Embed Size (px)
Citation preview

Recombinant engineering of gene function:
mutagenesisI. Why mutagenize?II. Random mutagenesis, mutant
selection schemesIII. Site-directed mutagenesis,
deletion mutagenesisIV. Engineering of proteinsV. Alterations in the genetic code
Lecture for Bio FMIPA UB, by Fatchiyah

Uses for mutagenesis
• Define the role of a gene--are phenotypes altered by mutations?
• Determine functionally important regions of a gene (in vivo or in vitro)
• Improve or change the function of a gene product
• Investigate functions of non-genes, eg. DNA regions important for regulation

Protein engineering-Why?
• Enhance stability/function under new conditions– temperature, pH, organic/aqueous
solvent, [salt]• Alter enzyme substrate specificity• Enhance enzymatic rate• Alter epitope binding properties

Enzymes: Biotech Cash Crops

From Koeller and Wang, “enzymes for chemical synthesis”, Nature 409, 232 - 240 (2001)
Obtaining useful enzymes

Random mutagenesis
• Cassette mutagenesis with “doped”oligos
• Chemical mutagenesis– expose short piece of DNA to mutagen,
make “library” of clones, test for phenotypes
• PCR mutagenesis by base misincorporation– Include Mn2+ in reaction– Reduce concentration of one dNTP

Random mutagenesis by PCR: the Green Fluorescent Protein
Screen mutants

Cassette mutagenesis (semi-random)
Strands synthesized individually, then annealed
Allows random insertion of any amino acid at defined positions
Translation of sequence

Random and semi-random mutagenesis: directed evolution
• Mutagenize existing protein, eg. error-prone PCR, doped oligo cassette mutagenesis
-- and/or --Do “gene shuffling”(Creates Library)
• Screen library of mutations for proteins with altered properties– Standard screening: 10,000 - 100,000
mutants– Phage display: 109 mutants

Gene shuffling: “sexual PCR”

Gene shuffling
For gene shuffling protocols you must have related genes in original pool: 1) evolutionary variants, or 2) variants mutated in vitro
Shuffling allows rapid scanning through sequence space:faster than doing multiple rounds of random mutagenesis and screening

Shuffling of one gene mutagenized in two ways

Gene shuffling--cephalosporinase from 4 bacteria
Single gene mutagenesis
Multiple gene shuffling

Screening by phage display: create library of mutant proteins fused to M13
gene III
Human growth hormone: want to generate variants that bind to hGH receptor more tightly
Random mutagenesis

Phage display:production of recombinant phage
The “display”

Phage display: collect tight-binding phage
The selection

Types of Mutagenesis• PCR Based Methods
– Site-directed mutagenesis– Mismatched mutagenesis– 5’ add on mutagenesis– Cassette Mutagenesis
• Insertional Mutagenesis– Trasposon mutagenesis
• In vivo Mutagenesis– Direct Mutagenesis

Mismatched Mutagenesis
• Similar to Site-Directed Mutagenesis
• But only focuses on a single amino acid
• Important when trying to determine a particular missense mutation in known gene of a disease.
• Or when just trying to evaluate the contributation of the single amino acid to the function of the protein.

Gene or cDNA is cloned into M13 vector
Screen for mutant
Use of M13 allows for single strand recombinant DNA recovery

5’ add on Mutagenesis
• Involves adding on a new sequence or chemical group to the 5’end of a PCR product
• This involves a particular way of designing the primers:– 3’ end of the primer matches the sequence of PCR
product.– 5’ end contains the novel sequence
• Suitable restriction site• Addition of a functional sequence (promoter sequence)• Modified nucleotide that contains labeled group, biotinlyated,
or fluorophore.

Uses and Limitations• PCR based methods are
useful in making specific mutations in the DNA
• Which is useful when studying different aspects of protein function
• With PCR based methods it is hard to replicate the mutated DNA…in order for replication to occur super competent cells must be used and are expensive!
• Screening can be tedious, usually requires sequencing to confirm if mutation occurred.

