Embed Size (px)
Citation preview

Reactive Nitrogen Species Reactivities with Nitrones: Theoretical andExperimental StudiesKevin M. Nash,† Antal Rockenbauer,§ and Frederick A. Villamena*,†,‡
†Department of Pharmacology and ‡Davis Heart and Lung Research Institute, College of Medicine, The Ohio State University,Columbus, Ohio 43210, United States§Institute of Molecular Pharmacology, Research Center for Natural Sciences, H-1025 Budapest, Pusztaszeri 59, Hungary
*S Supporting Information
ABSTRACT: Reactive nitrogen species (RNS) such as nitrogen dioxide(•NO2), peroxynitrite (ONOO
−), and nitrosoperoxycarbonate (ONOOCO2−)
are among the most damaging species present in biological systems due to theirability to cause modification of key biomolecular systems through oxidation,nitrosylation, and nitration. Nitrone spin traps are known to react with freeradicals and nonradicals via electrophilic and nucleophilic addition reactions andhave been employed as reagents to detect radicals using electron paramagneticresonance (EPR) spectroscopy and as pharmacological agents against oxidativestress-mediated injury. This study examines the reactivity of cyclic nitrones suchas 5,5-dimethylpyrroline N-oxide (DMPO) with •NO2, ONOO
−, ONOOCO2−,
SNAP, and SIN-1 using EPR. The thermochemistries of nitrone reactivity withRNS and isotropic hfsc's of the addition products were also calculated at thePCM(water)/B3LYP/6-31+G**//B3LYP/6-31G* level of theory with andwithout explicit water molecules to rationalize the nature of the observed EPR spectra. Spin trapping of other RNS such as azide(•N3), nitrogen trioxide (•NO3), amino (•NH2) radicals and nitroxyl (HNO) were also theoretically and experimentallyinvestigated by EPR spin trapping and mass spectrometry. This study also shows that other spin traps such as 5-carbamoyl-5-methyl-pyrroline N-oxide, 5-ethoxycarbonyl-5-methyl-pyrroline N-oxide, and 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide can react with radical and nonradical RNS, thus making spin traps suitable probes as well as antioxidants against RNS-mediated oxidative damage.
■ INTRODUCTION
It has become clear that reactive nitrogen species (RNS) alongwith reactive oxygen species (ROS) have been implicated in thepathogenesis of various diseases1−6 or as key mediators in cellsignaling7 and immune response.8 Nitrosative stress (The term“nitrosative stress” is a misnomer as commonly used in theliterature because most manifestations of which are not due tonitrosation reaction but will be referred to in general as stresscaused by RNS.) results from a myriad of biomolecularmodifications9−15 caused by RNS such as the paramagneticnitric oxide (•NO), nitrogen dioxide (•NO2), and thediamagnetic peroxynitrite (ONOO−). Nitric oxide is knownto be formed in vivo by nitric oxide synthase (NOS) from L-arginine and O2
16 or can be released directly by syntheticmolecules such as S-nitroso-N-acetyl-D,L-penicillamine(SNAP).17 The NO-generating function of hemoglobin as anS-nitrosothiol synthase using nitrite as a substrate has beenproposed.18 Nitric oxide is a free radical that serves as anintracellular messenger and vasodilator, and its toxicity isgenerally limited to its reaction or oxidation to more highlyreactive species such as ONOO− and •NO2 (Scheme 1).19
Peroxynitrite is formed from the addition reaction of •NOwith superoxide (O2
•−) at a diffusion-controlled rate.20,21
Peroxynitrite is known to exist in the relatively stable cis-
conformation or to gain a proton to form peroxynitrous acid(ONOOH, pKa = 6.8). Like O2
•−, ONOO− is capable ofreacting with protein active sites containing Cu, Zn, sulfhydryl,and Fe−S clusters to cause nitration and protein cleavage,resulting in enzyme deactivation.22−25 Currently, indirectmethods of ONOO− detection are limited by their sensitivityand specificity, which include fluorescence,26−28 electro-chemistry,29 tyrosine, tryptophan, and DNA nitration,30,31 andindirect radical detection via electron paramagnetic resonance(EPR) spectroscopy.32 It should be noted, however, thatdetection of nitration is not exclusively an indication of
Received: December 4, 2011Published: July 9, 2012
Scheme 1. Formation of RNS from •NO
Article
pubs.acs.org/crt
© 2012 American Chemical Society 1581 dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−1597

