Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Degradative and synthetical studies in the Erythrina alkaloidseries
Author(s): Merchant, Jaysukhlal Ranchoddas
Publication Date: 1953
Permanent Link: https://doi.org/10.3929/ethz-a-000089729
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2215
DEGRADATIVE AND
SYNTHETICAL STUDIES IN THE ERYTHRINA
ALKALOID SERIES
A Thesis submitted to
The Swiss Federal Institute of Technology
Zurich
for the Degree of Doctor of Technical Science
by
JAYSUKHLAL RANCHODDAS MERCHANT
(M.Sc.,Ph.D.)
Citizen of India
Accepted on the recommendation of
Prof. Dr.V.Prelogand
Prof. Dr. L. Ruzicka
1953
Juris-Verlag, Zurich

Leer - Vide - Empty

I wish to express my gratitude to
Prof. Dr. L. Ruzicka
for giving me the opportunity and the facilities
for research work in his laboratories.
My sincere thanks are due to
Prof. Dr. V. Prelog
for his valuable help, encouragement and keen
interest throughout the course of this work.
Leer - Vide - Empty

TABLE OF CONTENTS
INTRODUCTION 7
THEORETICAL PART 10
EXPERIMENTAL PART 39
A) Degradative work on the Erythrina alkaloids 39
The von Braun degradation of dihydroerysotrine 39
The Hofmann degradation of apoerysopine 44
B) Synthetical work 48
Synthesis of l-benzyl-6,7-dimethoxy-l,2,3,4-tetrahydroisoquinoline .48
Attempted synthesis of 5-aza-4',5'-dimethoxy-3-phenyl-
benzocycloheptene 51
Synthesis of 4,5-dimethoxydiphenyl-2,2'-dicarboxylic acid 58
Synthesis of 2,3'-diethyl-4,5-dimethoxy-2'-dimethylaminobiphenyl . .63
Synthesis of unsymmetrical biaryls by the Ullmann reaction....
70
Studies in the Pictet-Spengler synthesis of tetrahydroisoquinoline
derivatives and related compounds 78
SUMMARY 83

Leer - Vide - Empty

INTRODUCTION
The Erythrina alkaloids are a group of compounds which occur in the
plants of the genus Erythrina. Some 51 species of this genus have been
investigated, and in about 25 cases the alkaloids present have been
identified. There are to date 10 known Erythrina alkaloids, and with the
exception of the two erythroidines, the molecules of all seem to possess
a common carbon-nitrogen skeleton. These substances are of interest
medicinally, because they have a strong curare-like physiological activityand could therefore find use in surgery and in the relief of convulsive
states like epilepsy.The problem of elucidating the structure of the Erythrina alkaloids
attracts the organic chemist, because this structure apparently has features
seldom encountered in nature. Of further interest is the fact that these alka¬
loids are all tertiary nitrogen bases, while in most other compounds havingcurare activity, the nitrogen is in the quaternary form.
Our knowledge of the occurrence and the distribution of the Erythrinaalkaloids and the basic experimental evidence available relative to their
structure are largely due to the contributions made by K. Folkers and his
collaborators1 over the last fourteen years. A few papers on the subjecthave also been published by other groups of workers 2.
Investigations about the structure of the Erythrina alkaloids have been
carried out in this laboratory during the past few years by different
workers 3 whose contributions have considerably advanced our knowledgeof the chemistry of these alkaloids.
1 a) Am. Soc. 59, 1580 (1937); 61, 1232, 3053 (1939); 62, 436, 1673, 1677 (1940);63, 1544 (1941); 64, 1892, 2146 (1942); 66, 1083 (1944), 71, 875 (1949);72, 1833, 5579 (1950); 73, 333, 589 (1951).
b) T. A.Henry, «The Plant Alkaloids*, p. 386—93 (1949).c) R. H. Manske and H. L. Holmes, «The Alkaloids*, Vol. //, p. 499—510 (1952).
2 R. A. Gentile and R. Labriola, J. Org. Chem. 7, 136 (1942); V. Deulofeu et al.,Soc. 1939, 1841; J. Org. Chem. 12, 486 (1947); R. A. Labriola, V. Deulofeu and
B. Berinzaghi, J. Org. Chem. 16, 90 (1951); V. Deulofeu, Ber. 85, 620 (1952);V.Prelog et al., Helv. 32, 453 (1949); 34, 1601, 1969 (1951); V. Boekelheide et
al., Science, 109, 627 (1949) Am. Soc. 72, 2062 (1950); 73, 2286 (1951); 74,1866, 2637 (1952).
3 V.Prelog, K.Wiesner, H.G.Khorana and G.W.Kenner, Helv.32, 453 (1949);M.Carmack, B. C. McKusick and V.Prelog, Helv. 34, 1601 (1951); G. W.
Kenner, H. G. Khorana and V.Prelog, Helv. 34, 1969 (1951).
7

The chief source of material for these investigations have been the seeds
of Erythrina abyssinica Lam. which are available to this laboratory from
Belgian Congo, due to the latter's use there as shadow plants for the coffee
plantations. The principal alkaloids present in this Erythrina species are
erythraline and erysodine.
Erythraline has the molecular formula C18H19O3N and contains an
aromatic nucleus, two reducible double bonds, a methylenedioxy group
attached to the aromatic ring and an aliphatic methoxyl group. The nitro¬
gen atom in erythraline is tertiary and is common to two rings. There is no
N-methyl or C-methyl group present in erythraline. Erythraline methiodide
is oxidised by permanganate in acetone to hydrastic acid N-methylimide;fusion of erythraline with potassium hydroxide gives indole. Erysodine
possesses the molecular formula C18H21O3N and like erythraline, contains
an aromatic ring, two reducible double bonds, an aliphatic and an aro¬
matic methoxyl group, and a phenolic hydroxyl group. The nitrogen atom
in erysodine is tertiary and has no methyl group attached to it. Oxidation
of erysodine with permanganate followed by methylation gives m-hemi-
pinic acid N-methylimide. Recently4, indole has been isolated in 26 %
yield by the alkaline fusion of erysodine. Upon the basis of the above
data, as well as from biogenetical considerations, Folkers and his colla¬
borators 5 proposed the following formulae for the Erythrina alkaloids.
R—R'=H erysopineR+R-CH2 erythralineR or R'=H erysovineR or R'=CH3 erysodine
During the course of investigations, carried out in this laboratory, about
the structure of the Erythrina alkaloids, a large amount of experimentalevidence was obtained which could not be satisfactorily accounted for
on the basis of the formulae, proposed by Folkers and his collaborators,
for these alkaloids. This led to a consideration of other possible structures
for the Erythrina alkaloids and further degradative work on them seemed
to be necessary. Moreover, to verify the validity of the structures assigned
4 K. Folkers, F. Koniuszy and J. Shavel, jr., Am. Soc. 73, 589, (1951).5 K. Folkers, F. Koniuszy and J. Shavel, Am. Soc. 64, 2146 (1942).
XJ
8

to the different degradation products, their synthesis was found to be
equally essential. The present work describes the various attempts made
to elucidate the structure of the Erythrina alkaloids by both these
methods.
9

THEORETICAL PART6
With a view to investigate the nature of the carbon-nitrogen skeleton
present in the Erythrina alkaloids, dihydroerysotrine (dihydroerysodine
methyl ether) was subjected to a von Braun degradation. It was observed
that dihydroerysotrine, C19H25O3N, when heated with cyanogen bromide
in chloroform solution, formed an oily bromcyanamide, which on re¬
duction with lithium aluminium hydride gave a basic substance isolated
from the reaction mixture as a crystalline picrate, C24H24O9N4, of m. p.
235—36 °. A similar treatment of erysotrine and tetrahydroerysotrine was
found to give no well-defined products. The free base regenerated from
its picrate was a crystalline, optically inactive compound of m. p. 90—92°
and possessed the molecular composition C18H21O2N. It contained two
OCH3 but no N-CH3 or C-CH3 groups. The presence of a secondary amino
group was indicated by an active hydrogen determination and the for¬
mation of a crystalline acetyl derivative; the pK. value of 8.33 was
indicative of the non-aromatic nature of basic nitrogen. The U. V. ab¬
sorption spectrum (fig. 1, curve 1) showed a band characteristic of a
dimethoxybenzene derivative. The I. R. absorption spectrum (fig. 3,
curve 1) could add little to the above information, except indicating the
presence of a 1, 2, 4, 5 tetrasubstituted and a 1,2 disubstituted benzene
nuclei.
6 The roman numbers in brackets refer to the compounds represented in the
flow-sheets.
10

Flow-sheet 1)
S
CO
o
1
e
e
o
11

On the basis of the general structure proposed by K. Folkers et al.' for
the Erythrina alkaloids, it was thought possible that the von Braun
degradation product, C18H21O2N was a benzylisoquinoline derivative
(XII) which could be formed from dihydroerysotrine as shown below:
OCH,
(XII)
Although a large number of benzylisoquinoline derivatives are known
in literature, compound (XII), namely, l-benzyl-6, 7-dimethoxy-l, 2, 3,
4-tetrahydroisoquinoline has not been described and was therefore
synthesised for purposes of comparison. The different stages of the
synthesis of (XII) are represented in the flow-sheet 3. The starting
material was homoveratrylamine (IX) which was prepared in about 70 %
yield by the reduction of 3,4-dimethoxy-(3-nitrostyrene with lithium
aluminium hydridee. This method was found to be very suitable for the
preparation of small batches of (IX), but due to the low solubility of the
nitrostyrene in ether or tetrahydrofuran, a large scale preparation of (IX)
was found to be impractical by this procedure. However, 30 to 40 g batches
of (IX) were conveniently prepared by the reduction of homoveratronitrile
with Raney nickel in ION methanolic ammonia, as described in the
«Organic Synthesis» °.
' K. Folkers, F.Koniuszy and J. Shavel, Am. Soc. 64, 2146 (1942); 73, 589 (1951).8 F.Ramirez and A. Burger, Am. Soc. 72, 2781 (1950); M. Erne and F.Ramirez.Helv. 33, 912 (1950).
9 23, p. 72—73 (1943).
12

Flow-sheet 3)
e
oe
0x
91
s
"T3
o
IS.
03
13

Condensation of (IX) with phenylacetyl chloride in the presence of
alkali gave N-phenylacetyl-homoveratrylamine (X) which on cyclisationwith phosphorus oxychloride at 110° yielded l-benzyl-6, 7-dimethoxy-3,
4-dihydroisoquinoline (XI). By the reduction of (XI) with zinc and
sulphuric acid, the required l-benzyl-6, 7-dimethoxy-l, 2, 3, 4-tetrahydro-
isoquinoline was obtained in fairly good yield. The latter compound was
an oil but readily gave a crystalline picrate of m. p. 162-63 °, which is
about 70° lower than the melting point of the picrate of the degradation
product obtained from dihydroerysotrine. A comparison of the I. R.
absorption spectra (fig. 3, curves 1 and 2) showed them to be two
distinctly different substances.
Since it was found difficult to reconcile this and other experimentalevidence, collected by different workers in this laboratory, with the
skeleton formula proposed by K. Folkers et al.10 for the Erythrina alka¬
loids, other possible structures for these alkaloids, e. g. (B) were con¬
sidered. On the basis of such a consideration, the formation of the von
Braun degradation product, C18H21O2N, from dihydroerysotrine, could
be explained as follows:
CH:
CHs
\
cao-^xF*—J*
(B)
CHsO
« XH2.-CH2CH30S^Y \
N-CN
/'CH— CH*
OCHs
CH30 -fx^CH*-CH2
( >CH30-\)\CH— CH*
6(C)
10 K. Folkers, F. Koniuszy and J. Shavel, Am. Soc. 64, 2146 (1942).
14
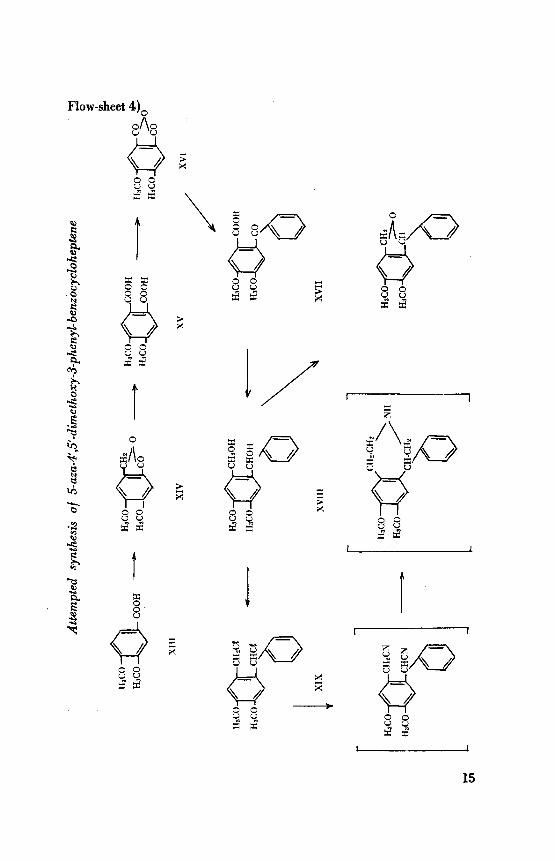
Flow-sheet 4)Q0A0
>x
e
8-
ooN
e
•o
te
-ca.
ff>
«o
-e
a,
S
a a
>
o =-L/0
>
O
>X
X X
^ o
33 XU (J
&o oo u
/O o S / \
gA_y

To test this hypothesis, the synthesis of the compound (C), namely,
5-aza-4', 5'-dimethoxy-3-phenyl-benzocycloheptene was attempted as re¬
presented in the flow-sheet 4. The starting material, veratric acid (XIII)
was obtained by the oxidation of veratraldehyde with silver nitrate in
alkaline medium". This oxidising agent was found to be superior to
potassium permanganate, since it gave not only a better yield but a purer
quality of (XIII). Chloromethylation of the latter, according to Edwards
et al.", with formaldehyde and hydrochloric acid yielded m-meconine
(XIV), which was oxidised by potassium permanganate in alkaline so¬
lution to m-hemipinic acid (XV). The anhydride (XVI) was readilyobtained from the acid by boiling it with acetic anhydride. A Friedel-
Crafts reaction of (XVI) with benzene in the presence of anhydrousaluminium chloride gave a mixture of probably demethylated and methy¬lated o-benzoylbenzoic acids, as was indicated by a ferric chloride co¬
louration. However, methylation of this mixture with methyl iodide, in
acetone solution, in the presence of anhydrous potassium carbonate, readily
gave the methyl ester of 4,5-dimethoxy-2-benzoylbenzoic acid (XVII).
The ester (XVII) was reduced by lithium aluminium hydride to the
corresponding diol (XVIII). The latter was found to be very susceptibleto acids, and in the presence of small traces of sulphuric acid readilyunderwent cyclisation with the elimination of a molecule of water to a
dihydrofuran derivative. Treatment of the diol (XVIII) with thionylchloride in chloroform solution gave the dichloride (IXX). Conversion
of this dichloride to the corresponding dinitrile was attempted under varied
experimental conditions, but in all cases, only oily mixtures containing
0.1 to 0.9 % nitrogen were obtained. The use of non-basic metallic cyanideslike cuprous, mercuric and silver cyanides also seemed to be of no avail,
because the desired dinitrile could not be prepared. The reason of this
failure appears to be probably due to side reactions like dehalogenationwhich often take place during the treatment of alkoxybenzyl or benzo-
hydryl halides with metal cyanides13.
A second method which was attempted for the synthesis of the com¬
pound (C) is described in the flow-sheet 5.
11 Rf. C.J. Lintner and L. M. Parks, J. Am. Pharm. Ass. 37, 39—40 (1948). C. A.
42, 3749 (1948).12 G. A. Edwards, W. H. Perkin jun. and F. W. Stoyle, Soc. 1925, 198.
13 D. T. Mowry, Chem. Revs. 42, 189 (1948).
16

-a
XXII
HOOC-H2C/vN.SO2.C0H4.CH!
CH2-CH2
/
H3CO—Kv
—(T
H3CO
CO-CH2
NTs
^H3CO-
\H,
C0f^
YCH.CH.
XXI
XX
ROOC-H2C/N
H.HCP
H3C0
—^/y
CH>-CH»
ROOC-H2C
NH
-u
CH2-CH2
.xs.
HjCO
H3CO
"co:
fiP
er3
5-az
a-4'
,5'-
dime
thox
y-3-
phen
yl-b
enzo
cycl
ohep
tene
ofsy
nthe
sis
Attempted

Homoveratrylamine (IX) was condensed with bromoethyl acetate at
room temperature, when homoveratrylglycine ethyl ester (XX) was
obtained. It was characterised by the preparation of a picrate. If however,
the condensation between (IX) and chloroethyl acetate was carried out
in hot alcohol solution in the presence sodium acetate, the product obtained
was found to be quite different from (XX), since the mixed melting pointof the picrates of the two compounds showed an appreciable depression.From the analytical results, the product formed in the presence of sodium
acetate appears to be probably a diketopiperazine derivative. Hydrolysisof (XX) with concentrated hydrochloric acid gave homoveratrylglycine(XXI), isolated as the hydrochloride. To facilitate the ring closure of
(XXI) to a seven-membered ring system, the amino group in (XXI) was
protected by the preparation of a p-toluenesulphonyl derivative (XXII).
Cyclisation of the latter to get the compound (C) was attempted, usingstannic chloride ,4 and 85 % ortho phosphoric acid " as the dehydrating
agents, but in both cases, no well-defined products could be isolated.
However, other reagents like anhydrous aluminium chloride 16or hydrogen
fluoride " might prove useful for this cyclisation.While this synthesis was in progress, new and convincing experimental
evidence about the probable structure of the Erythrina alkaloids was
obtained by other workers in this laboratory 18, which completely altered
the existing concept about the structure of these alkaloids. Hence further
attempts to synthesise the seven-membered ring derivative (C) were
discontinued.
According to the new probable structure for the Erythrina alkaloids,
suggested 18 mainly as a result of the reinterpretation of the formation of
apoerysopine and its Hofmann degradation products (described on p. 21),the von Braun degradation base was believed to contain a nine-membered
ring system as represented in the flow-sheet 1. A compound with such a
structure is not easy to synthesise, but it appeared likely that on oxidation
with potassium permanganate it would easily break down 19to yield 4,5-
dimethoxydiphenyl-2,2'-dicarboxylic acid (XXIX) which was therefore
synthesised for comparison.
M Cf. W. S. Johnson and H. J. Glenn, Am. Soc. 71, 1092 (1949).15 Cf. R. C. Gilmore, jr., and W. J. Horton, Am. Soc. 73, 1411—14 (1951).16 Cf. W.S.Johnson, E. L. Woroch, and B. G. Buell, Am. Soc. 71, 1901 (1949).17 Cf. W.S.Johnson, «Organic Reactions*, 2, 157 (1944).18 M. Carmack, B. C. McKusick and V. Prelog, Helv. 34, 1601 (1951).19 Cf. K. W. Bently and R. Robinson, Soc. 1952, 947.
18

