Embed Size (px)
Citation preview

Rheumatology E-learning
University of Szeged
Department of Rheumatology and Immunology

Overview
„Prototype” autoimmune disease
The most common and variable systemic
autoimmune connective disease
Female:male: 10:1
Prevalence: 1:2000
Typical age at onset: 20-30 years
More prevalent among people with African
and Hispanic origin

Disease course, prognosis Course: fluctuating.
Provoking factors (e.g. sunshine, infection, drug): exacerbationor flare or relapse
After treatment: remission
Active disease causes acuta organ failure (nephritis, cerebritis, arthritis, severe skin laesions, respiratory failure etc.) and mayeven cause death. Furthermore, it leads to irreversible damage(e.g. renal failure, deforming arthritis, lung fibrosis, fingernecrosis, etc.)
The aim of patient care: 1. prevent relapses 2. detect diseaseactivity as early as possible (e.g. proteinuria, synovitis, pericardial rub, subtle cognitive decline), and treat effectively
Prognosis: 10 year survival now is higher than 90%
But: nephritis occurs in 40-50%, and end-stage renal failuredevelops still in about 10-15% of affected patients
Irreversible damage (related to both disease and drug/corticosteroid!/ accumulates, and quality of life is still poor

Joint involvement in SLE
Articular (polyarthritis / polyarthralgia)
Most common manifestation (70-80% of patients)
Symmetric, involving mostly the small joints
Fingers, wrists > shoulders > feet, knees, elbows
Joint pain, tenderness, swelling and reduced mobility
Usually there is less swelling and shorter morning
stiffness than in rheumatoid arthritis
Pain is often severe
In contrast with rheumatoid arthritis, there is no
pannus, no cartilage and bone destruction (= non-
erosive on radiograph)
But may be deforming in many cases

SLE arthritis
Deformities look like those in
RA (ulnar deviation, swan
neck, etc.)
But: they are not fixed, but
reducible, even hypermobile
due to ligamentous laxity:
Jaccoud’s arthropathy:

Skin involvements
Very common (> 50%)
Acute: photosensitive rash, butterfly
erythema, small vessel vasculitis (purpura or
urticarial)
Subacute: SCLE (subacute cutaneous LE)
Chronic: discoid LE, livedo reticularis,
panniculitis
Photosensitive rash, butterfly erythema and
SCLE are provoked by ultraviolet light
(sunshine)

SLE – butterfly erythema
Erythematous plaque over the nose and the cheeks, but the
nasolabial folds are spared

SLE – photosensitive dermatitis

SLE – photosensitive rash

Subacute
cutaneous
lupus
Predilection sites: arms, upper torso

Severe subacute cutaneous LE
provoked by sun-exposure
Note also the Jaccoud’s arthropathy

SLE – discoid laesion

Discoid lupus erythematosus

SLE – digital vasculitis

Haematological involvement
Anaemia
autoimmune, Coombs-positive haemolytic
autoimmune myelopathy (decreased
haematopoesis)
non-haemolytic (anaemia of the chronic disease)
Leukopenia, lymphopenia
Thrombocytopenia

Haemolytic anaemia in SLE
Normocytic, Coombs-positive, reticulocyte counthigh, Fe norm/high, LDH high, bilirubin high, haptoglobin low
Chronic or acute haemolytic crisis Warm antibodies – IgG – phagocytosis –
extravascular sequestration in the spleen Cold antibodies – IgM – bind between 4-15 C –
complement activation – intravascularhaemolysis

Leukopenia in SLE
WBC: < 4000/ul
Lymphocyte: <1500
Activity sign! – if WBC decreases, lupus flare may be suspected
Anti-lymphocyte antibodies, accelerated apoptosis
If a lupus patient presents with fever, otherinflammatory symptoms: leukopenia helps in thedifferentiation from infection vs lupus flare
Must be differentiated from myelosuppressive adverseeffects of immunosuppressive drug (e.g. azathioprin, methotrexate, mycophenolate mofetil)

