Embed Size (px)
Citation preview

Redox Properties of Structural Fe in Clay Minerals. 1. ElectrochemicalQuantification of Electron-Donating and -Accepting Capacities ofSmectitesChristopher A. Gorski,† Michael Aeschbacher,‡ Daniela Soltermann,§,‡ Andreas Voegelin,†,‡
Bart Baeyens,§ Maria Marques Fernandes,§ Thomas B. Hofstetter,*,†,‡ and Michael Sander*,‡
†Eawag, Swiss Federal Institute of Aquatic Science and Technology, Dubendorf, Switzerland‡Institute of Biogeochemistry and Pollutant Dynamics (IBP), ETH Zurich, Zurich, Switzerland§Laboratory for Waste Management, Paul Scherrer Institut (PSI), Villigen, Switzerland
*S Supporting Information
ABSTRACT: Clay minerals often contain redox-active struc-tural iron that participates in electron transfer reactions withenvironmental pollutants, bacteria, and biological nutrients.Measuring the redox properties of structural Fe in clay mineralsusing electrochemical approaches, however, has proven to bedifficult due to a lack of reactivity between clay minerals andelectrodes. Here, we overcome this limitation by using one-electron-transfer mediating compounds to facilitate electrontransfer between structural Fe in clay minerals and a vitreouscarbon working electrode in an electrochemical cell. Using thisapproach, the electron-accepting and -donating capacities(QEAC and QEDC) were quantified at applied potentials (EH)of −0.60 V and +0.61 V (vs SHE), respectively, for four naturalFe-bearing smectites (i.e., SWa-1, SWy-2, NAu-1, and NAu-2) having different total Fe contents (Fetotal = 2.3 to 21.2 wt % Fe)and varied initial Fe2+/Fetotal states. For every SWa-1 and SWy-2 sample, all the structural Fe was redox-active over the tested EHrange, demonstrating reliable quantification of Fe content and redox state. Yet for NAu-1 and NAu-2, a significant fraction of thestructural Fe was redox-inactive, which was attributed to Fe-rich smectites requiring more extreme EH-values to achieve completeFe reduction and/or oxidation. The QEAC and QEDC values provided here can be used as benchmarks in future studies examiningthe extent of reduction and oxidation of Fe-bearing smectites.
INTRODUCTION
The Fe2+/Fe3+ redox couple is an important redox buffer in theenvironment that affects biogeochemical element and nutrientcycling and controls the partitioning and redox transformationsof organic and inorganic contaminants.1−5 Much of the Fe inthe lithosphere is present as a structural component of clayminerals (i.e., phyllosilicates),1 where it can participate in avariety of electron transfer reactions. Structural Fe2+ in clayminerals may reduce heavy metals, radionuclides, and organiccontaminants, altering their mobility, (bio)availability, andtoxicity.6−12 Structural Fe3+, in contrast, may act as a terminalelectron acceptor for Fe-reducing bacteria, providing amechanism by which structural Fe2+ can be (re)generated.13
The structural Fe redox state also affects the physicochemicalproperties of clay minerals, which can include the cationexchange capacity and the swelling capacity.14−18 Thesechanges can alter the fate and (bio)availability of redox-inactivecontaminants and nutrients (e.g., K+, Ca2+)18 as well as dictatethe viability of clay minerals in engineered systems (e.g., claymineral backfill in radioactive waste repositories).19
Developing a thorough understanding of clay mineral redoxproperties in the context of biogeochemistry has, however,proven to be challenging. Structural Fe reduction and oxidationis coupled to structural rearrangements in the clay minerallattice that occur to maintain charge balance and accommodatethe atomic size difference between Fe3+ and Fe2+.20−25 As aresult, clay mineral redox reactions are dependent on both theclay mineral properties (i.e., the total Fe content, the Febinding environment, the layer charge, and mineral structure)and the solution chemistry (e.g., pH and the reductant oroxidant used). In addition to the complexity of clay minerals,methodological limitations have also impaired our under-standing of clay mineral redox properties at a mechanistic level.Previous experimental approaches have been unable to directlyquantify the number of electrons transferred to and from a claymineral over the course of a reaction under well-defined
Received: May 20, 2012Revised: July 16, 2012Accepted: July 24, 2012Published: July 24, 2012
Article
pubs.acs.org/est
© 2012 American Chemical Society 9360 dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−9368
solution conditions. Instead, changes in the redox states ofstructural Fe were inferred from spectroscopic measurementsand/or labor-intensive acidic dissolution studies.9,13,26−29 Otherstudies monitored the reduction of probe compounds to assessthe reactivity of structural Fe in the clay minerals.6−8,11,30−32
While such approaches provide information on the relativereactivity of structural Fe in different clay minerals, results fromthese studies may be dependent on the experimental conditionsused. To broaden our understanding of clay mineral redoxproperties, the measurement of thermodynamic values is clearlydesirable. To arrive at such values, novel experimentalapproaches are required that allow for a direct quantificationof electron transfer to and from structural Fe as a function ofsolution conditions and redox potential (i.e., EH).Our group has recently demonstrated direct quantification of
the number of electrons transferred to and from natural organicmatter (NOM) samples using mediated electrochemicalreduction (MER) and oxidation (MEO).33−37 MER andMEO rely on the use of one-electron-transfer mediatingcompounds (i.e., mediators) to facilitate electron transferbetween the sample of interest and a working electrode (WE)(Scheme 1). In this approach, an electrochemical cellcontaining a pH-buffered solution is set to a constant EH-value while the current is measured over time. In the presenceof a mediator in redox equilibrium with the WE, knownamounts of a sample are added to the electrochemical cell,resulting in current responses that can be integrated to directlydetermine the number of electrons transferred to or from the
WE. This approach has several unique features that would bebeneficial to studying the redox properties of structural Fe inclay minerals: (i) the number of electrons transferred to andfrom the clay mineral can be directly quantified, (ii) theexperiments can be conducted under well-controlled solutionand EH conditions, (iii) the effect of solution pH on thestructural Fe redox properties can be readily elucidated becausethe electron transfer to and from the selected mediators isunaffected by pH, and (iv) the analysis can be performed onthe time scale of minutes to hours.The goal of this work was therefore to evaluate the
applicability of MER and MEO to Fe-bearing clay mineralsand to determine the electron-accepting capacities (QEAC) andelectron-donating capacities (QEDC) of clay minerals underwell-defined solution conditions and EH-values. To this end, weadapted the MER and MEO approach to quantify QEAC andQEDC values at strongly reducing (EH = −0.60 V) and stronglyoxidizing (EH = +0.61 V) redox potentials, respectively, for fournatural Fe-bearing smectites (i.e., SWa-1, SWy-2, NAu-1, andNAu-2). These smectites were selected because they have beenstudied extensively in the past and because their structural Fecontent varies considerably, ranging from 2.3 (SWy-2) to 21.2(NAu-1) wt % Fe (Table 1). The minerals were characterizedusing 57Fe Mossbauer spectroscopy to confirm their purity andto explore relationships between QEAC and QEDC values and thelocal coordination environment of structural Fe atoms. Theresults from this study build the foundation for the companionpaper, in which electron transfer to and from structural Fe in
Scheme 1. Mediated Electrochemical Reduction and Oxidation of SWa-1
Table 1. Fe Contents and Electron Accepting and Donating Capacities (QEAC and QEDC, respectively) of Natural Fe-BearingClay Minerals As Determined by MER and MEO at EH = −0.60 V and EH = +0.61 V, Respectively
Fe content electron capacity [mmole−/gclay]
clay mineral wt % mmolFe/gclay redox treatment QEAC QEDC Qtotal redox-active Fe [%]a
SWa-1 12.6 ± 0.1 2.26 ± 0.02 native 2.21 ± 0.01 0.00 ± 0.00 2.21 ± 0.01 97.8 ± 1.0dithionite-reduced 0.05 ± 0.01 2.24 ± 0.02 2.29 ± 0.03 101.4 ± 1.6partially reduced 1.46 ± 0.07 0.76 ± 0.03 2.22 ± 0.08 98.6 ± 3.6partially re-oxidized 1.71 ± 0.02 0.44 ± 0.12 2.15 ± 0.12 95.3 ± 5.4
SWy-2 2.3 ± 0.2 0.41 ± 0.04 native 0.40 ± 0.04 0.00 ± 0.00 0.40 ± 0.04 96.5 ± 13.6dithionite-reduced 0.00 ± 0.00 0.39 ± 0.01 0.39 ± 0.01 94.5 ± 9.6
NAu-1 21.2 ± 1.1 3.80 ± 0.20 native 3.11 ± 0.10 0.00 ± 0.00 3.11 ± 0.10 81.8 ± 5.0dithionite-reduced 0.68 ± 0.12 2.37 ± 0.16 3.06 ± 0.20 80.5 ± 6.8
NAu-2 19.2 ± 0.3 3.44 ± 0.05 native 3.41 ± 0.14 0.00 ± 0.00 3.41 ± 0.14 99.3 ± 4.2dithionite-reduced 0.24 ± 0.01 2.55 ± 0.02 2.78 ± 0.03 81.0 ± 1.5
aTotal standard deviations in Fe content and Qtotal were calculated from at least five and four replicate measurements, respectively.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689361

SWa-1 is explored as a function of EH.38 Together, the
companion papers illustrate the applicability of MER and MEOto assess the redox properties of Fe in solid-phase samples.
MATERIALS AND METHODSMaterials. Section S1 of the Supporting Information
contains a complete list of chemicals used in this study.Ferruginous smectite (SWa-1), Na-rich montmorillonite (SWy-2), and two nontronites (NAu-1 and NAu-2) were purchasedfrom the Source Clay Minerals Repository (Purdue University,West Lafayette, IN). Literature unit cell formulas of the clayminerals are provided in Table S2. ABTS (2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid), 99%) and diquat (1,1′-ethylene 2,2′-bipyridyl dibromide, DQ, 100%) were purchasedfrom Sigma-Aldrich (St. Louis, MO). Triquat (1,1′-tri-methylene 2,2′-bipyridyl dibromide, TQ) was synthesizedfollowing an established method39 and was recrystallized inMeOH, as described in the Section S2. NMR, high-resolutionmass spectrometry (thermoexactive MS), and cyclic voltam-metry were used to confirm the identity and high purity of thetriquat (Section S2). All aqueous solutions were prepared usingnanopure water (resistivity, σ > 18 MΩ·cm) (NanopureDiamond Water System). All experiments were carried out in apH 7.5 buffered solution (0.1 M NaClO4, 0.01 M MOPS, pKa =7.2).Anaerobic Conditions. All experiments were conducted in
an anaerobic glovebox (<0.1 ppm O2) with an N2 atmosphere(Unilab 2010, M.Braun GmbH, Germany). Plastic andglassware were evacuated overnight in the exchange chamberand allowed to equilibrate in the glovebox atmosphere forseveral days prior to use. All aqueous solutions and methanolwere purged with 99.999% Ar gas for 2 h prior to transferringthem into the glovebox.Clay Mineral Purification, Size Fractionation, and
Treatment. Clay minerals were purified, Na+-saturated, andsize-fractionated according to a previously described method.40
Clay minerals were broken up by milling and added to 1 MNaClO4. The suspensions were stirred for 3 h and then allowedto settle. The supernatant was discarded, and the bottle wasrefilled with fresh 1 M NaClO4. This process was repeatedthree times. Aliquots of the clay mineral suspension werecentrifuged at 600 g for 7 min, with the ≤0.5 μm size fractionremaining in the supernatant. The supernatant was decantedand saved, and the centrifuge bottle was refilled with DI water,shaken, and recentrifuged. This process was repeated until thesupernatant remained clear after centrifugation. The fine clayfraction (i.e., the decanted suspension from centrifugation) wasthen flocculated by the addition of 1 M NaClO4. Thissuspension sat undisturbed overnight, and then the supernatantwas removed and discarded. To remove any loosely bound Feand Fe (oxyhdr)oxide impurities, a portion of this claysuspension was acidified with HNO3 to pH 3.5 and stirredfor 1 h, with the pH monitored and readjusted as needed. Thesuspension was then centrifuged and resuspended in 1 MNaClO4 at neutral pH. The final clay mineral concentration wastypically in the range of 5−10 g L−1.Dithionite-reduced clay minerals were generated inside the
glovebox following standard procedures.6,41 An aliquot of theclay mineral stock suspension (75 mL) was brought into theglovebox after purging with Ar. A 1 M NaHCO3 (23.4 mL)solution and 0.3 M Na3-citrate (1.6 mL) solution were thenadded to the clay mineral suspension. This suspension was thenmixed and heated to 70 °C, at which point sodium dithionite
was added (3 times the clay mineral mass in suspension), andthe suspension was stirred at 70 °C overnight. A portion of thesuspension was then taken and put into a prewashed and drieddialysis tube (MWCO 12400 Da, Sigma Aldrich), and added to1 L of deoxygenated 0.1 M NaClO4. The suspension wasequilibrated for at least 8 h, at which point the dialysis tubingwas placed into fresh NaClO4 buffer. This process was repeatedfour times. Native clay mineral samples were produced in anidentical manner, except that no sodium dithionite was addedto the citrate−bicarbonate buffer. Partially reoxidized SWa-1was obtained by purging dithionite-reduced SWa-1 with airoutside of the glovebox for approximately 18 h, at which pointit was purged with Ar for 2 h and transferred back into theglovebox.The clay mineral concentration was determined gravimetri-
cally by drying the suspension at 105 °C while accounting forthe NaClO4. The Fe content of the clay minerals was measuredaccording to an established acidic digestion method30 adaptedfrom earlier procedures,42,43 using ferrous ethylenediammo-nium sulfate tetrahydrate as the Fe standard.
