Embed Size (px)
Citation preview

May 29-31, 20132013 MFDS, Global
Biopharmaceutical ForumSeoul, Korea
Duu-Gong, Ph.D.Director, Global Regulatory Consulting/Senior ConsultantPPD, Rockville, Maryland, USA
Regulatory Considerations on Quality by Design During the Development of
Biopharmaceuticals

Agenda
• Introduction• Concept of Quality by Design• QbD Guidance and Guideline• QbD and Biopharmaceuticals• Regulatory Issues and Challenges• Conclusion

Quality by Design Terminology
• Quality by Design (QbD): A systematic approach to development that begins with predefined product profiles and emphasizes product and process understanding and process control based on sound science and quality risk management.
• Quality Target Product Profile (QTPP): A summary of the quality characteristics of a DP or DS that ensure the desired quality linking safety and efficacy of the drug product.
• Critical Quality Attribute (CQA): A physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
• Design Space: The multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality..
• Control Strategy: A planned set of controls based on the current product and process understanding .The controls can include parameters and attributes of drug substance and drug product, materials and components; facility and equipment operating conditions; in-process controls; finished product specifications.
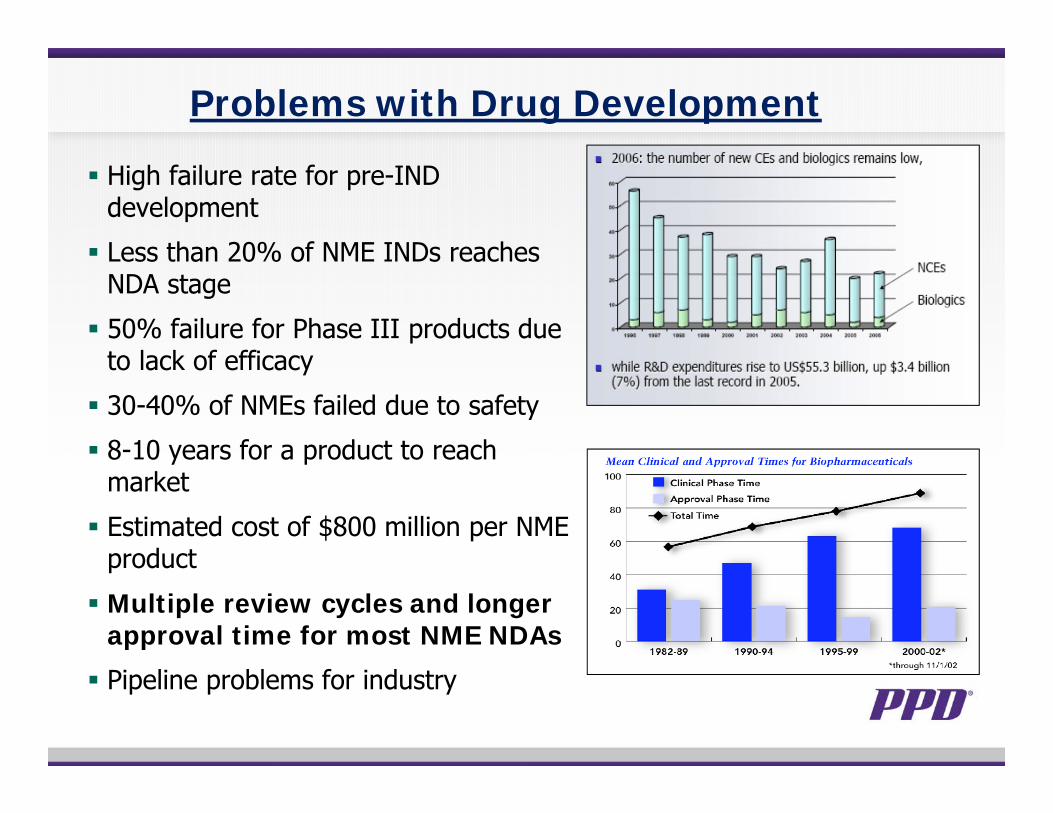
Problems with Drug Development
High failure rate for pre-IND development
Less than 20% of NME INDs reaches NDA stage
50% failure for Phase III products due to lack of efficacy
30-40% of NMEs failed due to safety
8-10 years for a product to reach market
Estimated cost of $800 million per NME product
Multiple review cycles and longer approval time for most NME NDAs
Pipeline problems for industry

Decline in New Biological Product Approvals
• Pre-clinical studies− Lack of animal models or failure to investigate pharm/tox in animals.
• Phase 1/2:− Animal model fails to predict response in human.− Unexpected adverse events; Immunogenicity − Lack of clinically relevant biological activity − MOA in human not fully evaluated and understood.− Appropriate endpoints not sufficiently investigated..
• Phase 2/3: Clinical Issues− Insufficient dose ranging studies; wrong dose− Unexpected adverse events with longer exposure
• Newer products are more complex with difficult safety and manufacturing and quality issues .

