Embed Size (px)
Citation preview

THEO CHEM
Journal of Molecular Structure (Theochcm) 338 (1995) 347-362 ELSEVIER
Relativistic effects of p-block molecules
Abstract
Density functional calculations including relativistic as well as nonlocal exchange and correlation corrections have been performed on group 14 molecules MO. MH,, MCI, (M = C, Si, Ge. Sn, Pb). Good bond lengths, and reasonable bond energies and force constants have been obtained. The dipole moments of the MO series are quite good, and dp/dR of CO is close to the experimental results. Relativistic corrections are important for the heavier molecules (M = Ge, Sn. Pb). Fractional relativistic changes of bond energies, bond lengths and force constants are nearly exactly proportional to Z2 (Z: heavy nuclear charge). The origin of the relativistic changes has been analyzed: the change upon bond formation of the valence orbitals near the heavy nucleus is decisive.
1. Introduction
The p-block elements form a chemically rich region. these elements showing greater variation in properties than those of the s and d blocks. In this paper. we choose the five group 14 elements M = C, Si, Ge, Sn. Pb to investigate the electronic structures of three series of molecules: MO, MHJ and MC14. The heavy elements are strongly influ- enced by relativistic effects, many properties chan- ging from the nonrelativistic approximation (nr) to the more realistic relativistic approach (r) as the square of their atomic numbers Z. For instance. the fractional relativistic contraction or expansion of different AOs [l] is approximately
6r r’ - r”’ -= r r’
x (Z/c,’
where the proportionality factor is of order i 1.
* Corresponding author
c = 137a.u. is the velocity of light. For Pb with Z = 82. (Z/c)’ = 0.36. So, when investigating heavy elements, the relativistic corrections must be considered.
Since the relativistic effects are small for the lighter elements of the periodic table, first order perturbation theory has been a popular way to correct for relativistic effects. Although second- order perturbation theory for molecules has been reported [z-5]. it has only been used for very light molecules. The most important first-order effects for valence electrons are the relativistic mass-velo- city variation (MV). the Darwin correction to the potential (DW), the change of nuclear screening by the relativistically modified atomic cores (DVC) and the spin-orbit interaction of an electron moving in the screened nuclear potential (SO). Dyall and Taylor reported [6] for the group 14 tetrahydrides MH4 that the bond lengths are well predicted by first-order perturbation theory (FOPT). He also reported that FOP7 (where also
0166-1X0/95/$09.50 J 1995 Elsewer Science B.V. All rights reserved SSDI 0166-1280(95)0106’-1

the core electrons were treated by FOPT) over- estimate the relativistic corrections to the harmonic frequencies.
Density functional (DF) calculations using the ADF program system [7-91 have been performed here on group 14 molecules. The atomic cores have been treated at the DiraccSlater level and are then frozen and transferred to the molecules. The mole- cular valence electrons have been treated at the relativistic first-order perturbation level in the field of the ,fu//?. relativistic atomic ewes. Different exchange-correlation functionals for the valence electrons have been tested. The average deviations between theoretical and experimental bond lengths, bond energies and force constants are within 0.02A. 0.5 eV and 0.5 N cm ’ , respectively. depending somewhat on the chosen exchange- correlation potential. Relativistic corrections to molecular spectroscopic constants are in good agreement with all-electron Dirac-HartreeeFock results [6, lo]. Spin-orbit splittings of p-orbitals are close to experimental data.
After having proven the reliability of the calcula- tions, the origin of the relativistic corrections to atomic and molecular properties has been analyzed. The atomic orhitul riotle sttwturr
creates the atomic spatial shell structure which is not easily perceivable in the total density. Here we denote the atomic spatid shells as K-, L-, M-. shells. The outer R-values of the shells corre- sponding to the average positions of the nodes of HartreeeFockkSlater AOs [ 1 l] of different /-values are presented in Table 1. Concerning the atoms, we remember [12] that the dominant relativistic contri- butions come from the inner shells.
Table I Outer radii of atomic shell!, in XL.
Atom K L M N 0
c 0.49 St 0 15 0.70 Ge 0.06 0.24 0 x2 Sn 0.04 0. I4 0.40 1.04 Pb 0.02 0 OY IO.21 0.49 1.16
0 0.17 Cl 0.12 0 54
This paper is organized as follows. Methods and calculation details are given in section 2. In section 3. the calculated molecular spectroscopic constants and relativistic corrections are discussed. In section 4. relativistic effects in group 14 atoms and mole- cules are analyzed in more detail. At last, some conclusions are given in section 5, concerning both the performance of DF approaches for heavy systems and the relativistic effects in such systems.
2. Details of calculation and interpretation methods
2. I. Calculations
All calculations were carried out with the Density-Functional (DF) [13,14] Program Package ADF developed in Amsterdam by Baer- ends et al. [7], and extended the relativistic first- order perturbation method by Snijders and Baer- ends [8] and to first-order relativistic correction to the bond energy by Ziegler et al. [9]. The analysis of the spatial origin of relativistic corrections to atomic and molecular orbitals and to bond ener- gies was implemented by us.
In the present work, we use (1) the simple local spin density (LSD) Xa approach of Slater (DF(S)) [ 151. (2) we included the correlation corrections of VWN [ 161 and SPP [ 171 (DF(C)) and (3) the “full” correction by the additional nonlocal (NL) gradient exchange correction [18] (DF(F)). With these three levels of methods, 15 molecules were treated.
As mentioned before the frozen core approxima- tion has been used. The inner atomic core orbitals are calculated by numerical Hartree- and Diracc Fock-Slater methods [19]. The numerically accu- rate orbitals, nonrelativistic and relativistic ones, are then frozen and transferred to the atomic and molecular calculations in the nonrelativistic and relativistic cases.
To obtain accurate results, triple-zeta ST0 basis sets [20] have been used for the valence shells, extended by single-zeta STOs for the inner core wiggles [21] and double-zeta polarization func- tions (C 3d. Si 3d. Ge 4d. Sn 5d, Pb 6d, H 2p, 0 3d. Cl 3d). The exponents are given in Table 2.
In order to analyze the physical origin of relati- vistic effects, the distributions of the mass-velocity
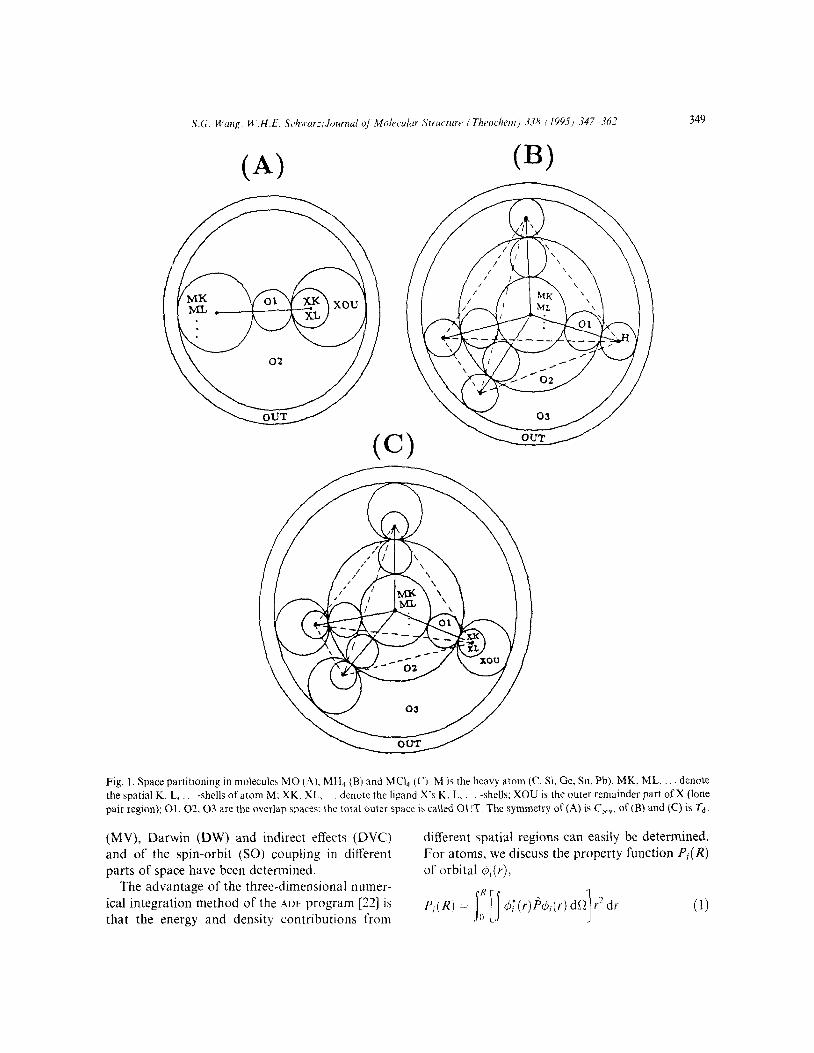
S.G. Wang, U’.H.E. Schwur;,Journul q/ i~tolecuiur Srrucrur~ ( Throthem) 338 i 19951 347- 362 349
Fig. 1. Space partitioning in molecules MO (A). MH4 (B) and MCI4 (C). M is the heavy atom (C. Si. Ge, Sn, Pb). MK. ML. denote the spatial K. L. -shells of atom M; XK. XL, denote the ligand X’s K, L. -shells; XOU is the outer remainder part ofX (lone
pair region); 01, 02,03 are the overlap spaces: the total outer space is called OUT. The symmetry of (A) is C,,, of(B) and (C) is rd.