Cassette Mutagenesis
• Used to introduce multiple mutations into the DNA sequence
• Uses blunt ended DNA for insertion site of mutation
• Where mutation is inserted a 3 base pair direct terminal repeat is created
• The mutagenic codon cassette has two head to head SapI sites allowing for removal of all DNA except for mutation.

Targeted codon removed using restriction enzyme that creates blunt cut
SapI digestion creates 3’ overhang allowing for ligation.
Ligation, which creates final mutation

Uses and Limitations
• Typically used for protein structure but possibly used for gene function
• Less expensive than site directed mutagenesis to create several mutations, because there is no need for primers
• Requires the SapI restriction enzyme cut sites, and other cut sites flanking the target region for removal of DNA
• Works best when target region is contained in a small DNA fragment

Transposon Mutagenesis
• Transposon: a piece of short DNA that replicates by inserting into other pieces of DNA (plasmids, chromosomes, etc…)
• Useful for studying gene function because when the transposon moves into different location in the DNA it may cause a disruption in a gene or a set of genes.
• Transposons also have many useful properties for mutagenesis:– Cause clean mutations– Can be random or specific mutations– Typically encode for antibiotic resistance or some other
advantageous gene.– Can use a transposon that inserts at a high frequency– When used in bacteria it causes selectable phenotypes– Recognize specific sequence that is ~2-12 base pairs long

Uses and Limitations
• Primary use is for the study of gene function, though can be used to create gene fusions
• Usually easy to see a change in phenotype due to gene knockout
• Because the transposon inserts at a specific sequence, helps in determining where insertion occurred
• Not useful in large plasmids because many recognition sites could be contained in the a single plasmid
• Suicide vectors are used, though some may have limited replication, so further screening is needed

Site-directed mutagenesis:
primer extension method
Drawbacks:
-- both mutant and wild type versions of the gene are made following transfection--lots of screening required, or tricks required to prevent replication of wild type strand
-- requires single-stranded, circular template DNA

Alternative primer extension mutagenesis techniques

“QuikChangeTM” protocol
Advantage: can use plasmid (double-stranded) DNA
Destroys the template DNA (DNA has to come from dam+ host

Site-directed mutagenesis: Mega-primer
method
Megaprimer needs to be purified prior to PCR 2
Allows placement of mutation anywhere in a piece of DNA
A
B
Wild type template
First PCR
Second PCR

Domain swapping using “megaprimers” (overlapping PCR)
N- -C
Mega-primer
PCR 1
PCR 2
Domains have been swapped
Template 1
Template 2

PCR-mediated deletion mutagenesis
Target DNA
PCR products
Oligonucleotide design allows precision in deletion positions

Directed mutagenesis
• Make changes in amino acid sequence based on rational decisions
• Structure known? Mutate amino acids in any part of protein thought to influence activity/stability/solubility etc.
• Protein with multiple family members? Mutate desired protein in positions that bring it closer to another family member with desired properties

An example of directed mutagenesis
T4 lysozyme: structure known
Can it be made more stable by the addition of pairs of cysteine residues (allowing disulfide bridges to form?) without altering activity of the protein?

T4 lysozyme: a model for stability studies
Cysteines were added to areas of the protein in close proximity--disulfide bridges could form

More disulfides, greater stabilization at high T
Bottom of bar: melting temperature under reducing condtions
Top of bar: Melting temperature under oxidizing conditions
Green bars: if the effects of individual S-S bonds were added together

Stability can be increased - but there can be a cost in activity

The genetic code• 61 sense codons, 3 non-sense (stop) codons• 20 amino acids
• Other amino acids, some in the cell (as precursors to other amino acids), but very rarely have any been added to the genetic code in a living system
• Is it possible to add new amino acids to the code?• Yes...sort of
Wang et al. (2001) “Expanding the genetic code” Science 292, p. 498.