Pitfalls with Quality Issues
• Product development: Safety issues are not properly addressed, e.g. viral safety and
immunogenicity. Products are not adequately characterized. Formulation and storage conditions are not properly selected Product stability is not well-established.
• Manufacturing: Optimal production system is not used. Robust manufacturing process is not developed. Constant process changes cause delay. Frequent recalls, warning letters and penalties cause loss of
consumer confidence
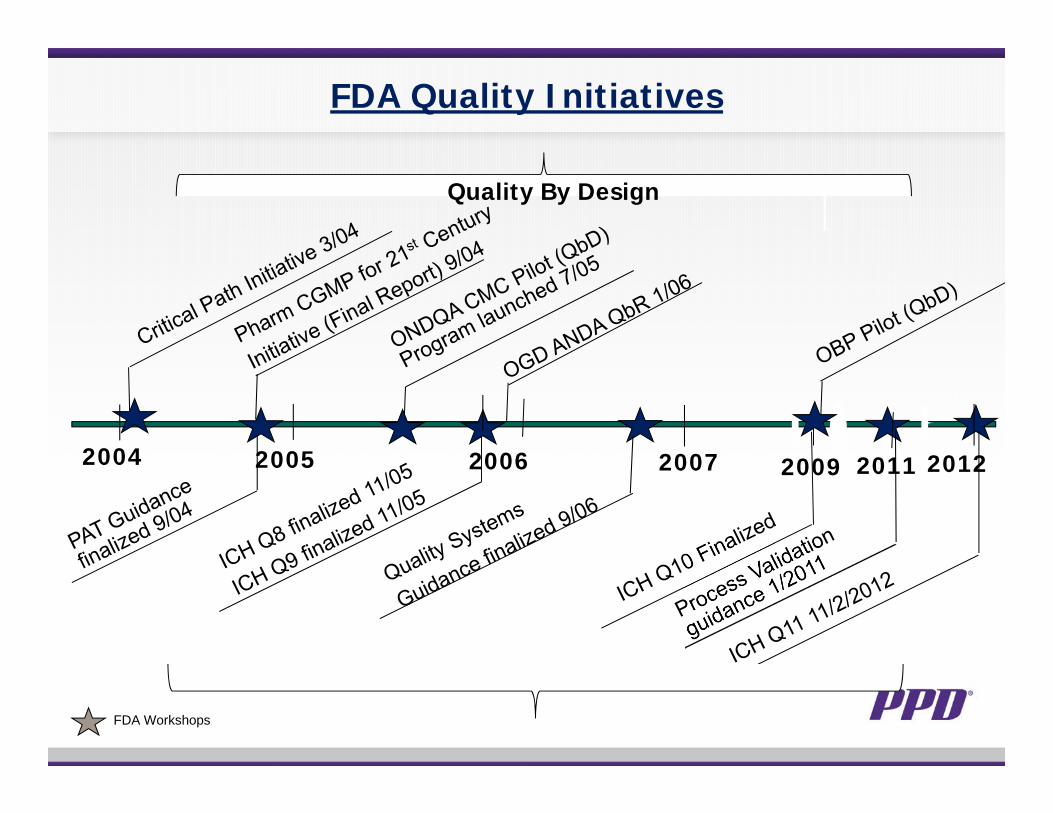
2004 2005 2006 2007
FDA Workshops
FDA Quality Initiatives
2011 2012
Quality By Design
2009