peroxynitrite production and that the nitrating agent, NO2, canoriginate from other routes.33 Attempts to trap •NO and O2
•−
concurrently using Fe(DTCS)2 and various nitrones, respec-tively, only yielded an individual spectrum of •NO, O2
•−, andHO• adducts due to the varying rates of reaction of theseradicals with the spin traps.34 One emerging method ofdetection is the selective reaction of ONOO− with boronatescoupled with fluorescence spectroscopy.35
One relevant mechanism for ONOO−/ONOOH decay is itshomolytic cleavage through •ONO···O•− and •ONO···•OHintermediates (Scheme 2).36 For ONOOH, the rate of radical
cleavage has been reported to be 0.35 ± 0.03 s−1, with about30% •OH and •NO2 release at pH < 5 via escape from thesolvent cage. The rate constant for ONOOH isomerization tonitric acid (HNO3) was found to be 1.1 ± 0.1 s−1.37 Hydrolysisof •NO2 results in the formation of NO2
− and NO3−.37,38 For
ONOO−, the rate of radical cleavage has been reported at≈10−6 s−1, with negligible •NO2 and O•− release.39 While thelow rate of homolytic cleavage of ONOO− makes the reactiontrivial, ONOO− is known to react with dissolved CO2 to formnitrosoperoxycarbonate (ONOOCO2
−), an oxidizing speciesthat undergoes homolytic cleavage to form 30% CO3
•− and•NO2.
40,41 The decay of ONOOCO2− and ONOOH has been
shown to vary depending on the ability of the solvent to holdthe intermediate species in the solvent cage and is thereforedependent on the viscosity of solvent.42 Peroxynitrite is astrong nucleophile and has been shown to cause β-scission ofcarbonyl groups,43,44 where acyl- and H-spin adducts have beenobserved using EPR spin trapping.45,46 Peroxynitrite hasrecently been shown to form peroxynitrate (O2NOO
−) atneutral pH through a combination of ONOO− and ONOOHto form O2NOOH and nitrite (NO2
−).47 •NO2 is also knownto dimerize into N2O4 at moderate concentrations, whicheffectively hides its radical character.48
Nitrogen trioxide radical (•NO3) is an important oxidant inthe troposphere and has been considered as a major oxidant fora variety of unsaturated gas-phase organic species. Thereactivity of •NO3 radical with organic compounds occurs viahydrogen atom abstraction or addition to a double bond with arate constant range of 105−107 and 5 × 107 M−1 s−1,respectively.49 The azide anion binds to many heme proteinsand metalloenzymes inhibiting their activities. It is also a potent
inhibitor of the mitochondrial electron transport chain byblocking cytochrome c oxidase and preventing ATP hydrol-ysis.50 The azidyl radical (•N3) has been generated in solution,and their formation was observed using a spin trappingtechnique via oxidation of azide anion by peroxidases in thepresence of H2O2,
51 succinate-driven respiration in azide-inhibited rat brain submitochondrial particles,52 or via oxidationof azide anion in cadmium sulfide and zinc oxide suspensions.53
Ammonia is one of the most important trace gases in theatmosphere. Activation of NH3 in aqueous solution leads to theformation of a reactive amidogen (•NH2) radical, which canlater react with a variety of molecular species such as O2, aminoacids, and melanins.54 Nitroxyl (HNO) (pKa = 11.4) is the one-electron reduction product of •NO and has been shown toregulate cellular function with unique pharmacological proper-ties, specifically to cardiovascular diseases.55
Although EPR has been used to detect RNS in biologicalsystems with hydroxylamine redox probes [e.g., 1-hydroxy-2,2,6,6-tetramethyl-4-oxo-piperidine (TEMPONE-H)],56 theinterpretation of the observed signal due to the formation ofnitroxide is complicated by the fact that the nitroxide can alsobe formed from a variety of other reactions.57 In this study, weexplored the reactivities of various RNS using EPR spintrapping. The spin trapping technique unambiguously identifiesfree radicals via formation of a persistent radical adduct fromthe addition reaction of a radical to a nitrone spin trap.58,59