(MV), Darwin (DW) and indirect effects (DVC) and of the spin-orbit (SO) coupling in different parts of space have been determined.
The advantage of the three-dimensional numer-
different spatial regions can easily be determined. For atoms, we discuss the property function P;(R) of orbital G,(Y),
ical integration method of the ADF program [22] is Pi(R) = &(r)kb;(r) dR r’dr I
(1) that the energy and density contributions from

where p can be the MV. DW. DVC or SO operator
[121. Concerning relativistic corrections to the bond
energy, we divide the molecular volume into several spatial regions: atomic core shells, overlap regions and the outer space. The space division of the molecules is shown in Fig. 1. The first- order correction to the bond energy -D = AE =
E Mel - E~tom\ can then be written as
-6D = AE’ = J (I&, - I&v + I&~ + I?;, )
+ AE&(,z) + AE;&)] (2)
Here and in the following. L!, will be used for bond effects, and 0 for relativistic effects. &!iB is the nonrelativistic change of density from the superim- posed atoms to the molecule. L!&’ is divided into several spatial partial contributions, coming from the nth subspace among the PZ ones.
2.2. Intrrprelutims
A comment is appropriate here [33]. In quantum mechanics. observables are given as integrals over the whole space. Upon integral transformation. the physically observable values of the integrals do not change, though the contributions from different parts of space may be changed arbitrarily. Concerning the energy.
E = (&Q) = E(oo) = E. 1
p(r) . dr’
means that in the standard representation the energy comes from that part of space where there is most electron density, i.e. for valence electrons from the valence shells. This holds both for the relativistic and nonrelativistic energies, and accord- ingly for the relativistic corrections, too.
The valence electrons are fast and behave rela- tivistically when they are near to the nucleus. Although the probability of valence electrons being near the nucleus is very small, its dynamics, that is the value. slope and curvature of the inner
tail of the valence orbital is changed significantly. This relativistic orbital change 60 = 4’ near the nucleus propagates to the outer valence shell, there resulting in the change of the nonrelativistic energy contributions, see the right side of the follwing first-order equation:
hE = E’ = (@‘fi”cjo) + (q”I?~o) + (@“I?oq+)
= E’ J Gp(r).dr’
=.I' ~".(ff'- E0)&dr3 outer region
(3) The first and third brackets in Eq. (3) depending on the relativistic change 0’ of the wavefunction, yield large, oscillating contributions in the inner shells which, for intermediate normalization, sum to zero over the whole space. Therefore, we decided to investigate only the behavior of the middle bracket
r;E = (@~~fi’@~) (4)
which contains the valence density @I’$‘, being large in the outer valence shell. and the relativistic correction of the Hamiltonian fi’, whose action is largest in the inner core. While the dominant contribution to the relativistic energy change is collected in the valence shell in Eq. (3) it is collected in the core in Eq. (4).
Since 8’ contains products of Hermitian opera- tors, for instance of the form 2 . j, we make use of the equality
(oOl.~blQO) = (Ac$“p~o)
if the right side is computationally easier to handle or numerically more stable. Again, contributions from different regions of space are changed thereby. For instance, since the standard form of the Darwin operator, (V’ I’), is numerically ill behaved at the nucleus, we make use of
(oO’(V;’ V)l@O) = -2(GqVV~Vd0)
Whereas the left side gets its major contributions from inside the nucleus, the right side gets signifi- cant contributions also from the nuclear surround- ings.
Intuitive explanations of quantum mechanical phenomena must be based on sound quantum

Table 2
S.G. Wang. U’ H.E. Schwurz: Journal of. Molecular Structurr i Thewhem) 338 (1995) 347-362 351
Slater orbital exponents for valence orbitals of H. C. 0, Si. Cl, Ge, Sn and Pb
H C 0 Si Cl Ge Sn Pb
Is 1.58 Is 5.40 Is 7.36 Is 11.9 Is 13.95 Is 16.95
0.92 2s 4.60 2s 7.58 2s 4.50 2s 5.65 2s 13.95 0.69 2.10 2.88 2P 5.15 2P 6.70 3 14.10
2P 1.50 I .28 1.72 3s 2.85 3s 3.30 3s 5.95
0.75 2p 2.94 3J 4.0x 1.85 2.30 3p 6.15 I .48 2.08 1.20 1.60 3d 9.20 0.82 1.12 3P 1.85 3P 2.85 4.80
3d 2.21 3d 3.10 1.20 2.05 2.50 1.15 1.50 0.75 1.20 4s 3.15
3d 1.22 3d 2.45 1.95
0.64 1.65 1.25 4P 2.35
1.35
0.80 4d 1.38
0.75
1s 33.20
2s 18.15
7-P 21.55 3s 11.65
3P 11.20 3d 12.70 4s 5.95
4P 5.85 4d 5.65
3.70 2.30
5s 3.25 2.10
1.35
5P 2.45 1.45
0.90 5d 1.50
0.75
Is
2s
2P 3s
3P 3d 4s
4P 4d 4f 5s
5P 5d
6s
6~
6d
48.15 30.30
34.85 20.6 20.3
24.2 11.8 12.0
11.4 9.19 6.90
6.35 6.00
3.80 2.35 3.20
2.10 1.45 2.75
1.65 1.00 1.68
0.85
mechanical formalism. Since the explicit represen- tation of this formalism is not unique, there obviously exist different, equivalent complemen- tary explanation schemes; for instance those in terms of shell-contributions to the energy, which are nonobservable quantities. Nonobservables (such as orbitals, for instance) form a most impor- tant ingredient of understanding chemistry.
3. Calculated spectroscopic constants and their relativistic corrections
The calculated bond lengths R,, atomization energies D, = -AE, and totally symmetric force constants k, are displayed in Tables, 3. 5 and 6.
3.1. Monoxides
The monoxides are well known experimentally. Many calculations have been performed on this series of molecules. Besides CO and SiO, calcu- lations on GeO, SnO and PbO have recently been
reported [24&26]. Most later ones are based on the effective core pseudopotential (ECP or relativistic RECP) approximations at the SCF, MCSCF, SDCI or CASSCF levels. Some ab initio calcula- tions used first-order perturbation theory for the relativistic corrections, others used the Dirac- Hartree-Fock approach.