The Desired State
A maximally efficient, agile, flexible pharmaceutical manufacturing sector that reliably produces high-quality drug products without extensive regulatory oversight
A mutual goal of industry, society, and regulator
Janet Woodcock, October 2005)
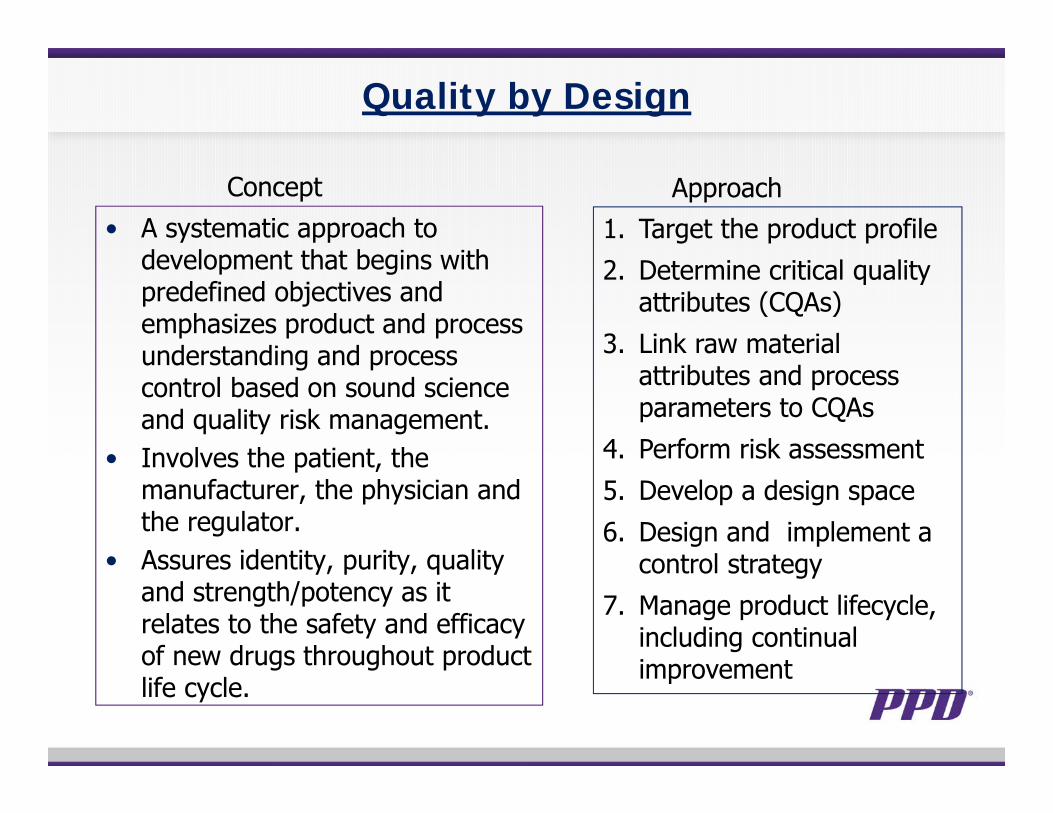
Quality by Design
• A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control based on sound science and quality risk management.
• Involves the patient, the manufacturer, the physician and the regulator.
• Assures identity, purity, quality and strength/potency as it relates to the safety and efficacy of new drugs throughout product life cycle.
1. Target the product profile2. Determine critical quality
attributes (CQAs)3. Link raw material
attributes and process parameters to CQAs
4. Perform risk assessment5. Develop a design space6. Design and implement a
control strategy7. Manage product lifecycle,
including continual improvement
Concept Approach
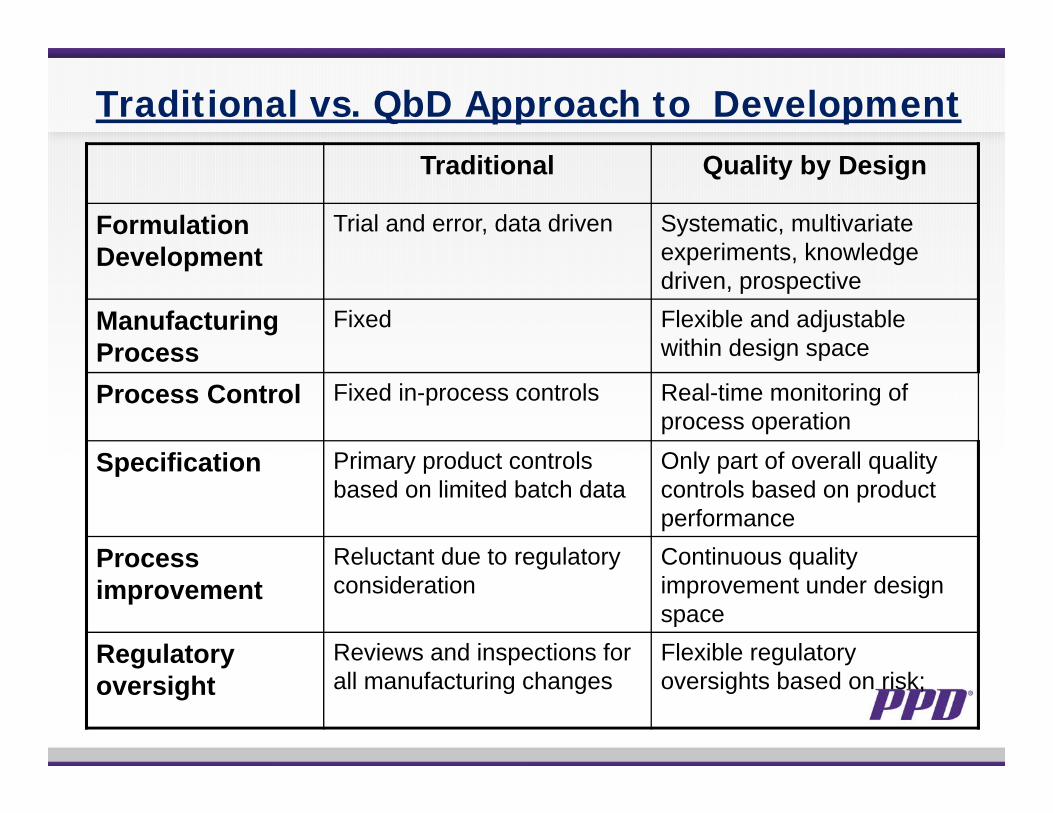
Traditional vs. QbD Approach to DevelopmentTraditional Quality by Design
Formulation Development
Trial and error, data driven Systematic, multivariate experiments, knowledge driven, prospective
Manufacturing Process
Fixed Flexible and adjustable within design space
Process Control Fixed in-process controls Real-time monitoring of process operation
Specification Primary product controls based on limited batch data
Only part of overall quality controls based on product performance
Process improvement