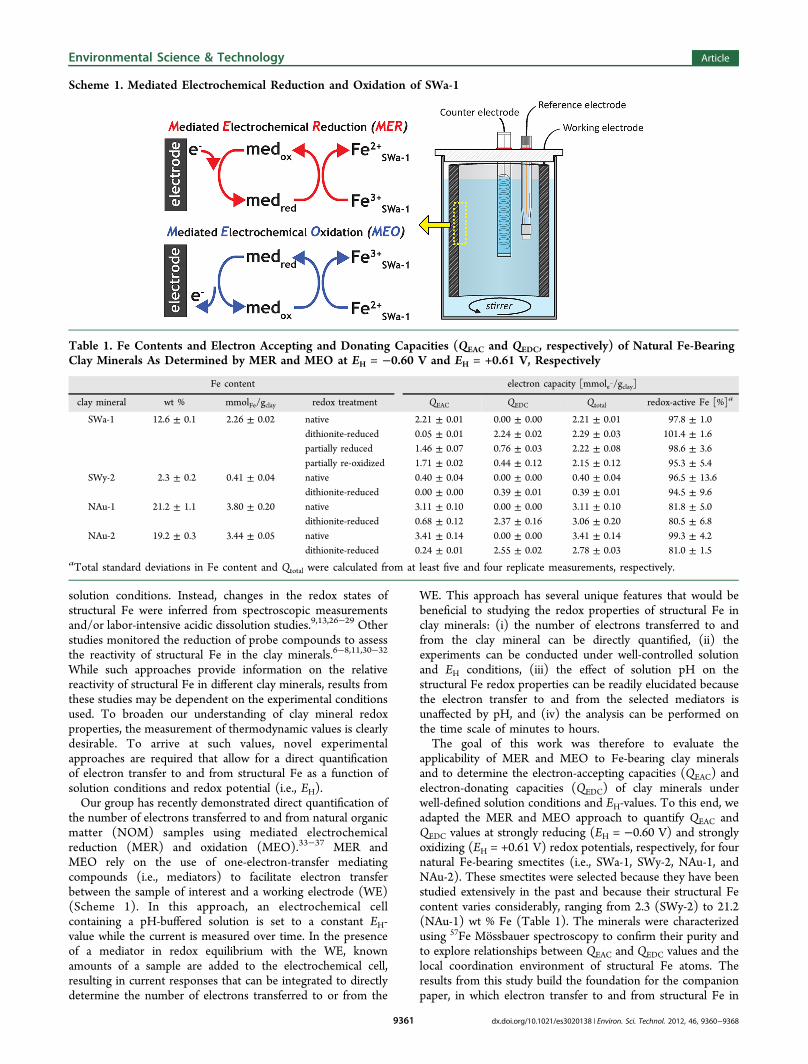
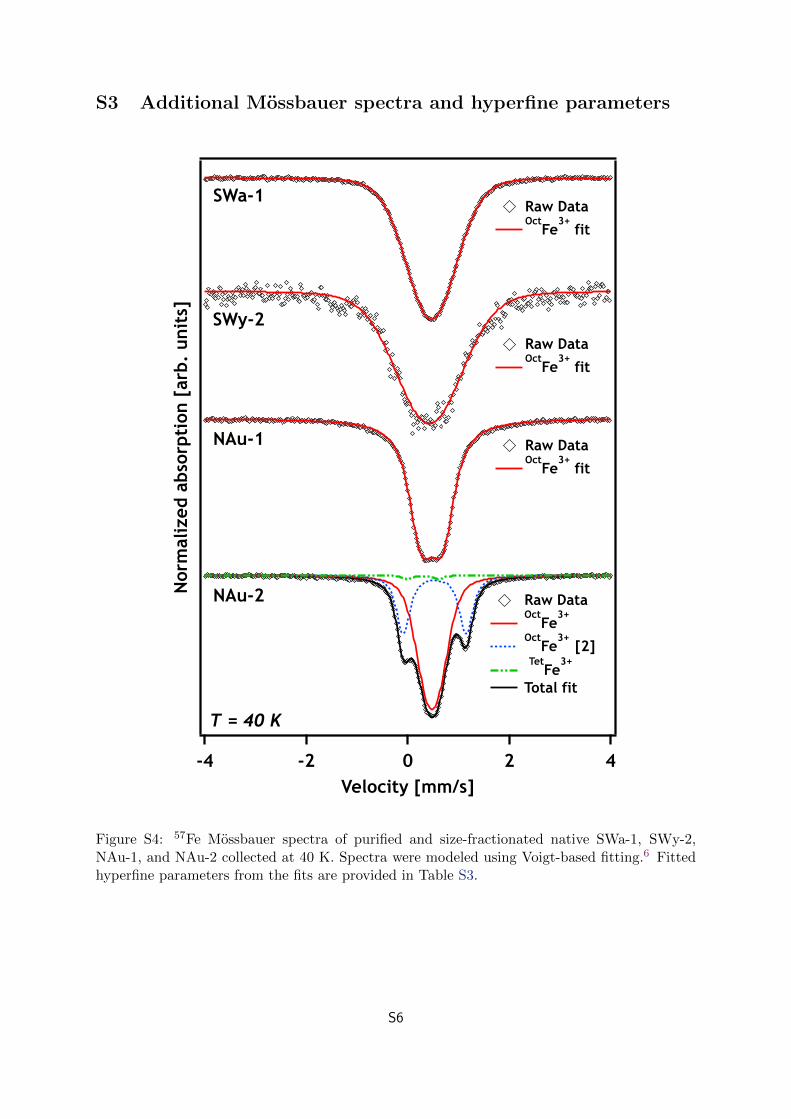
57Fe Mossbauer Spectroscopy. Samples for Mossbaueranalysis were filtered, dried, and ground in the anaerobicglovebox prior to analysis. Measurements were made using apreviously described setup.44 Spectra were fit using thecommercial software Recoil (Ottawa, Canada) and a Voigt-based model (fit parameters shown in Table S3).45 57FeMossbauer spectra were collected for each smectite at 13 Kafter the purification procedure to determine the structuralcoordination and oxidation state of the Fe (Figure 1). Spectracollected at 40 K were consistent with 13 K spectra (FigureS4). All spectra were characteristic of Fe3+ in clay minerals, withno indication of any Fe2+ phases or Fe3+ (oxyhydr-)oxideimpurities (detection limit ≈1−2% Fe).46 The fitted hyperfineparameters of SWa-1, SWy-2, and NAu-1 indicated that theminerals contained only cis-octahedral Fe3+ (OctFe3+; i.e., thetwo binding hydroxyl groups were adjacent to one another).The NAu-2 spectrum was more complex: ≈2% of the structuralFe3+ was tetrahedrally coordinated (TetFe3+) and a secondoctahedral Fe3+ site (i.e., OctFe3+ [2], 32% of area) wasobserved. The OctFe3+ [2] site has previously been interpretedas both trans-octahedral Fe3+ (i.e., two hydroxyl groupsopposite each other)47−51 and distorted cis-octahedral Fe3+
near a tetrahedral Fe3+ atom28,52,53 (Figure 1).Electrochemistry. Electrochemical experiments were con-
trolled with a 630D electrochemical analyzer (CH Instruments,Austin, TX). Potentials were measured versus an Ag/AgClreference electrode but are reported versus the standardhydrogen electrode (SHE). MER and MEO experimentswere carried out in electrochemical cells containing 80 mL ofpH 7.5 buffer, a cylindrical vitreous carbon working electrode(WE), a Pt wire counter electrode separated from the WEcompartment by a glass frit, and an Ag/AgCl referenceelectrode (all electrodes from Bioanalytical Systems, UK)(Scheme 1). Cyclic voltammetry (CV) experiments wereconducted in a 8−10 mL solution of buffer containing a 3.0mm diameter glassy carbon disk working electrode, a platinumwire counter electrode, and an Ag/AgCl reference electrode (allelectrodes from Bioanalytical Systems Inc.). CV scans weredone at 10 mV·s−1 over a potential range of approximately ±0.3V of the expected EH
0 -values. CV was used to measure the redoxpotentials (EH
0 ) of diquat (DQ2+/DQ•+, −0.35 V), triquat(TQ2+/TQ•+, −0.54 V), and ABTS (ABTS2−/ABTS•−, +0.70
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689362
V). A CV of triquat is shown in Figure S3, and CVs of diquatand ABTS can be found in the companion paper.38
To quantify the electron-accepting capacities (QEAC, mole−/gclay mineral) of the smectites, MER was performed by spikingTQ2+ into the electrochemical cell (10 μmol) after the WE waspre-equilibrated at EH = −0.60 V. At this EH, TQ
2+ was reducedto TQ•+ at the WE, resulting in a reductive current response.After an initial fast increase, the current began to decrease andleveled off at a stable baseline value upon attainment of redoxequilbriation between the TQ2+/TQ•+ redox couple and theWE. Small volumes (10−100 μL) of the clay mineralsuspensions were then spiked into the electrochemical cell.Electron transfer from TQ•+ to structural Fe3+ formed TQ2+,which was rapidly re-reduced at the WE, resulting in reductivecurrent peaks (Scheme 1). Peak integration yielded the numberof electrons transferred to the WE (eq 1):
∫=qF
I t1
dt
t
1
2
(1)
where q is the number of electrons transferred in moles, F is theFaraday constant (96 485 C/mol), and t is time. Peakintegration was performed using Igor Pro software (Wave-metrics, Lake Oswego, OR). The electron-donating capacity(QEDC, mole−/gclay mineral) was measured by MEO with anexperiment setup similar to MER, except that ABTS2− wasoxidized to ABTS•− at the WE, which was polarized to EH =+0.61 V. Electron transfer from structural Fe2+ in spiked clay
minerals resulted in the reduction of ABTS2− to ABTS•− andsubsequent re-oxidation of ABTS2− at the WE, resulting inoxidative current peaks that were integrated to quantify q(Scheme 1).
