, L. Pelkmans, Image-basedtranscriptomics in thousands of single human cells atsingle-molecule resolution. Nat. Methods 10, 1127–1133 (2013).doi: 10.1038/nmeth.2657; pmid: 24097269
16. J. M. Levsky, S. M. Shenoy, R. C. Pezo, R. H. Singer, Single-cellgene expression profiling. Science 297, 836–840 (2002).doi: 10.1126/science.1072241; pmid: 12161654
17. E. Lubeck, L. Cai, Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 9,743–748 (2012). doi: 10.1038/nmeth.2069; pmid: 22660740
18. M. J. Levesque, A. Raj, Single-chromosome transcriptionalprofiling reveals chromosomal gene expression regulation.Nat. Methods 10, 246–248 (2013). doi: 10.1038/nmeth.2372;pmid: 23416756
19. E. Lubeck, A. F. Coskun, T. Zhiyentayev, M. Ahmad, L. Cai,Single-cell in situ RNA profiling by sequential hybridization.Nat. Methods 11, 360–361 (2014). doi: 10.1038/nmeth.2892;pmid: 24681720
20. J. Harrow et al., GENCODE: The reference human genomeannotation for The ENCODE Project. Genome Res. 22,1760–1774 (2012). doi: 10.1101/gr.135350.111; pmid: 22955987
21. T. K. Moon, Error Correction Coding: Mathematical Methods andAlgorithms (Wiley, New York, ed. 1, 2005).
22. B. J. Beliveau et al., Versatile design and synthesis platform forvisualizing genomes with Oligopaint FISH probes. Proc. Natl.Acad. Sci. U.S.A. 109, 21301–21306 (2012). doi: 10.1073/pnas.1213818110; pmid: 23236188
23. Y. E. Murgha, J.-M. Rouillard, E. Gulari, Methods for thepreparation of large quantities of complex single-strandedoligonucleotide libraries. PLOS ONE 9, e94752 (2014).doi: 10.1371/journal.pone.0094752; pmid: 24733454
24. A. Sanchez, I. Golding, Genetic determinants and cellularconstraints in noisy gene expression. Science 342, 1188–1193(2013). doi: 10.1126/science.1242975; pmid: 24311680
25. L. H. So et al., General properties of transcriptional timeseries in Escherichia coli. Nat. Genet. 43, 554–560 (2011).doi: 10.1038/ng.821; pmid: 21532574
26. M. Safran et al., GeneCards Version 3: The human geneintegrator. Database (Oxford) 2010, baq020 (2010).doi: 10.1093/database/baq020; pmid: 20689021
27. K. Dolinski, D. Botstein, Changing perspectives in yeastresearch nearly a decade after the genome sequence.Genome Res. 15, 1611–1619 (2005). doi: 10.1101/gr.3727505;pmid: 16339358
28. O. Padovan-Merhar, A. Raj, Using variability in gene expressionas a tool for studying gene regulation. Wiley Interdiscip. Rev.Syst. Biol. Med. 5, 751–759 (2013). doi: 10.1002/wsbm.1243;pmid: 23996796
29. M. B. Eisen, P. T. Spellman, P. O. Brown, D. Botstein, Clusteranalysis and display of genome-wide expression patterns.Proc. Natl. Acad. Sci. U.S.A. 95, 14863–14868 (1998).doi: 10.1073/pnas.95.25.14863; pmid: 9843981
30. N. Gehlenborg et al., Visualization of omics data forsystems biology. Nat. Methods 7 (suppl.), S56–S68 (2010).doi: 10.1038/nmeth.1436; pmid: 20195258
31. M. Ashburner et al., Gene ontology: Tool for the unification ofbiology. Nat. Genet. 25, 25–29 (2000). doi: 10.1038/75556;pmid: 10802651
32. H. Yoshida et al., KIAA1199, a deafness gene of unknownfunction, is a new hyaluronan binding protein involved inhyaluronan depolymerization. Proc. Natl. Acad. Sci. U.S.A. 110,5612–5617 (2013). doi: 10.1073/pnas.1215432110; pmid: 23509262