Our DF bond lengths agree quite well with the experimental data (Table 3). Among the three DF approaches, DF(S) gives the best values for R, the average error being only +O.Ol A. Dyall’s DHF [25] results are too short by 0.03 A on the average of GeO, SnO and PbO, which is not unexpected for ab initio Fock-type calculations. DF(C) and espe- cially DF(F) yield R-values too long by O-0.04 and 0.06 A respectively. Becke’s NL exchange correc- tion in general tends to overestimate the bond lengths [27]. The comparatively small relativistic contractions of the bond lengths are a little bigger than the more reliable DHF results of Dyall: GeO (-0.003 A), SnO (-0.0074 A). PbO (-0.0146 A)
~251. The force constants of the present density func-
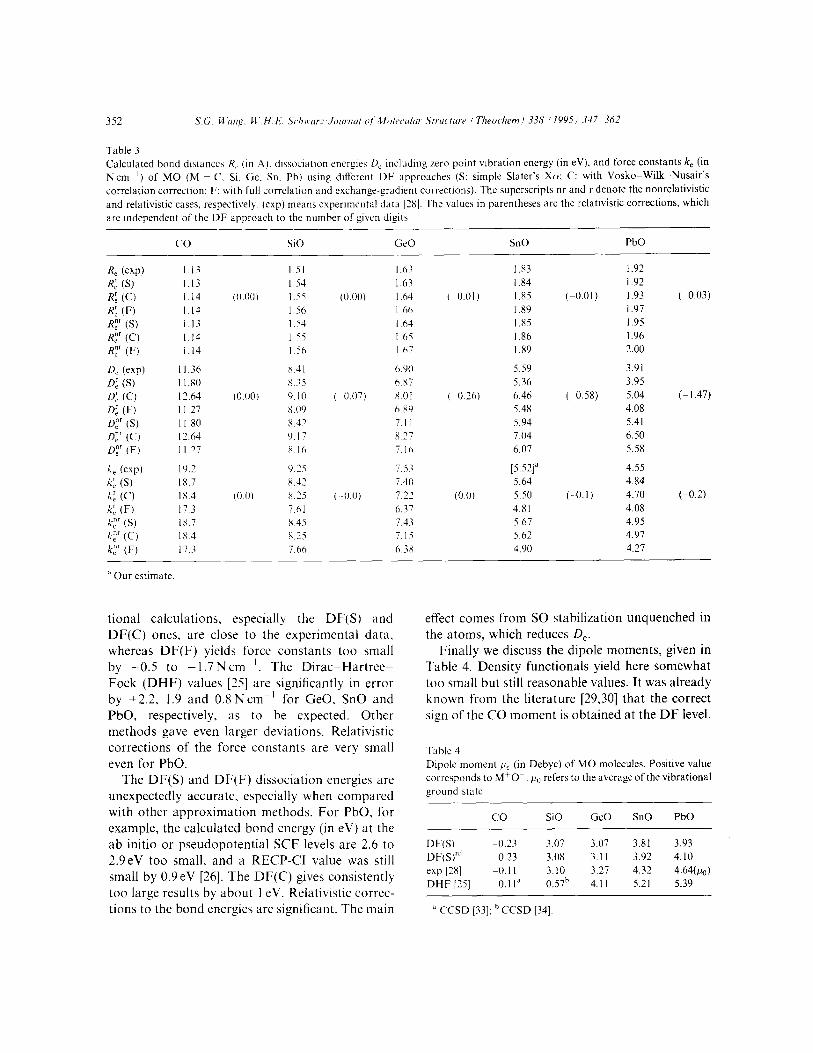
Table 3 Calculated bond distances R, (in .A). dissoclatlon encrg~s II, Including zero point vibration energy (in eV), and force constants k, (in
Ncm-‘) of MO (M = C. SI. Ge, Sn. Pb) using different DF approaches (S: simple Slater’s Xa; C: with Vosko-Wilk-Nusair’s correlation correction; F: uith full correlation and exchanee-eradient corrections). The superscripts nr and r denote the nonrelativistic _ c and relativistic cases, respectively. (cxp) means experimental data [2X]. The values in parentheses arc the relativistic corrections, which
are independent of the DF approach to the number of given digits
co SiO tie0 SnO PbO
R, (ev) 1.13
R: (9 1.13 R: CC) 1.14 R: (F) 1.14 R,“’ (S) 1.13 R:’ CC) 1.14 R:’ (F) 1.14
4 (ev) 11.36 D: (S) 11.80
D: (Cl 11.64 D: (F) 11 27 0:’ (S) 11 80
0:’ (0 12.64 0,“’ (F) 11.27
X, (exp) 19.2
k: 6) 1x.7
ii: CC) IX.4
k: (F) 17.3 k:’ (S) 1x.7 k:’ CC) 18.4 k,“’ (F) 17 3
(0.00)
(0 00)
10 0)
I.51
1.54 1.55 1.56
1.54 I 55 1 56
x.41 8.35
9.10 x.09 x.42
Y 17 X.16
Y 25 x.42 x.25
7.61 8.45 x.25
7.6h
(0.00)
(-0.07)
( -0.0)
1.63 1.83
1.63 1.84 1.64 ( (1.01) 1.85 I 6h I .89
I.64 1.85 I 65 I .86 1.67 1.89
6 YO 5.59 (x87 5.36
x.01 ( 026) 6.46 6.X”, 5.48 1.11 5.94
x 17 7.04 7.16 6.07
7 i7 8 ._. 7.30 7.22 (0.0)
6.37 I 43 7 15
b 3 x
[5.52]” 5.64 5.50
4.51 5.67 5.62
4.90
a Our estimate.
tional calculations. especially the DF(S) and DF(C) ones, are close to the experimental data, whereas DF(F) yields force constants too small by -0.5 to -1.7 N cm ‘. The Dirac-Hartree- Fock (DHF) values [25] are significantly in error by +2.2. 1.9 and 0.8 N cm -’ for GeO. SnO and PbO, respectively, as to be expected. Other methods gave even larger deviations. Relativistic corrections of the force constants are very small even for PbO.
The DF(S) and DF(F) dissociation energies are unexpectedly accurate, especially when compared with other approximation methods. For PbO, for example, the calculated bond energy (in eV) at the ab initio or pseudopotential SCF levels are 2.6 to 2.9eV too small. and a RECP-CI value was still small by 0.9eV [26]. The DF(C) gives consistently too large results by about 1 eV. Relativistic correc- tions to the bond energies are significant. The main
(-0.01)
( 0.58)
(m-0.1)
1.92 1.92 I .93 (-0.03) 1.97 I .95 1.96
2.00
3.91
3.95 5.04 (- 1.47) 4.08
5.41 6.50 5.58
4.55
4.84 4.70 (-0.2)
4.08 4.95 4.97 4.27
effect comes from SO stabilization unquenched in the atoms, which reduces D,.
Finally we discuss the dipole moments, given in Table 4. Density functionals yield here somewhat too small but still reasonable values. It was already known from the literature [29,30] that the correct sign of the CO moment is obtained at the DF level.
Table 4 Dipole moment /Lo (in Debye) of MO molecules. Positive value corresponds to M+O-. p0 refers to the average of the vibrational
ground state
DFIS)
DF(S)“’
exp [2X] DHF [25]
co SiO GeO SnO PbO
-0.23 3.07 3.07 3.81 3.93 PO.23 3.08 3.11 3.92 4.10
-0.11 3.10 3.21 4.32 4.64(/Q) -0.1 Ia 0.57h 4.11 5.21 5.39
d CCSD [33]: h CCSD [34]
-- MCSCF
1.7 1.8 1.9 2.0 2.1 2.2 2.3 2.4 2;
R/(a.u.)
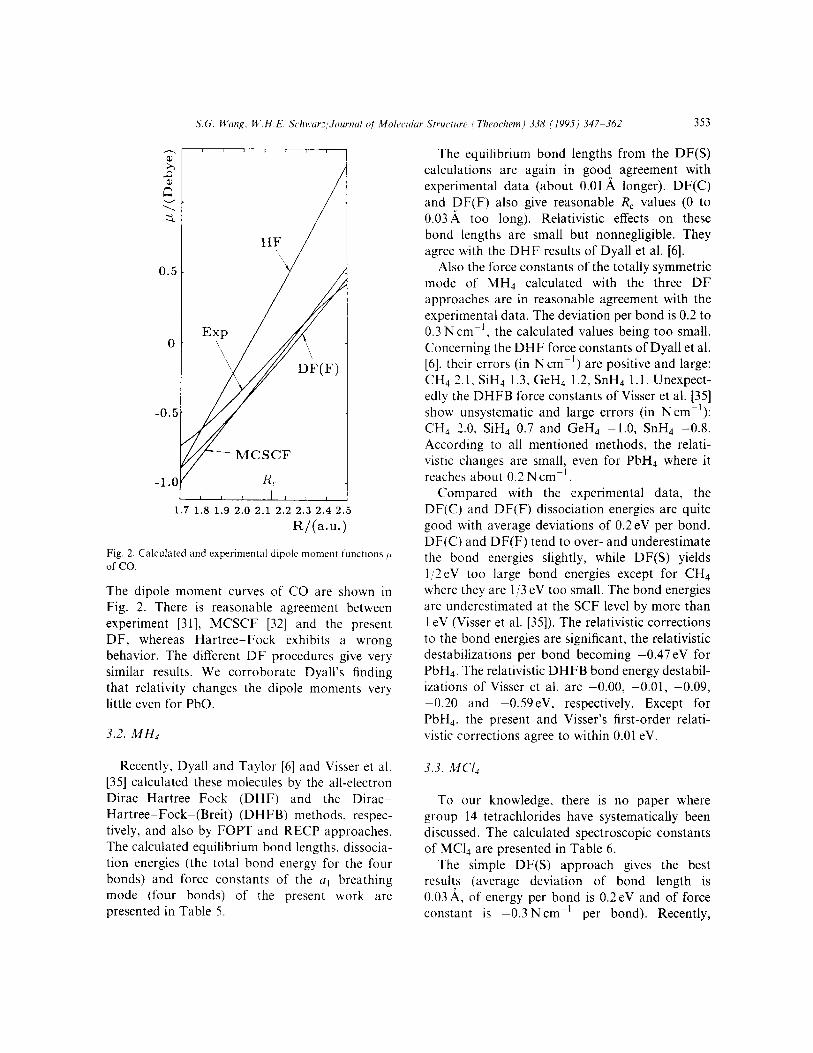
Fig. 2. Calculated and experimental dipole moment functions IL of co.
The dipole moment curves of CO are shown in Fig. 2. There is reasonable agreement between experiment [31], MCSCF [32] and the present DF, whereas Hartree-Fock exhibits a wrong behavior. The different DF procedures give very similar results. We corroborate Dyall’s finding that relativity changes the dipole moments very little even for PbO.