Furthermore, spin adducts have also been shown to be formedvia nucleophilic addition reaction to nitrones and subsequentoxidation to the paramagnetic adduct (Forrester−Hepburnmechanism), as demonstrated by others,60,61 making thistechnique appropriate to study reactive nonradical species aswell. Another path is that of inverted spin trapping as proposedby Eberson62 where one-electron oxidation of nitrone yields thenitrone radical cation [nitrone]•+ and its subsequent addition toa nucleophile (Nu−) forms the nitrone-Nu spin adduct.Nitrones have been used for the detection of RNS63 andhave been shown to trap decomposition products and tertiaryradicals formed from ONOO−.64 In this study, we alsocomputationally investigated the thermodynamics of RNSreaction to nitrones with the aim to rationalize the nature ofthe EPR spectral data obtained from the reactions. Studies onthe reaction of RNS with nitrones are important, not only forthe purpose of RNS detection but also to partly provide arationale for the protective properties of nitrones against RNS-mediated cellular toxicity.65
■ EXPERIMENTAL PROCEDURESComputational Study. An initial conformational search of the
RNS, ROS, and spin traps and their respective adducts was carried outusing Spartan 04 at the MMFF level. Density functional theory(DFT),66 at the B3LYP/6-31G* level of theory, was employed in thisstudy to determine the optimized geometry, and each yielded noimaginary vibrational frequency. All calculations were performed withGaussian 0367 at the Ohio Supercomputer Center. A scaling factor of0.980668 was used for the zero-point vibrational energy (ZPE)corrections for all of the B3LYP/6-31G* geometries. The effects ofsolvation on the gas-phase calculations were also investigated by usingthe PCM,69,70 and the spin and charge densities were obtained fromnatural population analysis (NPA) approach,71 at the PCM/B3LYP/6-31+G** level of theory. Optimization was also performed for adductswith two explicit water molecules for prediction of hyperfine splittingconstants at the PCM(water)/B3LYP/6-31+G**//B3LYP/6-31G*.Negligible spin contamination of 0.75 < ⟨S2⟩ < 0.76 was obtained forall of the minima. Using the B3LYP/6-31G* optimized structures, an
Scheme 2. Proposed Decomposition and Reactions ofONOO− and Their Respective ΔG298K,rxn (in kcal/mol)Calculated at the PCM/B3LYP/6-31+G**//B3LYP/6-31G*Level of Theory
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971582

additional calculation was performed for RNS/ROS shown in Table 1at the G3(MP2) level of theory, which is known to perform better
than DFT for calculations involving enthalpies of formation, ionizationpotentials, electron affinities, and proton affinities.72,73
General EPR Experiments. EPR measurements were taken atroom temperature with microwave power 10 mW, modulationamplitude 1 G, receiver gain 1.0 × 105, scan time 21.4 s, timeconstant 42.0 s, and sweep width 120 G. Solvents used include DMSOor a 10 mM potassium phosphate buffer (pH 7.0) containing 100 μMdiethylenetriaminepentaaceticacid (DTPA). All solutions werebubbled with nitrogen gas prior to irradiation. The sample cells usedwere 50 μL quartz or glass capillary tubes for UV or non-UVirradiation experiments, respectively. For UV irradiation experiments,the sample cell was irradiated with a Spectroline low-pressure mercuryvapor lamp with 0.64 cm × 5.4 cm dimensions and a 254 nmwavelength. The spectrum simulation was carried out by an automaticfitting program.74 All hyperfine splitting constants are in gauss.
Peroxynitrite Trapping. Peroxynitrite (≥90% in 0.3 M NaOH)was commercially obtained with nitrite/nitrate as impurities and wasstored at −86 °C. The peroxynitrite concentration was determinedspectrophotometrically at 302 nm. In a typical experiment, solutionscontain 20 mM 5,5-dimethyl-pyrroline N-oxide (DMPO) and 8−12mM ONOO− in 70% (v/v) DMSO in PBS. For inert atmosphereconditions, solvents [e.g., 70% (v/v) DMSO in PBS] were first purgedwith argon before making the DMPO and ONOO− solutions. ForONOO− in 100% water, ONOO− was prepared according to themethod previously described.75 One milliliter of aqueous solutions of0.7 M HCl and 0.6 M H2O2 was extruded simultaneously with 1 mL of0.6 M NaNO2 using 1 mL plastic syringes into a stirred ice-cold 0.3mL solution of 3 M NaOH. The excess H2O2 was removed withMnO2. The concentration of ONOO
− solution was calculated to be 15mM based on an extinction coefficient of 1700 M−1 cm−1 at 302 nm.For ONOOCO2
− generation, a solution of DMPO in DMSO waspurged with argon and subsequently bubbled with CO2. A stock argon-purged DMSO solution of ONOO− was then added to the DMPOsolution to make a 70% (v/v) DMSO−30% PBS final solution.