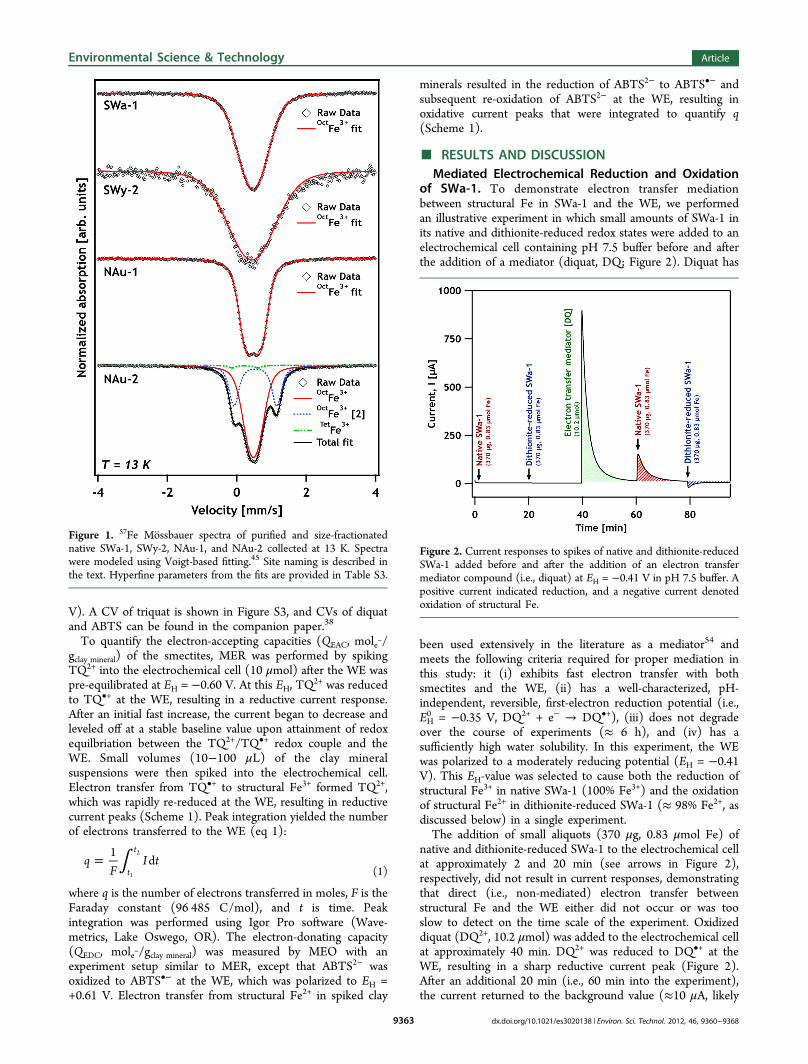
RESULTS AND DISCUSSIONMediated Electrochemical Reduction and Oxidation
of SWa-1. To demonstrate electron transfer mediationbetween structural Fe in SWa-1 and the WE, we performedan illustrative experiment in which small amounts of SWa-1 inits native and dithionite-reduced redox states were added to anelectrochemical cell containing pH 7.5 buffer before and afterthe addition of a mediator (diquat, DQ; Figure 2). Diquat has
been used extensively in the literature as a mediator54 andmeets the following criteria required for proper mediation inthis study: it (i) exhibits fast electron transfer with bothsmectites and the WE, (ii) has a well-characterized, pH-independent, reversible, first-electron reduction potential (i.e.,EH0 = −0.35 V, DQ2+ + e− → DQ•+), (iii) does not degrade
over the course of experiments (≈ 6 h), and (iv) has asufficiently high water solubility. In this experiment, the WEwas polarized to a moderately reducing potential (EH = −0.41V). This EH-value was selected to cause both the reduction ofstructural Fe3+ in native SWa-1 (100% Fe3+) and the oxidationof structural Fe2+ in dithionite-reduced SWa-1 (≈ 98% Fe2+, asdiscussed below) in a single experiment.The addition of small aliquots (370 μg, 0.83 μmol Fe) of
native and dithionite-reduced SWa-1 to the electrochemical cellat approximately 2 and 20 min (see arrows in Figure 2),respectively, did not result in current responses, demonstratingthat direct (i.e., non-mediated) electron transfer betweenstructural Fe and the WE either did not occur or was tooslow to detect on the time scale of the experiment. Oxidizeddiquat (DQ2+, 10.2 μmol) was added to the electrochemical cellat approximately 40 min. DQ2+ was reduced to DQ•+ at theWE, resulting in a sharp reductive current peak (Figure 2).After an additional 20 min (i.e., 60 min into the experiment),the current returned to the background value (≈10 μA, likely
Figure 1. 57Fe Mossbauer spectra of purified and size-fractionatednative SWa-1, SWy-2, NAu-1, and NAu-2 collected at 13 K. Spectrawere modeled using Voigt-based fitting.45 Site naming is described inthe text. Hyperfine parameters from the fits are provided in Table S3.
Figure 2. Current responses to spikes of native and dithionite-reducedSWa-1 added before and after the addition of an electron transfermediator compound (i.e., diquat) at EH = −0.41 V in pH 7.5 buffer. Apositive current indicated reduction, and a negative current denotedoxidation of structural Fe.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689363
due to minor H+ reduction), indicating that the diquat hadreached redox equilibrium with the WE.Native and dithionite-reduced SWa-1 (i.e., 370 μg, 0.83 μmol
Fe) were then added to the cell at approximately 60 and 80min, respectively, in amounts identical to those added beforethe diquat spike (Figure 2). In the presence of diquat, theaddition of native SWa-1 resulted in a reductive current peak(red shaded area in Figure 2). The reductive current responseindicated that diquat facilitated electron transfer from the WEto the SWa-1 to an extent that reduction was detectable at thetime scale of the experiment (Scheme 1). The addition ofdithionite-reduced SWa-1 resulted in an oxidative peak (blueshaded area in Figure 2), demonstrating that diquat facilitatedelectron transfer from the structural Fe2+ in the dithionite-reduced SWa-1 to the WE (Scheme 1). This finding suggestedthat potentials lower than −0.41 V were needed to fully reducethe structural Fe3+ to Fe2+ in SWa-1 and hence to quantify thetotal electron-accepting capacity of SWa-1.Electron-Accepting and -Donating Capacities of SWa-
1. Similar experiments to the one in Figure 2 were performedto quantify the electron-accepting capacity (QEAC, mole−/gSWa‑1), electron-donating capacity (QEDC, mole−/gSWa‑1), andtotal electron-exchanging capacity (Qtotal = QEAC + QEDC) ofSWa-1 samples. These experiments differed from the previousexperiment in that strongly reducing (EH = −0.60 V) andstrongly oxidizing (EH = +0.61 V) potentials were applied tothe WE, requiring mediators with different EH
0 -values near theapplied EH. We chose triquat (EH
0 = −0.54 V) and ABTS (EH0 =
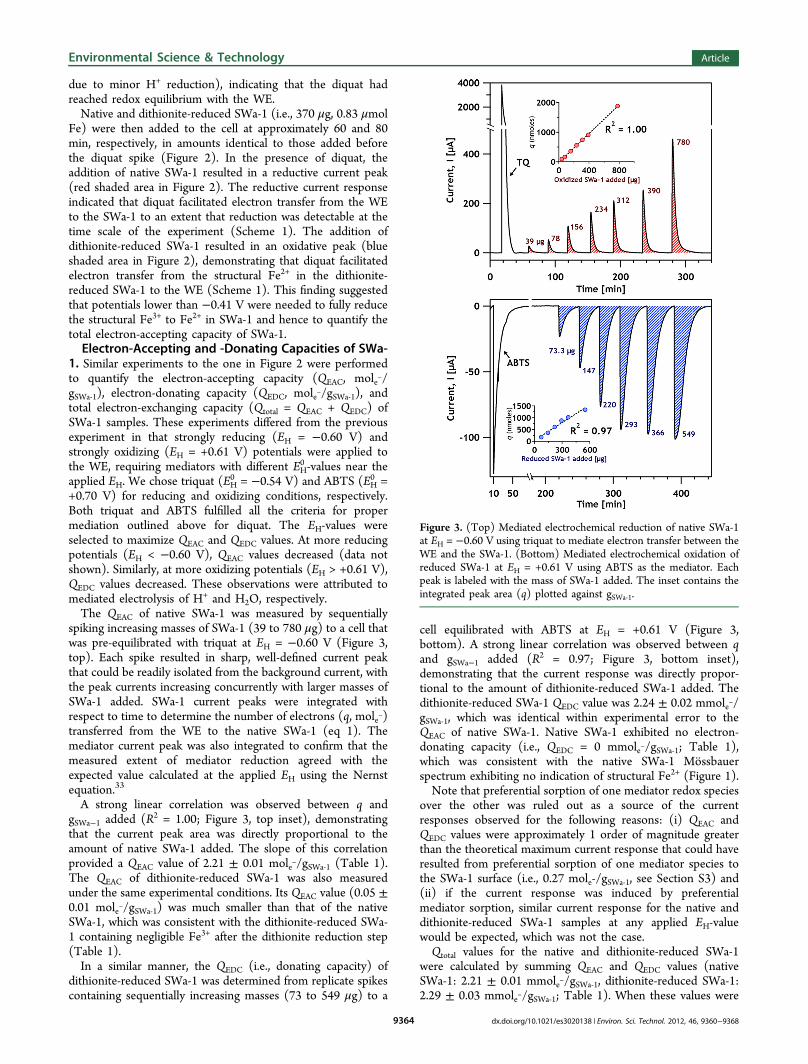
+0.70 V) for reducing and oxidizing conditions, respectively.Both triquat and ABTS fulfilled all the criteria for propermediation outlined above for diquat. The EH-values wereselected to maximize QEAC and QEDC values. At more reducingpotentials (EH < −0.60 V), QEAC values decreased (data notshown). Similarly, at more oxidizing potentials (EH > +0.61 V),QEDC values decreased. These observations were attributed tomediated electrolysis of H+ and H2O, respectively.The QEAC of native SWa-1 was measured by sequentially
spiking increasing masses of SWa-1 (39 to 780 μg) to a cell thatwas pre-equilibrated with triquat at EH = −0.60 V (Figure 3,top). Each spike resulted in sharp, well-defined current peakthat could be readily isolated from the background current, withthe peak currents increasing concurrently with larger masses ofSWa-1 added. SWa-1 current peaks were integrated withrespect to time to determine the number of electrons (q, mole−)transferred from the WE to the native SWa-1 (eq 1). Themediator current peak was also integrated to confirm that themeasured extent of mediator reduction agreed with theexpected value calculated at the applied EH using the Nernstequation.33
A strong linear correlation was observed between q andgSWa−1 added (R2 = 1.00; Figure 3, top inset), demonstratingthat the current peak area was directly proportional to theamount of native SWa-1 added. The slope of this correlationprovided a QEAC value of 2.21 ± 0.01 mole−/gSWa‑1 (Table 1).The QEAC of dithionite-reduced SWa-1 was also measuredunder the same experimental conditions. Its QEAC value (0.05 ±0.01 mole−/gSWa‑1) was much smaller than that of the nativeSWa-1, which was consistent with the dithionite-reduced SWa-1 containing negligible Fe3+ after the dithionite reduction step(Table 1).In a similar manner, the QEDC (i.e., donating capacity) of
dithionite-reduced SWa-1 was determined from replicate spikescontaining sequentially increasing masses (73 to 549 μg) to a
cell equilibrated with ABTS at EH = +0.61 V (Figure 3,bottom). A strong linear correlation was observed between qand gSWa−1 added (R2 = 0.97; Figure 3, bottom inset),demonstrating that the current response was directly propor-tional to the amount of dithionite-reduced SWa-1 added. Thedithionite-reduced SWa-1 QEDC value was 2.24 ± 0.02 mmole−/gSWa‑1, which was identical within experimental error to theQEAC of native SWa-1. Native SWa-1 exhibited no electron-donating capacity (i.e., QEDC = 0 mmole−/gSWa‑1; Table 1),which was consistent with the native SWa-1 Mossbauerspectrum exhibiting no indication of structural Fe2+ (Figure 1).Note that preferential sorption of one mediator redox species
over the other was ruled out as a source of the currentresponses observed for the following reasons: (i) QEAC andQEDC values were approximately 1 order of magnitude greaterthan the theoretical maximum current response that could haveresulted from preferential sorption of one mediator species tothe SWa-1 surface (i.e., 0.27 mole-/gSWa‑1, see Section S3) and(ii) if the current response was induced by preferentialmediator sorption, similar current response for the native anddithionite-reduced SWa-1 samples at any applied EH-valuewould be expected, which was not the case.Qtotal values for the native and dithionite-reduced SWa-1
were calculated by summing QEAC and QEDC values (nativeSWa-1: 2.21 ± 0.01 mmole−/gSWa‑1, dithionite-reduced SWa-1:2.29 ± 0.03 mmole−/gSWa‑1; Table 1). When these values were
Figure 3. (Top) Mediated electrochemical reduction of native SWa-1at EH = −0.60 V using triquat to mediate electron transfer between theWE and the SWa-1. (Bottom) Mediated electrochemical oxidation ofreduced SWa-1 at EH = +0.61 V using ABTS as the mediator. Eachpeak is labeled with the mass of SWa-1 added. The inset contains theintegrated peak area (q) plotted against gSWa‑1.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689364
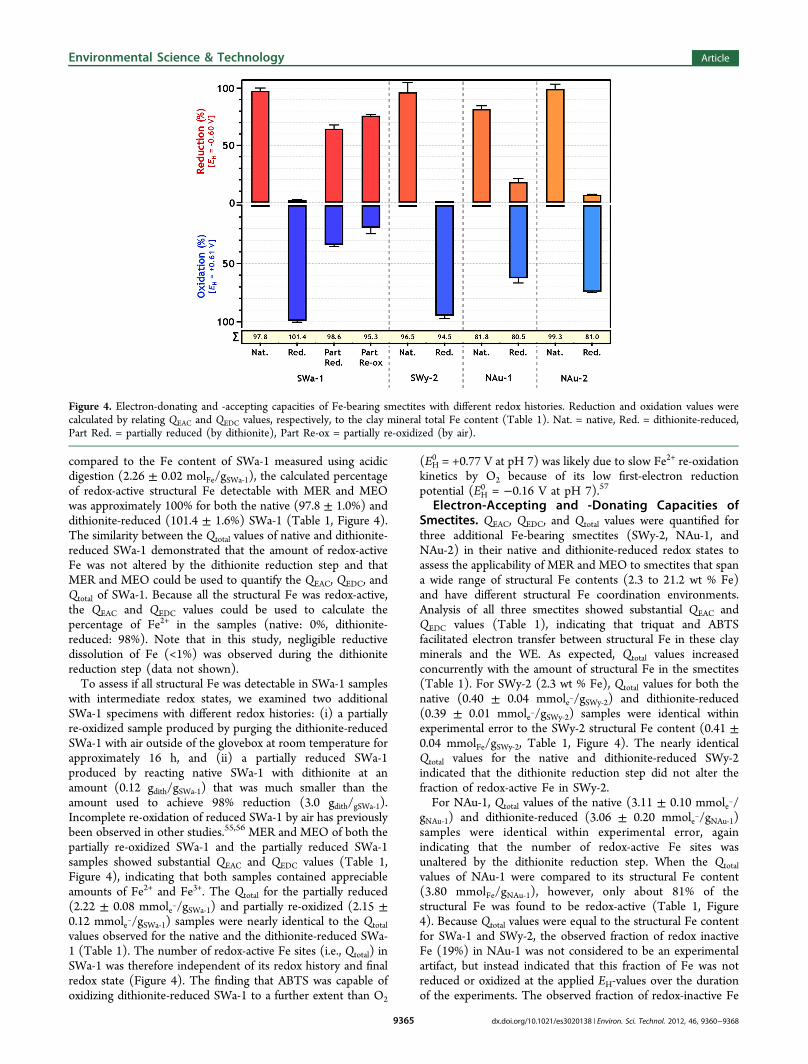
compared to the Fe content of SWa-1 measured using acidicdigestion (2.26 ± 0.02 molFe/gSWa‑1), the calculated percentageof redox-active structural Fe detectable with MER and MEOwas approximately 100% for both the native (97.8 ± 1.0%) anddithionite-reduced (101.4 ± 1.6%) SWa-1 (Table 1, Figure 4).The similarity between the Qtotal values of native and dithionite-reduced SWa-1 demonstrated that the amount of redox-activeFe was not altered by the dithionite reduction step and thatMER and MEO could be used to quantify the QEAC, QEDC, andQtotal of SWa-1. Because all the structural Fe was redox-active,the QEAC and QEDC values could be used to calculate thepercentage of Fe2+ in the samples (native: 0%, dithionite-reduced: 98%). Note that in this study, negligible reductivedissolution of Fe (<1%) was observed during the dithionitereduction step (data not shown).To assess if all structural Fe was detectable in SWa-1 samples
