Embed Size (px)
Citation preview
doi.org/10.26434/chemrxiv.12213914.v1
Redox Neutral and Acid-Free Minisci C-H Alkylation of HeteroarenesEnabled by Dual Photoredox/bromide Catalysis in Micellar SolutionsMarila S. Santos, Martyna Cybularczyk-Cecotka, Burkhard Koenig, Maciej Giedyk
Submitted date: 29/04/2020 • Posted date: 30/04/2020Licence: CC BY-NC-ND 4.0Citation information: Santos, Marila S.; Cybularczyk-Cecotka, Martyna; Koenig, Burkhard; Giedyk, Maciej(2020): Redox Neutral and Acid-Free Minisci C-H Alkylation of Heteroarenes Enabled by DualPhotoredox/bromide Catalysis in Micellar Solutions. ChemRxiv. Preprint.https://doi.org/10.26434/chemrxiv.12213914.v1
Microstructured aqueous solutions were employed to engage non-activated alkyl bromides in thevisible-light-promoted C‑H functionalization of heteroarenes. The reactive carbon-centered alkyl radicals weregenerated by merging the photoredox approach, bromide anion co-catalysis and spatial pre-aggregation ofreacting species in the mixture. The presented methodology allowed obtaining alkylated heteroarenes withoutstoichiometric radical-promoters, in acid-free conditions and using blue LEDs as the light source.
File list (2)
download fileview on ChemRxivGiedyk et al.pdf (1.08 MiB)
download fileview on ChemRxivGiedyk et al SI.pdf (3.52 MiB)

1
Redox neutral and acid-free Minisci C-H alkylation of heteroarenes enabled by
dual photoredox/bromide catalysis in micellar solutions
Marilia S. Santos,b† Martyna Cybularczyk-Cecotka,a† Burkhard Königb and Maciej Giedyk*a
a Institute of Organic Chemistry Polish Academy of Sciences Kasprzaka 44/52, 01-224 Warsaw, Poland b Institute of Organic Chemistry, Faculty of Chemistry and Pharmacy, University of Regensburg, Universitätsstraße 31, 93053
Regensburg, Germany. † equal contribution
E-mail: [email protected]
Abstract
Microstructured aqueous solutions were employed to engage non-activated alkyl bromides in the visible-
light-promoted C-H functionalization of heteroarenes. The reactive carbon-centered alkyl radicals were
generated by merging the photoredox approach, bromide anion co-catalysis and spatial pre-aggregation
of reacting species in the mixture. The presented methodology allowed obtaining alkylated heteroarenes
without stoichiometric radical-promoters, in acid-free conditions and using blue LEDs as the light source.
1. Introduction
C-H alkylation of heteroarenes, known as Minisci reaction, is a well-established synthetic tool for
C(sp2)-C(sp3) bond formation.1 It enables direct, late-stage modification of aromatic heterocycles, which
are omnipresent structural motifs in various natural products, pharmaceuticals and agrochemicals.2 The
Minisci reaction involves generation of carbon-centered alkyl radicals and their addition to an
electron-deficient heteroaromatic ring, which is accompanied by a formal loss of the hydrogen atom.
Various precursors of alkyl radicals have been employed in the Minisci reaction including amino acids,3
aldehydes,4–6 ketones,7 carboxylic acids,8–10 alkyltrifluoroborate salts,11 pyridinium salts,12,13 boronic
acids,14,15 diazonium salts,16 peroxides,17–19 alkyl halides20–27 etc. Among them, alkyl bromides are of
particular synthetic potential, as they are readily available and inexpensive starting materials. However,
the cleavage of the relatively strong C−Br bond in alkyl bromides, which must occur in the course of the
process, presents a major challenge. As a result, only few variants of the Minisci reaction exploiting
non-activated alkyl bromides have been reported so far.
Fig. 1. Strategies for C-H alkylation of heteroarenes with non-activated alkyl bromides.
The established strategies to overcome the challenge of C-Br bond activation involve the use of high
temperatures, strong UV-light irradiation or the addition of stoichiometric amounts of silyl radical-promoters
(Fig. 1). Accordingly, in 2012 Hu et al. developed an efficient method for alkylation of benzoxazoles with
secondary alkyl halides.22 The majority of presented syntheses were realized using alkyl iodides, but few

2
examples with bromides have also been reported. The reaction was performed at elevated temperatures,
and with the use of copper-based catalyst. One year later, Fu et al. demonstrated the cross-coupling of
non-activated secondary and tertiary alkyl bromides with pyridine N-oxides in the presence of palladium
catalyst and phosphine ligand.23 The method was further developed by Zhou et al., who extended the
palladium-catalyzed Minisci reaction on a broad variety of heteroarenes, including indole- and pyridine
derivatives.24 The photocatalytic alternative towards the activation of alkyl bromides have been presented
by McCallum and Barriault.25 Using gold complexes as catalysts and UV light as the energy source they
performed Minisci reactions with various non-activated bromoalkanes and heteroarenes and supported
their studies by the detailed investigations of photophysical and electrochemical properties of the
photocatalyst.26 Minisci reactions with non-activated alkyl bromides were also investigated by the groups
of Wang, ElMarrouni and Xu, who capitalized on the joined action of the photocatalyst, acid, silyl radical-
promoters and visible light irradiation.27–29
While these pioneering methods are of unquestionable value, the need for mild, redox-neutral catalytic
methods of C-H alkylation of heteroarenes with alkyl bromides still remains. In order to address this
challenge we resorted to photocatalysis in aqueous structured solutions. We exploited the pre-aggregation
of the reacting species and merged it with the autocatalytic role of bromide anions, which were generated
in situ from the starting material.30 This allowed facilitating the C-C coupling of non-activated alkyl bromides
with heteroarenes without stoichiometric radical-promoters, in acid-free conditions and with commercial
blue LEDs as the light source.
2. Results and discussion
The redox potential of typical photocatalysts in their excited state, including strongly reducing Ir-species,
precludes the direct single-electron-transfer (SET) to alkyl bromides (-2.29 V vs. SCE for
1-bromooctane)31. However, catalytic species of a much higher reducing power can be generated via the
reductive quenching of the catalyst followed by the subsequent excitation with a second visible-light-
photon.30,32–34 Although the typically used reductive quenchers include tertiary amines, Hantzsch esters,
alcohols, ascorbate anions etc.,35 it has recently been shown that the efficient quenching of excited
Ir-complexes can also be achieved using simple halide anions, leading to Ir(II)-species and halide
radicals.36–40 We decided to test, if Br- anions, which are released upon the single-electron-reduction and
fragmentation of alkyl bromides, can be recycled and used as mediators in the Minisci reaction - quench
the excited Ir(III)-photocatalyst and thus promote the generation of alkyl radicals.
Unique properties of structured solutions of surfactants, combined with operationally simple preparation,
render them advantageous media for chemical reactions such as biocatalysis,41 polymerizations,42
transition-metal catalyzed cross-coupling reactions,43 and organocatalytic transformations44. Recent
reports show that they may also play a vital role in photocatalysis.30,45–48 From the viewpoint of the designed
Minisci reaction, structured aqueous solution could provide the necessary pre-association of the starting
materials and the photocatalyst, improve the kinetics of the reaction and thus eliminate the harsh reaction
conditions or stoichiometric additives, including radical promotors and acids.
In order to test the working hypothesis, we subjected bromocyclohexane (2a) to the reaction with lepidine
(1a) in the presence of Ir(dtbby)(ppy)2PF6 (4) as photocatalyst, in aqueous solution of surfactant and under
irradiation with blue LEDs (Table 1). We were pleased to see that the reaction proceeded and the desired
coupling product 3a was formed in 31% yield (entry 1). The reaction parameters were then optimized with
respect to the surfactant, photocatalyst, co-catalyst, time, as well as the ratio and concentration of reagents
(for full optimization studies see SI). The addition of the catalytic amount of NaBr facilitated the process
and increased the yield of compound 3a to 47% (entry 2). Further screening established CBr4 as a
co-catalyst of choice (entries 2-5). The applied conditions, which were called Procedure A, afforded the
full conversion of lepidine 1a and the desired product 3a in 91% yield. The alkylation occurred selectively
at position C2. Although the optimal reaction conditions involved 20 mol% of CBr4 and 42 hours of