Altering the genetic code

Why add new amino acids to proteins?
• New amino acid = new functional group• Alter or enhance protein function
(rational design)• Chemically modify protein following
synthesis (chemical derivitization)– Probe protein structure, function– Modify protein in vivo, add labels and
monitor protein localization, movement, dynamics in living cells

How to modify genetic code?Adding new amino acids to the code--must bypass
the fidelity mechanisms that have evolved to prevent this from occurring
2 key mechanisms of fidelity
• Correct amino acid inserted by ribosome through interactions between tRNA anti-codon and mRNA codon of the mRNA in the ribosome
• Specific tRNA charged with correct amino acid because of high specificity of tRNA synthetase interaction
• Add new tRNA, add new tRNA synthetase

tRNA charging and usage
Charging: (tRNA + amino acid + amino acyl-tRNA synthetase)
Translation:(tRNA-aa + codon/anticodon interaction + ribosome)

• Chose tRNAtyr, and the tRNAtyr synthetase (mTyrRS) from an archaean (M.jannaschii)--no cross-reactivity with E. coli tRNAtyr and synthetase
• Mutate m-tRNAtyr to recognize stop codon (UAG) on mRNA
• Mutate m-TyrRS at 5 positions near the tyrosine binding site by doped oligonucleotide random mutagenesis
• Obtain mutants that can insert O-methyl-L-tyrosine at any UAG codon


Outcome
• Strategy allows site specific insertion of new amino acid--just design protein to have UAG stop codon where you’d like the new amino acid to go
• Transform engineered E. coli with plasmid containing the engineered gene
• Feed cells O-methyl tyrosine to get synthesis of full length gene
Utility of strategy
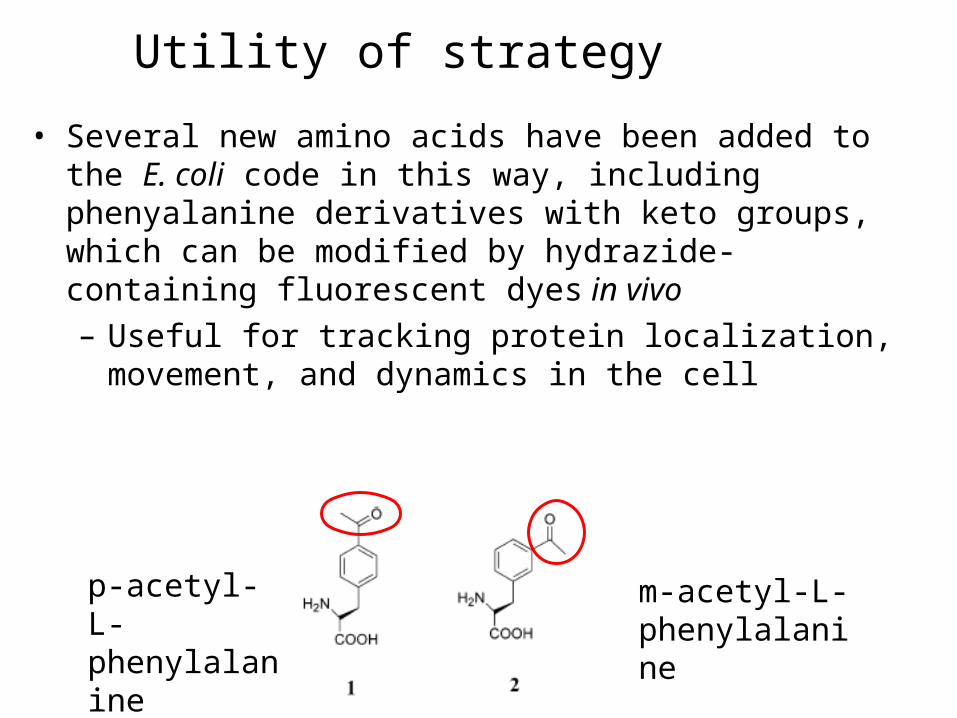
• Several new amino acids have been added to the E. coli code in this way, including phenyalanine derivatives with keto groups, which can be modified by hydrazide-containing fluorescent dyes in vivo– Useful for tracking protein localization,
movement, and dynamics in the cell
p-acetyl-L-phenylalanine
m-acetyl-L-phenylalanine

Some questions:
• What are the consequences for the cell with an expanded code?
• Do new amino acids confer any kind of evolutionary advantage to organisms that have them? (assuming they get a ready supply of the new amino acid…)
• Why do cells have/need 3 stop codons????





























