Embed Size (px)
Citation preview

Ua
BA
a
ARRAA
KDMGdPO
1
mpaoacioff
marhec
h0
Sensors and Actuators B 240 (2017) 631–639
Contents lists available at ScienceDirect
Sensors and Actuators B: Chemical
journa l homepage: www.e lsev ier .com/ locate /snb
ltra-trace analysis of d-and l-aspartic acid applying one-by-onepproach on a dual imprinted electrochemical sensor
him Bali Prasad ∗, Swadha Jaiswal, Kislay Singhnalytical Division, Department of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, India
r t i c l e i n f o
rticle history:eceived 13 July 2016eceived in revised form 23 August 2016ccepted 7 September 2016vailable online 9 September 2016
eywords:ual imprinted polymerulti-walled carbon nanotubes
a b s t r a c t
An enantiomeric pair imprinted polymeric film, with embedded functionalized multiwalled carbon nano-tubes, was developed following the ‘surface grafting from’ approach onto a gold nanoparticles decoratedpencil graphite electrode. Double imprinting of chiral molecules in a single polymer motif, as a sensoryplatform, for the one-by-one evaluation of individual components of a racemic mixture, is a challengingtask. The underlying state-of-art proposed for this purpose is novel and if this works well, any two simplemolecules could be used as templates. In this work, a pencil graphite electrode was first dipped for theovernight in the aqueous suspension of gold nanoparticles. The electrode was then subjected to spincoating with a pre-polymerization mixture consisting a monomer (N-acryloyl-pyrrolidine-2,5-dione),templates (d- and l-Aspartic acid), a cross-linker (ethylene glycol dimethacrylate) in the presence of an
old nanoparticles- and l-Aspartic acidotassium ferricyanide probene-by-one approach
initiator (�,�′-azoisobutyronitrile). As aspartic acid isomers have been found to be electro-inactive, theirevaluation was feasible indirectly with the help of a potassium ferricyanide probe. The quantitation abil-ity of the proposed sensor, with differential pulse anodic stripping voltammetric transduction, was foundto be in the tune of 4.08 ng mL−1 (S/N = 3) for both the enantiomers in the real samples, which reportedlymanifest several chronic diseases at their stringent limits.
. Introduction
In nature, chirality of a molecule is as important as its chemicalakeup. Our body recognizes chirality in terms of their distinct
harmacological profiles. In many cases, one enantiomer is anctive pharmaceutical ingredient while the other can be benignr even toxic. Apart from their difficult isolation, the quantitativenalysis of enantiomers is a major issue from the standpoint of effi-acy and safety of drugs [1]. Although the most prevalent opticalsomers of amino acids are having L-configuration, the occurrencef D-configuration has also been found in human, both in the freeorm and bound in peptides or proteins, in high concentrations toulfil specific biological function.
Aspartic acid (Asp), being a non-essential amino acid, is aajor excitatory neurotransmitter in the central nervous system. d-
spartic acid (d-Asp) serves in the central brain region to cause the
elease of hormones, viz., luteinizing hormone, follicle-stimulatingormone, growth hormone, and sex hormone. According to the lit-rature [2], Asp concentration levels in cerebrospinal fluid (CSF)ould be correlated with a number of neurological disorders such as∗ Corresponding author.E-mail address: [email protected] (B.B. Prasad).
ttp://dx.doi.org/10.1016/j.snb.2016.09.031925-4005/© 2016 Published by Elsevier B.V.
© 2016 Published by Elsevier B.V.
Alzheimer’s (7.19 ± 2.53 ng mL−1), epilepsy (4.22 ± 2.40 ng mL−1),and lennox syndrome (4.21 ± 1.99 ng mL−1). In particular, l-Aspcan be a biomarker for lung cancer and head and neck can-cer manifested at 266 ng mL−1 and 930 ng mL−1, respectively inserum. Hence, analysis of d & l-Asp in CSF and serum mayprovide a suitable mean of diagnosis and possible treatment ofneuropsychiatric diseases. Numerous methods for the enantiose-lective analysis of d- or l-Asp have been reported namely, thinlayer chromatography [3] gas chromatography [4] high perfor-mance liquid chromatography [5], chiral ligand-exchange capillaryelectrophoresis [6], spectrophotometry [7], and fluorometry [8].However, these methods are time-consuming, solvent-usage inten-sive, and highly expensive. Although electrochemical detection canbe considered relatively very simple and elegant, it has been failedso far to reveal acute Asp levels that can cause the prognosis ofepilepsy and lennox syndrome at primitive stage [2]. Thus, a highlyselective and sensitive electrochemical system for Asp evaluationis called for.
Simply put, molecularly imprinted polymers (MIPs) are syn-
thetic receptors made with the signature of template(s) byarranging suitable monomer(s) around it, subsequently followedby polymerization in the presence of a cross-linker. The retrievalof template molecules generates molecular cavities in the poly-mer network, which are specific for the molecular recognition
6 d Actu
itfMMobtdcteMuameoopnfbedliaapcmu
hDftamNibaoetiloaswdpttruatbyDit
32 B.B. Prasad et al. / Sensors an
n terms of size and shape of the template. Molecular architec-ure in the imprinting world witnessed a very limited attentionor imprinting two or more targets (print molecules) in a single
IP format [9–18]. Memory sites for only a single compound inIP network might exert restriction in the detection of a group
f analytes present in samples. Although multi imprinting savesoth time and labor as compared to the traditional imprinting,he diffusion pathways of templates are often crisscrossed andisturbed during their recapture and/or stripping processes. Thisould be the reason that no attempt has been made so far forhe simultaneous/sequential analysis of chiral compounds. How-ver, with the advent of nanotechnology; an ultra thin layer ofIP on a solid substrate might improve the analyte diffusitivity
ninterruptedly. Such nano-structured materials can be exploiteds sensors to exhibit a high degree of success in the improve-ent of detection sensitivity and selectivity. Molecularly imprinted
lectrochemical sensors apparently combine the characteristicsf electrochemical detection and molecular imprinting technol-gy. The underlying state-of-art in fabricating a nanosensor in theresent work is typical in the sense that we have initially used goldanoparticles (AuNPs) decorated pencil graphite electrode (PGE)
or the subsequent surface modification with multiwalled car-on nanotubes (MWCNTs) interspersed doubly imprinted polymer,mploying the ‘surface grafting from’ approach. Herein, the embed-ed CNTs have advantages of enhanced electronic properties and a
arge edge plane/basal plane ratio to impart rapid electrode kinet-cs. On the other hand, AuNPs would effectively serve as “electronntennae” for channelling electron transport between the electrodend the electro-active species. Moreover, MWCNTs embedded thinolymer film artificially anchored onto AuNPs/PGE, in physical orhemical manner, may show much potentiality for the effectiveass transport with a facilitated egress-ingress of the test analyte,
nder the pool of imprinting and electrostatic effects [19].Double imprinting in itself is an arduous task with the appre-
ension of its success in the field of molecular recognition.espite the fact that chiral compounds exists in two enantiomeric
orms with the identical molecular formula, their different spa-ial conformations forming non-superimposable mirror imagesre advantageous to generate distinct D-specific and L-specificolecularly imprinted cavities, for enantioselective recognition.evertheless, the voltammetric peak separation of D- and L-
somers is not possible as a consequence of their identical redoxehaviour. The situation may further be critical when both isomersre electro-inactive in nature. In the present work, the model pairf analytes, d-Asp and l-Asp, selected for analysis is found to belectrochemically inactive. We have, thus, endeavoured for the firstime to resolve this problem by developing a protocol for doublemprinting of d-Asp and l-Asp in a single polymer motif, as out-ined in Scheme 1. In this work, we have proposed a novel methodf indirect enantioselective analysis of d- and l-Asp, with the help of
ferricyanide probe, using a single modified electrochemical sen-or. Accordingly, one has to saturate both of the imprinted cavitiesith the probe molecules and initially obtain the corresponding
ifferential pulse anodic stripping voltammetry (DPASV) signal inhosphate buffer (pH = 3). After washing the electrode with water,he magnitude of probe response remains unaltered. Further, forhe quantification of d-Asp, this electrode is exposed to l-Asp byeplacing the probe solution from corresponding cavities till sat-ration. The D-isomer cavities duly filled-in with probe solutionre now all set for the quantitative analysis of d-Asp. The elec-rode at this stage is regenerated by extraction of templates from
oth type of cavities. Similar procedure is adopted for the anal-sis of other isomer, l-Asp, using the same refreshed electrode.espite the fact that MIP-sensors for enantioselective analysis ofndividual isomers, hitherto, have been utilized two separate elec-rodes [2,20,21], the proposed method of one-by-one sensing on
ators B 240 (2017) 631–639
a single MIP-sensor could relatively be more sensitive, accurateand cost-effective. Furthermore, this sequential method of indirectanalysis can be extended to the determination of any pair of simplemolecules, whether electro-active or electro-inactive in nature.