3
irradiation, the efficient formation of the product 3a was observed already after shorter reaction time (52%
after 18 h, 78% after 24 h) or using lower co-catalyst loading (5 mol%) (entries 6 and 7, respectively).
Among various tested photocatalysts, the highest activity was achieved using Ir(dtbbpy)(ppy)2PF6 (4).
Other mediators proved inefficient (entries 8, 10-12), or provided the product 3a in low yield (entry 9).
Having catalyst and co-catalyst selected, we evaluated the influence of popular and readily available
surfactants. The superior performance of sulfate-based surfactants: sodium dodecyl sulfate (SDS) and
sodium lauryl oligoethylene glycol sulfate (SLES) was observed (entries 1 - 13), which is in agreement
with previous reports.30 The satisfactory 41% yield of the desired product 3a was also detected when
zwitterionic surfactant SB3-14 was used (entry 15). The application of anionic potassium dodecanate,
cationic dodecyltrimethylammonium chloride (DTAC) or non-ionic Triton X-100 led to less efficient product
3a formation (entries 14, 16, 17).
Table 1. Optimization studiesa
No. Co-catalyst Photocatalyst Time
[h]
Surfactant Yield
3ab [%]
1 - [Ir] 4 42 SDS 31
2 NaBr [Ir] 4 42 SDS 47
3 NBS [Ir] 4 42 SDS 19
4 CCl3Br [Ir] 4 42 SDS 85
5 CBr4 [Ir] 4 42 SDS 91
6 CBr4 [Ir] 4 24 SDS 78
7 CBr4 (5 mol%) [Ir] 4 42 SDS 48
8 CBr4 [Ir] 5 42 SDS 0
9 CBr4 [Ir] 6 42 SDS 16
10 CBr4 Ru(bpy)3PF6 (7) 42 SDS 0
11 CBr4 4CzIPn (8) 42 SDS 0
12 CBr4 Eosin Y (9) 42 SDS 0
13 CBr4 [Ir] 4 42 SLES 48
14 CBr4 [Ir] 4 42 C11H23CO2K 13
15 CBr4 [Ir] 4 42 SB3-14 41
16 CBr4 [Ir] 4 42 Triton X-100 24
17 CBr4 [Ir] 4 42 DTAC 16
aReaction conditions: lepidine 1a (0.1 mmol), bromocyclohexane 2a (0.2 mmol), surfactant (0.25 mmol), co-catalyst (20 mol%),
photocatalyst (3 mol%), water (5 mL), 40 °C, 451 nm, 42 h. bYields were calculated using GC analysis. n-Dodecane was used as
internal standard. [Ir] 4 - Ir(dtbbpy)(ppy)2PF6, [Ir] 5 - Ir(ppy)3PF6, [Ir] 6 - Ir[dF(CF3)(ppy)2](dtbby)PF6.
With the reaction conditions established, we next investigated the scope of the developed transformation
(Table 2). In general, secondary bromides 2a-2d provided higher yields of the desired products than
primary ones 2e-2l, which reflects higher thermodynamic stability of the intermediate radicals. However,
the precursors 2m-o of even more stabilized benzyl, tertiary or α-carbonyl radicals, proved unsuitable,
presumably due to the competing oxidation to carbocations and hydrolysis, which led to respective
alcohols. Several functional groups in the bromide moiety showed good compatibility with our procedure
such as free hydroxyl group (3h), primary (3k) and secondary amides (3l), chlorides (3g) or CF3 function
(3j). Additionally, the product 3i possessing a terminal double bond was also isolated. Alkyl bromides 2p
decorated with acetal groups proved unstable under the reaction conditions. Evaluation of the aromatic
coupling partners showed that the reaction is compatible not only with simple heterocycles such as lepidine
1a, phenantridine 1b and quinoline 1c, but also derivatives, which contain ester or cyano substituents.
4-Phenylpyridine 1d gave a mixture of mono- and disubstituted products 10d and di-10d, both of which
could be selectively isolated (38% and 40% respectively). In the case of nicotinonitrile 1e and
methylnicotinate 1f, the increase in the amount of alkyl bromide (from 3 to 5 equiv.) led to selective
formation of tri-substituted products 10e and tri-10f in very good yields. Alternatively, by keeping the
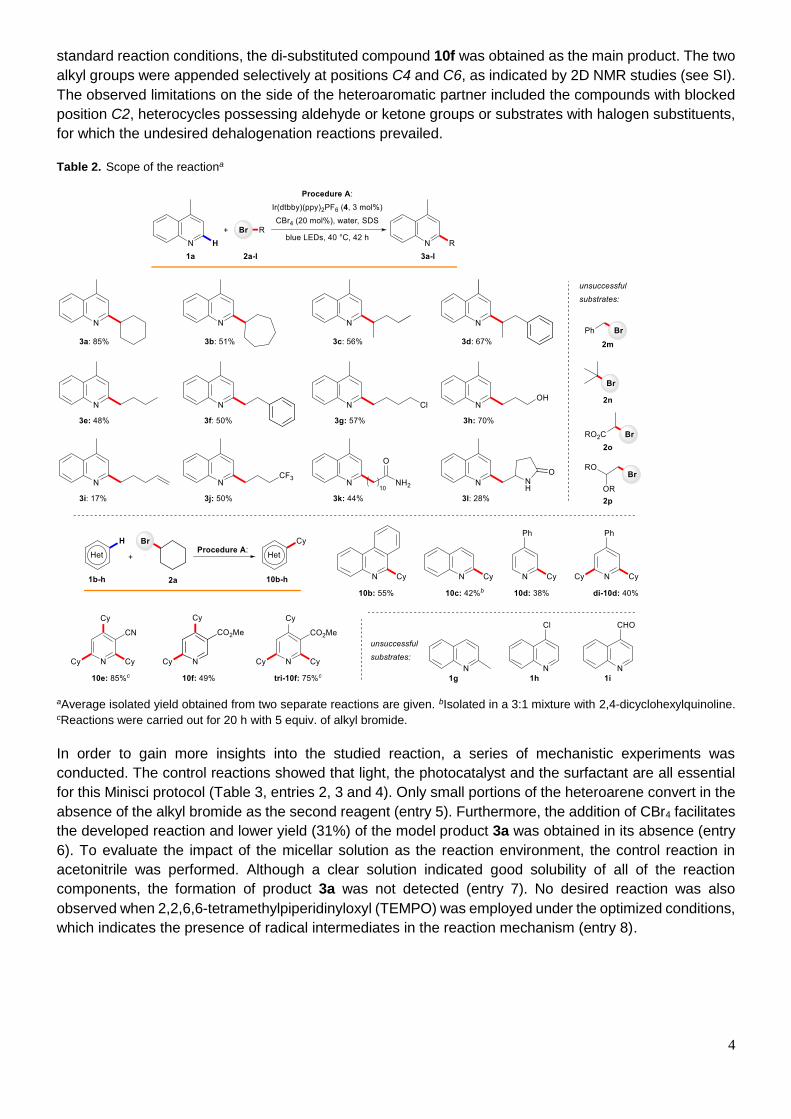
4
standard reaction conditions, the di-substituted compound 10f was obtained as the main product. The two
alkyl groups were appended selectively at positions C4 and C6, as indicated by 2D NMR studies (see SI).
The observed limitations on the side of the heteroaromatic partner included the compounds with blocked
position C2, heterocycles possessing aldehyde or ketone groups or substrates with halogen substituents,
for which the undesired dehalogenation reactions prevailed.
Table 2. Scope of the reactiona
aAverage isolated yield obtained from two separate reactions are given. bIsolated in a 3:1 mixture with 2,4-dicyclohexylquinoline.
cReactions were carried out for 20 h with 5 equiv. of alkyl bromide.
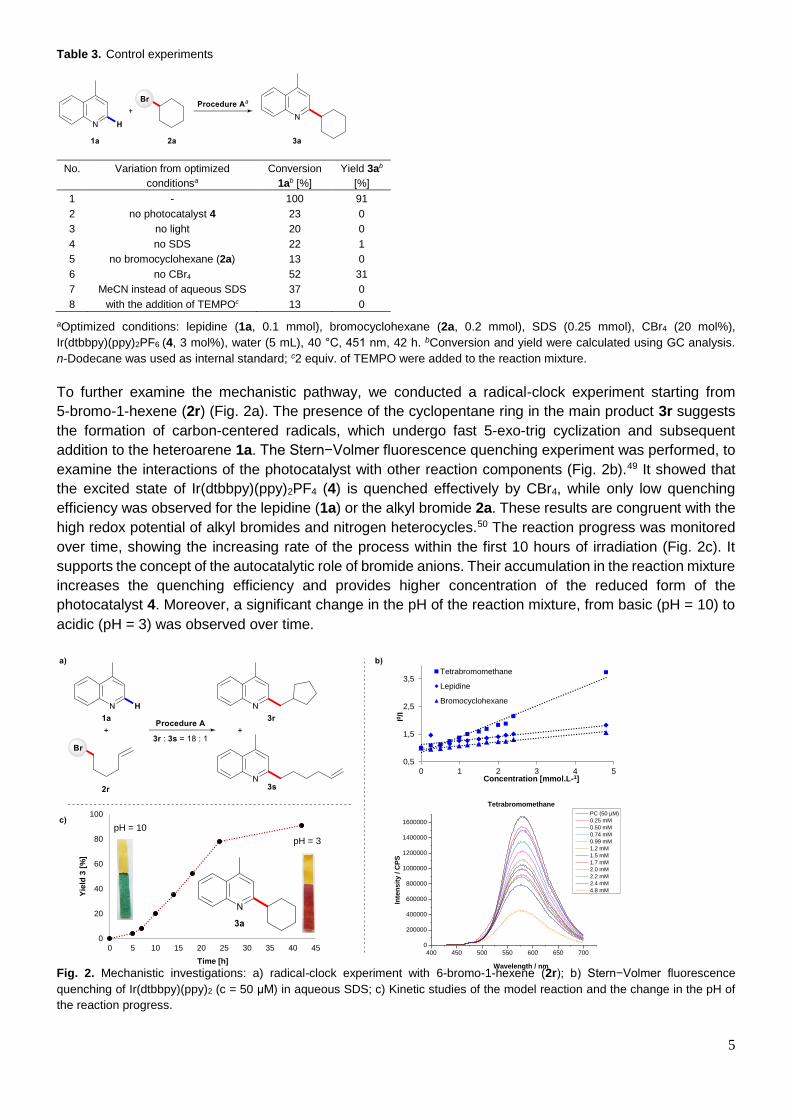
In order to gain more insights into the studied reaction, a series of mechanistic experiments was
conducted. The control reactions showed that light, the photocatalyst and the surfactant are all essential
for this Minisci protocol (Table 3, entries 2, 3 and 4). Only small portions of the heteroarene convert in the
absence of the alkyl bromide as the second reagent (entry 5). Furthermore, the addition of CBr4 facilitates
the developed reaction and lower yield (31%) of the model product 3a was obtained in its absence (entry
6). To evaluate the impact of the micellar solution as the reaction environment, the control reaction in
acetonitrile was performed. Although a clear solution indicated good solubility of all of the reaction
components, the formation of product 3a was not detected (entry 7). No desired reaction was also
observed when 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) was employed under the optimized conditions,
which indicates the presence of radical intermediates in the reaction mechanism (entry 8).
5
Table 3. Control experiments
No. Variation from optimized
conditionsa
Conversion
1ab [%]
Yield 3ab
[%]
1 - 100 91
2 no photocatalyst 4 23 0
3 no light 20 0
4 no SDS 22 1
5 no bromocyclohexane (2a) 13 0
6 no CBr4 52 31
7 MeCN instead of aqueous SDS 37 0
8 with the addition of TEMPOc 13 0
aOptimized conditions: lepidine (1a, 0.1 mmol), bromocyclohexane (2a, 0.2 mmol), SDS (0.25 mmol), CBr4 (20 mol%),
Ir(dtbbpy)(ppy)2PF6 (4, 3 mol%), water (5 mL), 40 °C, 451 nm, 42 h. bConversion and yield were calculated using GC analysis.
n-Dodecane was used as internal standard; c2 equiv. of TEMPO were added to the reaction mixture.
To further examine the mechanistic pathway, we conducted a radical-clock experiment starting from
5-bromo-1-hexene (2r) (Fig. 2a). The presence of the cyclopentane ring in the main product 3r suggests
the formation of carbon-centered radicals, which undergo fast 5-exo-trig cyclization and subsequent
addition to the heteroarene 1a. The Stern−Volmer fluorescence quenching experiment was performed, to
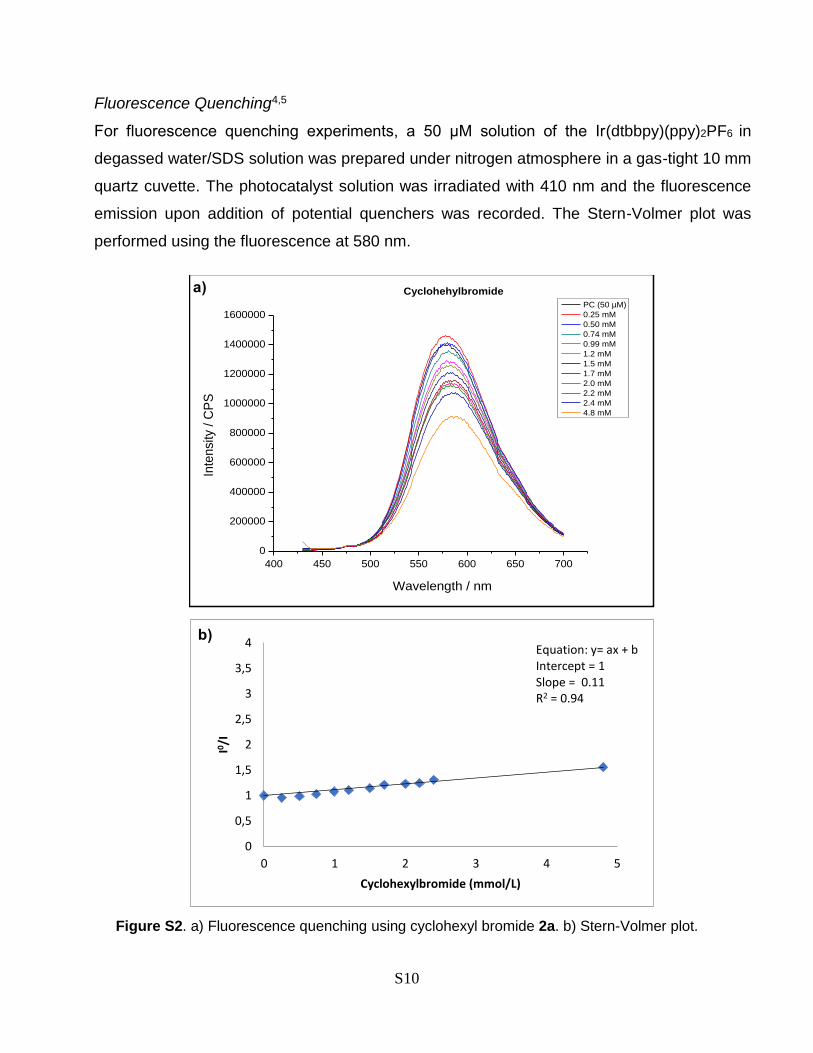
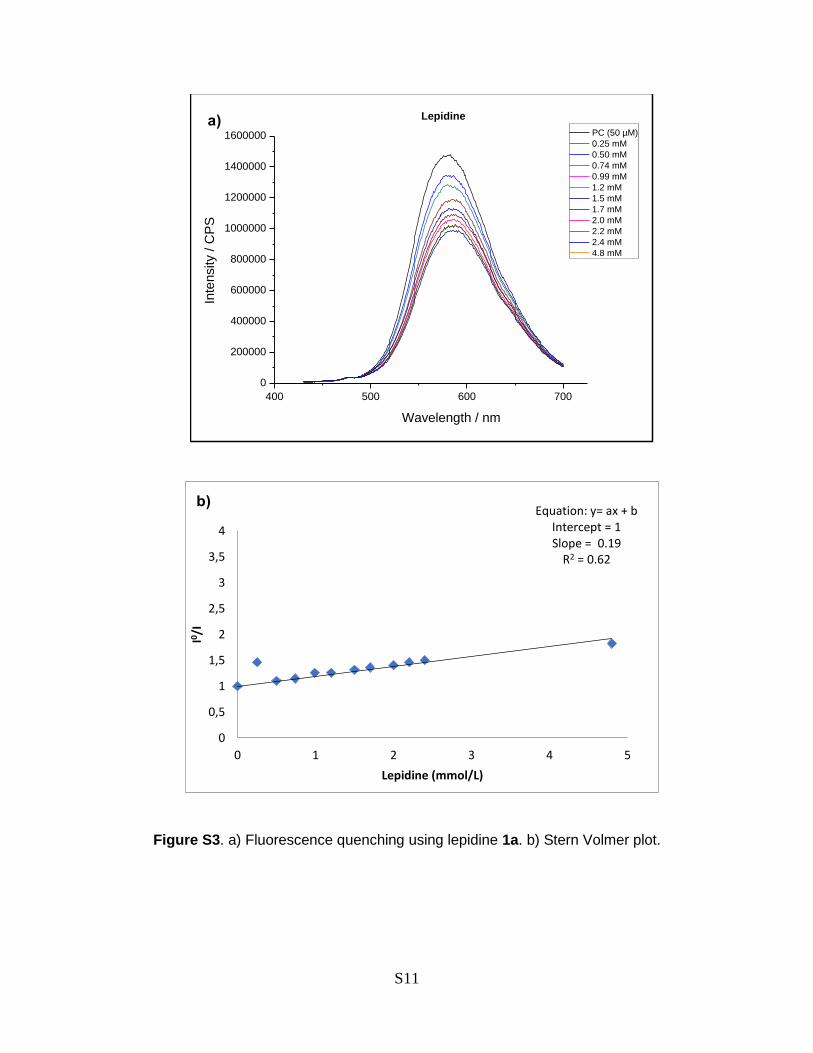
examine the interactions of the photocatalyst with other reaction components (Fig. 2b).49 It showed that
the excited state of Ir(dtbbpy)(ppy)2PF4 (4) is quenched effectively by CBr4, while only low quenching
efficiency was observed for the lepidine (1a) or the alkyl bromide 2a. These results are congruent with the
high redox potential of alkyl bromides and nitrogen heterocycles.50 The reaction progress was monitored
over time, showing the increasing rate of the process within the first 10 hours of irradiation (Fig. 2c). It
supports the concept of the autocatalytic role of bromide anions. Their accumulation in the reaction mixture
increases the quenching efficiency and provides higher concentration of the reduced form of the
photocatalyst 4. Moreover, a significant change in the pH of the reaction mixture, from basic (pH = 10) to
acidic (pH = 3) was observed over time.
Fig. 2. Mechanistic investigations: a) radical-clock experiment with 6-bromo-1-hexene (2r); b) Stern−Volmer fluorescence
quenching of Ir(dtbbpy)(ppy)2 (c = 50 μM) in aqueous SDS; c) Kinetic studies of the model reaction and the change in the pH of
the reaction progress.
0
20
40
60
80
100
0 5 10 15 20 25 30 35 40 45
Yie
ld 3
[%
]
Time [h]
pH = 3
pH = 10
0,5
1,5
2,5
3,5
0 1 2 3 4 5
I0/I
Concentration [mmol.L-1]
Tetrabromomethane
Lepidine
Bromocyclohexane
400 450 500 550 600 650 700
0
200000
400000
600000
800000
1000000
1200000
1400000
1600000
Wavelength / nm
Tetrabromomethane
Inte
nsit
y / C
PS
PC (50 µM)
0.25 mM
0.50 mM
0.74 mM
0.99 mM
1.2 mM
1.5 mM
1.7 mM
2.0 mM
2.2 mM
2.4 mM
4.8 mM

6
In accordance with these results, as well as the optimization studies, we propose a mechanism of the
developed Minisci reaction (Fig. 3). It has been firmly established that the excited [Ir] 4 photocatalyst (E1/2
= -1.51 V vs. SCE in acetonitrile)51 undergoes reductive quenching by Br- anions (E1/2 = +0.80 V vs. SCE
in DME).36–39 Consequently, a bromine radical and the reduced Ir(II)-complex are generated. The latter
species can undergo consecutive absorption of a second photon, resulting in the formation of a strongly
reducing form of the iridium-complex30,32 or a solvated electron.34 SET to alkyl bromide A followed by
fragmentation affords alkyl radical B and a bromide anion, which participates in subsequent catalytic
cycles. An addition of alkyl radical B to pyridinium salt C provides the radical cation D, able to undergo
hydrogen-atom-transfer (HAT) with an electrophilic bromine radical. As a result, the protonated form E of
the final product is produced. Additionally, the contribution of radical propagation through the interaction
of radical cation D with alkyl bromide A should also be considered.
Detailed mechanistic studies on the role of CBr4 co-catalyst are ongoing, but preliminary results suggest
that, through the photosensitized hydrolysis of CBr4, it may provide the starting concentration of bromide
anions at the early stage of the process. Although the light-induced reactivity of this compound is usually
associated with mesolytic bond cleavage,52–55 or homolytic dissociation to CBr3 and Br radicals,56,57 it has
been shown that in the aqueous conditions the photoinduced hydrolytic pathway to HBr prevails.58
Alternatively, the reduction of CBr4 by excited Ir(III)*-photocatalyst can be considered, leading to Br-, the
CBr3 radical and Ir(IV)-complex. The last two species may undergo SET to recover Ir(III) and produce CBr3
cation, which reacts with water to give the tribromomethanol and a proton. Finally, tetrabromomethane
may contribute to the overall reaction outcome through yet another catalytic mode. Due to the halogen
bonding with bromide anions,59,60 it may decrease their hydrophilic character, slow down the migration to
the water bulk and, consequently, render Br- more accessible to the excited Ir(III)* photocatalyst.
Fig. 3. Proposed mechanistic pathway.
To further examine the decisive role of the solution structuring, in particular the postulated
pre-arrangement of bromide anions, we investigated the reaction in the presence of the cationic surfactant
cetyltrimethylammonium bromide (CTAB) and catalytic amount of NaBr instead of CBr4. The positively charged
head of the surfactant would retain the bromide counter-anion through ion-pairing interactions and keep it in a
close distance to the interface, thus favoring the interaction with the photocatalyst. We were pleased to find,
that, under this condition, which were called Procedure B, the compound 3a was obtained in 88% yield (Table
4). Moreover, the use of CTAB as a sole source of bromide anions, without external NaBr added, also afforded
the desired product 3a in good yield (66%). Finally, we demonstrated that the Procedure B can be successfully

