Embed Size (px)
Citation preview

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 1/28
University of Cebu – BaniladCollege of Nursing
Cebu City
A Resource Unit on
COPD: CYSTIC
FIBROSISSubmitted by:
BSN 4 – L Group 35:Magalona, Roselle B.
Milallos, Mark Gilbert S.Mondido, Ryan Anthony P.
Monilar, Mary Grace B.Munda, Angela May O.
8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 2/28
Submitted to:
Ms. Ramelda G. Villarazo, RNClinical Instructor
University of Cebu – BaniladCollege of Nursing
Cebu City
A Resource Unit onCYSTIC FIBROSIS
GENERAL OBJECTIVES:After 90 minuteS of lecture – discussion the BSN $ - L Group 35 students will be able to acquire adequate knowledge, gain competen
skills and develop positive attitudes on the concept CYSTIC FIBROSIS.
SPECIFICOBEJECTIVES
CONTENTS METHODOLOGY
T.A RESOURCES
EVALON
After 90 minutes
of lecturediscussion theBSN 4 – L Group35 students will
be able to:
1. Discuss theoverview of
Cystic Fibrosis.
INTRODUCTION
Cystic fibrosis (also known as CF or mucoviscidosis) is a commonhereditary disease which affects the entire body, causing progressivedisability and often, early death. The name cystic fibrosis refers to thecharacteristic scarring (fibrosis) and cyst hairy within the pancreas, firstrecognized in the 1930s. Difficulty breathing is the most serious symptomand results from frequent lung infections that are treated, though notcured, by antibiotics and other medications. A multitude of othersymptoms, including sinus infections, poor growth, diarrhea, andinfertility result from the effects of CF on other parts of the body.
CF is caused by a mutation in a gene called the cystic fibrosis transmembrane conductance regulator (CFTR). This gene helps createsweat, digestive juices, and mucus. Although most people without CFhave two working copies of the CFTR gene, only one is needed to preventcystic fibrosis. CF develops when neither gene works normally. Therefore,CF is considered an autosomal recessive disease.
8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 3/28
2. Define related
terms.
CF is most common among Caucasians and Ashkenazi Jews; onein 25 people of European descent carry one gene for CF. Approximately30,000 Americans have CF, making it one of the most common life-shortening inherited diseases. Individuals with cystic fibrosis can bediagnosed prior to birth by genetic testing or in early childhood by asweat test. There is no cure for CF, and most individuals with cysticfibrosis die young — many in their 20s and 30s from lung failure.Ultimately, lung transplantation is often necessary as CF
Cystic fibrosis is a genetic disorder that affects the digestive systemand lungs. It causes children to be more likely to have infections in theirlungs repeatedly. This happens because there is a disruption in thenormal function of the body’s epithelial cells. These cells linepassageways in the liver, lungs, pancreas, reproductive, and digestivesystems. The skin’s sweat glands are also made up of epithelial cells.Patients with cystic fibrosis continuously have an excessive buildup of mucus in the lungs and passageways. Cystic fibrosis poses a seriousthreat to patients because of the excessive mucus buildup in the lungs. The mucus is very thick and can create the perfect environment forbacteria to develop and grow, especially pseudomonas aeruginosa, whichcan cause severe life-threatening infections or nosocomial infections in
persons with cystic fibrosis. A cystic fibrosis sufferer has defective CFTRs(cell membranes), and their immune system is unable to offer properimmune response to fight such infections. Therefore, these infections canbe deadly.
A baby with cystic fibrosis usually gets this infection by the age of 3. By the age of 10, the infection has become chronic in many patients.Many cystic fibrosis sufferers do not live to be in their 40s.
Another danger of cystic fibrosis is maldigestion due to a poor-working pancreas. This can lead to symptoms of steatorrhea andmalabsorption. The stools will become greasy and large, and may evenhave visual droplets of fat. With this condition, the patient might have adifficult time gaining weight.
People who have CF produce a defective version of a protein calledCFTR (Cystic Fibrosis Transmembrane Conductance Regulator). This isresponsible for transport of salts and water across cell membranes. Thismeans that in certain parts of the body, the secretions lack water,becoming sticky.

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 4/28
3. Review the
Anatomy andPhysiology of the
RespiratorySystem.
DEFINITION OF TERMS
1. Epithelial cells – cells that cover the surface of the body and line itscavities. Epithelial cells help to protect or enclose organs; some producemucus or other secretions. Certain types of epithelial cells have tiny hairscalled cilia, which help remove foreign substances2. Fibrous Tissue – Tissue composed of bundles of collagenous whitefibers between which are rows of connective tissue cells. It is simplytermed as scar.3. Mucus – is a slippery secretion produced by, and covering, mucous membranes. It is a viscous colloid containing antiseptic enzymes (such aslysozyme) and immunoglobulins that serves to protect epithelial cells inthe respiratory, gastrointestinal, urogenital, visual, and auditory systemsin mammals; the epidermis in amphibians; and the gills in fish. Mucusalso contains mucins, produced by goblet cells in the mucous membranesand submucosal glands, and inorganic salts suspended in water. Theaverage human body produces about a liter of mucus per day.3. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)– is a cAMP-regulated chloride channel that resides in the apicalmembrane of many epithelial cells. Channel opening requires
phosophorylation of serine residues in an intracellular regulatory domainby protein kinase A and as the binding and hydrolysis of ATP byintracellular nucleotide binding domains. Besides conducting the chlorideion, CFTR also regulates the function of other membrane proteins, directlyor indirectly, notably the outwardly rectifying chloride channel and theepithelial sodium channel. The disease cystic fibrosis is caused bymutations in CFTR, which can result in defective protein production,defective processing and degradation in the endoplasmic reticulum, ordefective channel pore properties or gating properties.4. Pseudomonas aeruginosa – is a common bacterium which cancause disease in animals and humans. It is found in soil, water, skin flora and most man-made environments throughout the world. It thrives not
only in normal atmospheres, but also with little oxygen, and has thuscolonized many natural and artificial environments. It uses a wide rangeof organic material for food; in animals, the versatility enables theorganism to infect damaged tissues or people with reduced immunity. The symptoms of such infections are generalized inflammation andsepsis. If such colonization’s occur in critical body organs such as the

