Embed Size (px)
Citation preview

Ribosome-inactivating Proteins: Ricin and Related Proteins, First Edition. Edited by Fiorenzo Stirpe and Douglas A. Lappi.
© 2014 John Wiley & Sons, Inc. Published 2014 by John Wiley & Sons, Inc.
134
9 Updated Model of the Molecular Evolution of RIP GenesWilly J. Peumans1, Chenjing Shang2, and Els J. M. Van Damme2
1 Aalst. Belgium2 Department of Molecular Biotechnology, Ghent University, Belgium
Introduction
Though numerous RIPs from diverse origin have intensively been studied for several decades, the molecular evolution and overall phylogeny of genes/proteins with a RIP domain is still poorly understood. Previous attempts to corroborate this issue allowed tracing quite accurately the phylo-genetic relationships within a few subsets of RIPs, but did not provide a complete picture, the main reason being that most analyses were confined to entries from protein databases. Such an approach might be suitable for many other proteins but is, for the time being, inefficient when applied to RIPs, for two main reasons. First, the protein databases cover only a relatively small fraction of all cur-rently identified RIP genes. Second, the sequence of many annotated RIPs is incorrect. This incon-venience applies particularly to putative/hypothetical RIPs identified in completed genomes of especially grass species. Automated annotation programs often fail to extract correct sequences due to the occurrence of (i) pseudogenes with truncated open reading frames (ORFs), (ii) genes inter-rupted by a transposable element, and (iii) genes with one or more introns. The low performance of the automated annotation systems is not due to the programs themselves but can be ascribed to a lack of (i) sufficient transcriptome sequences and (ii) (correct) sequences of homologous proteins. Apart from erroneously annotated sequences, the NCBI protein database suffers from what can be called “database pollution.” A group working on “Cloning and Sequence Analysis of Six Novel Genes of Ribosome Inactivating Proteins from Different Plants” deposited five sequences (from the dicots Nicotiana tabacum and Senna occidentalis, and the monocots Agave sisalana, Alocasia mac-rorrhizos, and Musa acuminata) in the NCBI database that are virtually identical at the nucleotide level to part of the gene encoding β-luffin, a type 1 RIP from the Cucurbitaceae species Luffa acu-tangula. Taking into account that β-luffin shares only 90% sequence identity with its conspecific paralog α-luffin, it is highly improbable that nearly identical sequences occur in unrelated species, which implies that there must have been a contamination of materials. This rather unfortunate case of database pollution is not just anecdotal but has important consequences. For example, in a recent review on RIP1 the aforementioned “ghost sequences” cluster in a totally anomalous way in the Cucurbitaceae branch of the dendrogram, creating the wrong impression that the classical

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 135
Cucurbitaceae type 1 RIPs are highly conserved within the whole group of flowering plants. Closer observation reveals another important anomaly in the same dendrogram. Only three Oryza (rice) proteins are included, whereas a much larger number of RIP genes were identified in Oryza sativa (japonica).2 Evidently, the omission of all but three rice RIPs results in a highly biased dendrogram that does not allow the drawing of definitive conclusions with respect to the overall phylogeny and evolution of RIP genes. This certainly does not imply that all previous attempts to unravel the phy-logeny and evolution of the RIP domain are worthless. On the contrary, they provide a good starting point for a renewed approach whereby care is taken to include an as complete as possible set of correct sequences.
Apart from the bias inherent in the use of an incomplete set of sequences, and the discrepancies resulting from the inclusion of incorrect or ghost sequences, most attempts to corroborate the molec-ular evolution of RIPs were primarily focused on a sequence-based phylogenetic analysis and paid too little attention to the genetic events – in a broad sense – that shaped the overall composition and taxonomic distribution of the RIP gene family. Evidently this is rather unfortunate because RIP gene evolution cannot be uncoupled from other aspects like RIP gene expression and the “general biology” of the expressed proteins (including topogenesis and intracellular location, activity, and function). Therefore, the present contribution aims to discuss the evolution of the RIP gene family against the background of our current knowledge of both the genes and the corresponding protein products. After a brief summary of some issues of RIP research that provide crucial information for what concerns the outlines of the evolution of the gene family, an overview is given of the most important novel insights generated by a comprehensive in silico analysis of genome and transcrip-tome data. This analysis (i) allowed updating of the RIP gene family in terms of domain architecture, (ii) provided sequence-based evidence for the generation of at least two different lineages of plant type [A] RIPs from chimeric forms, and (iii) revealed marked differences between the RIP gene families found in the completed plant genomes. The latter information generated novel insights into the phylogeny of plant RIPs, which in turn allowed the building of an updated model for the evolu-tion of the plant RIP gene family and in a next phase of the whole RIP family including those found in bacteria, fungi, and insects.
Important Issues to be Considered in View of the Evolution of RIP Genes
Prior to elaborating the details of the (sequence-based) phylogeny and evolution, a few aspects of the “general biology” of RIPs are discussed that might be relevant to identifying (some of) the driving forces and molecular mechanisms that shaped the eventual form of the present-day RIP gene family.
The RIP Domain is not Ubiquitous in Plants
The obvious absence from many plant genomes (see Chapter 2) demonstrates that the RIP domain is certainly not ubiquitous in plants, which in turn implies that modern RIPs are not required for normal plant growth and development but are merely accessory proteins involved most probably in some defense-related processes. Accordingly one can expect that: (i) there is no strong selective pressure on the RIP genes, which in turn might result in a high mutation rate, (ii) functional RIP genes can readily be converted into pseudogenes, and (iii) RIP genes can be lost from a genome without major consequences for the species.