2. Experimental
2.1. Chemicals and reagents
All chemicals were of analytical reagent grade, and used withoutfurther purification. Demineralized triple distilled water (con-ducting range 0.06–0.07 × 10−6 S cm−1) was used throughout theexperiment. Succinimide (pyrrolidine-2,5-dione), acryloyl chlo-ride, potassium ferricyanide, trisodium citrate, and chloroauric acid(HAuCl4·H2O) were purchased from Loba Chemie (Mumbai, India).All solvents, dimethylsulphoxide (DMSO), tetrahydrofuran (THF),triethylamine (TEA), and ethanol, were procured from SpectrochemPvt. Ltd. (Mumbai, India). Ethylene glycol dimethacrylate (EGDMA),MWCNTs (internal diameter 2–6 nm, outer diameter 10–15 nm,length 0.2–10 �m, and purity >90%), �,�′-azoisobutyronitrile(AIBN), d-Asp, and l-Asp were obtained from Aldrich (Steinheim,Germany). All interferents studied were purchased from Fluka(Steinheim, Germany). The supporting electrolyte used was phos-phate buffer solution (pH 3.0, ionic strength 0.01 M). Standardstock solutions of d-Asp and l-Asp (500 �g mL−1) and potassiumferricyanide (33 �g mL−1) were prepared in water. All workingsolutions were prepared by diluting stock solution with water.Human blood serum and CSF were obtained from the Institute ofMedical Science, Banaras Hindu University (Varanasi, India) andkept in refrigerator below −4 ◦C, before use. The pharmaceuticalsample analyzed was astymin hepa (Tablets India Ltd., Chennai,India). Pencil rods (2B grade, 2 mm diameter, 5.0 cm length) werepurchased from Hi Par, Camlin Ltd. (Mumbai, India). The PGEwas used for modification with MIP because it is better in termsof higher electrochemical activity, commercial availability, goodmechanical stability, low cost, low background current, and widepotential window as compared to other solid electrodes (Pt, Au, Pd,Ag, glassy carbon, etc.) [22,23]. Furthermore, PGE possesses severalgraphite pores into which MIP film could be physically adsorbed,with firm adherence as compared to glassy carbon electrode andother solid electrodes.
2.2. Apparatus
DPASV and cyclic voltammetry (CV) were performed on aportable potentiostat �-Stat 200 (Drop Sens S.L. Oviedo, Spain),which was connected via USB to a computer with measurementsoftware Drop View (DropSens). The electrochemical cell was con-sisted of MIP-AuNPs@PGE, platinum wire, and Ag/AgCl (3.0 M KCl)as working, counter, and reference electrodes, respectively. FT-IR(KBr) spectra were recorded on Perkin Elmer (model-L1600300Spectrum TWO LITA), Llantrisant, UK. Surface morphologies ofcoatings were studied using scanning electron microscope (SEM)[JEOL, JSM model-840A (Netherlands)] and atomic force micro-scope (AFM) [using a NT-MDT microscope, NT-MDT Co. (Russia), inthe semi-contact mode]. A spin coater (ACE → 200, Dong Ah Tech,Seoul, South Korea) was used for the electrode modification. Allexperiments were carried out at 25 ± 1 ◦C.
2.3. Synthesis of AuNPs and functionalized MWCNTs
AuNPs were prepared following the known recipe [24]. In short,2.5 mL of 1% tri-sodium citrate was added to 100.0 mL of boiling0.01% HAuCl4 solution. The prepared AuNPs were stored in dark andrefrigerated at approximately −4 ◦C. For evidence, AuNPs showeda characteristic absorption at �max of about 520 nm.

B.B. Prasad et al. / Sensors and Actuators B 240 (2017) 631–639 633
nt of D
kwAfi
2
(r0wTww
2d
itoflp(dl(pw
Scheme 1. Schematic developme
MWCNTs were functionalized as MWCNTs-COOH following thenown procedure [25]. For this, MWCNTs (0.5 g) were oxidizedith 60 mL of concentrated nitric acid solution at 100 ◦C for 12 h.fter cooling to room temperature, the solid MWCNTs-COOH wereltered out, washed, and finally dried in vaccum.
.4. Synthesis of monomer
Monomeric precursor, N-acryloyl pyrrolidine-2,5-dioneNAPD), was synthesized, following a known method [26], byeacting succinimide (25 mmol) and acryloyl chloride (25 mmol) at◦C in THF, in the presence of TEA (25 mmol). The reaction mixtureas maintained at 0 ◦C with stirring for 3 h. After evaporation of
HF, a crude product was obtained. This was generously washedith water to remove the triethylamine-acid salt and recrystallizedith ethanol.