7
implemented to obtain alkylated heterocycles 3b, 3h and 10d from other aliphatic bromides and heteroarenes
in good efficiency.
Table 4. The C-H alkylation of heteroarenes using cationic surfactant with bromide counter ion.
aYields were calculated using NMR analysis with 1,3,5-trimethoxybenzene as an internal standard. bThe reaction was performed
in the absence of NaBr.
3. Conclusions
In summary, we have developed a new photocatalytic procedure for Minisci-type coupling of heteroarenes
with various alkyl bromides, which exploits the combination of photoredox catalysis with bromide anion
catalysis. With the use of micellar solution as the reaction media, it is possible to carry out the reaction in
mild, aqueous conditions, with no need for external oxidant or stoichiometric radical promoter. The
coupling products were obtained in the absence of equimolar amounts of acid, a requirement for standard
Minisci protocols. The external additives are simple and cost-efficient and they were used in catalytic
amounts. The obtained optimization data and mechanistic experiments highlight the critical importance of
microstructuring and pre-organization of the components in the reaction mixture.
4. Conflicts of interest
There are no conflicts to declare
5. Acknowledgements
We gratefully acknowledge funding from the National Science Centre, Poland
(SONATA 2018/31/D/ST5/00306), Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP
(2018/20956-5 and 2017/03120-8) and the German Science Foundation (DFG, KO 1537/18-1) for the
financial support.
TOC

8
6. Notes and references
1. R. S. J. Proctor and R. J. Phipps, Angew. Chemie Int. Ed., 2019, 2–36. 2. K. C. Majumdar and S. K. Chattopadhyay, Eds., Heterocycles in Natural Product Synthesis, Wiley-VCH Verlag GmbH & Co.
KGaA, Weinheim, Germany, 2011. 3. D. N. Mai and R. D. Baxter, Org. Lett., 2016, 18, 3738–3741. 4. K. Matcha and A. P. Antonchick, Angew. Chemie - Int. Ed., 2013, 52, 2082–2086. 5. Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856–3865. 6. Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512–5516. 7. P. Liu, W. Liu and C. J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321. 8. W.-F. Tian, C.-H. Hu, K.-H. He, X.-Y. He and Y. Li, Org. Lett., 2019, 21, 6930–6935. 9. F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579. 10. J. Kan, S. Huang, J. Lin, M. Zhang and W. Su, Angew. Chemie - Int. Ed., 2015, 54, 2199–2203. 11. G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855. 12. F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chemie - Int. Ed., 2017, 56, 12336–12339. 13. X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 8718–8721. 14. G. X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412. 15. I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–
13196. 16. D. Xue, Z. H. Jia, C. J. Zhao, Y. Y. Zhang, C. Wang and J. Xiao, Chem. - A Eur. J., 2014, 20, 2960–2965. 17. D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chemie - Int. Ed., 2014, 53, 4802–4806. 18. F. Minisci, C. Giordano, E. Vismara, S. Levi and V. Tortelli, J. Am. Chem. Soc., 1984, 106, 7146–7150. 19. F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 4411–4416. 20. N. B. Bissonnette, M. J. Boyd, G. D. May, S. Giroux and P. Nuhant, J. Org. Chem., 2018, 83, 10933–10940. 21. P. Nuhant, M. S. Oderinde, J. Genovino, A. Juneau, Y. Gagné, C. Allais, G. M. Chinigo, C. Choi, N. W. Sach, L. Bernier, Y. M.
Fobian, M. W. Bundesmann, B. Khunte, M. Frenette and O. O. Fadeyi, Angew. Chemie - Int. Ed., 2017, 56, 15309–15313. 22. P. Ren, I. Salihu, R. Scopelliti and X. Hu, Org. Lett., 2012, 14, 1748–1751. 23. B. Xiao, Z. J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616–619. 24. X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. S. Zhou, Angew. Chemie, 2014, 126, 13791–13795. 25. T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758. 26. C. D. McTiernan, M. Morin, T. McCallum, J. C. Scaiano and L. Barriault, Catal. Sci. Technol., 2016, 6, 201–207. 27. J. Dong, X. Lyu, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2019, 10, 976–982. 28. J. J. Perkins, J. W. Schubert, E. C. Streckfuss, J. Balsells and A. ElMarrouni, European J. Org. Chem., 2019, 4, 2–10. 29. R. Chang, J. Fang, J.-Q. Chen, D. Liu, G.-Q. Xu and P.-F. Xu, ACS Omega, 2019, 4, 14021–14031. 30. M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47. 31. P. W. Jennings, D. G. Pillsbury, J. L. Hall and V. T. Brice, J. Org. Chem., 1976, 41, 719–722. 32. T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D.
Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658. 33. I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science (80-. )., 2014, 346, 725 LP – 728. 34. F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chemie, , DOI:10.1002/ange.201915762. 35. D. Petzold, M. Giedyk, A. Chatterjee and B. König, European J. Org. Chem., 2020, 2020, 1193–1244. 36. P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087. 37. T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370. 38. Z. Wang, X. Ji, T. Han, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2019, 361, 5643–5647. 39. Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159. 40. M. Zidan, A. O. Morris, T. McCallum and L. Barriault, European J. Org. Chem., , DOI:10.1002/ejoc.201900786. 41. M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J. Y. Berthon and B. H. Lipshutz, Nat. Commun.,
2019, 10, 1–10. 42. I. Capek and T. Kocsisova, Des. Monomers Polym., 2011, 14, 327–345. 43. S. Handa, B. Jin, P. P. Bora, Y. Wang, X. Zhang, F. Gallou, J. Reilly and B. H. Lipshutz, ACS Catal., 2019, 9, 2423–2431. 44. L. Baxová, R. Cibulka and F. Hampl, J. Mol. Catal. A Chem., 2007, 277, 53–60. 45. M. Bu, G. Lu, J. Jiang and C. Cai, Catal. Sci. Technol., 2018, 8, 3728–3732. 46. T. Kohlmann, C. Kerzig and M. Goez, Chem. – A Eur. J., 2019, 25, 9991–9996. 47. C. Kerzig and M. Goez, Chem. Sci., 2016, 7, 3862–3868. 48. R. Naumann, F. Lehmann and M. Goez, Angew. Chemie Int. Ed., 2018, 57, 1078–1081. 49. M. H. Gehlen, J. Photochem. Photobiol. C Photochem. Rev., 2020, 42, 100338. 50. H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723. 51. C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363. 52. M. J. W. Taylor, W. T. Eckenhoff and T. Pintauer, Dalt. Trans., 2010, 39, 11475–11482. 53. M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – A Eur. J., 2012, 18, 7336–7340. 54. M. N. C. Balili and T. Pintauer, Dalt. Trans., 2011, 40, 3060–3066. 55. T. Hou, P. Lu and P. Li, Tetrahedron Lett., 2016, 57, 2273–2276. 56. Q. Kong, M. Wulff, J. H. Lee, S. Bratos and H. Ihee, J. Am. Chem. Soc., 2007, 129, 13584–13591. 57. S. Tripathi, S. N. Singh and L. D. S. Yadav, RSC Adv., 2016, 6, 14547–14551. 58. C. Zhao, X. Lin, W. M. Kwok, X. Guan, Y. Du, D. Wang, K. F. Hung and D. L. Phillips, Chem. - A Eur. J., 2005, 11, 1093–1108. 59. G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601. 60. T. Brinck and A. N. Borrfors, J. Mol. Model., , DOI:10.1007/s00894-019-4014-7.

download fileview on ChemRxivGiedyk et al.pdf (1.08 MiB)

S1
Supporting Information
Redox-neutral and acid-free Minisci C-H alkylation of heteroarenes
enabled by dual photoredox/bromide catalysis in micellar solutions
Marilia S. Santos,b† Martyna Cybularczyk-Cecotka,a† Burkhard Königb and Maciej Giedyk*a
aInstitute of Organic Chemistry Polish Academy of Sciences Kasprzaka 44/52, 01-224 Warsaw, Poland bInstitute of Organic Chemistry, Faculty of Chemistry and Pharmacy, University of Regensburg, Universitätsstraße 31,
93053 Regensburg, Germany. † equal contribution
E-mail: [email protected]
Table of Contents
GENERAL INFORMATION ....................................................................................................................... 2
EXPERIMENTAL SECTION ..................................................................................................................... 3
General Procedure for Minisci Reaction mediated by CBr4 (Procedure A) ....................................... 3
General Procedure for Minisci Reaction mediated by NaBr (Procedure B) ...................................... 4
Optimization studies ................................................................................................................................... 5
Mechanistic experiments ........................................................................................................................... 8
Characterization data of the compounds .............................................................................................. 13
REFERENCES .......................................................................................................................................... 23
NMR SPECTRAL DATA .......................................................................................................................... 24

S2
GENERAL INFORMATION
All reagents and solvents were purchased from commercial suppliers and used without
further purification unless otherwise stated or purified according to the procedures outlined
in Purification of Common Laboratory Chemicals.1 Thin-layer chromatography (TLC) was
performed on aluminum silica gel plates 60 F254 (MN TLC sheets ALUGRAM® Xtra SIL
G/UV254) and the visualization was accomplished by irradiation at 254 nm and staining
with the solution of ethanolic phosphomolybdic acid and potassium permanganate. All
aqueous solutions were prepared using distilled water. Saturated brine refers to an
aqueous saturated sodium chloride solution. All products were purified by
chromatography column of silica gel 60 M (40-63 µm, 230-440 mesh). NMR spectra were
recorded at room temperature in CDCl3 and (CD3)2CO using Varian 500 MHz, Varian 600
MHz, Bruker 500 MHz, Bruker 400 MHz, and Bruker Avance 300 MHz spectrometers.
Chemical shifts are reported relatively in δ-scale as parts per million (ppm) referenced to
the residual solvent peak (Chloroform: 7.26 ppm for 1H NMR, and 77.16 ppm for 13C NMR;
acetone: 2.05 ppm for 1H NMR and 29.84 and 206.26 for 13C NMR).2 Coupling constants
J are given in Hertz (Hz) and the following abbreviations were used for indicating signal
multiplicity: 1H NMR: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and the
respective combinations.
Gas chromatography coupled with a flame ionization detector (GC-FID) was performed
on an Agilent 7890 GC SystemGC and Shimadzu GCMS-QP2010 SE with helium as the
carrier gas. Capillary column: length: 30 m; diam.: 0.25 mm; film: 0.25 µm. High-resolution
mass spectra (HRMS) were recorded on Waters SYNAPT G2-S HDMS instrument using
electrospray ionization (ESI) with the time of flight detector (TOF).
Photocatalytic reactions were performed using TAK 120 photoreactor equipped with blue
LEDs (OSRAM OSLON® SSL 80 GD CS8PM1.14 LEDs (λ= 451 nm (± 15 nm), 800 mW
optical power), unless stated otherwise. The optical power of LEDs was determined using
FieldMaxII-TOTM laser power meter equipped with PM3 sensor (Figure S1).

S3
Figure S1. The reaction vials (10 mL crimp cap vials) were illuminated from the bottom with blue
LEDs and cooled from the side using a custom-made fan.
EXPERIMENTAL SECTION
General Procedure for Minisci Reaction mediated by CBr4 (Procedure A)
10 mL crimp vial, equipped with a magnetic stirring bar was charged with
Ir(dtbby)(ppy)2PF6 (2.7 mg, 3 mol%), SDS (72.1 mg, 0.25 mmol) and CBr4 (6.6 mg, 0.02
mmol). The vial was sealed and degassed via two pump-argon cycles, followed by
degassed water (5 mL) addition. The resulting mixture was degassed via five pump-argon
cycles, the heterocycle (0.10 mmol) and alkyl bromide (0.20 mmol) were added under
argon and the mixture was degassed again via two pump-argon cycles, keeping the
vacuum above 50 mbar. The reaction mixture was irradiated with 800 mW 451 nm LEDs
through the plane bottom side and stirred intensely for 42 h. The temperature was
maintained at 40 °C to 42 °C by cooling with the built-in cooling fan. Then the vial was
opened and the crude reaction mixture was transferred to a separatory funnel. Solution of
KHCO3 (1 M, 5 mL) and brine (20 mL) were added and the mixture was extracted with
AcOEt (3 x 20 mL). Combined organic fractions were washed with fresh brine, dried over
Na2SO4, filtrated and concentrated in vacuo. A crude product was purified by flash column
chromatography on silica gel.

S4
General Procedure for Minisci Reaction mediated by NaBr (Procedure B)
10 mL crimp vial, equipped with a magnetic stirring bar was charged with
Ir(dtbby)(ppy)2PF6 (2.7 mg, 3 mol%), CTAB (91.1 mg, 0.25 mmol) and NaBr (2.1 mg, 0.02
mmol). The vial was sealed and degassed via two pump-argon cycles, followed by
degassed water (5 mL) addition. The resulting mixture was degassed via five pump-argon
cycles, the heterocycle (0.10 mmol) and alkyl bromide (0.20 mmol) were added under
argon and the mixture was degassed again via two pump-argon cycles, keeping the
vacuum above 50 mbar. The reaction mixture was irradiated with 800 mW 451 nm LEDs
through the plane bottom side and stirred intensely for 42 h. The temperature was
maintained at 40 °C to 42 °C by cooling with the built-in cooling fan. Then the vial was
opened and the crude reaction mixture was transferred to a separatory funnel. Solution of
NaHCO3 (1 M, 5 mL) and brine (20 mL) were added and the mixture was extracted with
AcOEt (3 x 20 mL). Combined organic fractions were washed with fresh brine, dried over
Na2SO4, filtrated and concentrated in vacuo.