33. D. A. Lauffenburger, A. F. Horwitz, Cell migration: A physicallyintegrated molecular process. Cell 84, 359–369 (1996).doi: 10.1016/S0092-8674(00)81280-5; pmid: 8608589
34. T. A. Rapoport, Protein translocation across the eukaryoticendoplasmic reticulum and bacterial plasma membranes.Nature 450, 663–669 (2007). doi: 10.1038/nature06384;pmid: 18046402
35. C. H. Jan, C. C. Williams, J. S. Weissman, Principles of ERcotranslational translocation revealed by proximity-specificribosome profiling. Science 346, 1257521 (2014). doi: 10.1126/science.1257521; pmid: 25378630
SCIENCE sciencemag.org 24 APRIL 2015 • VOL 348 ISSUE 6233 aaa6090-13
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

36. J. B. Lawrence, R. H. Singer, Intracellular localization ofmessenger RNAs for cytoskeletal proteins. Cell 45, 407–415(1986). doi: 10.1016/0092-8674(86)90326-0; pmid: 3698103
37. L. A. Mingle et al., Localization of all seven messenger RNAs forthe actin-polymerization nucleator Arp2/3 complex in theprotrusions of fibroblasts. J. Cell Sci. 118, 2425–2433 (2005).doi: 10.1242/jcs.02371; pmid: 15923655
38. H. Babcock, Y. M. Sigal, X. Zhuang, A high-density 3Dlocalization algorithm for stochastic optical reconstructionmicroscopy. Opt. Nanoscopy 1, 6 (2012). doi: 10.1186/2192-2853-1-6; pmid: 25431749
39. L. Zhu, W. Zhang, D. Elnatan, B. Huang, Faster STORMusing compressed sensing. Nat. Methods 9, 721–723 (2012).doi: 10.1038/nmeth.1978; pmid: 22522657
40. H. P. Babcock, J. R. Moffitt, Y. Cao, X. Zhuang, Fastcompressed sensing analysis for super-resolution imagingusing L1-homotopy. Opt. Express 21, 28583–28596 (2013).doi: 10.1364/OE.21.028583; pmid: 24514370
41. S. W. Hell, Microscopy and its focal switch. Nat. Methods 6,24–32 (2009). doi: 10.1038/nmeth.1291; pmid: 19116611
42. B. Huang, H. Babcock, X. Zhuang, Breaking thediffraction barrier: Super-resolution imaging of cells. Cell 143,1047–1058 (2010). doi: 10.1016/j.cell.2010.12.002;pmid: 21168201
43. Q. Xu, M. R. Schlabach, G. J. Hannon, S. J. Elledge, Design of240,000 orthogonal 25mer DNA barcode probes. Proc. Natl.Acad. Sci. U.S.A. 106, 2289–2294 (2009). doi: 10.1073/pnas.0812506106; pmid: 19171886
44. C. Camacho et al., BLAST+: Architecture and applications. BMCBioinformatics 10, 421 (2009). doi: 10.1186/1471-2105-10-421;pmid: 20003500
45. C. Trapnell et al., Differential gene and transcript expressionanalysis of RNA-seq experiments with TopHat and Cufflinks.Nat. Protoc. 7, 562–578 (2012). doi: 10.1038/nprot.2012.016;pmid: 22383036
46. I. Dunham et al., An integrated encyclopedia of DNA elementsin the human genome. Nature 489, 57–74 (2012).pmid: 22955616
47. J.-M. Rouillard, M. Zuker, E. Gulari, OligoArray 2.0: Design ofoligonucleotide probes for DNA microarrays using athermodynamic approach. Nucleic Acids Res. 31, 3057–3062(2003). doi: 10.1093/nar/gkg426; pmid: 12799432
48. M. Batish, A. Raj, S. Tyagi, in Methods in Molecular Biology,J. E. Gerst, Ed. (Humana Press, Totowa, NJ, 2011), vol. 714,pp. 3–13.