136 RIBOSOME-INACTIVATING PROTEINS
Occurrence and Abundance of RIP Domains in Plants
RIPs are not associated with (a) particular tissue(s) but are found in virtually all plant parts. Both the distribution over different tissues and the abundance are highly variable depending on the species. This atypical behavior indicates that the expression of RIP genes is under the control of different promotors. Moreover, the expression of (slightly) different homologs in different tissues of the same species might be indicative for the occurrence of gene families resulting from gene dupli-cation events.
Differences in Topogenesis Among Plant RIPs
Most plant RIPs studied thus far are synthesized on the rough endoplasmic reticulum (RER) and follow the secretory pathway. However, a few RIPs from grass species (barley, wheat, maize) are synthesized on free ribosomes in the cytoplasm and presumably reside in the nucleocytoplasmic compartment. Based on this observation it can be expected that the acquisition/loss of targeting sequences (signal peptide, vacuolar retention signal) played an important role in RIP gene evolution.
Non-Plant RIPs
A survey of the data published on bacterial RIPs yields three important indications with respect to the evolution of the corresponding gene family within the prokaryotes. First, the (documented) occurrence of RIP genes is very limited (in terms of taxonomical distribution). Second, the recent identification and characterization of a type [A] RIP from Streptomyces coelicolor that is more closely related to some plant RIPs than to the Shiga toxins demonstrates that bacterial RIPs do not form a monophyletic gene family.3 Third, the Shiga toxin genes are located in the genome of lambdoid prophages, which behave as highly mobile genetic elements and play a role in horizontal gene transfer.4
To date, no information has been reported that could be relevant for the evolution of the RIP gene family in fungi and insects. The only, but highly useful, information concerns the very limited (in terms of taxonomical distribution) occurrence of the RIP domain in both taxa (see Chapter 2).
Dissecting RIP Genes and RIP Gene Families by in silico Analysis of Genome and Transcriptome Data
The RIP Gene Family Revised in Terms of Domain Architecture
As pointed out in Chapter 2, the in silico identification of several novel forms urges a revision of the classic subdivision (in terms of Shiga toxins and type 1, type 2, and type 3 RIPs) and adoption of a novel classification system based primarily on the domain architecture of the RIP genes. Following this criterion, two main subclasses can be distinguished. A first subclass comprises those genes that consist exclusively of a RIP domain either, or not, accompanied by short N- and/or C-terminal extensions. Genes in which the RIP domain is fused to an unrelated (identified or unidentified) domain are considered chimeric genes. This subdivision based on the structure of the genes has the advantage that it can be applied without knowledge of the final protein products. For the sake of

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 137
clarity, the definition of a RIP gene applies also to prokaryotic polycistronic genes (genes in which a single promotor directs the synthesis of an mRNA that can be translated into multiple proteins). For example, dicistronic genes encoding a Shiga toxin are classified here as chimeric genes (even though the final toxins are assembled from polypeptides translated from two distinct mRNAs).
A schematic overview of the different types of RIP genes identified thus far is summarized in Table 9.1. The novel nomenclature/classification presented in this table takes into account the evo-lutionary diversity (which is discussed in detail in the next sections) of especially the heterogeneous group of type [A] RIPs, and whenever applicable refers to the previous classification in terms of type 1, type 2, and type 3 RIPs. Plant type [A] RIPs are now subdivided in [AΔB], [AΔX], and [Au]. The terms [AΔB] and [AΔX] refer to the fact that these type [A] RIPs originated from type [AB] and type [AX] chimers by deletion (Δ)of the B and X-domain, respectively. Type [A] RIPs, from unknown evolutionary origin, are classified in the [Au] subgroup, which does not refer to a single phylogenetic line but groups all type [A] RIPs other than [Ac] and [Ax]. To distinguish the fungal [AX] and [AΔX] homologs from their plant counterparts they are referred to as [AXf] and [AΔXf], respectively. Since the evolutionary origin of bacterial RIPs as well as their phylogenetic relation-ships are still poorly understood, a definitive classification can hardly be made. Shiga and Shiga-like toxins are indicated as type [As]-[Bs] RIPs (the superscripts referring to Shiga). Bacterial RIPs corresponding to the single A chain of the Shiga toxins are classified as type [As] RIPs. All other bacterial RIP domains are referred to as [A] without any further specification.
Table 9.1 Overview of documented domain architectures of RIP genes and genes derived thereof
by a domain deletion.*
Plants
[AΔB]** Type [A] derived from type [AB] RIP (most classical type 1 RIPs)
[AΔx] Type [A] derived from type [AX] RIP (some classical type 1 RIPs; e.g., muscarin)
[Au] Type [A] from undefined origin (e.g., maize RIP b-32; described as type 3 RIP)
[AB] Chimer with C-terminal ricin-B domain (type 2 RIPs)
[AC] Chimer with C-terminal (unknown) C domain (JIP 60; type 3 RIP)
[AD] Chimer with C-terminal (unknown) D domain
[AX] Chimer with C-terminal (unknown) X domain
[APM41] Chimer with C-terminal peptidase M41domain
[APC19] Chimer with C-terminal peptidase C19 domain
Fungi
[AΔxf] Fungal homolog of plant [AΔx] RIP
[AXf] Fungal homolog of chimeric plant [AX] RIP
Insects
[AI] Chimer with C-terminal (unknown) I domain
Bacteria
[A] Bacterial homolog of plant type [A] RIP (Streptomyces coelicolor RIPs)
[As] Type [A] equivalent to Shiga toxin A-subunit
[As]-[Bs]° Shiga and Shiga-like toxins
[As]-[U]° Chimer with unknown C-terminal domain
[As]-[P]-[W]° Chimer with PMT-C3 and unknown C-terminal domain
[AM] Chimer with C-terminal (unknown) M domain
*Part of this table is also presented in Chapter 2. For a schematic representation of the domain
architectures, see Figures 2.2, 2.5, 2.6, and 2.7 of this chapter.
**Domain (s) between square brackets are synthesized on a single mRNA
°Polycistronic gene

138 RIBOSOME-INACTIVATING PROTEINS
An important remark to be made is that Table 9.1 presents only an overview of the currently iden-tified domain architectures and certainly does not claim to be complete or final. On the contrary, one can expect that as soon as more genomes are completed, novel RIP gene architectures will be iden-tified (especially in plants).
Sequence-Based Evidence for the Generation of Type [A] RIPs from Type [AB] and [AX] Chimers
In a previous review, it was suggested on the basis of a phylogenetic analysis that most type 1 RIPs found in dicots are derived from type [AB] RIPs.5 Now, detailed comparisons of genomic sequences confirm this hypothesis. Two cases nicely illustrate how a type [AB] RIP gave rise to type [AΔB] with a different subcellular location. First, one of the type [AΔB] RIPs from Jatropha curcas has a C-terminus that closely resembles the C-terminus (including the QxW signature) of the Euphorbiaceae type [AB] RIPs. Sequence alignment with a conspecific type [AB] pseudogene (with a frame shift near the C-terminus of the A domain) revealed that the type [A] sequence can be perfectly aligned with the type [AB] sequence from which the entire B domain, except a short sequence at both the N- and C-terminus, is deleted (Figure 9.1). Second, sequence alignment of a
Signal peptide
A domain B domain[AB] pseudogene
Major deletion
Major deletion
Major deletion
Out of frame
StopOut of frame: signal peptide lost
[AB] gene
[AB] gene
[AΔB] gene
[AΔB] gene
[AΔB] protein
[AΔB] protein with
C–terminal QxW
[ΔAB] gene
[ΔAB] protein
Jatropha curcas
Malus domestica
Sambucus nigra
Figure 9.1 Documented domain deletion events that illustrate the conversion of an [AB] chimer into an [AΔB] and [ΔAB] gene.