.5. Fabrication of MIP with dispersed MWCNTs-COOH on AuNPsecorated PGE (MIP-AuNPs@PGE)
The fabrication protocol of MIP-AuNPs@PGE sensor is shownn Scheme 1. PGE was first dipped overnight in the AuNPs solu-ion. AuNPs were physically adsorbed on the graphite surfacef PGE [27]. The AuNPs@PGE so obtained was dried under theow of nitrogen. For the preparation of MIP-AuNPs@PGE, a pre-olymer mixture was prepared which contained a monomerNAPD, 0.4 mmol, 1.0 mL DMSO), d- and l-Asp (0.1 mmol each
issolved in 1.0 mL DMSO), an initiator (AIBN, 0.003 g), and a cross-inker (EGDMA, 2.0 mmol). To this mixture, an optimized amount20 �L) of functionalized MWCNTs (0.005 g MWCNTs-COOH sus-ended in 800 �L DMSO) was added. The whole content was purgedith N2 gas for 10 min, and 15.0 �L of this was spin coated on to
ual imprinted MIP-AuNPs@PGE.
the surface of AuNPs decorated PGE at 2600 rpm for 30 s. Herein,the dispersed MWCNTs-COOH may help for the firm adherenceof MIP-film onto PGE via aromatic �-� interactions between thecarbon nanotubes and the graphite layers [28–30]. This modifiedelectrode is subjected to the free radical polymerization at 70 ◦Cfor 4 h. Template molecules were finally retrieved from the so pro-duced MIP-adduct, by immersing the modified electrode into 0.1 MNaOH plus 0.1 M phosphate buffer (1:2, v/v) mixture for 1 h. Thetemplate removal could be easier from the surface imprinted sitesas obtained by the ‘surface grafting from approach’. The completeremoval of all template molecules was ensured by the gradualincrease of probe response until a maximum is attained. A non-imprinted polymer modified electrode (NIP-AuNPs@PGE) was alsomade in the identical manner as stated above, but in the absence oftemplate (d-Asp and l-Asp) molecules in the pre-polymer mixture.
2.6. Voltammetric procedure
For electrochemical measurements, MIP-AuNPs@PGE wasimmersed into a cell containing 10.0 mL of 0.01 M phosphate buffer(pH 3.0), in the presence of potassium ferricyanide (33.0 �g mL−1,50.0 �L). Before recording CV and DPASV runs, the probe moleculeswere accumulated at the electrode surface in the form of electricaldouble layer consisting an array of K+ and [Fe(CN)6]3− [31] at −0.4 Vfor 150 s. Here, [Fe(CN)6]3− was instantly reduced at accumula-tion potential −0.4 V. The reduced form [Fe(CN)6]4− was scannedfor CV within the potential window −0.6 to +0.6 V in the anodic
stripping mode. Similarly, DPASV runs were recorded from −0.6 to+0.4 V at a scan rate of 10 mV s−1 applying pulse amplitude (25 mV),pulse time (50 ms) and step potential (5 mV). Note that the modifiedelectrode was found not to be responsive, when it was anodicallycharged which restricted the formation of an electrical double layer.
6 d Actu
Afrsd
fwtodcress
rTda
3
3
cMCfhtawfiattletMeoptaktbt[roeltehadb[i
34 B.B. Prasad et al. / Sensors an
fter water-washings, molecular cavities remained occluded witherricyanide probe solution that identically responded to initial cur-ent for the oxidative stripping, [Fe (CN)6]4− → [Fe (CN)6]3−. At thistage, the dual imprinted electrode was exclusively saturated with-Asp until a constant DPASV current (I) was attained, without any
urther decrease of probe response. Now, cavities specific to l-Aspere set free to recapture test analyte and simultaneously release of
he commensurate amount (proportional to l-Asp concentration)f probe responding decreased DPASV current (I′). Accordingly, theifference in probe oxidation current (�I = I − I′) versus l-Asp con-entration profile was obtained. Finally, the modified electrode wasegenerated by retrieving both templates for the next use for d-Aspstimation. This was carried out following the similar manner astated above for l-Asp. For this, molecular cavities for l-Asp wereaturated and d-Asp was evaluated on the same electrode.
Since dissolved oxygen present in the cell did not affect the cur-ent response, any deaeration of the cell content was not necessary.he limit of detection (LOD) was calculated as three times the stan-ard deviation for the blank measurement in the absence of targetnalyte divided by the slope of the calibration plot.
. Results and discussion
.1. Polymer characteristic
Traditional MIP@PGE did not respond satisfactorily for ferri-yanide probe (80 ng mL−1) owing to the insulating nature of acrylicIP-film (Fig. 1, curve b). However, the corresponding MWCNTs-
OOH dispersed MIP@PGE revealed 1.3 times higher current (curve) as compared to MIP@PGE. The MIP-AuNPs@PGE revealed muchigher current with better electronic transmission (curve g) fromhe surface imprinted binding sites to the electrode, even in thebsence of CNTs. The electron transport was drastically improvedhen MIP used was duly dispersed with MWCNTs-COOH in thelm texture and modified over AuNPs@PGE surface (curve h). As
matter of fact, this current height (curve h) was realized justwice than the MIP film (without MWCNTs-COOH) (curve g) andhrice than traditional MIP (curve b). This showed that cumu-ative effect of AuNPs and MWCNTs-COOH imparted significantlectro-conductivity to the MIP film with the channelized electronransport from the recognition sites to the electrode. Furthermore,
IP-AuNPs film (with dispersed MWCNTs-COOH) had shown thelectrocatalytic property to some extent to decrease ferrocyanidexidation overpotential at −0.18 V vs Ag/AgCl (curve h) as com-ared to bare PGE (curve c) and AuNPs@PGE (curve e). Duringhe fabrication of MIP membrane at AuNPs@PGE, the carboxyliccid groups at the CNT entrance provide an electrostatic ‘gate-eeper’ effect on ionic transport providing an exciting opportunityo dramatically enhance the mass diffusion through CNT core. Also,eing electronically conductive, MWCNTs-COOH may localize elec-ric field at CNT tips to perform electrochemical transformation19]. On the other hand, AuNPs in between the porous PGE and theedox sites of the MIP membrane might serve as “nanomediators”r “electronic bridges” to trap charge and fortify the channelizedlectron transport [32]. The AuNPs electronic bridges help estab-ishing a fast mediated electron transfer between the redox sites ofhe immobilized MIP film and the electrode. Note that the directlectron transfer between the MIP (with MWCNTs-COOH) and PGEad demonstrated a restricted probe current (curve f), without
ny reduction of overpotential. This is because of the fact that theirect electron transfer requires a short distance (less than 15–20 Å)etween the redox centre of the MIP film and the electrode surface33]. This indicated that AuNPs had exclusive role in this study tonduce electrocatalytic behaviour to the MIP film.ators B 240 (2017) 631–639
In support of improved electrode kinetics of MIP (withMWCNTs)/AuNPs@PGE as discussed above, we have calculatedelectron-transfer rate constants (k) for the redox process withprobe solution (80 ng mL−1) at bare/modified PGEs, with the helpof following Laviron equation [34], implicating the CV run recordedat a scan rate of 0.1 V s−1 [Fig. 1 (inset)]:
logk = ̨ log (1 − ˛) + (1 − ˛) log ̨ − log
(RT
nFv
)− ˛ (1 − ˛)nF�Ep/2.3RT (1)
where � is the electron-transfer coefficient, F is the Faradayconstant, � the scan rate (Vs−1), R the gas constant, T the tem-perature, and n is the number of electron transfer. For n = 1, the� value could be obtained from the slope (2.303RT/(1 − �)nF)of Ep vs. log � plot. The effect of scan rate on CV runsis shown in Fig. 1 (inset), exclusively for MIP(with MWC-NTs)/AuNPs@PGE. We have also undertaken the similar studywith other bare/modified PGEs (Fig. not shown). From the cor-responding Ep vs. log � plots, the estimated values of � and kare: 0.72 and 0.44 × 10−2 (MIP@PGE), 0.73 and 0.48 × 10−2 (barePGE), 0.72 and 0.88 × 10−2 (MWCNTs@PGE), 0.70 and 1.24 × 10−2
(AuNPs@PGE), 0.72 and 2.20 × 10−2 (MIP(withMWCNTs)@PGE),0.75 and 3.24 × 10−2 MIP(without MWCNTs)/AuNPs @PGE, 0.78and 9.24 × 10−2 s−1 (MIP(with MWCNTs)/AuNPs @PGE). Thefractional � value indicates the quasi-reversible behaviour of ferri-cyanide probe on each electrode studied. The deviance from idealreversibility of probe may be attributed to the difficulty in strippingof reduced ferricyanide, under the influence of electrical doublelayer formed at Eacc = −0.4 V. Nevertheless, the relatively high kvalue, which supports the improved electron-transfer kinetics ofthe proposed sensor, is due to the cumulative contribution of MWC-NTs and AuNPs toward augmenting the electron transport.