Reluctant due to regulatory consideration
Continuous quality improvement under design space
Regulatory oversight
Reviews and inspections for all manufacturing changes
Flexible regulatory oversights based on risk;
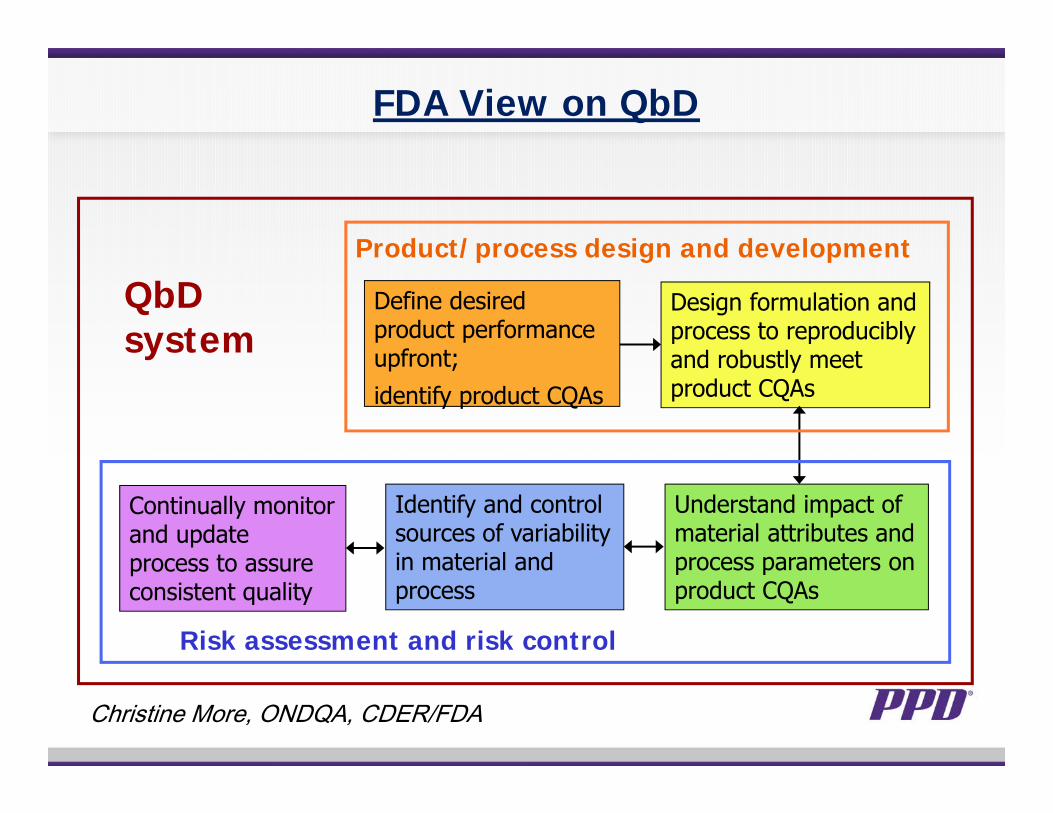
FDA View on QbD
Define desired product performance upfront;identify product CQAs
Design formulation and process to reproducibly and robustly meet product CQAs
Understand impact of material attributes and process parameters on product CQAs
Identify and control sources of variability in material and process
Continually monitor and update process to assure consistent quality
Risk assessment and risk control
Product/process design and development
QbD system
Christine More, ONDQA, CDER/FDA
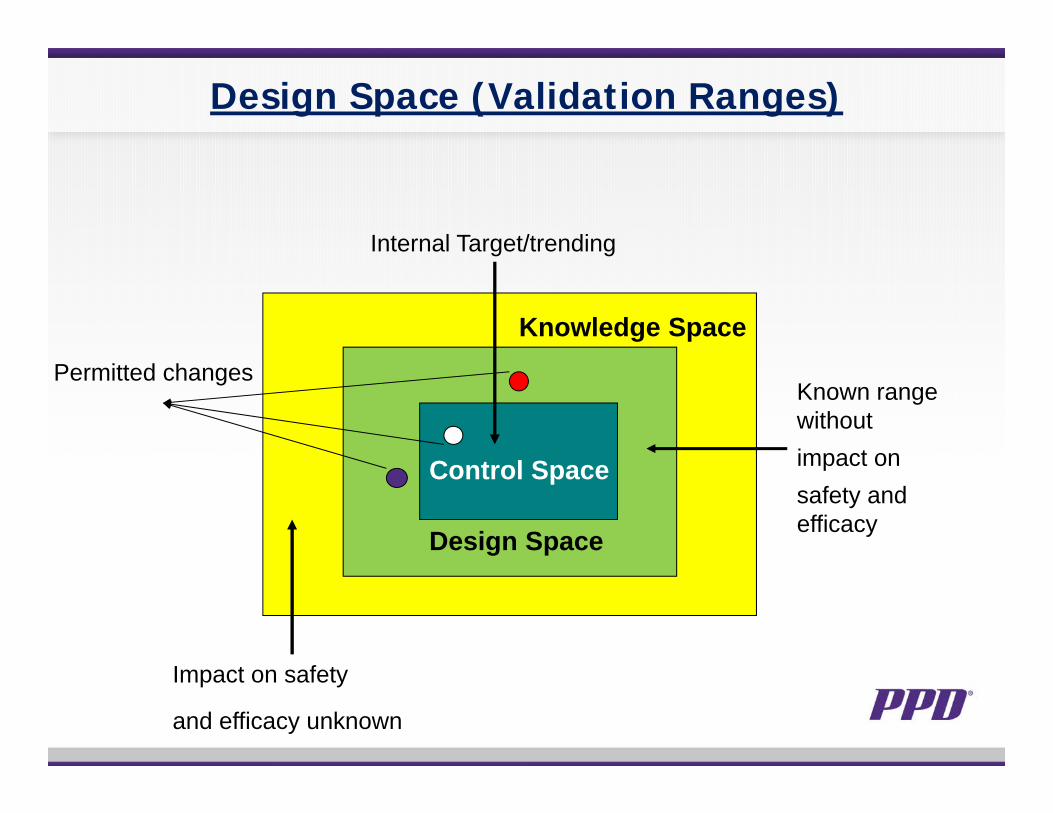
Design Space (Validation Ranges)
Control Space
Design Space
Knowledge Space
Impact on safety
and efficacy unknown
Internal Target/trending
Known range withoutimpact onsafety and efficacy
Permitted changes
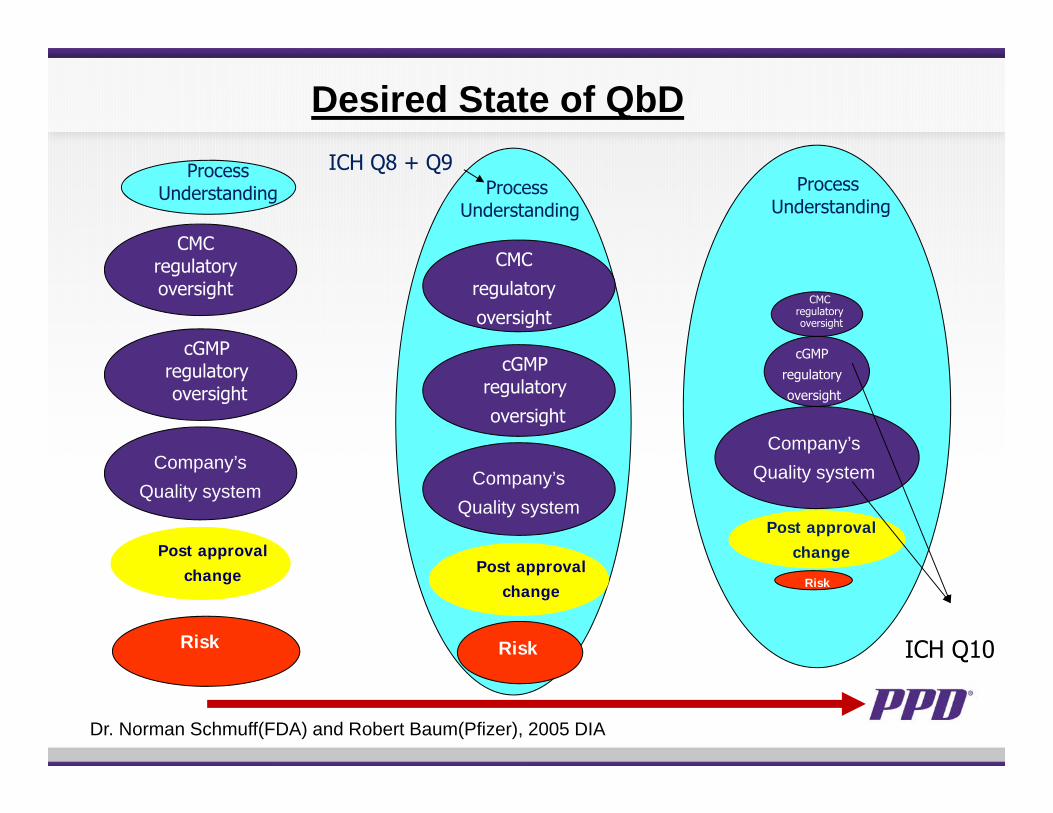
ICH Q10
Dr. Norman Schmuff(FDA) and Robert Baum(Pfizer), 2005 DIA
Desired State of QbD
Process Understanding
Risk
CMC regulatory oversight
Company’sQuality system
cGMP regulatory oversight
Post approval change
Post approval change
Process Understanding
Risk
CMC regulatory oversight
Company’sQuality system
cGMP regulatory oversight
Process Understanding
ICH Q8 + Q9
CMC regulatory oversight
Company’sQuality system
cGMP regulatory oversight
Post approval change
Risk