Thrombocytopenia in SLE
May be chronic and mild-to-moderate (plateletcount > 50 G/L) – treatment may not be necessary
In other cases, it may be acute and critical (plateletas low as < 5 G/L) – immediate agressiveimmunosuppression is necessary
Anti-platelet (GP IIb/IIIa complex) antibodies
Fc-receptor-mediated phagocytosis in the spleen
Diff. dg.: Antiphospholipid syndrome
Drug adverse effect
Infection
Thrombotic thrombocytopenic purpura

Bone marrow histology from an SLE
patient with fatal pancytopenia
Megakariocyte hyperplasia
CD8-positive cytotoxic lymphocytic
infiltration

Serositis
Pleuritis, pericarditis, ascites
Pleuritis (prevalence in SLE: 30-40%)
Sharp chest pain exacerbated by inspiration,
cough
Dyspnea
Coughing
Physical exam:
○ Pleuritic rub
○ Dullness on percussion, diminished respiratory
sounds on auscultation

Pericarditis
Prevalence in SLE: 30-40%
Symptoms Dull or sharp retrosternal pain
Increasing dyspnea
Physical finding Pericardial rub (synchronous with heart sounds)
Increased heart dullness with percussion
Diminished heart sounds with auscultation
If severe: dilated jugular veins, cyanosis, dyspnea, signs of heart failure
Diagnosis: ECG, echocardiography
If severe: emergency. Achieve quick dg and diff dg with physical exam, ECG and echocardiography, and administer corticosteroid. If not, pericardium drainage with puncture or surgical fenestration is performed, but with appropriate dg it can usually be prevented

Lupus nephritis
Acute or chronic glomerulonephritis
Prevalence in SLE: 40-50%!
Key determinant of prognosis, organ damage
and quality of life in SLE
Progresses to end-stage renal disease in
about 10% of affected patients
Frequently diagnosed late – urinalysis on
every patient visit!

Lupus nephritis - symptoms
Nephritis syndrome
Proteinuria (>0,2 g/die)
Microscopic haematuria (dysmorphous red blood cells)
Leukocyturia („sterile pyuria”)
Cellular (red blood cell or white blood cell) casts
Hypertension
Renal function impairment
Nephrotic syndrome
Proteinuria > 3,5 g/die
Hypalbuminaemia
Hyperlipidaemia
Oedema
Nephroso-nephritis syndrome
Asymptomatic proteinuria or microscopic haematuria

Lupus nephritis – clinico-
pathological classification

Dysmorphous red blood cell
Red blood cells with small „ear” or „bud”
indicating that they have passed through the
glomerulus. They always indicate glomerular
disease! Simple but vital differentiation from
lower urinary tract origin.
„Mickey mouse red
blood cell

Red blood cell cast

Diffuse proliferative lupus GN
Courtesy of Prof. Éva Kemény Éva (Dept of Pathology)

Membranous lupus nephritis
(ISN/RPN class V )
Courtesy of Prof. Kemény Éva (Dept
of Pathology)

Vascular involvement is SLE
Raynaud’s phenomenon
Livedo reticularis
Arterial and venous thrombosis, pulmonary
embolism (see: Antiphospholipid syndrome -
APS),
Vasculitis
Accelerated atherosclerosis (APS)

Raynaud’s phenomenon
Transient white (one-phase), white and blue (two-
phase – the most common) or white, blue and red
(three-phase) discolouration of the fingers
Often associated with pain, numbness – this may be
the presenting complaint, rather than the
discolouration
In severe cases: ulceration, gangrene may develop
(especially in the most severe forms of Raynaud’s, i.e.
in systemic sclerosis, systemic vasculitis or
antiphospholipid syndrome