•NO2 Generation. (a) A solution containing 20 mM nitrone and300 mM NaNO2 in PBS was transferred to a quartz capillary tube andwas UV photolyzed in the EPR cavity according to previousprocedure.76 (b) DMSO was bubbled with argon for 15 min, andthen, •NO gas was bubbled through the solvent for 15 min. A 25 μL of•NO-saturated DMSO solution was then added to a 25 μL solution of50 mM nitrone in DMSO.
Table 1. Free Energies of Reduction, ΔGaq,298K (in kcal/mol), of Selected RNS and ROS at the PCM(Water)/B3LYP/6-31+G**//B3LYP/6-31G* and PCM(Water)/G3(MP2)//B3LYP/6-31G* (in Parentheses) Levels ofTheory
entry ΔG298K,red
ONOOCO2− + 2e−/NO2
− + CO32− −242.5 (−236.5)
N2O4 + 2e−/2NO2− −240.3 (−232.1)
cis-ONOOH + 2e−/HO− + NO2− −237.1 (−230.3)
trans-ONOOH + 2e−/HO− + NO2− −235.6 (−231.8)
trans-ONOO− + 2e−/O2− + NO2− −170.5 (−158.0)
cis-ONOO− + 2e−/O2− + NO2− −166.3 (−153.9)
•NO3 + e−/NO3− −152.3 (−155.0)
NO+ + e−/•NO −146.6 (−140.7)DMPO•+ + e−/DMPO −139.7 (−147.8)•N3 + e−/N3
− −124.1 (−121.0)•NO2 + e−/NO2
− −120.4 (−116.8)•OH + e−/HO− −118.9 (−120.0)CO3
•− + e−/CO32− −103.3 (−115.9)
N2O4 + e−/N2O4•− −102.0 (−115.3)a
HO2• + e−/HO2
− −97.2 (−97.0)cis-ONOOH + e−/cis-ONOOH•− −86.5 (−78.6)•NH2 + e−/NH2
− −85.9 (−89.9)trans-ONOOH + e−/trans-ONOOH•− −83.3 (−80.0)b•NO + e−/3NO− −82.6 (−69.0)ONOOCO2
− + e−/ONOOCO2•2− −81.7 (−68.1)
trans-ONOO− + e−/trans-ONOO•2− −62.8 (−50.0)cis-ONOO− + e−/cis-ONOO•2− −55.7 (−45.9)b
O2•− + e−/O2
2− −53.7 (−49.6)aCalculation using NO2
− and •NO2 as products for G3(MP2) onlyand could be an overestimate. bEnergies for cis-ONOOH•− and trans-ONOO•2− were used for the G3(MP2) calculation only.
Figure 1. Optimized geometries showing bond lengths (in Å), charge and spin densities (e) (in parentheses), and free energies of •NO2 addition toDMPO at the PCM/B3LYP/6-31+G**//B3LYP/6-31G* level of theory.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971583
Spin Trapping Using SNAP. The final aqueous solution contains100 mM nitrone and 65 mM SNAP. For inert atmosphere conditions,solutions of nitrone and SNAP were argon purged prior to theiraddition to prevent •NO2 formation. An aliquot of 70 μL wastransferred to capillary tube, and the sample was irradiated in the cavityusing visible light while acquiring EPR spectra.Generation of •N3,
•NH2,•NO3, and HNO. PBS (pH 7.4)
solutions of NaN3, NH2OH, or HNO3 (100 mM) in the presence of0.2% H2O2 and 50 mM DMPO were UV irradiated to afford the
corresponding radical adducts. Spin adducts from HNO were carriedout using Ar purged Angeli's salt solution (120 mM) in distilled waterin the presence of DMPO (100 mM) and subsequent acidificationwith HNO3 to give a final acid concentration of 20 mM.