ICH Guidance and QbD
Pharm. QualitySystems (Q10)
Pharm. Develop. (Q8) & QbD(Q8R)
DesiredState
RiskManagement
(Q9)
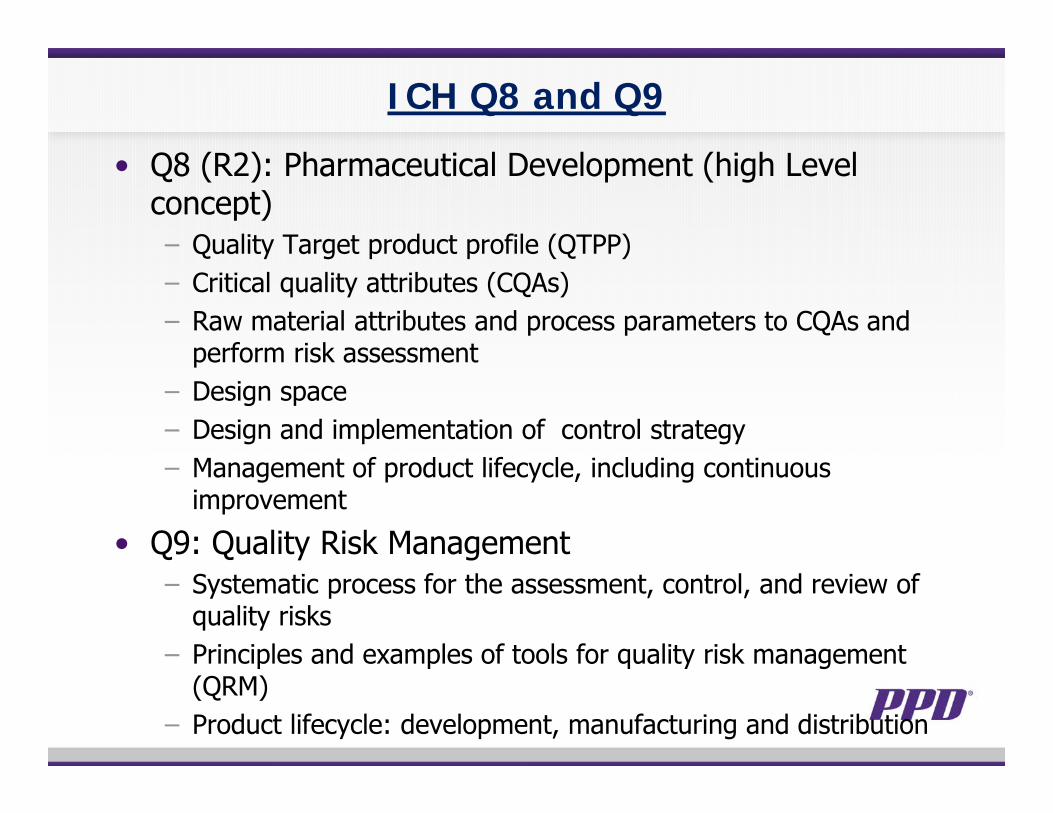
ICH Q8 and Q9
• Q8 (R2): Pharmaceutical Development (high Level concept)− Quality Target product profile (QTPP)− Critical quality attributes (CQAs)− Raw material attributes and process parameters to CQAs and
perform risk assessment− Design space− Design and implementation of control strategy− Management of product lifecycle, including continuous
improvement
• Q9: Quality Risk Management− Systematic process for the assessment, control, and review of
quality risks − Principles and examples of tools for quality risk management
(QRM)− Product lifecycle: development, manufacturing and distribution

