Embed Size (px)
Citation preview

Chapter 1
Review of Literature
~~----"",'-'"-;,,": '~.' ,"< ... '. "k £,J
----------- ---

Chapter 1
1.1 Introduction
1.1.1 Candida albicans - as a commensal and opportunistic pathogen
The yeast Candida albicans exists as a harmless commensal in virtually all humans along the
mucosal surfaces of gastrointestinal and urinogenital tracts and, to a lesser extent, on the skin
and oral cavity. Their dominance and spread to other tissues is kept in check by the immune
system of the host as well as the competing microflora that restrict their proliferation.
However, under conditions when the immune system of the host is compromised (for
example, as a result of cancer chemotherapy, human immunodeficiency virus [HIV] infection
or in neonates and the elderly) or when the competing microflora are eliminated (for
example, after antibiotic treatment), C. albicans takes advantage of the situation by
colonizing and invading host tissues. This can result in a range of conditions including
painful superficial infections, such as vaginitis in otherwise healthy women, severe surface
infections of the mouth and esophagus in HIV patients, and life-threatening blood stream
infections among vulnerable intensive care patients (especially those undergoing cancer
chemotherapy or immunosuppressive therapy following organ or bone marrow transplant
procedures). Although HIV patients frequently suffer from recurring oral candidiasis and
sometimes die from advanced oesophageal colonization, superficial infections (such as
thrush and vaginitis) of mucosal tissues are usually not life threatening. However, if the
organism gains access to the blood stream, by invasion of host tissues or through
contamination of indwelling catheters, the infection can progress to invasive fungal growth in
the kidney, heart, brain or other organs than can often be fatal.
1.1.2 Candidiasis - a rising medical threat
The incidence of C. albicans infection is escalating significantly on a global scale. This is
due in part to an increase in the number of individuals immunocompromised by disease, such
as AIDS patients, that are steeply on the rise. It also has to do with an ever-increasing
number of patients undergoing suppressive therapies, in the form of chemotherapy for
treatments against diseases such as cancer, or for organ transplantation and severe bums.
Concomitantly, there has also been an increase in hospital acquired Candida infections. This
has been especially due to extensive invasive surgical procedures, and the growing use of

Drug Class Target! Mechanism of action Adverse Effects
Amphotericin Poly Interact with membrane ergosterol! Acute infusion reactions, cardiac arrest, encephalopathy,
ene increase cell membrane neuropathy, GI upset, renal damage, liver failure, marrow
permeability injury, thrombophlebitis, hearing loss, visual impairment,
rash -
Nystatin Poly Binds to sterols! Increase cell Rash, GI upset, tachycardia, bronchospasm, facial
ene membrane permeability swelling, myalgia
Fluconazole Azole Inhibit ergosterol biosynthesis! GI upset, dizziness, angioedema, anaphylaxis, seizures,
interfere with the lanosterol exfoliative dermatitis, marrow and hepatic injury
demethylase enzyme
Itraconazole Azole Inhibit ergosterol biosynthesis! Liver damage, GI upset, rash, headache, dizziness,
interfere with the lanosterol marrow suppression, hemolysis, respiratory irritation,
demethylase enzyme erectile dysfunction, hypokalemia, hypertension, edema,
hepatitis, hallucinations
Ketoconazole Azole Inhibit ergosterol biosynthesis! Liver damage, GI upset, depression, itching, headache,
interfere with the lanosterol dizziness, marrow suppression, hemolysis
de methylase enzyme
Voriconazole Azole Inhibit ergosterol biosynthesis! Transient visual disturbances, edema, GI upset, rash,
interfere with the lanosterol sepsis, respiratory disorder, elevated hepatic enzymes
demethylase enzyme
Caspofungin Echino Target the cell wall! inhibit the Phlebitis, headache, GI upset, rash, marrow injury,
candin fungal b-(1,3) glucan synthase myalgia, edema, fever
complex leading to glucan cell wall
depletion and osmotic instability
Flucytosine Pyrimidi Targets DNA and RNA synthesis Myocardial toxicity; psychosis; neuropathy; nausea;
ne vomiting; hepatic, renal, and marrow injury; colitis;
respiratory arrest
Terbinafine Allyl Targets fungal cell membrane! Anorexia, dyspepsia, nausea, mild abdominal pain,
amine disrupts squalene oxidase diarrhea, arthralgia, myalgia
Table 1 Existing antifungal drugs used for the treatment of fungal infections

Chapter 1
prosthetic devices and vascular catheters together with treatment with corticosteroids and
broad-spectrum antibiotics[Groll et aI, 1998]. Over the last 20 years, C. albicans infection
has been by far the most frequently isolated fungus in immunocompromised patients
associated with mucosal and deep-tissue infections. It is also the leading cause for
nosocomial fungal infection worldwide. Candida albicans accounts for more than 50% of
cases with candidiasis [Sullivan et al., 2004].
1.1.3 Need for new antifungals
Another reason for the growing relevance of C. albicans infections is that, although many
antifungal compounds do exist [Bossche et al., 1995; Bennett, 1996], these drugs are often
limited in their use because of their toxicity and side effects [Georgopapadakou and Walsh,
1994]. The currently available antifungal agents are small in number and belong to only a
limited number of different chemical classes [Table 1]. One reason for this slow progress is
that, like mammalian cells, fungi are eukaryotes, and thus agents that inhibit protein, RNA,
and DNA synthesis in fungi have great potential for toxicity in the human host. An additional
problem that began to surface about a decade and half ago is the emergence of clinical strains
resistant to the existing antifungal drugs [Powderly et ai., 1994, Denning, 1995, Boschman et
al., 1998]. The usefulness of the azoles (like itraconazole and fluconazole), which are
presently the antifungal drugs of choice, has diminished in recent years owing to the
increasing incidence of resistance, a complex phenomenon that involves several molecular
mechanisms [White et ai., 1998].
1.1.4 Search for novel drug targets
All of the aspects above have contributed to the growing demand for the identification of
more potential drug targets and development of new drugs with novel mechanisms of action
to meet the acute requirements of antifungal therapy today. Ideally these drug targets should
be conserved across pathogenic fungi, but absent in humans, so that broad spectrum
antifungals with minimum side effects can be developed. Identification of drug targets have
been an area of active research, especially in pharmaceutical companies, spurring more
interest in Candida albicans biology and pathogenesis [Georgopapadakou and Walsh, 1996].
This in turn has lead to a more concerted effort in identifying genes that can serve as
2

Chapter 1
potential drug targets, which fall in two classes based on their function: essential genes that
are necessary for survival and growth of the pathogen per se, and virulence genes that are
important for its survival in the host and for the progression of the disease.
1.2 C. albieans as a model fungal pathogen
1.2.1 Diversity of C albicans as a commensal and pathogen
Apart from the prevalence of candidemia caused by C. albieans, it is also important to
underscore its versatility as a commensal and opportunistic pathogen. A crucial component
of this versatility is its ability to survive as a commensal in several anatomically distinct sites,
each with its own specific set of environmental pressures. This ability means that the
spectrum of diseases caused by C. albieans exceeds that of most other commensal
microorganisms. Only Psuedomonas auriginosa rivals C. albieans in its versatility as a
commensal. Moreover, in an immunocompromised individual, C. albieans can infect
virtually all organs once it is disseminated into the bloodstream. It is believed that it regulates
its catabolic and anabolic pathways during nutrient limitation or surplus, and survive the
nutrient limitation and competition among bacteria and fungi that result in the elimination of
less adapted microorganisms. For example, many of the Candida-specific genes (as
compared with S. eerevisiae) encode catabolic proteins, that enable it to exist as a successful
commensal and opportunistic pathogen. It is also believed that many among them are
important for pathogenesis and establishment of the diseased state [De Backer et aI., 2000).
1.2.2 Virulence factors of Candida albicans C. albieans as a fungal pathogen assumes greater significance due to the large repertoire of
genes implicated in its virulence and progression of disease. A virulence gene can be defined
as one whose deletion affects the in vivo virulence of the microorganism in some way,
regardless of the method designed for its quantification, and whose genetic reintroduction
restores virulence, thus obeying the criteria that comprises the so-called Koch molecular
postulates [Falkow, 1988]. What are the factors that lead to this opportunistic pathogen
becoming invasive and pathogenic? Host factors undoubtedly play a role, as
immunocompromised patients, such as those suffering from AIDS or undergoing
3

Chapter 1
chemotherapy, are frequently stricken with serious Candida infections, often systemic in
nature. Yet C. albieans certainly has its own strategies that allow it to maintain itself within
the body and become an aggressive pathogen under favourable conditions. The identification
of those strategies has become a major research goal, not only in order to understand and
possibly control Candida infection per se, but also as a means of gaining insights into the
general ways in which other fungal pathogens might interact with their human hosts. It has
been observed that several virulence factors identified in C. albieans have homologues in
other pathogenic fungal species as well [Braun et at., 2005].
Fungal virulence is a complex process that requires the expression of multiple genes at
different stages and different sites of infection. These virulence determinants are expressed
uniquely in infected tissues and interact directly with the host, and are distinct from those
molecules that serve a purely 'housekeeping' function that is equally essential for the fungus
in vitro and in vivo. Nevertheless, the overall virulence of C. albieans (i.e. its ability to cause
disease) depends upon two kinds of traits: survival traits and virulence traits. Survival traits
permit the organism to divide in the host environment; an example is the ability to synthesize
metabolites that are necessary for growth but is scarce in the in vivo milieu, such as
pyrimidines. Virulence traits permit the organism to invade new tissues, to evade phagocytic
cells, and to cause symptoms of infection. It include factors that contribute to adhesion to
host tissues, secretion of proteases and other hydro lases, reversible alteration of yeast cell
morphology (phenotypic switching), and dimorphism [Navarro-Garcia et al., 2001].
1.2.2.1 Adhesion to host tissues
Adherence of C. albieans to host cells is seen as an essential early step in the establishment
of disease. C. albieans possess an array of molecules (encoded by genes from the ALS and
PMT family, HWP 1, INTI etc) for efficient colonization of diverse niches in the human host,
thus contributing to its versatility as a commensal and pathogen. In addition, C. albieans can
also adhere to the surfaces of medical devices and form biofilms, which results in an increase
in candidemia and antifungal resistance related to catheter insertion. There is a positive
association between the degree of virulence and the ability to form biofilms [Chandra et at.,
2001].
4

Yeast cells • Cell density >106 cells ml-1
• Growth below 30°C
• pH 4.0
Pseudohyphae • pH 6.0, 35°C
• Nitrogen-limited growth on solid medium (SLAD)
Hyphae • Serum,>34°C
• Lees medium, 37°C
• pH 7.0, 37°C
Other filament-inducing conditions • Spider medium
• Engulfment by macro phages
• Mouse kidneys
• Growth in agar matrix
• Iron deprivation
• Anoxia
• n-acetyl glucosamine
Table 2 Conditions that induce yeast - hyphal switch in C. albicans
[From Berman & Sudbery, 2002]