19
XX
XX
4>
XX
XX>
X
XX
>
xx
a
"Si
S
J"
eIkIN
1
©
Icu
6)Flow-sheet

The synthesis of this acid was carried out according to the scheme
represented in the flow-sheet 6. The starting material, namely, 6-nitro-
veratraldehyde (XXIII) was prepared by the nitration of veratraldehyde
according to Cassaday and Bogert20. Condensation of (XXIII) with
sodium phenylacetate gave a-phenyl-4,5-dimethoxy-2-nitrocinnamic acid21
(XXIV) which was reduced to the corresponding amino acid (XXV) byammoniacal ferrous sulphate solution as described byMosettig and Burger22for similar compounds. The formation of 2,3-dimethoxyphenanthrene-9-
carboxylic acid (XXVI) from (XXV) was found to proceed with better
yields than reported by Pschorr et al.21 if the diazotisation of (XXV) was
carried out in a suspension of tetrahydrofuran with isoamyl nitrite and
sulphuric acid23. Decarboxylation of (XXVI) to 2,3-dimethoxyphenan-threne (XXVII) could be effected in excellent yields by the use of quino-line and copper powder. Oxidation of (XXVII) with chromic acid
in acetic acid solution gave 2,3-dimethoxy-9, 10-phenanthraquinone
(XXVIII) which was further oxidised with hydrogen peroxide in alkaline
medium to the required 4,5-dimethoxydiphenyl-2,2'-dicarboxylic acid
(XXIX).The dimethyl ester of the latter was a crystalline compound ofm. p.
117—18° and was characterised by its U. V. and I. R. absorption spectra.
However, contrary to expectations, the oxidation of the von Braun
degradation product with aqueous potassium permanganate did not givethe diphenic acid (XXIX) synthesised, but another product (III A) which
from analytical results appears to be a homologue of (XXIX), that is,
4,5-dimethoxydiphenyl-2,2'-diacetic acid.24. A possible synthesis of the
latter by means of an Arndt-Eistert reaction with the diphenic acid
(XXIX) should prove to be of value in establishing the structure of the
Erythrina alkaloids.
As mentioned before, further studies in the degradation of the Erythrinaalkaloids afforded very valuable and convincing experimental evidence
which altered all the older concepts about their structure and led to the
consideration of a new structural formula for these alkaloids. It was
observed by different workers in this laboratory25 that erythraline and
20 J. T. Cassaday and M. T. Bogert, Am. Soc. 61, 2462 (1939).21 R. Pschorr and W. Buckow, Ber. 33, 1829 (1900).22 E. Mosettig and A. Burger, Am. Soc. 52, 2993 (1930).23 Refer 20 above.
24 Cf. D. S. Tarbell, H. R. Frank and P. E. Fanta, Am. Soc. 68, 502 (1946).25 M. Carmack, B. C. McKusick and V. Prelog, Helv. 34, 1601 (1951).
20

erysodine on treatment with 3.2 N hydrochloric acid at 90°, eliminated
a molecule of methanol and were converted into apoerythraline, C17H15O2Nand apoerysodine, C17H17O2N respectively. Furthermore, vigourous treat¬
ment of erythraline, erysodine, and the two «apo» compounds with 48%
hydrobromic acid resulted in the formation of the same base, apoerysopine,C16H15O2N, which differed considerably from apoerythraline and apoery¬
sodine not only in the physical properties like specific rotation and pK^value, but showed entirely different U. V. and I. R. absorption spectra. For
these reasons, the structural changes which accompany the transformation
of the different alkaloids into apoerysopine formed the basis of the formu¬
lation of a new skeleton formula for the Erythrina alkaloids, shown below.
R—R'=H erysopineR+R'=CH2 erythraline
I R or R'=H erysovineI R or R'—CH3 erysodine
The formation of apoerysopine from the Erythrina alkaloids by similar
treatment with hydrobromic acid was also reported by K. Folkers and his
coworkers26. The latter studied the Hofmann degradation of apoerysopineand obtained products to which they assigned structures on the basis of
the skeleton formula previously suggested by them for the Erythrinaalkaloids. As the final product of the Hofmann degradation, a tertiary
K. Folkers, F. Koniuszy and J. Shavel, Am. Soc. 73, 589 (1951).
21

aromatic amine, C20H23O2N was isolated which was designated des-
dimethyl apoerysotrine and is represented as shown below in (F).
CHjO-^"
OfaO-*^
CH-CH-;
N(CHj).
ch, |rCHi
(F) (C)
On the basis of the new structure proposed by M. Carmack et al." for
apoerysopine, its Hofmann degradation products can be represented as
described in the flow-sheet 2. According to this formulation, the second
stage degradation product, namely, des-dimethyl apoerysotrine has the
structure shown in (G) above. The latter compound on catalytic hydro-
genation gives a tetrahydro derivative which in terms of the new formula
should be represented as a biphenyl derivative (VIII). A synthesis of
this biphenyl appeared feasible. It was therefore attempted and is
described later.
As tetrahydro-des-dimethyl apoerysotrine was required for comparison
purposes with the synthetical preparation, the Hofmann degradation of
apoerysopine (IV) was repeated. The first stage of the degradation was
carried out with dimethyl sulphate and 30 % potassium hydroxide as
described by Folkers et al.28, but as reported by these authors, the degra¬dation product, namely, des-methyl apoerysotrine could not be induced
to crystallise even after keeping it for two months in a refrigerator.
However, catalytic hydrogenation readily converted it into dihydro-des-
methyl apoerysotrine (V), a crystalline, optically inactive base, having
one C-CH3 group and melting at 96—97 °. The I. R. absorption spectra of
des-methyl apoerysotrine and dihydro-des-methyl apoerysotrine are
shown in fig. 4 (curves 5 and 6). The U. V. absorption spectrum of the
latter was found to be in accord with the hindered structure (V) ** and
differed from what would be predicted for the dihydro derivative of
Folkers' formula which has two benzene rings which are not in con¬
jugation.
27 M. Carmack, B. C. McKusick and V. Prelog, Helv.34, 1601 (1951).28 K. Folkers, F.Koniuszy and J. Shavel, Am. Soc. 73, 589 (1951).29 Cf. A.J.Manson, Z.Valenta and K. Wiesner, Chem. and Ind. 33, 805 (1952).
22

Flow-sheet 2)
e•(4
a.o
bo
a.e
eo
•Ki
1fee
ece
133
23

The second stage of Hofmann degradation on des-methyl apoerysotrine
and its dihydro derivative (V) with dimethyl sulphate and alkali, failed
to give well-defined products. However, des-dimethyl apoerysotrine, des¬
cribed by Folkers et al.30 was obtained in a small yield, if the degradation
was carried out by pyrolizing the quaternary ammonium base obtained
from the methiodide of des-methyl apoerysotrine, by treatment with moist
silver oxide. A similar method of degradation, when applied to dihydro-
des-methyl apoerysotrine, gave a 66 %, yield of dihydro-des-dimethyl
apoerysotrine (VII). The latter was a crystalline compound of m.p.
85—86° and was characterised by its U. V. and I.R. (fig. 4, curve 6)
absorption spectra. Catalytic hydrogenation of (VII) readily gave the
tetrahydro-des-dimethyl apoerysotrine (VIII) described by Folkers. This
compound had m. p. 57—58° and on Kuhn-Roth determination gave 1.3
equivalents of acetic acid indicating the presence of two C-CH3 groups
rather than one. The U. V. and the I. R. absorption spectra (fig. 2, curve 2
and fig. 3, curve 3) seem to support the structure (VIII) for tetrahydro-
des-dimethyl apoerysotrine.An additional, important experimental evidence conforming the struc¬
ture (IV) for apoerysopine was furnished by B. C. McKusick31- in this
laboratory, who obtained a crystalline indole derivative (VI) by the
dehydrogenation of dihydro-des-methyl apoerysotrine (V) with palladiumat 275°. The indole structure for (VI) was indicated by a consideration
of its U. V. absorption spectrum (fig. 2, curve 1), by the fact that it was
neutral and by its giving a positive test with Ehrlich reagent in the cold.
The I. R. absorption spectrum (fig. 4, curve 7) also indicated the presence
of an indole derivative.
To supplement this analytical evidence for the structure (V) for apoery¬
sopine with more definite facts, the synthesis of its degradation product,
namely, tetrahydro-des-dimethyl apoerysotrine (VIII) was attempted.Since the latter is represented as 2,3'-diethyl-4,5-dimethoxy-2'-dimethyl-
aminobiphenyl, the general methods described for the synthesis of the
unsymmetrical biaryls were tried. The two common methods32, namely, the
Gomberg-Bachmann diazo reaction method and the nitrosoacetylaminemethod were attempted with model compounds, but they proved unsuccess¬
ful since the desired biphenyl derivatives could not be obtained.
30 K. Folkers, F. Koniuszy and J. Shavel. Am. Soc. 73, 589 (1951).31 Unpublished work.32 Rf. W. E. Bachmann and R. A. Hoffman, «Organic Reactions*, //, 224 (1944).
24

N3
(XLI)
C.H,
NHCOCH!
(\L)
HsCO
NHCOCH,X\XI\
HsCO
H.CO^V/^
-A-f)
H3C0
C;H
I\H_
.OH,
CjH„
NOj
C.H,
XXX
h,coJLi
*
aco-^J
)—^S^V-QHs
HsCO
CiH,
—f^\-
HsCO
\X
s*
JJ—NHj
HjCO_L
C'H*
-t^\-
HjCO
2,3'
-die
thyl
-4,5
-dim
etho
xy-2
'-di
meth
ylam
inob
iphe
nyl
ofSynthesis

According to the third method33, the Ullmann reaction between 4-ethyl-5-iodoveratrol and o-iodonitrobenzene was attempted in the presence of
copper powder at 230—40°. 4-Ethyl-5-iodoveratrol (XXX) was obtained
by the direct iodination of 4-ethylveratrol in alcohol solution in the
presence of mercuric oxide. The position of iodine in (XXX) was proved
by also preparing it from the known 5-amino-4-ethylveratrol (XXXI) by
treating the diazonium salt of the latter with potassium iodide. In the
above Ullmann reaction, the symmetrical 2,2'-dinitrobiphenyl (XXXII)
evidently formed by the self-condensation of o-iodonitrobenzene, was the
only biphenyl that could be isolated. However, by carrying out the reaction
of 4-ethyl-5-iodoveratrol with o-bromonitrobenzene, the possibility of the
symmetrical biaryl formation was minimised, since the order of reactivityof the halogens in the Ullmann reaction is I > Br > CI33. By controlling the
reaction temperature at 230—40°, the required 2-ethyl-4,5-dimethoxy-2'-
nitrobiphenyl (XXXIII) was obtained in moderate yield. It was charac¬
terised by its I. R. absorption spectrum (fig. 5, curve 11). Catalytic re¬
duction of (XXXIII) gave the corresponding amino biphenyl (LI) which
on methylation with dimethyl sulphate and alkali yielded 2'-dimethylamino-
2-ethyl-4,5-dimethoxybiphenyl (LII).
Following the preparation of this model compound, the synthesis of
2,3'-diethyl-4,5-dimethoxy-2'-dimethylaminobiphenyl was carried out si¬
milarly as described in the flow-sheet 7.
Ullmann reaction of 4-ethyl-5-iodoveratrol (XXX) with 3-bromo-l-
ethyl-2-nitrobenzene (XXXVIII) in the presence of copper powder at
240° gave 2,3'-diethyl-4,5-dimethoxy-2'-nitrobiphenyl (XXXIX). The
starting material for the preparation of 3-bromo-l-ethyl-2-nitrobenzene was
4-acetamido-l-ethyl-2-nitrobenzene which was obtained according to the
method described by Brady et al.34 Nitration of the latter compound with
fuming nitric acid gave a 70%. yield of 4-acetamido-l-ethyl-2,3-dinitro-benzene (XXXIV), whereas nitration in the presence of sulphuric acid,
as described by the above authors, led to the formation of, mainly, the
isomeric 2,5-dinitro derivative. The influence of the medium of nitration
on the position assumed by the nitro group has also been observed byScott and Robinson35 during the nitration of similar compounds. This
33 Rf. P. E. Fanta, Chem. Revs., 38, 139 (1946).34 O. L. Brady, J. N. E. Day and P. S. Allam, Soc. 1928, 980.
35 J. Scott and R. Robinson, Soc. 1922, 844.
26

interesting behaviour might be due to the fact that whilst in nitration in
sulphuric acid medium the nitronium ion [NO2]+ is the active nitrating
agent, nitrations with fuming nitric acid have all the characteristics of a
radical type reaction, although the exact nature of the attacking radical
has not been established36.
4-Acetamido-l-ethyl-2,3-dinitrobenzene (XXXIV) was hydroylsed with
45 % sulphuric acid to 4-amino-l-ethyl-2,3-dinitrobenzene as described by
Brady et al.34. As it has been reported in literature37 that chlorine or
bromine sometimes replaces a nitro group in the ortho position to a
diazonium group, the diazotisation of 4-amino-l-ethyl-2,3-dinitrobenzene
was carried out in the presence of hydrobromic acid with a view to obtain
3-bromo-l-ethyl-2-nitrobenzene (XXXVIII) directly. However, contrary
to expectations, a crystalline compound of the composition CsrLrC^BnN
was formed which was assigned the structure 3,4-dibromo-l-ethyl-2-nitro-benzene (XXXV)3839.
3-Bromo-l-ethyl-2-nitrobenzene (XXXVIII) was therefore prepared bythe Sandmeyer reaction with 3-amino-l-ethyl-2-nitrobenzene (XXXVII),
the latter being obtained by the reduction of l-ethyl-2,3-dinitrobenzene
(XXXVI) with stannous chloride as described by Kondo and Uyeo40. The
position of the halogen in 3-bromo-l-ethyl-2-nitrobenzene was proved by
reducing it to 2-amino-3-bromo-l-ethylbenzene according to Kondo and
Uyeo40 and oxidising the acetyl derivative of the latter with aqueous
potassium permanganate, when the known 2-acetylamino-3-bromobenzoicacid was obtained.
The unsymmetrical biphenyl (XXXIX) prepared by the Ullmann re¬
action of 4-ethyl-5-iodoveratol (XXX) and 3-bromo-l-ethyl-2-nitrobenzene
(XXXVIII) wascatalyticallyreducedto2'amino-2,3'-diethyl-4,5-dimethoxy-
biphenyl (XL). Methylation of the latter with dimethyl sulphate and alkali
gave the required 2,3'diethyl-4,5-dimethoxy-2'-dimethylaminobiphenyl
(XLI). It was a crystalline compound of m. p. 56—58° and formed a
crystalline picrolonate of m. p. 175—76°. The U. V. and the I.R. absorption
spectra of (XLI) are shown in the figs. 2 and 3 respectively (curves 3
and 4).
36 L. P. Hammett, «Physical Organic Chemistry», p. 313, McGraw-Hill Book Com¬
pany, New York (1940).37 N. Kornblum, «Organic Reactions*, //, 271—72 (1944).38 Cf. R.Meldola and J. V. Eyre, Soc.8i, 988 (1902).39 Cf. L. A. Elson, C. S. Gibson and J. D. A. Johnson, Soc. 1929, 2741.40 H. Kondo and S. Uyeo, Ber. 70, 1092—93 (1937).
27

Although the biphenyl (XLI), and tetrahydro-des-dimethyl apoery-
sotrine (VIII) had identical melting points, their mixed melting point was
depressed appreciably. Moreover, the melting points of the picrolonates of
both differed by about 20 °. A comparison of the I. R. absorption spectra
(fig. 3, curves 3 and 4) of the two compounds confirmed their non-identity.
However, it was interesting to note that their U. V. absorption spectra
(fig. 2, curves 2 and 3) showed the same absorption maximum, indicatingthat the chromophore system in both was the same. Furthermore, althoughthe I. R. absorption spectra of the two compounds were quite distinct, still
in both of them was indicated the presence of a 1, 2, 4, 5 tetrasubstituted
and a 1, 2, 3 trisubstituted benzene nuclei.
From these considerations two things become apparent, firstly, that the
present structure (V) for apoerysopine should be seriously reconsidered
and secondly, the method of synthesis of the biphenyl (XLI) should be
more carefully examined. The analytical evidence supporting the structure
(V) for apoerysopine is too strong to be easily disregarded. In the
synthesis, however, one ambiguity perhaps exists. Although the structures
of the two components of the Ullmann reaction are proved, the positionof the biaryl bond formation is not definitely known. It has been
observed41 that in Ullmann reactions the coupling usually takes place at
the positions occupied by the halogens, but since the exact mechanism of
the reaction is uncertain, the alternative possibility cannot be completelyoverlooked.
Possibilities of both an ionic and a free radical mechanism 42 43 " for
the Ullmann reaction have been considered, the ionic mechanism at some
stage involving a nucleophilic attack on the carbon atom. On the basis of
the nucleophilic mechanism, it appeared possible that in the Ullmann
reaction in question, the so-called "cine substitution"44, where the entering
group does not occupy the position vacated by another, might have
occurred.
During the course of the synthesis of 2,3'-diethyl-4,5-dimethoxy-2'-di-
methylamino biphenyl (XLI), it appeared to be of interest to study the
nature of the biaryls formed in the Ullmann reaction between two different
41 P. E.Fanta, Chem. Revs. 38, 139 (1946).42 W. S. Rapson and R. G. Shuttleworth, «Nature», 747, 675 (1941).43 Cf. L. N. Fergusson, Chem. Revs. 50, 50 (1952).44 J.F. Bunnett and R. E. Zahler, Chem. Revs. 49, 382—395 (1951).
28