Differential diagnosis of
Raynaud’s phenomenon• Primary Raynaud’s: no underlying disease. Vasomotor
lability, vegetative dystonia, requires no specific
treatment
• Secondary Raynaud’s: there is an underlying disease
involving the circulatory system
○ Systemic connective tissue autoimmune disease (systemic
sclerosis, SLE, systemic vasculitis, antiphospholipid syndrome,
Sjögren’s syndrome – but: in rheumatoid arthritis: almost never)
○ Thoracic outlet syndrome (compression)
○ Hypothyreoidism, hyperthyreoidism
○ Smoking, atherosclerosis, diabetes mellitus
○ Hyperviscosity (paraproteinaemia, polycythaemia vera)

Raynaud’s phenomenon

Digital ischaemia

Livedo reticularis
www.dermquest.com
If transient, provoked by cold: probably normal
If fixed: severe microcirculatory abnormality – e.g. antiphospholipid
syndrome – in this case, it may herald a subsequent vascular
catastrophy, e.g stroke

Leukocytoclastic vasculitis
Leukocytic
infiltration, vessel
wall necrosis,
degradation of
granulocytes
Clinically presents
as purpura or
urticarial vasculitis
Frequent in SLE,
Sjögren’s and
small-vessel
vasculitides
(ANCA-associated
or Henoch-
Schönlein)

Purpura

Anti-phospholipid syndrome
Arterial or venous thrombosis, repeated spontaneousabortion or intrauterine death caused by autoantibodies thatpromote thrombosis (antiphospholipid antibodies)
Primary or secondary (associated with SLE, RA, infection ormalignancy)
Acute symptoms: Stroke, myocardium infarction, bowelinfarction, hypadrenia, obliterative atherosclerosis, gangraene, deep venous thrombosis, pulmonary embolism.
Chronic symptoms: Dementia, focal neurological signs, epilepsy, valvular heart disease, infertility, premature birth.
Catastrophic anti-phospholipid syndrome – simultaneousthrombosis in more than two organs
Antibodies: anti-cardiolipin, anti-beta2-GPI, lupusanticoagulant (prolonged activated partial thromboplastintime)
Must be suspected in every case with early vascular events, even if autoimmune symptoms are not present!

Pulmonary involvement
Acute:
lupus pneumonitis (e.g. lymphocytic interstitialpneumonitis - LIP, organizing pneumonia - OP)
diffuse alveolitis
diffuse pulmonary haemorrhage
Chronic
fibrosing alveolitis (non-specific interstitialpneumonitis – NSIP)
pulmonary hypertension
„shrinking lung” syndrome

Acute lupus pneumonitis
Progressive dyspnea
Coughing
Fever
Often accompanied by pleuritis – pleuritic
chest pain
Bilateral infiltrates on radiography and lung
CT

Lymphocytic interstitial pneumonitis

Diffuse pulmonary haemorrhage
Haemorrhagic alveolitis, alveolar capillaritis
Acute, severe alveolar capillary wall damage
Immune complex-deposition
Acute dyspnea, coughing, rapid anaemia, haemoptoe
Mortality: 40-80%
Iv. methylprednisolone, iv. cyclophosphamide, plasmapheresis

Bilateral alveolar patchy infiltrates
(alveolar filling)
On histology: immune-complex
deposition in the perialveolar
interstitium


Central nervous system in SLE
Anti-phospholipid-mediated
Focal, ischaemic laesions
Stroke, transverse myelitis, chronic cognitive
decline
Non-anti-phospholipid-mediated
Diffuse cerebral dysfunction („lupus-cerebritis”)
Psychosis, altered mental status, epilepsy,
headache

SLE – further organ
involvements Endocarditis, myocarditis
Fever, low-grade fever
Weight loss, chronic fatigue syndrome
Myositis
Oral ulceration
Secondary Sjögren’s syndrome
Retinal vasculitis, episcleritis
Autoimmune hepatitis
Amenorrhaea, infertility, repeated spontaneous
abortion, intrauterine death, premature birth