17O Isotopic Labeling Studies. O-17-labeled nitrogen dioxide,•N17O2, was prepared according to the procedure shown above by
mixing saturated DMSO solutions of •NO and O2 (28%17O atom) in
DMSO.
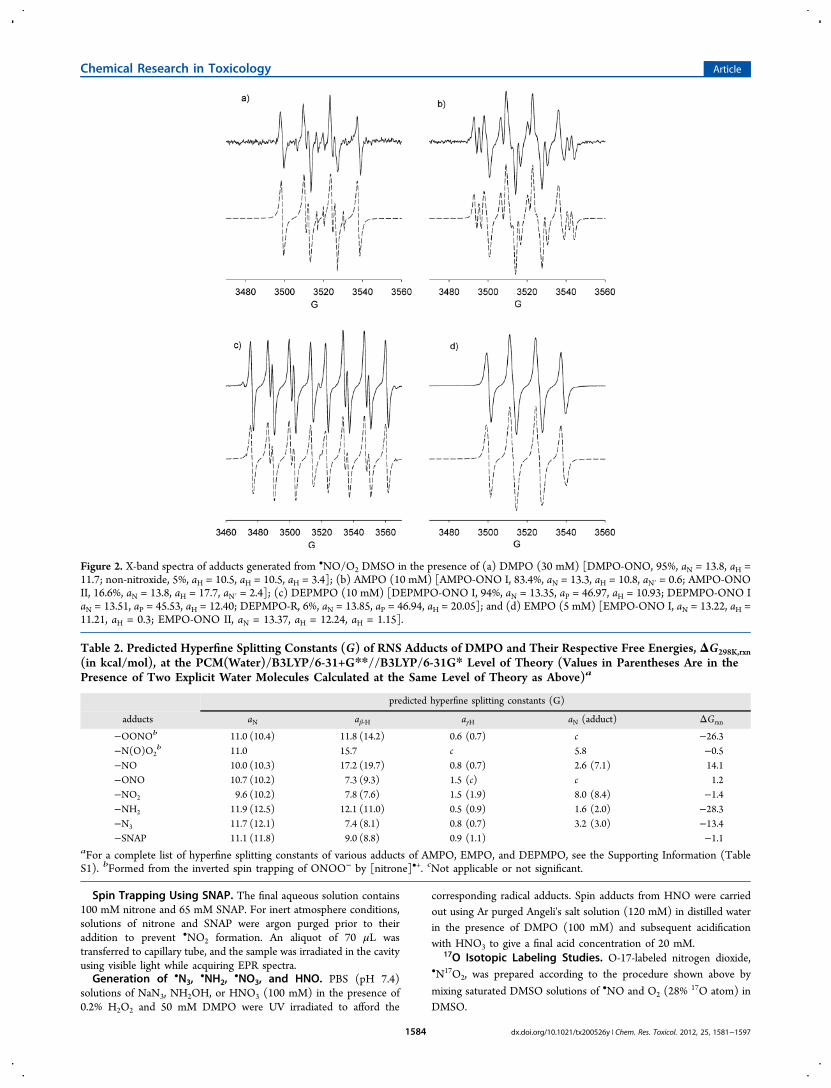
Figure 2. X-band spectra of adducts generated from •NO/O2 DMSO in the presence of (a) DMPO (30 mM) [DMPO-ONO, 95%, aN = 13.8, aH =11.7; non-nitroxide, 5%, aH = 10.5, aH = 10.5, aH = 3.4]; (b) AMPO (10 mM) [AMPO-ONO I, 83.4%, aN = 13.3, aH = 10.8, aN′ = 0.6; AMPO-ONOII, 16.6%, aN = 13.8, aH = 17.7, aN′ = 2.4]; (c) DEPMPO (10 mM) [DEPMPO-ONO I, 94%, aN = 13.35, aP = 46.97, aH = 10.93; DEPMPO-ONO IaN = 13.51, aP = 45.53, aH = 12.40; DEPMPO-R, 6%, aN = 13.85, aP = 46.94, aH = 20.05]; and (d) EMPO (5 mM) [EMPO-ONO I, aN = 13.22, aH =11.21, aH = 0.3; EMPO-ONO II, aN = 13.37, aH = 12.24, aH = 1.15].