Flow-sheet 8)
o
a
z z
3
e
5
V/o o
5
oz
•-& 11 V
ss °o z
^>-"A^
= 9u z
29

halogen derivatives. The results of these investigations are described in
the flow-sheet 8.
The Ullmann reaction of 4-ethyl-5-iodoveratrol (XXX) with 2-iodo-6-
nitrotoluene (XLIII) was attempted under the usual experimental condi¬
tions, but no reaction seemed to occur, since the original components were
obtained back. A similar reaction of m-iodonitrobenzene with 4-ethyl-5-iodoveratrol gave only the symmetrical 3,3'-dinitrobiphenyl (XLIV).When 4-iodoveratrol (XLV) and m-iodonitrobenzene were used as
the two components for the Ullmann reaction, the unsymmetrical biphenyl
(XLVII) as well as both the symmetrical biphenyls (XLIV) and (XLVI)
were obtained. The small amount of 3,3'-dinitrobiphenyl (XLIV) formed
in the reaction was separated by fractional crystallisation and the 3,4-
dimethoxy-3'-nitrobiphenyl (XLVII) and the 3,3', 4,4'- tetramethoxybi-
phenyl (XLVI) were recovered by a chromatographic separation. The
unsymmetrical biphenyl (XLVII) was characterised by its I.R. absorption
spectrum (fig. 5, curve 9).
To compare the biphenyl (XLVII) with 3.4-dimethoxy-2'-nitrobiphenyl,an Ullmann reaction between 4-iodoveratrol and o-bromonitrobenzene
was carried out, when the desired biaryl (XLVIII) was obtained. It was
characterised by its I.R. absorption spectrum (fig. 5, curve 10). Catalyticreduction of (XLVIII) gave 2'-amino-3,4-dimethoxybiphenyl (XLIX)
which was deaminated with alkaline formaldehyde to the known 3,4-dime¬
thoxybiphenyl (L), thus proving the position of the biaryl bond in
(XLIX).
The new skeleton formula suggested by M. Carmack et al.46 for the Ery-thrina alkaloids presents very interesting features from the biogenetical
standpoint and a glance at it reveals the relationship to (3-(3. 4-dihydroxy-
phenyl) ethylamine and 3, 4-dihydroxyphenylalanine, which are believed
to be the precursors in the biogenesis of a large number of importantalkaloids. However, very little seems to be known about the properties of
compounds having ring systems similar to the Erythrina alkaloids and it
therefore appeared to be of interest to prepare such compounds syntheti¬
cally.A perusal of literature showed that one simple method of synthesizing
compounds similar in structure to the Erythrina alkaloids is by means of
« W. M.Whaley and T. R. Govindachari, «Organic Reactions*, VI, 151 (1951)."6 M. Carmack, B. C. McKusick and V. Prelog, Helv. 34, 1601 (1951).
30

the Pictet-Spengler reaction45. This reaction has been frequently used for
the preparation of tetrahydroisoquinoline derivatives by the reaction of
P-phenylethylamines with aldehydes under strongly acid conditions. An
extension of this method, described by Hahn and his co-workers " involved
the condensation of P-(3,4-dihydroxyphenyl)ethylamine with pyruvic acid
derivatives under physiological conditions as shown below:
o
HC-COOH
ICHa
IC«Hj
ho\a/nhHOOC CHsCnHs
Following the method described by Hahn et al.47 the condensation of
homoveratryl and homopiperonyl amines with pyruvic acid was attemptedunder different physiological conditions, but the desired tetrahydroiso¬
quinoline derivatives could not be obtained. Drastic conditions of reaction
also proved unsuccessful. It therefore appeared that the use of the more
reactive (}-(3,4rdihydroxyphenyl)ethylamine was necessary for these reac¬
tions48 ".
The flow-sheet 9 represents the different compounds prepared by the appli¬cation of the Pictet-Spengler reaction. ^-(3,4-Dihydroxyphenyl)ethylamine(LIII) was obtained by the demethylation of homoveratrylamine with
hydrobromic acid in acetic acid solution. Condensation of the hydro-bromide of (LIII) with pyruvic acid was carried out under physiologicalconditions as described by Hahn et al., when l-carboxy-6,7-dihydroxy-l,
methyl-1, 2, 3, 4-tetrahydroisoquinoline (LIV) was formed. It was charac¬
terised by its I.R. absorption spectrum (fig. 6, curve 13) and the pre¬
paration of the methyl ester. Under exactly similar conditions, the reac¬
tion of (LIII) with a-ketobutyric acid resulted in the formation of 1-
" G. Hahn and K. Stiehl, Ber. 69, 2627 (1936).48 Cf. G.Hahn and O. Schales, Ber. 68, 24 (1935).
49 Cf. E.Spath, F.Kuffner and F.Kesztler, Ber. 69, 378 (1936).
hotV^HO -\i) NH'
31
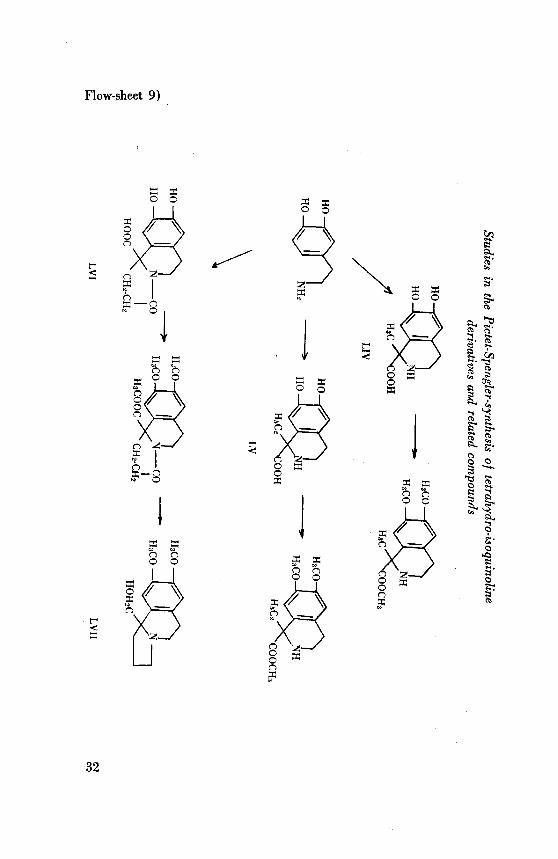
Flow-sheet 9)
r<
r<
C/3
ea*
a-
3-
re o
re
re <J
S re
1 ^
3-re
re
o
3•ao
§ I-
re
Ca
o>«sex.
3o
are
32

carboxy-l-ethyl-6, 7-dihydroxy-l,2,3,4-tetrahydroisoquinoline(LV) which
on methylation with diazomethane gave the methyl ester of the dimethylether. The I.R. absorption spectrum of (LV) is shown in fig. 6 (curve 14).
As a further extension of the Pictet-Spengler reaction, the use of a-
ketoglutaric and a-ketoadipic acids was attempted50. Under physiological
conditions, no reaction of [3-(3,4-dihydroxyphenyl)ethylamine (LIIl) was
foundtotake place with either. However, if an aqueous solution of the amine
and a-ketoglutaric acid at pH 1 was heated at 100 ° for 6 hours, a lactam
of the structure (LVI) was formed. The latter was found to be a crystalline
compound of m. p. 255—58° and was characterised by its U.V. absorption
spectrum. The methylated product obtained by the treatment of (LVI) with
diazomethane was reduced with lithium aluminium hydride, when a benzo-
pyrrocoline derivative (LVII) was formed. It was characterised by its
U.V. and I.R. absorption spectra (fig. 6, curve 15) and the preparationof a crystalline picrate.
It appeared that a similar reaction of the amine (LIIl) with a-ketoadipicacid would give the corresponding lactam, but contrary to expectations,
no condensation occurred and the a-keto acid was recovered unchanged.
To test its general applicability, the Pictet-Spengler reaction of the
amine (LIIl) with other ketone derivatives like (a) cyclohexanone-2-ethyl-
carboxylate (b) 2-methoxycyclohexanone (c) cyclohexanone-2-acetic acid
and (d) 1,2-cyclohexanedione was attempted. Both, under physiologicalconditions as well as at higher temperatures, no condensation occurred in
any case, and the starting materials were recovered unchanged. Apparently,
^-ketone derivatives do not seem to react under the conditions of the Pictet-
Spengler reaction.
50 G. Hahn and K. Stiehl, Ber. 69, 2627 (1936)
33

Figure 1
JS
1/
/ .1
i
iJO 300 280 260 21,0
*—Wavelength m /«w
i.,0
3S
Figure 2
s y
//A
ij
—.—1 ,—
300 2B0 ?«0 HO* Wavelength in mu
120
34

SQoo 3$oo zooo tepo goo ilpo mo tooo 900 ago
^Amff«*^^1 1 1 1 ,
1 1 1 1 1 1,
1 1,.
5 * 1 i f » » » « o a W * m
SOM 3000 HOC tepo y OfiO "fO HOC fO IPO Tft or'
Figure 3
35

Curve S
5 pe 30K 7000 »00 1400 aoo 1f)9 »0*» tpo ioc TOO cm
ta¬ _-~~~V\ /%lc-
~s~\ / s a/\ A /*
V»- \ w i/l /if \Kt'\as¬
nA*iJl^V'V W \I
lo¬ V'
\*J1' U*» u
SOW .1000 2000 WO" MOO ««?
Currt? 6
too TOO
SOOO 3000 2000 1600 1^)0 1200 ttpo tqoo 900 MOO
HP
Curve 7
700 cm-*
SOOO 3000 1000 WOO WOO ttOO J»C WOO 900 ttOO
Curve 8
700 an-f
I i 1 1 -I r-
3 4 5 6 7a
Tigure k
36

Curve $
3Q00 1000 1600 ty>0 1200 HpO 1QQQ 900 100 TOO t*-f
3000 2000 1600 MOO 1200 WO WOO 900 *00 700 a»-f
Curvm 10
»too 3000 2900 moo Mo 200 WO 1000 900 .00 wo OB-J
XP- >"^\ _/»/v\Af\nw- fS n fl wni. i\(ftif I fill rvATswto-
_/^\/
k in (V 1 V \l s.
«-
r*f
1y K
K v
30-
|-1 —1 1 —i——1 1 -^—r-
Curve 12
f <00 9000 2000 itpo iuo ttoo mo tqoo 900 #00 TOO «r*
$0-
to-^n^~"VVy""\jnyV^\40- V III
20-
i——i—i—
If
T 1 i "1 i i— 1
Tiqurs 5
37

Curve 1
5wo 30Q0 ifco lyi n» na> tm goo toe too wo
Curve H
Curve tt
Ticpure S
* A
38

EXPERIMENTAL PART51
Isolation of erythraline and erysodine from
Erythrina abyssinica Lam.
About 960 g of the extract (obtained from about 3.5 kg of the seeds of
Erythrina abyssinica Lam. by extraction first with petroleum ether and
then with methanol) was worked up as described by V. Prelog et al52. The
mixture of alkaloids (20 g) was dissolved in chloroform and chromato-
graphed over 630 g of neutral alumina (activity II—III). With 2.5 1 of
chloroform, 7.89 g of a pale yellow liquid was eluted, which after treatment
with methanolic hydrobromic acid and crystallisation gave 5.37 g of
erythraline hydrobromide of m. p. 245—46° (reported52 m. p. 246°).With 7.2 1 of chloroform-methanol (100 : 0.5), 7.12 g of a yellowish solid
was eluted, which after one crystallisation from alcohol gave 4.11 g of
colourless erysodine, m. p. 207—09° (reported52 m. p. 2041—05°).
The von Braun degradation of dihydroerysotrine
Dihydroerysodine (I)
Three and a half grams of erysodine was hydrogenated in alcohol
solution over 1.5 g of 5 % palladium-barium carbonate catalyst as des¬
cribed by V. Prelog et al. It was crystallised from ethyl acetate in colourless
needles of m. p. 212—14° (reported52 m. p. 212—14°). By exhaustive
reworking of the mother liquors, there was obtained in all 2.6 g (74 %
yield) of pure dihydroerysodine.
[«] 55 - + 238 ° ( + 4 •) (c - 0.78 in alcohol)
51 All melting points are corrected. The specific rotations were determined with
a tube of one decimeter in length. The U. V. absorption spectra were taken in
alcoholic solution with a Beckman spectrophotometer. The I. R. absorptionspectra were taken in Nujol-mull with a Baird double-beam spectrophotometer.The pKi values were determined by potentiometric titration with a glass elec¬
trode in 80 %> methylcellosolve solution.52 V. Prelog, K.Wiesner, H. G. Khorana and G. W. Kenner, Helv. 32, 453 (1949).
39

Dihydroerysotrine (II)
A solution of 2.6 g of dihydroerysodine in 26 c. c. of absolute methanol
was treated with 70 c. c. of a 2 % solution of diazomethane in ether. The
mixture was kept in a refrigerator for two days and on the third day,
30 c. c. more of the diazomethane solution was added. After keeping for
two more days in the refrigerator, the solution was evaporated to drynessunder vacuum, when 2.7 g (quantitative yield) of a pale yellow, viscous
oil was obtained. It was used for the von Braun degradation without
further purification. For analysis it was twice distilled in a sublimation
tube at 0.001 mm (block temperature 110—15 °).
3.739 mg subst gave 9.885 mg C02 and 2.764 mg H20
Calcd. for C19H25O3N C 72.35 H 7.99 %
Found C 72.15 H 8.27 %
The U. V. absorption spectrum showed bands with a maximum at
284 mju, loge - 3.54 and at 228 m,M, loge =- 3.91.
The picrate after three crystallisations from methanol melted at
175—76° (in an evacuated capillary). For analysis it was dried for 24
hours at 60 ° under high vacuum.
3.795 mg subst gave 7.674 mg CO2 and 1.777 mg H2O
2.880 mg subst required 4.860 c. c. of 0.02 N Na2S203
Calcd. for C25H28O10N4 C 55.14 H 5.18 30CH3 17.10 %
Found C 55.18 H 5.24 OCH3 17.46 %
Preparation of the base C18H21O2N (III) by the von Braun
degradation of dihydroerysotrine
To a cooled solution of 2.3 g of dihydroerysotrine in 35 c. c. of drychloroform53 was added dropwise a solution of 1.801 g of cyanogen
bromide54 in 35 c. c. of dry chloroform. After being allowed to stand for
ten minutes at room temperature, the mixture was gently boiled under
reflux in an oil bath at 75 ° for two hours. The solution was cooled and
Freshly distilled over phosphorus pentoxide.Prepared according to «Organic Synthesisji, Coll. Vol. //, p. 150 (1948).
40

the chloroform removed under vacuum at room temperature. The residue
was again treated with 10 c. c. of dry chloroform and the latter removed
under vacuum. This operation was repeated with two more 5 c. c. portionsof dry chloroform. The brownish viscous liquid left behind was dissolved
in 70 c. c. of chloroform and the solution extracted with two 35 c. c.
portions of 1.2 N hydrochloric acid. The acid layer was again washed
with two 10 c. c. portions of chloroform. The combined chloroform extracts
were washed with 30 c. c. of water and dried over anhydrous sodium
sulphate. Distillation of chloroform under vacuum at room temperature
gave 3.1 g of a tan coloured gummy residue. The residue was freed of last
traces of chloroform by twice dissolving it in 5 c. c. of dry tetrahydro-
furan55 and evaporating to dryness under vacuum. The residue was
dissolved in 50 c. c. of dry tetrahydrofuran, and 2.6 g of powdered lithium
aluminium hydride was added. Evolution of heat occurred and the solution
became almost colourless. The mixture was boiled under reflux for two
hours and allowed to stand overnight. Next day, the solution was refluxed
for two more hours and allowed to cool. To the cooled solution was added
dropwise with stirring 10 c.c. of icewater and 110 c. c. of a 20% solution of
sodium potassium tartrate. The mixture was then extracted with five
100 c.c. portions of chloroform. The combined chloroform extracts were
washed with 140 c. c. of water and dried over anhydrous sodium sulphate.
Distillation of chloroform gave about 2.1 g of a tan coloured gum. It was
distilled in a large sublimer at 0.01 mm (bath temperature 160—85 °)
when 1.56 g of a thick yellow oil was obtained. It was dissolved in about
30 c.c. of alcohol and the solution filtered to remove a little insoluble
material. To the hot filtrate was added a saturated solution of 1 g of picric
acid in alcohol. On cooling, the solution precipitated crystals of the picrate.
These were filtered and boiled with about 25 c.c of alcohol to remove a
small amount of tarry material. The yield of the crude picrate was 1.6 g.
It was crystallised from about 400 c.c. of absolute alcohol, giving 1.057 g
of yellow prisms, m. p. 235—36° (in an evacuated capillary) with pre¬
vious shrinking over four degrees. Concentration of the mother liquor gave
130 mg more of same picrate. In all there was obtained 1.187 g (32 %
yield) of the pure picrate, m. p. 235—36 °. For analysis the substance was
recrystallised from alcohol and dried for 16 hours at 90° under high
vacuum.
Distilled first over sodium and then over lithium aluminium hydride.
41

3.550 mg subst gave 7.325 mg CO2 and 1.530 mg H2O
3.173 mg subst gave 0.317 c.c N2 (21 °/730 mm)
3.655 mg subst required 4.322 c.c of 0.02N Na2S203
Calcd. for C24H24O9N4 C 56.25 H 472 N 10.93 2 OCH3 12.00 %
Found C 56.31 H4.82 N 11.14 2 0CH3 12.23%
The substance contained no N-CH3 group.
The above crystallised picrate (260 mg) was treated with chloroform and
saturated aqueous lithium hydroxide solution. The aqueous solution was
several times extracted with chloroform. The combined chloroform extracts
were washed with fresh lithium hydroxide solution and water and dried
over anhydrous sodium sulphate. Distillation of the solvent gave 172 mg
of a yellowish liquid which crystallised spontaneously after several hours
at room temperature. Sublimation at 125—30 ° and 0.001 mm gave directly
a white crystalline solid of m. p. 90—92° (in an evacuated capillary).
3.747 mg subst gave 10.463 mg C02 and 2.508 mg H20
4.108 mg subst gave 0.187 c.c.N2 (21°/723 mm)
5.248 mg subst gave 0.566 c. c. CH4 at 22.5 °/718 mm
0.494 c. c. CH4 at 0 °/760 mm
Calcd. for C18H21O2N C 76.29 H 7.47 N 4.94 «H>» 0.36 %
Found C 76.20 H 7.48 N 5.03 «H» 0.42 %
There was no C-CH3 group present in the substance.
pKA 8.33; [a]^5 = 0.0° ( + 1 °) (c = 0.709 in alcohol)
The U. V. absorption spectrum is shown in fig. 1, curve 1.
The I. R. absorption spectrum is shown in fig. 3, curve 1.
The acetyl derivative of the above base was prepared by boiling 40 mg
of it in benzene solution with 1 c. c. of acetic anhydride for ten minutes.
The solvent and the acetic anhydride were removed under vacuum. The
oily residue was dissolved in chloroform and the chloroform solution
washed with 1.2 N hydrochloric acid, sodium bicarbonate and water. After
drying and distilling off chloroform, about 40 mg of a yellowish, viscous
oil was obtained which solidified on keeping. For analysis it was twice
sublimed under high vacuum (block temperature 130—40°). It had m. p.
141—43 °.
42