3.2. MH4
Recently, Dyall and Taylor [6] and Visser et al. [35] calculated these molecules by the all-electron Dirac-HartreeeFock (DHF) and the Diracc HartreeeFockk(Breit) (DHFB) methods, respec- tively, and also by FOPT and RECP approaches. The calculated equilibrium bond lengths, dissocia- tion energies (the total bond energy for the four bonds) and force constants of the al breathing mode (four bonds) of the present vvork are presented in Table 5.
The equilibrium bond lengths from the DF(S) calculations are again in good agreement with experimental data (about 0.01 A longer). DF(C) and DF(F) also give reasonable R, values (0 to 0.03 A too long). Relativistic effects on these bond lengths are small but nonnegligible. They agree with the DHF results of Dyall et al. [6].
Also the force constants of the totally symmetric mode of MH4 calculated with the three DF approaches are in reasonable agreement with the experimental data. The deviation per bond is 0.2 to 0.3Ncm-‘, the calculated values being too small. Concerning the DHF force constants of Dyall et al. [6]. their errors (in Ncm-‘) are positive and large: CH4 2.1, SiH4 1.3, GeH4 1.2, SnH4 1.1. Unexpect- edly the DHFB force constants of Visser et al. [35] show unsystematic and large errors (in Ncm-‘): CH, 2.0, SiH4 0.7 and GeH, -1.0, SnH4 -0.8. According to all mentioned methods, the relati- vistic changes are small, even for PbH4 where it reaches about 0.2 Ncm-‘.
Compared with the experimental data, the DF(C) and DF(F) dissociation energies are quite good with average deviations of 0.2eV per bond. DF(C) and DF(F) tend to over- and underestimate the bond energies slightly, while DF(S) yields l/2 eV too large bond energies except for CH4 where they are l/3 eV too small. The bond energies are underestimated at the SCF level by more than I eV (Visser et al. [35]). The relativistic corrections to the bond energies are significant, the relativistic destabilizations per bond becoming -0.47eV for PbH4. The relativistic DHFB bond energy destabil- izations of Visser et al. are -0.00, -0.01, -0.09, -0.20 and -0.59eV, respectively. Except for PbH4, the present and Visser’s first-order relati- vistic corrections agree to within 0.01 eV.
3.3. MC/~
To our knowledge, there is no paper where group 14 tetrachlorides have systematically been discussed. The calculated spectroscopic constants of MCI4 are presented in Table 6.
The simple DF(S) approach gives the best results (average deviation of bond length is 0.03 A, of energy per bond is 0.2eV and of force constant is -0.3 Ncm-’ per bond). Recently,
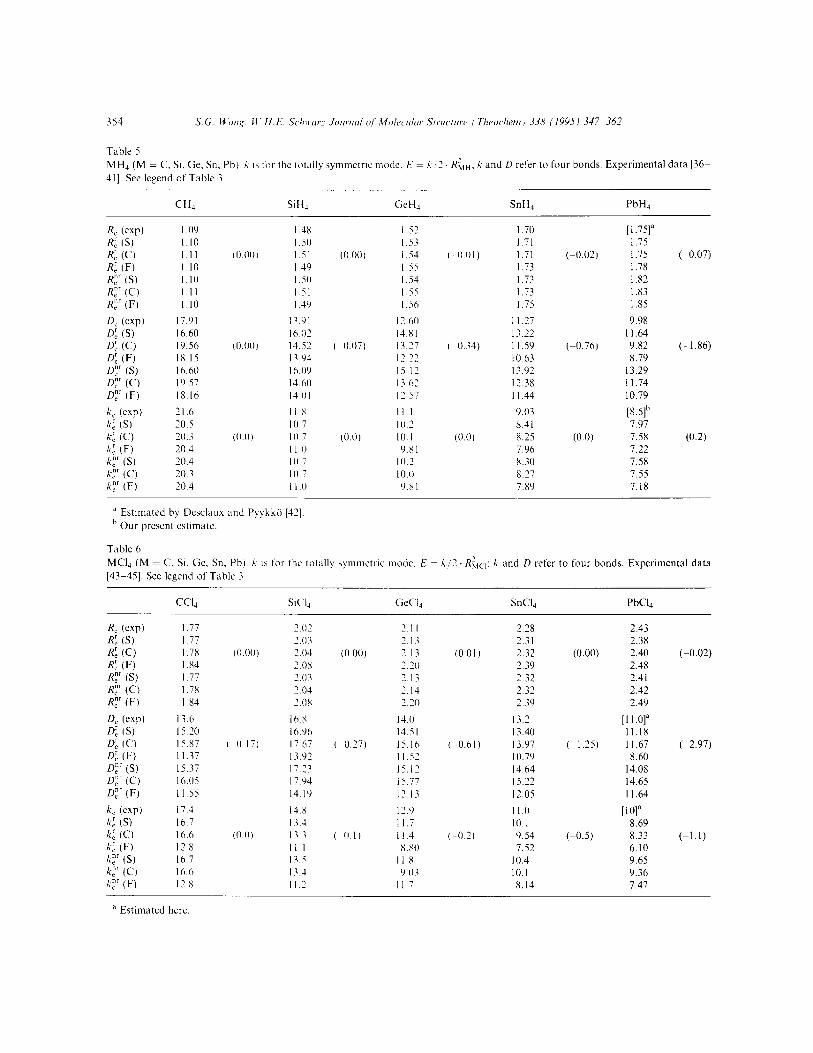
Table 5
MH4 (M = C. Si. Ge. Sn. Ph) X 15 for the rotally symmetric mode. E = X 2. RLH. k and LI refer to four bonds. Experimental data [36-
411. See legend of Table 3
CHJ SiH 4 GeH, SnH, PbH4
R, (expl R: 6) R: (0 R: (F) R:’ (S) R:’ (C) R,“’ (F)
4 (exp) n: (S) a (0 0; (F)
Dzr G) 0;’ (C)
Dzr (F)
k, (exp) I;: (SI !i: (C)
A: (F) her (S) k:’ (C)
C’ (F)
1.09 I.10 I.11 I I (I I.10 I.1 I 1.10
Ii.91 16.60 19.56 IX.15 16.60 19.57 IS.16
21.6 20.5 20.3 20.4 20.4 20.3 70.4
I 4x I 57 1.70 I 50 I.53 I.71
(0.001 I51 (0.00) I 53 I 0 0 I ) 1.71 (-0.02) I 49 I.55 1.73 I 50 I 54 1.73 151 I.55 1.73 I 49 I 56 1.75
13 91 12 60 I I.27 I6 02 14.X1 13.22
(0.00) 14.57 ( 0.07) 13.17 I 0.34) I I.59 (-0.76) Ii !I3 12.27 10.63 I6 09 15 12 13.92 14 60 I’62 12.38 14 01 12 5: Il.44
II R I I.1 9.03 IO 7 IO 2 8.41
lO.0) IO 7 (0.0) IO. I (0.01 X.25 (0.0) II 0 Y.Xl 7.96 IO 7 10.2 8.30 10.7 IO.0 8.27 I I .o 9.x1 7.89
[l.75]a 1.75 1.75 (-0.07) 1.78 1.82 1.83 1 .x5
9.98 II.64 9.82 (-1.86) 8.79
13.29 II.74 10.79
[8S]b 7.97 7.58 (0.2) 7.22 7.58 7.55 7.18
” Estimated by Desclaux and Pyykkii [42].
h Our present estimate.
Table 6 MCI4 (M = C, SI. Ge. Sn. Pb) X 1s for the totally s>mmctrlc mode. E = h /7. Ri,cl: h and D refer to four bonds. Experimental data
[43-~451. See legend of Table 3
ccl4 SICI, GeCl, SnCl, PbCL,
R, (exp) 1.77 2.02 2.1 I 3.28 2.43 R: (S) I 77 2 03 2.13 2.31 2.38 R: CC) I 7x (0.00) 2.04 (0.001 2 13 (0.01) 2.32 (0.00) 2.40 (-0.02) R; F) I.84 2 ox 2.20 2.39 2.48 R:’ (S) I 77 7.03 2 Ii 2.32 2.41 R:’ (0 1.78 2 03 2.14 2.32 2.42 R:’ (F) I 83 2.08 2 20 2 39 2.49
D, (expl 13.6 I6 s 14.0 13.2 [I I .oy D: (S) 15.20 16.96 14.51 13.40 11.18 D: (Cl 15.87 ( 0 17) I? 67 ( -0.27) 15.16 ( 0.61) 13.97 (-1.25) 11.67 (-2.97) D: (F) II.37 13 92 I I.52 10.79 8.60 0:’ (S) 15.37 17.2.3 15.12 14.64 14.08 DE” (C) 16.05 17.94 15.77 IS.22 14.65 0,“’ (F) I I.55 14.19 12.13 12.05 II.64
h, (exp) 17.3 14.x 12.Y 1 I.0 [IO]” x-: (9 16.7 I?.4 Il.7 IO.1 8.69 hi (C) 16.6 IO 0) I? 7 ( 0.1) I I.4 ( -0 2) 9.54 (-0.5) 8.33 (-1.1) k: (F) I2 8 II I x x0 7.52 6.10 k,“’ (S) I6 7 I? 5 11.x 10.4 9.65 k:’ (C) 16.6 Ii -I Y 03 10.1 9.36 k:’ (F) 12.8 II 2 I I.7 X.14 7.47
” Estimated here.