ICH Q10 and Q 11
• Q10 (R2): Pharmaceutical Quality System− Describes GMP systems that facilitate establishment and
maintenance of a state of control for process performance and product quality
− Facilitates continual improvement− Applies to drug substance and drug product throughout product
lifecycle
• Q11: Development and Manufacturing of Drug substance− Manufacturing process development: traditional vs enhanced vs
combination approaches.− Drug substance critical quality attributes for biotech products− Knowledge of mechanism of actions and biological
characterization and assessment of risk for some quality attributes.
− Selection of starting materials and source materials− Control Strategy: materials, in-process, and drug substance

FDA New FDA Process Validation Guidance
• New definition of process validation: 3 validation lots vs. flexible approach based on process understanding
• Stage 1 – Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities (critical process parameters).
• Stage 2 – Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
• Stage 3 – Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control.
• Understanding the impacts and controls of variations
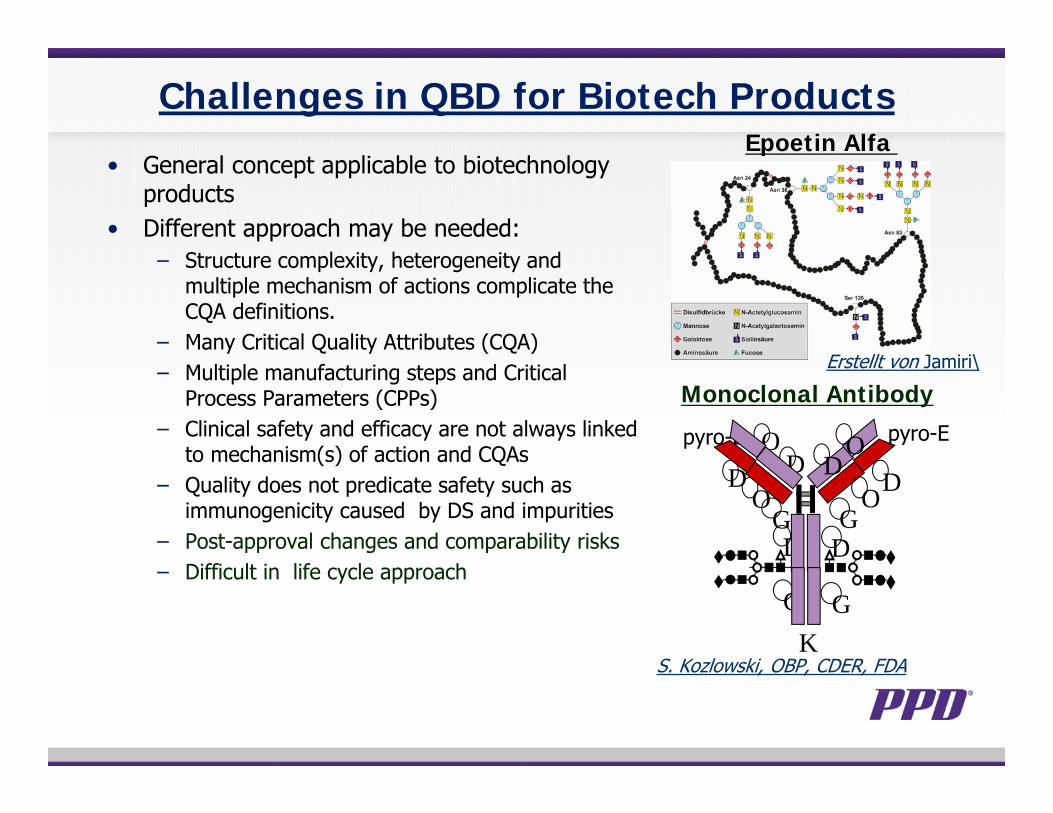
Challenges in QBD for Biotech Products
• General concept applicable to biotechnology products
• Different approach may be needed:− Structure complexity, heterogeneity and
multiple mechanism of actions complicate the CQA definitions.
− Many Critical Quality Attributes (CQA)− Multiple manufacturing steps and Critical
Process Parameters (CPPs)− Clinical safety and efficacy are not always linked
to mechanism(s) of action and CQAs− Quality does not predicate safety such as
immunogenicity caused by DS and impurities− Post-approval changes and comparability risks− Difficult in life cycle approach
Epoetin Alfa
K
pyro-E OD
G
GD
OD
O
O pyro-E
D
D
D
G
G
Monoclonal Antibody
S. Kozlowski, OBP, CDER, FDA
Erstellt von Jamiri\
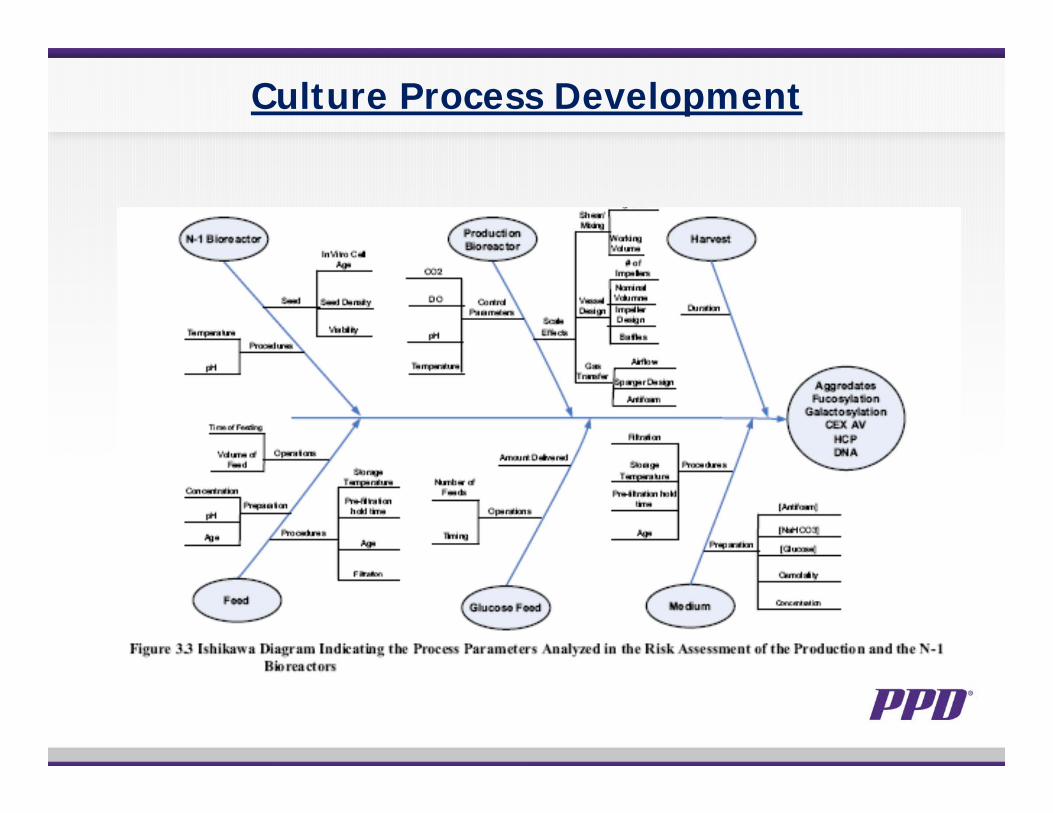
Culture Process Development