Chapter 1
1.2.2.2 Secretion of proteinases and other hydrolases
C. albieans possess 9 different genes for secreted aspartyl proteinases(SAP 1-9), the
proteolytic activity of which has been associated with tissue invasion [Monod et al., 1998].
The expression of SAPs has also been observed in murine macrophages after phagocytosis of
C. albieans cells[Borg et at., 1990]. These observations suggest that proteolytic activity of
Saps is important for the virulence of C. albieans. A specific inhibitor of acid proteinases,
pepstatin, blocks the events occurring during early invasion of C. albieans in murine skin and
modulates the course of experimental candidiasis in mice [Ruchel et at., 1990]. The
disruption of SAP1, SAP2 and SAP3 show attenuated virulence in a mouse model [Hube et
al., 1997]. The triple disruption of SAP4-6 genes is less virulent than the respective
homozygous single mutants. The product of SAP2 is required for disease development in a
murine and vaginitis model. SAP5 is found to be expressed at high amounts throughout the
course of systemic infection in mice [Staib et at., 2000].
Among phospholipases that hydrolyse one or more ester linkages of glycerophopsholipids,
phospholipase B has the major activity in C. albieans. Strains isolated from blood produce
higher level of phospholipases than commensal strains. Cells producing less phospholipases
are less virulent than strains producing high phospholipases in a murine model, suggesting
that phospholipases may be virulence factors [Ghannoum, 2000].
1.2.2.3 Yeast to hyphal transition
The ability of C. albieans to switch between yeast and hyphal forms is now established to be
important for virulence from independent studies. Conditions that contribute to this switching
in vitro are listed in Table 2. Many of the genes implicated in the signal transduction
pathways that regulate morphogenesis in C. albieans (CPH1, EFP 1, CRK1, RlMIOl, INTI,
CHKl, TUP 1 etc.) have been found to be crucial for virulence in animal models [Braun and
Johnson, 1997; Gale et at., 1998; Calera and Calderone, 1999; Chen et al., 2000; Davis et
at., 2000; etc]. These findings, as well as the fact that both forms are found at infection sites,
suggest that only strains that can produce both filamentous and yeast form cells are capable
of penetrating vital organs and proliferate sufficiently to kill the host.
5

Chapter 1
1.2.2.4 Phenotype switching
The colonies of C .albicans can switch among different phenotypes including smooth, rough,
star, striped, hat, irregular wrinkle, and fuzzy at high frequency (10-4 to 10-1) [Slutsky et aI.,
1985]. But, the most well studied switching phenomenon in this organism is the white (raised
colonies with round-ovoid cells) to opaque (flat and gray colonies with elongatedlbean
shaped cells) switching. Though opaque phase cells have higher capability to colonize the
skin in a cutaneous model and higher frequency for mating than do white-phase cells [Soli,
1997; Miller and Johnson, 2002], the latter is more virulent in a systemic animal model
[Kvaal et aI., 1999]. However, the basic mechanism of phenotype switching and, the
involvement of this switching in the virulence of C. albicans is not yet clear.
1.2.3 Virulence genes as potential drug targets
It is now possible to identify molecular targets that are essential for the fungus to produce
disease. The identification of these unique, important molecular pathobiological functions
could translate into the use of these molecular targets for the design and development of new
antifungal drugs [PIa et al., 1996]. A fungal infection is simply an accidental encounter in its
life cycle, but the fungus must survive in the host to cause disease. Therefore, more subtle
factors such as the ability to survive and/or grow within the host are essential to the parasite's
ability to produce disease [Navarro-Garcia et aI., 2001].
It has been claimed that virulence determinants may not necessarily be good targets because
altering their functions may not result in a fungicidal effect, a desirable effect in patients in
which immune deficiencies are responsible for the underlying infections, as occurs with
AIDS patients and opportunistic fungal infections. However, targeting virulence may have
the advantage of being highly specific, both in terms of fungal species and the actual stage of
the pathogenic process inhibited. It may therefore lead to the development of potential
inhibitory drugs with very limited side-effects as they are less likely to be conserved in
humans, unlike essential genes. Moreover, exposure to drugs against targets that are not
essential for survival per se do not put the pathogen under as much a stringent selection
6

Chapter 1
pressure as against an essential target, which in tum limits the possibility of emergence of
drug resistance.
1.2.4 Genetic tractability of C. albicans
The infection process itself is still not well understood and many Candida genes involved in
pathogenesis have to be yet identified or further characterized. Fortunately, fed by the
progress made in the molecular genetic analysis strategies for S. cerevisiae, powerful
molecular methods for transformation, gene expression and gene disruption in C. albicans
have been developed in recent years. Molecular analysis of gene function is also easier than
that of other fungal pathogens (such as Histoplasma capsulatum, Cryptococcus neoformans
or pathogenic Aspergillus species) due to higher efficiency of transformation and
homologous recombination as well as molecular tools available for genetic analysis
(discussed later), even though there is still a requirement for new strategies to carry out
broad-based genetic studies.
1.2.5 Genome sequence of C. albicans To a significant extend, genome wide studies in C. albicans has been and will be facilitated
by the release of full diploid sequence of this fungus [Jones et al., 2004]. It was the
culmination of more than ten years of work, and represents a landmark in the history of
Candida research. From the genome sequence of C. albicans that contains 16 million base
pairs, 33% larger than that of S. cerevisiae, it could be gleaned that homologous
chromosomes show substantial divergence, and many genes are represented as two
distinctive alleles. Recently, a human-curated annotation of the C. albicans genome was
published based on the version 19 computational assembly [Braun et aI., 2005], that mapped
and identified 6,354 genes within the genome. The mean protein coding length of 1,439 bp
(480 aa) was found to be almost identical to what has been observed in S. cerevisiae and S.
pombe, while the gene density stood at one gene per 2,342 bp. Short descriptions for all gene
products were provided by annotators, usually based on sequence similarity. A total of 1,218
(19.2%) genes encoded unique proteins with no significant homo logs in the sequence
databases, a percentage almost identical to that observed in the current version of the S.
cerevisiae annotation [Kellis et aI., 2003]. An additional 819 (12.9%) gene products
7

Species Length (Mb) Number of Genes
Mean Coding Length (bp)
Gene Densitt
Coding Percent Introns Unique
Proteinsb Reference
C a/bicans ! ~~ v_ •
14.88 _ .6J~_ "]i3§~-" - ~~'~42~ ' .. , ._ ~]l:S~~~ __ ~'~i..i..4 ~.-:.~i.:.J;;.;31~:;:..;.::(:1=9:2.;;.;.;%},--" _~-l 5. cerevisiae 12.16 5,726 lA85 2,124 69.9% 272 tl04 (19.1%) 16 [E~~Q~_~ _._,'_'_., __ ~1~:46~-~"- _"_ 4;ffl .~~_-f;426- ._~_~ .. _"~- -l:~~-_ ~ - -=~::-::l~=-%--'·~=--=10':"':'34-~68=J-':'~(,J~4%~),-~11'-' -, N. crassa 38.64 10,082 1;673 3;832 43.6% 171139 41140 (41%) 18 ri[iib~atdl' _ ... . .'":- ,!~.~8 ~. _. . '~~ __ -~·.1~-·_·. -_ -"~lil-_--.. ''''''''6?,-.9%''''''''. ---M-, --,-od---" --""""l~-~---' Kluyveromyces iactis 1Q.63 5,329 1,383 1,995 71.6% nd nd 128 p~¥!y~~y~is~1fa.ns_enii ~:_-_~~i2j" - ..... -~"~- 6i~" :.~ ~-~l§!_· ... ~'~-~-~-.~_-'.'" ~ _--7,9-;2%---;f-:cld:,---·-,:.ng,---"J ....... ·~~---; Yarrowla lipolytlca 20.50 6,703 1,428 3,058 46.3% nd nd 128 &ryptot2C~S neof~rmqns".-"-_·if.~ .'." .... '6!?,7~-' .- .. -.19~ - -. -~?I~ '. . ."~ J~.:$~"_ . ~,3~2L_ ~.",~3j=%I __ -,-1"","£9_--,
Table 3 Features of C. albicans genome compared to other completed fungal genomes aNumber of base pairs in ge nome divided by number of genes.
bNumber and proportion of proteins with no significant similarity to known proteins. nd, not determined. [From Braun et al., 2005]

Chapter 1
exhibited significant similarities to other proteins of unknown function [Braun et ai., 2005].
The latest assembled sequence of the genome (Assembly 20) is now available at the Candida
Genome Database (CGD) [http://www.candidagenome.org/].
It is envisaged that comparative genomic analysis will highlight genes that contribute to C.
alMeans survival and its fitness as a human commensal and pathogen. Comparison with
other fungal genomes have already permitted the identification of numerous fungus-specific
genes that are absent from the human genome and whose products might be targeted for
antifungal therapy. The results of these efforts will thus ensure that the Candida research
community has uniform and comprehensive genomic information for medical research, and
for the development of functional genomic tools as well as for future diagnostic and
therapeutic applications. Salient traits of the C. albieans genome with respect to some of the
already sequenced fungal genomes is compared in Table 3.
1.2.6 Differences with respect to S .cerevisiae Analysis of the C. albieans genomic sequence [http://www.candidagenome.org/] also
indicates that many of its genes lack close S. eerevisiae homologs. This has been supported
by molecular genetics studies that have also revealed many differences between the two
organisms in terms of specific gene functions [Liu, 2001]. Though S. eerevisiae has often in
the past been used as a model organism for predicting targets in Candida alMeans, many
recent studies suggest the essential role of S. eerevisiae genes can neither be reliably
extrapolated directly to C. alMeans [E.g. Nagahashi et al ., 1998; Kelly et al ., 2000] nor vice
versa [E.g. Mio et al ., 1997; Lussier et al ., 1998]. In part, this is probably because these two
fungi diverged approximately 800 million years ago and therefore likely show important
evol utionary divergence in their gene complements [Heckman et al ., 2001]. In fact, using the
computer algorithm BLAST at P<1 *10-2°, approximately 40% of C. albieans genes was
found to have no identifiable S. eerevisiae homologue. Thus, it is perhaps not surprising, that
these two fungi differ in their capacity to buffer deleterious mutations, resulting in different
sets of essential genes [Hartman et al ., 2001]. Genomic approaches performed using a
nonpathogenic organism, such as S. eerevisiae, thus limit the ability to study pathogenesis
and combat fungal disease. Preferably, phenotypic analysis performed in C. alMeans permits
8