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 5/28
lungs, the urinary tract, and kidneys, the results can be fatal.5. Nosocomial Infection – are infections which are a result of treatmentin a hospital or a healthcare service unit, but not secondary to thepatient’s original condition. Infections are considered nosocomial if theyfirst appear 48 hours or more after hospital admission or within 30 daysafter discharge.
THE ANATOMY AND PHYSIOLOGY OF THE RESPIRATORY SYSTEm
FUNCTIONS:1. Gas exchange. The respiratory system allows oxygen from the air
to enter the blood and carbon dioxide to leave the blood and enterthe air.
2. Regulation of blood pH. The respiratory system can alter blood pHby changing blood carbon dioxide levels.
3. Voice production. Air movement past the vocal cords makes soundand speech possible.
4. Olfaction. The sensation of smell occurs when airborne moleculesare drawn into the nasal cavity.
5. Innate immunity. The respiratory system provides protection
against some microorganisms by preventing their entry into thebody and by removing them from respiratory surfaces.

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 6/28
UPPER RESPIRATORY TRACT:
Nose The nose consists of the external nose and the nasal cavity. The
external nose is the visible structure that forms a prominent feature of the face. Most of the external nose is composed of hyaline cartilage. Thenasal cavity extends from the nares to the choane. The nares or nostrilsare the external openings of the nose, and the choane are the openingsinto the pharynx. The nasal septum is a partition dividing the nasal cavityinto right and left parts.Paranasal sinuses are air-filled spaces within bone. The maxillary, frontal,ethmoidal, and sphenoidal sinuses are named after the bones in which
they are located. They reduce the weight of the skull, produce mucus,and influence the quality of the voice by acting as resonating chambers. The nasolacrimal ducts, which carry tears from the eyes, also open intothe nasal cavity.
Pharynx

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 7/28
4. Trace thePathophysiology
of the diseaseprocess.
Refer to appendix
The pharynx is the common passageway of both the respiratoryand digestive systems. The nasopharynx is the superior part off thepharynx. It is located posterior to the choanae and superior to the softpalate. The uvula is the posterior extension of the soft palate. Theposterior part of the nasopharynx contains the pharyngeal tonsil. The oropharynx extends from the uvula to the epiglottis, and the oralcavity opens into the oropharynx. The palatine tonsils are located in thelateral walls near the border of the oral cavity and the oropharynx. Thelingual tonsil is located on the surface of the posterior part of the tongue.
The laryngopharynx passes posterior to the larynx and extends from thetip of the epiglottis to the esophagus.
LOWER RESPIRATORY TRACT
Larynx The larynx is located in the anterior throat, and it is continuously
superiorly with the pharynx and inferiorly with the trachea. The larynxconsists of an outer casing of nine cartilages that are connected to oneanother by muscles and ligaments. Three of the nine cartilages areunpaired, and six of them form three pairs. The largest cartilage is theunpaired thyroid cartilage, or the Adam’s apple. The most inferiorcartilage of the larynx is the unpaired cricoid cartilage. The third unpairedcartilage is the epiglottis. The epiglottis helps prevent swallowedmaterials from entering the larynx. The six paired cartilages consist of three cartilages on either side of the posterior part of the larynx. The topcartilage on each side is the cuneiform cartilage, the middle cartilage isthe corniculate cartilage, and the bottom cartilage is the arytenoidcartilage. Two pairs of ligaments extend from the posterior surface of the thyroidcartilage to the paired cartilages. The superior pair forms the vestibularfolds, or false vocal cords, and the inferior pair composes the vocal folds,or true vocal cords.
Trachea The trachea, or windpipe, is a membranous tube that consists of
connective tissue and smooth muscle, reinforced with 16-20 C-shapedpieces of cartilage.

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 8/28
1 for thediagram.
Bronchi The trachea divides into the left and right main (primary) bronchi,
each of which connects to a lung. The left main bronchus is morehorizontal than the right main bronchus because it is displaced by theheart. The right main bronchus is more vertical and is in direct line withthe trachea.
Lungs The lungs are contained within the thoracic cavity. Each lung is
surrounded by a separate pleural cavity. Each pleural cavity is lined witha serous membrane called the pleura. The pleura consist of parietal andvisceral pleura. The parietal pleura, which lines the walls of the thorax,diaphragm, and mediastinum, is continuous with the visceral pleura,which covers the surface of the lung. Between the parietal and visceralpleurae is filled with a small volume of pleural fluid produced by thepleural membranes.
The lungs are the principal organs of respiration. Each lung is cone-shaped, with its base resting on the diaphragm and its apex extendingsuperiorly to a point about 2.5 cm above the clavicle. The right lung hasthree lobes called the superior, middle, and inferior lobes. The left lunghas two lobes called the superior and inferior lobes. Each lobe is dividedinto bronchopulmonary segments separated from each other byconnective tissue septa. There are 9 bronchopulmonary segments in theleft lung and 10 in the right lung.
The main bronchi branch many times to form the tracheobronchialtree. Each main bronchus divides into lobar bronchi as they enter theirrespective lung. The lobar (secondary) bronchi, two in the left lung andthree in the right lung, conduct air to each lobe. The lobar bronchi in turngive rise to segmental (tertiary) bronchi, which extend to thebronchopulmonary segments of the lungs. The bronxhi continue tobranch many times, finally giving rise to bronchioles. The bronchioles alsosubdivide numerous times to give rise to terminal bronchioles, which then
subdivide into respiratory bronchioles. Each respiratory bronchiolesubdivides to form alveolar ducts and open into alveoli, which are smallair sacs. The alveolar ducts end as two or three alveolar sacs, which arechambers connected to two or more alveoli. There are about 300 millionalveoli in the lungs.
The lungs have two lymphatic supplies. The superficial lymphatic