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 139
type [AΔB] and a type [AB] RIP gene from Malus domestica demonstrated that after deletion of the C-terminus of the A domain and the entire B-domain, minus the last amino acids at the C-terminus, both sequences can readily be aligned at the nucleotide level. However, due to the introduction of two frame shifts, the type [AΔB] RIP lacks the signal peptide as well as the C-terminus of the type [AB] RIP (Figure 9.1). As pointed out previously for Sambucus nigra, a type [AB] can also be con-verted into a gene encoding a ricin-B lectin chain only through the deletion of the entire A domain (minus the N-terminus) and the linker between the A and B domains (Figure 9.1).6
Similar domain deletion events can be traced for type [AX] genes. Alignment of cDNAs from Elaeocarpus photiniifolius revealed that a type [AΔX] sequence can be aligned perfectly with a con-specific type [AX] sequence from which the entire X domain, minus a short sequence at both the N- and C-terminus, is deleted (Figure 9.2). Due to a frame shift, the type [AΔX] RIP lacks the C-terminal part of the type [AX] RIP. A domain deletion event, whereby an [AX] gene was con-verted into an [X] gene by deletion of the entire A domain except the first 8 N-terminal amino acid residues, could be traced in the cacao (Theobroma cacao) genome (Figure 9.2).
At present, no examples can be identified of type [A] RIP genes that might be derived from type [AC], [AD], or [AP] chimers. However, it cannot be excluded that some grass type [Au] RIP gene(s) evolved from one of these chimers through a deletion of the C-terminal domain.
Besides domain deletion events, type [A] genes can also be generated by “truncation” of [AB], [AX], and possibly also [AC] and [AD] chimers through “more simple” genetic events, like the introduction of a stop codon or a frame shift or the insertion of a transposable element. However, it is not always clear whether the truncated genes behave as pseudogenes or control the expression of a corresponding truncated protein.
Out of frame
Stop
Deletion of most of the A domain
[AX] gene
[AX] gene
[AΔx] gene
[ΔAX] gene
Truncated [AX] gene
(expressed)
Truncated [AΔx] protein
[AΔx] protein
[ΔAX] protein
Elaeocarpus photiniifolius
Theobroma cacao
Populus trichocarpa
A domain X domain
Deletion of most of the X domain
Figure 9.2 Documented domain deletion events that illustrate the conversion of an [AX] chimer into an [AΔX] and [ΔAX] gene.

140 RIBOSOME-INACTIVATING PROTEINS
One particular example of such a truncated gene deserves some special attention. The genome of Populus trichocarpa possesses an [AX] gene that lacks the second half of the X domain. It is ques-tionable whether this gene is expressed in P. trichocarpa because not a single EST can be found in the transcriptome. However, two cDNAs, which were sequenced at both the 5′ and 3′ end (Populus tremula x Populus tremuloides cDNA clone UR108TF08 and Populus nigra cDNA clone PnFL2-100_P09) comprise both an A and a C-domain, and hence correspond to genuine RIP [AX] orthologs. One possible explanation for this discrepancy is that P. tremula x Populus tremuloides and P. nigra possess a functional [AX] gene that is present in a truncated (and possibly inactive) form in P. trichocarpa.
Survey of RIP Genes and RIP Gene Families in Completed Plant Genomes
Since the number of identified genes largely surpasses that of the RIP proteins studied thus far (see Chapter 2) a reconstruction of the outlines of the molecular evolution should be based preferentially on a phylogenetic analysis of the RIP genes/gene families found in the completed genomes. Therefore, a comprehensive analysis was made of the RIP gene complement in all completed plant genomes. The results of this analysis are schematically presented in Figure 9.3 except for the grass genomes. For the latter species the data are still too preliminary for an in-depth phylogenetic anal-ysis. However, one grass species namely rice (Oryza sativa) will be discussed below in some detail because of the unique complexity and plasticity of the Oryza RIP gene family.
An overview of the data presented for the dicots and the monocot Phoenix dactylifera (Figure 9.3) allows the drawing of several important conclusions. First, the RIP gene family in flowering plants is very heterogeneous with respect to both complexity (in terms of gene number) and heterogeneity (in terms of domain architectures). Even with a single family there are striking differences between individual species (e.g., Euphorbiaceae). Second, pseudogenes lacking an ORF due to nonsense mutations, frame shifts, or insertion of transposable elements are quite common. In some instances it is difficult to figure out whether a gene is a pseudogene or a gene encoding a truncated protein. For example, the Populus trichocarpa genome comprises an (expressed) gene that corresponds to a typical [AX] gene but lacks almost the entire X domain. Third, the number of loci harboring (mul-tiple) RIP domain(s) is highly variable. Most loci comprise a single RIP gene, but there are also loci with a more or less complex set of (duplicated) genes (Figure 9.3). For example, most Ricinus communis RIP genes are located in clusters. Similarly, all four Prunus persica [AΔB] RIP sequences are tandemly arrayed in a single locus. Fourth, the obvious heterogeneity in gene number and domain architecture argues for a rather complex overall evolution of the plant RIP gene family. Fifth, all grass genomes apparently harbor a complex set of RIP genes. Unfortunately, the annotation of these genes is still very preliminary and incomplete (in the sense that many genes are not recognized as RIP genes and numerous others are attributed an incorrect sequence). To get an accurate idea about the RIP complement of at least one grass species a detailed (manual) comparative analysis was made of the genomes of Oryza sativa (indica and japonica) and O. glaberrima.
The Oryza Genome Harbors a very Extended RIP Gene Family
A total of 56 different loci were identified in the combined genomes of O. sativa and O. glaberrima that harbor a RIP gene or pseudogene. Analysis of the sequences yielded, for 44 genes, at least one ortholog comprising a coding sequence (CDS) covering a full size RIP-protein. For four other genes