with intermediate redox states, we examined two additionalSWa-1 specimens with different redox histories: (i) a partiallyre-oxidized sample produced by purging the dithionite-reducedSWa-1 with air outside of the glovebox at room temperature forapproximately 16 h, and (ii) a partially reduced SWa-1produced by reacting native SWa-1 with dithionite at anamount (0.12 gdith/gSWa‑1) that was much smaller than theamount used to achieve 98% reduction (3.0 gdith/gSWa‑1).Incomplete re-oxidation of reduced SWa-1 by air has previouslybeen observed in other studies.55,56 MER and MEO of both thepartially re-oxidized SWa-1 and the partially reduced SWa-1samples showed substantial QEAC and QEDC values (Table 1,Figure 4), indicating that both samples contained appreciableamounts of Fe2+ and Fe3+. The Qtotal for the partially reduced(2.22 ± 0.08 mmole−/gSWa‑1) and partially re-oxidized (2.15 ±0.12 mmole−/gSWa‑1) samples were nearly identical to the Qtotalvalues observed for the native and the dithionite-reduced SWa-1 (Table 1). The number of redox-active Fe sites (i.e., Qtotal) inSWa-1 was therefore independent of its redox history and finalredox state (Figure 4). The finding that ABTS was capable ofoxidizing dithionite-reduced SWa-1 to a further extent than O2
(EH0 = +0.77 V at pH 7) was likely due to slow Fe2+ re-oxidation
kinetics by O2 because of its low first-electron reductionpotential (EH
0 = −0.16 V at pH 7).57
Electron-Accepting and -Donating Capacities ofSmectites. QEAC, QEDC, and Qtotal values were quantified forthree additional Fe-bearing smectites (SWy-2, NAu-1, andNAu-2) in their native and dithionite-reduced redox states toassess the applicability of MER and MEO to smectites that spana wide range of structural Fe contents (2.3 to 21.2 wt % Fe)and have different structural Fe coordination environments.Analysis of all three smectites showed substantial QEAC andQEDC values (Table 1), indicating that triquat and ABTSfacilitated electron transfer between structural Fe in these clayminerals and the WE. As expected, Qtotal values increasedconcurrently with the amount of structural Fe in the smectites(Table 1). For SWy-2 (2.3 wt % Fe), Qtotal values for both thenative (0.40 ± 0.04 mmole−/gSWy‑2) and dithionite-reduced(0.39 ± 0.01 mmole−/gSWy‑2) samples were identical withinexperimental error to the SWy-2 structural Fe content (0.41 ±0.04 mmolFe/gSWy‑2, Table 1, Figure 4). The nearly identicalQtotal values for the native and dithionite-reduced SWy-2indicated that the dithionite reduction step did not alter thefraction of redox-active Fe in SWy-2.For NAu-1, Qtotal values of the native (3.11 ± 0.10 mmole−/
gNAu‑1) and dithionite-reduced (3.06 ± 0.20 mmole−/gNAu‑1)samples were identical within experimental error, againindicating that the number of redox-active Fe sites wasunaltered by the dithionite reduction step. When the Qtotalvalues of NAu-1 were compared to its structural Fe content(3.80 mmolFe/gNAu‑1), however, only about 81% of thestructural Fe was found to be redox-active (Table 1, Figure4). Because Qtotal values were equal to the structural Fe contentfor SWa-1 and SWy-2, the observed fraction of redox inactiveFe (19%) in NAu-1 was not considered to be an experimentalartifact, but instead indicated that this fraction of Fe was notreduced or oxidized at the applied EH-values over the durationof the experiments. The observed fraction of redox-inactive Fe
Figure 4. Electron-donating and -accepting capacities of Fe-bearing smectites with different redox histories. Reduction and oxidation values werecalculated by relating QEAC and QEDC values, respectively, to the clay mineral total Fe content (Table 1). Nat. = native, Red. = dithionite-reduced,Part Red. = partially reduced (by dithionite), Part Re-ox = partially re-oxidized (by air).
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689365

in NAu-1 was consistent with previous reports that have shownthat clay minerals become progressively more “difficult” toreduce biologically as the structural Fe content is increased.58
We attribute this trend to higher Fe content clay minerals beingincapable of making the necessary structural rearrangementsneeded to maintain a charge balance during reduction due tothe formation of trioctahedral domains (i.e., vacancy-lesssections in the octahedral sheets). Whether or not the redox-inactive fraction of Fe would be accessible with MER and MEOperformed at more extreme EH-values could not be investigatedbecause of reasons provided earlier. This question may also beof limited environmental relevance, as such extreme EH-valueswould not be expected in aqueous systems at circumneutral pHvalues.A substantial QEAC value was detected for the dithionite-
reduced NAu-1 (0.68 ± 0.12 mmole−/gNAu‑1, Table 1, Figure 4),indicating that the dithionite-reduced sample contained aconsiderable amount of structural Fe3+ that could be reducedby TQ•+. This meant that a portion of the structural Fe3+
remained unreduced during the dithionite reduction step and/or it was subsequently re-oxidized. Inadvertent oxidation of thedithionite-reduced NAu-1 by O2 after the dithionite reductionwas unlikely, as (i) QEAC values were reproducible in replicatedithionite-reduced batches of NAu-1 (data not shown), (ii) thedithionite reduction step was performed in an anaerobicglovebox (<0.1 ppm O2), with samples remaining sealed in theglovebox until use, and (iii) SWa-1 and SWy-2 did not re-oxidize in the glovebox during storage. Oxidation of the NAu-1by H+ was thermodynamically possible (H+/H2; EH
0 = −0.33 Vat pH 7),57 although it remains unclear why NAu-1 wouldreduce H+ on the time scale of days while SWa-1 and SWy-2would not. During the reduction step, the standard amount ofdithionite was used (i.e., 3.0 gdith/gNAu‑1),
41 which providedapproximately a 10-fold excess of SO2
•− radicals to structural Featoms in NAu-1. Therefore a lack of adequate amounts ofdithionite seemed unlikely, although NAu-1 containedsubstantially more structural Fe3+ than SWa-1 and SWy-2(Table 1). From a thermodynamical perspective, measurementsof the EH
0 of the dithionite radical at pH 7 range from −0.47 to−0.66 V.59−61 Based on these values, it seems plausible that theapplied potential (EH = −0.60 V) reduced structural Fe inNAu-1 to a larger extent than dithionite.For NAu-2, the Qtotal of the native sample (3.41 ± 0.14
mmole−/gNAu‑2) was significantly higher than that of thedithionite-reduced sample (2.78 ± 0.03 mmole−/gNAu‑2),indicating that the fraction of total Fe that was redox-activedecreased as a result of the dithionite reduction step. Severallines of evidence suggested that the decrease in the fraction ofredox-active Fe did not result from an experimental artifact orfrom loss of structural Fe during the dithionite reduction step:(i) the results were reproducible across replicate batches ofnative and dithionite-reduced NAu-2 (data not shown), and (ii)negligible Fe2+ dissolution (<1% of Fetotal) occurred during thedithionite reduction step, as confirmed by both aqueous Fe2+
measurements of the reduced suspension and acidic digestionof the dithionite-reduced NAu-2.Instead, the Qtotal value observed for the dithionite-reduced
NAu-2 sample indicated that a substantial fraction of structuralFe (≈18%) was no longer accessible by MER and MEO, whichcould be due to either (i) the structural Fe3+ and Fe2+ requiringa lower or higher EH to be reduced or oxidized, respectively,and/or (ii) the electron transfer rate to or from these Fe sitesbeing kinetically limited relative to the time scale of the
measurements. In the companion paper,38 we observed thatreduction of SWa-1 using dithionite resulted in permanentstructural alterations that affected the EH-value distribution ofFe sites in SWa-1. Similar irreversible changes could haveaffected the thermodynamics or kinetics of electron transfer toa fraction of structural Fe in NAu-2.Note that the dithionite-reduced NAu-2 had a substantial
QEAC value (0.24 ± 0.01 mmol-/gNAu-2, Table 1, Figure 4),indicating that the sample contained structural Fe3+. The samerationale used to explain the source of Fe3+ in the dithionite-reduced NAu-1 can be used for the dithionite-reduced NAu-2.Also note that unlike the other smectites investigated, NAu-2contained tetrahedral Fe3+ (≈2%, Figure 1). Whether or nottetrahedral Fe3+ is redox-active has been a topic of debate in theliterature.19,26,28,51,56,62−64 While nearly all the structural Fe wasreduced in NAu-2 (99.3 wt %), the standard deviation of themeasurement (4.2 wt %) was larger than the amount oftetrahedral Fe in the NAu-2, making it impossible to definitivelyconclude that the tetrahedral Fe3+ was reduced.