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 9/28
5. Know the signsand symptoms of
the diseasespecifically in the
RespiratorySystem.
vessels are deep to the visceral pleura and function to drain lymph fromthe superficial lung tissue and the visceral pleura. The deep lymphaticvessels follow the bronchi and function to drain lymph from the bronchiand associated connective tissues.
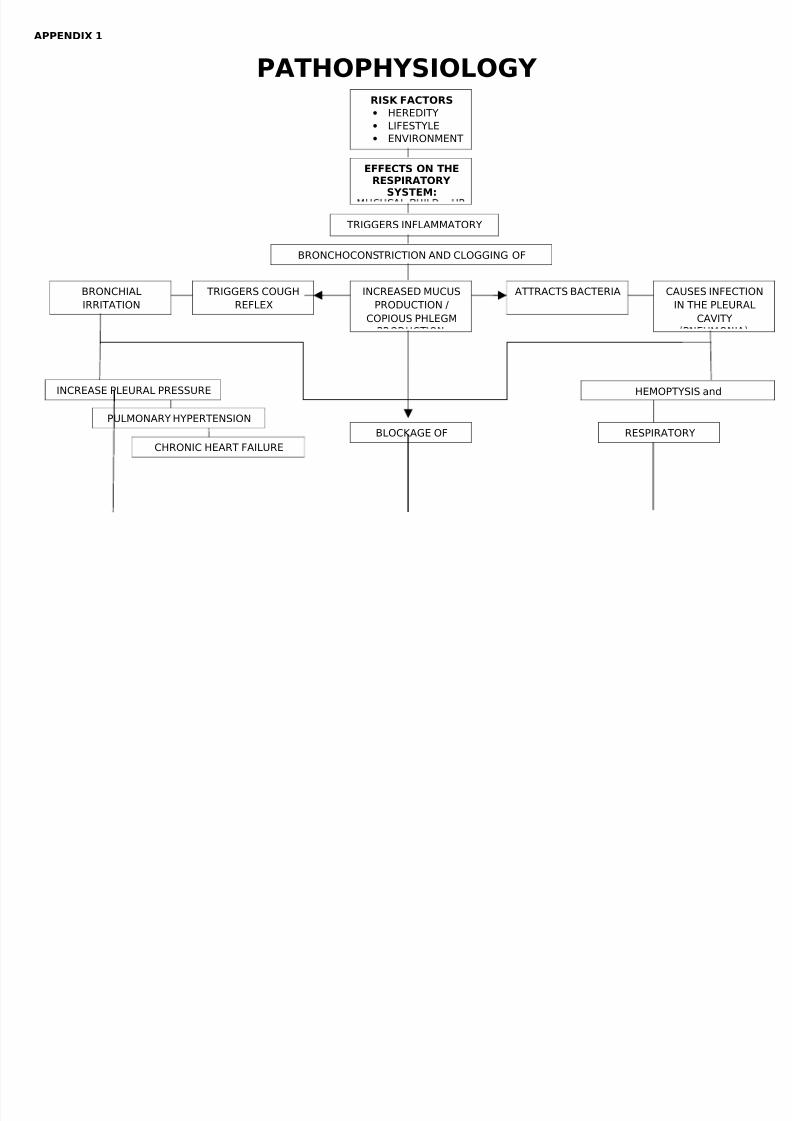
THE PATHOPHYSIOLOGY OF CYCTIC FIBROSIS IN THERESPIRATORY SYSTEM
There are several mechanisms by which mutations cause problems
with the CFTR protein. ΔF508, for instance, creates a protein that doesnot fold normally and is degraded by the cell. Several mutations, whichare common in the Ashkenazi Jewish population, result in proteins thatare too short because production is ended prematurely. Less commonmutations produce proteins that do not use energy normally, do not allowchloride to cross the membrane appropriately, or are degraded at a fasterrate than normal. Mutations may also lead to fewer copies of the CFTRprotein being produced.
The protein created by this gene is anchored to the outer membrane of cells in the sweat glands, lungs, pancreas, and otheraffected organs. The protein spans this membrane and acts as a channel connecting the inner part of the cell (cytoplasm) to the surrounding fluid. This channel is primarily responsible for controlling the movement of chloride from inside to outside of the cell; however, in the sweat ducts itfacilitates the movement of chloride from the sweat into the cytoplasm.When the CFTR protein does not work, chloride is trapped inside the cellsin the airway and outside in the skin. Because chloride is negatively charged, positively charged cations cross into the cell because they areaffected by the electrical attraction of the chloride ions. Sodium is themost common ion in the extracellular space and the combination of sodium and chloride creates the salt, which is lost in high amounts in thesweat of individuals with CF. This lost salt forms the basis for the sweattest.
How this malfunction of cells in cystic fibrosis causes the clinicalmanifestations of CF is not well understood. One theory suggests that thelack of chloride exodus through the CFTR protein leads to theaccumulation of more viscous, nutrient-rich mucus in the lungs thatallows bacteria to hide from the body's immune system. Another theoryproposes that the CFTR protein failure leads to a paradoxical increase in