Chapter 1
relevant conclusions to be drawn with respect to the biology of a human fungal pathogen that
are applicable throughout the drug discovery process (e.g. target identification, target
validation and high throughput screening).
1.2.7 Animal models for localised and systemic candidiasis
It is imperative to have appropriate animal models so that genes implicated in virulence can
be studied in vivo. The use of animal models to reproduce the infection under controlled
conditions is essential for the confirmation of virulence. Virulence is determined typically
with a murine model of disseminated candidiasis, although other reproducible animal models
have been described including those of rat vaginal and oral candidiasis [Ghannoum et ai.,
1995]. In the mouse model of hematogenously inoculated, systemic C. aibicans infection, it
has been possible to adopt an analytical approach to infection, and to evaluate the biological
significance of different measures of susceptibility and resistance. The most commonly used
parameters are mortality, fungal burden in the tissues, and the severity and extent of tissue
damage. However, these different outcomes reflect substantially different aspects of the
yeast/host interaction. The various measures of disease and host resistance are defined as
follows. (a) Mortality is often regarded as an index of 'overall' susceptibility, but it is
important to recognize that death is a consequence of the failure of a critical organ system.
This is not always associated with overwhelming infection throughout the body. (b)
Quantitation of the infectious burden in different organs or tissues is a direct measure of the
efficiency of the host's Candida cidal or Candida static effector mechanisms. (c) The severity
of lesions in the tissues represents the interaction between the proliferating yeast and the host
response against it. This includes immunopathology directly or indirectly attributable to the
activities of the host's effector cells [Ashman, 1997].
1.2.7.1 Role of kidney in systemic candidiasis
There is a well-documented correlation between mortality and colony counts in the infected
kidney [Marquis et aI., 1988; Ashman et ai., 1996]. However, this relationship does not
appear to hold true in other organs, such as the brain [Ashman et ai., 1993]. It is also seen
that C. aibicans grows and multiplies maximally in mice kidneys. It can, therefore, be
inferred that the kidney is the critical organ system in Candida infection; and independent
9

Tools Properties/comments Selectable markers URA3 Selection: uril:::line protttrophy, counterselection
on 5-FOA; Ura- cells have reauced virulence
1-151 Selection: histidine prototrophy
ARG4 Selection: argil1ine Iprttotrophy fMH3 Wikt-typeaUele effective ool~ at high copy.
resistant alleles function at slng'le copy and hcrnol~y with e~dog~,ous copy reduces targete Integration effaency
~AU -cassette that carries URA3 , anked by 5' and 3' POrtiOIilS '~ioclooing
PC~Vlillediated transfOlmation for arginine prototfqp~ followed by selection fur
~600 bp of overlaplof ARG4 Irecombinatiorl betw'een the URA3 frag ments ,(while maimaining selecton for u.ra+ cells), yields some Bolates in whiCh both cQpies ·of the gere of interest have been disrupted; mitotic recombination might make homo~ous sequen::es distal to the insertblil site
Promoters ADH1 High levels dexpressbn
ACT1 High levels d expressbn; stronger than ADHl GALl Induced ~ 10-12-fdkt with galactose. repressed
with glucose; 3- 4-fold weakervhanACTl PCKl Induced on succinate or, at higher levels (up to
1 OO-fold), with casamino acids (a cd digests d casein treated to eliminate or reduce vitamins); repressed with glucose
MAL2 Induced - 3- 4-fold by maltose and sucrose. ,repressed by glucose
MET3 Repressed up to 85-fold in the presence of methbnine andror cysteine
Tetracydine~ulatall>le f scherichia cOO tetR fused to' ap4 (SaccharO'i1?)'ces
~ to 500-fold~pression; requires two components . i etR al1d TetO) inserted in the gencrne; a Iladk of
cerevfsfael activatbn dama in; promoter to hcrnology t.o the C. aibicansgenome improves the be regulated contaillS tetO binding site frequency with which non-homologous recombilil!ltibn
generates the desired ,integrants
Heteroiogousremrter genes ~~lactis LAC4 'l)3-ga IBctosidase)
Does not \/\Ork well as a single copy
Streptococcus cf:ttrmophflus facZ Expressbn levels much higher than those of 03-ga lactosidasel LAC4 in K lactis; no C. a/l:icans homdogue
Renilla renfformis !Iuciferase Ca 11 be detected at lOW" !Ievels of ed'ression; no C. a/bicans hcrnologue: no CU cooons
Aequore~ victooa GFP Codon optimized for use in C. alb.fcans ModWIed GFPs, YFPs an::! CYPs Codon0r.!mized and available in cassettes for
gene rep acement or fLBioo protein constructbn through PCR-mediated transformation
FlpJ FRT in mo expression system Flp reccrnbinase driven br. a test promoter is used to excise a ma rker f a nked by FRT s~es; the timillg of marker exdsbn reflects the time when the test promoter was first active
Table 4 Molecular tools commonly used in the study of C. albicans [Adopted from Berman & Sudbery, 2002]

Chapter]
studies confirm that the immediate cause of death after systemic challenge is acute fungal
pyelonephritis [Ashman et aI., 1996]. Hence fungal burden is most often determined by
recovering cells from infected kidney, although quantitation of infectious burden has been
estimated from infected spleen and liver as well.
1.3 Strategies and tools to study gene function in C. albicans
Gene function analysis in any organism requires adequate strategies and molecular tools such
as appropriate strains, marker genes, efficient transformation system and efficient
homologous recombination machinery, to name a few. Molecular tools commonly employed
in the functional analysis of genes in C. albieans is listed in Table 4. Most of the strategies to
study gene function in C. albieans have been heavily borrowed from those that was
developed for S. eerevisiae. Nevertheless, C. albieans genetics is not as simple and straight
forward, owing to following limitations.
1.3.1 Technical difficultiesllimitations of C. albicans genetics
C. albieans is an obligate diploid under natural conditions. Though C. albieans strains that
were subtly altered at the mating-type-like (MTL) locus were shown to mate to form
tetraploid strains under specialised, laboratory conditions [Hull, 2000; Magee et aI., 2000], as
yet there is no evidence that this organism can sporulate. Hence both the alleles of a gene
have to be disrupted to obtain a functionally mutant phenotype. This has been the major rate
limiting step that has hampered the progress in understanding the various factors required for
survival and pathogenicity of C. albieans. Another limitation contributed by the obligate
diploid and sporulation deficiency status of C. albieans is the lack of a direct method to
determine essentiality of genes. Currently, it is largely determined by an inability to generate
homozygous mutants. Moreover, transformation efficiency in C. albieans is much lower in
comparison with S. eerevisiae [De Backer et aI., 1999; Walther et aI., 2003], and the lower
frequency of homologous recombination make it necessary to render long flanking regions of
homology to achieve targeted integration. Also, episomal vectors in C. albieans are highly
unstable, leading to wide variations in copy number from cell to cell and rapid loss of the
plasmid in the absence of selection pressure [Cannon et aI., 1992; PIa et aI., 1995], due to
10

Chapter 1
lack of a stable, robust origin of replication such as that of 211 plasmid of S. cerevisiae. C.
albicans also has a non-canonical codon usage, where CUG, a codon used by many other
organisms with high frequency, codes for serine instead of leucine [Santos and Tuite, 1995]
so that it becomes necessary for heterologous genes to be modified to comply with the C.
albicans genetic code before they can be expressed in this organism. These characteristics
make C. albicans intractable to many of the molecular genetic analysis amenable to
Saccharomyces cerevisiae.
1.3.1.1 Variations at genome and gene level
Many C. albicans strains, especially from clinical settings, are aneuploid [Magee and Magee,
1987; Hughes et aI., 2000; Rustchenko-Bulgac, 1991; Selmecki et aI., 2005], and
consequently or otherwise, some genes are present in more than two copies [Gow et al.,
1994; Wysong et aI., 1998]. Therefore, after two rounds of gene disruption, a third disruption
should be carried out in such cases to achieve a true knockout strain. Multiple rounds of
disruption in tum potentially increases the incidence of ectopic mutations such as trisomy
[Chen et al., 2003; Rustchenko et aI., 1994, Rustchenko et aI., 2003; Selmecki et al., 2006],
chromosome abnormalities [Rustchenko et aI., 1994; Rustchenko et aI., 1997; Selmecki et
aI., 2005] or transformation induced mutations. Apart from these genome level variations,
there may also be intra-allelic variation within a gene [Saparito-Irwin et aI., 1995], or in the
regulatory region [Bretagne at ai, 1997; Staib et aI., 2002], thereby leading to variation in
gene expressIOn.
1.3.2 Strains and selection markers to study gene function
An important aspect of gene characterization is the use of an appropriate microbial host strain
for gene disruption experiments. Different strains of C. albicans have been used, but the most
popular has been the use of CAI-4, a strain deleted for URA3 gene obtained from a
pathogenic clinical isolate SC5314 [Fonzi and Irwin, 1993], the strain whose genome was
subsequently sequenced. Although the use of a single strain for genetic studies clearly
facilitates a comparison of research results from different independent laboratories, it also
poses a serious problem, because all experiments are done in one specific genetic background
and the choice of host strains is limited. In C. albicans, a selection marker that could be
11