For developing imprinted network, different template-template-monomer molar ratios (1:1:1, 1:1:2, 1:1:3, 1:1:4)were attempted to explore an optimum stoichiometry of theMIP-adduct complex. The maximum development of DPASVdiminishing current (�I) of both the analytes was obtained whentemplate-template-monomer ratio of 1:1:2 was used for the poly-merization at 70 ◦C for 4 h. Insofar as the cross-linker optimizationis concerned, any amount of cross-linker more than 2.0 mmolrevealed a decrease in the �I owing to excessive cross-linkingto block the analyte diffusitivity into respective molecular cavi-ties. Templates from MIP-adduct were retrieved by 0.1 M NaOHplus 0.1 M phosphate buffer (v/v 1:2), in a sufficient duration of60 min, under dynamic condition [For details on stoichiometry ofMIP-adduct and optimization of polymerization conditions, videSupporting data Section S.1 and Fig. S1].
3.2. Spectral and surface characterization
FT-IR (KBr) spectra (Fig. S2) of monomer (NAPD), template (Asp),MIP-adduct, and MIP (template-free), were comparatively studiedto propose a tentative binding mechanism between monomer andtemplate (Scheme 1). Accordingly, MIP possesses two distinct spa-tial patterns of d-Asp and l-Asp cavities: one in which � − aminogroup ( NH3
+) is in the plane and readily accessible for hydrogenbonding with the host, whereas the other isomer carries aminogroup out of the plane and is not available for hydrogen bond-ing, under steric compression. In the present instance, specificityof these molecular cavities is not primarily dependent on theirshapes, but also on their respective chemical affinities for the selec-tive analyte binding, under the impact of phenomenal imprintingeffect. The complexation between the monomer and template(s)
via hydrogen bondings was indicated by the downward shifts oftheir respective key bands participating in the adduct formation[For details on IR characteristics, vide Supporting data Section S.2].The SEM image of AuNPs@PGE (Fig. 2A) shows somewhat non-uniform distribution of highly packed and aggregated AuNPs on the

B.B. Prasad et al. / Sensors and Actuators B 240 (2017) 631–639 635
Fig. 1. DPASV response of 80 ng mL−1 [Fe(CN)6]3−(accumulated in the reduced form as [Fe(CN)6]4−at −0.4 V) at (a) NIP (with MWCNTs)/AuNPs@ PGE, (b) MIP@PGE, (c) barePGE, (d) MWCNTs@PGE, (e) AuNPs@PGE, (f) MIP (with MWCNTs) @ PGE, (g) MIP (without MWCNTs)/AuNPs@ PGE, and (h) MIP (with MWCNTs)/AuNPs@ PGE. Inset showsCV for probe (80 ng mL−1) recorded in anodic stripping mode at different scan rates (a → e): 10, 50, 100, 200, 500 mV s−1 at MIP (with MWCNTs)/AuNPs@ PGE.
(D) NI
ehAvtac
Fig. 2. SEM images: (A) AuNPs, (B) MIP-adduct, (C) MIP,
lectrode surface. In contrast, the MIP adduct-AuNPs@PGE (Fig. 2B)as a relatively compact and rigid structure with clearly visibleuNPs aggregation within the polymer matrix; MWCNTs are non-
isible in this compact film. Interestingly, upon templates retrieval,he MIP-AuNPs@PGE surface revealed pores of different depths andpertures with dispersed MWCNTs in the nanofilm (Fig. 2C), inontrast to the corresponding NIP-based electrode surface havingP, and (E) side view of MIP- AuNPs films at PGE surfaces.
almost no pores (Fig. 2D). Fig. 2E displays the side view of MIP-AuNPs@PGE which suggests the film thickness to be about 95 nm.
Surface morphologies were further supported from AFM (three-
dimensional) images, recorded under semi-contact mode, for MIPadduct-AuNPs@PGE (Fig. S3A) and MIP-AuNPs@PGE (Fig. S3B). Thisalso revealed MIP coatings on the electrode surface with thicknessof 94.5 nm [For details on AFM morphology, vide Supporting dataSection S.3].