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 10/28
6. Understand
and know theMedical
Management of the disease.
sodium and chloride uptake, which, by leading to increased waterreabsorption, creates dehydrated and thick mucus. Yet another theoryfocuses on abnormal chloride movement out of the cell, which also leadsto dehydration of mucus, pancreatic secretions, biliary secretions, etc. These theories all support the observation that the majority of thedamage in CF is due to blockage of the narrow passages of affectedorgans with thickened secretions. These blockages lead to remodelingand infection in the lung, damage by accumulated digestive enzymes inthe pancreas, blockage of the intestines by thick faeces, etc.
The lungs of individuals with cystic fibrosis are colonized andinfected by bacteria from an early age. These bacteria, which oftenspread amongst individuals with CF, thrive in the altered mucus, whichcollects in the small airways of the lungs. This mucus leads to theformation of bacterial microenvironments known as biofilms that aredifficult for immune cells and antibiotics to penetrate. Viscous secretionsand persistent respiratory infections repeatedly damage the lung bygradually remodeling the airways which makes infection even moredifficult to eradicate.
Over time, both the types of bacteria and their individualcharacteristics change in individuals with CF. In the initial stage, commonbacteria such as Staphylococcus aureus and Hemophilus influenzae colonize and infect the lungs. Eventually, however, Pseudomonas aeruginosa (and sometimes Burkholderia cepacia) dominates. Oncewithin the lungs, these bacteria adapt to the environment and developresistance to commonly used antibiotics. Pseudomonas can developspecial characteristics that allow the formation of large colonies, knownas "mucoid" Pseudomonas, which are rarely seen in people that do nothave CF.
SYMPTOMATOLOGY
The hallmarks of cystic fibrosis are salty tasting skin, normal
appetite but poor growth and poor weight gain, excess mucus production,frequent chest infections and coughing/shortness of breath. Males can beinfertile due to congenital absence of the vas deferens. CF symptomsoften appear in infancy and childhood, with meconium ileus being atypical finding in newborn babies. As the child grows, he or she will haveto exercise to release mucus stuck to the alveoli. Cilial epithilial cells in

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 11/28
the patient have a mutated protein that instead of creating the right resinthat is used to prevent the alveoli from collapsing, it makes a thickerresin, mucus. Poor growth is a hallmark of CF. Children with CF typicallydo not gain weight or height at the same rate as their peers, andoccasionally are not diagnosed until investigation is initiated for poorgrowth. The causes of growth failure are multi–factorial and includechronic lung infection, poor absorption of nutrients through thegastrointestinal tract, and increased metabolic demand due to chronicillness.
Lung disease results from clogging the airways due to mucosabuild-up and resulting inflammation. Inflammation and infection will causeinjury and structural changes to the lungs, leading to a variety of symptoms. In the early stages, incessant coughing, copious phlegm production, and decreased ability to exercise are common. Many of thesesymptoms occur when bacteria that normally inhabit the thick mucusgrow out of control and cause pneumonia. In later stages of CF, changesin the architecture of the lung further exacerbate chronic difficulties inbreathing. Other symptoms include coughing up blood (hemoptysis),changes in the major airways in the lungs (bronchiectasis), high blood pressure in the lung (pulmonary hypertension), heart failure, difficultiesgetting enough oxygen to the body (hypoxia), and respiratory failurerequiring support with breathing masks such as bilevel positive airway pressure machines or ventilators. In addition to typical bacterialinfections, people with CF more commonly develop other types of lungdisease. Among these is allergic bronchopulmonary aspergillosis, in whichthe body's response to the common fungus Aspergillus fumigatus causesworsening of breathing problems. Another is infection withMycobacterium avium complex (MAC), a group of bacteria related totuberculosis, which can cause further lung damage and does not respondto common antibiotics.
Mucus in the paranasal sinuses is equally thick and may also causeblockage of the sinus passages, leading to infection. This may cause
facial pain, fever, nasal drainage, and headaches. Individuals with CF maydevelop overgrowth of the nasal tissue (nasal polyps) due toinflammation from chronic sinus infections. These polyps can block thenasal passages and increase breathing difficulties.
GENERAL MEDICAL MANAGEMENT

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 12/28

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 13/28
Lung Transplanatation Lung transplantation often becomes necessary for individuals with
cystic fibrosis as lung function and exercise tolerance declines. Althoughsingle lung transplantation is possible in other diseases, individuals withCF must have both lungs replaced because the remaining lung mightcontain bacteria that could infect the transplanted lung. A pancreatic orliver transplant may be performed at the same time in order to alleviateliver disease and/or diabetes.Lung transplantation is considered whenlung function declines to the point where assistance from mechanical
devices is required or patient survival is threatened. This point typicallyoccurs when lung function declines to approximately 20 to 30 percent,however there is a small time frame when transplantation is feasible asthe patient must be healthy enough to endure the procedure.
Lung Volume ReductionAs noted earlier, the lungs of an emphysematous patient become
hyperinflated with air spaces that contribute little to gas exchange andthe resulting widened chest is difficult for the patient to expand furtherwhen attempting to inhale. By removing lung tissue around dead airspace, surgery can sometimes reduce the patient's work of breathing.In lung volume reduction surgery, 20% to 30% of the lung volume isremoved from both sides of the chest. As a result, survivors can usuallyexercise more than they could before the surgery. Those patients whohave mainly upper-lung emphysema also have an increased lifespan afterthis surgery. For other COPD patients, however, longevity is not increasedand it may even be shortened. The major postoperative complication of lung volume reduction surgery issignificant continuing air leakage from the lungs into the chest. Operativemortality rates are from 4% to 10% in hospitals providing the procedure.
Bullectomy
Selective removal of individual large empty air spaces (bullae) cansometimes be carried out using a thoracoscope. In patients withemphysema, bullae are usually a few centimeters in diameter.Occasionally, however, bullae can be huge, taking up as much as a thirdof the chest space. These giant bullae squeeze the healthier lung tissueand compress the adjacent blood vessels. By removing giant bullae, the