Chapter 1
selected for and counter-selected against would be ideal since the same marker could be used
for the construction of both heterozygous and homozygous disruption mutants. This has
made URA3, the selection marker for CAI-4 strain and possessing traits mentioned above, the
most widely used selection marker in C. albieans. It is however not ideal for functional
analysis of virulence genes, since this gene is an important virulence determinant in itself. It
is now clearly recognized that differences in expression level of URA3 brought about by its
site of integration within the genome can modulate virulence in this pathogen [Lay et aI.,
1998; Sundstrom et al., 2002]. Hence there is a need to develop selection cassettes devoid of
URA3 to study genes involved in virulence. Moreover, 5-FOA based selection of Ura' cells
have been found to induce ectopic chromosomal abnormalities [Wellington et al., 2006].
Another popular strain to study gene function has been BWP]7 [Enloe et al., 2000], a His
Ari strain derived from SC5314, in which the two different marker genes can be
conveniently used to disrupt the two alleles of a gene. However, this strain has a part of
chromosome V deleted distal to the HIS] loci, acquired during the process of HIS] deletion
[Forche et al., 2004]. Noble and Johnson [2005] have recently made strains using auxotropic
markers that do not have a role in virulence, such as HIS], ARG4 and LEU2, as well as
URA3, in different permutations and combinations, to meet the growing demands for
generating double disruptants in C. albieans. Marker genes from C. albieans, as well as from
heterologous hosts such as C. dubliensis and C. maltosa have been made use of to minimise
non-targeted integration of selection cassettes in the above strains [Noble and Johnson,
2005]. Dominant selection markers such as the MPA R [Kohler et al., 1997; Staib et aI., 1999]
and NATJ/SATJ [Shen at aI, 2005; Reuss et al., 2004], that confers resistance to
mycophenolic acid and nourseothricin, respectively, have also been introduced; these
markers can be used for construction of mutants in wild type background, including that of
clinical strains.
1.3.3 Selection cassettes for gene deletion/disruption
Selection marker cassettes have been developed in C. albieans to meet the requirement of
disrupting/deleting both alleles of a gene. This has necessitated the use of markers that can be
recycled, which in turn require mechanisms by which it can be evicted from the genome.
12
Parental all~QS ~ABC XYZ~
---1 ABC XYZ r--~'t~>~ ____ ------------------____ ~~.~> I A B IliSb I U RAJ I hlsq I y Z I
Transform with Urn-blast9r I disrupoon cassette ..
~ABC XYZ~
---II A B I JisG I URAJ I hisG I y Z 1-1 --L-----J _ _ _________ ~=-------------........ ---I HetQrozygcus
---1 ABC
--------11 A B I hisG I 5-FOAselaction for
U ra recombinants I
URA 3
disruption strain (Ura T)
XYZ r--
-------iIAB I fist ! YZII-------
~ABC XYZ~ \ j'Heterozygcus r'-' ---, ___ ------------------___ r---"'....," disruption strain I A B I liSa I U RAJ I hlsb I Y Z I (Urn T)
Transform v ith Urn-blaster I disruptbn cassette •
-------------1IAB I ~G I Y ZI~ ------
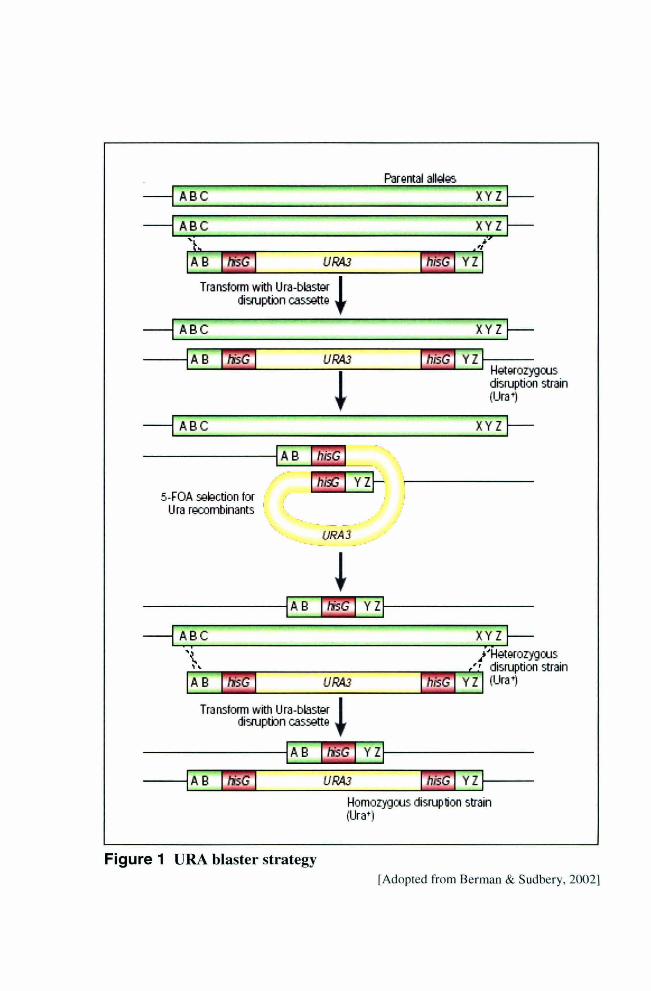
Figure 1 URA blaster strategy
URA3 I hisG I YZ I-I--Homozygcus disrup tion strain (UraT
)
[Adopted from Berman & Sudbery, 2002]

f.,..t .. Ie •• Ura -
second .11 ...
Homologous recom bina tion
KA
SIN G L E A LLE LE K N OCK OUT
__________ ~I~p~~----------------
Figure 2a URA flipper cassette [From de Backer el af. , 2000]
first ...... Ura +
.econd ......
first •• Ie •• Ura-
second .11 ...
P N SeII
~ 1 i .-- caFLP
-'-aL~~---c========~~ FR T M A L 2p ACT I I FRT
Figure 2b SAT! flipper cassette with FLP recombinase under control of MAL2 promoter [From Reub et al., 2004 ]

Chapter I
This has led to the development of many strategies for the generation of homozygous
mutants in this obligate diploid organism.
1.3.3.1 URA blaster strategy
The URA-blaster strategy [Fonzi and Irwin, 1993] for sequential disruption of both copies of
a gene in C. albicans is the most extensively and successfully used gene inactivation system
for this fungus. A hisG- URA3-hisG cassette (consisting of a C. albicans URA3 gene flanked
by Salmonella typhimurium hisG direct repeats) is used as the core construct to disrupt a
gene by insertional inactivation. Insertion is achieved by homologous recombination between
the long gene-specific regions flanking the cassette and a homologous copy of the gene in the
genome. Subsequent intrachromosomal homologous recombination between the hisG repeats
in the cassette leads to loss of the URA3 marker, and one can select for Ura - revertants on 5-
fluoroorotic acid (5-FOA) containing medium. The same disruption cassette can then in
principle be used for disruption of the second allele [Fig. I] .
1.3.3.2 Flipper cassettes
A refinement of the URA-blaster strategy makes use of the Flp recombinase of the 2~
plasmid of S. cerevisiae [Sadowski , 1995] in which Staib and coworkers [\999] have
engineered a version of the FLP recombinase gene that conforms to the peculiar C. albicans
genetic code. Their gene disruption scheme use a marker gene flanked by short, 34-bp direct
repeat regions (FRT sites) that act as recognition sites for the Flp recombinase. After
integration of the marker cassette (using long flanking regions of homology) at the desired
chromosomal region, excision of the marker is accomplished by inducing Flp recombinase
production , the expression of which is under the control of a regulatable promoter (such as
SAP2 or MAL2 promoters) present within the same gene disruption construct, leaving behind
a single FRT site [Fig. 2a]. Originally, URA 3 was used as the marker gene in the flipper
cassette [Morschhauser et aI. , \999] . Dominant delection markers such as MPA R [Staib et al.,
\999] and SATl [Reuss el aI. , 2004] has been later introduced as part of flipper casettes in
order to generate mutations in wild type strains as well [Fig. 2b].
\3
First ampified de.letbn cassette
J AI HIS? [ z l ----i ABC X-Y ·z I------1 ABC XYZ~
Select for HisT. screen for I heterozygous de.letion strains ..
Second am plified de.l«ion cassette Se.lect for ArgT . screen for I
het€4"ozygous de.letion suai ns ..
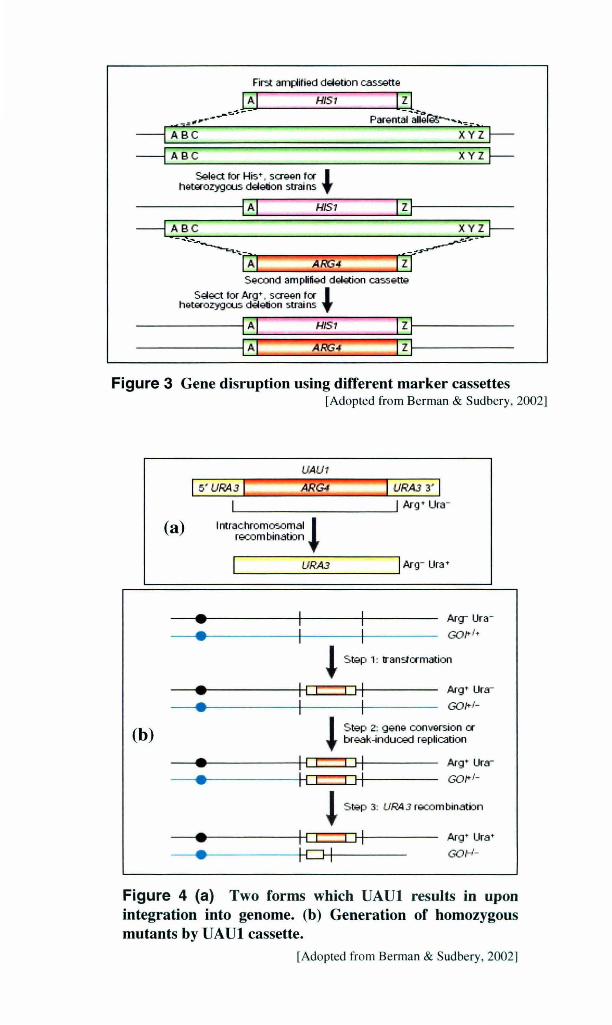
Figure 3 Gene disruption using different marker cassettes [Adopted from Berman & Sudbery, 2002]
UAU?
15' URA3! ~ ARG4 ~ ! URA3 3' I I I Arg ' Ura-
(a) I ntrachromosomal l recombinatbn
I URA.3 I Arg- Ura T
• I I Arg- Ura-
• I I GO,./· l Step 1: transtcrmation
• II I- I II ArgT Ura-
• I I GOftl -
(b) l Step 2: gene conversion or break-induced replication
• II I I II ArgT Ura-
• II I I II GO,.I-l Step 3: URA 3 recom binatbn
• II I- I II ArgT Ura'
• 101 GOr-'-
Figure 4 (a) Two forms which UAUI results in upon integration into genome. (b) Generation of homozygous mutants by UAUI cassette.
[Adopted from Berman & Sudbery, 2002]

Chapter 1
Although the overall scheme is conceptually similar to the URA blaster disruption, there are
several advantages of this system, as well as alternative uses that render it an invaluable
strategy in C. albicans molecular biology. First, it increases the frequency of
intrachromosomal recombination by ~10,000-fold. Consequently, one can use markers for
which no positive counter-selection exists in gene disruptions. Second, this system can also
be used to detect whether a specific gene is expressed under certain experimental conditions
(such as those that might exist during infection). In this case, the FLP recombinase gene
would be placed under the control of the regulatory regions of the gene to be tested, as has
already been described for genes of the SAP family [Staib et aI., 1999; Staib et aI., 2000].
1.3.3.3 One step gene disruption
Using different marker cassettes, homozygous disruption mutants can be generated after a
single transformation by co-transforming two different auxotropic markers in an appropriate
background strain, as can be seen schematically in Fig. 3. The main advantage of these
systems is that they normally require less time to achieve gene deletion, because they do not
require the recovery of the initial auxotrophy after first disruption and because it is possible
to disrupt both alleles of a gene in a single transformation event [Negredo et al., 1997].
Although this is a significant improvement from the general gene disruption strategy and
could be used to identify knockout strains more rapidly, there are technical limitations caused
by the low probability (product of the frequencies of integrative transformation) of obtaining
the desired transformant. Isolation of strains in which both copies of a gene are disrupted
may be possible only if a predictable and easy-to-check phenotype is expected.
Another strategy for disrupting both alleles of a gene after a single transformation was
developed by Mitchell and co-workers [2000]. This involved the use of UAUI cassette that
could select for Ura + Arg + double segregants in which both alleles of a gene would be
disrupted. The UAUI cassette comprises of 3 segments: a ura383'region, an intact ARG4
gene, and a ura385' region. The ura383' and ura385' regions are non-functional since they
lack the 3'and 5' region of the gene, respectively, but share a 530 bp homologous region that
upon recombination releases ARG4 and forms an intact, functional URA3 gene. Hence UA UI
integration into genome can result in two forms: a UA UI state in which only the ARG4 gene
14