355
Kaupp and Schleyer [46] reported results on PbCI,, by the RECP approach. At the HF level. the bond length of PbC& is 2.38 A\. just as our DF(S) result. Remarkably the 0 experimen- tal R, is larger. 2.43A. DF(C) performs compar- able to DF(S), whereas DF(F) yields large bond lengths (AR = 0.05 to 0.1 I A). small bond ener- gies (deviation about -0.6eV per bond) and force constants (deviation about - 1 N cm ’ per bond). We estimate the experimentally unknown values of PbC14tobeD,~lleVandk,~10Ncm ‘.Atthe QCISD level and using the experimental atomic SO splitting, Kaupp and Schleyer’s dissociation energy of PbC& is about 8.5 eV. as low as our DF(F) result of 8.6 eV.
1.6
1.2
c .
2
2
0.4
0
i
Relativity does not influence the bond lengths while it decreases the bond energies significantly because of spin-orbit stabilization of the atoms, Contrary to MH1, relativity also decreases the force constants notably.
1
C Si Ge Sn Pb
Fig. 3. Calculated valence-p-A0 SO splitting and Condon-
Shortley SO parameter for the ground configurations of group 14 atoms
4. Analysis of the relativistic effects
4.1. Relativistic corrections of’c~tom.s
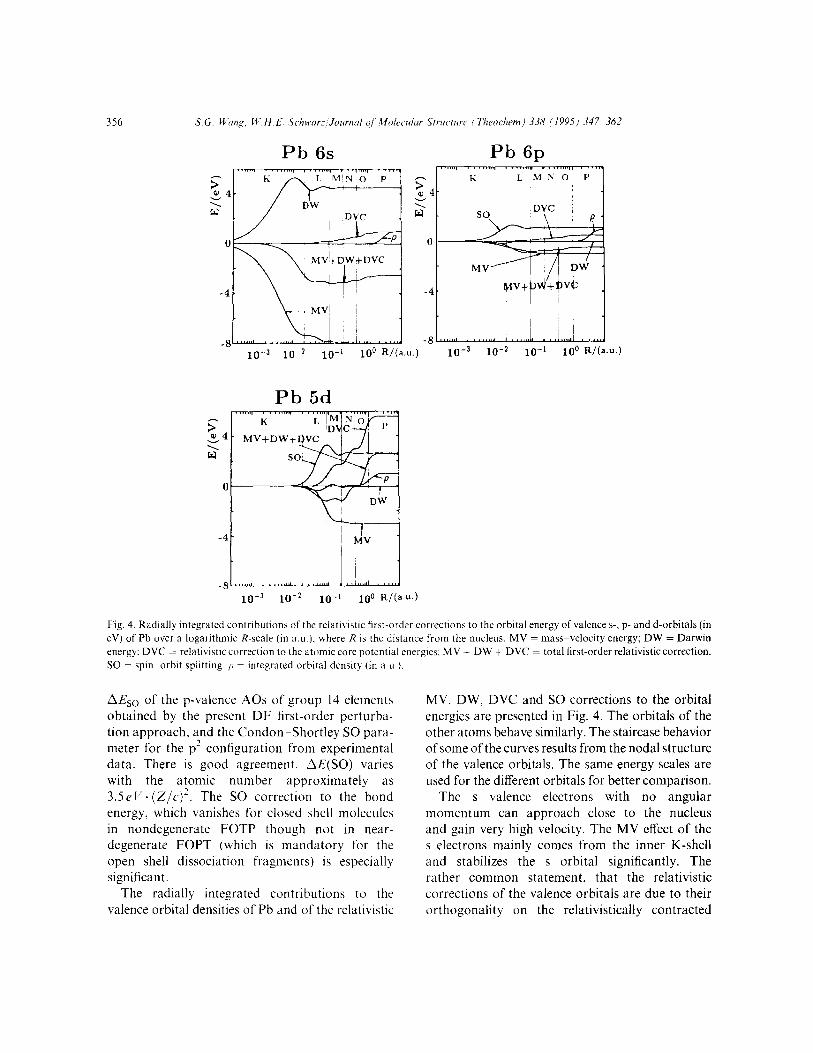
The well known relativistic effects of atoms are contraction and stabilization of s,,? and p,;? orbi- tals and expansion and destabilization of d and f orbitals. The nonrelativistic and relativistic atomic valence orbital energies and their diagonal first- order relativistic corrections are presented in Table 7 for the Pb atom.
tals due to the relativistic reduction of the kinetic energy. For s orbitals, MV is the most important correction. For p orbitals from the same shell, it is an order of magnitude smaller. The Darwin (DW) effect cancels about 55% of the MV stabilization of the s orbitals and nearly vanishes for other orbitals. The indirect DVC effect is important for d orbitals, due to s and pI/? core orbital contraction and thereby increased shielding of the nuclear charge. The situation is very similar to the valence orbitals of the other atoms.
The mass-velocity (MV) effect stabilizes all orbi- I > 0 orbitals are split by the relativistic spin-
orbit coupling (SO). Fig. 3 shows the SO splitting
Table 7
Nonrelatiwtic (e:‘) and relativistic (6:) energies (m eV) of atomic valence orbitals of Pb. and relativistic first-order diagonal contribu- tions. 12, = occupation number: r\~, = relativistx change of orbital energy: MV = mass velocity term: DW = Darwin term; SO = spin- orbit shift: DVC = change of valence electrons’ mteraction energy due to relativrstlc density change of core electrons
Orbital ‘I, nr
F, 4 r’f, Diagonal 1st order contributions
MV DW DVC so
5d 312 4 -23.x4 21.23 2.61 3.02 0.04 5.58 PI.57 5dS:? 6 18 71 5.13 1.05 b,2 2 -x.1)2 IO.59 1.67 8.1 I 4.63 0.98 0.00
6~1~ 2 3.1 I -3.37 -0.36 0.96 0.01 0.52 PO.73
6~39 ’ ‘I -.- 0.90 0.37
356
Pb 6s Pb 6p L “iv N 0” P
-8 .U.) 10-3 10-Z 10-l loo R/(a
Pb 5d
10-3 10-Z 10-l 100 R/(a.u.)
Fig. 4. Radially integrated contributions of the relatwstic first-order correctlons to the orbital energy of valence s-. p- and d-orbitals (in eV) of Pb over a logarithmic R-scale (in a.~.), where R is the distance from the nucleus. MV = mass-velocity energy; DW = Darwin energy: DVC = relativistic correction to the atomic core potential energies: MV - DW + DVC = total first-order relativistic correction.
SO = spin-orbit splitting. 0 = integrated orbital density (in d u.).
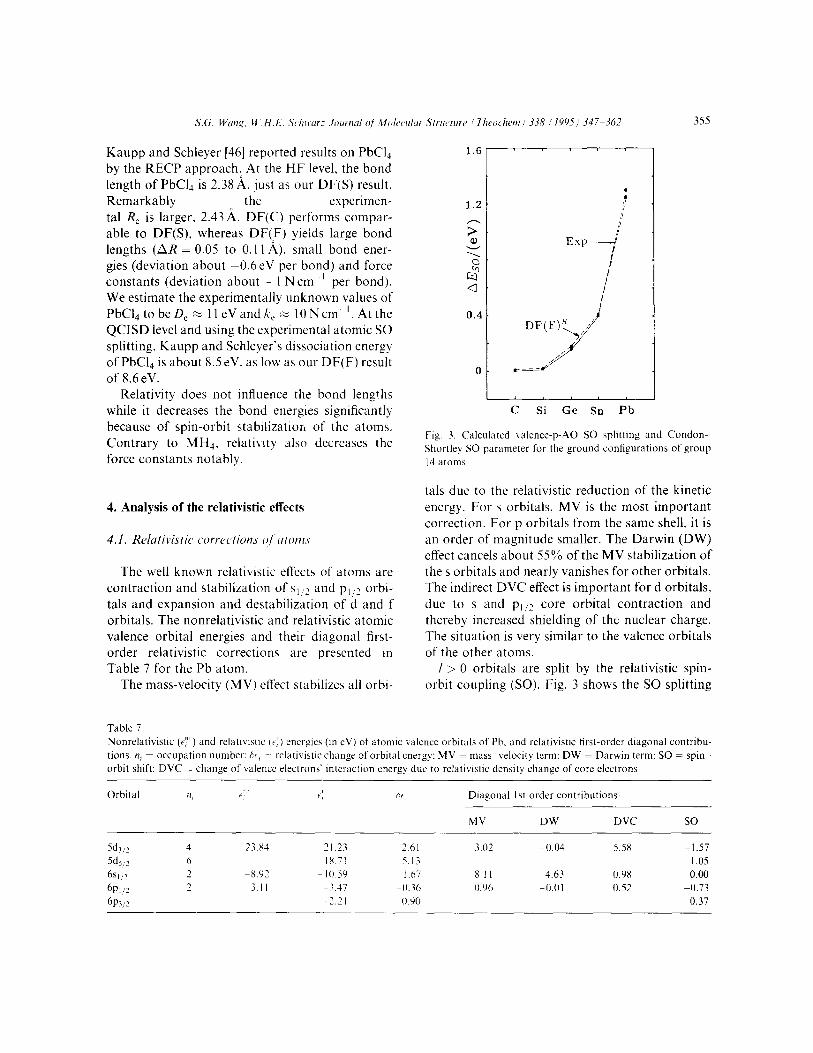
LIE,, of the p-valence .4Os of group 14 elements obtained by the present DF first-order perturba- tion approach, and the Condon-Shortley SO para- meter for the p’ configuration from experimental data. There is good agreement. AE(S0) varies with the atomic number approximately as 3.5ek’. (Z/c)‘. The SO correction to the bond energy, which vanishes for closed shell molecules in nondegenerate FOTP though not in near- degenerate FOPT (which is mandatory for the open shell dissociation fragments) is especially significant.