Methodological Implications. MER and MEO is a novelapproach that can (i) directly quantify the electron-acceptingand -donating capacities of clay minerals at set EH-values and(ii) discriminate between total Fe contents measured by acidicdigestion and redox-active Fe across a wide EH range (≈1.2 V).This approach has now been successfully applied to humicsubstances, natural organic matter,33−37 and Fe-bearing clayminerals and can potentially be applied to several otherenvironmentally relevant, redox-active phases. Several studieshave demonstrated that mediator compounds similar to theones used here rapidly transfer electrons to and from Fe andother metal (oxyhdr-)oxides.65−69 There is therefore a greatpotential to characterize the redox properties of manyenvironmentally relevant phases that have been elusive totraditional experimental approaches, including minerals,amorphous phases, and complexes. Note, however, that manyimportant environmental redox reactions are thought to beirreversible, such as the uptake of Fe2+ by Fe oxides,67,70,71
which may complicate interpretation.While strongly reducing and oxidizing EH-values were used in
this study to maximize the extent of reduction and oxidation ofstructural Fe in clay minerals, the EH dependence of electrontransfer to and from Fe may also be quantified, provided thatsuitable mediators are available. In the companion paper, wedemonstrate how this can be done by utilizing a large set ofmediators having EH
0 -values that lie within the EH range used inthis study (−0.60 V < EH < +0.61 V). Using this approach, weare able to examine the extent of Fe reduction (i.e., QEAC) andoxidation (i.e., QEDC) at environmentally relevant EH-values andthus to assess the electron-accepting and -donating behavior ofan Fe-bearing clay mineral.
ASSOCIATED CONTENT
*S Supporting InformationThe chemicals used in this study, literature unit cell formulasfor the clay minerals investigated, details regarding the synthesisand purity of triquat (TQ), additional Mossbauer spectra andtabulated Mossbauer hyperfine parameters, a TQ sorptionisotherm on SWa-1, and an estimation of the current responseexpected if one species of TQ had sorbed preferentially overthe other. This material is available free of charge via theInternet at http://pubs.acs.org.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689366

AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected]; [email protected].
NotesThe authors declare no competing financial interest.
ACKNOWLEDGMENTSThe authors thank Timonthy Pasakarnis and Michelle M.Scherer (University of Iowa) for Mossbauer spectroscopymeasurements, Olivier Leupin (Nagra, Switzerland) for fruitfuldiscussions, and P.J. Alaimo (Seattle University) for help withtriquat synthesis. This work was supported by the SwissNational Science Foundation (grant no. 200021-129476/1 toM.S and T.B.H.) and Nagra (project nos. 7246 and 9009).
REFERENCES(1) Amonette, J. E. Iron redox chemistry of clays and oxides:Environmental applications. CMS Workshop Lect. 2002, 10, 89−147.(2) Elsner, M.; Schwarzenbach, R.; Haderlein, S. B. Reactivity ofFe(II)-bearing minerals toward reductive transformation of organiccontaminants. Environ. Sci. Technol. 2003, 38, 799−807.(3) White, A.; Peterson, M. Reduction of aqueous transition metalspecies on the surfaces of Fe(II)-containing oxides. Geochim.Cosmochim. Acta 1996, 60, 3799−3814.(4) Cornell, R.; Schwertmann, U. The Iron Oxides - Structure,Properties, Reactions, Occurence and Uses 2003, p 664.(5) Jickells, T. D.; et al. Global iron connections between desert dust,ocean biogeochemistry, and climate. Science 2005, 308, 67−71.(6) Hofstetter, T. B.; Schwarzenbach, R. P.; Haderlein, S. B.Reactivity of Fe(II) species associated with clay minerals. Environ. Sci.Technol. 2003, 37, 519−528.(7) Hofstetter, T. B.; Neumann, A.; Schwarzenbach, R. P. Reductionof nitroaromatic compounds by Fe(II) species associated with iron-rich smectites. Environ. Sci. Technol. 2006, 40, 235−242.(8) Fredrickson, J.; Zachara, J.; Kennedy, D.; Kukkadapu, R.;McKinley, J.; Heald, S.; Liu, C.; Plymale, A. Reduction of TcO4
− bysediment-associated biogenic Fe(II). Geochim. Cosmochim. Acta 2004,68, 3171−3187.(9) Kukkadapu, R. K.; Zachara, J. M.; Fredrickson, J. K.; McKinley, J.P.; Kennedy, D. W.; Smith, S. C.; Dong, H. Reductivebiotransformation of Fe in shale-limestone saprolite containing Fe(III)oxides and Fe(II)/Fe(III) phyllosilicates. Geochim. Cosmochim. Acta2006, 70, 3662−3676.(10) Bishop, M. E.; Dong, H.; Kukkadapu, R. K.; Liu, C.; Edelmann,R. E. Bioreduction of Fe-bearing clay minerals and their reactivitytoward pertechnetate (Tc-99). Geochim. Cosmochim. Acta 2011, 75,5229−5246.(11) Neumann, A.; Hofstetter, T. B.; Skarpeli-Liati, M.;Schwarzenbach, R. P. Reduction of polychlorinated ethanes andcarbon tetrachloride by structural Fe(II) in smectites. Environ. Sci.Technol. 2009, 43, 4082−4089.(12) Yang, J.; Kukkadapu, R. K.; Dong, H.; Shelobolina, E. S.; Zhang,J.; Kim, J. Effects of redox cycling of iron in nontronite on reduction oftechnetium. Chem. Geol. 2012, 291, 206−216.(13) Dong, H.; Jaisi, D. P.; Kim, J.; Zhang, G. Microbe-clay mineralinteractions. Am. Mineral. 2009, 94, 1505−1519.(14) Gates, W.; Winkinson, H.; Stucki, J. Swelling properties ofmicrobially reduced ferruginous smectite. Clays Clay Miner. 1993, 41,360−364.(15) Shen, S.; Stucki, J.; Boast, C. Effects of structural iron reductionon the hydraulic conductivity of Na-smectite. Clays Clay Miner. 1992,40, 381−386.(16) Stucki, J.; Low, P.; Roth, C.; Golden, D. Effects of oxidation-state of octahedral iron on clay swelling. Clays Clay Miner. 1984, 32,357−362.
(17) Wu, J.; Low, P.; Roth, C. Effects of octahedral-iron reductionand swelling pressure on interlayer distances in Na-nontronite. ClaysClay Miner. 1989, 37, 211−218.(18) Khaled, E.; Stucki, J. Iron oxidation − State effects on cationfixation in smectites. Soil Sci. Soc. Am. J. 1991, 55, 550−554.(19) Anastacio, A. S.; Aouad, A.; Sellin, P.; Fabris, J. D.; Bergaya, F.;Stucki, J. W. Characterization of a redox-modified clay mineral withrespect to its suitability as a barrier in radioactive waste confinement.Appl. Clay Sci. 2008, 39, 172−179.(20) Drits, V. A.; Manceau, A. A model for the mechanism of Fe3+ toFe2+ reduction in dioctahedral smectites. Clays Clay Miner. 2000, 48,185−195.(21) Manceau, A.; Drits, V. A.; Lanson, B.; Chateigner, D.; Wu, J.;Huo, D.; Gates, W. P.; Stucki, J. W. Oxidation-reduction mechanism ofiron in dioctahedral smectites: II. Crystal chemistry of reducedGarfield nontronite. Am. Mineral. 2000, 85, 153−172.(22) Stucki, J. W.; Roth, C. B. Oxidation-reduction mechanism forstructural iron in nontronite. Soil Sci. Soc. Am. J. 1977, 41, 808−814.(23) Lear, P. R.; Stucki, J. W. Role of structural hydrogen in thereduction and reoxidation of iron in nontronite. Clays Clay Miner.1985, 33, 539−545.(24) Favre, F.; Stucki, J.; Boivin, P. Redox properties of structural Fein ferruginous smectite. A discussion of the standard potential and itsenvironmental implications. Clays Clay Miner. 2006, 54, 466−472.(25) Stucki, J. W.; Bailey, G. W.; Gan, H. Oxidation-reductionmechanisms in iron-bearing phyllosilicates. Appl. Clay Sci. 1996, 10,417−430.(26) Schaefer, M. V.; Gorski, C. A.; Scherer, M. M. Spectroscopicevidence for interfacial Fe(II)−Fe(III) electron transfer in a claymineral. Environ. Sci. Technol. 2011, 45, 540−545.(27) Kostka, J. E.; Haefele, E.; Viehweger, R.; Stucki, J. W.Respiration and dissolution of iron(III)-containing clay minerals bybacteria. Environ. Sci. Technol. 1999, 33, 3127−3133.(28) Jaisi, D. P.; Kukkadapu, R. K.; Eberl, D. D.; Dong, H. Control ofFe(III) site occupancy on the rate and extent of microbial reduction ofFe(III) in nontronite. Geochim. Cosmochim. Acta 2005, 69, 5429−5440.(29) Heron, G.; Crouzet, C.; Bourg, A.; Christensen, T. Speciation ofFe(II) and Fe(III) in contaminated aquifer sediments using chemical-extraction techniques. Environ. Sci. Technol. 1994, 28, 1698−1705.(30) Neumann, A.; Hofstetter, T. B.; Lussi, M.; Cirpka, O. A.; Petit,S.; Schwarzenbach, R. P. Assessing the redox reactivity of structuraliron in smectites using nitroaromatic compounds as kinetic probes.Environ. Sci. Technol. 2008, 42, 8381−8387.(31) Ilton, E. S.; Haiduc, A.; Moses, C. O.; Heald, S. M.; Elbert, D.C.; Veblen, D. R. Heterogeneous reduction of uranyl by micas: Crystalchemical and solution controls. Geochim. Cosmochim. Acta 2004, 68,2417−2435.(32) Ilton, E.; Heald, S.; Smith, S.; Elbert, D.; Liu, C. Reduction ofuranyl in the interlayer region of low iron micas under anoxic andaerobic conditions. Environ. Sci. Technol. 2006, 40, 5003−5009.(33) Aeschbacher, M.; Sander, M.; Schwarzenbach, R. P. Novelelectrochemical approach to assess the redox properties of humicsubstances. Environ. Sci. Technol. 2010, 44, 87−93.(34) Aeschbacher, M.; Vergari, D.; Schwarzenbach, R. P.; Sander, M.Electrochemical analysis of proton and electron transfer equilibria ofthe reducible moieties in humic acids. Environ. Sci. Technol. 2011, 45,8385−8394.(35) Aeschbacher, M.; Graf, C.; Schwarzenbach, R. P.; Sander, M.Antioxidant properties of humic substances. Environ. Sci. Technol.2012, 46, 4916−4925.(36) Aeschbacher, M.; Brunner, S. H.; Schwarzenbach, R. P.; Sander,M. Assessing the effect of humic acid redox state on organic pollutantsorption by combined electrochemical reduction and sorptionexperiments. Environ. Sci. Technol. 2012, 46, 3882−3890.(37) Page, S. E.; Sander, M.; Arnold, W. A.; McNeill, K. Hydroxylradical formation upon oxidation of reduced humic acids by oxygen inthe dark. Environ. Sci. Technol. 2012, 46, 1590−1597.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689367