Chapter 1
is functional, and a URA3 form in which the ARG4 is evicted out and only the URA3 gene is
functional [Fig. 4a]. Here, the first allele of a target locus is disrupted by integrating the
UAUi cassette and selecting for the ARG4 marker. Then the naturally occurring Ura+, Arg+
segregants are selected that have presumably arisen through a combination of gene
conversion followed by intrachromosomal recombination between the direct URA3 repeat
regions at one allele [Fig. 4b].
Another advantage of using this ingenious strategy is the rapidity with which essential genes
can be identified. The inability to yield double segregants without the wild type copy among
a specified number of transformants would indicate that the gene is essential, which the
authors have termed as the homozygote trisome(HT) test [Enloe et ai., 2000]. However, the
essentiality of genes is judged based on negative evidence, and hence there is always an
element of uncertainty attached to it. Also, the use of URA3 as a marker gene renders it
unsuitable for virulence. Moreover, selection of events such as gene conversion that result in
the generation of double segregants could also lead to loss of heterozygosity beyond the point
of targeted integration. Hence the possibility of the phenotype arising out of an extraneous
mutation from a linked locus should be verified by conventional gene-disruption approaches
and by complementation of the mutation with a wild-type copy of the gene.
1.3.4 Generation of gene disruption/deletion constructs
1.3.4.1 Cloning based approach Conventionally, gene disruption constructs were generated by cloning it in a plasmid, making
use of cassettes such as URA blaster, URA flipper and UA Ui. Initially, the gene to be
disrupted was cloned in a vector, and the marker cassette subsequently inserted into the target
gene using suitable restriction sites. The disruption construct thus generated was then
transformed into C. albicans. This was done to have long flanking regions of homology to
bring about efficient targeted integration. More importantly, cassettes with direct repeats
could not be easily PCR amplified since the repeat regions tend to recombine with each other
resulting in a shorted amplified product without the selection marker. But these cloning
strategies suffered from the limitations that these methods were very tedious, time-
15

5' target '-. primer , ...•
5' vector················ .... -, -TD.I- .1 .... •••••••••••••••• 3' vector Disruption ...... ................ ,, __ ~ __ .I................ vector
1 3~;~~~ primer
5' target ~-..,.
.f"-_....,. 3' target
Figure 5 Microhomology mediated disruption [Adapted fro m Wilson et aI. , 1999]
Figure 6 Split marker mediated gene disruption [Adapted from de Hoogt el aI. , 2000]
Disruption Cassette
Wild Type
First disruption cassette
Second disruption cassette
Wild Type

Chapter I
consuming and inflexible, as they depended on the presence of suitable restriction sites in the
target gene.
1.3.4.2 peR based strategies
PeR-mediated gene disruption strategy that was previously developed for S. cerevisiae
[Baudin et al 1993, Wach et al 1994] was initially adapted for C. albicans by Wilson et al
[1999]. In this method, long oligonucleotides were synthesized as targets for recombination
and were used to amplify the selectable marker from a vector [Fig. 5]. C. albicans disruption
cassettes were generated with 50-60 bp identical to the genomic target sequence at each end
of the selectable marker. This contrasts with the much longer flanking regions (typically
several hundred base pairs) used in the cloning approaches described above. This is a rapid
method for gene disruption, but owing to a high frequency of non-homologous
recombination in C. alb icans , extensive screening has to be done to identify transformants
with integration at the targeted locus [Wilson et al. , 1999; Weig et al. , 200 I] . A rather
expensive alternative, which has recently been described [Gola et al. , 2003], is to use long
peR primers to introduce 100 bp of flanking homology to increase the frequency of targeted
integration.
Many peR-based methods that add long flanking homology regions with relatively short
primers have also been reported [e.g. Eberhardt and Hohmann, 1995 ; Lorenz et ai. , 1995 ;
Wach, 1996] . These methods involve PeR-mediated synthesis of long regions of homology
flanking the target gene to be disrupted, followed by fusion of these regions to the selection
cassettes. All these methods, including those that introduce flanking homologies as part of
primers, involve peR amplification of the selection cassettes. Thus, these methods cannot be
easily used with selection cassettes having direct repeats, which are widely used in C.
albicans, like the URA-blaster and UA UI.
To avoid the above limitation, the use of split marker cassettes with long flanking
homologies has been reported [de Hoogt et ai. , 2000]. In this approach, the split markers are
separately peR amplified as upstream and downstream constructs. These upstream and
downstream split marker constructs have an overlapping region within the marker gene that
16

5'~ US UA
~CAG5'
(5'ends of amplification primers)
ITOq_POifmmue
5' GTC(A) 3'
3'(A) CAG 5' Upstream flank
5'GTCGAC ___ ACTAea 3' 5' AGC ~ DA ~ 5' 3'CAGCTG TGATCA 5' DS
(Sol.! and Spe.l sites flanking the marker cassette)
!
~ JUU+Spd
5'TCGAC A J' J'G TGATC 5'
Cleaved marker cassette
(5'ends of amplification primers)
I Toq-PoIymerose
5'AGC (A) 3' 3'(A)TCG 5'
Downstream flank
I r.t ONA poifmerose dATP + dGTP dCTP+dTTP
~ K/enow IT .. ONA polfmeros.-dATP+dGTP
5' G 3' 5'TCGAC ACT 3' 5'AGC 3' 3' CAG 5' J'CTG TGATC 5' 3'G 5'
Trimmed 3' ends Partially filled-in 3' ends Trimmed 3' ends
Ligase
5' ---- GTCGAC---ACTAGC ____ 3' 3' CAG CTG TGATCG 5'
Disruption construct
Figure 7 ' Directional-ligation ' approach to introduce long flanks for targeted gene disruption
[Adapted from Taneja el ai , 2004]

Chapter I
allow it to recombine with each other in vivo upon co-transformation, generating an intact
gene deletion construct in the process. The latter upon further recombination with
homologous target sequence within the genome get integrated into the target locus [Fig. 6]. It
has also been reported in case of C. neoformans that split marker cassettes result in a higher
frequency of targeted integration in comparison with intact cassettes [Fu et al. , 2006] ,
probably by recruiting the machinery involved in homologous recombination to the target
locus.
To circumvent the limitations associated with cloning and PCR based approaches, Vibha et al
[2004] also came up with a strategy in which the upstream and downstream flanking regions
of homology that are PCR amplified separately are directionally ligated to the selection
marker cassette(released from a vector) using the dinucleotide-sticky-end-ligation (DISEC)
approach [Dietmaier et aI. , 1993], as schematically represented in Figure 7. The ligation mix
is then used as such to transform C. albicans for gene disruption. Using this strategy,
disruption constructs with marker cassettes having direct repeats as well as long flanking
regions of homology can be generated that would result in efficient targeted integration .
1.3.5 Gene expression strategies
An important way of studying the function of a gene is to alter its expression. So far, this
approach has not found general application in C. albicans, mainly because there are no
efficient ways to block gene expression completely. Although gene disruption techniques are
essential tools for the analysis of gene function , a major problem arises in verifying the
essentiality of a specific gene in this diploid asexual organism. This has encouraged the
development and use of vectors that allow conditional expression of genes for studying
terminal phenotypes (even under conditions in which the gene is not expressed) and to
analyze the effect of gene overexpression on cell physiology [e.g. Mendoza el aI. , 1999].
Such approaches are also useful for functional analysis of changes in expression, which may
cause a detectable phenotype in nonessential genes.
17
Wild type allele
'-------'~ •
Wild type allele Wild type transcripts
I Firsl/r=formU/ion 10 durupl Ine /irsl o/Iele
Non functional transcript
MI Wild type allele
~--------I~ Wild type transcript
•
I Second troniformotion to bring the second allele under the control 0/0 regulatable promoter
Disrupted a llele
Non functional transcript
Regulatable
pro. L....-_R_eg_UI_at_e_d _al_le_le_----J~ Regulatable transcript
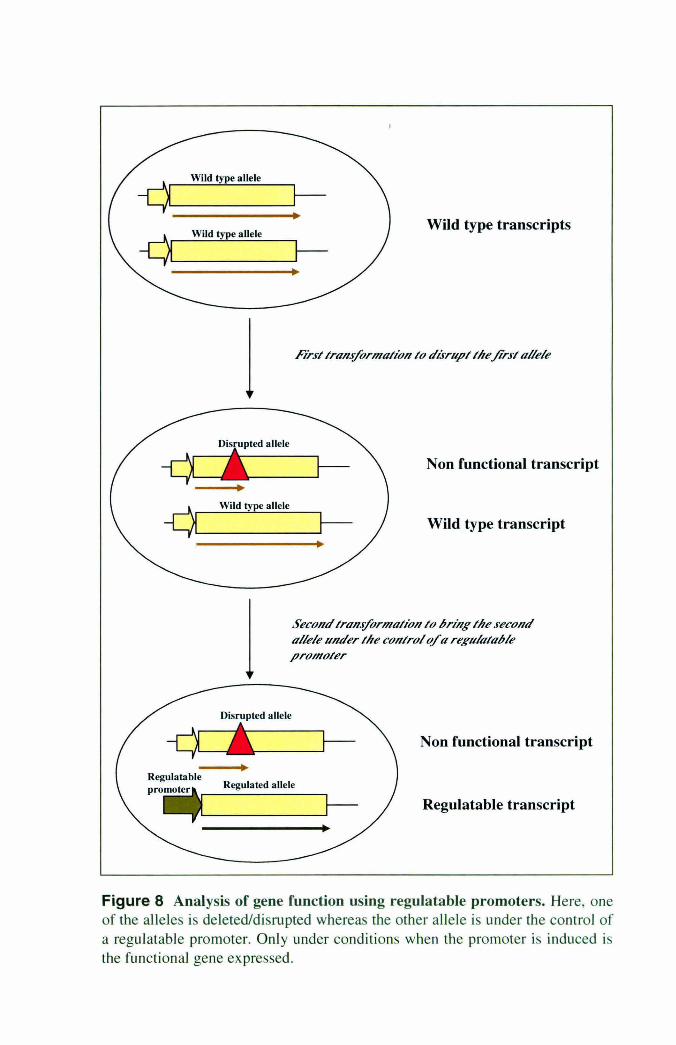
Figure 8 Analysis of gene function using regulatable promoters. Here, one of the alleles is deleted/disrupted whereas the other allele is under the control of a regulatable promoter. Only under conditions when the promoter is induced is the functional gene expressed.