49. A. R. Buxbaum, B. Wu, R. H. Singer, Single b-actin mRNAdetection in neurons reveals a mechanism for regulating itstranslatability. Science 343, 419–422 (2014). doi: 10.1126/science.1242939; pmid: 24458642
50. I. Rasnik, S. A. McKinney, T. Ha, Nonblinking and long-lastingsingle-molecule fluorescence imaging. Nat. Methods 3,891–893 (2006). doi: 10.1038/nmeth934; pmid: 17013382
51. X. Shi, J. Lim, T. Ha, Acidification of the oxygen scavengingsystem in single-molecule fluorescence studies: In situ sensingwith a ratiometric dual-emission probe. Anal. Chem. 82,6132–6138 (2010). doi: 10.1021/ac1008749; pmid: 20583766
ACKNOWLEDGMENTS
We thank H. Babcock for technical advice and help withinstrumentation and B. Bintu for aid in error analysis. This workwas in part supported by the National Institutes of Health. K.H.C.acknowledges a National Science Scholarship from the Agency forScience, Technology and Research of Singapore. A.N.B. acknowledgessupport by the Damon Runyon Foundation postdoctoral fellowship.J.R.M. acknowledges support from the Helen Hay Whitney Foundationpostdoctoral fellowship. S.W. acknowledges support from Jane CoffinsChild Foundation postdoctoral fellowship. X.Z. is a Howard HughesMedical Institute investigator. RNA-seq data were deposited underthe accession number GSE67685. MERFISH data are available athttp://zhuang.harvard.edu/merfish. X.Z., K.H.C., A.N.B., J.R.M.,and S.W. are inventors on a patent applied for by Harvard Universitythat covers the MERFISH method described here; X.Z., J.R.M., andA.N.B. are inventors on a patent applied for by Harvard University thatcovers the probe synthesis method described here.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/348/6233/aaa6090/suppl/DC1Figs. S1 to S9Tables S1 to S5
5 January 2015; accepted 19 March 2015Published online 9 April 2015;10.1126/science.aaa6090
aaa6090-14 24 APRIL 2015 • VOL 348 ISSUE 6233 sciencemag.org SCIENCE
RESEARCH | RESEARCH ARTICLEon S
eptember 10, 2020
http://science.sciencem
ag.org/D
ownloaded from

Spatially resolved, highly multiplexed RNA profiling in single cellsKok Hao Chen, Alistair N. Boettiger, Jeffrey R. Moffitt, Siyuan Wang and Xiaowei Zhuang
originally published online April 9, 2015DOI: 10.1126/science.aaa6090 (6233), aaa6090.348Science
, this issue p. 10.1126/science.aaa6090Sciencein a single cell. Such large-scale detection allows regulatory interactions to be analyzed at the transcriptome scale.
simultaneously imaged 100 to 1000 RNA specieset al.combinatorial labeling with error-robust encoding schemes, Chen however, the numbers of RNA species that can be simultaneously measured by smFISH has been limited. Usingfluorescence in situ hybridization (smFISH) experiments quantify the copy number and location of mRNA molecules;
The basis of cellular function is where and when proteins are expressed and in what quantities. Single-moleculeMultiplexed RNA imaging in single cells
ARTICLE TOOLS http://science.sciencemag.org/content/348/6233/aaa6090
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2015/04/08/science.aaa6090.DC1
REFERENCES
http://science.sciencemag.org/content/348/6233/aaa6090#BIBLThis article cites 49 articles, 14 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Copyright © 2015, American Association for the Advancement of Science
on Septem
ber 10, 2020
http://science.sciencemag.org/
Dow
nloaded from








![1 arXiv:2008.00217v1 [cs.CV] 1 Aug 2020 · E cient Adversarial Attacks for Visual Object Tracking Siyuan Liang 1;2, Xingxing Wei4, Siyuan Yao;2, and Xiaochun Cao1;2;3 1 Institute](https://img.pdfslide.us/doc/110x75/600185f28bc71c2a3c69fa4a/1-arxiv200800217v1-cscv-1-aug-2020-e-cient-adversarial-attacks-for-visual-object.jpg)