6 d Actuators B 240 (2017) 631–639
3
cftp(HtpaeAc(recLnd
sopcdooaLttid
cdACtimagltmml
3
wn(a(AtvanisM
Fig. 3. (A) DPASV response of ferricyanide probe (168 ng mL−1) on MIP-AuNPs@PGEwith l-Asp saturated (67 ng mL−1) cavities: (b-l) spiking with different d-Aspconcentrations: 0.0, 3.89, 9.58, 13.72, 17.29, 23.21, 29.32, 33.25, 40.68, 50.18,66.23 ng mL−1; curve ‘a’ represents initial response with ferricyanide probes dulyfilled in both D- and L-cavities of MIP-AuNPs@PGE, and curve ‘m’ represents cur-rent response of ferricyanide probe on NIP-AuNPs@PGE [operating conditions:Eacc−0.4 V, tacc 150 s, pH 3.0, and scan rate 10 mV s−1 (for ferricyanide probe)]. (B)DPASV response of ferricyanide probe (168 ng mL−1) on MIP-AuNPs@PGE with d-Aspsaturated (67 ng mL−1) cavities: (b-l) spiking with different l-Asp concentrations 0.0,
−1
36 B.B. Prasad et al. / Sensors an
.3. Electrochemical studies
First and foremost, all the operating conditions of electro-hemical analysis were optimized in aqueous conditions usingerricyanide with MIP-AuNPs@PGE. Accordingly, the accumula-ion potential (Eacc), the accumulation time (tacc), and pH of thehosphate buffer, for [Fe(CN)6]3− probe, were obtained as −0.4 Vversus saturated Ag/AgCl), 150 s, and 3.0, respectively (Fig. S4).erein, both potassium and ferricyanide ions occupy and fill up
he imprinted cavities of d-Asp and l-Asp in an open circuit. Therobe gets effectively diffused to the electrode surface, under theccumulation potential effect at −0.4 V, and arranged there as anlectrical double layer under the pool of electrostatic interactions.t this potential, [Fe(CN)6]4−, the reduced form of probe, was anodi-ally oxidized to respond DPASV signal in phosphate buffer solutionpH 3). This practice was always carried out in the beginning toecord the initial signal of ferricyanide probe that was unaltered,ven after water-washings. This meant cavities remained practi-ally filled-in with probe solution in the open circuit. At this stage,-specific cavities were presaturated with l-Asp (67 ng mL−1) tillo reduction of DPASV current occurred and then subjected to the-Asp measurement. For this, the electrode is exposed to d-Aspolution maintained at pH 3.0, (Fig. S4) for 120 s accumulation in anpen circuit. This electrode was immersed into the cell containinghosphate buffer. The existing probe molecules in d-Asp imprintedavities started diffusing to the electrode under potentiostatic con-ition which subsequently stripped off to give rise to a diminishedxidation current. This process was continued with the additionf d-Asp and measured the reduction in current (Fig. 3A) till itttained an optimum decrease. At this stage, both D-specific and-specific cavities are now completely saturated. For the sequen-ial analysis of l-Asp, the electrode was refreshed by retrieving bothemplates and used as such following the above procedure. Accord-ngly, now d-Asp specific cavities were blocked by saturating with-Asp (66 ng mL−1) and then analysis with L-specific cavities wasarried out (Fig. 3B). Herein, the effective surface area is reducedue to blocking of imprinted sites with the hydrogen bonded d-sp (or l-Asp), leading to the decrease in the DPASV (Fig. 3) orV response (Fig. S5). In this work, DPASV was preferred to CV forhe quantitative analysis because of its relatively high sensitivityn sufficient time scale of voltammetric measurement. The �I so
easured could be related to the concentration (C) of analyte inccordance with the regression equations potrayed in Table 1. Theradually diminished current of probe with the increase of ana-yte concentration attained a constant above 67 ng mL−1 for bothhe isomers, due to binding sites saturation. The non-imprinted
odified electrode revealed insignificant �I response for probeolecules upon analyte spiking (Fig. 3) which indicated an excel-
ent imprinting phenomenon.
.4. Interference studies and sensor endurance
Interference studies were performed for both the enantiomersith some structurally related co-existing interferents which are
ormally found in biological fluids. These were: glutamic acidGlu), tryptophan (Trp), proline (Pro), cysteine (Cys), glycine (Gly),sparagine (Asn), glutamine (Gln), phenylalanine (Phe), histidineHis), malic acid (Mal), and thyroxine (Thy). The present MIP-uNPs@PGE was slightly responsive (Fig. 4c), without subjecting
he electrode to water washing treatment, for some of the indi-idual interferents. This response could be termed as non-specific
nd false-positive. In fact, such non-specific contribution was firstoticed on the corresponding NIP-AuNPs@PGE for some of thenterferents (Fig. 4a). This contribution could easily be mitigatedimply by water washings (n = 2, 0.5 mL) (Fig. 4b). Therefore, theIP electrode was also subjected to similar washing treatment as
3.99, 7.89, 10.79, 19.78, 24.12, 31.67, 35.57, 43.52, 55.78, 66.12 ng mL ; curve ‘a’ rep-resents initial response with ferricyanide probe duly filled in both D- and L-cavitiesof MIP-AuNPs@PGE, and curve ‘m’ represents current response of ferricyanide probeon NIP-AuNPs@PGE [operating conditions same as above].
a safeguard against false-positives. In a parallel work with binarymixtures of the template and interferent(s) concomitantly presentin clinically relevant concentration ratio, the MIP-AuNPs@PGEshowed an exclusive response for the template in question (Fig. 4dand e) in the quantitative manner by means of stereochemicalselectivity. There was virtually no cross reactivity between thetarget and the interferent(s) i.e, D-specific MIP-AuNPs@PGE couldnot respond l-Asp and vice-versa. Any molecule that is smaller(Pro, Cys, Gly, Gln), larger (Glu, Trp, Thy, Phe, His) and similar(Mal, Asn) in size than d- and l-Asp could not be detected onthe proposed sensor. This reflects substrate-selective imprintingeffect in the present instance. Although the smaller interferentsmay have an equal opportunity to reach the binding sites but theystill mismatch with molecular cavities in terms of chemical affin-ity. Interferences were also examined in real samples (Fig. notshown) which revealed similar behaviour as observed in aqueoussample. Note that, any probe like entities present in the real sam-
ples may affect the voltammetric measurements. However, sucheffect was found to be largely obviated under the massive sampledilution effect, and therefore all results were found to be quantita-tive (100%) in this study. Imprinting factors (� = �MIP-AuNPs@PGE/�iNIP-AuNPs@PGE) for both the templates (d- and l-Asp) were found as
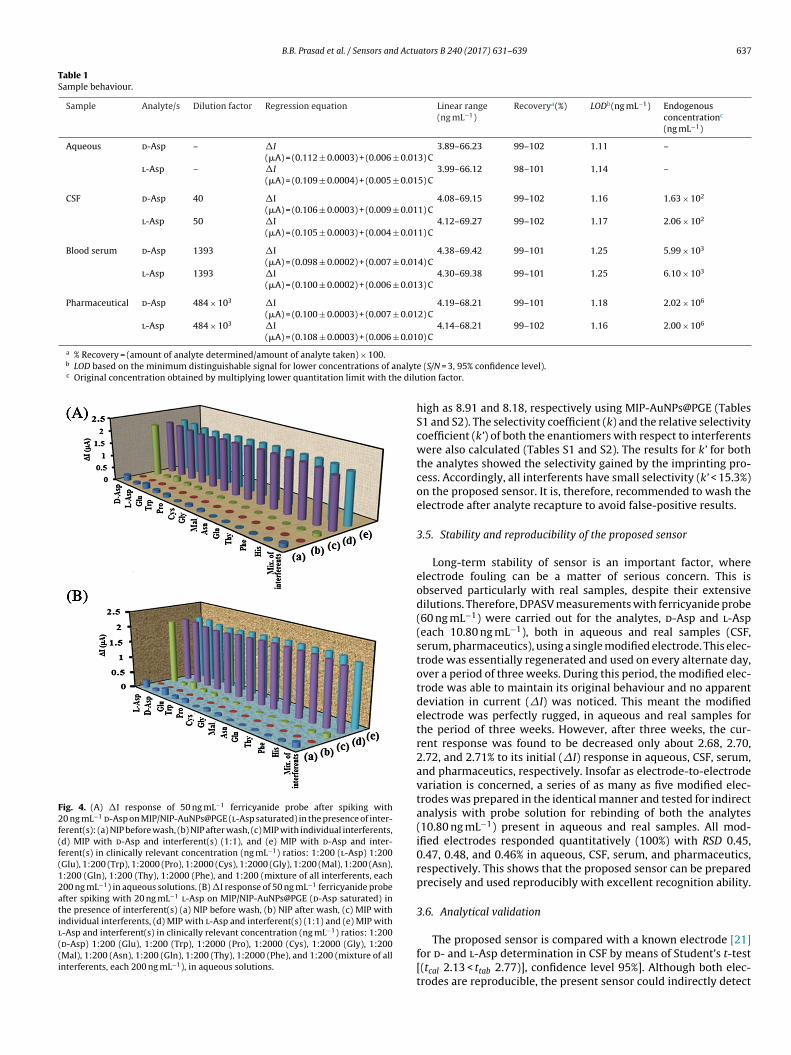
B.B. Prasad et al. / Sensors and Actuators B 240 (2017) 631–639 637
Table 1Sample behaviour.