Biotech Quality Related to Safety and Efficacy
• Focus on the drug substance CQAs and CPPs• Biological activity and efficacy• Glycosylation and PK/PD• Immunogenicity of active ingredient and
safety/efficacy• Impurity and clinical safety• Interaction with other CQA, e.g. glycoforms and
biological activities
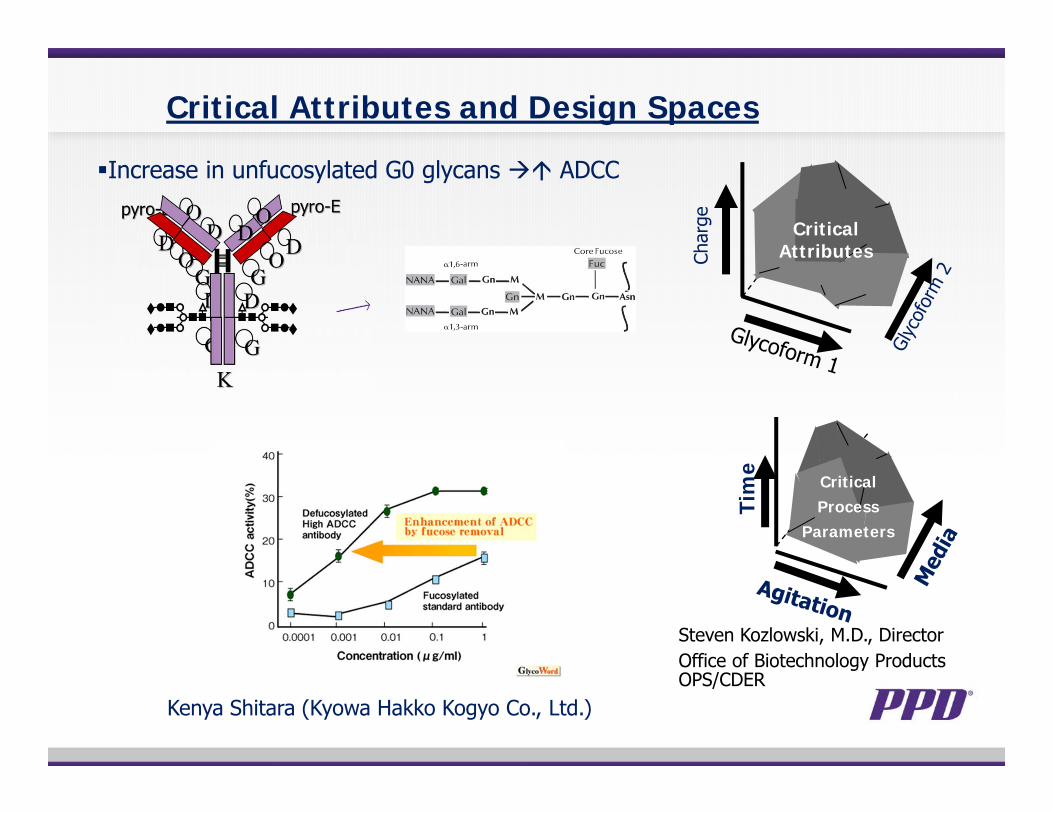
Critical Attributes and Design Spaces
Char
ge Critical Attributes
Tim
e
Critical Process
Parameters
Steven Kozlowski, M.D., DirectorOffice of Biotechnology Products OPS/CDER
Increase in unfucosylated G0 glycans ADCC
Kenya Shitara (Kyowa Hakko Kogyo Co., Ltd.)
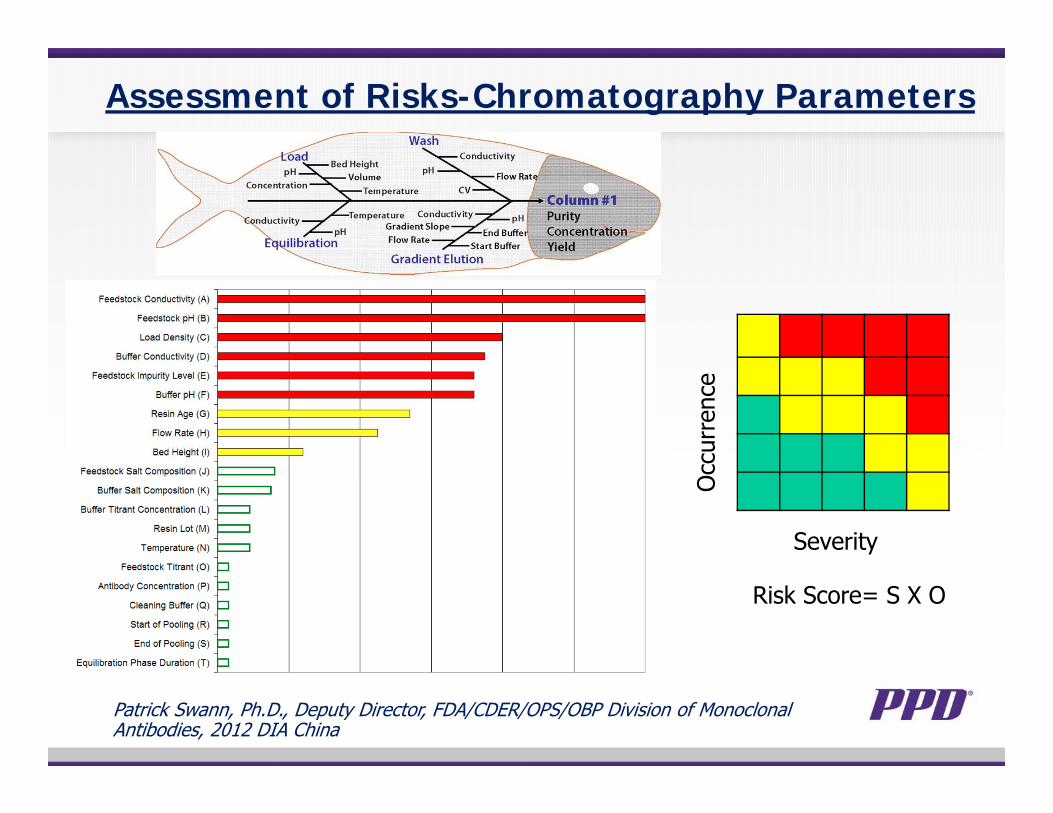
Assessment of Risks-Chromatography Parameters
Patrick Swann, Ph.D., Deputy Director, FDA/CDER/OPS/OBP Division of Monoclonal Antibodies, 2012 DIA China
Occ
urre
nce
Severity
Risk Score= S X O