3.720 mg subst gave 10.119 mg CO2 and 2.490 mg H2O
3.879 mg subst gave 0.160 c.c. N2 (23°/722 mm)
Calcd. for C20H23O3N C 73.82 H 7.12 N4.30 %
Found C 74.22 H 7.49 N 4.52 %
Oxidation of the base C1SH21O2N with aqueous potassium permanganate
To 15 c. c. of water stirred by means of a «Vibro Mixer» was added a
solution of 127 mg of the once distilled base in 1 c. c. of benzene. The
benzene was removed by warming the mixture and blowing air into it.
On cooling, the base separated out as a yellowish, white solid. To this
well-stirred suspension of the base in water was added dropwise a solution
of 955 mg of potassium permanganate in 15 c. c. of water. The mixture
was then stirred continuously at 90° for twenty hours. Sulphur dioxide
was passed through the mixture till a clear solution was obtained. It was
extracted with five 10 c. c. portions of chloroform and the combined
chloroform extracts washed with water. After drying and distilling off
chloroform, 40 mg of a semi-solid material was obtained. It was dissolved
in methanol and treated with 5 c. c. of a 2 % solution of diazomethane in
ether, when evolution of nitrogen occurred. Removal of ether under
vacuum gave a yellow oil. It was dissolved in chloroform and the chloro¬
form solution washed successively with 1.2 N hydrochloric acid, aqueous
sodium carbonate solution, 5 % sodium hydroxide solution and water.
Distillation of chloroform gave 32 mg of a pale yellow oil. It was dissolved
in benzene and the benzene solution passed through 1.2 g of neutral
alumina (act. II—III). With 20 c.c. of benzene, 23 mg of a yellowish,viscous oil was eluted. Elution with ether, chloroform and methanol gave
in all 6 mg of a gum which was discarded. The oil obtained with benzene
was twice distilled in a sublimation tube at 0.001 mm (block temperature
110—20°), and analysed.
3.818 mg subst gave 9.352 mg CO2 and 2.036 mg H2O
3.732 mg subst required 12.155 c. c. of 0.02 N Na2S203
Calcd. for C20H22O6 C 67.02 H6.19 40CH3 34.62%
Found C 66.85 H5.97 OCH3 33.69%
The substance did not contain any nitrogen.
From the analytical results it appears that the compound is probably
4,5-dimethoxydiphenyl-2,2'-diacetic acid dimethylester (HI A).
43

The Hofmann degradation of apoerysopine
Apoerysopine (IV)
A solution of 3.618 g of erysovine M in 33 c. c. of 48 % hydrobromicacid was heated under reflux in a 110—20° oil bath for 95 minutes. The
solution was under an atmosphere of carbon dioxide during heating. On
cooling, the dark coloured solution was diluted with 300 c. c. of water,
filtered to remove resinous material and extracted with three 130 c. c.
portions of chloroform. The chloroform extracts were discarded and the
aqueous solution adjusted to pH 7—8 with 50 % aqueous suspension of
sodium bicarbonate. It was then extracted with seven 130 c. c. portionsof chloroform. The combined chloroform extracts were washed with
150 c. c. of water and dried over anhydrous magnesium sulphate. Distil¬
lation of chloroform gave 3 g of a brownish, partly crystalline residue.
It was sublimed at 0.005 mm (bath temperature 165—90°) giving 2.02 g
of a yellowish crystalline solid. Crystallisation from a mixture of absolute
alcohol and cyclohexane gave 1.362 g (44 % yield) of colourless apoery¬
sopine of m. p. 171—72 ° (reported57 58m. p. 172—73 °; 169—70 °).
Dihydro-des-methyl apoerysotrine (V)
Apoerysopine (500 mg) was treated with dimethyl sulphate and 30 %
potassium hydroxide as described by Folkers et al.58 The crude productof the Hofmann degradation was sublimed at 0.001 mm (bath temperature
165—80 °) giving 430 mg of a pale yellow oil which could not be induced
to crystallise even after keeping for two months in a refrigerator. (Folkers
et al.59 describe a m. p. of 72—73° for this substance). A small amount
of this oil was redistilled and submitted for U. V. and I. R. absorption
spectra determinations.
The U. V. absorption spectrum showed absorption bands with a ma¬
ximum at 294 mfi, loge = 3.87 and at 264 mft. logs = 4.21.
The I. R. absorption sectrum is shown in fig. 4, curve 5.
66 I am indebted to Dr. K. Folkers for a gift of erysovine.57 M. Carmack, B. C. McKusick and V. Prelog, Helv. 34, 1610 (1951).58 K. Folkers, F.Koniuszy and J. Shavel jr., Am. Soc. 73, 591 (1951).59 K. Folkers, F.Koniuszy and J. Shavel jr., Am. Soc. 73, 592 (1951).
44

The above oil (410 mg) was hydrogenated in 25 c. c. of methanol over
77 mg of platinum catalyst. Evaporation of the filtrate left 398 mg of a
white solid. Crystallisation from dilute methanol gave 382 mg (65 %
yield) of colourless, crystalline dihydro-des-methyl apoerysotrine of m. p.
94—95 °. An analytical sample, prepared by two recrystallisations from
cyclohexane followed by sublimation at 0.004 mm (block temperature102—04 °) melted at 96—97 °. It gave a scarlet colour with alcoholic ferric
chloride solution and was soluble in 5 % hydrochloric acid.
PKA ~ 3.5
3.441 mg subst gave 9.662 mg C02 and 2.380 mg H20
3.933 mg subst gave 0.174 c. c. N2 (23 °/725 mm)
4.019 mg subst required 7.691 c. c. of 0.02 N N2S2O3 (OCH3)and 3.741 c. c. of 0.02 N Na2S203 (N-CH3)
5.790 mg subst required 1.274 c. c. of 0.01 N alkali
Calcd. for C19H23O2N C 76.73 H 7.80 N4.71 OCH3 20.88
NCH3 5.06 and CCH3 5.06 %
Found C 76.65 H 7.73 N4.87 OCH3 19.80
NCH3 4.66 and CCH3 3.31 %
The U. V. absorption spectrum in 0.01 N hydrochloric acid showed a
band with a maximum at 288 m/n, logs — 3.8. The I. R. absorption
spectrum is shown in fig. 4, curve 6.
The picrolonate after two crystallisations from alcohol melted at
169—71° with decomposition (in an evacuated capillary).
4.228 mg subst gave 9.592 CO2 and 2.170 mg H2O
Calcd. for C29H31O7N5 C 62.02 H 5.56 %
Found C 61.91 H 5.74 %
l-Methyl-7-(l-ethyl-3,4-dimethoxyphenyl) indole (VI)
An intimate mixture of 25 mg of dihydro-des-methyl apoerysotrine and
9.0 mg of 10 % palladium on carbon was placed in a sublimation tube.
Air was swept from the tube by adding small pieces of dry ice and the
end was then drawn out to a capillary. The mixture was heated to 275 °
in thirty minutes and held at 275—80° for additional thirty minutes.
During heating, a colourless oil sublimed up the tube. The tube was re¬
moved from the sublimation block and at once sealed to keep out air
during cooling. The oil obtained above solidified on keeping. It was
45

crystallised from a small amount of methanol and the crystals resublimed
at 0.01 mm (block temperature 115—25°). The sublimed compound had
m. p. 119—20°. It was insoluble in 5 % hydrochloric acid and gave
instantaneously a deep magenta colour with Ehrlich reagent.
3.806 mg subst gave 10.758 mg C02 and 2.434 mg H2O
3.970 mg subst gave 0.176 c. c. N2 (21 °/728 mm)
Calcd. for C19H21O2N C 77.26 H 7.17 N 4.73 %
Found C 77.14 H7.16 N4.93 %
The U. V. absorption spectrum is shown in fig. 2, curve 1.
The I. R. absorption spectrum is shown in fig. 4, curve 1.
Dihydro-des-dimethyl apoerysotrine (VII)
A solution of 103 mg of dihydro-des-methyl apoerysotrine in 2 c. c. of
dry acetone was boiled gently under reflux with 2,5 g of methyl iodide
for two hours. On cooling, the yellow solution was evaporated to drynessunder vacuum. The solid residue was treated with 3 c. c. of water and
the mixture shaken with freshly precipitated silver oxide (from 250 mg
of silver nitrate) at 60° for fifteen minutes. The aqueous solution was
filtered and the filtrate evaporated to dryness under vacuum giving 92 mg
of a partially solid residue. The latter was sublimed (using a «cold finger*)at 0.01 mm (bath temperature 170°) giving 82 mg of a white crystallinesolid. One crystallisation from dilute methanol gave 72 mg (66 %. yield)of colourless needles of m. p. 83—84 °. For analysis the twice crystallisedsubstance was sublimed under high vacuum (block temperature 110°).
It had m. p. 85—86 °.
3.752 mg subst gave 10.626 mg CO2 and 2.715 mg H20
3.473 mg subst gave 0.148 c. c. N2 (21 °/725 mm)
3.063 mg subst required 5.794 c.c. of 0.02N Na2S203 (OCHs)
and 4.922 c. c. of 0.02 N Na2S203 (N-CHs)
Calcd. for C20H25O2N C 77.13 H8.09 N4.50 2 0CH3 19.94
2 N-CH3 9.66 %
Found C 77.29 H8.10 N4.72 OCH3 19.57
N-CHs 8.05 %
PKA ~ 3.5
46

The U. V. absorption spectrum showed three bands with maxima at
320 m//, loge = 3,3, 280 m/x, logs — 3,9 and 240 m/z, loge = 4,33.
The I. R. spectrum is shown in fig. 4, curve 8.
Tetrahydro-des-dimethyl apoerysotrine (VIII)
Fifty-eight milligrams of dihydro-des^dimethyl apoerysotrine was hy-
drogenated in 6 c. c. of alcohol over 9 mg of platinum catalyst. The
theoretical amount of hydrogen was absorbed in 15 minutes. Evaporationof the filtrate gave 55 mg of a colourless oil which crystallised from a
small amount of methanol on keeping in a refrigerator. It had m. p.
55—56 °. For analysis it was twice distilled at 0.001 mm in a sublimation
tube (block temperature 85—90 °) when it solidified having m. p. 57—58 °.
(Folkers et al. 6°
report m.p. 58—59°).
3.768 mg subst gave 10.598 mg C02 and 2.930 mg H2O
2.815; 7.354 mg subst required 1.115; 3.262 c. c. of
0.01 N alkali (Kuhn-Roth)
Calcd. for C20H2-O2N C 76.64 H 8.68 2C-CH3 9.6%
Found C 76.76 H 8.70 C-CH3 5.95; 6.67%
The U. V. absorption spectrum is shown in fig. 2, curve 2.
The I. R. absorption spectrum is shown in fig. 3, curve 3.
The picrolonate, crystallised from methanol, melted at 152—53 ° with
decomposition (in an evacuated capillary). For analysis it was dried for
48 hours at 70 ° under high vacuum.
3.399 mg subst gave 7.736 mg CO2 and 1.777 mg H2O
Calcd. for C30H35O7N5 C 62.38 H6.ll %
Found C 62.11 H5.85%
Des-dimethyl apoerysotrine
A solution of 40 mg of des-methyl apoerysotrine in 2 c. c. of dry acetone
was boiled gently under reflux with 2 g of methyl iodide for two hours.
On cooling, the yellow solution was evaporated to dryness under vacuum.
The oily residue was treated with about 3 c. c. of water and the mixture
shaken with freshly precipitated silver oxide (from 200 mg of silver
nitrate) at 60° for twenty minutes. The aqueous solution was filtered and
60 Am. Soc. 73, 589 (1951).
47

the filtrate evaporated to dryness under vacuum, giving about 30 mg of
a gummy residue. The latter was sublimed (using a «cold finger») at
0.01 mm (bath temperature 180°) when 20 mg of a yellow, viscous oil
was obtained. A refrigerated solution of this oil in methanol gave 14 mg
of crystalline material, m. p. 86—92 °. Recrystallisation of this substance
from methanol followed by sublimation at 0.001 mm (block temperature
120—30°) gave pure des-dimethyl apoerysotrine, m. p. 98—99° (Folkers
et al. " report m. p. 97.5—98 °).
3.570 mg subst gave 10.151 mg C02 and 2.412 mg H20
Calcd. for CaoHasC^N C 77.64 H 7.49 %
Found C 77.60 H 7.56 %
Synthesis of l-benzyl-6,7-dimethoxy-l, 2,3,4-tetrahydro-isoquinoline
Homoveratrylamine (IX)
Small amounts of homoveratrylamine were obtained by the reduction
of 3,4-dimethoxy-(3-nitrostyrene with lithium aluminium hydride62.To a well stirred mixture of 12 g of lithium aluminium hydride and
900 c. c. absolute ether was added by Soxhlet extractor technique, 13.5 g
of 3,4-dimethoxy-^-nitrostyrene" over a period of 96 hours. The flask
was cooled well and 800 c. c. of ice-cold 2 N sulphuric acid were added
dropwise with stirring. The aqueous layer was separated and its pH
adjusted to 6 with solid lithium carbonate. The solution was heated to
boiling and the precipitated aluminium hydroxide filtered through «celite»
and washed well with hot water to remove any adsorbed substance. The
clear filtrate and washings were heated and whilst hot, mixed with a
concentrated solution of 18 g of picric acid in hot alcohol. Upon standing
overnight, 21,3 g of the picrate was obtained. One crystallisation from
dilute alcohol gave 20.5 g (77% yield) of orange crystals of m. p.
165—67° (reported04 m. p. 165°; 165—67°).
61 Am. Soc. 73, 589 (1951).b2 Cf. F. Ramirez and A. Burger, Am. Soc. 72, 2781 (1950); M. Erne and F. Ramirez,
Helv. 33, 912 (1950).63 Prepared according to L. C. Raiford and D. E. Fox, J. Org. Chem., 9, 172 (1944).64 K. Kindler, W. Peschke and E.Brandt, Ber. 68, 2244 (1935); E.Kaufman,
E. Eliel and J. Rosenkranz, Ciencia (Mex). 7, 136—37 (1946), C.A.41, 2398
(1947).
48

3.632 mg subst gave 6.246 mg CO2 and 1.461 mg H2O
3.340 mg subst gave 0.416 c. c. N2 (20°/720 mm)
Calcd. for C16H18O9N4 C 46.83 H4.42 N 13.66 %,
Found C 46.93 H4.50 N 13.75 %
A solution of 20 g of the picrate in 640 c. c. of boiling water was mixed
with 140 c. c. of concentrated hydrochloric acid. The picric acid which
precipitated on cooling was filtered and the filtrate extracted with nitro¬
benzene and then with ether. The aqueous solution was evaporated to
dryness under vaccum when tan coloured crystals of the hydrochloridewere obtained. Crystallisation from methanol-ethyl acetate gave 10.1 g
(71 % yield based on the nitrostyrene) of colourless crystals of m. p.
153—55° (reported65 m. p. 154—55°; 152—56°).
The free amine obtained from the above hydrochloride distilled
between 155—57 °at 12 mm as a colourless oil.
For large quantities the amine was prepared by the catalytic reduction
of homoveratronitrile66 in 10 N methanolic ammonia as described in
«Organic Synthesis 6,».
N-Phenylacetyl-homoveratrylamine (X)
To a solution of 800 mg of homoveratrylamine hydrochloride in 3 c. c.
water was added 8 c. c. of 10 %. potassium hydroxide followed by 0.5 c. c.
of freshly distilled phenylacetyl chloride, the latter being added dropwiseand with stirring. After stirring further for ten minutes and keeping for
an hour, a white solid was obtained. Two crystallisations from dilute acetic
acid or benzene gave 910 mg of colourless needles of m. p. 107—08 °. For
analysis the substance was sublimed under high vacuum (block tem¬
perature 105°).
3.670; 1.165 mg subst gave 9.672; 3.088 mg C02 and
2.262; 0.736 mg H20
1.634 mg subst gave 0.070 c. c. N2 (19°/720 mm)
Calcd. for C18H21O3N C 72.22 H7.07 N4.68 %.
Found C 71.92 H6.87 N4.74%
72.34 7.07
65 C. Mannich and W. Jacobsohn, Ber. 43, 196 (1910); E.Kaufman et al„ Ciencia
(Mex). 7, 136—37 (1946); C.A. 41, 2398 (1947).66 Prepared, according to A. E. Bide and P. A. Wilkinson, J. Soc. Chem. Ind. 64,84—5 (1945); C.A.39, 3527 (1945).
67 Vol. 23, 72—74 (1943).
49