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 14/28
remaining lung tissue can re-expand and some of the circulation will berestored. As with lung volume reduction surgery, a major postsurgicalcomplication of bullectomy is persistent air leakage
Gene Therapy Gene therapy and has been explored as a potential cure forcystic fibrosis. Ideally, gene therapy attempts to place a normal copy of the CFTR gene into affected cells. Transferring the normal CFTR gene intothe affected epithelium cells would result in the production of functional
CFTR in all target cells, without adverse reactions or an inflammationresponse. Studies have shown that to prevent the lung manifestations of cystic fibrosis, only 5–10% the normal amount of CFTR gene expression isneeded. Multiple approaches have been tested for gene transfer, such asliposomes and viral vectors in animal models and clinical trials. However,both methods were found to be relatively inefficient treatment options. The main reason is that very few cells take up the vector and express thegene, the treatment has little effect. Additionally, problems have beennoted in cDNA recombination, such that the gene introduced by thetreatment is rendered unusable.
Preventive Measures
Cystic fibrosis is a genetic disorder that cannot be prevented.But people who have cystic fibrosis can help prevent more serious healthproblems such as lung infections by:
• Keeping immunizations current. Children who have cysticfibrosis should have all the recommended immunizations inaddition to pneumococcal and flu shots. For more information onthe recommended schedule of immunizations for children, see thetopic Immunizations.• Not smoking and avoiding secondhand smoke.• Using airway clearance techniques, such as postural drainage
and chest percussion.• Eating nutritious, high-calorie foods.• Having regular checkups and frequent tests, and followingtheir treatment plan.• Seeking care from a cystic fibrosis specialty treatment center,

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 15/28
if possible.
In addition, parents of a newborn who has been diagnosed with cysticfibrosis should care for the baby at home and avoid placing him or her inday care for at least 6 months to 1 year, if possible.
LABORATORY FINDINGS The key chemistry values for a person with COPD are the levels of
blood gases—oxygen and carbon dioxide—and the pH of the blood.
BLOOD OXYGEN LEVELS The severity of a patient's COPD can be estimated by the degree
that the blood gases deviate from normal. In the early stages of thedisease the amount of oxygen in arterial blood is usually within normallimits. Oxygen concentration in arterial blood is measured as its partialpressure (PaO2), and a normal oxygen partial pressure (or oxygen tension)is 80 to 100 mm Hg. As COPD worsens, the PaO2 can drop below 60 mmHg; this level signals respiratory distress to the brain, and it stronglyactivates the respiratory centers. When the PaO2 is below 60 mm Hg, aperson hyperventilates in an attempt to reverse the hypoxemia bybreathing in more oxygen. Unfortunately, hyperventilation due to
hypoxemia expels too much carbon dioxide from the bloodstream, andthis causes respiratory alkalosis, a pH imbalance.
In later stages of COPD, even with a PaO2 <60 mm Hg, the patientdoes not have the energy to hyperventilate and carbon dioxide builds upin the blood. Now the hypoxemia is accompanied by hypercapnia (excessblood carbon dioxide) and the patient develops chronic respiratoryacidosis, an ominous sign (Sharma, 2006; Swadron & Mandavia, 2006).
Arterial Blood GasesEarly in the course of COPD, arterial blood gases do not need to be
checked regularly. However, an early set of baselines values can be aused as a comparison to measure the degree of change brought on duringacute exacerbations, such as when the patient gets a serious respiratoryinfection.
Pulse OximetryAccurately measuring a person's blood oxygen tension requires

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 16/28
drawing arterial blood and testing it in a laboratory. Pulse oximetry is aquick and noninvasive alternative tool. A pulse oximeter has a small probethat can be clipped onto a patient's finger or earlobe. Usingmeasurements of transmitted light, the oximeter determines the percentof the patient's hemoglobin (Hb) that is saturated with oxygen.
Pulse oximeters are not as accurate as direct oxygen tensionmeasurements from arterial blood gases, and the percent of hemoglobinsaturation measured by an oximeter is not the same as a person's PaO 2.Nonetheless, the two values are related. A person with a normal PaO2 (80–
100 mm Hg) will have an Hb saturation of ≥96%; a person withhypoxemia of 60 mm Hg will have an Hb saturation of about 86%.
RED BLOOD CELLSRoutine blood analyses are not needed to manage most cases of
COPD. Some people with severe COPD produce excess red blood cells(polycythemia), apparently in response to their chronic hypoxia. Thisleads to hematocrit readings of >52% in men (normal is 43–52%) and>48% in women (normal is 37–48%).
ALPHA1-ANTITRYPSIN LEVELSPatients who develop emphysema at an early age (younger than 40
years old) and nonsmokers of any age who develop emphysema areusually tested for their blood levels of the enzyme alpha1-antitrypsin(AAT). Deficiency of this enzyme makes a person unusually susceptible toemphysematous COPD, and the patient and family should be educatedabout this genetic disease. It is sometimes possible to treat AAT-deficiency with replacement doses of the enzyme.
IMAGING STUDIES The most commonly used images for evaluating and managing
COPD are chest x-rays and computed tomography (CT) scans.
CHEST X-RAYSCOPD is a disease defined by functional problems, specifically, the
restriction or obstruction of airflow in the lungs. Breathing measurementsare better diagnostic indicators of the disease than are chest x-rays.Chest x-rays are important, however, for ruling out other causes of airwayobstruction, such as mechanical obstruction, tumors, infections, effusions,