S5
Optimization studies
The reactions were carried out according to the general procedure.
Table S1. Evaluation of reaction conditions – part I: preliminary screening
Noa Additive Water [mL]
Photocatalyst loading [mol%]
Surfactant Time [h]
Atm. Bromide
2a [equiv.]
Additive [mol%]
Yieldb [%]
1 NaBr 3 1 SLES 19 Ar 2 20 12
2 NaBr + DIPEA 3 1 SLES 19 Ar 2 20 each 21
3 NaBr + H2Asc 3 1 SLES 19 Ar 2 20 each 0
4 NaBr 5 1 SLES 19 Ar 2 20 18
5 NaBr 5 3 SLES 19 Ar 2 20 24
6 TBAB 5 3 SLES 19 Ar 2 20 21
7 NaBr 5 3 SLES 19 Ar 2 20 24
8 NaBr 5 3 SLES 42 Ar 2 20 34
9 NaBr 5 3 SDS 42 Ar 2 20 47
10c NaBr 5 3 SDS 42 Ar 2 20 24
11 NaBr 5 3 SDS 42 Air 2 20 32
12 NaBr 5 3 SDS 42 Ar 1.2 20 13
13 NaBr 5 3 SDS 42 Ar 3 20 19
14 NaBr 5 3 SDS 42 Ar 2 10 17
15 NaBr 5 3 SDS 42 Ar 2 50 31
16 NaBr 5 3 SDS 42 Ar 2 100 48
17 NaBr 5 3 SDS 42 Ar 2 250 51
18 NaBr 3 3 SDS 42 Ar 2 20 23
19 NaBr 5 2 SDS 42 Ar 2 20 24 aReaction conditions: lepidine 1a (0.1 mmol), cyclohexylbromide 2a (0.2 mmol), Ir(ppy)2(dtbby)PF6, surfactant (0.25
mmol), additive, water, 40 °C, 451 nm (800 mW – optical power). bYields and conversions calculated from GC
measurements using dodecane as an internal standard. cViolet LEDs (400 nm, 160 mW measured optical power) were
used instead of blue LEDs (451 nm, 800 mW measured optical power).
S6
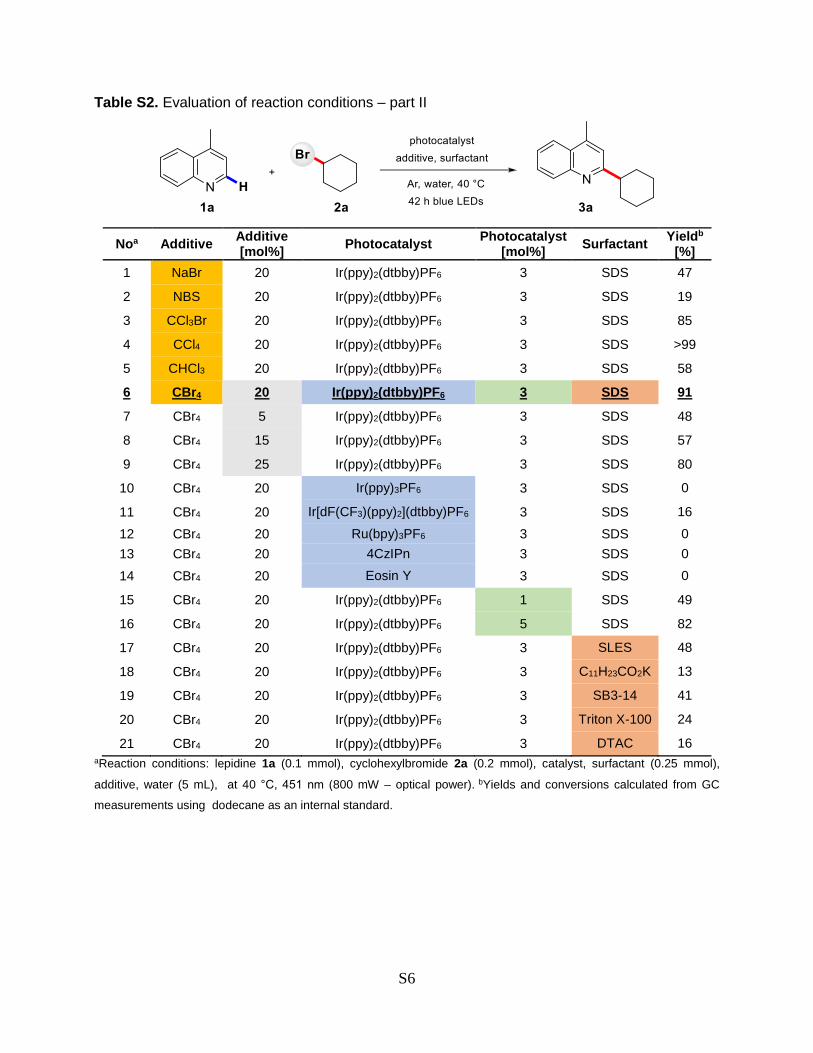
Table S2. Evaluation of reaction conditions – part II
Noa Additive Additive [mol%]
Photocatalyst Photocatalyst
[mol%] Surfactant
Yieldb [%]
1 NaBr 20 Ir(ppy)2(dtbby)PF6 3 SDS 47
2 NBS 20 Ir(ppy)2(dtbby)PF6 3 SDS 19
3 CCl3Br 20 Ir(ppy)2(dtbby)PF6 3 SDS 85
4 CCl4 20 Ir(ppy)2(dtbby)PF6 3 SDS >99
5 CHCl3 20 Ir(ppy)2(dtbby)PF6 3 SDS 58
6 CBr4 20 Ir(ppy)2(dtbby)PF6 3 SDS 91
7 CBr4 5 Ir(ppy)2(dtbby)PF6 3 SDS 48
8 CBr4 15 Ir(ppy)2(dtbby)PF6 3 SDS 57
9 CBr4 25 Ir(ppy)2(dtbby)PF6 3 SDS 80
10 CBr4 20 Ir(ppy)3PF6 3 SDS 0
11 CBr4 20 Ir[dF(CF3)(ppy)2](dtbby)PF6 3 SDS 16
12 CBr4 20 Ru(bpy)3PF6 3 SDS 0
13 CBr4 20 4CzIPn 3 SDS 0
14 CBr4 20 Eosin Y 3 SDS 0
15 CBr4 20 Ir(ppy)2(dtbby)PF6 1 SDS 49
16 CBr4 20 Ir(ppy)2(dtbby)PF6 5 SDS 82
17 CBr4 20 Ir(ppy)2(dtbby)PF6 3 SLES 48
18 CBr4 20 Ir(ppy)2(dtbby)PF6 3 C11H23CO2K 13
19 CBr4 20 Ir(ppy)2(dtbby)PF6 3 SB3-14 41
20 CBr4 20 Ir(ppy)2(dtbby)PF6 3 Triton X-100 24
21 CBr4 20 Ir(ppy)2(dtbby)PF6 3 DTAC 16
aReaction conditions: lepidine 1a (0.1 mmol), cyclohexylbromide 2a (0.2 mmol), catalyst, surfactant (0.25 mmol),
additive, water (5 mL), at 40 °C, 451 nm (800 mW – optical power). bYields and conversions calculated from GC
measurements using dodecane as an internal standard.

S7
Table S3. Evaluation of reaction conditions – part III
Noa Lepidine 1a [mmol]
Bromocyclohexane 2a [mmol]
Water [mL]
Time [h] Yieldb
[%]
1 0.1 0.2 5 42 91
2 0.1 0.2 3 42 37
3 0.1 0.1 5 42 26
4 0.2 0.1 5 42 traces
5 0.1 0.2 5 24 78
6 0.1 0.2 5 18 52
7 0.1 0.2 5 14 35
8 0.1 0.2 5 10 20
9 0.1 0.2 5 7 8
10 0.1 0.2 5 5 4 aReaction conditions: lepidine 1a, bromocyclohexane 2a, SDS (0.25 mmol), CBr4 (20 mol%), Ir(dtbbpy)(ppy)2PF6 (3
mol%), water, 40 °C, 451 nm, 42 h. bYield was calculated using GC analysis. n-Dodecane was used as internal standard.
Table S4. Control experiments
Noa Variation from optimized conditionsa Conversion 1ab [%]
Yieldb [%]
1 - 100 91
2 no photocatalyst 23 0
3 no light 20 0
4 no SDS 22 1
5 no bromocyclohexane (2a) 13 0
6 no CBr4 52 31
7 MeCN instead of aqueous SDS 37 0
8 with the addition of TEMPOb 13 0
aReaction conditions: lepidine 1a (0.1 mmol), bromocyclohexane 2a (0.2 mmol), SDS (0.25 mmol), CBr4 (20 mol%),
Ir(dtbbpy)(ppy)2PF6 (3 mol%), water (5 mL), 40 °C, 451 nm, 42 h. bConversion and yield were calculated using GC
analysis. n-Dodecane was used as internal standard; c2 equiv. of TEMPO were added to the reaction mixture.

S8
Mechanistic experiments
Radical clock reaction
The radical clock reaction described in the results and discussion section was first
analysed by GC-MS. The chromatogram of a crude reaction mixture revealed the
presence of two products, whose mass corresponds to the alkylated heteroarenes 3r and
3s. The product 3r was then isolated and the 1H NMR spectrum confirmed its structure
(consistent with the literature data3) thus proving the radical mechanism of the reaction.
GC-MS of a crude reaction mixture:
60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 2900
5
10
15
20
25
30%
157
170
184
11577143
12822489
19798277 297210 29025424670 230
60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 3000.0
2.5
5.0
7.5
%157
115
77142 182128
22419616791
21010270
297233 284244 256 270

S9
1H NMR of isolated 3r:
S10
Fluorescence Quenching4,5
For fluorescence quenching experiments, a 50 µM solution of the Ir(dtbbpy)(ppy)2PF6 in
degassed water/SDS solution was prepared under nitrogen atmosphere in a gas-tight 10 mm
quartz cuvette. The photocatalyst solution was irradiated with 410 nm and the fluorescence
emission upon addition of potential quenchers was recorded. The Stern-Volmer plot was
performed using the fluorescence at 580 nm.
Figure S2. a) Fluorescence quenching using cyclohexyl bromide 2a. b) Stern-Volmer plot.
400 450 500 550 600 650 700
0
200000
400000
600000
800000
1000000
1200000
1400000
1600000
Wavelength / nm
Inte
nsity / C
PS
PC (50 µM)
0.25 mM
0.50 mM
0.74 mM
0.99 mM
1.2 mM
1.5 mM
1.7 mM
2.0 mM
2.2 mM
2.4 mM
4.8 mM
Cyclohehylbromide
0
0,5
1
1,5
2
2,5
3
3,5
4
0 1 2 3 4 5
I0 /I
Cyclohexylbromide (mmol/L)
Equation: y= ax + bIntercept = 1Slope = 0.11R2 = 0.94
S11
Figure S3. a) Fluorescence quenching using lepidine 1a. b) Stern Volmer plot.
400 500 600 700
0
200000
400000
600000
800000
1000000
1200000
1400000
1600000
Inte
nsity /
CP
S
Lepidine
Wavelength / nm
PC (50 µM)
0.25 mM
0.50 mM
0.74 mM
0.99 mM
1.2 mM
1.5 mM
1.7 mM
2.0 mM
2.2 mM
2.4 mM
4.8 mM
0
0,5
1
1,5
2
2,5
3
3,5
4
0 1 2 3 4 5
I0 /I
Lepidine (mmol/L)
Equation: y= ax + bIntercept = 1Slope = 0.19
R2 = 0.62

S12
Figure S4. a) Fluorescence quenching using CBr4. b) Stern Volmer plot.
0
0,5
1
1,5
2
2,5
3
3,5
4
0 1 2 3 4 5
I0/I
Tetrabromomethane (mmol/L)
Equation: y= ax + bIntercept = 1Slope = 0.50
R2 = 0.95

S13
Characterization data of the compounds
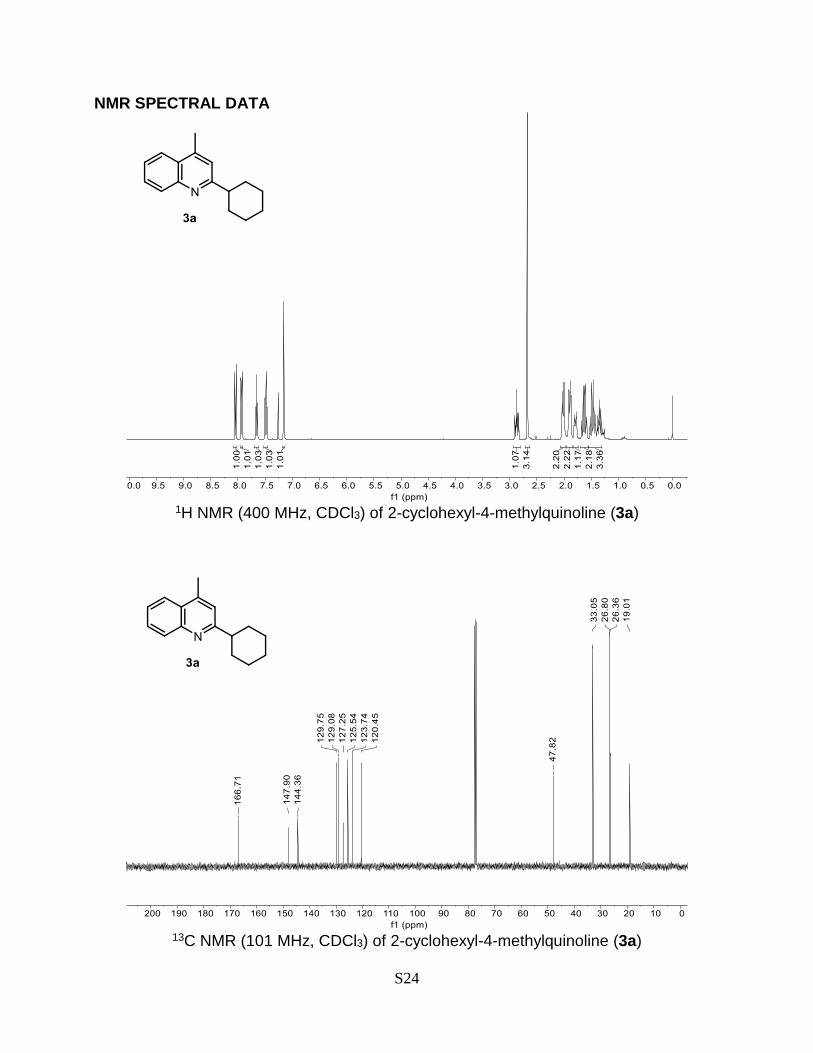
2-Cyclohexyl-4-methylquinoline (3a)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
cyclohexyl bromide 2a (32.6 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt:n-hexane 0-10% + 0.2% Et3N) to give a product as a pale yellow
oil. Isolated yield = 85% (19.2 mg). TLC (AcOEt/n-hexane 10%): Rf = 0.50. 1H NMR (400
MHz, CDCl3) δ 8.05 (dd, J = 8.4, 1.2 Hz, 1H), 7.94 (dd, J = 8.3, 1.3 Hz, 1H), 7.66 (ddd, J
= 8.3, 6.8, 1.4 Hz, 1H), 7.48 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.16 (d, J = 1.1 Hz, 1H), 2.87
(tt, J = 12.0, 3.5 Hz, 1H), 2.68 (d, J = 0.9 Hz, 3H), 2.06 – 1.98 (m, 2H), 1.89 (dt, J = 12.7,
3.3 Hz, 2H), 1.79 (dtd, J = 12.0, 3.4, 1.7 Hz, 1H), 1.63 (qd, J = 12.4, 3.2 Hz, 2H), 1.54 –
1.25 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 166.7, 147.9, 144.4, 129.8, 129.1, 127.3,
125.5, 123.7, 120.5, 47.8, 33.1, 26.8, 26.4, 19.0. The obtained spectral data is consistent
with the literature reports.6
2-cycloheptyl-4-methylquinoline (3b)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
cycloheptyl bromide 2b (35.4 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt:DCM:n-hexane 1:1:98 + 0.2% Et3N) to give a product as a
colorless oil. Isolated yield = 51% (12.2 mg). TLC (AcOEt:DCM:n-hexane 1:1:8): Rf = 0.63. 1H NMR (400 MHz, CDCl3) δ 8.04 (dd, J = 8.5, 1.3 Hz, 1H), 7.93 (dd, J = 8.3, 1.4 Hz, 1H),
7.66 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.48 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.13 (d, J = 1.1
Hz, 1H), 3.04 (tt, J = 10.4, 3.5 Hz, 1H), 2.67 (d, J = 1.0 Hz, 3H), 2.11 – 1.99 (m, 2H), 1.93
– 1.57 (m, 10H).13C NMR (101 MHz, CDCl3) δ 168.3, 147.6, 144.6, 129.7, 129.1, 127.2,
125.5, 123.7, 120.5, 49.8, 35.3, 28.2, 27.7, 19.0. The obtained spectral data is consistent
with the literature reports.6