1 -Benzyl-6,7-dimethoxy-3,4-dihydroisoquinoline (XI)
A mixture of 500 mg of N-phenylacetyl-homoveratrylamine, 3 c. c. drytoluene and 1 c. c. of phosphorus oxychloride was boiled under reflux for
two hours. The brownish yellow solution contained an oil in suspensionwhich solidified on cooling. The reaction mixture was diluted with light
petroleum ether and the supernatent liquid decanted. The residue was
dissolved in a small amount of alcohol, the solution made alkaline and
poured into ice-cold water with stirring, when a light brown solid se¬
parated. The crude material weighed 390 mg and had m. p. 80—84 °. It
was used for the next step without further purification. For analysis the
substance was reprecipitated from acid solution and twice sublimed under
high vacuum (block temperature 100 °); it had m. p. 84—85 °.
3.678 mg subst gave 10.350 mg C02 and 2.256 mg H20
Calcd. for CwHioCbN C 76.84 H 6.81 %
Found C 76.79 H 6.86 %
The picrate after two crystallisations from methanol-acetic acid had
m. p. 179—81 °. For analysis it was dried for 48 hours at 90 ° under
high vacuum.
3.787 mg subst gave 7.806 mg CO2 and 1.411 mg H2O
3.370 mg subst gave 0.341 c.c. N2 (20°/720 mm)
Calcd. for C24H22O9N4 C 56.47 H 4.34 N 10.98 %
Found C 56.25 H4.17 N 11.17 %
The hydroiodide was obtained by the addition of potassium iodide to
a solution of the base in dilute hydrochloric acid. After two crystallisationsfrom water, it melted at 196—97 °. For anal) sis it was dried for 72 hours
at 90 ° under high vacuum.
3.682 mg subst gave 7.089 mg CO2 and 1.678 mg H2O
Calcd. for C18H20O2NI C 52.82 H4.93 %
Found C 52.55 H 5.11 %
l-Benzyl-6,7-dimethoxy-l, 2, 3, 4-tetrahydroisoquinoline
To a solution of 300 mg of l-benzyl-6,7-dimethoxy-3,4-dihydroisoquino-line in 10 c. c. of N/2 sulphuric acid was added about five drops of 10 %
copper sulphate solution and 3 g of pure zinc dust. The mixture was heated
on a steam-bath for three hours with occasional shaking. The solution was
50

filtered, the filtrate made alkaline with 10 % sodium hydroxide and
extracted several times with ether. The combined ether extracts were
washed with water and dried over sodium sulphate. On distilling off ether,120 mg of a thick, colourless oil was obtained. For analysis it was twice
distilled in a sublimation tube at 0.01 mm (block temperature 135—40 °).
3.680 mg subst gave 10.294 mg C02 and 2.454 mg H20
Calcd. for G8H2i02N C 76.29 H 7.48 %
Found'
C 76.34 H 7.45 %
PKA 7.79
The U. V. absorption spectrum is shown in fig. 1, curve 2.
The I. R. spectrum is shown in fig. 3, curve 2.
The picrate after three crystallisations from alcohol had m. p. 162—3 °.
For analysis it was dried for 48 hours under high vacuum at 90 °.
3.794 mg subst gave 7.820 C02 and 1.600 mg H20
3.041 mg subst gave 0.346 c.c. N2 (20V720 mm)
Calcd. for C24H2409N4 C 56.25 H4.72 N 10.93%
Found C 56.25 H 4.72 N 10.79 %,
The hydroiodide prepared as before and three times crystallised from
water had m. p. 209—10 °. For analysis it was dried for 96 hours at 80 °
under high vacuum.
3.712 mg subst gave 7.158 mg CO2 and 1.822 mg H20
Calcd. for Ci8H2202NI C 52.56 H 5.39 %.
Found C 52.62 H 5.49 %
Attempted synthesis of 5-aza-4', 5*-elimethoxy - 3 - phenyl-benzocycloheptene
Veratric acid (XIII)
It was obtained by the oxidation of veratraldehyde with silver nitrate in
alkaline medium68.
To 400 c.c of water were added with vigourous stirring 50 g (1.25 mole)
of sodium hydroxide and 33.2g (0.2 mole) of veratraldehyde69. The tem¬
perature of the mixture was at that point about 55°. With continued
68 Rf. C.J. Lintner and L. M. Parks, J.Am. Pharm. Ass. 37, 39—40 (1948); C. A.
42, 3749 (1948).69 Prepared according to G. Barger and R. Silberschmidt, Soc. 1928, 2924.
51

agitation, a solution of 34 g (0.2 mole) of silver nitrate in 150 c.c. of water
previously warmed to 55 °was added. Silver oxide was momentarily
formed as a granular brownish black powder, but at the same moment
reaction set in, the temperature rose to 85 * and the silver oxide was de¬
colourised with the production of fluffy, spongy, metallic silver. The
reaction mixture was filtered hot and washed with water. The alkaline
filtrate was acidified with sulphur dioxide and after cooling, the white
precipitate was filtered, washed with water and dried. The crude material
weighed 31 g (91% yield) and had m. p. 179—80° (reported70 m. p. 181°).
It was used for the preparation of m-meconine without further purification.
m-Meconine (4,5-dimethoxyphthalide) (XIV)
It was prepared according to Edwards et al.71 by the chloromethylationof veratric acid with formaldehyde and concentrated hydrochloric acid. It
was obtained in 29% yield as colourless needles on crystallisation from
dilute alcohol, m.p. 155—57° (reported71 m.p. 155—57°).
Oxidation of m-meconine to m-hemipinic acid (XV)
m-Hemipinic acid was obtained in 69 % yield by the following modifi¬
cation of the method given by Edwards et al.71.
m-Meconine (18g) was dissolved in 180 c.c of 10 % potassium hydro¬xide by slight warming on a water-bath. The flask was cooled in ice-water
and a solution of 21.6 g of potassium permanganate in 360 c. c. water was
added dropwise with stirring. The reaction mixture was heated on a
steam-bath for 90 minutes with frequent shaking. The precipitated man¬
ganese dioxide was filtered off and washed with hot water. The filtrate
was acidified whilst hot with concentrated hydrochloric acid. On cooling,
crystals of m-hemipinic acid separated. Crystallisation from dilute
methanol gave 14.5 g of colourless needles of m. p. 184—85 ° (reported 72
m.p. 174—75°; 179—82° depending on the rate of heating).
3.750 mg subst gave 7.294 mg C02 and 1.510 mg H20
Calcd. for CwHioOe C 53.10 H4.46%
Found C 53.08 H4.51 %
*> G. Goldschmiedt, Mon. 6, 379 (1885).71 G. A. Edwards, W. H. Perkin jun. and F. W. Stoyle, Soc. 1925, 198.72 Beilstein Vol. X, p. 552 (1927).
52

m-Hemipinic anhydride (XVI)
A mixture of 12 g of powdered, dry m-hemipinic acid and 50 c. c. of
acetic anhydride was heated at 100—110 ° for two hours. Upon standing
overnight, the clear solution deposited 10.1 g (91 % yield) of large colour¬
less prisms. These were collected, washed several times with dry ether,
and dried in a vacuum desiccator. It had m. p. 175—76° (reported '3
m. p.
166—7°; 175°).
4,5-Dimethoxy-2-benzoylbenzoic acid methyl ester (XVII)
To a suspension of 10 g of finely powdered m-hemipinic anhydride in
200 c. c. of dry thiophene free benzene was added 25 g of anhydrousaluminium chloride in small portions at a time with vigourous stirring.The colour of the mixture gradually changed to dark red and fumes of
hydrogen chloride were slowly evolved. It was boiled under reflux for
six hours. On cooling, ice and 50 c. c. of dilute hydrochloric acid (1:1)
were added. The excess of benzene was removed under vacuum and the
solid residue filtered and washed with water. It was dissolved in 2 N
sodium hydroxide and reprecipitated by addition of hydrochloric acid.
The crude material weighed 7.8 g and gave a green colouration with ferric
chloride solution. After two crystallisations from dilute alcohol it melted
at 167—83 ° showing it to be a mixture.
A mixture of 6.4 g of the above substance dissolved in 200 c. c. of dry
acetone, 12 g of anhydrous potassium carbonate and 20 g of methyl iodide
was boiled under reflux for 12 hours. The solution was filtered and the
acetone removed by distillation. The solid residue was washed with water
and crystallised from dilute alcohol giving 5.2 g of colourless needles of
m. p. 142—3° (reported74 m. p. 110—111°). For analysis the substance
was three times crystallised from dilute alcohol and dried for 36 hours at
80 ° under high vacuum.
3.744 mg subst gave 9.342 mg C02 and 1.848 mg H20
3.748 mg subst required 11.233 c. c. of 0.02 N Na2S203
Calcd. for CnHioOs C 67.98 H 5.37 3 OCH3 30.99 %
Found C 68.09 H 5.52 OCH3 30.99 %
73 G. Goldschmiedt, Mon. 6, 380 (1885); A. N. Meldrum and P. H. Parikh, Proc.
Indian Acad. Sci. 1 A, 437—39 (1935), C. A. 29, 3324 (1935).74 A. Oliverio, Gazz. chim. ital. 64, 139-48 (1934) C. A. 28, 4727 (1934).
53

The 2,4-dmitrophenylhydrazone was prepared by boiling the alcoholic
solution of the keto-ester with a solution of 2,4-dinitrophenylhydrazine
containing a few drops of concentrated hydrochloric acid for two hours.
The product was crystallised from glacial acetic acid, m. p. 235—37 °. For
analysis it was dried for 72 hours at 90 ° under high vacuum.
3.155 mg subst gave 0.330 c. c. N2 (21°/716 mm)
Calcd. for C23H20O8N4 N 11.66 %
Found N 11.44 %
(2 Hydroxymeihyl-4,5-dimethoxydiphenyl) methanol (XVIII)
To a well stirred mixture of 4 g of lithium aluminium hydride in 100
c. c. absolute ether was added dropwise and with stirring a solution of
1.5 g of 4,5-dimethoxy-2-benzoylbenzoic acid methyl ester in 360 c. c.
of ether during the course of two hours. With continued stirring, the
reaction mixture was boiled under reflux for two hours. The flask was
cooled well and 250 c. c. of ice-cold water was added dropwise with
stirring. The ether layer was separated and the aqueous solution extracted
with three 100 c. c. portions of ether. The combined ether extracts were
washed with water, and dried over sodium sulphate. On distillation of
ether, 1.27 g (93 % yield) of a white solid was obtained. Crystallisationfrom a mixture of benzene and petroleum ether gave 1.2 g of crystals of
m. p. 95—97 °. For analysis the substance was distilled under high vacuum
(block temperature 175—80° at 0.02 mm) when it again solidified. It had
m. p. 97—98 °.
3.649 mg subst gave 9.370 mg CO2 and 2.156 mg H2O
5.776 mg subst gave 1.094 c. c. CH4 at 19 °/722 mm
0.972 c.c. CH4 at 0°/760 mm
Calcd. for GeHisCh C 70.05 H 6.61 2 «H» 0.74 %
Found C 70.08 H 6.61 «H» 0.76 %
If in the above reduction, the reaction mixture was decomposed with
a little of sulphuric acid, then the product obtained was a yellowish,viscous oil which did not solidify, even after being carefully chromato-
graphed over alumina (activity II—III) and distilled. The analysis and
the I. R. spectrum of this oil, which was twice distilled at 0.01 mm (block
temperature 175—80°) for the purpose, showed it to be a furane deri¬
vative obtained by the loss of a molecule of water from the above diol.
54

3.820 mg subst gave 10.545 C02 and 2.252 mg H20
Calcd. for CieHieOs C 74.98 H6.29%
Found C 75.33 H 6.60 %
(2 Chloromethyl-4,5-dimethoxydiphenyl) methyl chloride (IXX)
A solution of 77 mg of the above diol in 2 c. c. of dry chloroform was
gently boiled under reflux with 0.3 c. c. of thionyl chloride for 90
minutes. The excess of chloroform and thionyl chloride were removed
under vacuum. The oily residue was dissolved in dry benzene and the
latter removed under vacuum. This operation was repeated with two more
5 c. c. portions of dry benzene when 82 mg of a pink coloured oil was
obtained. It was dissolved in absolute ether and the ether extract quicklywashed with a little ice-cold water. The ether solution was dried over
sodium sulphate and the ether distilled off giving 72 mg of an almost
colourless oil. It was distilled in a sublimation tube at 0.01 mm (block
temperature 140°—42°). The tube was sealed under vacuum and givenfor analysis.
5.046 mg subst gave 4.486 mg AgClCalcd. for C16H16O2CI2 CI 22.79 %
Found CI 21.99%
The conversion of the above dichloride into a dinitrile was attemptedunder different experimental conditions. In all cases, only mixtures
containing 0.1 to 0.9 % nitrogen were obtained; the pure dinitrile could
not be prepared.
Homoveratrylglycine ethyl ester (XX)
To a well stirred solution of 15.03 g (0.083 mole) of homoveratrylaminein 70 c. c. of absolute ether at 0 °
was added dropwise a solution of 7.0 g
(0.042 mole) of bromoacetic ester in 20 c. c. of absolute ether during the
course of 20 minutes. During addition, the hydrobromide of the originalamine separated. After further 10 minutes of stirring, the reaction mixture
was allowed to stand overnight at room temperature. Next day, the homo¬
veratrylamine hydrobromide (about 9 g) was collected and washed several
times with ether. It was crystallised from a mixture of methanol and ethyl
acetate, m. p. 179—80 °.
55

A small amount of this hydrobromide when treated with sodium picratein dilute alcoholic solution gave a crystalline picrate which was identical
with that obtained from homoveratrylamine as found by a mixed melting
point determination.
4.530 mg subst gave 0.207 c. c. N> (18 °/722 mm)
Calcd. for CioHi602NBr N5.34%
Found N 5.09 %
The ethereal filtrate and washings from above were washed with two
50 c. c. portions of water and dried over sodium sulphate. On distilling off
ether, 7.5 g of a yellowish oil was obtained. It was distilled at 151—53 °
and 0.03 mm giving 3.7 g (16 % yield) of a colourless oil.
For analysis the oil was redistilled in a sublimation tube at 0.001 mm
(block temperature 122—26°) 75.
3.790 mg subst gave 8.625 mg C02 and 2.612 mg H2O
3.699 mg subst gave 0.181 c. c. N2 (23 °/729 mm)
Calcd. for C14H21O4N C 62.90 H 7.92 N 5.24 %
Found C 62.10 H7.71 N5.41 %
The picrate after crystallisation from alcohol was obtained in the form
of yellow needles, m. p. 157—58 °. For analysis it was dried for 48 hours
at 75 ° under high vacuum.
3.868 mg subst gave 6.859 mg CO2 and 1.687 mg H2O
4.069 mg subst gave 0.420 c. c. N2 (20°/723 mm)
Calcd. for C20H24O11N4 C 48.39 H 4.87 N 11.29 %
Found C 48.39 H4.88 N 11.44 %
Homoveratrylglycine hydrochloride (XXI)
Three grams of homoveratrylglycine ethyl ester was mixed with 5 c. c.
of concentrated hydrochloric acid and the mixture heated on a water-bath
for two hours. On cooling, white needles of the hydrochloride separated.These were filtered and washed with a little glacial acetic acid and then
with dry ether. Three crystallisations from glacial acetic acid gave 1.8 g
of colourless needles of m. p. 199—200 °. For analysis it was dried for
48 hours at 80° under high vacuum.
During this distillation traces of a white substance were formed which mayaccount for the low value of carbon.
56

3.849 mg subst gave 7.386 mg C02 and 2.269 mg H20
Calcd. for C12H18O4NCI C 52.27 H 6.58 %
Found C 52.37 H 6.60 %
N-Tosyl-homoveratrylglycine (XXII)
To a solution of 840 mg of homoveratrylglycine hydrochloride in 8 c. c.
of 10 % potassium hydroxide was added with stirring 660 mg of
p-toluenesulphonyl chloride in ether solution. The mixture was shaken
well and the ether was removed by heating on a water-bath. The alkaline
solution was acidified with 2 N hydrochloric acid when a white solid
separated. One crystallisation from a mixture of benzene and petroleumether gave 652 mg of colourless crystals. For analysis the substance was
three times crystallised and dried for 48 hours at 70 ° under high vacuum.
It had m. p. 136—38°.
3.685 mg subst gave 7.845 mg CO2 and 1.975 mg H2O
3.174 mg subst gave 0.100 c.c. N2 (23°/729 mm)
Calcd. for C19H23O6NS C 58.00 H 5.89 N3.56%
Found C 58.10 H6.00 N3.48%
Cyclisation of the above N-tosyl derivative (to obtain a seven-membered
ring) was attempted with stannic chloride and with a solution of phos¬
phorus pentoxide in 85% orthophosphoric acid, but no well defined
substance could be isolated in either case.
Condensation of homoveratrylamine and chloroacetic ester in the
presence of sodium acetate
A solution of 588 mg (3.25 m mole) of homoveratrylamine in 2 c.c.
of alcohol, 405 mg (3.3 m mole) of chloroacetic ester and 279 mg (3.25 m
mole) of dry sodium acetate were mixed together and kept at room tem¬
perature for 16 hours. The mixture was boiled under reflux for twenty
minutes and filtered from the inorganic residue. From the filtrate, the
alcohol was removed under vacuum and the residue taken up in ether.
The ether extract was treated with 2 N hydrochloric acid and the acid
layer separated. The latter was cooled to 0° and neutralised with 2N
sodium hydroxide to a pH of about 8. The alkaline solution was extracted
with ether and the ether layer washed with water. After drying and
57

distilling off ether, 460 mg of an oil was obtained. A small amount of it
was dissolved in alcohol and treated with an alcoholic solution of picricacid. As nothing separated on keeping, the above solution was concen¬
trated under vacuum to a small volume when an oil was obtained. On
addition of ether, yellow crystals separated. These were crystallised from
a mixture of chloroform and ether. Three more crystallisations gave yellowneedles of m. p. 162—63 °. A mixed melting point with the picrate of
homoveratrylglycine ethyl ester showed a depression of 10 °. It was dried
for 76 hours at 80 ° under high vacuum and analysed.
3.760 mg subst gave 6.568 mg CO2 and 1.398 mg H2O
3.859 mg subst gave 0.446 c.c. N2 (23°/720 mm)
Calcd. for C36H36O20N8 C 48.00 H4.03 N 12.44 %
Found C 47.67 H4.16 N 12.62 %
The analysis of this picrate agrees with that of a dipicrate (addition
product) of the diketopiperazine that might have been formed in the above
reaction.
Synthesis of4,5 dimethoxydiphcnyl- 2,2'-dicarboxylic acid
6-Nitroveratraldehyde (XXIII)
It was obtained by the nitration of veratraldehyde 76 according to the
modified method of Cassaday and Bogert77. It was crystallised from
alcohol in yellow needles of m. p. 132—33° (reported78 m. p. 133.5—
34.5°).
Qs-Phenyl-2-nitro-4,5-dimethoxycinnamic acid (XXIV)
It was prepared according to Pschorr and Buckow79, but their method
was modified as follows:
A mixture of 21 g of 6-nitroveratraldehyde, 16 g of powdered anhydroussodium phenylacetate (dried at 130° for 6 hours), 90 c.c. of acetic an¬
hydride and 1 g of anhydrous zinc chloride was heated in an oil-bath at
76 Prepared according to G. Barger and R. Silberschmidt, Soc. 1928, 2924.77 J. T. Cassaday and M. T. Bogert, Am. Soc. 61, 2462 (1939).
E. B. Marr and M. T. Bogert, Am. Soc. 57, 1329 (1935).'• R. Pschorr and W. Buckow, Ber. 33, 1829—31 (1900).
58