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 17/28
7. Understand,learn and
practice theNursing
Management of the disease.
or interstitial lung diseases. In acute exacerbations of COPD, chest x-raysare used to look for pneumothorax, pneumonia, and atelectasis (collapseof part of a lung) (Wise, 2007).
At the severe stage, COPD produces a number of changes visible inchest x-rays. When the disease includes significant emphysema, the chestis widened, the diaphragm flattened, and the lung fields have fainter andfewer vascular markings. The heart may appear long, narrow, and verticaland the airspace behind the heart can be enlarged.
When severe COPD includes significant chronic bronchitis, chest x-
rays have a "dirty" look. There are more vascular markings, morenonspecific bronchial markings, and the walls of the bronchi look thickerthan normal when viewed end-on. Often, the heart appears enlarged(Swadron & Mandavia, 2006).
COMPUTED TOMOGRAPHY (CT) SCANSComputed tomography scans, especially high-resolution scans, are
better than chest x-rays at resolving the details of lung abnormalitiescaused by COPD. These scans are also used to more definitiveidentification of diseases such as tumors or infections that may becomplicating a patient's COPD. Late in the disease, CT scans are used toevaluate COPD patients who are to be treated surgically.
LUNG FUNCTION TESTSPulmonary function tests can quantify the severity of airway
obstruction in COPD. When COPD is diagnosed, baseline pulmonaryfunction values should be recorded. Later tests can then be usedobjectively to follow the progression of the disease and the effectivenessof treatments (Gold, 2005a). For COPD, the two general classes of breathing tests are (1) measurements of effective lung volumes, and (2)measurements of airflow rates and airflow volumes.
LUNG VOLUMESIn COPD, airway obstruction makes it difficult to empty the lungs
fully. The residual air merely takes up space. The air that remains behindkeeps the lungs hyperinflated even after a complete exhalation, making itmore difficult for a patient to pull in sufficient air for the next full breath.As a result, the total air volume of the lungs often increases while the

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 18/28
volume of air actually breathed in and out decreases. The effective volume of air is called the vital capacity (VC), which
is the largest volume of air that can be exhaled after a full inhalation. Thevital capacity is measured by having a patient take as large a breath aspossible and then exhale as quickly and forcefully as possible. The volumeof exhaled air is the vital capacity. When measuring vital capacity thisway, the result is also called the forced vital capacity (FVC) (Wanger &West, 2005).
AIRFLOW RATES The airway obstruction of COPD slows the movement of air in the
lungs. This slowing can be measured directly. Measurements of the rate of air movement during breathing are called spirometric measurements.Specifically, spirometry records the volume of air exhaled in a definedperiod of time (Miller et al., 2005).A full-function laboratory spirometry apparatus gives detailed reports on arange of characteristics of a patient's lung ventilation (NIH, n.d.c). The most common spirometric measurement used to characterize apatient's COPD is the one-second forced expiratory volume (FEV1). This is the maximum amount of air that a patient can breathe out in thefirst second of a forced exhalation after having taken a full breath.
EVALUATING AIRWAY OBSTRUCTIONPeople with normal lungs can expel most of the air in their lungs
within 1 to 2 seconds. The amount of air forcefully exhaled in the firstsecond (the FEV1) is about 3/4 of the vital capacity (the FVC) of a normalperson.If someone could exhale their entire vital capacity in 1 second, theirFEV1/FVC would be 1.00. A normal person has an FEV1/FVC between 0.70and 0.80; in other words, a person with normal lungs can exhale between70% and 80% of their vital capacity in the first second. This ratio,FEV1/FVC (the percent of the vital capacity that can be exhaled in onesecond), declines as a person ages, but even elderly people will haveFEV1/FVC >0.70 if their lungs are normal.
In COPD, airway obstruction restricts the rate of exhaling and peoplewith COPD cannot get a normal amount of air out of their lungs in one

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 19/28
second. People with COPD have FEV1/FVC <0.70. When a person has anFEV1/FVC <0.70 and a history of >20 pack-years of smoking, they can begiven a presumptive diagnosis of COPD (Wagner & West, 2005).
RANKING THE SEVERITY OF COPD The basic stages of COPD are termed mild, moderate, severe, and
very severe. All stages of COPD have an abnormally low one-secondexhaled percent of vital capacity (FEV1/FVC <0.70). The specific stage of COPD is then determined by how much the disease has reduced the
maximal airflow below normal; this is measured by comparing thepatient's FEV1 to the predicted value for their age, gender, height, andweight (Swadron & Mandavia, 2006; Wise, 2007)
STAGING OF COPD
Stage Severity FEV1/FVC
Stage I Mild FEV1/FVC <0.70 and FEV1 ≥80% predictedvalue*
Stage II Moderate FEV1/FVC <0.70 and 50% ≤FEV1 <80%predicted value*
StageIII
Severe FEV1/FVC <0.70 and 30% ≤FEV1 <50%predicted value*
StageIV
Very Severe FEV1/FVC <0.70 and FEV1 <30% predicted
*Predicted FEV1 values adjusted for a person's age, gender, height, andweight can be calculated from published equations (Pel legr ino et a l . ,2005).
Source: Modified from Rabe et al . , 2007.
NURSING MANAGEMENT