Sample Analyte/s Dilution factor Regression equation Linear range(ng mL−1)
Recoverya(%) LODb(ng mL−1) Endogenousconcentrationc
(ng mL−1)
Aqueous d-Asp – �I(�A) = (0.112 ± 0.0003) + (0.006 ± 0.013) C
3.89–66.23 99–102 1.11 –
l-Asp – �I(�A) = (0.109 ± 0.0004) + (0.005 ± 0.015) C
3.99–66.12 98–101 1.14 –
CSF d-Asp 40 �I(�A) = (0.106 ± 0.0003) + (0.009 ± 0.011) C
4.08–69.15 99–102 1.16 1.63 × 102
l-Asp 50 �I(�A) = (0.105 ± 0.0003) + (0.004 ± 0.011) C
4.12–69.27 99–102 1.17 2.06 × 102
Blood serum d-Asp 1393 �I(�A) = (0.098 ± 0.0002) + (0.007 ± 0.014) C
4.38–69.42 99–101 1.25 5.99 × 103
l-Asp 1393 �I(�A) = (0.100 ± 0.0002) + (0.006 ± 0.013) C
4.30–69.38 99–101 1.25 6.10 × 103
Pharmaceutical d-Asp 484 × 103 �I(�A) = (0.100 ± 0.0003) + (0.007 ± 0.012) C
4.19–68.21 99–101 1.18 2.02 × 106
l-Asp 484 × 103 �I(�A) = (0.108 ± 0.0003) + (0.006 ± 0.010) C
4.14–68.21 99–102 1.16 2.00 × 106
a % Recovery = (amount of analyte determined/amount of analyte taken) × 100.b LOD based on the minimum distinguishable signal for lower concentrations of analytc Original concentration obtained by multiplying lower quantitation limit with the dilu
Fig. 4. (A) �I response of 50 ng mL−1 ferricyanide probe after spiking with20 ng mL−1
d-Asp on MIP/NIP-AuNPs@PGE (l-Asp saturated) in the presence of inter-ferent(s): (a) NIP before wash, (b) NIP after wash, (c) MIP with individual interferents,(d) MIP with d-Asp and interferent(s) (1:1), and (e) MIP with d-Asp and inter-ferent(s) in clinically relevant concentration (ng mL−1) ratios: 1:200 (l-Asp) 1:200(Glu), 1:200 (Trp), 1:2000 (Pro), 1:2000 (Cys), 1:2000 (Gly), 1:200 (Mal), 1:200 (Asn),1:200 (Gln), 1:200 (Thy), 1:2000 (Phe), and 1:200 (mixture of all interferents, each200 ng mL−1) in aqueous solutions. (B) �I response of 50 ng mL−1 ferricyanide probeafter spiking with 20 ng mL−1
l-Asp on MIP/NIP-AuNPs@PGE (d-Asp saturated) inthe presence of interferent(s) (a) NIP before wash, (b) NIP after wash, (c) MIP withindividual interferents, (d) MIP with l-Asp and interferent(s) (1:1) and (e) MIP withl-Asp and interferent(s) in clinically relevant concentration (ng mL−1) ratios: 1:200(d-Asp) 1:200 (Glu), 1:200 (Trp), 1:2000 (Pro), 1:2000 (Cys), 1:2000 (Gly), 1:200(Mal), 1:200 (Asn), 1:200 (Gln), 1:200 (Thy), 1:2000 (Phe), and 1:200 (mixture of allinterferents, each 200 ng mL−1), in aqueous solutions.
e (S/N = 3, 95% confidence level).tion factor.
high as 8.91 and 8.18, respectively using MIP-AuNPs@PGE (TablesS1 and S2). The selectivity coefficient (k) and the relative selectivitycoefficient (k’) of both the enantiomers with respect to interferentswere also calculated (Tables S1 and S2). The results for k’ for boththe analytes showed the selectivity gained by the imprinting pro-cess. Accordingly, all interferents have small selectivity (k’ < 15.3%)on the proposed sensor. It is, therefore, recommended to wash theelectrode after analyte recapture to avoid false-positive results.
3.5. Stability and reproducibility of the proposed sensor
Long-term stability of sensor is an important factor, whereelectrode fouling can be a matter of serious concern. This isobserved particularly with real samples, despite their extensivedilutions. Therefore, DPASV measurements with ferricyanide probe(60 ng mL−1) were carried out for the analytes, d-Asp and l-Asp(each 10.80 ng mL−1), both in aqueous and real samples (CSF,serum, pharmaceutics), using a single modified electrode. This elec-trode was essentially regenerated and used on every alternate day,over a period of three weeks. During this period, the modified elec-trode was able to maintain its original behaviour and no apparentdeviation in current (�I) was noticed. This meant the modifiedelectrode was perfectly rugged, in aqueous and real samples forthe period of three weeks. However, after three weeks, the cur-rent response was found to be decreased only about 2.68, 2.70,2.72, and 2.71% to its initial (�I) response in aqueous, CSF, serum,and pharmaceutics, respectively. Insofar as electrode-to-electrodevariation is concerned, a series of as many as five modified elec-trodes was prepared in the identical manner and tested for indirectanalysis with probe solution for rebinding of both the analytes(10.80 ng mL−1) present in aqueous and real samples. All mod-ified electrodes responded quantitatively (100%) with RSD 0.45,0.47, 0.48, and 0.46% in aqueous, CSF, serum, and pharmaceutics,respectively. This shows that the proposed sensor can be preparedprecisely and used reproducibly with excellent recognition ability.