The radially integrated contributions to the valence orbital densities of Pb and of the relativistic
MV. DW, DVC and SO corrections to the orbital energies are presented in Fig. 4. The orbitals of the other atoms behave similarly. The staircase behavior of some of the curves results from the nodal structure of the valence orbitals. The same energy scales are used for the different orbitals for better comparison.
The s valence electrons with no angular momentum can approach close to the nucleus and gain very high velocity. The MV effect of the s electrons mainly comes from the inner K-shell and stabilizes the s orbital significantly. The rather common statement, that the relativistic corrections of the valence orbitals are due to their orthogonality on the relativistically contracted

S.C. Uhng. W’.H.E. S~hwur~~Journal of Moleculur Strwturr (Theochem) 338 (1995) 347-362 357
Table 8 Nonrelativistic Mulliken populations of PbO. PbHj. and PbCl,
Pb 5d
6s
6~ Charge
HIS, 02s, C13s
H2p. 02p, C13p 03d, C13d Charge
PbO
10.04 1.88
1.22 0.86
1.97
4.85 0.04
-0.86
PbH?
10.04 1.45
2.29 0.22
1.06 0.00
-0.06
PbC14
10.24 1.02
1.37 1 37
1.99
5.32 0.03
m-o.34
inner core orbitals, has been proven to be wrong [21,47,48]. Because of the centrifugal force the dominant MV contributions for the p orbital come from the outer part of the K- and from the L-shell; for the d orbital from the L- and M-shells.
The Darwin energies are mainly contributed by the nuclear potential, and the electronic potential contribution can be neglected. The dominant DW contributions are created in the inner part of the K- shell. The behavior of the radially integrated SO contributions for I > 0 orbitals is similar to that
of the DW effect for s orbitals, but is shifted outwards with increasing centrifugal force as in the case of the MV effect.
The so-called “indirect effect” DVC is caused by the relativistic change of the core potential. Rela- tivity contracts the s and p orbitals and expands the less core-penetrating d and f orbitals. There results an overall increase of the electronic shielding of the nuclear attraction, being of different magnitude for different valence orbitals [12,23,47,48]. The less a valence electron pene- trates the contracted inner s and p shells and the more it penetrates the expanded d and f shells, the larger is the destabilizing DVC effect (see Fig. 4).
The orbitals of the other atoms behave similarly.
4.2. Relativistic corrections of molecules
Chemical bonding can both be strengthened or weakened relativistically. The bond lengths can be contracted or expanded. In order to understand the trends of the relativistic corrections, the Mulliken populations of PbO, PbH, and PbCl, are presented
-5
5 Y UY
-15
-20
-25
4*c1 PbGW’ PbClg(NR)’ PbC&(R)
=%==‘, \ 1t1
3P \ 4tl ,--~=:::::~z’I:~
--.---- \ . . --- 2e ‘.,rjs %,,,) y---1---- ---- --~
--_ -. .I ‘\
7. ’ 3tz at.2 &I,) ( ..’ , -‘- ( ’ 2al ‘x. 2” ‘-- I \
’ \ 2al ,’ \ PI/I)
I , \ /’ i
, ! , /’
! %E,,,, /
/’ , ’ 2tz %“,,,I -------------i----- --- 3s
------____ ----L---- la1 Wm) la1
-- le(u,,,)
Fig. 5. Orbital energy level diagram of PbCI, (NR) and (R) denote nonrelativistic and relativistic orbitals.

in Table 8. The orbital energy diagram of PbCl, is shown in Fig. 5.
The Pb5d and Pbha A0 populations are changed only a little in the PbO molecule. while the Pb6p A0 is depopulated. The change of populations with decreasing interatomic distance around R, corresponds, in chemical terms. to M&; 4 01 . with dMs/dR = 0.09 a.u.. dMpjdR = 0.22 a.~. and dMd/dR = 0.07a.u.. for the heavier oxides. That is the slight depopulation of the relativisti- tally stabilized MS upon bond contraction acts weakly repulsive, that of the relativistically desta- bilized Md acts weakly attractive. while Mp has only a small influence, together yielding a small dSE/dR = 0.02a.u. for Pb. Therefore there is little relativistic bond change from the MV. DW and DVC terms (small hAE, small 6R, z -dbE/dR.k-‘, also small Csh-). The SO effect in closed shell molecules with no nearby empty MOs is also small. But SO stabilizes the Pb atom signifi- cantly, thus the SO effect decreases the bond energy by about - 1.5 eV.
Strong sp-promotional hybridization occurs in PbH,. The depopulation of Pbbs contributes to relativistic destabilization of the molecule, yielding SD, = - 1.9 eV. The force constant again remains unchanged. while the bond length is slightly contracted. Namely. upon R-reduction around R, there occurs already some increase of Pbs population and decrease of Pbd population. due to the strong Pb H overlaps (at R,. dPbs, dR M 0.07, dPbd,‘dR = +O. 13). The largest popula- tion change at R, occurs for Pbp (dPbp/dR = ~ I .O) which, however, does not contribute much to the relativistic energy derivative. It has been explained recently why relativistic corrections do not follow Badger’s or Gordy’s rules. which state that bond energy decrease - force constant decrease N bond length increase [3.49]. Namely, there is in general no proportionality between hE, hAE and their derivatives.
Concerning PbCl,. the four Cl ligands depopu- late both Pb6p and Pb6s. the latter being strongly stabilized by the relativistic MV effect (see Figs. 3 and 5). Accordingly. the bond energy is consider- ably reduced. Shortening of R around R, reduces the Pbs population further. and also the Pbd popu- lation (Pbp is increased). So as in the monoxides.
there is only very little bond length contraction in the tetrachlorides. Concerning the bond energy, about -0.15 eV molecular destabilization comes from the relativistic stabilization of the chlorine atoms. mainly due to SO coupling.