110—120° for five hours with occasional shaking of the mixture. On
cooling, the reaction mixture was poured into 500 c. c. of ice-water and
after the initial cloudiness had settled, the clear solution was decanted
from the brown resinous products. These were brought into solution by
boiling with water (2—3 litres) and with the addition of concentrated
ammonia. The solution was filtered through «celite» and the clear filtrate
was acidified with hydrochloric acid, when a yellow precipitate was ob¬
tained. It was crystallised from alcohol, when 16.2 g (43 % yield) of
yellow needles were obtained, m. p. 217—19° (reported80 m. p. 219).
«-Phenyl-2-amino-4-5-dimethoxycinnamic acid81 (XXV)
Five grams of the nitro acid (XXIV) was dissolved in 10 c. c. of 25 %
ammonia and 30 c. c. of water. The solution was heated to boiling and
poured with stirring into a suspension of ferrous hydroxide prepared from
35 g of hydrated ferrous sulphate in 105 c. c. water and 20 c. c. of 25 %
ammonia. The mixture was heated on a steam-bath for two hours with
frequent shaking and filtered through «celite». The residue together with
the «celite» was boiled out three times with dilute ammonia and the com¬
bined ammoniacal filtrates were acidified with glacial acetic acid. The
amino acid which separated as yellow crystals was collected and washed
several times with water. One crystallisation from alcohol gave 3.1 g (68 %
yield) of yellow, flaky needles of m. p. 185—88 °. After three more crystal¬
lisations and drying for 36 hours at 70° under high vacuum, the melting
point was raised to 193—95° (reported80 m.p. 209°).
2.262 mg subst gave 5.604 mg C02 and 1.188 mg H2O
Calcd. for C17H17O4N C 68.21 H 5.73 %
Found C 67.61 H 5.88 %
Although the above substance was not analytically pure, it was pure enough
for further work.
80 R.Pschorr and W. Buckow, Ber. 33, 1829—31 (1900).81 Cf. E. Mosettig and A. Burger, Am. Soc. 52, 2988—93 (1930).
59

2,3-Dimethoxyphenanthrene-9-carboxylic acid (XXVI)
A better yield than reported82 of 2,3-dimethoxyphenanthrene-9-car-
boxylic acid was obtained by the following procedure83.One gram of the amino acid XXV was dissolved in 25 c. c. of tetra-
hydrofuran and 0.5 c. c. of concentrated sulphuric acid was added to the
solution with stirring. A white precipitate of the sulphate separated and
50 c. c. more of tetrahydrofuran was added to keep the whole mixture in
suspension. With cooling, 0,7 c. c. of isoamyl nitrite (previously washed
with sodium bicarbonate solution, followed by a saturated sodium chloride
solution and dried over sodium sulphate) was added and the mixture was
stirred for 90 minutes at room temperature.
To a solution of 3.5 g of sodium hypophosphite in 4 c. c. of water there
was added some freshly prepared copper powder 84 and the mixture just
heated to boiling. After cooling to 50°, the diazotized amino acid was
added to it with stirring, while the temperature was kept at 40—50 °. The
mixture was then heated on a water-bath at 60—70 ° for fifteen minutes,
whereby most of the tetrahydrofuran distilled over. The residue was
poured into water containing sufficient ammonia to ensure solution of
the acid. The filtered solution was acidified with IN hydrochloric acid,
when brownish crystals were obtained. One crystallisation from alcohol
(charcoal) gave 568 mg (60 % yield) of colourless needles of m. p.
269—70°, beginning to get brown at 260° (reported85 m. p. 270°).
For analysis the substance was three times crystallised from alcohol and
dried for 36 hours at 80 ° under high vacuum.
3.632 mg subst gave 9.657 mg CO2 and 1.719 mg H2O
Calcd. for C17H14O4 C 72.33 H5.00%
Found C 72.56 H 5.30 %
2,3-Dimethoxyphenanthrene (XXVII)
2,3-Dimethoxyphenanthrene was obtained in 85 % yield by carryingout the decarboxylation of the acid with quinoline and copper powder
as follows:
82 R.Pschorr and W. Buckow, Ber. 33, 1829—31 (1900).63 Cf. J. T. Cassaday and M. T. Bogert, Am. Soc. 61, 2462—63 (1939).84 Prepared according to L. Gatterman, Ber. 23, 1219 (1890).85 R. Pschorr and W. Buckow, loc. cit. ref. 82.
60

A solution of 511 mg of the acid in 3 c. c. of freshly distilled quinolineand about 50 mg of copper powder were boiled under reflux for 90
minutes. On cooling, the mixture was dissolved in ether and the ether
extract washed successively with three 30 c. c. portions of 2 N hydro¬chloric acid, 2 N sodium hydroxide and water. After drying and distillingoff ether, 389 mg of a brownish solid was obtained. One crystallisationfrom dilute alcohol (charcoal) gave 366 mg of colourless plates of m. p.
130—31° (reported86 m. p. 131°).For analysis the substance was recrystallised and sublimed under high
vacuum (block temperature 120°).
3.814 mg subst gave 11.245 mg C02 and 2.059 mg H2O
Calcd. for C16H14O2 C 80.65 H 5.92 %
Found C 80.46 H6.04%
2,3-Dimethoxy-9,10-phenanthraquinone (XXVIII)
To a solution of 152 mg of 2,3-dimethoxyphenanthrene in 2.5 c. c. of
glacial acetic acid was added dropwise with stirring a solution of 303 mg
of chromic acid in about three drops of water and 0.5 c. c. of acetic acid.
A little evolution of heat occurred and a red substance began to separate.
After heating the mixture for twenty minutes on a water-bath, the red
substance was filtered and washed with acetic acid and water. It was
crystallised from glacial acetic acid when 62 mg of red needles of m. p.
304° were obtained (reported86 m.p. 304°).For analysis the substance was three times crystallised from glacial
acetic acid and dried for 72 hours at 80 ° under high vacuum.
3.650 mg subst gave 9.565 mg CO2 and 1.467 mg H2O
Calcd. for C16H12O4 C 71.63 H 4.51 %
Found C 71.51 H4.50%
4,5-Dimethoxydiphenyl-2,2'-dicarboxylic acid (XXIX)
To a suspension of 45 mg of 2,3-dimethoxy-9,10-phenanthraquinone in
1 c. c. of methanol was added 0.12 c. c. of 30 % hydrogen peroxide. With
continued stirring, 0.2 c. c. of 2 N sodium hydroxide was added to the
88 R.Pschorr and W. Buckow, Ber. 33, 1829—31 (1900).
61

above suspension during an interval of 10 minutes. Since the solid did not
completely dissolve on warming, more hydrogen peroxide (0,7 c. c. in all)
was added in 0.1 c. c. portions at a time during the course of 15 minutes.
One c. c. more of methanol was added and the mixture boiled under reflux
for one hour. All the substance gradually went into solution and a faint
yellow clear solution was obtained. Most of the methanol was removed
by distillation and the resulting solution was filtered through «celite».
The filtrate was acidified with 2N hydrochloric acid and extracted with
chloroform. The chloroform extract was washed with water and dried
over sodium sulphate. Evaporation of chloroform left 30 mg of a colour¬
less solid. Crystallisation from chloroform-methanol gave colourless prisms
of m. p. 210—12°. For analysis the substance was sublimed under high
vacuum (block temperature 180—90°). It had m.p. 215—16°.
2.468 mg subst gave 5.747 mg CO2 and 1.039 mg H2O
Calcd. for CwHuOe C 63.57 H4.67%
Found C 63.55 H4.71%
The dimethyl ester obtained by methylation with diazomethane in ether
solution was crystallised from a mixture of benzene and petroleum ether.
After drying for 48 hours at 60 ° under high vacuum, it had m. p.
118—19°.
3.717 mg subst gave 8.895 mg C02 and 1.827 mg H20
2.617 mg subst required 9.478 c. c. of 0.02 N N2S2O3
Calcd. for Ci8Hi806 C 65.44 H 5.49 40CH3 37.56 %
Found C 65.31 H 5.50 OCH3 37.47%
The U. V. absorption spectrum showed absorption bands with a maxi¬
mum at 296 mjA, loge = 3.84 and at 224 ra.fi, loge = 4.60.
The I. R. spectrum showed bands at about 875 cm-1 and at 772 cm-1
indicating the presence of 1, 2, 4, 5 tetra-substituted and 1,2 disubstituted
benzene nuclei respectively.
62

Synthesis of 2,3'-(lictlijl-4,5-diinethoxj2'- dimethylaminobiphenyl
4-Ethyl-5-iodoveratrol (XXX)
To a well-stirred solution of 11.2 g of 4-ethylveratrol87 in 30 c. c. of
absolute alcohol were added alternately 17.0 g of powdered iodine and
10.2 g of yellow mercuric oxide during one hour. After stirring for two
more hours, the reaction mixture was filtered and the alcohol removed
from the filtrate by distillation. The residue was dissolved in ether and
the ether solution washed successively with 20 % sodium thiosulphatesolution, 2 N sodium hydroxide and water. After drying and distillingoff ether, 12.7 g of a yellow crystalline solid was obtained. Two crystalli¬sations from dilute methanol gave 10.8 g (55 % yield) of colourless
prisms of m. p. 58—60 °. For analysis the substance was sublimed under
high vacuum (block temperature 55°). The sublimed substance melted
at 60—61 °.
4.073 mg subst gave 6.180 mg CO2 and 1.700 mg H2O
Calcd. for C10H13O2I C 41.11 H4.49%
Found C 41.41 H 4.67 %
5-Amino-4-ethylveratrol (XXXI)
It was obtained in 93 % yield by reducing 4-ethyl-5-nitroveratrol8S with
hydrogen in alcohol solution in the presence of platinum catalyst (10 %).
Its acetyl derivative was crystallised from water in colourless needles
of m. p. 147 ° (reported88 m. p. 147 °).
3.660 mg subst gave 8.648 mg CO2 and 2.504 mg H20
Calcd. for Ci2Hi703N C 64.48 H 7.65 %
Found C 64.55 H 7.68 %
87 Prepared according to G. Barger and R. Silberschmidt, Soc. 1928, 2925.88 Prepared according to F. E. King and P. L'Ecuyer, Soc. 1937, 430—31.
63

4-Ethyl-5-iodoveratrol from 5-amino-4-ethylveratrol
A solution of 500 mg of 5-amino-4-ethylveratrol in 0.5 c. c. of concen¬
trated hydrochloric acid and 2 c. c. of water was cooled to —5 ° and
diazotised with a solution of 153 mg sodium nitrite in 2 c. c. of water.
The diazo solution was treated with a solution of 558 mg of potassiumiodide in 1.5 c. c. water, added dropwise and with stirring, the temperature
being always kept below 0° during addition. After standing for fifteen
minutes at 20°, the reaction mixture was shaken up with ether and the
ether solutionS9 washed successively with water, 2 N sodium hydroxide,2 N hydrochloric acid and water. After drying and distilling off ether,
30 mg of a dark coloured oil was obtained. It was sublimed at 0.01 mm
using the «cold finger* apparatus (bath temperature 55—60 °) when a
yellow solid was obtained. Recrystallisation from dilute methanol gave
colourless crystals of m. p. 58—59 °. Its melting point when mixed with
4-ethyl-5-iodoveratrol obtained above showed no lowering.
Ullmann-reaction of 4-ethyl-5-iodoveratrol with o-iodonitrobenzene:
2,2'-dinitrobiphenyl (XXXII)
A mixture of 2.346 g (0.008 mole) of 4-ethyI-5-iodoveratrol and 2.998 g
(0.012 mole) of o-iodonitrobenzene91 was heated in a graphite bath to
180 ° and 2.45 g of copper powder 9°was added to it in small quantities
at a time with stirring during an interval of 10 minutes. The temperature
of the mixture was gradually raised and kept between 235—40° for
30 minutes with frequent stirring. On cooling, the brown mass was
extracted with three 25 c. c. portions of hot alcohol and filtered. The filtrate
after boiling with animal charcoal was concentrated to a small volume,
when 1.066 g of yellow needles of m. p. 116—20° were obtained. Two
more crystallisations from ligroin gave 970 mg of pale yellow crystals of
m.p. 122—23° (reported82 m.p. 124°). The substance was sublimed
under high vacuum (block temperature 87—92°) and analysed.
89 An appreciable amount of a sticky, orange-coloured, and ether insoluble material
formed in the reaction was rejected.91 Prepared according to F. Ullmann, Ber. 29, 1880 (1896).90 The copper powder used for all Ullmann reactions was thrice washed with dry
ether before u?e.
92 F. Ullmann and J. Bielecki, Ber. 34, 2176 (1901).
64

3.991 mg subst gave 8.630 mg CO2 and 1.197 mg H20
Calcd. for C12H8O4N2 C 59.02 H 3.30 %
Found C 59.01 H 3.36 %
No other biphenyl could be isolated from the mother liquors.
Ullmann-reaction of 4-ethyl-5-iodoveratrol with o-bromonitrobenzene:
2-ethyl-4,5-dimethoxy-2'-nitrobiphenyl (XXXIII)
A mixture of 4.4 g (0.015 mole) of 4-ethyl-5-iodoveratrol and 2.04 g
(0.01 mole) of o-bromonitrobenzene'3 was heated in a graphite bath to
165—70 ° and 4.6 g of copper powder was added to it in small quantities
at a time with stirring during an interval of fifteen minutes. The tem¬
perature of the mixture was gradually raised and kept at 230—40° for
40 minutes with frequent stirring. On cooling, the brown mass was
extracted with three 40 c. c. portions of hot alcohol and filtered. The
filtrate after treatment with animal charcoal and concentration gave 1.5 g
of yellow crystals. One crystallisation from methanol gave 1.32 g of yellow
needles of m. p. 72—74 °. The mother liquor from above after removal of
alcohol was fractionally distilled at 0.01 mm (bath temperature up to
155°) in a Craig coloumn. The fractions collected at different temperatures
consisted of the starting materials and their mixtures. The brownish
residue in the distillation flask after two crystallisations from methanol
(charcoal) gave further 250 mg of the above biphenyl of m. p. 72—-74°.
For analysis the substance was three times crystallised from ligroin and
dried for 24 hours at 30 ° under high vacuum, m. p. 73—74 °.
3.692 mg subst gave 9.050 mg CO2 and 1.992 mg H2O
4.340 mg subst gave 0.198 c. c. N2 (21 °/731 mm)
Calcd. for C16H17O4N C 66.88 H5.96 N4.87%
Found C 66.89 H6.04 N5.10%
4-Acetamido-l-ethyl-2,3-dinitrobenzene (XXXIV)
A better yield than reported94 of 4-acetamido-l-ethyl-2,3-dinitrobenzene
was obtained by the following modified method.
93 Prepared according to F. Ullmann, Ber. 29, 1880 (1896).94 0. L. Brady, J. N. E. Day, and P. S. Allam, Soc. 1928, 980.
65

Thirty grams of 4-acetamido-l-ethyl-2-nitrobenzene95 was added in
small portions at a time to 100 c. c. of fuming nitric acid (d 1.5), the
temperature being kept below 20 ° during addition. After thirty minutes,
the mixture was poured rapidly with vigourous stirring into 3 litres of
ice-cold water. The yellow precipitate obtained was quickly filtered and
washed with a large excess of ice-cold water. It was dried at room tem¬
perature and dissolved in 120 c. c. of hot glacial acetic acid. On cooling,25 g (68 % yield) of fine yellow needles separated, m. p. 140—42 ° (re¬
ported95 m.p. 143°).
It was hydrolysed with 45 % sulphuric acid to 4-amino-l-ethyl-2,3-
dinitrobenzene, crystallised from dilute alcohol in golden yellow needles
of m.p. 120—21° (reported95 m.p. 121.6°).
Diazotisation of 4-amino-l-ethyl-2,3-dinitiobenzene in the presence
of alcohol and hydrohromic acid*": 3,4-dibromo-l-ethyl-2-nitrobenzene (XXXV)
A suspension of 2.4 g of finely powdered 4-amino-l-ethyl-2,3-dinitro-benzene in 35 c. c. of hydrobromic acid (d 1.324) and 18 c. c. of alcohol
was treated below 0 ° with a solution of 1.96 g of sodium nitrite in 3.5 c. c.
of water. The mixture was heated on a water-bath. When the temperature
of the mixture reached 20 °, nitrogen and acetaldehyde were freely evolved;
at 50 °, the liquid had a deep red colour; at 70 °, the colour of the solution
changed to orange yellow and a dark coloured heavy oil separated. It was
extracted with ether and the ether solution washed with 2 N sodium hy¬droxide and water. After removal of ether, the dark oily residue left
behind was distilled at 10 mm. The fraction boiling between 122—37 °
was a yellow oil and weighed 300 mg. It was twice distilled and submitted
for analysis which showed it to be a mixture. The residue in the distillation
flask weighed 1.7 g and solidified on cooling. Two crystallisations from
methanol (charcoal) gave colourless crystals of m. p. 81—83 °. For
analysis, it was five times crystallised from methanol and sublimed under
high vacuum (block temperature 50°). The sublimed substance had
m.p. 83—84°.
95 Prepared according to O. L. Brady, J. N. E. Day, and P. S. Allam, Soc. 1928, 980.96 Cf. L. A. Elson, C. S. Gibson, and J. D. A. Johnson, Soc. 1929, 2741 for a similar
reaction with 4-amino-2,3-dinitrotoluene.
66