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 141
only ortholog(s) with a CDS corresponding to a protein that is slightly truncated (as compared to the most closely related paralog/homolog) at either the N- or C-terminus but still covering a complete RIP domain, could be identified. In seven other cases no ortholog(s) with a (nearly) complete CDS could be retrieved. However, a complete CDS could be “reconstructed” by either a small change in
Clustered genes
Signal peptide
Poaceae species:Further details are required toestablish the relationshipsbetween the Poaceae RIP genes
Brachypodium distachyon: 15 Au; 1[AC]; 1 [AP]Oryza sativa: >30 Au; 3[AC]; 1 [AP]Setaria italica: >5 Au; 2[AC]Sorghum bicolor: 14 Au; 1 [AB]Zea mays: 7 Au; 2 [AB]; 1[AC]; 1 [AD]
Phoenix dactylifera*
Citrullus lanatus*
Cucumis sativus
Prunus persicaMalus domestica*
Populus trichocarpa Cannabis sativa*
Ricinus communis
Jatropha curcas*Manihot esculenta
Gossypium raimondii Theobroma cacao
Cucumis melo*
Aquilegia coerulea*
Pseudogene
* Assembly does not allow tracing possible RIP linkage
[AΔx][AX][AB] [ΔAX][AΔB] [ΔAB]
Figure 9.3 Schematic representation of the RIP genes found in completed plant genomes.

142 RIBOSOME-INACTIVATING PROTEINS
the nucleotide sequence (e.g., insertion/deletion/substitution of a single nucleotide to remove a frame shift or convert a stop codon in a sense codon) or removal of a transposable element. Due to the absence of a CDS, the latter genes can be considered pseudogenes.
The rice RIP gene family can be subdivided in three main groups on the basis of the domain architecture of the protein products. Fifty-one genes encode proteins in which, besides the A domain, no other con-served domain can be recognized, and accordingly can be classified as a type [A] RIP. However, it should be emphasized that in many of these presumed type [A] RIPs the A domain is accompanied by short or middle long (up to 100 amino acid residues) extensions at the N- and/or C-terminus. Four other genes definitely display a chimeric domain architecture (3 [AC] and one [AP] form).
At present the phylogeny of the Oryza RIP gene family has not been worked out in detail. However, a preliminary dendrogram (Figure 9.4) gives a good idea of (i) the complexity of the gene family, (ii) the occurrence of multiple clades, and (iii) the distribution all over the genome. For the sake of clarity, the identification of the genes is based on the position of the genes on the chromosomes and the domain architecture of the protein products. The dendrogram shown in Figure 9.4 differs quite substantially from that reported by Jiang et al. (2008), who made a genome-wide survey of the RIP domain in Oryza sativa, but confirms that genome-wide and tandem duplications are at the basis of the large RIP gene number.
Similar analyses indicate that the Sorghum bicolor, Zea mays, Brachypodium distachyon, and Setaria italica RIP gene families are less complex. In contrast, the RIP gene families in Hordeum vulgare and Triticum aestivum might be of a complexity comparable to that of Oryza. Irrespective of the remaining uncertainty, one can reasonably conclude that the complexity of the RIP gene family in Poaceae species exceeds that found in dicots and date palms. Moreover, it seems that the evolution of the RIP gene family within the Poaceae followed a pathway well distinct from that of the dicots and the non-Poaceae monocots.
New Insights in the Overall Phylogeny of Plant RIPs
Genome and transcriptome sequence information deposited in the databases after completion of the phylogenetic analysis reported in our previous review,5 revealed that the evolutionary model pro-posed at that time needs to be updated and refined. To highlight the improvements, it is summarized here to what extent the major issues of our previous model built from the sequences available until 2009 still hold true or have to be updated or revised.
Issue 1: it was proposed that an ancestor of modern seed plants developed the RIP domain at least 300 million years (Myr) ago. This conclusion still holds true.
Issue 2: it was proposed that an ancestral RIP domain gave rise to (i) a direct lineage of type 1 RIPs (named primary type 1 RIPs that are still found in many monocots and at least one dicot), and (ii) an ancestral type 2 RIP through fusion with a duplicated (prokaryotic) ricin-B domain.
The presumed direct lineage of type 1 RIPs might still exist but is not represented by the so-called primary type 1 RIP. Until recently, the only identified member of this lineage within the dicots was a presumed type 1 RIP from Populus sp. Now it appears that closely related type [A] proteins are expressed in several other dicots (e.g., Fagus sylvatica, Cannabis sativa, Elaeocarpus photiniifolius). Moreover, several species (e.g., cannabis, cacao, Elaeocarpus photiniifolius) express one or more chimeric proteins consisting of an N-terminal domain sharing high sequence similarity with the “primary type 1 RIP” fused to an unknown C-terminal domain. Though these chimeric proteins are to a certain extent reminiscent of the jasmonate responsive type [AC] RIP

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 143
Oryg03.A8Oryg03.A11Oryg03.A6Oryg03.A9Oryg03.A12Oryg03.A10Oryg03.A13Oryg03.Aps2Oryg03.A7
Oryg02.A1Oryg04.A1
Oryg10.A4
Oryg10.A1Oryg10.A2Oryg10.A3Oryg03.A1Oryg03.A3Oryg03.Aps1Oryg03.A5Oryg03.A2Oryg03.A4Oryg07.A3Oryg01.A1Oryg01.A2Oryg07.Aps1Oryg07.A4Oryg07.A5Oryg11.A1Oryg11.A2
Oryg12.A1Oryg04.A2
Oryg08.A4Oryg08.A3Oryg08.A1Oryg08.A2Oryg02.A2Oryg02.A3Oryg12.A3
Oryg11.AC3Oryg11.AC1Oryg11.AC2
Oryg11.A4
Oryg11.A3
Oryg11.A5Oryg12.A2
Oryg12.A5Oryg12.A6
Oryg05.A1Oryg09.A1Oryg01.AP1Oryg04.A3
Oryg07.A2
Oryg12.A4
Figure 9.4 Phylogenetic tree of the rice RIP gene family. Genes are indicated by their relative position on the chromosomes.
Dendrogram was rendered using Mega5.7