SLE – laboratory changes
Elevated ESR, but CRP is typically normal
Cytopenia (anaemia, leukopenia, thrombopenia)
Proteinuria, haematuria
Antinuclear antibody (ANA)
Anti-double-stranded DNA antibody (anti-dsDNA)
Extractable nuclear antibody (ENA) anti-SSA/Ro, anti-SSB/La, anti-U1RNP, anti-Sm
Anti-phospholipid antibodies (anti-cardiolipin, anti-beta2-glycoprotein 1), lupus anticoagulant …
Anti-nucleosome, anti-C1q (nephritis!)
Decreased complement-3 (C3), C4

ANA – homogenous pattern
Direct immunofluorescence: autoantibodies that bind to cell nucleus are
visualized by radiolabelled antibodies – screening method, and the exact
autoantigen-specificity is determied by ELISA

ANA – speckled pattern

ANA – nucleolar pattern

ANA – anti-centromere positivity

SLE – pathogenesis I.
Impaired apoptosis-
regulation
Complement-receptor 1
polymorphism
Immune-complex clearance ↓
Increased
apoptosis (UV
light, infection,
drugs)
INCREASED SUPPLY
OF ANTIGEN
(CHROMATIN)
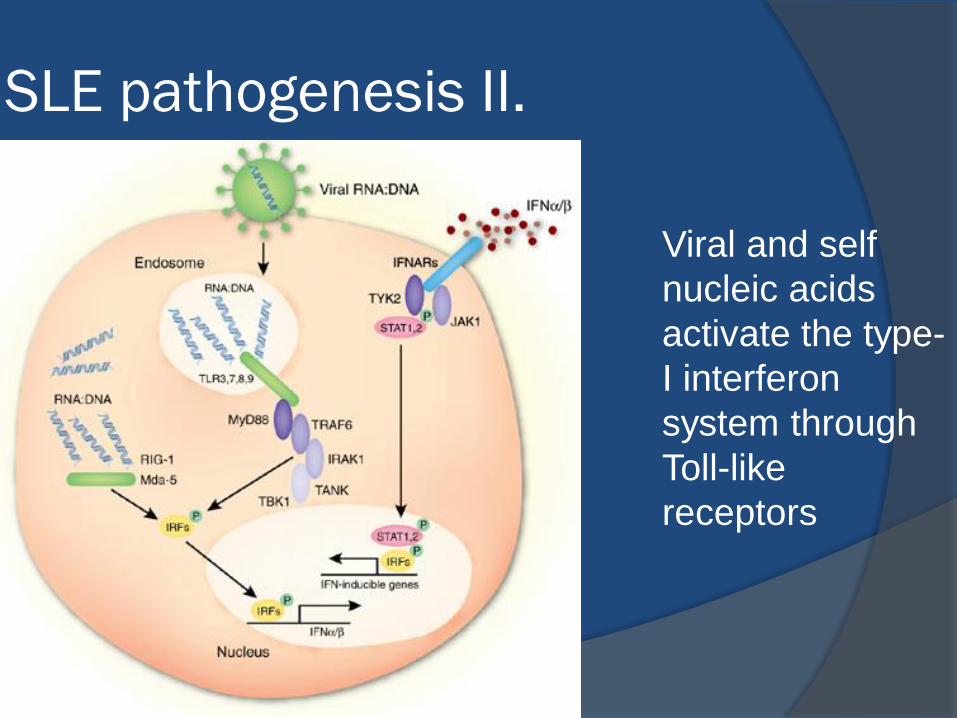
SLE pathogenesis II.
Viral and self
nucleic acids
activate the type-
I interferon
system through
Toll-like
receptors