S14
4-methyl-2-(pentan-2-yl)quinoline (3c)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
2-bromopentane 2c (30.2 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt:DCM:n-hexane 1:1:98 + 0.2% Et3N) to give a product as a
colorless oil. Isolated yield = 56% (12.0 mg). TLC (AcOEt:DCM:n-hexane 1:1:8): Rf = 0.63. 1H NMR (400 MHz, CDCl3) δ 8.07 (dd, J = 8.5, 1.2 Hz, 1H), 7.94 (dd, J = 8.4, 1.3 Hz, 1H),
7.66 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.49 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.14 (d, J = 1.1
Hz, 1H), 3.07 (q, J = 7.1 Hz, 1H), 2.69 (d, J = 0.9 Hz, 3H), 1.86 – 1.76 (m, 1H), 1.72 –
1.59 (m, 1H), 1.44 – 1.31 (m, 4H), 1.30 – 1.16 (m, 1H), 0.90 (t, J = 7.3 Hz, 3H).13C NMR
(101 MHz, CDCl3) δ 167.1, 147.8, 144.5, 129.7, 129.1, 127.2, 125.6, 123.8, 120.4, 42.8,
39.5, 21.1, 20.9, 19.0, 14.4. The obtained spectral data is consistent with the literature
reports.6
4-Methyl-2-(1-phenylpropan-2-yl)quinoline (3d)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
2-bromo-1-phenylpropane 2d (39.8 mg, 0.2 mmol). The crude product was purified by
flash chromatography (AcOEt:DCM:n-hexane 1:1:98 + 0.2% Et3N) to give a product as a
pale yellow oil. Isolated yield = 67% (17.5 mg). TLC (AcOEt:DCM:n-hexane 1:1:8): Rf =
0.62. 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.5, 1H), 7.95 (dd, J = 8.4, 1.4 Hz, 1H),
7.69 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.51 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.27 – 7.15 (m,
5H), 7.11 (d, J = 1.1 Hz, 1H), 3.38 (h, J = 7.0, 1H), 3.26 (dd, J = 13.5, 6.3 Hz, 1H), 2.92
(dd, J = 13.5, 8.5 Hz, 1H), 2.67 (d, J = 1.0 Hz, 3H), 1.35 (d, J = 6.9 Hz, 3H).13C NMR (126
MHz, CDCl3) δ 165.9, 147.3, 144.6, 140.7, 129.6, 129.4, 129.2, 128.3, 127.2, 126.0,
125.7, 123.7, 121.0, 44.5, 43.0, 20.1, 18.9. The obtained spectral data is consistent with
the literature reports.7

S15
2-Butyl-4-methylquinoline (3e)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
1-butyl bromide 2e (27.4 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt:n-hexane 5-10% + 0.2% Et3N) to give a product as a colorless
oil. Isolated yield = 48% (9.6 mg). TLC (AcOEt/n-hexane 30%): Rf = 0.66. 1H NMR (400
MHz, CDCl3) δ 8.04 (d, J = 8.5 Hz, 1H), 7.95 (dd, J = 8.3 Hz, 1H), 7.66 (ddd, J = 8.4, 6.8,
1.4 Hz, 1H), 7.49 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.14 (s, 1H), 2.96 – 2.88 (m, 2H), 2.68
(s, 3H), 1.84 – 1.73 (m, 2H), 1.49 – 1.40 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (101
MHz, CDCl3) δ 163.0, 148.0, 144.3, 129.6, 129.2, 127.0, 125.6, 123.8, 122.3, 39.2, 32.4,
23.0, 19.0, 14.2. The obtained spectral data is consistent with the literature reports.8
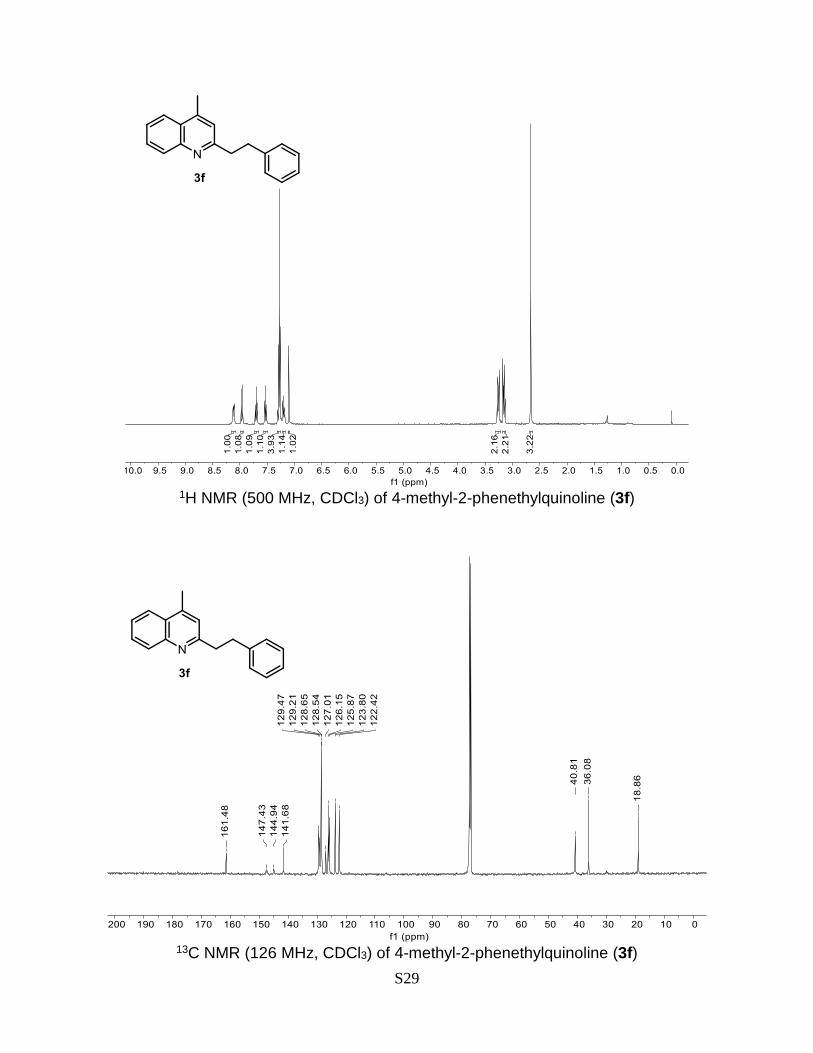
4-Methyl-2-phenethylquinoline (3f)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
2-(bromoethyl)benzene 2f (37.0 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt/n-hexane 0-5% + 0.5% Et3N) to give a product as a colorless oil.
Isolated yield = 50% (12.4 mg). TLC (AcOEt/n-hexane 20%): Rf = 0.49. 1H NMR (500
MHz, CDCl3) δ 8.11 (d, J = 8.3, 1H), 7.95 (dd, J = 8.3, 1.5 Hz, 1H), 7.69 (ddd, J = 8.4, 6.8,
1.4 Hz, 1H), 7.51 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.30 – 7.23 (m, 4H), 7.21 – 7.16 (m, 1H),
7.09 (d, J = 1.2 Hz, 1H), 3.25 (dd, J = 9.6, 6.0 Hz, 2H), 3.14 (dd, J = 10.3, 6.4, 2H), 2.65
(d, J = 1.0 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 161.5, 147.4, 144.9, 141.7, 129.5, 129.2,
128.7, 128.5, 127.0, 126.2, 125.9, 123.8, 122.4, 40.8, 36.1, 18.9. The obtained spectral
data is consistent with the literature reports.9

S16
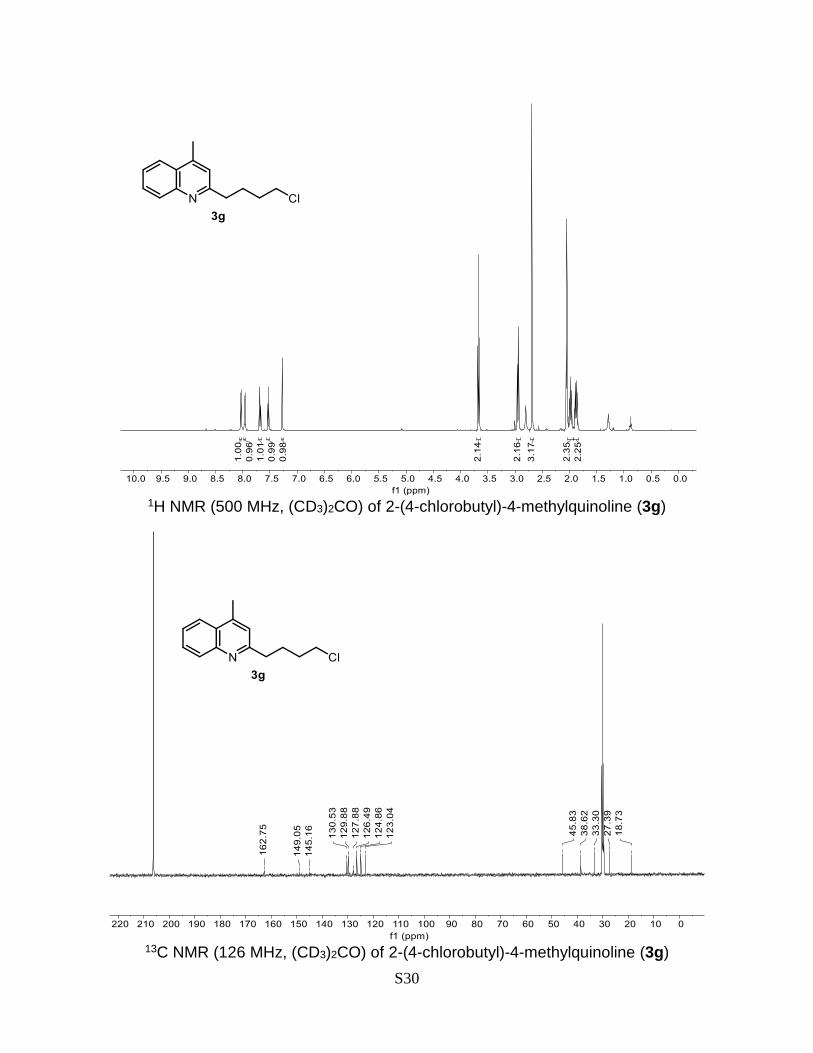
2-(4-chlorobutyl)-4-methylquinoline (3g)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
1-bromo-4-chlorobutane 2g (34.3 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt/n-hexane 2-8% + 0.2% Et3N) to give a product as a yellow oil.
Isolated yield = 57% (13.4 mg). TLC ((AcOEt/n-hexane 20%): Rf = 0.4. 1H NMR (500 MHz,
(CD3)2CO) δ 8.04 (dd, J = 8.3, 1.4 Hz, 1H), 7.96 (dd, J = 8.3, 1.3 Hz, 1H), 7.96 (ddd, J =
8.4, 6.8, 1.4 Hz, 1H), 7.53 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.28 (d, J = 1.1 Hz, 1H), 3.69 (t,
J = 6.6 Hz, 2H), 2.94 (t, J = 7.5 Hz, 2H), 2.69 (d, J = 1.0 Hz, 3H), 1.95 – 2.02 (m, 2H), 1.84
– 1.91 (m, 2H). 13C NMR (126 MHz, (CD3)2CO) δ 162.8, 149.1, 145.2, 130.5, 129.9, 127.9,
126.5, 124.9, 123.0, 45.8, 38.62, 33.3, 27.4, 18.7. HRMS (ESI) calculated for C14H18NCl
[M+H]+: 234.1050, found: 234.1050.
2-(3-hydroxypropyl)-4-methylquinoline (3h)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
3-bromopropanol 2h (28.6 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt/n-hexane 60-75% + 0.5% Et3N) to give a product as a yellow oil.
Isolated yield = 70% (14.1 mg). TLC (DCM/MeOH 7%): Rf = 0.22. 1H NMR (500 MHz,
CDCl3) δ 8.04 (dd, J = 8.5, 1.3 Hz, 1H), 7.95 (dd, J = 8.4, 1.5 Hz, 1H), 7.68 (ddd, J = 8.4,
6.9, 1.5 Hz, 1H), 7.52 (ddd, J = 8.3, 6.9, 1.4 Hz, 1H), 7.17 (s, 1H), 4.32 (br s, 1H), 3.77 (t,
J = 5.8 Hz, 2H), 3.17 – 3.06 (t, J = 6.9 Hz, 2H), 2.68 (s, 3H), 2.13 – 2.04 (m, 2H). 13C NMR
(126 MHz, CDCl3) δ 161.7, 146.7, 145.6, 129.7, 128.7, 126.8, 126.0, 123.8, 122.6, 62.5,
36.2, 31.3, 18.8. The obtained spectral data is consistent with the literature reports.11