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 20/28
PHYSICAL EXAMINATIONA patient with mild COPD may have few signs of the disease,
especially when sitting quietly. In contrast, the physical exam of a personwith severe COPD can be diagnostic (Shapiro et al., 2005; Swadron &Mandavia, 2006).
GENERAL APPEARANCE
Most COPD patients especially with significant emphysema are
typically thin with a barrel-shaped chest. They tend to breathe throughpursed lips, and they sit leaning forward in a "tripod position"; this is aposture that widens the chest as much as possible by supporting theupper body on the elbows or the extended arms.
The tripod position. Patient leans forward, resting on elbowsorhands, in an effort to expand the chest and ease breathing.(Illustration by Jason M. McAlexander, MFA. Copyright © 2007Wild Iris Medical Education.)
In contrast, patients with significant chronic bronchitisare typically of normal weight or overweight. They have a
productive cough and may be cyanotic. At rest, their rate of respirations is high, often more than 20 breaths per minute.
Patients with the chronic bronchitis form of COPD who are hypoxemic mayhave a clouded consciousness, making them dull and irritable.
WEIGHTObesity can worsen the symptoms of COPD. On the other hand,
many COPD patients—especially patients with the emphysematous formof COPD—are cachectic and underweight, with muscle wasting. In thesecases, nutritional therapy is an important part of their treatment.
CHEST
A COPD patient with chronic bronchitis but little emphysema may have anormal-sized chest. Significant emphysema, on the other hand, leads to awide, barrel-shaped chest with a flattened diaphragm. In a patient withemphysema, the chest remains perpetually in the position of inhalation.

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 21/28
To take a new breath, therefore, emphysematous patients must expandtheir chests beyond the normal position of inhalation, and this requiresusing accessory respiratory muscles of the shoulder, neck, and back.
LUNGS The chest of an emphysematous patient is unusually resonant to
percussion and the breath sounds are distant. At the other end of thespectrum, the chest of a chronic bronchitis patient can have dull spotswhen percussed and it will be noisy with rales, rhonchi, and wheezing.
The common feature of all forms of COPD is airway obstruction,which worsens as the disease becomes more severe. A simple, directmeasure of airway obstruction is the time it takes a patient to blow out anentire lungful of air. A normal person has a forced expiratory time (FET) of <3 seconds. An FET of >4 seconds suggests obstruction. An FET of >6seconds indicates considerable airway obstruction, at the level of moderate-to-severe COPD.
HEARTCOPD can injure the heart in two major ways. First, the chronic
inflammatory state of COPD predisposes a person to develop coronaryartery disease; therefore, a heart history and physical examination should
probe for evidence of ischemic heart problems. Second, COPD can causepulmonary hypertension, which strains the right ventricle of the heart.Pulmonary hypertension makes the pulmonary component of the secondheart-sound louder. In addition, it can cause tricuspid valve insufficiency,which will be heard as a holosystolic murmur loudest along the left sternalborder. Pulmonary hypertension can cause right-sided heart failure (corpulmonale), and this will lead to jugular venous distension and edema of the legs and ankles.
LIFESTYLE ADDITIONS AND MODIFICATIONSMedications are the fundamental day-to-day tools for controlling the
symptoms of COPD, but there are also four key nonpharmaceutical stepsin the treatment of COPD (Shapiro et al., 2005; Stulbarg & Adams, 2005).
PATIENT EDUCATION Teach your patients about COPD. Explain that the disease causes

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 22/28
irreversible and progressive problems. Warn patients that they will haveepisodes in which the symptoms—difficulty breathing, wheezing,productive cough and tiredness—get worse for days or even weeks.
Assure patients that you will help them by ordering medicationsthat make breathing easier. Tell them there are a number of things theythemselves can do to slow the progression of the disease and to lessenthe number of acute exacerbations. The most important of these things isto stop smoking; although smoking has already damaged their lungs,continued smoking will increase the damage and will make their COPD
worsen more quickly.Explain to patients the importance of staying active. In addition, givethem practical suggestions that will help them to cope with the inevitablelimitations posed by COPD. For example, tell them:
• Slow the speed at which you do things, and stop and rest when youare tired. Don't push yourself.• Pace yourself and plan your activities for times when you have themost energy. You will feel best soon after you take yourbronchodilator medicines. Wait an hour after meals before you doactivities.• Sit on a chair or stool in the shower—don't stand. Likewise, sit whileyou shave, comb your hair, and brush your teeth.• Don't use products that are hard on the lungs; for example, hairsprays, spray-on deodorants, or strong perfumes.• Use the exhaust fan in your kitchen to make it less likely that youwill breathe smoke and cooking vapors.• Wear slip-on shoes so you don't have to bend over to tie laces.• Make sure your occupation does not require more physical exercisethan you can actually do. Consider setting smaller goals at work andallowing more time to finish tasks.• Find out how to get a daily air pollution report, and don't go out ondays with moderate or severe pollution.• Ask people not to smoke in your home or work area. (ALA, 2007)
STOP SMOKINGIn the United States, smoking starts in the teenage years: 90% of
adult smokers began smoking before the age of 18. More than a quarterof high school students and 1 in 10 middle school students smoke(Ranney et al., 2006).

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 23/28
Most patients with COPD have a long smoking history and many will stillbe smoking when they are under medical care. Currently, the only way tochange the course of COPD is for the patient to stop smoking. No matterhow old they are and no matter how long they have been smoking, COPDpatients benefit from quitting.COPD is an insidious disease, and it develops over many years before itcompromises people enough that they go to a doctor. The disease isalready active and destructive by the time that it is diagnosed, andtreatment should be aggressive from the beginning. From day one,
strongly urge your patients to stop smoking.Nicotine is powerfully addictive. In addition, the smoking ritual fills manybasic psychological needs. When doctors merely tell patients to stopsmoking, their patients succeed over the long-term only 5% of the time.Although simply advising smokers to quit is rarely effective, healthcareprofessionals are less likely to offer smoking cessation assistance withtheir advice (CDC, 2007).Long-term success rates of greater than 20% to 40% can be achieved bycomprehensive programs that include behavioral therapy andmedications. Successful smoking intervention programs begin by setting aquitting date with the patient. They then maintain continued contact withthe patient to provide medication, counseling, support, advice, and a
modicum of social pressure. For specific recommendations, the reportTreating Tobacco Use and Dependence: Clinical Practice Guidelines canbe downloaded from the United States Surgeon General's website athttp://www.surgeongeneral.gov/tobacco. The pharmacologic aspect of smoking cessation programs attempts toease the effects of nicotine withdrawal. Smokers who need their firstcigarette within a half-hour of getting up in the morning are likely to behighly addicted to nicotine. When these people stop smoking theybecome anxious, irritable, easily angered, easily tired, and depressed. Their sleep is disrupted and they have difficulty concentrating. Withdrawaleffects happen during the first 2 to 3 weeks after quitting. To lessen withdrawal symptoms, nicotine can be taken directly. Nicotine
replacements are available as gum, lozenges, transdermal patches,inhalers, and nasal sprays. These should be used on a regular scheduleand also prn (as needed for cigarette cravings) for about two weeks, andthen the doses reduced gradually.Antidepressants (notably, bupropion and nortriptyline) have been shown