Self-perpetuating circle of immune-
complex-mediated IFN-α activation
and autoantibody production
Rönnblom L, Arthritis Rheum 2006;54:408
Key players: plasmocytoid
dendritic cells, interferon-
alpha, autoantibody-
producing B-cells and plasma
cells, Th2, Th17, and to
lesser extent, Th1 cells
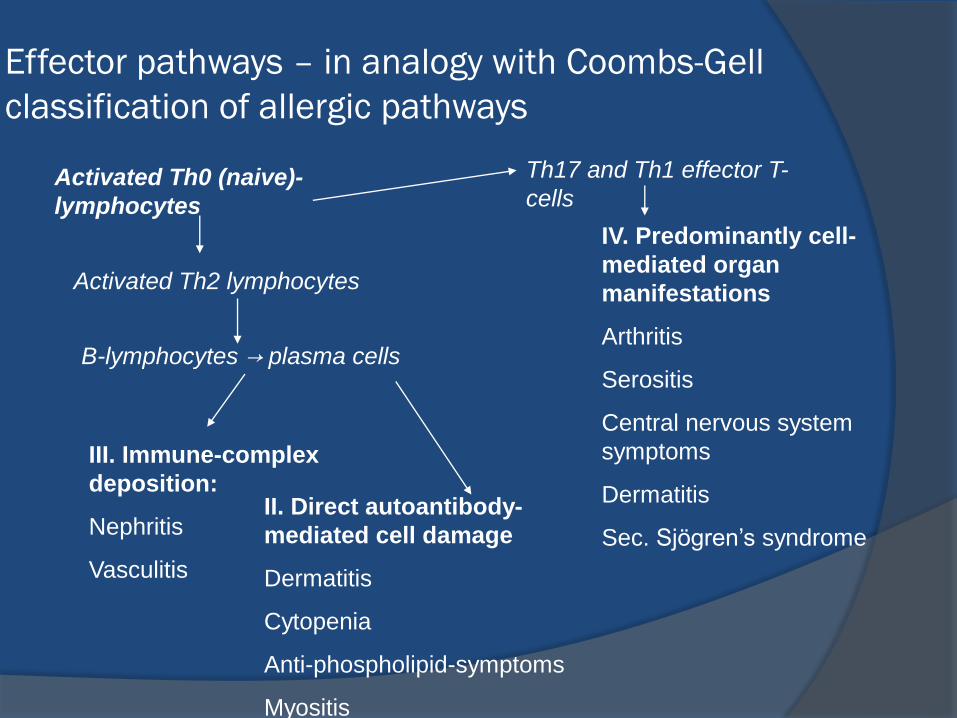
Effector pathways – in analogy with Coombs-Gell
classification of allergic pathways
Activated Th0 (naive)-
lymphocytes
Th17 and Th1 effector T-
cells
B-lymphocytes → plasma cells
Activated Th2 lymphocytes
III. Immune-complex
deposition:
Nephritis
Vasculitis
II. Direct autoantibody-
mediated cell damage
Dermatitis
Cytopenia
Anti-phospholipid-symptoms
Myositis
IV. Predominantly cell-
mediated organ
manifestations
Arthritis
Serositis
Central nervous system
symptoms
Dermatitis
Sec. Sjögren’s syndrome


Management of SLE
Lifestyle patient education: Avoid sunshine – always use suncream when leaving home
Oral anticoncipient: to be avoided in anti-phospholipid positivepatients and during active disease
Aim: control of flares, and preservation of remission
During flares: corticosteroids and otherimmunosuppressants – intensity is determined by theseverity of flare (low-medium-high dose corticosteroid + methotrexate, cyclophosphamide, rituximab, intravenousimmunoglobulin, plasmapheresis, etc.)
After disease has become quiescent: tapering of corticosteroids, use of maintenance immunosuppression(hydroxychloroquine + less potent immunosuppressantse.g. azathioprin, methotrexate)

Treatment
Mild cases (mild skin or joint involvement): NSAID, topical skin treatment, hydroxy-chloroquine
Chloroquine, hydroxi-chloroquine Originally: antimicrobial agent against malaria
Proven to reduce the frequency and severity of flares(current guidelines: lifelong antimalarial to every lupuspatient!)
Vitamin D Effective immunomodulatory agent: reduces
autoimmunity, promotes antimicrobial defense
Prevents osteoporosis – mandatory if corticosteroidsare administerd