QBD Activities of Biotechnology Products
• Industry CMC Biotech Working group− Case Study in Bioprocess Development drafted by 7 pharma
companies and published by ISPE (10/30/09)− Another case study by EFPIA in Europe
• FDA− ICH Q8, Q9, Q10 and Q11 guidance and implementation− FDA pilot program on biotech products (2009)− MaPPs for CMC review− Design of Experiments training− EMA-FDA joint pilot QbD review

FDA Biotech QbD Pilot Applications
• Applications Accepted in QbD Pilot Program− 6 Original Applications: 5 Monoclonal Antibodies and 1 Fc Fusion
Protein− 4 Post-approval Supplements: 2 Monoclonal Antibodies, 1
Therapeutic Protein, 1 multi-product− One with site transfers; Working closely with Compliance
• OBP QbD Pilot Meetings: − 23 meetings held with applicants − More were in planning (2009)
• Results− Pilot programs for QbD submissions have been completed and
the information collected for future implementation within FDA.
Patrick Swann, Ph.D.,Deputy Director, US-FDA, CDER, OPS, OBP, Division of Monoclonal Antibodies; The 4th DIA China

Common Issues in QbD Applications
• Critical Quality Attributes• Modeling • Design Space
− Factor choices (e.g. raw materials)− Impact of assay variability− Viral clearance− Linkage to other steps − Claims for scale in design space− Protocols as part of a design space− CPPs alone do not define a design space-assurance of quality− Limited parameter used
• Control Strategy-FDA, CDER, OPS, OBP• Risk Assessment Monoclonal Antibodies
Patrick Swann, Ph.D.,Deputy Director, US-FDA, CDER, OPS, OBP, Division of Monoclonal Antibodies; The 4th DIA China

Practical Questions on QBD
• When and where does a QBD program start?• What is considered a QBD biotech application?• Will Q8, Q9 and Q10 increase regulatory requirements?• How much data are needed for a QBD application?• How does one build QbD technical information under
CTD submissions?• How to manage the products approved under traditional
and QbD system within the same company?• To be or not to be in the QbD games for a small startup
companies?

QBD and Biosimilar Development
• Advances in manufacturing science and Quality-by-Design approaches may facilitate production processes that can better match a reference product’s fingerprint (2012 FDA Biosimilar Guidance)
• Biosimilar development process Select candidate product and reference product (desired or target
product profile). Define the characteristics (critical quality attributes) and ranges and
lot-to-lot variations of the reference product from multiple lots and multiple years (design space).
Establish the tests and acceptance criteria for comparability testing. Process development to produce DS meeting the targeted ranges of the
biosimilar products (DoE, process understandings). Manufacturing the DS under robust process with scale adequate for
quality and pre-clinical and no-clinical comparability testing (risk assessments).
•

Conclusion
• ICH Q8, Q9, Q10 and Q11 also provide guidelines for implementation Biotech Quality-by-Design drug development.
• Either traditional or QbD approaches will be acceptable in US, but Q8 to Q11 should be applied together for QbD.
• QBD principles for drugs applies, but for biotech products, complex structure, quality attributes, manufacturing process present more challenges to define the design space and implement QbD.
• The process may start with the use of pre-clinical and clinical data to identify critical quality attributes with links to safety and efficacy and the studies on unit operations.
• A QbD system for biotech products may need to be developed in incremental steps through different phases of development.





