Table 2. Predicted Hyperfine Splitting Constants (G) of RNS Adducts of DMPO and Their Respective Free Energies, ΔG298K,rxn(in kcal/mol), at the PCM(Water)/B3LYP/6-31+G**//B3LYP/6-31G* Level of Theory (Values in Parentheses Are in thePresence of Two Explicit Water Molecules Calculated at the Same Level of Theory as Above)a
predicted hyperfine splitting constants (G)
adducts aN aβ‑H aγH aN (adduct) ΔGrxn
−OONOb 11.0 (10.4) 11.8 (14.2) 0.6 (0.7) c −26.3−N(O)O2
b 11.0 15.7 c 5.8 −0.5−NO 10.0 (10.3) 17.2 (19.7) 0.8 (0.7) 2.6 (7.1) 14.1−ONO 10.7 (10.2) 7.3 (9.3) 1.5 (c) c 1.2−NO2 9.6 (10.2) 7.8 (7.6) 1.5 (1.9) 8.0 (8.4) −1.4−NH2 11.9 (12.5) 12.1 (11.0) 0.5 (0.9) 1.6 (2.0) −28.3−N3 11.7 (12.1) 7.4 (8.1) 0.8 (0.7) 3.2 (3.0) −13.4−SNAP 11.1 (11.8) 9.0 (8.8) 0.9 (1.1) −1.1
aFor a complete list of hyperfine splitting constants of various adducts of AMPO, EMPO, and DEPMPO, see the Supporting Information (TableS1). bFormed from the inverted spin trapping of ONOO− by [nitrone]•+. cNot applicable or not significant.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971584
Mass Spectral Studies. (a) GC-MS analysis was carried out usinga positive ion electron impact ionization (EI) detection. Tenmicroliters of the CH3Cl fraction was injected to the column at aninitial temperature of 40 °C with a ramp of 20 °C min−1 up to amaximum of 250 °C. MS detection was conducted at a 200 °C ionsource temperature, electron energy of 70 eV, and scan speed of1.6584 scans s−1. In a typical experiment for DMPO-NO2 adductformation, 1 μL of pure DMPO (∼10 M) was added to a 25 μL of O2-saturated DMSO solution followed by 25 μL of NO-saturated DMSOsolution. Seventy microliters of CH3Cl was then added, the resultingmixture was vigorously mixed, and 10 μL of the bottom CH3Clfraction was injected into the column. The procedure was repeated forDMPO alone and O2/NO in the absence of DMPO. For DMPO-OONO, ONOO− was prepared according to the method previouslydescribed.75 A 70 μL aliquot solution of ONOO− (15 mM) was addedto a 1 μL of pure DMPO followed by 70 μL of CH3Cl. The resultingmixture was vigorously mixed, and 10 μL of the bottom CH3Clfraction was injected into the column. The procedure was repeated forDMPO alone and ONOO− solution in the absence of DMPO. ForGC-MS analysis of DMPO adducts generated from ONOOCO2
−,SNAP, •NO3,
•NH2,•N3, and HNO, procedures for their generation
as mentioned above were followed except that distilled water was usedinstead of PBS. (b) Time-of-flight (TOM) mass spectral analysis wascarried out on DMPO-ONOO system only since solution containingDMSO from the generation of DMPO-NO2 is not compatible for thistype of analysis. To 70 μL of freshly prepared aqueous solution ofONOO− (15 mM) was added 1 μL of pure DMPO. The resultingsolution (70 μL) was directly infused into the spectrometer. Allmeasurements were done in triplicate.