S17
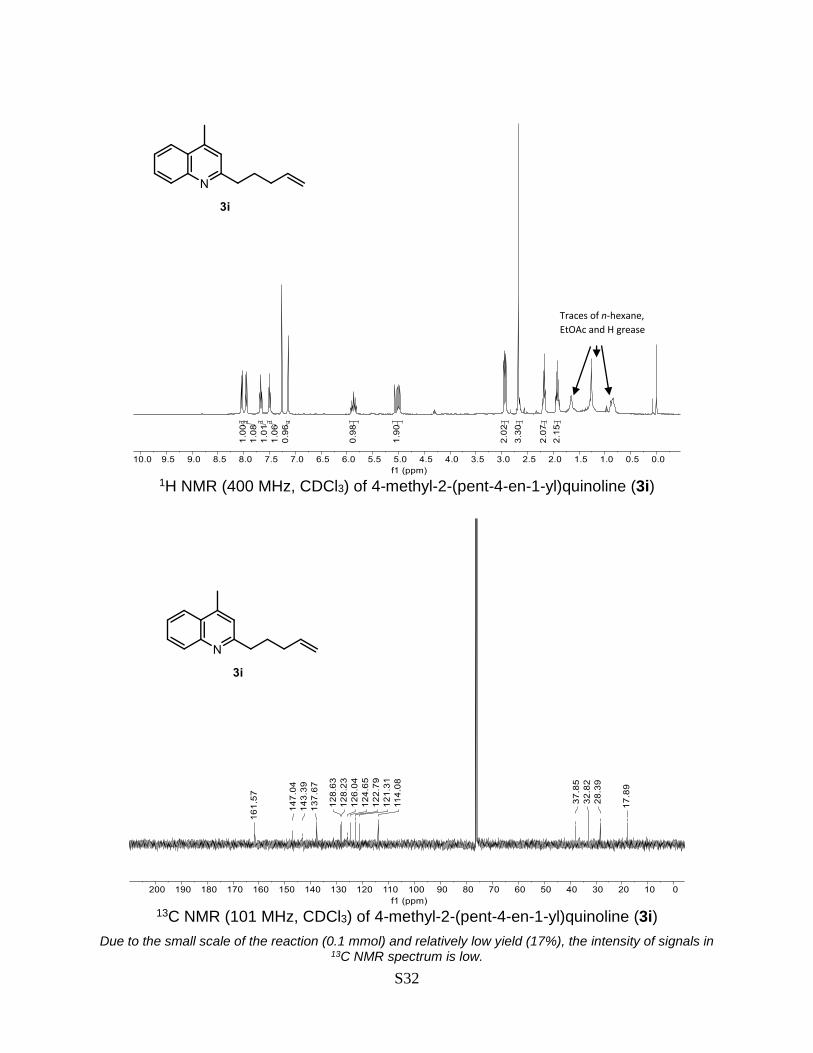
4-Methyl-2-(pent-4-en-1-yl)quinoline (3i)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
5-bromo-1-pentene 2i (31.3 mg, 0.2 mmol). The crude product was purified by flash
chromatography (AcOEt:DCM:n-hexane 1:1:98% + 0.2% Et3N) to give a product as a
colorless oil. Isolated yield = 17% (3.6 mg). TLC (AcOEt:DCM:n-hexane 1:1:8): Rf = 0.41. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.67 (ddd,
J = 8.4, 6.8, 1.5 Hz, 1H), 7.50 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.16 – 7.10 (m, 1H), 5.92 –
5.82 (m, 1H), 5.11 – 4.93 (m, 2H), 3.00 – 2.88 (m, 2H), 2.68 (d, J = 1.0 Hz, 3H), 2.22 –
2.13 (m, 2H), 1.98 – 1.85 (m, 2H).13C NMR (101 MHz, CDCl3) δ 161.6, 147.0, 143.4,
137.7, 128.6, 128.2, 126.0, 124.7, 122.8, 121.3, 114.1, 37.9, 32.8, 28.4, 17.9. The
obtained spectral data is consistent with the literature reports.10
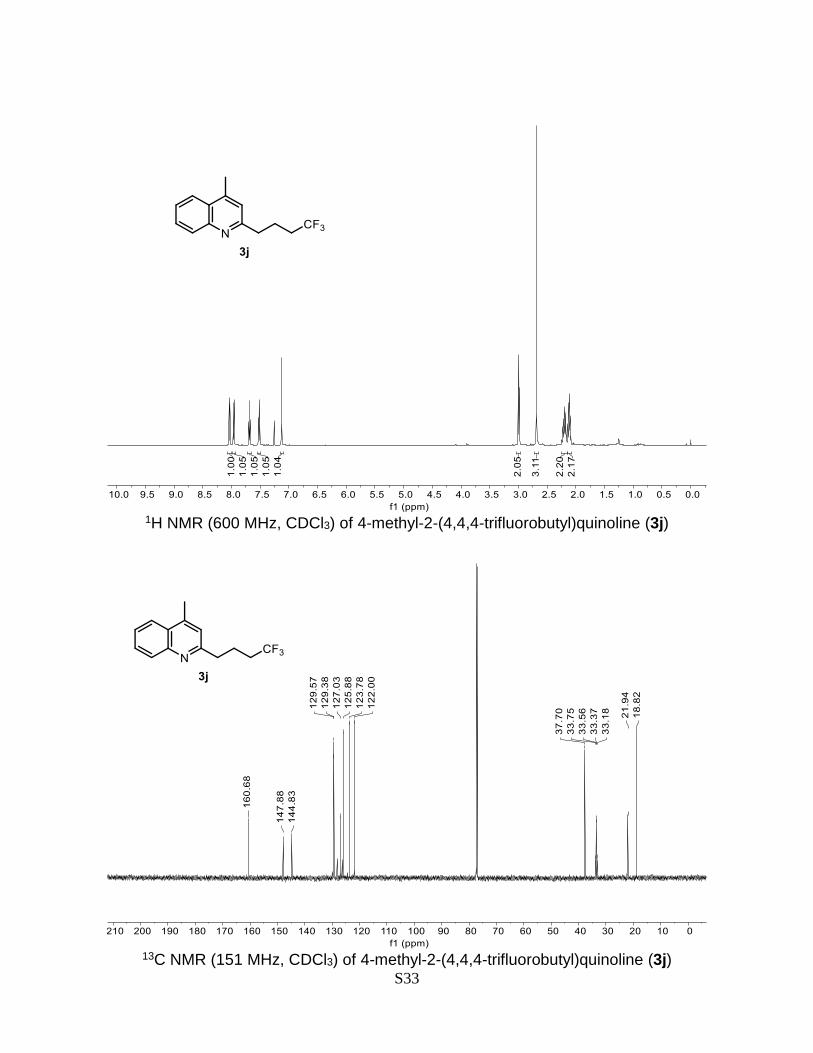
4-methyl-2-(4,4,4-trifluorobutyl)quinoline (3j)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
4-bromo-1,1,1-trifluorobutane 2j (39.3 mg, 0.2 mmol). The crude product was purified by
flash chromatography (AcOEt/n-hexane 2% + 0.2% Et3N) to give a product as a colorless
oil. Isolated yield = 50% (12.6 mg). TLC (AcOEt/n-hexane 20%): Rf = 0.48. 1H NMR (600
MHz, CDCl3) δ 8.04 (dd, J = 8.5, 1.3 Hz, 1H), 7.96 (dd, J = 8.3, 1.4 Hz, 1H), 7.69 (ddd, J
= 8.3, 6.8, 1.4 Hz, 1H), 7.52 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.13 (s, 1H), 2.99 (t, J = 7.6
Hz, 2H), 2.68 (s, 3H), 2.25 – 2.15 (m, 2H), 2.15 – 2.08 (m, 2H). 13C NMR (151 MHz,
CDCl3) δ 160.7, 147.9, 144.8, 129.6, 129.4, 128.2, 127.0, 126.4, 125.9, 123.8, 122.0,
37.7, 33.5 (q, J = 28.6 Hz), 21.9, 18.8. The obtained spectral data is consistent with the
literature reports.7

S18
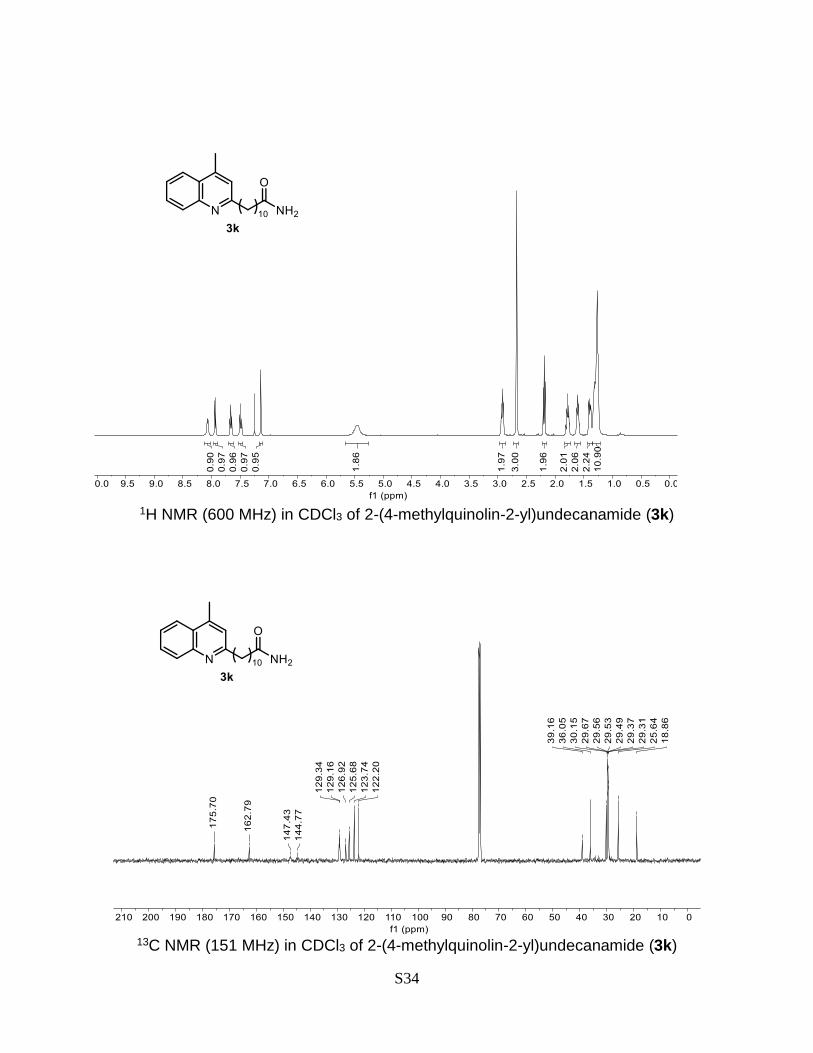
2-(4-methylquinolin-2-yl)undecanamide (3k)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
11-bromoundecanamide 2k (54.4 mg, 0.2 mmol). The crude product was purified by flash
chromatography (DCM/MeOH 2% + 0.2% Et3N) to give a product as a white solid. Isolated
yield = 44% (14.4 mg). TLC (DCM/MeOH 10%): Rf = 0.43. 1H NMR (500 MHz, CDCl3) δ
8.08 (d, J = 8.5 Hz, 1H), 7.95 (dd, J = 8.4, 1.4 Hz, 1H), 7.67 (ddd, J = 8.4, 6.7, 1.4 Hz,
1H), 7.50 (t, J = 7.6 Hz, 1H), 7.15 (s, 1H), 5.49 (s, 2H), 2.93 (t, J = 7.9 Hz, 2H), 2.68 (s,
3H), 2.20 (t, J = 7.6 Hz, 2H), 1.88 – 1.75 (m, 2H), 1.67 – 1.57 (m, 2H), 1.41 (dd, J = 10.3,
5.4 Hz, 2H), 1.36 – 1.22 (m, 10H). 13C NMR (151 MHz, CDCl3) δ 175.70, 162.79, 147.43,
144.77, 129.34, 129.16, 126.92, 125.68, 123.74, 122.20, 39.16, 36.05, 30.15, 29.67,
29.56, 29.53, 29.50, 29.37, 29.31, 25.64, 18.86. HRMS (ESI) calculated for C21H30N2O
[M+H]+: 327.2436, found: 327.2428.
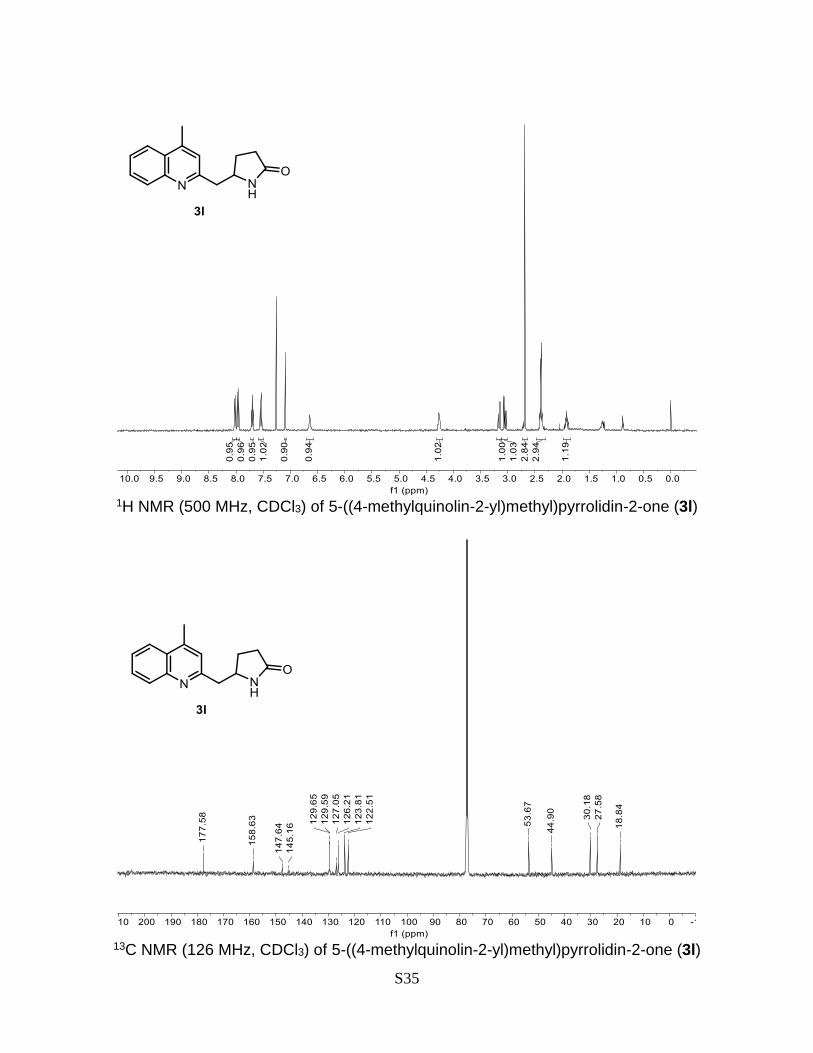
5-((4-methylquinolin-2-yl)methyl)pyrrolidin-2-one (3l)
Prepared according to the general Procedure A from lepidine 1a (14.3 mg, 0.1 mmol) and
(R)-5-(bromomethyl)-2-pyrrolidinone 2l (37.0 mg, 0.2 mmol). The crude product was
purified by flash chromatography (DCM/MeOH 2% + 0.2% Et3N) to give a product as a
colorless oil. Isolated yield = 28% (6.7 mg). TLC (DCM/MeOH 10%): Rf = 0.5. 1H NMR
(500 MHz, CDCl3) δ 8.02 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.70 (ddd, J = 8.3,
6.6, 1.4 Hz, 1H), 7.54 (td, J = 7.4, 7.0, 1.2 Hz, 1H), 7.09 (s, 1H), 6.64 (s, 1H), 4.26 (m,
1H), 3.16 (dd, J = 15.0, 3.7 Hz, 1H), 3.05 (dd, J = 15.1, 9.6 Hz, 1H), 2.69 (s, 3H), 2.46 –
2.31 (m, 3H), 1.98 – 1.85 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 177.6, 158.6, 147.6,
145.2, 129.7, 129.6, 127.1, 126.2, 123.8, 122.5, 53.7, 44.9, 30.2, 27.6, 18.8. HRMS (ESI)
calculated for C15H16N2O [M+H]+: 241.1341, found: 241.1334.