(38) Gorski, C. A.; Klupfel, L.; Voegelin, A.; Sander, M.; Hofstetter,T. B. Redox properties of structural fe in smectites. 2. Electrochemicaland spectroscopic characterization of electron transfer irreversibility inSWa-1. Environ. Sci. Technol. 2012, DOI: 10.1021/es302014u.(39) Homer, R. F.; Tomlinson, T. E. Stereochemistry of the bridgedquaternary salts of 2,2′-bipyridyl. J. Chem. Soc. 1960, 2498−503.(40) Baeyens, B.; Bradbury, M. H. A mechanistic description of Niand Zn sorption on Na-montmorillonite part I: Titration and sorptionmeasurements. J. Contam. Hydrol. 1997, 27, 199−222.(41) Stucki, J. W.; Golden, D. C.; Roth, C. B. Preparation andhandling of dithionite-reduced smectite suspensions. Clays Clay Miner.1984, 32, 191−197.(42) Stucki, J. W. The quantitative assay of minerals for iron(2+) andiron(3+) ions using 1,10-phenanthroline: II. A photochemical method.Soil Sci. Soc. Am. J. 1981, 45, 638−41.(43) Amonette, J. E.; Templeton, J. C. Improvements to thequantitative assay of nonrefractory minerals for Fe(II) and total Feusing 1,10-phenanthroline. Clays Clay Miner. 1998, 46, 51−62.(44) Larese-Casanova, P.; Scherer, M. M. Fe(II) sorption onhematite: New insights based on spectroscopic measurements. Environ.Sci. Technol. 2007, 41, 471−477.(45) Rancourt, D. G.; Ping, J. Y. Voigt-based methods for arbitrary-shape static hyperfine parameter distributions in Mossbauer spectros-copy. Nucl. Instrum. Methods Phys. Res., Sect. B 1991, B58, 85−97.(46) Dyar, M. D.; Schaefer, M. W.; Sklute, E. C.; Bishop, J. L.Mossbauer spectroscopy of phyllosilicates: Effects of fitting models onrecoil-free fractions and redox ratios. Clay Miner. 2008, 43, 3−33.(47) Besson, G.; Bukin, A. S.; Dainyak, L. G.; Rautureau, M.;Tsipurskii, S. I.; Tchoubar, C.; Drits, V. A. Use of diffraction andMoessbauer methods for the structural and crystallochemicalcharacterization of nontronites. J. Appl. Crystallogr. 1983, 16, 374−383.(48) Cashion, J. D.; Gates, W. P.; Thomson, A. Mossbauer and IRanalysis of iron sites in four ferruginous smectites. Clay Miner. 2008,43, 83−93.(49) Goodman, B. A.; Russell, J. D.; Fraser, A. R.; Woodhams, F. W.D. A Mossbauer and I.R. spectroscopic study of the structure ofnontronite. Clays Clay Miner. 1976, 24, 53−59.(50) Cashion, J. D.; Gates, W. P.; Riley, G. M. Origin of the twoquadrupole doublets in NAu-1 nontronite. J. Phys.: Conf. Ser. 2010,217, 012065.(51) Russell, J. D.; Goodman, B. A.; Fraser, A. R. Infrared andMossbauer studies of reduced nontronites. Clays Clay Miner. 1979, 27,63−71.(52) Jaisi, D. P.; Dong, H.; Plymale, A. E.; Fredrickson, J. K.; Zachara,J. M.; Heald, S.; Liu, C. Reduction and long-term immobilization oftechnetium by Fe(II) associated with clay mineral nontronite. Chem.Geol. 2009, 264, 127−138.(53) Luca, V. 57Fe Mossbauer spectroscopic study of structuralchanges during dehydration of nontronite: Effect of differentexchangeable cations. Clays Clay Miner. 1991, 39, 478−489.(54) Fultz, M. L.; Durst, R. A. Mediator compounds for theelectrochemical study of biological redox systems: A compilation. Anal.Chim. Acta 1982, 140, 1−18.(55) Komadel, P.; Madejova, J.; Stucki, J. W. Reduction andreoxidation of nontronite: Questions of reversibility. Clays Clay Miner.1995, 43, 105−10.(56) Neumann, A.; Petit, S.; Hofstetter, T. B. Evaluation of redox-active iron sites in smectites using middle and near infraredspectroscopy. Geochim. Cosmochim. Acta 2011, 75, 2336−2355.(57) Schwarzenbach, R. P.; Gschwend, P. M.; Imboden, D. M.Environmental Organic Chemistry, 2nd ed.; John Wiley & Sons,Hoboken, NJ, 2003.(58) Seabaugh, J.; Dong, H.; Kukkadapu, R.; Eberl, D.; Morton, J.;Kim, J. Microbial reduction of Fe(III) in the fithian and muloorinaillites: Contrasting extents and rates of bioreduction. Clays Clay Miner.2006, 54, 67−79.(59) Watt, G. D.; Burns, A. Thermochemical characterization ofsodium dithionite, flavin mononucleotide, flavin-adenine dinucleotide
and methyl and benzyl viologens as low-potential reductants forbiological-systems. Biochem. J. 1975, 152, 33−37.(60) Jellinek, K. Concenring the electrolytic potential of hydro-sulphite reactions. Z. Elektrochem. Angew. Phys. Chem. 1911, 17, 157−176.(61) Mayhew, S. G. The redox potential of dithionite and SO2− fromequilibrium reactions with flavodoxins, methyl viologen and hydrogenplus hydrogenase. Eur. J. Biochem. 1978, 85, 535−547.(62) Neumann, A.; Sander, M.; Hofstetter, T. B. In Aquatic RedoxChemistry; Tratnyek, P. G., Grundl, T. J., Haderlein, S. B., Eds.;American Chemical Society: Washington, DC, 2011; Vol. 1071;Chapter 18, pp 361−379.(63) Dong, H.; Kostka, J. E.; Kim, J. Microscopic evidence formicrobial dissolution of smectite. Clays Clay Miner. 2003, 51, 502−512.(64) Jaisi, D.; Dong, H.; Morton, J. Partitioning of Fe(II) in reducednontronite (NAu-2) to reactive sites: Reactivity in terms of Tc(VII)reduction. Clays Clay Miner. 2008, 56, 175−189.(65) Mulvaney, P.; Denison, L.; Grieser, F.; Cooper, R.; Sanders, J.V.; Meisel, D. Radiation-induced dissolution of colloidal manganeseoxides. J. Colloid Interface Sci. 1988, 121, 70−80.(66) Mulvaney, P.; Grieser, F.; Swayambunathan, V.; Meisel, D.Electron transfer to iron(III) oxide colloids. Proc. - Electrochem. Soc.1988, 88, 81−89.(67) Mulvaney, P.; Cooper, R.; Grieser, F.; Meisel, D. Chargetrapping in the reductive dissolution of colloidal suspensions ofiron(III) oxides. Langmuir 1988, 4, 1206−1211.(68) Buxton, G. V.; Rhodes, T.; Seller, R. M. Radiation-induceddissolution of colloidal haematite. Nature 1982, 295, 583−585.(69) Buxton, G. V.; Rhodes, T. Seller, R. M. Radiation chemistry ofcolloidal haematite and magnetite in water. Reductive dissolution by(CH3)2C*OH radicals and FeIIEDTA. J. Chem. Soc., Faraday Trans.1983, 79, 2961−2974.(70) Cwiertny, D.; Handler, R.; Schaefer, M.; Grassian, V.; Scherer,M. Interpreting nanoscale size-effects in aggregated Fe-oxidesuspensions: Reaction of Fe(II) with Goethite. Geochim. Cosmochim.Acta 2008, 72, 1365−1380.(71) Mulvaney, P.; Grieser, F.; Meisel, D. In Kinetics and catalysis inmicroheterogeneous systems; Gratzel, M., Kalyanasundaram, K., Dekker,M., Eds.; Taylor & Francis, Inc.: New York, 1991; Vol. 38, pp 303−373.
Environmental Science & Technology Article
dx.doi.org/10.1021/es3020138 | Environ. Sci. Technol. 2012, 46, 9360−93689368

Supporting Information for
Environmental Science and Technology
Redox properties of structural Fe in clay minerals: 1.
Electrochemical quantification of electron donating and accepting
capacities of smectites
Christopher A. Gorski1, Michael Aeschbacher2, Daniela Soltermann2,3, Andreas Voegelin1,
Bart Baeyens3, Maria Marques Fernandes3, Thomas B. Hofstetter1,2,*, Michael Sander2,*
1Swiss Federal Institute of Aquatic Science and Technology, Eawag, Dubendorf,
Switzerland2Swiss Federal Institute of Technology, ETH Zurich, Zurich, Switzerland
3Paul Scherrer Institut (PSI), Villigen, Switzerland
∗Corresponding authors e-mail: [email protected] and
Contents
S1 Chemicals used 2
S2 Triquat synthesis and characterization 3
S3 Additional Mossbauer spectra and hyperfine parameters 6
S4 Triquat sorption data 8
References 11
S1
S1 Chemicals used
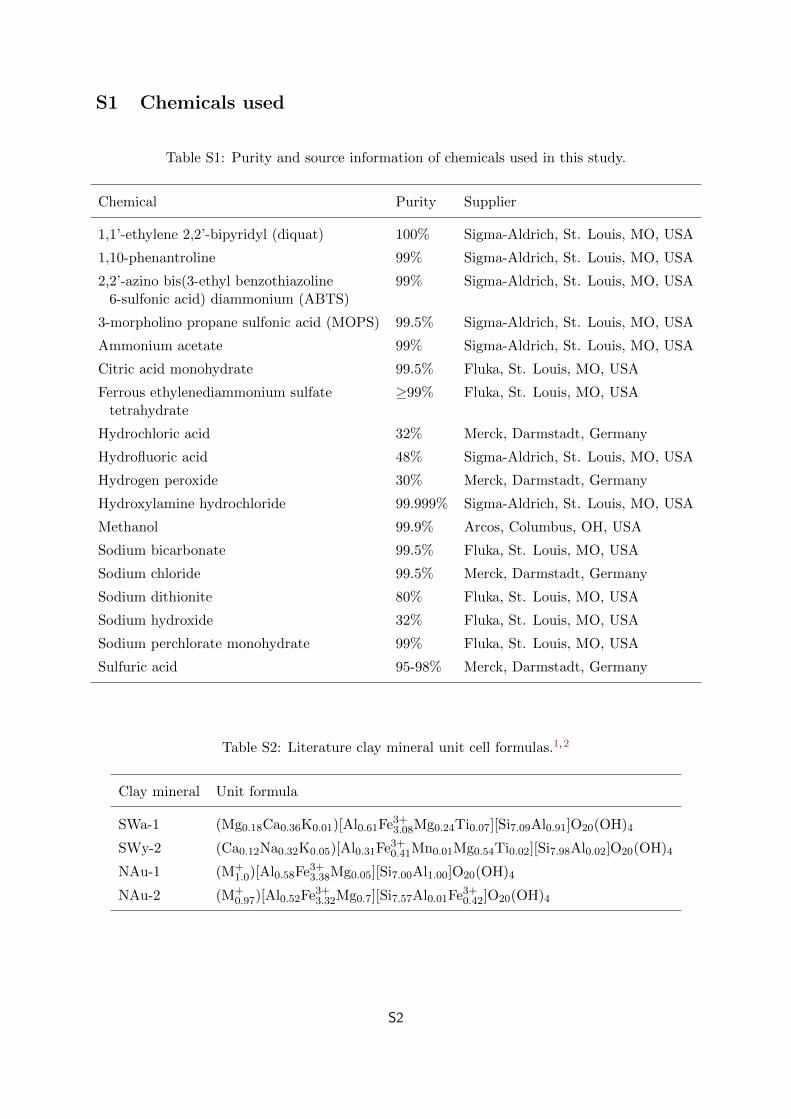
Table S1: Purity and source information of chemicals used in this study.