Chapter 1
1.3.5.1 Regulatable promoters
A straightforward approach to accurately attribute the role of a gene to a particular function
or phenotype, particularly its essentiality, is to bring it under the control of a regulatable
promoter. The presence/absence of phenotype when the promoter is induced as opposed to
when it is shut off would clearly indicate the role of the regulated gene in contributing to the
phenotype. The effect of unintented changes elsewhere in the genome contributing to the
phenotype can in this case be ruled out. In C. albicans, a few regulatable promoters have
been used to study function of genes by altering its expression, where one of the alleles is
deleted whereas the other allele is brought under the control of the regulatable promoter [Fig.
8]. Regulated gene expression with variable levels of background expression and induction
ratios has be achieved with the GAL1 [Gorman et al 1991], the PCK1 [Leuker et al 1997,
Rademacher et al 1998], the SAP2 [Morschhauser et al 1998], the MRP 1 [Jiang et al 1997],
the HEX1 [Cannon et al 1994], the MAL2 [Geber et al 1992, Brown et al 1996], the MET3
[Care et al 1999] or TET [Nakayama et at., 2000; Park et at., 2006; Roemer et at., 2003]
promoters. They are either promoter induble (as in case of MAL2, SAP2, PCK1, GAL2 and
TETpromoters) or promoter repressible (as in case of MET3 and reverse TETpromoters).
Tight regulation has been found with the MAL2, MET3 and TET promoters. The MAL2
promoter is induced (4-5 fold) when maltose is the sole carbon source while repressed in the
presence of glucose [Brown et at., 1996]. The MET3 promoter shows a ~85-fold range of
expression-using either GFP or URA3 as a reporter--dependent on the presence or absence
of either methionine or cysteine. In the presence of either amino acid, expression is reduced
to levels that are close to background [Care et al 1999]. In case of TET promoter based
activator/repressor expression system, tetracyclines control the ability of tetR-based activator
and repressor molecules to bind to tetO promoters in opposite ways (and hence a strain which
stably express the TET transactivator is required to make this promoter functional). These
combinations allow tight control of tetO-driven genes, both in a direct (tetracycline
repressible) and reverse (tetracycline-inducible) dual system. The tetracycline-repressible
system has been made use of to study genes implicated in virulence [Nakayama et at., 2000]
as well as in a genome wide GRACE approach (described below) to bring all C. albicans
genes under the control of the TET promoter [Roemer et at., 2003] with the ultimate goal of
18

Chapter 1
generating a complete genomic set of strains that can be conditionally repressed by treatment
with tetracycline. Recently, Park et al [2006] introduced a reverse tetracycline-inducible
promoter and transactivator system (conforming to the C. albicans genetic code) to regulate
gene expression (such as induction of gene expression under conditions where it is normally
not expressed) in this diploid organism.
1.3.5.2 Reporter genes
A good reporter gene is crucial to study gene function, protein localization, and gene
regulation. However, expression of heterologous genes in C. albicans has often proved
difficult, owing to the noncanonical genetic code in C. albicans [Santos and Tuite, 1995].
Although C. albicans genes are usually expressed in S. cerevisiae without difficulty, the
converse is not normally true [Kirsch et aI., 1984]. A second problem is the vehicle by which
a reporter gene is introduced into the test strain. Owing to possible cell-cell variations in copy
number of replicative plasmids in C. albicans, gene expression studies with this system has
remained difficult. The stability of chromosomally integrated reporter genes makes them
more suitable for the study of gene regulation than plasmid-based constructions.
The first reporter system described in C. albicans was based on a heterologous gene
encoding K. lactis B-galactosidase, which could be assayed in both intact cells and cell
extracts. Measurable expression of the LAC4 gene was observed only when it was present in
high copy numbers and not after integration into the genome [Leuker et al., 1992]. Another
system made use of C. albicans URA3, which turned out to be sensitive (a single copy per
cell could be detected) but required cell extracts and a suitable auxotrophic host strain for
analysis [Myers et al., 1993]. Measurable expression of R. reniformis luciferase after stable
chromosomal integration as a single copy has also been achieved in C. albicans [Srikantha et
aI., 1996]. Similar results have been achieved for genetically modified Aequorea victoria
GFP [Morschhauser et aI., 1998]. The R. reniformis luciferase gene, naturally lacking any
CTG codons, provides a sensitive bioluminescent reporter that is devoid of any background
in C. albicans. Inducible expression of luciferase was obtained with the GALl promoter,
constitutive luciferase expression was obtained with the EFla2 promoter, and phase-specific
expression could be demonstrated with WHll and OP4 promoters [Srikantha et aI., 1996].
19

Chapter 1
Morschhauser et al [1998] placed genetically engineered jellyfish GFP under control of the
C. albicans ACT1 promoter and showed efficient expression of the reporter after single-copy
integration into the genome. Substitution of a normal leucine-specific codon for the single
CTG codon in the GFP gene and introduction into the chromophore of mutations previously
identified to increase fluorescence [Cormack et ai., 1998] were necessary to confer a
fluorescence phenotype on the cells. The GFP reporter system offers the major advantage in
that gene induction can be monitored in living organisms at the level of single cells. Future
developments within this field will entail the introduction of nucleotide changes in the
chromophore to yield variants with different excitation or emission spectra, allowing for the
simultaneous detection of different proteins.
1.3.5.3 In vivo expression technology
In vivo expression technology (IVET) IS a promlsmg new technique with the ultimate
objective of identifying genes expressed in vivo within a natural or artificial host, thus
leading to their isolation and testing in any model system. This technique was originally
described for pathogenic bacteria [Mahan et ai., 1993; Slauch et ai., 1994] and was improved
with the use of antibiotic-based selection [Mahan et ai., 1995; 2000]. It relies on the
identification of in vivo expressed genes by constructing a promoter library with a suitable
reporter gene activity that can be monitored either in vivo or in vitro. Using IVET, gene
expression patterns during infection can be better understood leading to elucidation of the
complex interactions between a fungal pathogen and its host.
An elegant adaptation of the IVET system makes use of flipase (FLP) recombinase in C.
albicans [Staib et ai., 1999]. The FLP gene that encodes the site-specific FLP recombinase
was genetically modified for C. albicans and fused to the promoter of the SAP2 gene, one of
the secreted aspartic proteinases and a virulence factor of this organism. Expression of the
PSAP2-FLP fusion was monitored by FLP-mediated recombination of a dominant selectable
marker that conferred resistance to mycophenolic acid, which was flanked by direct repeats
of the FLP-recognition target. This system was extended to analyse the role of the secreted
aspartyl proteinase family during infection [Staib et aI., 2000] and should in theory be
adaptable to perform a broad screening of the C. albicans genome. An additional advantage
20

Chapter 1
of this system is that it allows the detection of transient expression patterns due to the
permanent (deletion) phenotype induced by FLP expression, thus circumventing one of the
limitations of the IVET strategy. Through this study, the expression of secreted aspartyl
proteinase (SAP) family proteins was reported at different stages of infection and localization
in the host [Staib et aI., 2000].
1.4 Large scale genetic analysis in C. albieans
1.4.1 Technicallimitations
Genome wide analyses of genes have been extensively applied in S. cerevisiae using its
powerful genetics, especially after its genome sequence was released in 1996. But it is yet to
take off in full measure in C. albicans, for several reasons. Firstly, one must be able to
culture the organism as a haploid or generate homozygotes conveniently for analysis of
recessive insertional mutations, both presently impossible for C. albicans. Secondly, there is
no convenient method available for random insertional mutagenesis, owing to a paucity of
functional transposable elements. Moreover, owing to the diploid status of C. albicans, such
a system would allow only heterozygous disruptions, often not leading to a specific
phenotype. The lack of a sexual cycle precludes the use of genetic crosses to render the
mutations homozygous. There are also reports of both stable and transient aneuploidy,
including chromosome loss induced and apparently selected for by particular growth
conditions leading to homozygosity of all of the rest of the genes in the affected
chromosome, thus yielding a strain that might be far from isogenic with the parent
[Rustchenko et al., 1994; Rustchenko et al., 1997; Se1mecki et ai., 2005]. In spite of these
technical difficulties, large scale forward genetic approaches, in which a given phenotype is
in hand and the genes whose products confer that phenotype is identified, is an invaluable
tool in the study of medically relevant fungi such as C. albicans. For example, a collection of
randomly mutagenized strains can be screened for a given phenotype, such as drug
resistance, yeast to hyphal transition defects, etc.
21