144 RIBOSOME-INACTIVATING PROTEINS
found in barley, the overall sequence similarity between both proteins is rather low, especially in the C-terminal domain. Therefore, these newly identified chimers are considered members of a novel type of RIPs, further referred to as the type [AX] RIP. Phylogenetic analyses leave no doubt about the common origin of the “primary type 1 RIPs” and the [A] domain of the [AX] chimers. Moreover, both the phylogeny and taxonomic distribution indicate that what we believed to rep-resent “primary type 1 RIPs” is in fact a second lineage of secondary type 1 RIP (further referred to as type [AΔX]) derived from type [AX] RIP through a domain deletion event (Figure 9.2). Besides in dicots, type [AΔX] RIPs and/or corresponding genes were also identified in monocots. Some of these monocot type [AΔX] RIP have been isolated and (partly) characterized like asparin 1 and 2 from Asparagus officinalis,8 the musarmins from Muscari armeniacum9 and charybdin from Charybdis maritima.10
At present, there are no data that challenge the proposed origin of an ancestral type 2 RIP gene through fusion of an A domain with a duplicated (prokaryotic) ricin-B domain.
Issue 3: the ancestral type 2 RIP gave rise to (i) a monophyletic line covering all modern type 2 RIPs, and (ii) different lines of “secondary” type 1 RIPs and ricin-B type lectins (through domain deletion events). Both conclusions remain valid. Moreover, sequence-based evidence was obtained for several independent domain deletion events (Figure 9.1).
Issue 4: at least three other domain fusions took place in the recent past within the family Poaceae, whereby type AC1 (type 3), type AC2, and type AD chimeric forms were generated. The pre-sumed origin within the family Poaceae of three other chimeric forms through recent independent domain fusions needs to be revised in view of the identification of the [AX] RIP lineage. Though the Poaceae [AC] and [AD] types do not share a high sequence identity with the [AX] RIP, the residual similarity might be indicative for a common origin. Accordingly, the Poaceae [AC] and [AD] types might have evolved directly from an early Poaceae [AX] form. Evolutionary events comparable to those observed for the dicot [AX] genes can explain the occurrence in some grasses of proteins with a C or D domain and, what is more important, might have given rise to still other lineages of secondary type 1 RIPs.
An additional but important novel insight concerns the identification in some grass species of chimeric RIPs with a C-terminal peptidase domain. In Oryza sativa the RIP domain is fused to a peptidase M41 domain (RIP [APM41]) and in Brachypodium distachyon and Triticum aestivum to a peptidase C19 domain (RIP [APC19]). Both the rice RIP [APM41] and wheat RIP [APC19] are expressed, indicating that these grass species do synthesize (cytoplasmic) proteins with a (putative) dual N-glycosidase/protease activity. At present, there is no experimental evidence in support of the pos-sible protease activity of the expressed proteins. However, since the canonical metal binding and catalytic HEXXH motif of the peptidase M41 family is conserved in rice RIP [APM41], and all four active site residues of the peptidase C19 domain are strictly conserved in wheat RIP [APC19], there is a reasonable chance that both RIP-protease chimers possess proteolytic activity. Evidently, the identification of these two novel previously unconceived domain architectures widens the range of possible biological activities/functions of plant RIPs and increases the complexity of the evolution of the plant RIP family.
An Updated Model of the Molecular Evolution of the Plant RIP Gene Family
The novel insights in the taxonomic distribution and overall phylogeny render our recently proposed model5 of the molecular evolution of the plant RIP gene family outdated. Part of the original scheme remains valid but three major novelties need to be incorporated in the new model: (i) the occurrence
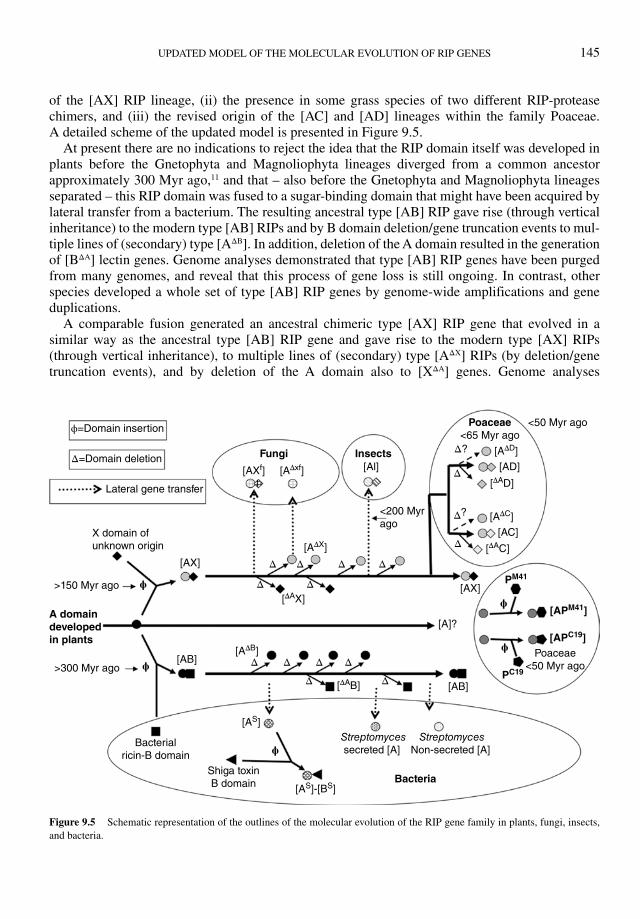
UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 145
of the [AX] RIP lineage, (ii) the presence in some grass species of two different RIP-protease chimers, and (iii) the revised origin of the [AC] and [AD] lineages within the family Poaceae. A detailed scheme of the updated model is presented in Figure 9.5.
At present there are no indications to reject the idea that the RIP domain itself was developed in plants before the Gnetophyta and Magnoliophyta lineages diverged from a common ancestor approximately 300 Myr ago,11 and that – also before the Gnetophyta and Magnoliophyta lineages separated – this RIP domain was fused to a sugar-binding domain that might have been acquired by lateral transfer from a bacterium. The resulting ancestral type [AB] RIP gave rise (through vertical inheritance) to the modern type [AB] RIPs and by B domain deletion/gene truncation events to mul-tiple lines of (secondary) type [AΔB]. In addition, deletion of the A domain resulted in the generation of [BΔA] lectin genes. Genome analyses demonstrated that type [AB] RIP genes have been purged from many genomes, and reveal that this process of gene loss is still ongoing. In contrast, other species developed a whole set of type [AB] RIP genes by genome-wide amplifications and gene duplications.
A comparable fusion generated an ancestral chimeric type [AX] RIP gene that evolved in a similar way as the ancestral type [AB] RIP gene and gave rise to the modern type [AX] RIPs (through vertical inheritance), to multiple lines of (secondary) type [AΔX] RIPs (by deletion/gene truncation events), and by deletion of the A domain also to [XΔA] genes. Genome analyses
ϕ=Domain insertion
Δ=Domain deletion
Lateral gene transfer
X domain ofunknown origin
[AX]
[A]?
[AB]
[AX]
[Al]Fungi
Bacteria
A domain
developed
in plants
Insects
[AB]
Shiga toxinB domain
Bacterialricin-B domain
Streptomycessecreted [A]
StreptomycesNon-secreted [A]
[AS]
[AS]-[BS]
>150 Myr ago
<50 Myr ago
Poaceae<50 Myr ago
Poaceae
<65 Myr ago
<200 Myrago
>300 Myr ago
ϕ
ϕ
ϕ
ϕ
ϕ
[AΔX]
[AΔD]
[AΔC]
[APM41]
PM41
[APC19]
PC19
[ΔAD]
[ΔAC]
[AD]
[AC]
[AΔxf][AXf]
[ΔAX]
[ΔAB]
[AΔB]
Δ
Δ Δ
Δ Δ ΔΔ Δ
Δ
Δ
Δ?
Δ
Δ
Δ
Δ
Δ
?
Figure 9.5 Schematic representation of the outlines of the molecular evolution of the RIP gene family in plants, fungi, insects,
and bacteria.