Chemical Purity Supplier
1,1’-ethylene 2,2’-bipyridyl (diquat) 100% Sigma-Aldrich, St. Louis, MO, USA
1,10-phenantroline 99% Sigma-Aldrich, St. Louis, MO, USA
2,2’-azino bis(3-ethyl benzothiazoline 99% Sigma-Aldrich, St. Louis, MO, USA6-sulfonic acid) diammonium (ABTS)
3-morpholino propane sulfonic acid (MOPS) 99.5% Sigma-Aldrich, St. Louis, MO, USA
Ammonium acetate 99% Sigma-Aldrich, St. Louis, MO, USA
Citric acid monohydrate 99.5% Fluka, St. Louis, MO, USA
Ferrous ethylenediammonium sulfate ≥99% Fluka, St. Louis, MO, USAtetrahydrate
Hydrochloric acid 32% Merck, Darmstadt, Germany
Hydrofluoric acid 48% Sigma-Aldrich, St. Louis, MO, USA
Hydrogen peroxide 30% Merck, Darmstadt, Germany
Hydroxylamine hydrochloride 99.999% Sigma-Aldrich, St. Louis, MO, USA
Methanol 99.9% Arcos, Columbus, OH, USA
Sodium bicarbonate 99.5% Fluka, St. Louis, MO, USA
Sodium chloride 99.5% Merck, Darmstadt, Germany
Sodium dithionite 80% Fluka, St. Louis, MO, USA
Sodium hydroxide 32% Fluka, St. Louis, MO, USA
Sodium perchlorate monohydrate 99% Fluka, St. Louis, MO, USA
Sulfuric acid 95-98% Merck, Darmstadt, Germany
Table S2: Literature clay mineral unit cell formulas.1,2
Clay mineral Unit formula
SWa-1 (Mg0.18Ca0.36K0.01)[Al0.61Fe3+3.08Mg0.24Ti0.07][Si7.09Al0.91]O20(OH)4
SWy-2 (Ca0.12Na0.32K0.05)[Al0.31Fe3+0.41Mn0.01Mg0.54Ti0.02][Si7.98Al0.02]O20(OH)4
NAu-1 (M+1.0)[Al0.58Fe3+3.38Mg0.05][Si7.00Al1.00]O20(OH)4
NAu-2 (M+0.97)[Al0.52Fe3+3.32Mg0.7][Si7.57Al0.01Fe3+0.42]O20(OH)4
S2

S2 Triquat synthesis and characterization
Tiquat (l,l’-trimethylene-2,2’-bipyridyl dibromide) was synthesized following an established method.3
2,2-bipyridyl (6 g = 0.04 mol) (ReagentPlus ≥ 99 %, Sigma-Aldrich) and 1,3-dibromopropane
(20 mL = 5.08 mol) (ReagentPlus ≥ 99 %; Sigma-Aldrich) were heated under reflux for 16 h.
The reaction mixture was cooled, and the formed solid filtered off and washed with acetone
(Chromasolv Plus, for HPLC, ≥ 99.9 %, Sigma-Aldrich) and diethylether (ACS reagent, ≥ 99.0
%, Sigma-Aldich).
The reaction product was dissolved in nanopure water (≥ 18 MΩ·cm; Barnstead NANOpure
System) and recrystallized in an Erlenmeyer flask by addition of ethanol (gradient grade, ≥ 99.8
%; Fluka).4 The recrystallization procedure was repeated twice. Figure S1 shows the triqaut
crystals obtained in the third and final recrystallization step. The final product was air dried
at 110 C for 4 h.
Figure S1: Triquat re-crystallized from a water/ethanol mixture.
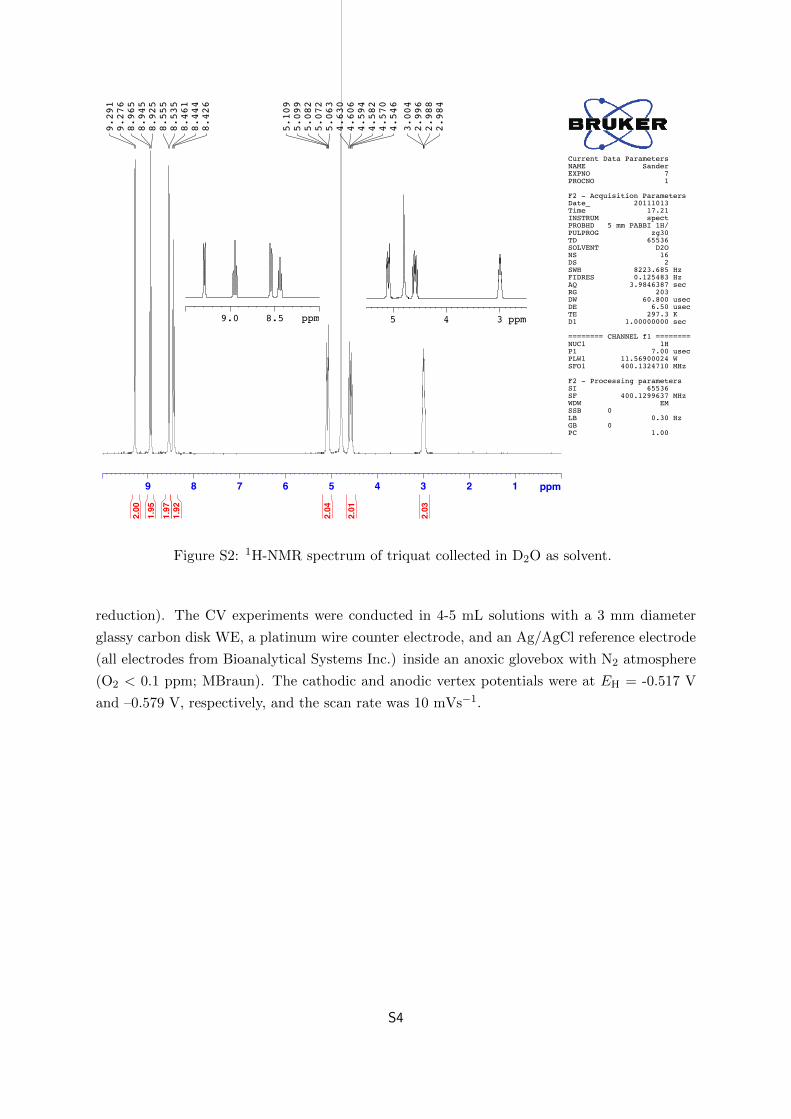
Figure S2 shows the H1 NMR spectrum of the purified triquat in D2O as solvent. The 1H
chemical shifts are: δ 9.28 (d; 2H), 8.95 (t, 2H), 8.54 (d, 2H), 8.44 (t, 2H), 5.08 (2H), 4.59 (2H),
2.99 (2H). These values correspond well to published 1H-NMR data.5
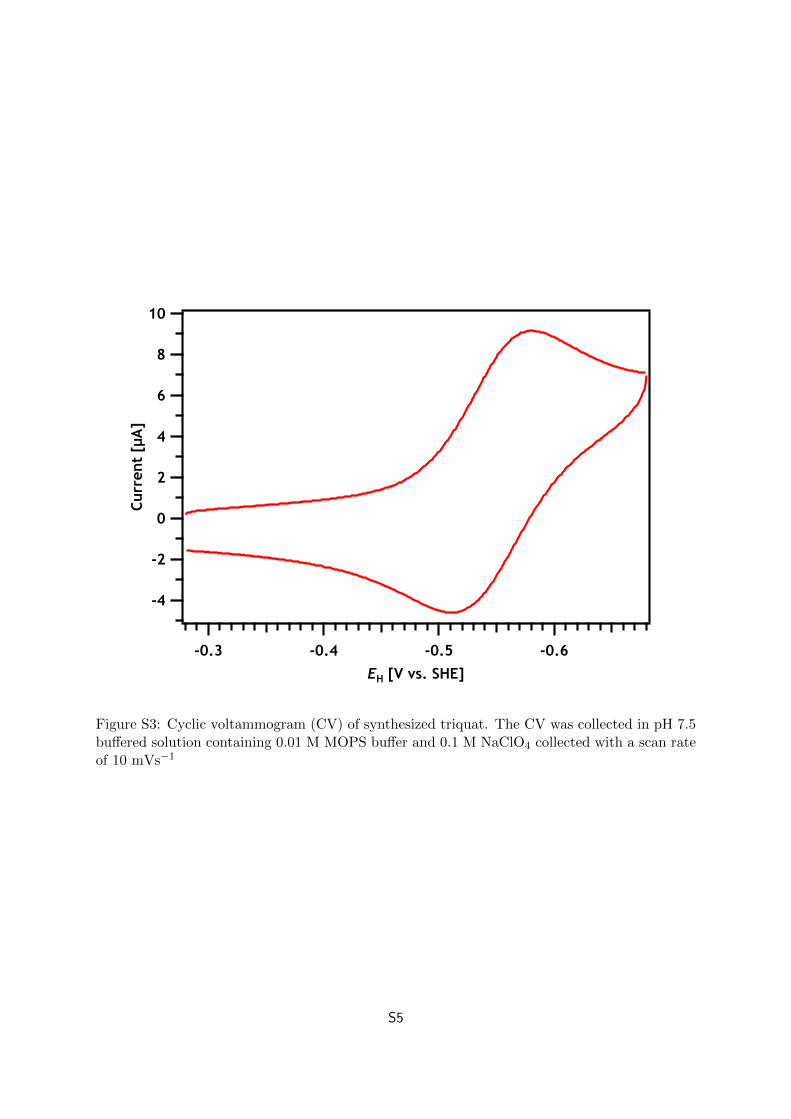
Figure S3 shows the cyclic voltammogram (CV) of the re-crystallized triquat (≈ 0.5 mM)
in pH 7.5 buffer (0.1 M NaClO4, 0.01 M MOPS; pH adjusted by addition of 1 M NaOH). The
CV shows a reversible, one electron reduction with reduction and oxidation waves centered at
EH = −0.54 V, in good agreement with the literature value of the standard reduction potential
E0H = –0.548 V at pH 103 (note that the first electron transfer to triquat is a pH-independent
S3
9 8 7 6 5 4 3 2 1 ppm
2.984
2.988
2.996
3.004
4.546
4.570
4.582
4.594
4.606
4.630
5.063
5.072
5.082
5.099
5.109
8.426
8.444
8.461
8.535
8.555
8.925
8.945
8.965
9.276
9.291
2.03
2.01
2.04
1.92
1.97
1.95
2.00
Current Data ParametersNAME SanderEXPNO 7PROCNO 1
F2 - Acquisition ParametersDate_ 20111013Time 17.21INSTRUM spectPROBHD 5 mm PABBI 1H/PULPROG zg30TD 65536SOLVENT D2ONS 16DS 2SWH 8223.685 HzFIDRES 0.125483 HzAQ 3.9846387 secRG 203DW 60.800 usecDE 6.50 usecTE 297.3 KD1 1.00000000 sec
======== CHANNEL f1 ========NUC1 1HP1 7.00 usecPLW1 11.56900024 WSFO1 400.1324710 MHz
F2 - Processing parametersSI 65536SF 400.1299637 MHzWDW EMSSB 0LB 0.30 HzGB 0PC 1.00
8.59.0 ppm 345 ppm
Figure S2: 1H-NMR spectrum of triquat collected in D2O as solvent.
reduction). The CV experiments were conducted in 4-5 mL solutions with a 3 mm diameter
glassy carbon disk WE, a platinum wire counter electrode, and an Ag/AgCl reference electrode
(all electrodes from Bioanalytical Systems Inc.) inside an anoxic glovebox with N2 atmosphere
(O2 < 0.1 ppm; MBraun). The cathodic and anodic vertex potentials were at EH = -0.517 V
and –0.579 V, respectively, and the scan rate was 10 mVs−1.
S4
10
8
6
4
2
0
-2
-4
Curr
ent
[µA
]
-0.6-0.5-0.4-0.3EH [V vs. SHE]
Figure S3: Cyclic voltammogram (CV) of synthesized triquat. The CV was collected in pH 7.5buffered solution containing 0.01 M MOPS buffer and 0.1 M NaClO4 collected with a scan rateof 10 mVs−1
S5
S3 Additional Mossbauer spectra and hyperfine parameters
Nor
mal
ized
abs
orpt
ion
[arb
. un
its]
-4 -2 0 2 4Velocity [mm/s]
SWa-1
SWy-2
NAu-1
NAu-2
Raw DataOct
Fe3+
fit
Raw DataOct
Fe3+
fit
Raw DataOct
Fe3+
fit
Raw Data
Oct
Fe3+
Oct
Fe3+ [2]
Tet
Fe3+
Total fit
T = 40 K
Figure S4: 57Fe Mossbauer spectra of purified and size-fractionated native SWa-1, SWy-2,NAu-1, and NAu-2 collected at 40 K. Spectra were modeled using Voigt-based fitting.6 Fittedhyperfine parameters from the fits are provided in Table S3.