3.736 mg subst gave 4.261 mg CO2 and 0.733 mg H2O
3.963 mg subst gave 0.170 c. c. N2 (20 °/730 mm)
3.741 mg subst gave 1.923 mg AgBrCalcd. for C8H7N02Br2 C 31.10 H2.28 N4.53 Br 51.73%
Found C 31.13 H2.20 N4.80 Br 51.40%
l-Ethyl-2,3-dinitrobenzene (XXXVI)
It was obtained by the diazotisation of 4-amino-l-ethyl-2,3-dinitro-benzene in absolute alcohol with fuming sulphuric acid according to
0. L. Brady et al.97. It was crystallised from dilute alcohol in faint yellowneedles of m. p. 57—58
° (reported 97m. p. 58.5 °).
3-Amino-l-ethyl-2-nitrobenzene (XXXV11)
It was prepared from l-ethyI-2,3-dinitrobenzene by reduction with
stannous chloride according to Kondo and Uyeo98. It was obtained as a
reddish oil distilling at 143—50° at 10 mm (reported98 b.p. 120—30°/
4 mm: m.p. 31—32°).
Its acetyl derivatives was crystallised from ether, m.p. 114—15° (re¬
ported98 m.p. 114°).
3.708 mg subst gave 7.838 mg CO2 and 1.919 mg H2O
Calcd. for C10H12O3N2 C 57.68 H 5.81 %
Found C 57.69 H 5.82 %
3-Bromo-l-ethyl-2-nitrobenzene (XXXV111)
It was prepared from 3-amino-l-ethyl-2-nitrobenzene by diazotisation in
24 % hydrobromic acid as described by Kondo and Uyeo '*. The product
distilled as a yellow oil at 130—37 °at 12 mm (reported99 b. p. 113/3 mm).
A part of this substance was reduced to 2-amino-3-bromo-l-ethylbenzenewith stannous chloride according to Kondo and Uyeo. The acetyl derivative
of the amine when crystallised from dilute methanol melted at 121—22 °
(reported99 m.p. 122°). This acetyl derivative was oxidised with a dilute
»' 0. L. Brady, J. N. E. Day, and P. S. Allam, Soc. 1928, 980.98 H. Kondo and S. Uyeo, Ber. 70, 1092—93 (1937).119 H. Kondo and S. Uyeo, ibid.
67

solution of potassium permanganate as described by the above authors
when 2-acetylamino-3-bromobenzoic acid of m. p. 212 °was obtained
(reported 10°m. p. 212 °).
Ullmann-reaction of 4-ethyl-5-iodoveratrol and 3-bromo-l-ethyl-2-nitrobenzene: 2,3'-diethyl-4,5-dimethoxy-2'-nitrobiphenyl (XXXIX)
A mixture of 2.2 g (0.0075 mole) of 4-ethyl-5-iodoveratrol and 1.16 g
(0.005 mole) of 3-bromo-l-ethyl-2-nitrobenzene was heated to 170° in a
graphite bath and 2.3 g of copper powder was added in small portions
during 10 minutes with stirring. The temperature of the mixture was
gradually raised and kept between 230—35 ° for forty-five minutes with
frequent stirring. The temperature of the mixture was not allowed to rise
above 240 °. On cooling, the brown mass was extracted with five 15 c. c.
portions of hot alcohol and filtered. The dark brown filtrate was boiled
with animal charcoal for fifteen minutes and filtered. The filtrate was
concentrated to a small volume and kept overnight in a refrigerator when
500 mg of yellow crystals were obtained. Two more crystallisations from
ligroin gave faint yellow prisms of m. p. 103—04'. For analysis the
substance was three times crystallised from ligroin and dried for 24 hours
at 70 ° under high vacuum, m. p. 104—05 °.
4.168 mg subst gave 10.489 mg C02 and 2.486 mg H2O
4.627 mg subst gave 0.199 c. c. N2 (22 °/730 mm)
Calcd. for C18H21O4N C 68.55 H6.71 N4.44%
Found C 68.68 H6.67 N4.77%
The I. R. absorption spectrum is shown in fig. 5, curve 12.
2'-Amino-2,3'-diethyl-4,5-dimethoxybiphenyl (XL)
A solution of 194 mg of the nitro-compound XXXIX in 30 c. c. of
alcohol was hydrogenated in the presence of 21 mg of platinum catalyst.The theoretical amount of hydrogen (42 c. c.) was absorbed in one hour.
On filtering and distilling off alcohol, 173 mg of a white crystalline solid
was obtained. Crystallisation from dilute methanol gave colourless needles
100 H. Burton, F. Hammond and J. Kenner, Soc. 1926, 1802—04.
68

of m. p. 92—94°. For analysis the substance was sublimed under highvacuum (block temperature 105 °), m. p. 96—97 °.
3.737 mg subst gave 10.370 mg C02 and 2.737 mg H2O
Calcd. for C18H23O2N C 75.75 H 8.12 %
Found C 75.73 H 8.20 %
The U. V. absorption spectrum showed a maximum at 288 mpi, loge =-
3.82.
The picrolonate, after crystallisation from alcohol, had m. p. 169—70 °.
For analysis it was dried for 24 hours at 70 ° under high vacuum.
4.931 mg subst gave 11.106 mg CO2 and 2.519 mg H2O
Calcd. for C28HS1O7N5 C 61.19 H 5.69 %
Found C 61.46 H5.72%
2,3'diethyl-4,5-dimethoxy-2'-dimethylaminobiphenyl (XLl)
To 135 mg of the amine (XL) were added alternately and with stirring223 mg of dimethyl sulphate and 0.1 c. c. of 30 % sodium hydroxide
during five minutes. After further addition of 0.04 c. c. of 30 % sodium
hydroxide, the mixture was extracted with ether and the ether solution
washed with two 3 c. c. portions of water. After drying and distilling off
ether, a yellow oil was left behind. It was heated with 1 c. c. of acetic
anhydride on a water-bath for five minutes, and the latter removed under
vacuum. The residue was taken up in ether and the ether solution extracted
with 15 c. c. of 2 N hydrochloric acid. The acid layer was separated and
neutralised with 2 N sodium hydroxide to pH 7—8. The basic solution
was extracted with ether and the ether solution washed with water. Distil¬
lation of ether gave 106 mg of a yellowish viscous oil. It was chromato-
graphed over 4 g of alumina (activity II—III). The substance eluted
with 100 c. c. of petroleum ether (b. p. 40—60°) was a colourless oil,
weighing 84 mg and solidified on keeping. It could be crystallised from
a very small amount of petroleum ether or from dilute methanol. It melted
at 55—57°. For analysis the substance was twice distilled under high
vacuum (85—90°/0.001 mm) when it solidified. The analytical samplehad m. p. 56—58. The mixed melting point with tetrahydro-des-dimethyl-
apoerysotrine was lower by 12 °.
69

3.862 mg subst gave 10.858 mg CO2 and 2.981 mg H20
Calcd. for C20H27O2N C 76.64 H 8.68 %
Found C 76.73 H 8.64 %
PKA ~ 3.5
The U. V. absorption spectrum is shown in fig. 2, curve 3.
The I. R. absorption spectrum is shown in fig. 3, curve 3.
The picrolonate after three crystallisations from alcohol had m. p. 175—6 °.
For analysis it was dried for 36 hours at 70 ° under high vacuum.
3.776 mg subst gave 8.610 mg CO2 and 2.020 mg H20
3.526 mg subst required 3.663 c. c. of 0.02 N NaaSaOs (OCH3)
and 3.770 c. c. of 0.02 N Na2S203 (N-CH3)
Calcd. for C30H35O7N5 C 62.38 H6.ll
2 OCH3 10.74 2 N-CH35.20 %
Found C 62.23 H 5.99
OCH3 10.75 N-CH35.36 %
Synthesis of unsymmetrically substituted biphenyls
by UHmann-reaction
Ullmann-reaction of 4-ethyl-5-iodoveratrol with m-iodonitrobenzene:
3,3'-dinitrobiphenyl (XLIV)
A mixture of 1.171 g (0.004 mole) of 4-ethyl-5-iodoveratrol and 1.5 g
(0.006 mole) of m-iodonitrobenzene101 was heated to 170° and 1.3 g of
copper powder was added in small portions at a time with stirring duringthe course of ten minutes. The temperature of the mixture was graduallyraised and kept between 220—30 ° for 30 minutes. The low temperature
was used to prevent the m-iodonitrobenzene reacting all with itself. After
extraction with hot alcohol, treatment with charcoal, and evaporating the
filtrate, 2.2 g of a yellow oil was obtained. It was dissolved in a small
amount of ligroin and the solution kept in a refrigerator for 48 hours. As
no solid separated, the oil was distilled undervacuum. The fraction distillingbetween 140—50 °
at 9 mm weighed 1,68 g and consisted of the originaliodo compounds. The reddish residue was dissolved in hot alcohol and the
solution kept in a refrigerator, when 15 mg of yellowish, fluffy, needles
101 Prepared according to P. Jacobson, F. K. Fertsch, and F. Heubach, Ann. 303,338, (1898).
70

separated. These were three times crystallised from ligroin and sublimed
under high vacuum (block temperature 130—40 °) when colourless crystalsof m. p. 204—05° were obtained (reported102 as orange to yellow crystalsof m. p. 200°; 197—98°).
4.540 mg subst gave 9.837 mg C02 and 1.421 mg H20
Calcd. for C12H8O4N2 C 59.02 H 3.30 %
Found C 59.13 H 3.50 %
The mother liquor from the residue (476 mg) was chromatographedover 15 g of alumina (activity II-III) but no other biphenyl derivative
could be isolated from the chromatogram fractions.
- In a second experiment, 1.256 g of 4-ethyl-5-iodoveratrol and 1.596 g
of m-iodonitrobenzene were heated together to 170—80° and 1.4 g of
copper powder was added. The temperature of the mixture was then keptat 240—45 ° for thirty minutes. Working up of the reaction mixture as
described before gave 1.6 of a yellow oil. It was chromatographed over
48 g alumina (activity I) but the original iodo compounds and the sym¬
metrical dinitrobiphenyl were the only substances which could be isolated
from the different fractions of the chromatogram.
In a third experiment, 3.032 g of 4-ethyl-5-iodoveratrol and 2.697 g of
m-iodonitrobenzene were heated together to 175 ° and 5 g of copper
powder was added. The mixture was then heated to 265 ° and kept there
for 45 minutes. After extraction with alcohol etc., 3 g of a yellow oil was
obtained. It was carefully chromatographed over 92 g of alumina (acti¬
vity II-III) eluting 25 c. c. fractions every time. In this case also, no other
substances could be isolated except the original iodo compounds and the
symmetrical dinitrobiphenyl.
4-lodoveratrol (XLV)
It was prepared by the iodination of veratrol according to the method
described by E. Ritchie103. It was obtained as a yellowish oil distillingbetween 142—44 °
at 9 mm.
1(>2 F.Ullmann and J. Bielecki, Ber. 34, 2177 (1901); Ph. Brunner and O.N.Witt,Ber. 20, 1028 (1887).
103 E.Ritchie, J. Proc. Roy. Soc. N. S. Wales, 78, 134-^10 (1945), C. A. 40, 876
(1946).
71

Ullmann-reaction of 4-iodoveratrol with m-iodonitrobenzene:
3,3'-dinitrobiphenyl (XL1V); 3,3'-4,4'-tetramethoxybiphenyl
(XLVI) and 3,4-dimethoxy-3'-nitrobiphenyl (XLVH)
A mixture of 2.112 g (0.008 mole) of 4-iodoveratrol and 2 g (0.008
mole) of m-iodonitrobenzene was heated to 170—80° and 2.3 g of copper
powder was added to it in small amounts at a time during an interval
of ten minutes. The temperature of the mixture was gradually raised and
kept between 225—40 ° for 30 minutes with occasional stirring of the
mixture. After extraction of the brown mass with hot alcohol, charcoaling,
and evaporating the filtrate, 2.022 g of a yellow oil was obtained. It was
dissolved in 25 c. c. of ligroin and the soluble part containing the original
iodo compounds (mostly 4-iodoveratrol) was rejected. The insoluble oily
residue was dissolved in the minimum amount of alcohol and kept in a
refrigerator when 20 mg of crystals separated. Recrystallisation from
ligroin gave light, colourless needles of m. p. 203—05 °. Its melting point
when mixed with 3,3'-dinitrobiphenyl showed no depression. The mother
liquor after removal of alcohol gave 1.383 g of a yellow oil. It was dissolved
in the minimum amount of benzene and chromatographed over 45 g
of neutral alumina (activity II-III) eluting 100 c. c. fractions every time.
The first 6 fractions eluted with petroleum ether (b. p. 40—60 °) gave
938 mg of an oil containing mainly m-iodonitrobenzene and a small
amount of 4-iodoveratrol. Fractions 7 and 8 eluted with a mixture of
petroleum ether and benzene (4:1) gave 178 mg and 171 mg respectively
of a yellow oil which solidified on keeping. Fractions 9 and 10 eluted also
with petroleum ether-benzene mixture (4:1) gave 46 mg and 19 mg of
a dark yellow solid respectively. Fractions 11 and 12 eluted with benzene
together gave 6 mg of a brownish tar from which no solid could be
isolated.
Fractions 9 and 10 were first worked up and were found to be mixtures
from which only the 3,3'-dinitrobiphenyl could be isolated in a small
yield.
Fraction 7 was dissolved in a small amount of methanol. On cooling,
yellow crystals of (XLVII) separated. Two crystallisations from ligroin
gave 80 mg of crystals of m. p. 93—95 °. For analysis it was three times
crystallised from ligroin and dried for 15 hours at 30 ° under high
72

vacuum. The analysis sample had m. p. 94—95 °. No pure substance could
be isolated from the mother liquor.3.662 mg subst gave 8.702 mg C02 and 1.681 mg H20
Calcd. for C14H13O4N C 64.86 H5.05%
Found C 64.85 H5.14%
The I. R. absorption spectrum is shown in fig. 5, curve 9. Its melting
point when mixed with 3,4-dimethoxy-2'-nitrobiphenyl showed a depres¬sion of 12 °.
Fraction 8 of the chromatogram was dissolved in the minimum amount
of methanol and the solution kept in a refrigerator, when a partly cry¬
stalline solid separated. One crystallisation from ligroin gave 120 mg of
faint yellow needles having m. p. 110—23 °. These were dissolved in 15 c. c.
of alcohol and hydrogenated over 10 mg of platinum catalyst. Only7—8 c. c. of hydrogen was absorbed. After filtering and evaporating the
solvent, 110 mg of a colourless solid was obtained. It was dissolved in
ether and the ether solution extracted with 2 N hydrochloric acid. The ether
and the acid layers were separated. From the ether layer, after distillation of
the solvent, 60 mg of colourless crystals were obtained. After two crystalli¬sations from methanol and three more from ligroin, 35 mg of colourless
crystals of m. p. 132—33 °were obtained (reported for 3,3'-4,4'-tetra-
methoxybiphenyl104, m. p. 133°). For analysis the substance was sublimed
under high vacuum (block temperature 110—20°).
3.848 mg subst gave 9.871 mg CO2 and 2.300 mg H2O
Calcd. for Ci6Hi804 C 70.05 H 6.61 %
Found C 70.01 H 6.69 %
The acid layer from the above experiment was neutralised with alkali
and the basic solution was extracted with ether. After drying and distillingoff ether, 32 mg of colourless crystals were obtained Three crystallisationsfrom dilute methanol gave 18 mg of colourless needles of m. p. 110—111 °.
After sublimation under high vacuum (block temperature 120°), the
substance melted at 111—112 °. Its melting point when mixed with the
3'-amino-3,4-dimethoxybiphenyl obtained by the reduction of 3,4-di-
methoxy-3'-nitrobiphenyl showed no depression.2.392 mg subst gave 6.424 mg CO2 and 1.367 mg H2O
Calcd. for C14H15O2N C 73.34 H 6.59 %
Found C 73.29 H6.39%
104 E.Ritchie, J. Proc. Roy. Soc. N. S. Wales, 78, 134—40 (1945); C. A. 40, 876
(1946).
73

Ullmann-reaction of 4-iodoveratrol with o-bromonitrobenzene:
3,4-dimethoxy-2'-nitrobiphenyl (XLVIII)
A mixture of 4.204 g (0.016 mole) of 4-iodoveratrolm and 2.393 g
(0.012 mole) of o-bromonitrobenzene was heated together to 170° and
4.6 g of copper powder was added to it with stirring during an interval
of 10 minutes. The temperature of the reaction mixture was graduallyraised and kept between 215—25 ° for 30 minutes. After cooling, the brown
mass was extracted with hot alcohol and the filtrate boiled with animal
charcoal. Evaporation of the filtrate gave 4.2 g of a yellow oil. It was
dissolved in a small amount of methanol and kept in a refrigerator when
crystals separated. These were collected and three times crystallised from
methanol giving 980 mg of yellow crystals of m. p. 98—99 °. For analysisthe substance was recrystallised from ligroin and dried for 36 hours at
30 ° under high vacuum.
3.850 mg subst gave 9.109 mg C02 and 1.782 mg H20
Calcd. for C14H13O4N C 64.86 H5.05%
Found C 64.57 H5.18%
The I. R. absorption spectrum is shown in fig. 5, curve 10.
2'-Amino-3,4-dimethoxybiphenyl (XLIX)
A solution of 910 mg of 3,4-dimethoxy-2'-nitrobiphenyl in 50 c. c. of
alcohol was hydrogenated over 135 mg of platinum catalyst. The theore¬
tical amount of hydrogen was absorbed in 90 minutes. Evaporation of the
filtrate gave 720 mg of a white solid. Three crystallisations from dilute
methanol gave 651 mg of colourless needles of m. p. 122—23 °. For analysisthe substance was sublimed under high vacuum (block temperature
110—20°).
3.980 mg subst gave 10.675 mg CO2 and 2.376 mg H20
Calcd. for C14H15O2N C 73.34 H 6.59 %
Found C 73.20 H6.68?^
105 E.Ritchie, J. Proc. Roy. Soc. N. S. Wales, 78, 134-40 (1945); C. A. 40, 876
(1946).
74

Deamination of 2'-amino-3,4-dimethoxybiphenyl to
3,4-dimethoxybiphenyl (L)
Deamination of the above biphenyl by diazotisation with sodium nitrite
and sulphuric acid in alcohol medium or in the presence of sodium hypo-
phosphite gave oily products from which no solid substance could be
isolated after chromatography. It was therefore carried out using the
alkaline formaldehyde method106.
A solution of 240 mg of the substance in 0.5 c. c. of concentrated hydro¬chloric acid, 0.5 c. c. of glacial acetic acid and 2 c. c. of water was chilled
in ice and diazotised in the usual manner with 96 mg of sodium nitrite in
about five drops of water. A solution of 1 g of sodium hydroxide in 6 c. c.
of water was prepared and placed with about 5 g of ice in a 50 c. c. flask.
With stirring, 0.75 c. c. of 38 % aqueous solution of formaldehyde was
added dropwise to the above alkali solution. The diazo solution was then
poured into the rapidly stirred alkaline solution of formaldehyde. Frothing
occurred and nitrogen was evolved. After all the diazonium chloride had
been added, the stirring was continued for five minutes, after which the
reaction mixture was allowed to stand for twenty minutes. A dark brown
paste was obtained. It was extracted with chloroform and the chloroform
solution washed twice with water. Distillation of chloroform gave a dark
red, pasty residue. It was dissolved in methanol and boiled with animal
charcoal for 15 minutes. Evaporation of the filtrate gave 20 mg of a yellowoil which solidified after three days. It was crystallised from a small
amount of methanol, but was still impure. It was therefore sublimed under
high vacuum (block temperature 70"), when 8 mg of colourless crystals
were obtained. Crystallisation from dilute methanol gave colourless
prisms of m. p. 69—70 °. Its melting point when mixed with 3,4-dimethoxy¬
biphenyl m (reported107
m. p. 70 °) showed no lowering.
The following biphenyl derivatives were prepared for a comparison of
their U. V. and I. R. absorption spectra with those of 2,3'-diethyl-4,5-
dimethoxy-2'-dimethylaminobiphenyl already described on page 69.
106 N. Kornblum, «Organic Reactions* Vol. II, p. 297 (1944).
107 Prepared according to S. E. Harris and W. G. Christiansen, J. Am. Pharm. Ass.,
23, 530—36 (1934); Chem. Zentr. II, 2217 (1934).
75