S19
.6-Cyclohexylphenanthridine (10b)
Prepared according to the general Procedure A from phenantridine 1b (17.9 mg, 0.1
mmol) and cyclohexyl bromide 2a (32.6 mg, 0.2 mmol). The crude product was purified
by flash chromatography (AcOEt:DCM:n-hexane 1:1:98 + 0.2% Et3N) to give a product as
a colorless oil. Isolated yield = 55% (14.4 mg). TLC (AcOEt:DCM:n-hexane 1:1:8): Rf =
0.63. 1H NMR (500 MHz, CDCl3) δ 8.66 (dd, J = 8.3, 1.2 Hz, 1H), 8.54 (dd, J = 8.2, 1.4
Hz, 1H), 8.32 (d, J = 8.3, 1H), 8.14 (d, J = 8.2, 1H), 7.81 (t, J = 7.6, 1H), 7.69 (dddd, J =
8.3, 6.7, 5.0, 1.2, 2H), 7.65 – 7.56 (m, 1H), 3.63 (td, J = 11.1, 10.6, 5.6 Hz, 1H), 2.14 –
2.03 (m, 2H), 2.03 – 1.90 (m, 4H), 1.90 – 1.79 (m, 1H), 1.65 – 1.51 (m, 2H), 1.51 – 1.38
(m, 1H).13C NMR (126 MHz, CDCl3) δ 165.4, 144.1, 133.2, 130.10, 130.07, 128.5, 127.2,
126.3, 125.8, 124.9, 123.5, 122.8, 122.0, 42.2, 32.4, 27.0, 26.5. The obtained spectral
data is consistent with the literature reports.6
2-cyclohexylquinoline (10c) and 2,4-dicyclohexylquinoline (S-di-10c)
Prepared according to the general Procedure A from quinoline 1c (12.9 mg, 0.1 mmol)
and cyclohexyl bromide 2a (81.5 mg, 0.5 mmol). The crude product was purified by flash
chromatography (AcOEt/DCM/n-hexane 3:3:93% + 0.2% Et3N) to give a mixture of
unseparable products: 2-cyclohexylquinoline (10c) and 2,4-dicyclohexylquinoline (S-di-
10c) at 3:1 ratio, as a colorless oil. Summarized isolated yield = 56% (23.0 mg). TLC
((AcOEt/n-hexane 20%): Rf = 0.78. 1H NMR (500 MHz, CDCl3) δ 8.13 – 7.99 (10c, m, 2H
+ S-di-10c, m 2H), 7.79 – 7.74 (10c, d, J = 8.1 Hz, 1H), 7.70 – 7.62 (10c, m, 1H + S-di-
10c, m, 1H), 7.53 – 7.44 (10c, m, 1H + S-di-10c, m, 1H), 7.36 – 7.30 (10c, d, J = 8.5 Hz,
1H), 7.22 (S-di-10c, s, 1H), 3.36 – 3.27 (S-di-10c, m, 1H), 3.01 – 2.86 (10c, m, 1H + S-
di-10c, m, 1H), 2.08 – 1.98 (10c, m, 2H + S-di-10c, m, 4H), 1.97 – 1.84 (10c, m, 2H + S-
di-10c, m, 5H), 1.83 – 1.75 (10c, m, 1H + S-di-10c, m, 1H), 1.70 – 1.42 (10c, m, 4H + S-
di-10c, m, 8H), 1.41 – 1.29 (10c, m, 1H + S-di-10c, m, 2H). 13C NMR (126 MHz, CDCl3)
δ 167.0 (10c + S-di-10c), 148.0 (10c, close to the baseline), 136.5 (10c), 129.4 (10c),

S20
129.1 (10c), 127.6 (10c), 127.1 (10c), 125.8 (10c + S-di-10c), 123.0 (S-di-10c), 119.7
(10c), 115.9 (S-di-10c), 47.8 (10c + S-di-10c), 39.3 (S-di-10c), 33.8 (S-di-10c), 33.0 (10c
+ S-di-10c), 27.1 (S-di-10c), 26.7 (10c + S-di-10c), 26.5 (S-di-10c), 26.3 (10c + S-di-
10c). Some aromatic carbon signals from S-di-10c are not visible in 13C NMR spectrum,
due to the small scale of the reaction and small amount of this compound in mixture. The
obtained spectral data is consistent with the literature reports.12
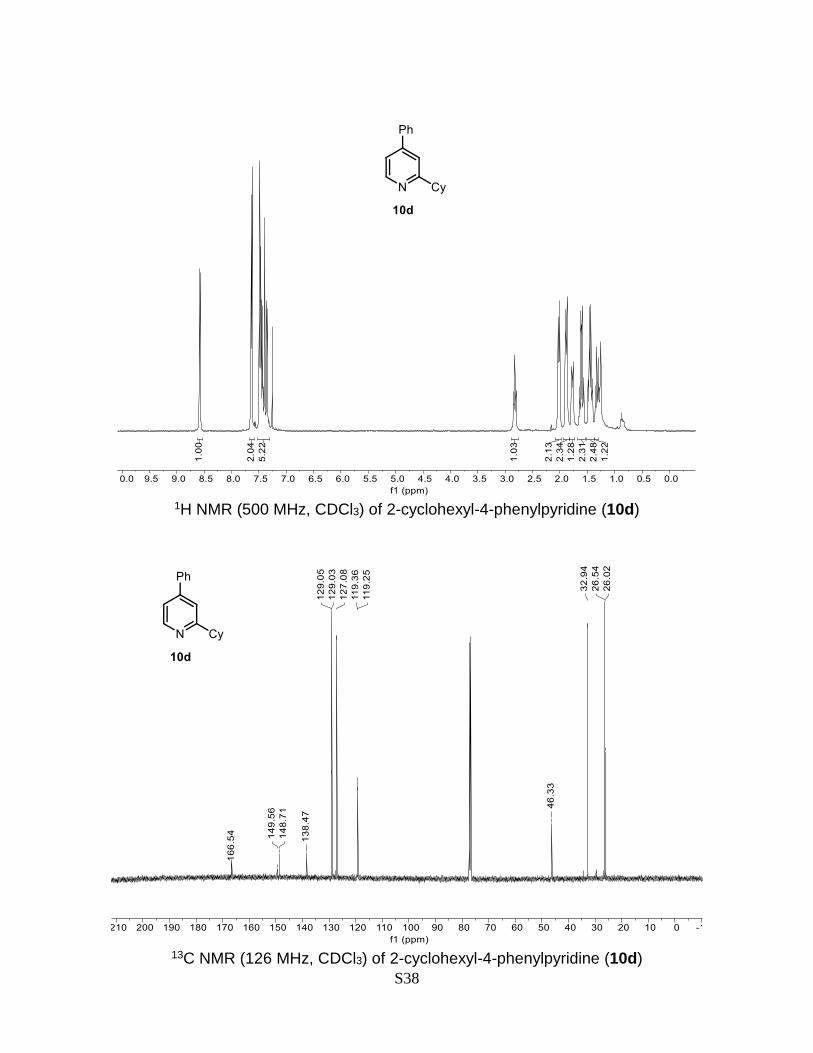
2-Cyclohexyl-4-phenylpyridine (10d)
Prepared according to the general Procedure A from 4-phenylpyridine 1d (15.5 mg, 0.1
mmol) and cyclohexyl bromide 2a (32.6 mg, 0.2 mmol). The crude product was purified
by flash chromatography (AcOEt/n-hexane 0-2% + 0,2% Et3N) to give a product as a
colorless oil. Isolated yield = 38% (9.0 mg). TLC (AcOEt/n-hexane 20%): Rf = 0.39. 1H
NMR (500 MHz, CDCl3) δ 8.58 (d, J = 5.2 Hz, 1H), 7.63 (d, J = 7.0 Hz, 2H), 7.53 – 7.31
(m, 5H), 2.82 (tt, J = 12.0, 3.5 Hz, 1H), 2.05 – 1.98 (m, 2H), 1.93 – 1.83 (m, 2H), 1.82 –
1.73 (m, 1H), 1.67 – 1.53 (m, 2H), 1.51 – 1.38 (m, 2H), 1.37 – 1.29 (m, 1H). 13C NMR (125
MHz, CDCl3) δ 166.5, 149.6, 148.7, 138.5, 129.1, 129.0, 127.1, 119.4, 119.3, 46.3, 32.9,
26.5, 26.0. The obtained spectral data is consistent with the literature reports.6
2,6-Dicyclohexyl-4-phenylpyridine (di-10d)
Prepared according to the general Procedure A from 4-phenylpyridine 1d (15.5 mg, 0.1
mmol) and cyclohexyl bromide 2a (32.6 mg, 0.2 mmol). The crude product was purified
by flash chromatography (AcOEt/n-hexane 0-2% + 0,2% Et3N) to give a product as a
colorless oil. Isolated yield = 40% (12.8 mg). TLC (AcOEt/n-hexane 20%): Rf = 0.83. 1H
NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.0 Hz, 2H), 7.46 (t, J = 7.3 Hz, 2H), 7.40 (t, J = 6.6
Hz, 1H), 7.17 (s, 2H), 2.80 – 2.64 (m, 2H), 2.08 – 1.98 (m, 4H), 1.92 – 1.82 (m, 4H), 1.81
– 1.71 (m, 2H), 1.62 – 1.40 (m, 8H), 1.37 – 1.29 (m, 2H). 13C NMR (101 MHz, CDCl3) δ

S21
166.2, 149.0, 139.6, 128.9, 128.5, 127.1, 116.0, 46.7, 33.2, 26.7, 26.2. The obtained
spectral data is consistent with the literature reports.6
2,4,6-Tricyclohexylnicotinonitrile (10e)
Prepared according to the general Procedure A from 3-pyridinecarbonitrile 1e (10.4 mg,
0.1 mmol) and cyclohexyl bromide 2a (81.5 mg, 0.5 mmol). Reaction was carried out for
20h. The crude product was purified by flash chromatography (DCM/n-hexane 0-10% +
0.2% Et3N) to give a product as a white solid. Isolated yield = 85% (29.8 mg). TLC
(AcOEt/n-hexane 20%): Rf = 0.77. 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 3.13 (tt, J =
11.5, 3.6 Hz, 1H), 2.90 (ddd, J = 11.5, 8.2, 3.3 Hz, 1H), 2.67 (tt, J = 11.8, 3.5 Hz, 1H),
1.96 – 1.63 (m, 16H), 1.60 – 1.20 (m, 14H). 13C NMR (101 MHz, CDCl3) δ 168.6, 167.8,
159.4, 116.0, 114.7, 103.9, 46.0, 44.1, 41.9, 32.3, 31.7, 31.1, 25.6, 25.6, 25.5, 25.3, 25.1.
HRMS (ESI) calculated for C24H34N2 [M+H]+: 351.2800, found: 351.2797.
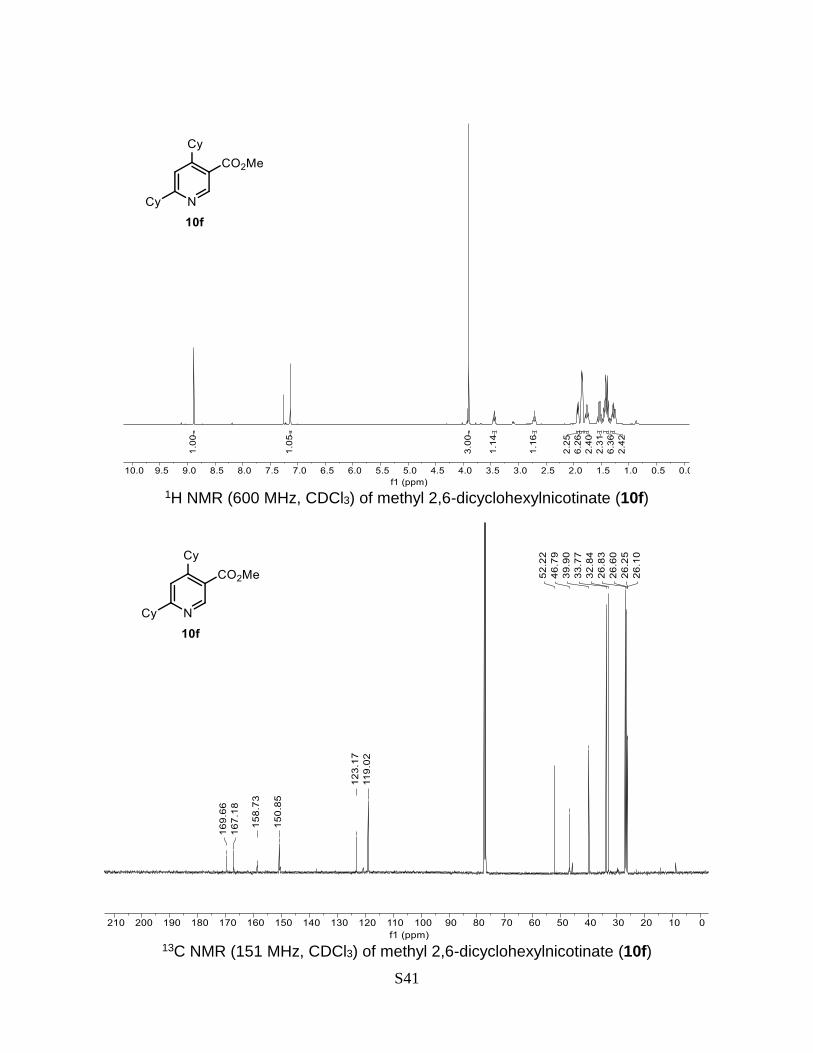
Methyl 2,6-dicyclohexylnicotinate (10f)
Prepared according to the general Procedure A from methyl nicotinate 1f (13.7 mg, 0.1
mmol) and cyclohexyl bromide 2a (32.6 mg, 0.2 mmol). The crude product was purified
by flash chromatography (AcOEt/n-hexane 2-5% + 0.2% Et3N) to give a product as a white
solid. Isolated yield = 49% (14.8 mg). TLC ((AcOEt/n-hexane 20%): Rf = 0.52. 1H NMR
(600 MHz, CDCl3) δ 8.89 (s, 1H), 7.14 (s, 1H), 3.90 (s, 3H), 3.44 (tt, J = 11.4, 2.9 Hz, 1H),
2.71 (tt, J = 12.0, 3.4 Hz, 1H), 1.93 (ddq, J = 12.2, 3.7, 2.1 Hz, 2H), 1.89 – 1.79 (m, 6H),
1.81 – 1.70 (m, 2H), 1.54 (qd, J = 12.6, 3.3 Hz, 2H), 1.49 – 1.35 (m, 6H), 1.33 – 1.23 (m,
2H). 13C NMR (151 MHz, CDCl3) δ 169.7, 167.2 (C=O), 158.7, 150.9, 123.2, 119.0, 52.2,
46.8, 39.9, 33.8, 32.8, 26.8, 26.6, 26.3, 26.1. HRMS (ESI) calculated for C19H27NO2
[M+H]+: 302.2120, found: 302.2116.