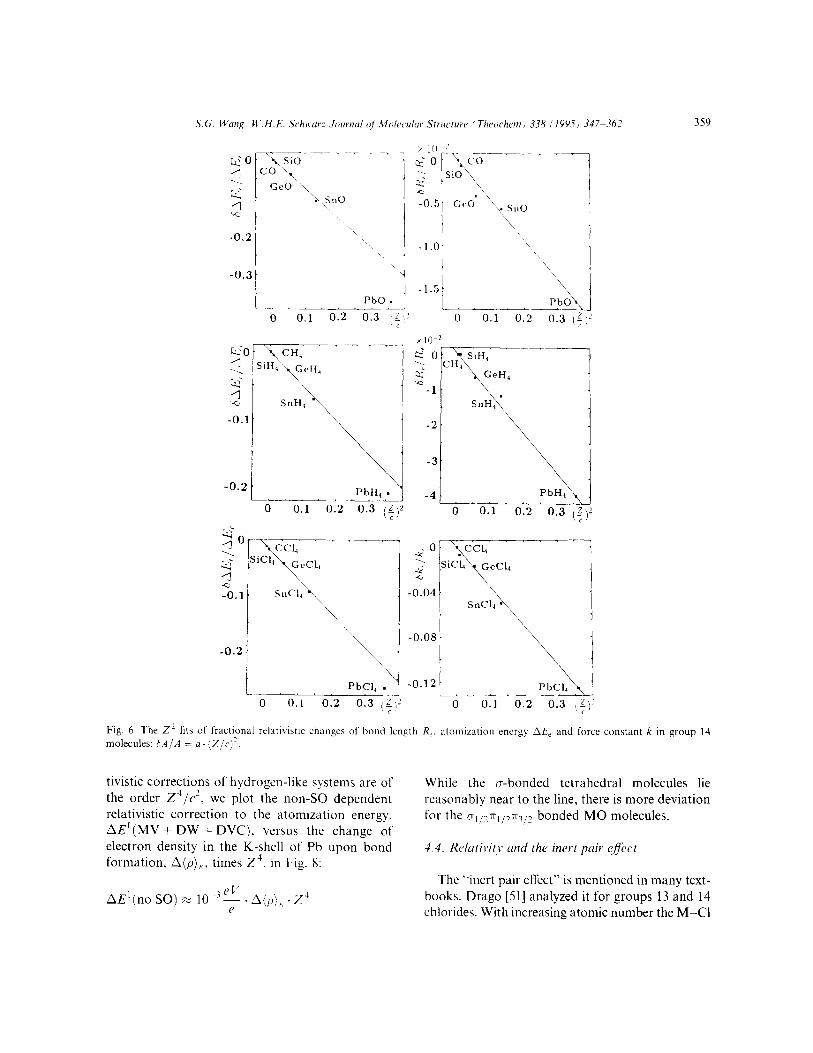
Relativistic corrections of atomic properties roughly scale as (Z/c)? [l]. Contrary to nonrelati- vistic properties A’, which are proportional to the squared cfltictive nuclear charges Z,?f, the relati- vistic corrections SA depend also on the full nuclear charge, roughly as 6A =: Z&, . Z,$ [21]. We find here (compare also Pyykkii [SO]) that within one homologous series, relativistic correc- tions of rnolecuhr constants also vary approxi- mately as Z2 (see Fig. 6) provided the corrections are not too small. The same trend of relativistic bond length changes can also be found from Dirac-Fock calculations [6,25,35]. In the case of MC&, bAE + 0.15 eV (corresponding to the Cl contributions) correlates with Z(M)*. Fig. 6 gives the impression that a (Z/c)4-term may become relevant for the very heavy molecules. It should be noted, however, that only the first-order hamil- tonian is used here for the direct relativistic effects of the valence electrons while the indirect DVC effect from the core electrons is treated at higher order.
4.3. The origin qf’relutiristic bond effects of group 14 molecules
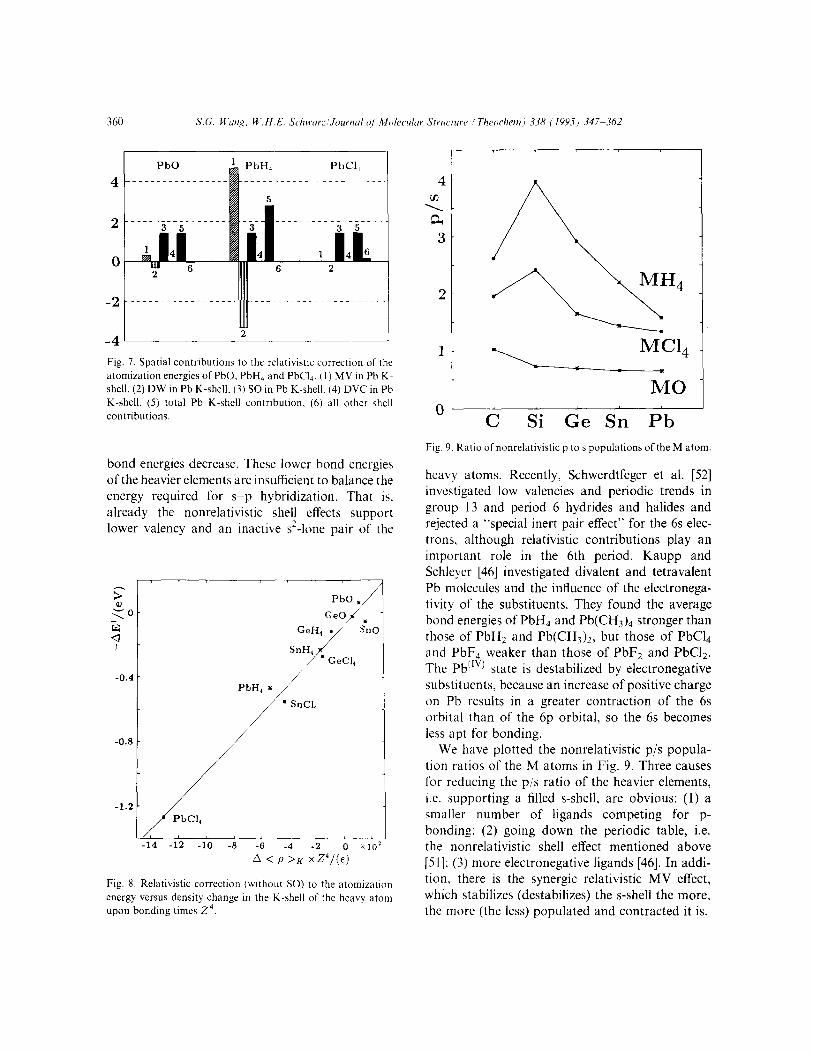
The spatial contributions of relativity to the atomization energies of the three Pb compounds are shown in Fig. 7. Only the K-shell of Pb contri- butes significantly, so the contributions from all the other shells are taken together; even the SO contri- bution to bonding mainly originates in the Pb K- shell. The situation in the other molecules is similar except that all effects are smaller, the lighter the atom M is.
The SO effect on bonding in these molecules is mainly related to population changes of Pb6pt12 and 6~~;~. Therefore, it is not related to the elec- tron density changes in the Pb K-shell. The MV + DW effect, however. is mainly due to Pb6s population changes and is expected to be related to density changes in the Pb K-shell. Since the poten- tial near the nucleus is hydrogen-like, and the rela-
359
t -0.08 t
Fig. 6. The Z’ fits of fractional rclatlwstlc changes of bond length R,. atomization energy AE, and force constant k in group 14
molecules: 0.4/A = a. [Z/cj’.
tivistic corrections of hydrogen-like systems are of the order Z”/c’, we plot the non-SO dependent relativistic correction to the atomization energy, AE’(MV + DW + DVC). versus the change of electron density in the K-shell of Pb upon bond formation, A(p),, times Z’, in Fig. 8:
While the o-bonded tetrahedral molecules lie reasonably near to the line, there is more deviation for the CJ , /?r, II~I ,, bonded MO molecules. ,A .,‘A
4.4. Rtllutivity und the inert pair c$ect
The “inert pair effect” is mentioned in many text-
AE’(no SO) = 10 3!? . A((,j, . zJ books. Drago [51] analyzed it for groups 13 and 14 e chlorides. With increasing atomic number the M-Cl
4
2
0
-2
-4 Fig. 7. Spatial contributions to the relativisttc correction of the
atomization energies of PbO. PbH? and PbQ. (I) MV in Pb K- shell, (2) DW in Pb K-shell, (3) SO in Pb K-shell. (4) DVC in Pb K-shell, (5) total Pb K-shell contribution. (6) all other shell
contributions
bond energies decrease. These lower bond energies of the heavier elements are insufficient to balance the energy required for ssp hybridization. That is. already the nonrelativistic shell effects support lower valency and an inactive ?-lone pair of the
PbH, x
I I (14
I -12 -10 -8 -6 -4 -2 0 XIOZ
A < p >K xZ”/(e)
Fig. 8. Relativistic correction (without SO) to the atomizatton
energy versus density change in the K-shell of the heavy atom upon bonding times Z4.
4-
-rz. CL
1
l- MCI, 1
MO O-
C Si Ge Sn Pb Fig. 9. Ratio of nonrclativistic p to s populations of the M atom.
heavy atoms. Recently, Schwerdtfeger et al. [52] investigated low valencies and periodic trends in group 13 and period 6 hydrides and halides and rejected a “special inert pair effect” for the 6s elec- trons, although relativistic contributions play an important role in the 6th period. Kaupp and Schleyer [46] investigated divalent and tetravalent Pb molecules and the influence of the electronega- tivity of the substituents. They found the average bond energies of PbH4 and Pb(CH3)4 stronger than those of PbH2 and Pb(CHJ)2, but those of PbC14 and PbF4 weaker than those of PbF2 and PbC12. The Pb”‘) state is destabilized by electronegative substituents, because an increase of positive charge on Pb results in a greater contraction of the 6s orbital than of the 6p orbital, so the 6s becomes less apt for bonding.
We have plotted the nonrelativistic p/s popula- tion ratios of the M atoms in Fig. 9. Three causes for reducing the p/s ratio of the heavier elements, i.e. supporting a filled s-shell, are obvious: (1) a smaller number of ligands competing for p- bonding; (2) going down the periodic table, i.e. the nonrelativistic shell effect mentioned above [51]; (3) more electronegative ligands [46]. In addi- tion, there is the synergic relativistic MV effect, which stabilizes (destabilizes) the s-shell the more, the more (the less) populated and contracted it is.

5. Summary and conclusions
The following conclusions can be drawn from our calculations.