Chapter 1
1.4.2 Transposon as tools for large scale forward genetic studies
Notwithstanding the dearth of naturally occurring transposons and its diploid genome,
transposons have been used to generate pools of random insertional heterozygous mutants in
C. alhicans, for various large scale forward genetic applications. Transposon mutagenesis has
many advantages over chemical mutagenesis including a high mutation frequency without
killing the organism, single hit mutations, the ability to recover the mutated gene after
mutagenesis, the ability to introduce selectable markers in strain construction, and use as a
portable region of homology for genetic manipulations [Kumar et aI., 2002]. This has led to
various strategies (briefly descibed below) in employing transposons to study different
phenotypes using the pool of heterozygous mutants.
Insertional mutagenesis by transposons have been facilitated by in vivo and in vitro
transposition. In S. cerevisiae, in vivo transposition has been achieved by shuttle mutagenesis
using Tn3 [Seifert et aI., 1986] and Tn 10 [Snyder et aI., 1986], using E. coli as a surrogate
host,. Here, the host DNA carried in a vector within E. coli is inserted with transposon
(supplied in trans), which carry yeast and bacterial marker genes. The mutagenized DNA is
then isolated and transformed into the host strain and integrated into the genomic DNA by
homologous recombination. In recent years, in vivo transposition has largely been replaced
by in vitro transposition, in which transposition reaction is carried out in a test tube using
transposon, the corresponding transposase and target DNA. The transposon inserted target
DNA is then introduced into the host by integrative transformation. The in vitro strategy has
advantages over in vivo transposition in that it results in higher transposition frequency and is
less biased for specific regions of the target molecule [Devine and Boeke, 1994].
1.4.2.1 Advances in transposon technology
Signature-tagged mutagenesis
Signature-tagged mutagenesis is a modification of transposon-mediated mutagenesis, in
which each transposon mutant is tagged with a different DNA sequence [Hensel et al., 1995].
This system has been used to identify virulence genes, using a mouse model of typhoid fever
by S. typhimurium. In this approach, pools of strains individually tagged with specific
oligonucleotides are used to infect an animal and, after a certain period of time, successfully
22
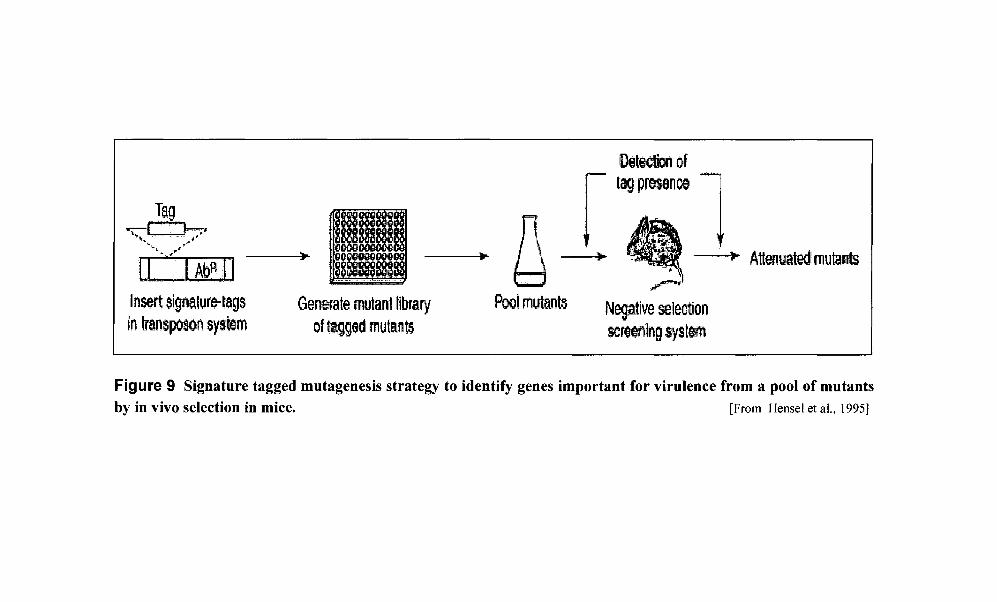
_Tag "f~~'
'I'i":':~j'~ 11 Insert signature-tags In Imnsposonsystem
Generate mutant library of tagg.ed mutants
Pool mutants
r Oetection of lag presence 1
=~~ Attenuated mutants ~
Negative selection screerlillg system
Figure 9 Signature tagged mutagenesis strategy to identify genes important for virulence from a pool of mutants by in vivo selection in mice. [From Hensel et aI., 1995]

Chapter 1
established cells are recovered and used as a DNA source for hybridization (and
identification) of unsuccessful clones [Fig. 9]. The strategy has been extensively used in
bacteria and, recently, also with some fungal pathogens. In the opportunistic fungus C.
glabrata (responsible for about 15% of mucosal and systemic candidiasis), Cormack and
coworkers used this strategy to screen for altered adherence to human epithelial cell cultures.
Mutants with enhanced or diminished adherence were selected and, from among them, a
surface adhesin (EPA1) was isolated. This protein probably encodes a glucan-cross-linked
cell wall protein that binds to host-cell carbohydrate, specifically recognizing asialo-Iactosyl
containing carbohydrates [Cormack et al. 1999]. Interestingly, disruption of the EPA I gene
had no discernible effect on virulence in a mouse model of gastrointestinal or vaginal
infection, emphasizing the importance of screening for genes relevant for in vivo growth. The
use of this system in Aspergillus fumigatus enabled Brown and coworkers to isolate mutant
strains that showed strong hybridization signals from the inocula but weak or absent
hybridization signals from fungi recovered from mouse lungs. One of the strains unable to
replicate in vivo was further characterized and found to have an insertion in the pabaA
promoter, which encodes para-amino-benzoic acid synthetase, an enzyme that catalyses a late
step in the biosynthesis of folate [Brown et aI., 2000]. This strategy was also used in C.
neoformans to isolate mutants that are either unable to proliferate or proliferate even better in
an animal host, as compared with the parental strain. Different mutations were found to
reside in the actin locus and, most importantly, the authors were able to identify reduced and
enhanced virulence mutants [Nelson et al., 2001].
In vitro mutagenesis
Castano et al [2003] described and characterized a method for in vitro insertional
mutagenesis of the yeast pathogen Candida glabrata using the bacterial transposon Tn 7,
which was used to mutagenize a C. glabrata genomic fosmid library. This method is of some
general interest because the generation of mutants requires only two steps: in vitro
mutagenesis by Tn7 followed by homologous recombination into the target genome (here C.
glabrata). The Tn7 transposon was modified to allow its use in C. glabrata and to facilitate
the recovery of DNA flanking insertion sites for mutants of interest. This method
demonstrates that it can be used in the efficient generation of thousands of randomly
23

Chapter 1
distributed insertion mutants, possessing an array of phenotypes. In theory, therefore, this
method can easily be applied to any organism with efficient homologous recombination.
Generation of homozygous mutant collection and test of essentiality
Davis et al. [2002] sought to identify essential genes and regulators of pH-dependent
filamentation, a trait linked to virulence. The authors took advantage of the UAUi cassette,
which permits selection of homozygous mutants from heterozygotes [Enloe et aI., 2000].
Heterozygous mutants occasionally undergo mitotic recombination or gene conversion to
give rise to homozygotes, which are selected because they express both alternate markers
encoded by UAUi. Heterozygous UAUi insertion mutants can express only one of the
markers. Mutations were made through insertions of a UAUi-marked transposon into a C.
albicans genomic library, and insertions in open-reading frames (ORPs) were identified by
sequencing. Of 253 orf::UAUi insertions transformed into C. albicans, 217 yielded an
average of 4-8 homozygous mutants after the UA Ui-homozygosity selection. On the basis of
C. albicans genome sequence assembly number 19, these insertions lie in 197 ORFs [Nobile
et aI., 2003]. Insertions into 36 genes failed to yield homozygous mutants. These putatively
essential genes include 20 with homologs that are essential in S. cerevisiae; only three
homo logs of essential S. cerevisiae genes were clearly nonessential in C. albicans. There
were also 12 genes whose S. cerevisiae homo logs are not essential, and four genes without a
clear S. cerevisiae homolog. These findings underscore the functional significance of non
conserved C. albicans genes and suggest that the spectrum of essential genes might be
broader in C. albicans than in S. cerevisiae.
1.4.3 Other advances in large scale genetic studies
1.4.3.1 REMI
Restriction enzyme-mediated integration (REM I) is a technique that involves random
integration of transforming DNA with a selectable marker, digested at the ends with an
appropriate restriction enzyme, into the host genomic DNA. The same enzyme is included in
the transformation mix and apparently enters the nucleus with the target DNA. where it
cleaves the genomic DNA at random sites to direct DNA integration. In C. albicans, the
MAL2 promoter fused to the URA3 gene was reported to permit a stable transformation, easy
24

_ Step '1 : Gene Replacernent and IBar Code Addition
HIS3
_ Step 2 : Co'nd'itional ,Express,lion by Promoter Replacement
----II I ISC11 HIS3
Figure 10 Identification of essential genes in Candida albicansby gene replacement and conditional expression (GRACETM). One allele of a target gene is precisely inactivated by targeted integration of a deletion cassette containing selectable marker (HISl). In a second transformation event using fungal marker B, the native promoter of the second allele of the candidate gene is replaced by a tetracycline controlled promoter. ['Bel' & 'Be2' represent the 2 bar codes that are introduced into the disruption cassette during peR amplification.
[From Roemer et aI., 2003]

Chapter 1
recovery of the integrated DNA and inducible gene expression [Brown et aI., 1996]. This
strategy was again used by the same group in their search for mutants defective in
filamentous growth and led to the isolation of the SEC14 gene from C. albicans. Although
this strategy has allowed the isolation of virulence factors in several plant pathogenic fungi
[Riggle et aI., 1997; Riggle and Kumamoto, 1998; Maier and Schafer, 1999], it has to be
borne in mind that it may often lead to transformants with partial, or even aberrant,
phenotypes that may be difficult to analyse.
1.4.3.2 GRACE
Roemer et al. [2003] set out to define C. albicans essential genes and to provide rational
criteria for their prioritization as prospective drug targets. Their strategy was to analyze a set
of C. albicans strains whose expression was tetracycline-regulated. Conditional expression
was achieved by deleting one allele and fusing a second allele to a tetracycline-repressible
promoter (PTET), creating a 'GRACE' (gene replacement and conditional expression) strain
[Fig. 10]. Essentiality was assessed by the sensitivity of a strain to tetracycline, which blocks
DNA-binding of the GRACE transcriptional activator, and also sensitivity to 5-FOA (5-
fluoro-orotic acid), which selects for loss of the GRACE activator gene cassette. This
collection of GRACE strains had 1152 genes under PTET regulation that included 823 genes
whose S. cerevisiae homologs are essential. The authors identified 567 essential C. albicans
genes in the entire collection. The astonishing finding is that only 61 % of C. albicans genes
that had essential S. cerevisiae homologs were also essential in C. albicans. Such modest
overlap between the C. albicans and S. cerevisiae essential gene sets might reflect a
substantial difference in gene or pathway function, or perhaps extensive gene duplication in
C. albicans. Constructing such a comprehensive C. albicans conditional mutant strain
collection should significantly accelerate functional annotation of the C. albicans genome to
aid both basic research and antifungal drug discovery.
1.4.4 DNA Microarrays
In a microarray, thousands of DNA spots representing genomic features (usually open
reading frames, or ORFs) are arrayed on a solid support, usually a glass microscope slide.
The DNA can be oligonucleotides, plasmids, genomic fragments or PCR products. mRNA
25

Chapter 1
populations that are derived from two different strains or under two different conditions are
enzymatically labeled with two different fluorescent dyes and are hybridized to the DNA
probes on the microarray. Induction (or repression) of each gene is determined by the
intensity ratio between the two fluorescent dyes that denote the relative quantity of RNA
transcript associated with each spot on the array. A key advantage of this technology is that
patterns of expression involving different conditions can be identified and thousands of genes
can be tracked simultaneously. The well-known obstacles to microarraying, mostly its cost
and complexity, have been reduced as the technology has become more widespread.
Commerical and academic software is easing the task of data analysis, though ascribing
biological meaning to array data remains a formidable challenge. An effort to create
Minimum Information About Microarray Experiments (MIA ME) standards to apply to
publication of array work has eased the problems encountered when comparing data from
different laboratories [Brazma et aI., 2001].
With the completion of the C. albicans genome-sequencing project, several investigators are
using DNA microarray technology to study gene expression on a genome wide scale, and
consequently, more than 30 papers detailing microarray experiments in C. albicans have
been published since then. The conditions investigated have ranged from growth on blood
after transfer from minimal medium [Fradin et aI., 2007] to changes in gene expression
associated with the acquisition of drug resistance [Cowen et aI., 2002; Rogers and Barker,
2002; Karababa et al., 2004]. Among other results, use of microarray technology has
identified the genes involved in yeast to hyphal transition [Nantel et aI., 2002], white to
opaque switching [Lan et aI., 2002], shown that Candida lacks a stress response found in S.
cerevisiae [Enjalbert et al., 2003] and looked at the patterns of gene expression in cells
lacking such important transcription factors as Nrg 1 p, Mig 1 p, and Tup 1 p [Murad et al.,
2001].
1.4.5 Approaches/strategies for genome wide studies in C. albicans Any comprehensive large scale genome wide study of C. albicans involving a library of
mutants has to consider the complication associated with the diploid genome status of this
yeast. Creating a 'GRACE' (gene replacement of one allele and conditional expression of the
26

No. of copies of Sensitivity to Strain Virulence inhibitors of target target gene
protein
____ .....Ji~
WT
Wild Type Two Normal Normal I Transformation
Heterozygous One Attenuated Hypersensitive mutant IS
Hetrozygote
Figure 11 Haploinsufficiency phenomenon as a phenotype to identify genes implicated in different processes.