S6
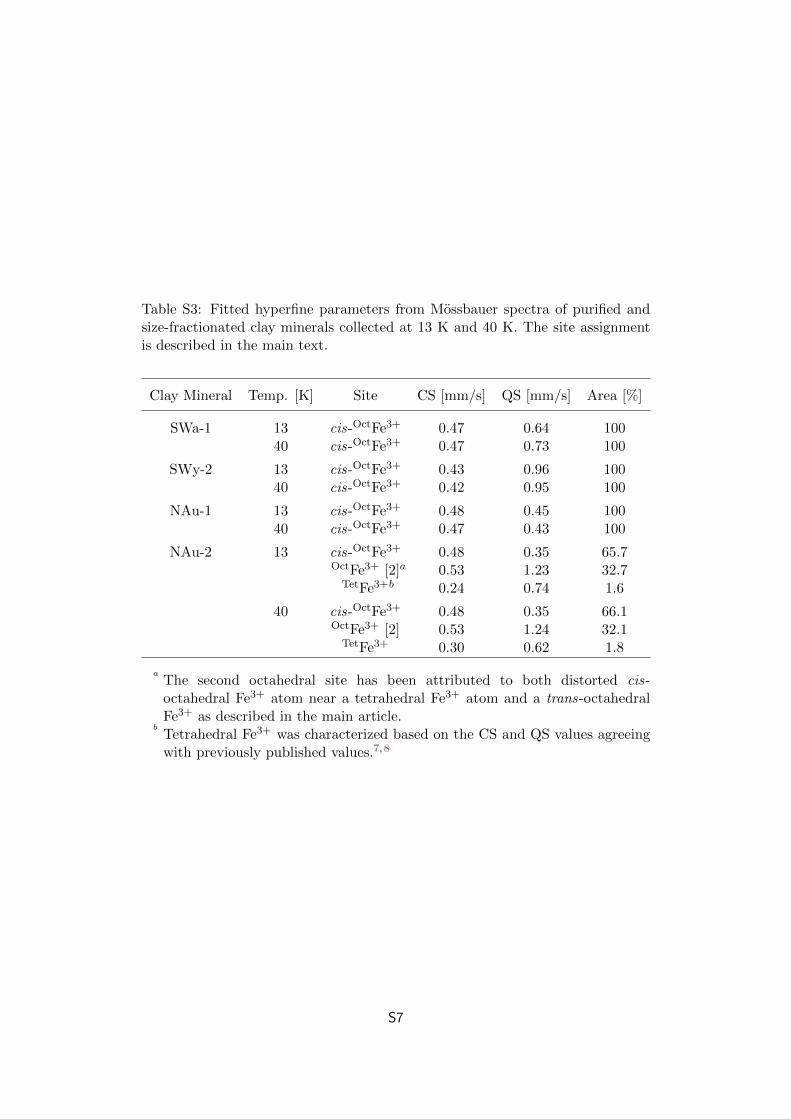
Table S3: Fitted hyperfine parameters from Mossbauer spectra of purified andsize-fractionated clay minerals collected at 13 K and 40 K. The site assignmentis described in the main text.
Clay Mineral Temp. [K] Site CS [mm/s] QS [mm/s] Area [%]
SWa-1 13 cis-OctFe3+ 0.47 0.64 10040 cis-OctFe3+ 0.47 0.73 100
SWy-2 13 cis-OctFe3+ 0.43 0.96 10040 cis-OctFe3+ 0.42 0.95 100
NAu-1 13 cis-OctFe3+ 0.48 0.45 10040 cis-OctFe3+ 0.47 0.43 100
NAu-2 13 cis-OctFe3+ 0.48 0.35 65.7OctFe3+ [2]a 0.53 1.23 32.7
TetFe3+b 0.24 0.74 1.6
40 cis-OctFe3+ 0.48 0.35 66.1OctFe3+ [2] 0.53 1.24 32.1
TetFe3+ 0.30 0.62 1.8
aThe second octahedral site has been attributed to both distorted cis-octahedral Fe3+ atom near a tetrahedral Fe3+ atom and a trans-octahedralFe3+ as described in the main article.
bTetrahedral Fe3+ was characterized based on the CS and QS values agreeingwith previously published values.7,8
S7
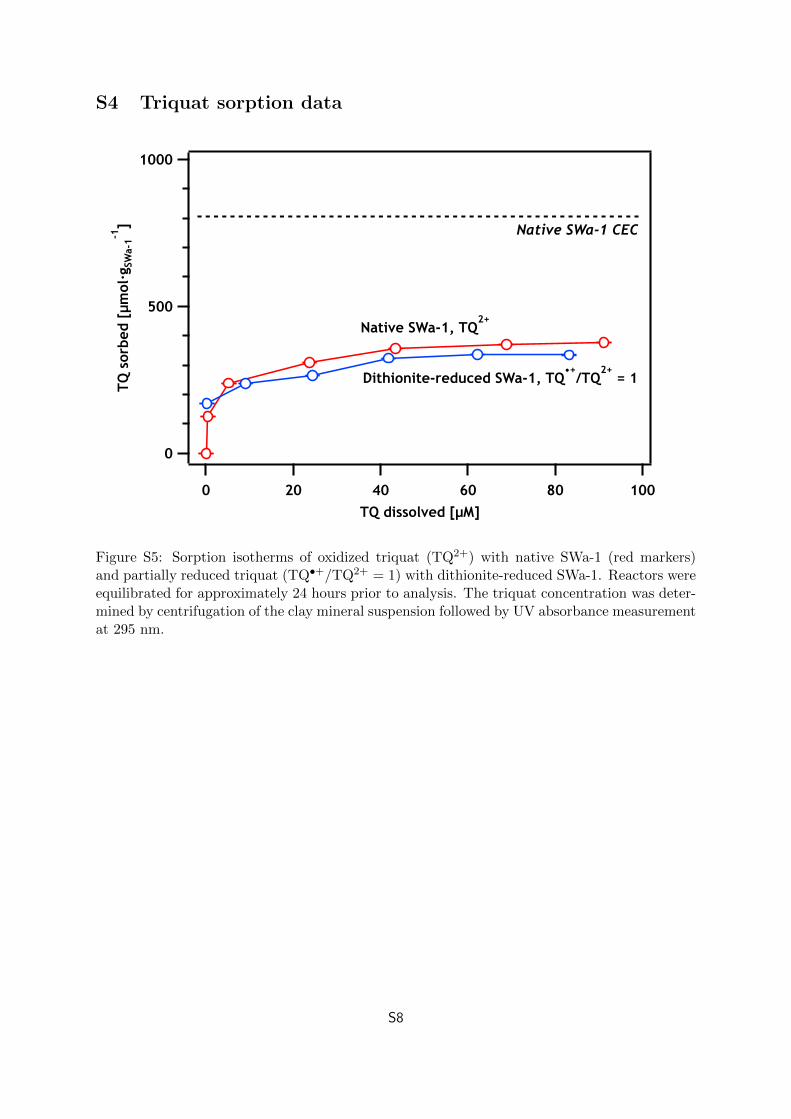
S4 Triquat sorption data
1000
500
0
TQ s
orbe
d [µ
mol·g
SWa-
1-1]
100806040200TQ dissolved [µM]
Native SWa-1 CEC
Native SWa-1, TQ2+
Dithionite-reduced SWa-1, TQ•+
/TQ2+
= 1
Figure S5: Sorption isotherms of oxidized triquat (TQ2+) with native SWa-1 (red markers)and partially reduced triquat (TQ•+/TQ2+ = 1) with dithionite-reduced SWa-1. Reactors wereequilibrated for approximately 24 hours prior to analysis. The triquat concentration was deter-mined by centrifugation of the clay mineral suspension followed by UV absorbance measurementat 295 nm.
S8

Calc. S1 Estimation of possible current response caused by preferential sorp-
tion of one triquat redox state over the other.
Amount of triquat added to the electrochemical cell:
TQtotal = 10 µmol (Calc. S1.1)
Redox conditions:
E0H(TQ) = −0.54 V (Calc. S1.2)
EH(applied) = −0.60 V (Calc. S1.3)
Calculated speciation of TQ:
EH = E0H − 0.0591 · log
[TQ•+
TQ2+
](Calc. S1.4)
TQ•+
TQ2+= 10.4 (Calc. S1.5)
TQ2+init = 0.88 µmol (Calc. S1.6)
TQ•+init = 9.12 µmol (Calc. S1.7)
Max TQ2+ sorption from Figure S5:
TQ2+max sorbed =
375 µmol
gclay(Calc. S1.8)
(1) Current response if TQ2+ preferentially sorbed with a 800 µg SWa-1 addition:
(A larger addition than ever used in experiments)
TQ2+new = 0.88 µmol − 375 µmol
gclay· 800 µgclay = 0.58 µmol (Calc. S1.9)
TQtotal,new = 0.58 µmol + 9.12 µmol = 9.70 µmol (Calc. S1.10)
Number of electrons needed to return to EH = –0.60 V (from Calc. S1.5):
10.4 · TQ2+ = (9.70 − TQ2+) (Calc. S1.11)
TQ2+equil,new = 0.85 µmol (Calc. S1.12)
S9

currenttheor. = TQ2+equil,new − TQ2+
new = 0.85 µmol − 0.58 µmol = 0.27 µmole− (Calc. S1.13)
(2) Current response if TQ•+ preferentially sorbed with a 800µg SWa-1 addition:
(A larger addition than ever used in experiments)
TQ•+new = 9.12 µmol − 375 µmol
gclay· 800 µgclay = 8.82 µmol (Calc. S1.14)
TQtotal,new = 0.88 µmol + 8.82 µmol = 9.70 µmol (Calc. S1.15)
Number of electrons needed to return to EH = –0.60 V (from Calc. S1.5):
TQ•+ = 10.4 · (8.82 µmol − TQ•+) (Calc. S1.16)
TQ•+equil,new = 8.84 µmol (Calc. S1.17)
currenttheor. = TQ•+equil,new − TQ•+
new = 8.84 µmol − 8.82 µmol = 0.02 µmole− (Calc. S1.18)
Electron transfer induced from adding native SWa-1 to the electrochemical cell (Table 1 in text):
QEAC =2210 µmol e−
gclay· (800 µgSWa−1) = 1.77 µmol e− (Calc. S1.19)
The measured current response for an addition of approximately 800 µg SWa-1 was approxi-
mately an order of magnitude larger than the expected current response if TQ2+ preferentially
sorbed and approximately two orders of magnitude larger than the expected current response
if TQ•+ preferentially sorbed.
S10

References
[1] H. Van Olphen and J.J. Fripiat. Data Handbook for Clay Materials and Other Non-Metallic
Minerals. Pergamon Press, Oxford and Elmsford, New York, 1979.
[2] John L. Keeling, Mark D. Raven, and Will P. Gates. Geology and characterization of two
hydrothermal nontronites from weathered metamorphic rocks at the uley graphite mine,
south australia. Clays Clay Miner., 48(5):537–548, 2000.
[3] R. F. Homer and T. E. Tomlinson. Stereochemistry of the bridged quaternary salts of
2,2’-bipyridyl. J. Chem. Soc., pages 2498–2503, 1960.
[4] R.T. Salmon and F.M. Hawkridge. The electrochemical properties of 3 dipyridinium salts
as mediators. J. Electroanal. Chem., 112:253–264, 1980.
[5] D. Wang, W.E. Crowe, R.M. Strongin, and M. Sibrian-Vazquez. Exploring the pH depen-
dence of viologen reduction by alpha-carbon radicals derived from HCy and Cys. Chem.
Commun., (14):1876–1878, 2009.
[6] D. G. Rancourt and J. Y. Ping. Voigt-based methods for arbitrary-shape static hyperfine
parameter distributions in Mossbauer spectroscopy. Nucl. Instrum. Methods Phys. Res.,
Sect. B, B58(1):85–97, 1991.
[7] M. D. Dyar. A review of Moessbauer data on trioctahedral micas: Evidence for tetrahedral
Fe3+ and cation ordering. Am. Mineral., 72(1-2):102–112, 1987.
[8] V. Luca. 57Fe Mossbauer spectroscopic study of structural changes during dehydration of
nontronite: Effect of different exchangeable cations. Clays Clay Miner., 39(5):478–489, 1991.
S11





