146 RIBOSOME-INACTIVATING PROTEINS
demonstrated that the [AX] RIP genes also have been, and are still being, purged from many genomes. Though both the [AB] and [AX] RIP lineages result from a comparable domain fusion event and follow a very similar evolutionary pathway there is – apart from the nature of the C-terminal domain – a very important difference. All type [AB] RIPs are synthesized on the RER and follow the secretory pathway, whereas all type [AX] RIPs are synthesized on free ribosomes and hence remain in the cytoplasmic/nucleoplasmic cell compartment. Evidently, this obvious difference in topogenesis and subcellular location profoundly affects the possible in planta activity of the proteins and strongly suggests that the [AB] and [AX] RIPs fulfill a completely different function. In general, the [AΔB] and [AΔX] RIPs retain the subcellular location of their respective par-ent chimers. However, there are exceptions. In Malus domestica the deletion of the A domain from an [AB] RIP is accompanied by a frame shift in the signal peptide resulting in an [AΔB] RIP with a cytoplasmic location (unpublished data). The reverse must have taken place to generate the vacuolar [AΔX] RIP found in Muscaria armenica and other Hyacynthaceae, but the presumed process of the introduction of a signal peptide cannot be traced in the available sequences.
At present, the origin of the ancestral [AX] RIP gene cannot accurately be located in time, but on the basis of the actual taxonomic distribution of the [AX] and [AΔX] RIPs, it predates the separation of the monocots and dicots (approximately 140–150 Myr ago12).
The identification of the [AX] RIP gene family also urges us to readdress the origin/evolution of the type [AC] and [AD] RIP genes, which both seem to be confined to the family Poaceae. Most probably these chimeric forms do not result – as was previously suggested5 – from two independent domain fusion events within the family Poaceae, but evolved directly from an early grass [AX] RIP. This explains why the [AC] and [AD] RIPs have the same subcellular location as the [AX] RIPs, and also accounts for the generation through deletion of the A domain of [CΔA] and [DΔA] genes in several grass genomes. Both [AC] and [AD] type RIPs share only a low sequence identity with the dicot and date palm [AX] RIP, but the residual similarity points towards a common origin. A pos-sible explanation is that – as can be concluded from the above described analysis of the Oryza RIP gene family – the RIP gene family evolved at an enhanced rate in Poaceae species. At present it is not clear whether deletion of the A domain from [AC] and [AD] RIP genes yielded [AΔC] and [AΔD], genes, respectively, but some of the Poaceae type [A] RIPs of unknown origin ([Au]) might belong to the latter types.
The large size and heterogeneity of the Poaceae RIP gene families, combined with the pro-nounced differences between individual species, render an accurate reconstruction of the evolution within this particular taxonomic group quite cumbersome. Unlike in dicots, the origin of the numerous Poaceae [Au] RIPs remains unclear. Most probably, some of these [Au] RIP genes resulted from a domain deletion in an [AC] or [AD] chimer, but it cannot be excluded that others descended in a direct way from the very first ancestral plant RIP gene (comprising a single RIP domain). If so, the latter [Au] RIPs can be considered primary type 1 RIPs that represent the eventual outcome of a RIP gene lineage that leads from the very first plant RIP gene to modern grasses, but was apparently discontinued in all other taxonomic groups. Hopefully, a thorough comparative phylogenetic anal-ysis of the RIP gene families in the currently completed grass genomes, and some additional genomes to be completed or released, will answer this critical question.
A final issue of the Poaceae RIP gene family concerns the [APM41] and [APC19] RIP-protease chimers. Since both peptidase domains are unrelated, the [APM41] and [APC19] RIP genes most prob-ably result from two independent domain fusion events. The fact that the currently documented occurrence of [APM41] and [APC19] RIP genes is confined to the BEP (Bambusoideae, Ehrhartoideae, and Pooideae) clade of the family Poaceae species, might indicate that the underlying domain fusion events took place in a subgroup of the grass family, but this is purely speculative. However, the