3.6. Analytical validation
The proposed sensor is compared with a known electrode [21]for d- and l-Asp determination in CSF by means of Student’s t-test[(tcal 2.13 < ttab 2.77)], confidence level 95%]. Although both elec-trodes are reproducible, the present sensor could indirectly detect

6 d Actu
btapoecd
dwtQctDsabatcdaspw
sdtp
4
afoil
tl
(bsdiapeta
A
gfa
A
t
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
38 B.B. Prasad et al. / Sensors an
oth enantiomers in the wide concentration range, with quanti-ation limit as low as 4.08 ng mL−1 requisite to diagnose epilepsynd lennox syndrome. Under optimized analytical conditions, theroposed sensor is validated for the selective and sensitive analysisf d-Asp and l-Asp in real samples (Table 1). CSF contains both thenantiomers. Insofar as other biological fluids are concerned, serumontains predominantly l-Asp with some non-detectable traces of-Asp whereas pharmaceutical sample contains only l-Asp. Foremonstrating the feasibility of analysis of both the templates,e have diluted the real samples with water so as to mitigate
he matrix effect and to move the analysis in the detection range.uantitative DPASV measurements with probe are shown in thease of a representative real sample (CSF) in which both enan-iomers are found to be concomitantly present (Fig. S6A and B).PASV runs with probe were also represented in diluted human
erum samples duly spiked with d-Asp and l-Asp isomers (Fig. S7And B). Upon massive dilution, all real samples approximated theirehaviours very close to the aqueous sample. This is evident fromlmost equivalent slopes of the respective linear calibration equa-ions (Table 1) which indicate negligible matrix complications. Thisould be the reason for applying similar operating analytical con-itions (Eacc , tacc , and pH) in real samples as those utilized withqueous sample analysis. In particular, since diluted (1393-fold)erum approximated its sample behaviour similar as aqueous sam-le, blood serum sample analysis could be accomplished at pH 3,ithout any effect of acidity and matrix complications.
The proposed MIP sensor was compared with other known sen-ors for d- and l-Asp determinations (Table S3). Accordingly, theetection ability of most of the known techniques was inferioro this work; and moreover, majority of the techniques were notroperly validated for the application in real samples.
. Conclusion
For the first time, we have contemplated the one-by-onepproach, using ferricyanide probe on a dual imprinted single PGE,or ultra-trace analysis of D- and L-enantiomers, prevalent togetherr alone, in real samples. This work merits special significance
n view of the fact that the enantioselective analysis of d- and-Asp is a challenge, because both the isomers have same oxida-ion potentials. The proposed MIP-AuNPs@PGE detected d-Asp and-Asp present together with detection sensitivities 1.16 ng mL−1
d-Asp) and 1.17 ng mL−1 (l-Asp), particularly in CSF, which cane useful in diagnosis of chronic diseases (epilepsy and lennoxyndrome) manifested at stringent limits. The sensor is repro-ucible, rugged, regenerable, and cost-effective showing excellent
mprinting effect (� = 8.91). This assures reliable results, withoutny cross-reactivity and false-positives, in clinical settings. The pro-osed indirect method of sequential analysis on a dual imprintedlectrochemical sensor may be considered novel and versatile inhe sense that any pair of simple molecules can be used as targetnalytes (templates).
cknowledgements
Authors thank University grant commission, New Delhi forranting a research fellowship to one of us (S.J). Instrumentalacilities procured from Banaras Hindu University are also greatlycknowledged.
ppendix A. Supplementary data
Supplementary data associated with this article can be found, inhe online version, at http://dx.doi.org/10.1016/j.snb.2016.09.031.
[
ators B 240 (2017) 631–639
References
[1] B.S. Sekhon, Enantioseparation of chiral drugs—an overview, Int. J. Pharm.Technol. Res. 2 (2010) 1584–1594.
[2] B.B. Prasad, A. Srivastava, M.P. Tiwari, Highly sensitive and selectivehyphenated technique (molecularly imprinted polymer solid-phasemicroextraction–molecularly imprinted polymer sensor) for ultra traceanalysis of aspartic acid enantiomers, J. Chromatogr. A 1283 (2013) 9–19.
[3] P.K. Katiyar, R.K. Wala, Chiral separation of aspartic acid by thin layerchromatography, Asian J. Chem. 22 (2010) 4945–4946.
[4] P.J. Van den Oetelaar, L.E. Van Beijsterveldt, J.R. Van Beckhoven, H.J. Hoenders,Detection of aspartic acid enantiomers by chiral capillary gaschromatography. Determination of in vivo racemisation and reduction ofmetal-induced background, J. Chromatogr. A 386 (1986) 135–143.
[5] H. Yu, Y.S. Ding, S.F. Mou, Some factors affecting separation and detection ofamino-acids by high performance anion-exchange chromatography withintegrated pulsed amperometric detection, J. Chromatogr. A 997 (2003)145–153.
[6] S. Wang, L. Fan, S. Cui, CE-LIF chiral separation of aspartic acid and glutamicacid enanatiomers using human serum albumin and sodium cholate as dualselectors, J. Sep. Sci. 32 (2009) 3184.
[7] H. Guan, P. Zhou, X. Zhou, Z. He, Sensitive and selective detection of asparticacid and glutamic aciod based on polythiophene-gold nanoparticlescomposites, Talanta 77 (2008) 319–324.
[8] Y.H. Deng, H. Wang, H.S. Zhang, Determination of amino acidneurotransmitters inhuman cerebrospinal fluid and saliva by capillaryelectrophoresis with laser-induced fluorescence detection, J. Sep. Sci. 31(2008) 3088–3097.
[9] C. Dai, J. Zhang, Y. Zhang, X. Zhou, Y. Duan, S. Liu, Removal of carbamazepineand clofibric acid from water using double templates- molecularly imprintedpolymers, Environ. Sci. Pollut. Res. 20 (2013) 5492–5501.
10] Y. Guo, T. Guo, A dual-template imprinted capsule with remarkably enhancedcatalytic activity for pesticide degradation and elimination simultaneously,Chem. Commun. 49 (2013) 1073–1075.
11] Y. Guo, R. Wang, W. Chi, S. Liu, H. Shi, T. Guo, One-step synthesis ofreactant-product-dualtemplate imprinted capsules as phosphotriesterasemimetic enzymes for pesticide elimination, RSC Adv. 4 (2014) 7881–7884.
12] T. Jing, Y. Wang, Q. Dai, H. Xia, J. Niuu, Q. Hao, S. Mei, Y. Zhou, Preparation ofmixed-templates molecularly imprinted polymers and investigation of therecognition ability for tetracycline antibiotics, Biosens. Bioelectron. 25 (2010)2218–2224.
13] A.C. Meng, J.J. Le Jeune, D.A. Spivak, Multi-analyte imprinting capability ofOMNiMIPs versus traditional molecularly imprinted polymers, J. Mol.Recognit. 22 (2009) 121–128.
14] B.B. Prasad, D. Jauhari, A. Verma, A dual-ion imprinted polymer embedded insol–gel matrix for the ultra trace simultaneous analysis of cadmium andcopper, Talanta 120 (2014) 398–407.
15] B.B. Prasad, D. Jauhari, A dual-template biomimetic molecularly imprinteddendrimer-based piezoelectric sensor for ultratrace analysis oforganochlorine pesticides, Sens. Actuators B: Chem. 207 (2015) 542–551.