S22
Table S5. 1H and 13C NMR correlation data for methyl 2,6-dicyclohexylnicotinate (10f)
Position δ 1H [ppm] δ 13C [ppm] HMBC correlations
C1 3.90 (3H, s) 52.2 C9
C2 8.89 (1H, s) 150.9 C3, C4, C5, C6, C8, C9
C3 - 169.7 C2, C5, C7
C4 - 123.2 C2, C5, C8
C5 7.14 (1H, s) 119.0 C3, C4, C7, C8, C9
C6 - 158.7 C2
C7 2.71 (1H, tt, J = 12.0, 3.4 Hz) 46.8 C3, C5
C8 3.44 (1H, tt, J = 11.4, 2.9 Hz) 39.9 C4
C9 - 167.2 C1, C2, C5
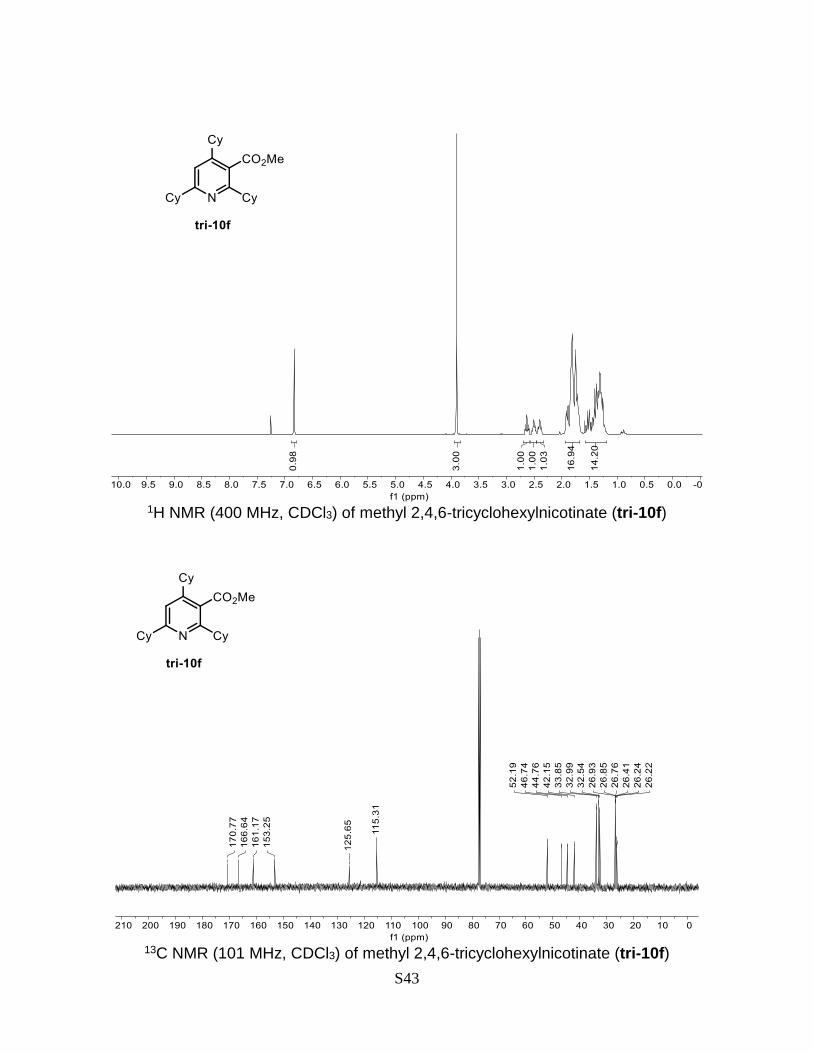
Methyl 2,4,6-tricyclohexylnicotinate (tri-10f)
Prepared according to the general Procedure A from methyl nicotinate 1f (13.7 mg, 0.1
mmol) and cyclohexyl bromide 2a (81.5 mg, 0.5 mmol). Reaction was carried out for 20h.
The crude product was purified by flash chromatography (AcOEt/n-hexane 1-3% + 0.2%
Et3N) to give a product as a white solid. Isolated yield = 75% (28.8 mg). TLC
((AcOEt/n-hexane 20%): Rf = 0.78. 1H NMR (400 MHz, CDCl3) δ 6.83 (s, 1H), 3.90 (s,
3H), 2.62 (ddt, J = 11.7, 7.1, 3.4 Hz, 1H), 2.55 – 2.45 (m, 1H), 2.44 – 2.34 (m, 1H), 1.94
– 1.68 (m, 16H), 1.58 – 1.21 (m, 14H).13C NMR (101 MHz, CDCl3) δ 170.8, 166.6, 161.2,
153.3, 125.7, 115.3, 52.2, 46.7, 44.8, 42.2, 33.9, 33.0, 32.5, 26.9, 26.85, 26.8, 26.4, 26.2,
26.2. HRMS (ESI) calculated for C25H37NO2 [M+H]+: 384.2903, found: 384.2900.

S23
REFERENCES 1. Armarego, W. L. F. & Chai, C. L. L. Purification of laboratory chemicals. Purif. Lab. Chem.
Fifth Ed. 1–609 (2003) doi:10.1016/B978-0-7506-7571-0.X5000-5. 2. Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR chemical shifts of common laboratory
solvents as trace impurities. J. Org. Chem. 62, 7512–7515 (1997). 3. Dong, J. et al. Visible-light-mediated Minisci C–H alkylation of heteroarenes with
unactivated alkyl halides using O 2 as an oxidant. Chem. Sci. 10, 976–982 (2019). 4. Berger, A. L., Donabauer, K. & König, B. Photocatalytic Barbier reaction – visible-light
induced allylation and benzylation of aldehydes and ketones. Chem. Sci. 9, 7230–7235 (2018).
5. Gehlen, M. H. The centenary of the Stern-Volmer equation of fluorescence quenching: From the single line plot to the SV quenching map. J. Photochem. Photobiol. C Photochem. Rev. 42, 100338 (2020).
6. Zhao, H. & Jin, J. Visible Light-Promoted Aliphatic C–H Arylation Using Selectfluor as a Hydrogen Atom Transfer Reagent. Org. Lett. 21, 6179–6184 (2019).
7. Li, Z., Wang, X., Xia, S. & Jin, J. Ligand-Accelerated Iron Photocatalysis Enabling Decarboxylative Alkylation of Heteroarenes. Org. Lett. 21, 4259–4265 (2019).
8. Zhang, L. & Liu, Z. Q. Molecular Oxygen-Mediated Minisci-Type Radical Alkylation of Heteroarenes with Boronic Acids. Org. Lett. 19, 6594–6597 (2017).
9. Tan, Z., Jiang, H. & Zhang, M. A novel iridium/acid co-catalyzed transfer hydrogenative C(sp3)-H bond alkylation to access functionalized N-heteroaromatics. Chem. Commun. 52, 9359–9362 (2016).
10. McCallum, T. & Barriault, L. Direct alkylation of heteroarenes with unactivated bromoalkanes using photoredox gold catalysis. Chem. Sci. 7, 4754–4758 (2016).
11. Zidan, M., Morris, A. O., McCallum, T. & Barriault, L. The Alkylation and Reduction of Heteroarenes with Alcohols Using Photoredox Catalyzed Hydrogen Atom Transfer via Chlorine Atom Generation. European J. Org. Chem. (2019) doi:10.1002/ejoc.201900786.
12. Sherwood, T. C., Li, N., Yazdani, A. N. & Dhar, T. G. M. Organocatalyzed, Visible-Light Photoredox-Mediated, One-Pot Minisci Reaction Using Carboxylic Acids via N-(Acyloxy)phthalimides. J. Org. Chem. 83, 3000–3012 (2018).
S24
NMR SPECTRAL DATA
1H NMR (400 MHz, CDCl3) of 2-cyclohexyl-4-methylquinoline (3a)
13C NMR (101 MHz, CDCl3) of 2-cyclohexyl-4-methylquinoline (3a)

S25
1H NMR (400 MHz, CDCl3) of methyl 2-cycloheptyl-4-methylquinoline (3b)
13C NMR (101 MHz, CDCl3) of methyl 2-cycloheptyl-4-methylquinoline (3b)

S26
1H NMR (400 MHz, CDCl3) of 4-methyl-2-(pentan-2-yl)quinoline (3c)
13C NMR (101 MHz, CDCl3) of 4-methyl-2-(pentan-2-yl)quinoline (3c)

S27
1H NMR (500 MHz) in CDCl3 of 4-methyl-2-(1-phenylpropan-2-yl)quinoline (3d)
13C NMR (126 MHz, CDCl3) of 4-methyl-2-(1-phenylpropan-2-yl)quinoline (3d)
Signals overlap
with residual CHCl3

S28
1H NMR (400 MHz, CDCl3) of 2-butyl-4-methylquinoline (3e)
13C NMR (101 MHz, CDCl3) of 2-butyl-4-methylquinoline (3e)
S29
1H NMR (500 MHz, CDCl3) of 4-methyl-2-phenethylquinoline (3f)
13C NMR (126 MHz, CDCl3) of 4-methyl-2-phenethylquinoline (3f)
S30
1H NMR (500 MHz, (CD3)2CO) of 2-(4-chlorobutyl)-4-methylquinoline (3g)
13C NMR (126 MHz, (CD3)2CO) of 2-(4-chlorobutyl)-4-methylquinoline (3g)

S31
1H NMR (500 MHz, CDCl3) of 2-(3-hydroxypropyl)-4-methylquinoline (3h)
13C NMR (126 MHz, CDCl3) of 2-(3-hydroxypropyl)-4-methylquinoline (3h)
S32
1H NMR (400 MHz, CDCl3) of 4-methyl-2-(pent-4-en-1-yl)quinoline (3i)
13C NMR (101 MHz, CDCl3) of 4-methyl-2-(pent-4-en-1-yl)quinoline (3i)
Due to the small scale of the reaction (0.1 mmol) and relatively low yield (17%), the intensity of signals in 13C NMR spectrum is low.
Traces of n-hexane,
EtOAc and H grease
S33
1H NMR (600 MHz, CDCl3) of 4-methyl-2-(4,4,4-trifluorobutyl)quinoline (3j)
13C NMR (151 MHz, CDCl3) of 4-methyl-2-(4,4,4-trifluorobutyl)quinoline (3j)
S34
1H NMR (600 MHz) in CDCl3 of 2-(4-methylquinolin-2-yl)undecanamide (3k)
13C NMR (151 MHz) in CDCl3 of 2-(4-methylquinolin-2-yl)undecanamide (3k)
S35
1H NMR (500 MHz, CDCl3) of 5-((4-methylquinolin-2-yl)methyl)pyrrolidin-2-one (3l)
13C NMR (126 MHz, CDCl3) of 5-((4-methylquinolin-2-yl)methyl)pyrrolidin-2-one (3l)

S36
1H NMR (500 MHz, CDCl3) of 6-cyclohexylphenanthridine (10b)
13C NMR (126 MHz, CDCl3) of 6-cyclohexylphenanthridine (10b)
Traces of n-hexane
and H grease

S37
1H NMR (500 MHz, CDCl3) of a mixture of 2-cyclohexylquinoline (10c) and 2,4-
dicyclohexylquinoline at ratio 3:1 (S-di-10c)
13C NMR (126 MHz, CDCl3) of a mixture of 2-cyclohexylquinoline (10c) and
2,4-dicyclohexylquinoline at ratio 3:1 (S-di-10c)
S38
1H NMR (500 MHz, CDCl3) of 2-cyclohexyl-4-phenylpyridine (10d)
13C NMR (126 MHz, CDCl3) of 2-cyclohexyl-4-phenylpyridine (10d)

S39
1H NMR (400 MHz, CDCl3) of 2,6-dicyclohexyl-4-phenylpyridine (di-10d)
13C NMR (101 MHz, CDCl3) of 2,6-dicyclohexyl-4-phenylpyridine (di-10d)

S40
1H NMR (400 MHz, CDCl3) of 2,4,6-tricyclohexylnicotinonitrile (10e)
13C NMR (101 MHz, CDCl3) of 2,4,6-tricyclohexylnicotinonitrile (10e)
S41
1H NMR (600 MHz, CDCl3) of methyl 2,6-dicyclohexylnicotinate (10f)
13C NMR (151 MHz, CDCl3) of methyl 2,6-dicyclohexylnicotinate (10f)

S42
HSQC spectrum (1H NMR: 600 MHz, 13C NMR: 151 MHz, CDCl3) of methyl 2,6-
dicyclohexylnicotinate (10f)
HMBC spectrum (1H NMR: 600 MHz, 13C NMR: 151 MHz, CDCl3) of methyl 2,6-
dicyclohexylnicotinate (10f)
S43
1H NMR (400 MHz, CDCl3) of methyl 2,4,6-tricyclohexylnicotinate (tri-10f)
13C NMR (101 MHz, CDCl3) of methyl 2,4,6-tricyclohexylnicotinate (tri-10f)

download fileview on ChemRxivGiedyk et al SI.pdf (3.52 MiB)





