Chapter 1
other allele) strain set of C. albicans, in which the expression of a particular gene in each
strain can be regulated, has been a big step in resolving this difficulty. Nevertheless, there is
a striking limitation for the GRACE strain report [Roemer et ai., 2003]. Though the claims
are based on a putative collection of 1152 strains, results from only 100 specific strains are
described. Since the GRACE report is not accompanied by comprehensive, accessible data,
the claims cannot be verified and the C. aibicans community is not fully benefited from the
work. Hence it becomes imperative to put strategies in place that can investigate genes that
are involved in various phenomenon, like progression of virulence, that is yet to be studied
on a genome wide scale. Two strategies described below, haploinsufficiency phenomenon
and antisense technology, seems to be promising approaches that can be exploited to generate
mutant libraries that can help resolve complex phenomenon such as host-pathogen
interaction and pathogenicity of C. aibicans.
1.4.5.1 Haploinsufficiency Phenomenon
Knocking out one of the alleles of a gene in a diploid organism may lead to an insufficient
concentration of the gene product to allow normal growth and division, especially if the
protein encodes essential function. Often, this loss of one functional copy of a gene produces
a noticeable phenotype, although this may be in a less severe form compared to that produced
by disrupting both alleles [Fig. 11]. Such a phenomenon that lead to a discernable phenotype
in a heterozygous mutant is termed as haploinsufficiency. Similarly, when the dosage of a
single gene (drug target) is lowered from two copies to one copy in diploid cells, it results in
drug-induced haploinsufficiency in which the heterozygote displays increased drug
sensitivity compared with wild-type strains. Giaever et al. [1999] pooled 233 "bar-coded"
heterozygous S. cerevisiae strains, grew them competitively in the presence of a drug whose
target in yeast cells was known, and successfully used DNA microarrays to identify the
heterozygous drug target strain whose growth was compromised in the presence of the drug,
thus revealing the promise this strategy holds.
Recently, a large-scale forward genetic analysis of filamentous growth was undertaken in C.
aibicans, taking advantage of haploinsufficiency and transposon mutagenesis [Uhl et ai.,
2003]. This loss-of-function screen set out to identify genes that govern the morphological
27

Chapter 1
transition between yeast (blastospores) and filamentous cells (hyphae and pseudohyphae).
Such genes have been a major focus of C. albicans research because many mutant strains that
are defective in this morphological transition have attenuated virulence in experimental
models [Liu et ai., 2001]. Here, the authors developed their screen based on the fact that
many genes that regulate filamentation display haploinsufficiency; in other words, loss of
just one allele causes a detectable phenotype [E.g. Liu et ai., 1994; Kohler et ai., 1996; Braun
and Johnson, 1997]. Random insertions were created through in vitro transposition of a
URA3-marked Tn7 based transposon into a C. albicans library. Transformation of the pooled
insertion library into C. albicans yielded a set of 18 000 heterozygous C. albicans mutant
strains. Screening the heterozygote collection identified 146 genes that are involved in the
yeast-hypha transition, including 39 putative filamentation regulators that lack S. cerevisiae
homologs. In addition, some mutants had defects in hypha formation that occurred
specifically in response to serum, an inducer to which C. albicans and not S. cerevisiae
responds. Because serum is encountered by C. albicans during infection, this response has
always been considered relevant to pathogenicity. This work provides clear evidence for the
functional significance of many unique C. albicans genes. In addition, it allows the dissection
of yeast-hypha regulatory pathways with a greater resolution than could be achieved by
relying solely on the S. cerevisiae model. More importantly, this work demonstrates that
haploinsufficiency profiling can be exploited as a valuable approach for genome scale
identification of genes involved in different processes in diploid organism such as C.
albicans.
1.4.5.2 Post-transcriptional gene silencing
A variety of RNA-mediated gene silencing methods that inhibit genes at the post
transcriptional level have been identified in different organisms. The most common
approaches involve the introduction of antisense RNA, double-stranded RNA (also called
RNA interference or RNAi mediated by short interfereing RNA or siRNA) or sense
transgenes (also called co-suppression in plants or quelling in fungi). These phenomena can
be collectively termed as post-transcriptional gene silencing (PTGS) and can serve as the
basis for controlling the expression of specific genes. These technologies have been the topic
28

Chapter 1
of a number of research studies aimed at understanding the underlying mechanism(s) and
also application in drug discovery, biomedicine, agriculture and functional genomics.
In C. albicans, like in S. cerevisiae, RNAi has not been observed, nor are the homologues of
molecular players involved in this mechanism present within the genome. (RNAi, on the
other hand, is a potent gene silencing mechanism in S. pombe.) Similarly, quelling, which has
been established and made use of in N. crassa for gene silencing, has not been reported in C.
albicans. This leaves the question of antisense strategy open for manipulation, though it has
yielded mixed results in S. cerevisiae, for which there are more potent large scale genetic
screens available.
Antisense Technology
Antisense strategy take advantage of antisense transcripts that are made in trans to silence
cognate gene expression at the post transcriptional level. It has been suggested that antisense
RNA can act within the nucleus or cytoplasm to interfere with multiple steps of
eukaryotic gene expression, resulting in degradation of the target mRNA or steric hindrance
of translation by the complementary binding of antisense RNA to its cognate mRNA, thereby
preventing the latter from translating into the gene product.
The genetic tractability of the fission yeast S. pombe and its amenability to antisense-RNA
mediated gene inhibition have made it an ideal model for studying factors that affect
antisense gene regulation in vivo [Arndt et aI., 1995; Raponi et aI., 2000]. Using a systematic
approach, it has been shown that gene inhibition is dependent on the concentration of
antisense RNA and that co-localization of antisense and target genes does not affect the level
of target gene suppression in this system. Additionally, the size of the antisense transcript,
and the region to which it is targeted, affects the efficacy of target gene inhibition [Raponi et
aI., 2000]. In addition, a unique dominant genetic screen has been established to identify
several host-encoded factors (antisense-enhancing sequences) that dramatically enhance
target gene silencing when co-expressed with antisense RNA [Arndt et aI., 2000], the role of
which is yet to be clearly understood.
In 2000, DeBacker et al. used C. albicans antisense cDNA libraries under the control of the
galactose-inducible promoter of the C. albicans GALl gene to screen for genes that are
29

Genomic { loc i
Genomic { loc i
Integration at GAL 1locus~
GAL1p ~ ----<¢==Jf--------~¢==JI------! + Galactose
Marker
Antisense RNA ~
--~¢==JI----
"--',-,Integration at Gene X locus
~ Gene X with endogenous promoter
I ~
L + Galactose
Antisense RNA + ~ Promoter interference ~
------c ... t ¢==J-----cJt------.---~ ~
Sense RNA
Promoter- less Gene X
Neutralisation of sense RNA by antisense RNA
~ 411
L Impaired growth if Gene X is critical
Figure 12 Antisense-based functional genomics approach for identifying genes critical for growth of C. olbicons. [From SangJard, 200 I]

Chapter 1
critical for growth in C. albicans. Homologous integration of the cloned antisense cDNAs
occured at the C. albicans endogenous GALl promoter or at the genomic locus
corresponding to the cDNA insert [Fig. 12]. In both cases, expression of an antisense RNA
complementary to the sense RNA is observed under inducible conditions and leads to the
formation of a double-stranded RNA, therefore resulting in gene inactivation. An additional
effect, referred to as promoter interference, is observed when integration occurs at the
genomic locus corresponding to the cDNA insert. In this case, the presence of two
convergent promoters flanking one allele of the gene leads to physical collision of the
transcription machineries working on opposite strands. A growth defect was observed in 198
of the 2000 transformants screened and 43% of them had a growth defect only in the
presence of the GALl inducer. Characterization of these transformants resulted III the
identification of 86 genes that are critical for C. albicans growth, of which 45 were of
unknown function and 33 did not have a homolog in other organisms [DeBacker et al.,
2000]. The limitations of this strategy was that the GAL I promoter that was used to regulate
gene expression in the antisense library is somewhat leaky in C. albicans. Moreover, such a
library cannot to be extended to screen for genes involved in virulence since URA3 that is
used as a marker gene in this library is an important virulence determinant in itself, and can
hence complicate such studies by modulating virulence by its positional effect. Nevertheless
this large scale study provided as a proof of principle that antisense strategy can be relied
upon to do genome wide forward genetic analysis in this diploid opportunistic pathogen.
30






![Atrophic Vaginitis Haefner.pptx [Read-Only]...9/17/2019 3 •pH: Vaginal pH in atrophic vaginitis is more than 4.5 with pH indicator strip or cotton swab inserted into the lateral](https://img.pdfslide.us/doc/110x75/5e615f0cd1cd2d1294685516/atrophic-vaginitis-read-only-9172019-3-aph-vaginal-ph-in-atrophic-vaginitis.jpg)




![Vaginitis: Making Sense of Over-the-Counter Treatment Optionsdownloads.hindawi.com/journals/idog/2007/097424.pdf · with a yeast vaginitis [9]. Further, women with a previous clinical](https://img.pdfslide.us/doc/110x75/5f08fb0d7e708231d424a82a/vaginitis-making-sense-of-over-the-counter-treatment-with-a-yeast-vaginitis-9.jpg)

