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 24/28
to help patients for whom nicotine replacement therapy has not worked.In 2006 a nicotine agonist, varenicline (Chantix), was approved by theFDA for anti-smoking therapy. Varenicline binds to nicotine receptors andprevents nicotine from activating the receptors while producing a smallerstimulant effect than nicotine.
PULMONARY REHABILITATIONPulmonary rehabilitation is the term for a group of techniques used
to improve patients' conditioning and ease their breathing difficulties.
Pulmonary rehabilitation is done as outpatient therapy. Some programscontinue for an extended time, but most run for a few weeks and thengive patients individualized instructions for continuing at home. Educationsessions are important parts of rehabilitation programs; in these sessions,patients and their families learn details about COPD and its treatment(Chesnutt et al., 2008).Pulmonary rehabilitation programs are tailored to the needs of eachindividual. Typically, they include graded aerobic exercise programs suchas regular sessions of walking or stationary bicycling three times weekly.A walking exercise program, for example, might begin with slow treadmillwalking for only a few minutes. The length and speed of the walkingwould then be increased gradually over 4 to 6 weeks. The goal would be
for the patient to walk for 20 to 30 minutes without needing to stopbecause of shortness of breath. At that point, the patient would beassigned a maintenance exercise program to be done at home.Rehabilitation sessions also include exercise routines to condition theupper body and exercises aimed at strengthening respiratory muscles.Breathing instruction teaches patients how to slow their rate of breathingby pursing their lips. Patients also learn how to give their upperrespiratory muscles a rest by using abdominal breathing techniquesinstead of chest breathing.Comprehensive pulmonary rehabilitation can reduce COPD patients'symptoms and increase the amount of exercise that patients can dowithout being stopped by dyspnea. It can also reduce the number of
hospitalizations for acute exacerbations, and improve the quality of patients' lives, both subjectively and objectively (Stulbarg & Adams,2005).
PHYSIOTHERAPY

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 25/28
It is important that COPD patients with significant chronic bronchitiskeep their airways clear. Coughing up sputum should be encouraged, andcough suppressants or sedatives should not be used routinely. Whenpatients cannot clear their secretions by coughing, postural drainage mayhelp (Stulbarg & Adams, 2005).Most people's lungs secrete extra mucus in response to inhaled irritants. To avoid stimulating excess secretion, COPD patients need to avoidsmoke-filled rooms and stay indoors during air pollution alerts. Home airconditioners and air filters are effective and helpful.
IMPROVED DIETOverweight COPD patients find improvement when they slim down.
Some COPD patients, however, have the opposite problem: they becomethin and malnourished. In part, cachexia results from the high-energy costof breathing with COPD. In addition, the chronic inflammatory stateunderlying COPD tends to put the body's metabolism into a catabolicstate. To help them maintain a healthy body weight, thin COPD patientsshould be given dietary counseling that includes specificrecommendations of meals that are nutritionally balanced and thatcontain sufficient calories to make up for the work of breathing (Stulbarg& Adams, 2005; Wise, 2007).
.

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 26/28
BIBLIOGRAPHY:
BOOKS:
Rod R. Seeley, Trent D. Stephens, and Philip Tate, ESSENTIALS OF ANATOMY AND PHYSIOLOGY .Copyright 2007 Edition. Mc-Graw Hill Education (Asia).
WEB SOURCES:
http://nursingcrib.com/nursing-notes-reviewer/cystic-fibrosis/
http://health.yahoo.com/respiratory-prevention/cystic-fibrosis-prevention/healthwise--hw188763.html
http://en.wikipedia.org/wiki/Cystic_fibrosis
http://ask.reference.com/web?qsrc=2417&o=10616&l=dir&q=pseudomonas+aeruginosa
http://www.medscape.com/medline/abstract/9759549?prt=true
http://en.wikipedia.org/wiki/Pseudomonas_aeruginosa
http://www.nursingceu.com/courses/217/index_nceu.html
APPENDIX 1
8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 27/28
APPENDIX 1
PATHOPHYSIOLOGY RISK FACTORS
• HEREDITY
• LIFESTYLE
• ENVIRONMENT
EFFECTS ON THERESPIRATORY
SYSTEM:
TRIGGERS INFLAMMATORY
CAUSES INFEC
IN THE PLEU
CAVITY
INCREASED MUCUS
PRODUCTION /
COPIOUS PHLEGM
BRONCHOCONSTRICTION AND CLOGGING OF
BRONCHIAL
IRRITATION
ATTRACTS BACTERIA TRIGGERS COUGH
REFLEX
HEMOPTYSIS andINCREASE PLEURAL PRESSURE
PULMONARY HYPERTENSIONRESPIRATORYBLOCKAGE OF
CHRONIC HEART FAILURE

8/8/2019 RESOURCE UNIT: CYSTIC FIBROSIS
http://slidepdf.com/reader/full/resource-unit-cystic-fibrosis 28/28