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 147
limited taxonomical distribution of [APM41] and [APC19] RIP genes within the completed plant genomes render a widespread occurrence comparable to that of the [AB] and [AX] lineages unlikely.
What is the Evolutionary Link Between Plant and Non-Plant RIPs?
Fungi
As described in more detail in Chapter 2, in silico analyses revealed the occurrence of genuine RIP genes in several fungal genomes. One species (Arthrobotrys oligospora) possesses a gene that is most closely related to plant type [AΔX] RIPs, whereas in ten others chimeric genes resembling the plant [AX] chimers were found. For both forms the sequence similarity between the fungal and plant RIPs amounts to approximately 50% (at the amino acid level) leaving no doubt that they are related evolutionary. This striking sequence similarity, as well as the very narrow taxonomic distri-bution of the fungal RIPs, are difficult to explain in terms of a classical vertical inheritance from a common ancestor that predates the separation of fungi and plants approximately 1200 Myr ago.13 A more likely – but still speculative – explanation is that the fungal RIP genes were acquired by lateral transfer from a plant (within a host–parasite relationship). Such a transfer could explain why RIP genes are confined to a dozen or so typical plant parasites. In principle, a single transferred plant [AX] gene could be the ancestor of all identified fungal RIP genes, but it is also possible that [AXf] and [AΔxf] resulted from two independent transfers.
Insects
The documented occurrence of RIP genes in insects is confined to the mosquitoes Culex quinque-fasciatus (southern house mosquito) and Aedes aegypti (yellow fever mosquito) who possess one and three type [AI] RIP genes, respectively (see also Chapter 2, Figure 2.7). At present, one can only speculate about the evolutionary origin of these insect RIP genes. Taking into consideration the very limited taxonomic distribution (and the absence of homologs from all other completed meta-zoan genomes) it is difficult to envisage how the [AI] RIP genes eventually ended up in the Culex quinquefasciatus and Aedes aegypti genomes by a classical vertical inheritance from a common ancestor that predates the separation of metazoa/fungi and plants approximately 1200 Myr ago.11 A more plausible, but also very speculative, explanation is that the small group of modern mosquito type [AI] RIP genes evolved from a gene that was acquired by an ancestral fly species by a lateral transfer from a plant. An argument in favor of this hypothesis is the observation that the [AI] RIP genes share the highest sequence similarity with a type [AC] RIP from Zea mays. If a horizontal RIP gene transfer took place, the absence of [AI] RIP genes from the completed genomes of Drosophila and other flies could be dated most likely after the mosquitoes branched off the same evolutionary tree as flies about 220 Mya.14
Bacteria
During the last few years, evidence has accumulated that the bacterial RIP gene family is not con-fined to the classical Shiga and Shiga-like toxins found in Enterobacteriaceae but comprises several other genes present in the genomes of species belonging to unrelated taxa (for more details see

148 RIBOSOME-INACTIVATING PROTEINS
Chapter 2 and Figure 9.5). At present, RIP genes can be identified in only 16 bacterial species. However, in spite of this limited number, the bacterial RIP gene family exhibits a marked heteroge-neity with respect to both taxonomic distribution and domain architecture. To “visualize” this het-erogeneity, a preliminary phylogenetic analysis was made of the bacterial RIP sequences and the dendrogram combined with both taxonomic and domain architecture data. The results of this approach (summarized in Figure 9.6) revealed that the bacterial RIP gene family comprises four distinct groups. A first group are the Shiga toxin genes and closely related genes found in (the genome of lambdoid prophages of) Gammaproteobacteria as well as in plasmid pBC201 of Burkholderia sp. CCGE1002. The second group comprises closely related homologs of the secreted Streptomyces coelicor type [A] RIP found in four different Streptomyces species as well as a [AM] chimeric gene from a Micromonospora species. Three non-secreted type [A] RIPs found in two
Actinobacteria
Gammaproteobacteria
Betaproteobacteria
Bacteroidetes
*Located on plasmid pBC201
*
StrcoA
StrliA
StrsoA
StrscA
StrlyA
StrMgA1
StrMgA2
EntclShA
EsccoShA2
EsccoShA1
ShidyShA1
RicgrShA
BurspShA
BuramAi
BuramAm
FlacoA
CitfrShA
MicmoShA
Figure 9.6 Schematic overview of the phylogeny, domain architecture and taxonomic distribution of the bacterial RIP genes.
Dendrogram was rendered using CLUSTALW.7

UPDATED MODEL OF THE MOLECULAR EVOLUTION OF RIP GENES 149
other Streptomyces species form a third group (which shares little sequence similarity with the Streptomyces coelicor type [A] RIP). Finally, a fourth group is represented by the [As]-[U] and [As]-[P]-[W] chimeric genes found in the genomes of Burkholderia ambifaria and Flavobacterium columnare, respectively.
An important conclusion to be drawn from Figure 9.6 is that obvious differences in sequence, domain architecture, and taxonomic distribution argue against a monophyletic structure of the bac-terial RIP gene family, which in turn implies that its molecular evolution might be quite complex. Evidently, the phylogeny and evolution of the bacterial RIP genes cannot be uncoupled from that of their eukaryotic, and especially plant, counterparts. Previous phylogenetic analyses of the whole protein family revealed that the bacterial RIP genes do not form a single cluster but are spread over at least three different branches.1 Moreover, the same group reported in a previous paper that the secreted Streptomyces coelicor type [A] RIP was more closely related to some plant RIPs than to any bacterial RIP.3 This observation actualizes the hypothesis proposed in a previous review5 that bacteria did not develop the RIP domain but acquired it through lateral gene transfer from a plant. One of the major arguments in favor of this hypothesis is the fact that RIP genes are confined to less than a score of bacterial species. However, the apparent occurrence of four different RIP gene line-ages in four distinct bacterial taxa is difficult to reconcile with a single lateral gene transfer. An alternative scenario might be that multiple lateral transfers took place of a plant RIP gene into a bacterium (Figure 9.5). Two independent transfers of a plant type [A] RIP gene into two distinct Streptomyces species can explain the occurrence of two distinct type [A] RIP gene lineages (with different subcellular location) within the genus Streptomyces. Similarly, the whole subfamily of Shiga toxins can have evolved from an ancestor of modern Enterobacteriaceae that acquired a plant type [A] RIP. Possibly, the plant gene was first incorporated into the bacterial chromosome (as it still is in e.g., Rickettsiella grylli) and in a later stage fused to the Shiga toxin B domain and was transferred into the genome of a lambdoid prophage.4 Once incorporated in the phage genome, the RIP [As]-Bs gene became part of the highly mobile genetic elements that play a role in horizontal gene transfer. Such a mobile genetic element might have mediated the transfer of the RIP domain into the Burkholderia ambifaria and Flavobacterium columnare genome, where it was eventually incorporated in the [As]- [U] and [As]-[P]-[W] genes, respectively.
Conclusions
Recently released genome and transcriptome sequence data have allowed updating and refining of the phylogeny and evolution of the RIP gene family. All evidence suggests that the RIP domain was developed in plants at least 300 Mya. In a next step, two independent fusions of the RIP domain with a duplicated ricin-B and a still unidentified X domain resulted in the generation of the [AB] and [AX] chimers, respectively. Both lineages played a determining role in the further evolution of the RIP gene family. Besides a vertical inheritance of the [AB] and [AX] chimers, domain deletion events yielded multiple lines of secondary [AΔB] and [AΔX] (type 1) RIP genes. During evolution of the Magnoliophyta the [AB], [AX], or both lineages were lost in numerous species, whereas other gene amplifications eventually shaped complex RIP gene families. At present the evolution of the plant RIP gene family is fairly well understood except for the Poaceae family. Species of the latter family possess RIP gene families of an unusual complexity in both gene number and domain architectures.
The identification in plants of the [AX] chimers also sheds a new light on the origin of RIP genes in fungi and insects. Though controversial, the most plausible explanation is that these genes were