Treatment II.
Cases of intermediate severity
E.g. serositis, cytopenia, marked skin or jointinvolvement, etc., but no involvement of vitalorgans (renal, pulmonary, central nervoussystem, cardiac)
During active disease: corticosteroid (12-64 mg methylprednisolon) + azathioprin, methotrexate, cyclosporine, mycophenolate mofetil
In remission: taper off corticosteroids, maintainother immunosuppressants for 3-5 years (+ antimalarial, Vitamin D3 lifelong)

Treatment III
Severe, life-threatening organ involvements(carditis, nephritis, systemic vasculitis, cerebralmanifestations): high-dose intravenouscorticosteroid (500 mg methylprednisolone) + iv. cyclophosphamide
Or: + rituximab (anti-CD20 monoclonal antibody)
In critical situations: plasmapheresis or iv. immunoglobulin
Maintenance: mycophenolate mofetil,
tacrolimus
cyclosporine (especially membranous nephritis, thrombocytopenia, myositis)
belimumab (monoclonal antibody to the cytokineB-lymphocyte stimulator (BLyS)

Rituximab (anti-CD20) – long-lasting B-cell
depletion

B-lymphocyta stimulator (BLyS) or B-
cell activating factor (BAFF)
Cancro et al J Clin Invest 2009
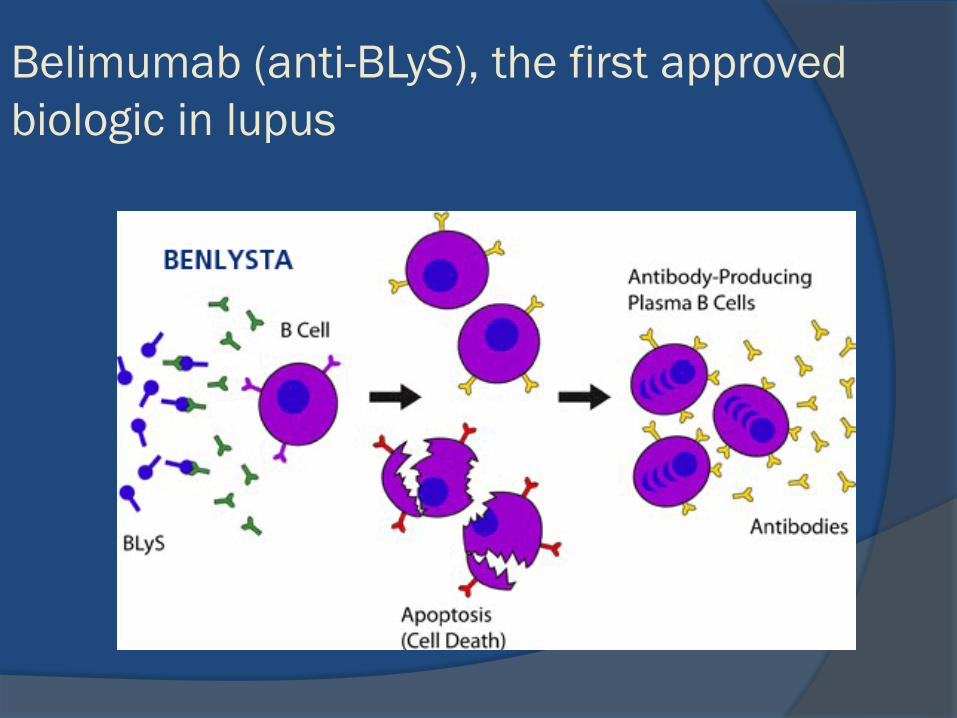
Belimumab (anti-BLyS), the first approved
biologic in lupus





