16] B.B. Prasad, D. Jauhari, Double-ion imprinted polymer @magneticnanoparticles modified screen printed carbon electrode for simultaneousanalysis of cerium and gadolinium ions, Anal. Chem. Acta 875 (2015) 83–91.
17] M.P. Tiwari, R. Madhuri, D. Kumar, D. Jauhari, Double imprinting in a singlemolecularly imprinted polymer format for the determination of ascorbic acidand dopamine, Adv. Mater. Lett. 2 (2011) 276–280.
18] J. Xin, X. Qiao, Z. Xu, J. Zhou, Molecularly imprinted polymer as sorbent forsolid-phase extraction coupling to gas chromatography for the simultaneousdetermination of trichlorfon and monocrotophos residues in vegetables, FoodAnal. Methods 6 (2013) 274–281.
19] M. Majumder, N. Chopra, B.J. Hinds, Mass transport through carbon nanotubemembranes in three different regimes: ionic diffusion and gas and liquid flow,ACS Nano 5 (2011) 3867–3877.
20] B.B. Prasad, I. Pandey, Electrochemically imprinted molecular recognitionsites on multiwalled carbon-nanotubes/pencil graphite electrode surface forenantioselective detection of d- and l-aspartic acid, Electrochem. Acta 88(2016) 24–34.
21] B.B. Prasad, A. Srivastava, M.P. Tiwari, Molecularly imprinted polymer-matrixnanocomposite for enantioselective electrochemical sensing of D- andL-aspartic acid, Mater. Sci. Eng. C 33 (2013) 4071–4080.
22] W. Gao, J. Song, W. Naiying, Voltammetric behavior and square-wavevoltammetric determination of trepibutone at a pencil graphite electrode, J.Electroanal. Chem. 576 (2016) 1–7.
23] H.M. Elqudaby, H.A.M. Hendawy, E.R. Souaya, G.G. Mohamed, G.M.G. Eldin,Utility of activated glassy carbon and pencil graphite electrodes forvoltammetric determination of nalbuphine hydrochloride in pharmaceuticaland biological fluids, Int. J. Electrochem. 2016 (2015) 1–9.
24] Y. Xiao, H.X. Ju, H.Y. Chen, Hydrogen peroxide sensor based on horseradishperoxidase-labeled Au colloids immobilized on gold electrode surface bycysteamine monolayer, Anal. Chim. Acta 39 (1999) 73–82.
25] B.B. Prasad, R. Madhuri, M.P. Tiwari, P.S. Sharma, Imprinting molecularrecognition sites on multi-walled carbon nanotubes surface forelectrochemical detection of insulin in real samples, Electrochim. Acta 55(2010) 9146–9156.

d Actu
[
[
[
[
[
[
[
[
[
Kislay Singh is currently pursuing Ph.D. at Banaras Hindu University (BHU) underthe supervision of Prof. Bhim Bali Prasad. She received her B.Sc. degree in 2011and M.Sc. degree in 2013 from BHU. She is recipient of UGC meritorious research
B.B. Prasad et al. / Sensors an
26] N. Ogata, C. Azuma, C. Itsubo, Radical- and photopolymerizations ofN-cycloalkyl acrylamides, J. Polym. Sci. 13 (1975) 1959–1962.
27] B.B. Prasad, D. Jauhari, M.P. Tiwari, Doubly imprinted polymernanofilm-modified electrochemical sensor for ultra trace simultaneousanalysis of glyphosate and gluphosinate, Biosens. Bioelectron. 59 (2014)81–88.
28] A. Erdem, H. Karadeniz, A. Caliskan, Single-walled carbon nanotubes modifiedgraphite electrodes for electrochemical monitoring of nucleic acids andbiomolecular interactions, Electroanalysis 21 (2009) 464–471.
29] B. Rezaei, S. Foroughi-Dehnavi, A.A. Ensafi, Fabrication of electrochemicalsensor based on molecularly imprinted polymer and nanoparticles fordetermination trace amounts of morphine, Ionics 21 (2015) 2969–2980.
30] V. Georgakilas, J.N. Tiwari, K. Christian Kemp, J.A. Perman, A.B. Bourlinos, K.S.Kim, R. Zboril, Noncovalent functionalization of graphene and graphene oxidefor energy materials, biosensing, catalytic, and biomedical applications,Chem. Rev. 116 (2016) 5464–5519.
31] B.B. Prasad, R. Singh, A new micro-contact imprinted l-cysteine sensor basedon sol?geldecorated graphite/multiwalled carbonnanotubes/goldnanoparticles composite modified sandpaper electrode, Sens.Actuators B: Chem. 212 (2015) 155–164.
32] S. Patil, S. Datar, N. Rekha, S.K. Asha, C.V. Dharmadhikari, Charge storage andelectron transport properties of gold nanoparticles decorating aurethane-methacrylate comb polymer network, Nanoscale 5 (2013) 4404.
33] C. Gutierrez-Sanchez, M. Pita, V. Dominguez, S. Shleev, A.L. De Lacey, Goldnanoparticles as electronic bridges for laccase-based biocathodes, J. Am.Chem. Soc. 134 (2012) 17212–17220.
34] E. Laviron, General expression of the linear potential sweep voltammogram inthe case of diffusionless electrochemical systems, J. Electroanal. Chem 101(1979) 19–28.
ators B 240 (2017) 631–639 639
Biographies
Bhim Bali Prasad is currently working as a professor of Analytical Chemistry inthe Banaras Hindu University (BHU), Varanasi, India. He has mentored 25 Ph.D.students and published 115 research papers in several reputed international andnational Journals. He received his B.Sc. degree in Chemistry in 1972 and M.Sc.degree in 1974 form BHU. He obtained his Ph.D. from BHU. He is a recipient ofseveral national and international awards for his research contributions in Analyt-ical Chemistry and nano-materials. His research interests include environmentalchemistry, chromatography, electroanalysis, and detection principle for chemicalanalysis, nano-technology, and development of biomimetic nano sensors usingmolecularly imprinted polymers for clinical, pharmaceutical and biological analysis.
Swadha Jaiswal is currently pursuing Ph.D. at Banaras Hindu University (BHU) underthe supervision of Prof. Bhim Bali Prasad. She received her B.Sc. degree in 2011and M.Sc. degree in 2013 from BHU, Varanasi. She is recipient of UGC Fellowship.Her research interest lies in the field of chemical sensor development, molecularlyimprinted polymers, and electro-analytical chemistry.
fellowship. Her research interest lies in the field of chemical sensors, molecularlyimprinted polymers, and electro-analytical chemistry.











![HISTORY Syllabus PAPER II Paper-II (Covering entire ... · 1 State Eligibility Test MP SET – 2018 [Code No. – 08] HISTORY Syllabus PAPER – II Note:- Paper-II (Covering entire](https://img.pdfslide.us/doc/110x75/5f6dac9bccaedd55362b9f04/history-syllabus-paper-ii-paper-ii-covering-entire-1-state-eligibility-test.jpg)

