150 RIBOSOME-INACTIVATING PROTEINS
acquired by a lateral transfer of an [AX] RIP gene from a plant. A similar conclusion holds true for the RIP genes found in bacteria, but in this case the polyphyletic nature of the RIP gene family requires multiple independent lateral gene transfers from a plant into a prokaryote.
Though the results of the present in silico analysis leave no doubt that the RIP gene family is far more heterogeneous than could be inferred from previous studies with purified proteins, there is an important caveat because sequence similarity does not necessarily imply a conservation of activity. It remains to be demonstrated that the newly identified RIP domains exhibit a catalytic activity comparable to that of the classical Shiga toxins or plant RIPs. However, taking into account the low sequence identity (< 25%) shared by proteins with a documented RIP activity, one can reasonably expect that the N-glycosidase domain “supports” a substantial sequence divergence. Structural analyses of Shiga toxins and plant type [AB], [AΔB], [AΔX], and [Au] RIPs confirmed that the overall three-dimensional fold of the respective RIP domains is – apart from significant differences in specific regions – comparable.15, 16 Accordingly, one can conclude that the overall structure of the RIP domain is quite well conserved in spite of the obvious sequence heterogeneity generated throughout the evolution of this extended protein family.
References
1. Reyes AG, Anné J, Mejía A. Ribosome-inactivating proteins with an emphasis on bacterial RIPs and their potential medical
applications. Future Microbiol. 2012;7:705–717.
2. Jiang SY, Ramamoorthy R, Bhalla R, et al. Genome-wide survey of the RIP domain family in Oryza sativa and their expres-
sion profiles under various abiotic and biotic stresses. Plant Mol Biol. 2008;67:603–614.
3. Reyes AG, Geukens N, Gutschoven P, et al. The Streptomyces coelicolor genome encodes a type I ribosome-inactivating
protein. Microbiology. 2010;156:3021–3030.
4. Herold S, Karch H, Schmidt H. Shiga toxin-encoding bacteriophages–genomes in motion. Int. J. Med. Microbiol. 2004;294:115–121.
5. Peumans WJ, Van Damme EJM. Evolution of plant ribosome-inactivating proteins. In: Lord JM, Hartley MR, (eds.) Toxic Plant Proteins. Plant Cell Monographs: Springer, 2010:1–26.
6. Van Damme EJM, Roy S, Barre A, et al. The major elderberry (Sambucus nigra) fruit protein is a lectin derived from a trun-
cated type 2 ribosome-inactivating protein. Plant J. 1997;12:1251–1260.
7. Tamura K, Peterson D, Peterson N, et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood,
evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739.
8. Bolognesi A, Barbieri L, Abbondanza A, et al. Purification and properties of new ribosome-inactivating proteins with RNA
N-glycosidase activity. Biochim Biophys Acta. 1990;1087:293–302.
9. Arias FJ, Antolín P, de Torre C, et al. Musarmins: three single-chain ribosome-inactivating protein isoforms from bulbs of
Muscari armeniacum L. and Miller. Int. J. Biochem. Cell Biol. 2003;35:61–78.
10. Touloupakis E, Gessmann R, Kavelaki K, et al. Isolation, characterization, sequencing and crystal structure of charybdin, a
type 1 ribosome-inactivating protein from Charybdis maritima agg. FEBS J. 2006;273:2684–2692.
11. Palmer JD, Soltis DE, Chase MW. The plant tree of life: an overview and some points of view. Am J Bot. 2004;91:1437–1445.
12. Chaw S, Chang C, Chen H, Li W. Dating the monocot-dicot divergence and the origin of core eudicots using whole chloro-
plast genomes. J Mol Evol. 2004;58:424–441.
13. Berbee ML, Taylor JW. Dating the molecular clock in fungi-how close are we? Fungal Biology Reviews. 2010;24:1–16.
14. Wiegmann BM, Trautwein MD, Winkler IS, et al. Episodic radiations in the fly tree of life. Proc Natl Acad Sci U S A. 2011;108:5690–5695.
15. Lee BG, Kim MK, Kim B, et al. Structures of the ribosome-inactivating protein from barley seeds reveal a unique activation
mechanism. Acta Cryst. 2012;D68:1488–1500.
16. Yang Y, Mak AN, Shaw PC, Sze KH. Solution structure of an active mutant of maize ribosome-inactivating protein (MOD)
and its interaction with the ribosomal stalk protein P2. J. Mol. Biol. 2010;395:897–907.










![Small-Molecule Inhibitor Leads of Ribosome-Inactivating ... · and resume the cationic Arg180 [10–13]. Small-moleculeinhibitorsof ricin and Shiga/Shiga-like toxins are sought for](https://img.pdfslide.us/doc/110x75/5ecf5fd8e42b0e45a3177c8b/small-molecule-inhibitor-leads-of-ribosome-inactivating-and-resume-the-cationic.jpg)


