2-Dimethylaminobiphenyl
It was obtained by the methylation of 2-aminobiphenyl with dimethyl
sulphate and alkali. The latter substance was prepared by the reduction of
2-nitrobiphenyl with sodium hydrosulphite as follows:
A solution of 2 g of 2-nitrobiphenyl108 in 100 c. c. of alcohol was mixed
with a solution of 7 g of sodium hydrosulphite in 32 c. c. of water, and the
mixture boiled under reflux for 90 minutes. On cooling, the solution was
filtered and most of the alcohol removed from the filtrate by distillation.
The remaining liquid was poured into cold water, when 1,3 g (76 % yield)of colourless crystals separated. Recrystallisation from dilute methanol
gave colourless prisms of m. p. 45—46° (reported108 m. p. 44—45.5°;
49°).
The 2-aminobiphenyl (1.27 g) was methylated110 with 3.4 g of dimethyl
sulphate in the presence of 2 c. c. of 30 % sodium hydroxide in exactly the
same way as described for the biphenyl on page 69, when 845 mg of a co¬
lourless oil was obtained. Treatment of this oil with an alcoholic solution of
picric acid gave a crystalline picrate which after three crystallisations from
alcohol had m. p. 186—89° (decomp.). For analysis it was dried for
48 hours at 70 ° under high vacuum.
4.079 mg subst gave 8.429 mg CO2 and 1.529 mg H2O
Calcd. for C20H18O7N4 C 56.34 H 4.26 %
Found C 56.39 H 4.19 %
The free base obtained from the picrate was distilled at 145—50° at
11 mm, n^°- 1.605.
3.895 mg subst gave 12.186 mg CO2 and 2.659 mg H2O
Calcd. for C14H15N C 85.23 H 7.66 %
Found C 85.38 H 7.64 %
The U. V. absorption spectrum showed bands with a maximum at
236 m,a, loge - 4.3; 268 mM, loge _ 3.7 and at 308 m/i, logs - 3.42.
108 Prepared according to H. France, I. M. Heilbron and D. H. Hey, Soc. 1940, 370.109 Beilstein, Vol. XII, p. 1317 (1929).110 Cf. A. H. Popkin, G. M. Perretta and R. Selig, Am. Soc. 66, 834 (1944).
76

2'-Amino-2-ethyl-4,5-dimethoxybiphenyl (LI)
A solution of 212 mg of 2-ethyl-4,5-dimethoxy-2'-nitrobiphenyl in 15 c. c.
of alcohol was hydrogenated in the presence of 30 mg of platinum catalyst.The theoretical amount of hydrogen (about 50 c. c.) was absorbed in 30
minutes. Evaporation of the filtrate gave 180 mg of an oil which solidified
on scratching. It was three times crystallised from dilute methanol in co¬
lourless crystals of m. p. 91—92 °. For analysis it was sublimed under
high vacuum (block temperature 110—20°).
3.595 mg subst gave 9.823 mg C02 and 2.425 mg H20
Calcd. for C16H19NO2 C 74.68 H 7.44 %
Found C 74.57 H 7.55 %
2-Ethyl-4,5-dimethoxy-2'-dimethylaminobiphenyl (LII)
The biphenyl (LI) (186 mg) was methylated in the usual manner with
288 mg of dimethyl sulphate in the presence of 0.25 c. c. of 30 % sodium
hydroxide, when 70 mg of a colourless oil was obtained. The picrate from
this oil after three crystallisations from ethyl acetate had m. p. 208—10 °
(decomp.). For analysis it was dried for 36 hours at 25° under high
vacuum.
4.351 mg subst gave 8.959 mg CO2 and 2.020 mg H2O
Calcd. for C24H26O9N4 C 56.03 H 5.09 %.
Found C 56.19 H 5.20 %
The free base obtained from the picrate was distilled in a sublimation
tube at 0.01 mm (block temperature 90—95 °).
Its I. R. absorption spectrum showed bands in the neighbourhood of
865 cm-1 and 755 cm'1 indicating the presence of 1, 2, 4, 5 tetrasubstituted
and 1,2 disubstituted benzene nuclei respectively.
77

Studies in the Pictet-Spengler synthesis of tetrahydro-
isoquinoline derivatives and related compounds
(S- (3,4-Dihydroxyphenyl)-ethylamine (hill)
It was obtained by the demethylation of homoveratrylamine with hydro-bromic acid.
A solution of 10 g of homoveratrylamine in 5 c. c. of glacial acetic acid
was boiled under reflux at 120 ° with 65 c. c. of a 33 % solution of hydro-bromic acid (in glacial acetic acid) for three hours. On cooling, crystalsof the hydrobromide separated. These were collected and washed with
dry ether. Concentration of the filtrate gave a further crop of the hydro¬bromide. It was crystallised from a mixture of alcohol and ethyl acetate
giving 10.5 g (78 % yield) of colourless plates of m. p. 211—212° (re¬
ported»"
m. p. 212 °).
The picrate obtained from this hydrobromide by treatment with sodium
picrate had m. p. 189—90 ° which is identical with that of the correspon¬
ding picrate in literature 112.
l-Carboxy-6,7-dihydroxy-l-methyl-l,2,3,4-tetrahydroisoquinoline (L1V)
A solution of 562 mg (2.4 millimoles) of (3- (3,4-dihydroxyphenyl)-ethy¬lamine hydrobromide and 247 mg (2.8 millimoles) of pyruvic acid in
2 c. c. of water was adjusted to pH 4 by the addition of a few drops of
concentrated aqueous ammonia. The reaction mixture was kept at 25 ° for
five days, after which it was concentrated to about 1 c. c. under vacuum.
The white crystalline product which separated was washed with water
and acetone. It weighed 437 mg (70 % yield based on pyruvic acid). It
was crystallised from hot water in colourless crystals of m. p. 230—35 °
(with decomposition). It turned yellowish on repeated crystallisations and
gave an intense green colouration with ferric chloride solution.
111 G. Barger and A. J. Ewins, Soc. 1910, 2257—58.112 E.Waser and H. Sommer, Helv. 6, 61 (1923).
78

For analysis the substance was three times crystallised from hot water
and dried for 72 hours at 90' under high vacuum. It melted at 238—41 °
with decomposition (in an evacuated capillary) (reported113 m.p.230-—35°with decomposition).
3.724 mg subst gave 8.097 mg C02 and 1.956 mg H2O
Calcd. for C11H13O4N C 59.18 H5.87%
Found C 59.34 H 5.88 %
The U. V. absorption spectrum showed absorption bands with a
maximum at 288 m/j,, loge = 3.54 and at 228 ra.fi, logs = 3.76.
The I. R. spectrum is shown in fig. 6, curve 13.
On treatment with diazomethane in methanol-ether suspension, the
methyl ester of the dimethyl ether was obtained. It was distilled at
0.001 mm (block temperature 110—120°) as a viscous, colourless oil.
3.700 mg subst gave 8.597 mg CO2 and 2.409 mg H20
Calcd. for C14H19O4N C 63.38 H 7.22 %
Found C 63.41 H 7.29 %
l-Carboxy-6,7-dihydroxy-l-ethyl-l,2,3,4-tetrahydroisoquinoline (LV)
A solution of 562 mg (2.4 millimoles) of (3-(3,4-dihydroxyphenyl)-ethy-lamine hydrobromide and 286 mg (2.8 millimoles) of a-ketobutyric acid
in 2 c. c. of water was adjusted to pH 4 by the addition of a few drops of
concentrated aqueous ammonia. After keeping the reaction mixture for
four days at 27 °, 322 mg of a white crystalline substance separated. The
mother liquor was concentrated to about 1 c. c. under vacuum, yieldingfurther 148 mg of the product. It was washed with acetone and water to
remove any unreacted substance. In all, there was obtained 470 mg of the
desired product, m. p. 240—44° with decomposition.
For analysis it was three times crystallised from hot water giving
slightly yellow crystals. These were dried for 24 hours at 150 ° under highvacuum. The pure substance melted at 243—44° with decomposition (in
an evacuated capillary).
3.620 mg subst gave 8.051 mg CO2 and 2.067 mg H20
Calcd. for C12H15O4N C 60.75 H6.37%
Found C 60.69 H 6.39 %
113 G. Hahn and K. Stiehl, Ber. 69, 2643 (1936).
79

The substance was insoluble in acetone, ether, benzene and chloroform
but soluble in alcohol. It gave an intense green colouration with ferric
chloride solution. The I. R. spectrum is shown in fig. 6, curve 14.
The methyl ester of the dimethyl ether was obtained as a colourless,
viscous oil. It was distilled at 0.001 mm (block temperature 100—-110°).
3.704 mg subst gave 8.732 mg C02 and 2.476 mg H2O
Calcd. for C15H21O4N C 64.49 H 7.58 %
Found C 64.33 H 7.48 %
3 Keto-4',5'-dihydroxy-hexahydro-7,8-benzopyrrocoline-9-carboxylic acid
(LVI)
A solution of 3.3 g (14,2 millimoles) of fS-(3,4-dihydroxyphenyl)-ethyl-amine hydrobromide in 7 c. c. of water was mixed with a solution of 2.4 g
(16.4 millimoles) of a-ketoglutaric114 acid in 20 c. c. water. The solution
was filtered and heated on a steam-bath for eight hours. The pH of the
solution before heating was about 1. On cooling, 835 mg of a colourless
product separated. It was filtered and washed with acetone and water.
The mother liquor on heating further for 6 hours deposited 1.1 g of the
same substance which was slightly greenish in colour. In all, 1,935g (45 %
based on a-ketoglutaric acid) of the substance was obtained. It darkened
at 215 ° and decomposed at 255—260 °. It was insoluble in ether, benzene,
chloroform, acetone and ethyl acetate, and sparingly soluble in alcohol and
water.
For analysis the substance was dissolved in 18% hydrochloric acid, the
solution boiled with animal charcoal and filtered. The filtrate on cooling
deposited slightly yellow crystals. The above procedure was twice repeatedwithout the addition of charcoal and the crystals washed with cold water
till free from acid. The compound was dried for 72 hours at 70 ° under
high vacuum, when it melted at 252—55 ° with decomposition (in an
evacuated capillary); (reported115 m.p. 255—60°)
3.724 mg subst gave 8.066 mg CO2 and 1.656 mg H2O
Calcd. for C13H13O5N C 59.31 H 4.98 %
Found C 59.10 H 4.98 %
114 Prepared according to «Organic Synthesis*, Vol.26, p. 42 (1947).115 G.Hahn and K. Stiehl, Ber. 69, 2645 (1936).
80

The U. V absorption spectrum in IN aqueous hydrochloric acid showed
absorption bands with a maximum at 288 mp, loge —- 3.58 and at 360 m«
loge = 2.96.
4',5'-Dimethoxy-9-hydroxymethyl-hexahydro-7,8-benzopyrrocoline (LVII)
A suspension of 160 mg of the finely powdered lactam in 10 c. c. of
ether and 4 c. c. of methanol was treated with an ethereal solution of
diazomethane (from 1.1 g of nitrosomethyl urea). Nitrogen was briskly
evolved and on keeping overnight, a clear yellow solution was obtained.
Distillation of the solvents left 180 mg of a yellow oil.
To a well stirred mixture of 300 mg of lithium aluminium hydridein 20 c. c. of absolute ether was added dropwise a solution of the above oil
in 10 c. c. of absolute ether and 5 c. c. of dry benzene. After stirring for
one and half hours the reaction mixture was boiled under reflux for two
hours. The flask was cooled well and 25 c. c. of ice-water was added drop-
wise with stirring. The ether layer was separated and the aqueous layer
was extracted with three 10 c. c. portions of ether. After drying and distill¬
ing off ether, 90 mg of a colourless oil was obtained. It was treated with
an alcoholic solution of picric acid when the picrate at once separated.After three crystallisations from alcohol, it melted at 205—06 °. For ana¬
lysis it was dried for 48 hours at 60 ° under high vacuum.
3.852 mg subst gave 7.192 mg C02 and 1.628 mg H20
Calcd. for C21H24O10N4 C 51.22 H 4.91 %
Found C 50.95 H 4.74 %
The free base obtained from the picrate was distilled at 0.005 mm in a
sublimation tube (block temperature 130—40°).
The U. V. absorption spectrum showed bands with a maximum at
284 mu, loge — 3.54 and at 224 m/j,, loge = 3.9.
The I. R spectrum is shown in fig. 6, curve 15.
81

Attempted condensation of $-(3,4-dihydroxyphenyl)-ethylamine hydro-
bromide with a-ketoadipic acid.
A solution of 472 mg (2 millimoles) of (3-(3,4-dihydroxyphenyl)-ethyl-amine hydrobromide in 1 c. c. of water was mixed with a solution of
362 mg (2.26 millimoles) of a-ketoadipic acid116 in 3 c. c. water. The
mixture was filtered and heated on a steam-bath for 16 hours. The pH of
of the solution before heating was about 2. The colour of the solution
changed to brown. The heating was continued for eight more hours and
the solution evaporated to dryness under vacuum, when a gummy, partially
crystalline residue was obtained. A small amount of it was dissolved in
glacial acetic acid and heated with a solution of 2,4-dinitrophenylhydrazinein acetic acid containing a drop of hydrochloric acid. On cooling, a yellow
solid separated. It was crystallised from ethyl acetate in yellow prisms
of m. p. 205—06 °. Its melting point when mixed with the 2,4-dinitrophenyl-
hydrazone of a-ketoadipic acid showed no depression.
4.185 mg subst gave 6.537 mg CO2 and 1.365 mg H2O
Calcd. for C12H12O8N4 C 42.36 H 3.56 %
Found C 42.63 H 3.65 %
The micro-analyses were carried out at this Institute by Mr. W. Manser
to whom I am greatly indebted.
The I. R. absorption spectra were taken by Mr. A. Hiibscher and inter¬
preted by Dr. Hs. H. Giinthard. The pK, values were determined byMr. L. C. Chopard. I gratefully acknowledge the assistance received from
them.
116 Prepared acording to H. Gault, Compt. rend. 148, 1114 (1909); Chem. Zentr. /,1978 (1909).
82

SUMMARY
1) Dihydroerysotrine (dihydroerysodine methyl ether) on treatment
with cyanogen bromide formed a bromcyanamide which on reduction
with lithium aluminium hydride gave a base C18H21O2N. On the basis of
the experimental evidence, available at the beginning of this work, about
the structure of the Erythrina alkaloids, this base was believed to be
l-benzyl-6,7-dimethoxy-1.2.3,4-tetrahydroisoquinoline or 5-aza-4',5'-dime-
thoxy-3-phenyI-benzocycIoheptene which were therefore required for com¬
parison purposes.
2) l-Benzyl-6,7-dimethoxy-l,2,3,4-tetrahydroisoquinoline was synthesi-sed (flow-sheet 3) and was found to be different from the base obtained
from dihydroerysotrine.
3) The synthesis of 5-aza-4',5'-dimethoxy-3-phenyl-benzocycloheptenewas attempted by two different methods (flow-sheets 4 and 5). Althoughsome steps of the synthesis were successfully worked out in both cases, the
final product was not obtained.
4) According to the new structure proposed for the Erythrina alka¬
loids, the base C18H21O2N obtained by the cyanogen bromide degradationis represented as having a nine-membered ring system (flow-sheet 1). It
was expected that the oxidation of such a ring system would yield 4,5-
dimethoxydiphenyl-2,2'-dicarboxylic acid, but instead of the latter, another
compound was obtained which is probably 4,5-dimethoxydiphenyl-2,2'-diacetic acid. 4,5-Dimethoxydiphenyl-2,2'-dicarboxylic acid was synthesi-sed for comparison (flow-sheet 6).
5) The Hofmann degradation of apoerysopine was carried out accor¬
ding to the flow-sheet 2 and new structures were assigned to the various
Hofmann degradation products. Des-methyl apoerysotrine, dihydro-des-
83

methyl apoerysotrine, dihydro-des-dimethyl apoerysotrine and tetrahydro-
des-dimethyl apoerysotrine were prepared. l-Methyl-7-(l-ethyl-3,4-dime-
thoxyphenyl) indole was obtained by the dehydrogenation of dihydro-des-
methyl apoerysotrine.
6) To compare it with tetrahydro-des-dimethyl apoerysotrine, the syn¬
thesis of 2,3'-diethyl-4,5-dimethox)'-2'-dimethylaminobiphenyl was carried
out (flow-sheet 7). The two compounds were found to be different.
7) A study of the synthesis of uns>mmetrical biaryls by the Ullmann
reaction was made (flow-sheet 8). 3,3'-Dinitrobiphenyl, 3,3'-4,4'-tetra-
methoxybiphenyl, 3,4-dimethoxy-3'-nitrobiphenyl and 3,4-dimethoxy-2'-
nitrobiphenyl were prepared.
8) Compounds similar in structure to the Erythrina alkaloids were ob¬
tained by the application of the Pictet-Spengler reaction (flow-sheet 9).
l-Carboxy-6,7-dihydroxy-l-methyl-l,2,3,4-tetrahydroisoquinoline, 1-car-
boxy-l-ethyl-6,7-dihydroxy-l,2,3,4-tetrahydroisoquinoline and 4',5'-dime-
thoxy-9-hydroxymethyl-hexahydro-7,8-benzopyrrocoline were synthesised.
84

CURRICULUM VITAE
I was born on 13th June 1922, at Bombay, the son of
Ranchoddas and Motiben of Bombay, India.
After attending the St. Xaviers High School at Bombay,
I studied at the Elphinstone College and the Royal Institute of
Science and obtained my B. Sc. degree in 1944 with Chemistry
and Physics as my optional subjects.
From 1944 to 1950, I carried out research work in organic
chemistry at the Royal Institute of Science, Bombay, from
where I received the M. Sc. and the Ph. D. degrees of the
Bombay University in 1947 and 1950 respectively.
From September 1950 until now, I have been working on
the present problem under the supervision of Prof. Dr. V.
Prelog at the Swiss Federal Institute of Technology.
Zurich. 1st December 1952.
85



![nanodiagnostics.ppt [Schreibgeschützt] [Kompatibilitätsmodus] · Sr + Benzyl Alcohol + Ti(OiPr) 4 SrTiO 3 Li + Benzyl Alcohol + Nb(OEt) 5 LiNbO 3 Ba,Sr + Benzyl Alcohol + Ti(OiPr)](https://img.pdfslide.us/doc/110x75/6059c0a096e778411b20081e/schreibgeschtzt-kompatibilittsmodus-sr-benzyl-alcohol-tioipr-4-srtio.jpg)


![Index [ ] · PDF fileacetylation 962–964 – one-pot 981 ... benzyl alcohol 822–823, 1177 benzylamines 155 – substituted 828 benzyl chloride 253](https://img.pdfslide.us/doc/110x75/5a85803e7f8b9a14748c25ad/index-962964-one-pot-981-benzyl-alcohol-822823-1177-benzylamines.jpg)






