(1) Density functional theory is a useful tool for investigating the electronic structures and chemical properties of heavy p-block molecules. Within the fully relativistic frozen core approximation, first- order relativistic perturbation corrections for the valence electrons, comprising the one-electron mass-velocity, Darwin, spin-orbit and indirect rela- tivistic core effects, give reasonable results.
(2) There is no overall improvement for different properties of the investigated heavy molecules. if recent density functional corrections are added to the simple Xo approach, as had been reported for the light second row molecules [53,54]. The average error of DF(S) in the present investigation is 2 pm for R,, 0.3 eV for the bond energies and 0.3 N cm-’ for the bond force constants.
(3) Mulliken population analysis reveals that the activity of the s electron of the central atom in the tetrahydrides and tetrachlorides decreases with increasing atomic number and with increasing the positive charge on the central atoms already at the nonrelativistic level.
(4) Relativistic spin-independent effects decrease the bond strengths with increasing the number and the electronegativity of the substituents due to the decrease of density in the heavy atomic K-shells.
(5) Relativistic SO coupling is similarly impor- tant in the three investigated series of molecules.
(6) For each series of these p-block molecules (MO, MHd, MC14), the fractional changes of bond energy. bond length and force constants due to relativistic corrections are quite well propor- tional to (Z/C)~. Also the SO splitting of the p- orbitals increases proportional to (Z/c)‘.
(7) While the bond energies are significantly reduced by relativity in all three series (roughly SD/D = 213. (Z/c)‘), a significant reduction of force constants occurs only for the tetrachlorides (6/z/k M 1.3. (Z/c)‘). Relativistic bond length changes are very small, and noticeable only for the tetrahydrides (bond contraction of hR/R z 0.1 . (Z/c)‘). Gordy’s and Badger’s rules are violated.
(8) The density changes from free atoms to mole-
cules in the heavy atomic K-shells are important for the spin-independent relativistic corrections. Spin-independent relativistic corrections to the bond energies are proportional to the K-shell density changes times the fourth power of the nuclear charges: AE’ = const . A(p). . Z4.
(9) The so-called inert pair effect results from synergic nonrelativistic shell effects, relativistic effects, bond polarizations and coordination number effects.
References
[II PI
PI
PI
[51 (61 [71
J.P. Desclaux. At. Data Nucl. Data Tables. 12 (1973) 311. A. Rutkowski. D. Rutkowska and W.H.E. Schwarz,
Theor. Chim. Acta, 84 (1992) 105. W. Kutzelnigg. R. Franke. E. Ottschofski and W. Klopper, in R. Broer, P.J.C. Aerts and P.S. Bagus (Eds.), New
Challenges in Computational Quantum Chemistry, Rijks Umversiteit. Groningen. 1993. W. Kutzelnigg. E. Ottschofski and R. Franke. J. Chem.
Phys. 102 (1995) 1740. 1752. R. Franke, Ph.D Thesis. Bochum (1994). K.G. Dyall and P.R. Taylor, J. Chem. Phys.. 95 (1991) 2583. E.J. Baerends. D.E. Ellis and P. Ros, Chem. Phys., 2 (1973)
31. [X] J.G. Snijders and E.J. Baerends, Mol. Phys.. 36 (1978)
1789
[9] T. Ziegler. J.G. Snijders and E.J. Baerends. J. Chem. Phys., 74 (1981) 1271.
[lo] Z. Barandiaran and L. Seijo, J. Chem. Phys. IO1 (1994) 4049.
[I I] F. Herman and S. Skillman, Atomic Structure Calcula- tions, Prentice-Hall. Englewood Cliffs, NJ. 1963.
[I’] W.H.E. Schwarz, E.M. van Wezenbeek. E.J. Baerends and J.G. Snijders, J. Phys. B. 22 (1989) 1515.
[Ii] P. Hohenberg and W. Kohn, Phys. Rev. A, 136 (1964) 864. [I31 W. Kohn and L.J. Sham, Phys. Rev. A, 140 (1965) 1133.
[ 151 J.C. Slater. Quantum Theory of Molecules and Solids. Vol. 4. McGraw-Hill. New York. 1974.
[ 161 S.H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 58
( 1980) 1200. [I 71 H. Stall, E. Golka and H. Prcuss, Theor. Chim. Acta., 55
(1980) 29.
[ 181 A.D. Becke. Phys. Rev. A. 38 (1988) 3098. [I91 A. Rosen and I. Lindgren. Phys. Rev.. 176 (1968) 114. [ZO] G.J. Snijdcrs. E.J. Baerends and P. Vcrnooijs. At. Data
Nucl. Data Tables. 26 (1982) 483. [21] P. Vernooijs, G.J. Snijders and E.J. Baerends, Slater Type
Basis Functions for the Whole Periodic System, Internal Report, Free University, Amsterdam. 198 1.
[22] P.M. Boerrigter. G. tc Veldt and E.J. Baerends, Int. J.
Quantum Chem., 33 (1988) 87.

[23] W.H.E. Schwarz, in 2. Maksic.(Ed.). Theoretical Models of Chemical Bonding Springer. Berlin, 1990. p. 595.
[24] K. Balasubramanian, Chem. Rev.. 89 (1989) 1801. [25] KG. Dyall, J. Chem. Phys.. 98 (1993) 2191. [26] 0. Matsuoka. L. Pisani and E. Clementi. Chem Phys.
Lett., 202 (1993) 13. [27] L. Fan and T. Ziegler, J. Chem. Phys., 94 (1991) 6057. [28] K.P. Huber and Cl. Herzber. Molecular Structure and
Molecular Spectra IV. Constants of Diatomic Molecules. Van Nostrand Reinhold, New York. 1979.
[29] M.P.C.M. Krijn and D. Feil, Chem. Phys. Lett.. 150 (1988)
45. [30] C.W. Murray, C.J Laming, N.C. Handy and R.D Amos.
Chem. Phys. Lett., 199 (1992) 551.
[31] L.A. Young and W.J. Eachus. J. Chem. Phys . 44 (1966) 4195.
[32] F.P. Billingsley II and M. Krauss. J. Chem. PhyJs.. 60 (1974)
4130. [33] L.A. Barnes, B. Lin and R. Lmdh, J Chem. Phys.. 98
(1993) 3972.
[34] V. Kelltj and A.J. Sadlej. J. Chem. Phys.. 98 (1993) 1345. [35] 0. Visser, L. Visscher. P.J.C. .4erts and W.C. Nieuwpoort.
Theor. Chim. Acta, Xl (1992) 405.
[36] D.J. Gray and A G. Robiette. Mol. Phys., 37 (1979) 1901 [37] Kagaku Benran. 3rd edn. Vol. II, Chemical Society of
Japan. 1984.
[38] R.C. Weast (Ed.). CRC Handbook of Chemistry and Physics. 68th edn.. CRC Press. Boca Raton. FL. 1987.
[39] F.E. Saalfeld. Inorg. Chem., 2 (lY63) 46. [40] S.R. Gunn and L.C. Green, J. Phys. Chem., 65 (1961) 779.
[4l] L. Brewer and G. Rosenblatt. Adv. High Temp. Chem., 2
(1969) 1. [42] J.P. Desclaux and P. Pyykko, Chem. Phys. Lett., 29 (1974)
534.
(431 M.W. Chase. J.L. Curnutt, H. Prophet, R.A. McDonald and A.N. Syverud, J. Phys. Chem. Ref. Data, 4 (1975) 1.
[44] K.F. Purcell and J.C. Kotz. Inorganic Chemistry, W.B. Saunders. New York, 1977.
[45] D.R. Lide (Ed.). CRC Handbook of Chemistry and Physics. 73th Edn.. CRC Press, Boca Raton, FL, 1992.
[46] M. Kaupp and P.v.R. Schleyer, J. Am. Chem. Sot., 115 (1993) 1061.
[47] S.J. Rose, I.P. Grant and N.C. Pyper, J. Phys. B 11 (1978) 1171.
[48] E.J. Baerends, W.H.E. Schwarz, P. Schwerdtfeger and J.G. Snijders. J. Phys. B 23 (1990) 3225.
[49] W.H.E. Schwarz. A. Rutkowski and S.G. Wang, Int. J.
Quantum Chemistry, in press (1995). (501 P. Pyykkii, Chem. Rev.. 88 (1988) 563.
[51] R.S. Drago, J. Phys. Chem., 62 (1958) 353. [52] P. Schwerdtfeger, G.A. Heath, M. Dolg and M.A. Bennett,
J. Am. Chem. Sot., 114 (1992) 7518.
[S3] B.C. Johnson, P.M.W. Gill and J.A. Pople, J. Chem. Phys.. 97 (1992) 7846.
[54] B.G. Johnson, P.M.W. Gill and J.A. Pople, J. Chem. Phys.,
98 (1993) 5612.





