■ RESULTS AND DISCUSSION
Redox Properties. Free energies (ΔG298K,red) of the variousRNS formation and decomposition pathways were computa-tionally studied at the PCM/B3LYP/6-31+G**//B3LYP/6-
31G* level of theory. Reduction free energies (ΔG298K,red) foreach of the species were calculated and are shown in Table 1.Three oxidizing ROS, O2
•−, HO2•, and •OH, were also
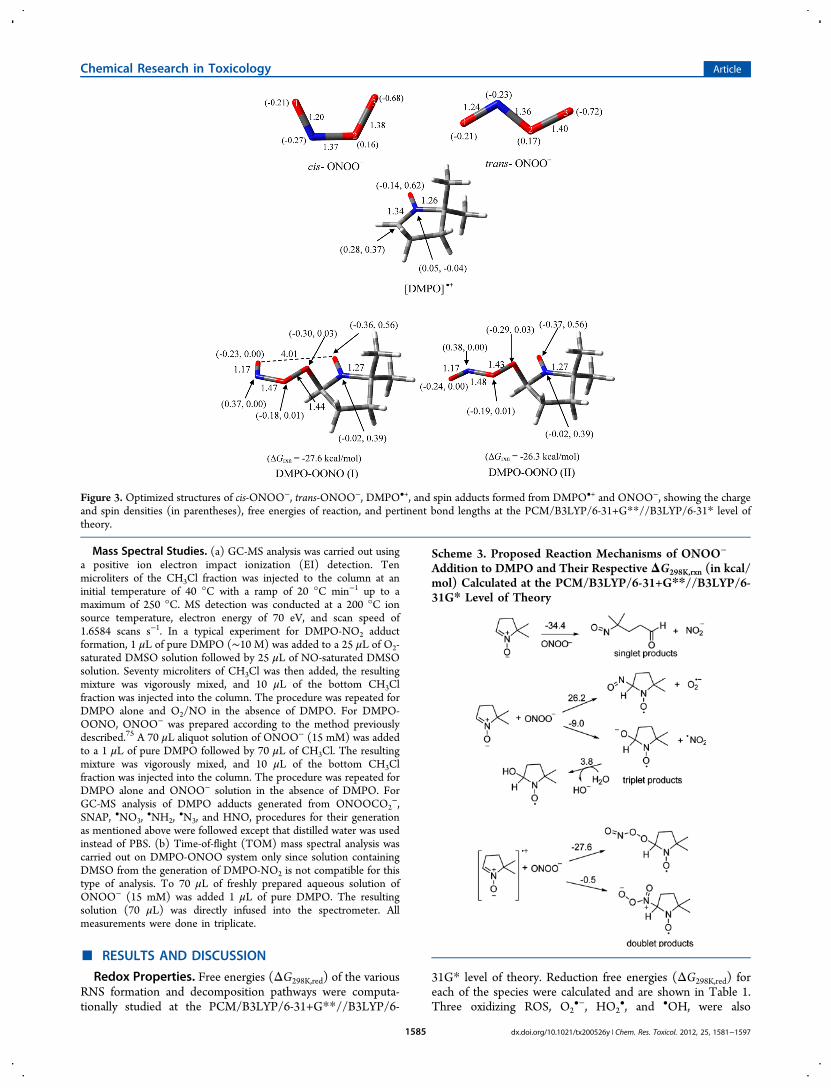
Figure 3. Optimized structures of cis-ONOO−, trans-ONOO−, DMPO•+, and spin adducts formed from DMPO•+ and ONOO−, showing the chargeand spin densities (in parentheses), free energies of reaction, and pertinent bond lengths at the PCM/B3LYP/6-31+G**//B3LYP/6-31* level oftheory.
Scheme 3. Proposed Reaction Mechanisms of ONOO−
Addition to DMPO and Their Respective ΔG298K,rxn (in kcal/mol) Calculated at the PCM/B3LYP/6-31+G**//B3LYP/6-31G* Level of Theory
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971585
included for comparison. For one-electron reduction reactions,the ΔG298K,red of •NO3 to NO3
− was found to be the mostfavorable, even more so than the ΔG298K,red of the highlyreactive •OH. Reduction of N2O4 resulted in an 18.4 kcal mol
−1
decrease in ΔG298K,red as compared to its monomeric form•NO2. cis-Peroxynitrite was found to be the least oxidizing ofthe RNS's studied (ΔG298K,red = −55.7 kcal/mol), and has freeenergy of reduction close to O2
•− (ΔG298K,red = −53.7 kcal/mol). Previous studies77 support our theoretical data showingthat trans-ONOO− is more oxidizing than cis-ONOO−, by 7.1kcal mol−1. In contrast to ONOO−, the protonated form, cis-ONOOH, is more oxidizing by 3.2 kcal mol−1 than its transanalogue, but in general, cis- or trans-ONOOH is moreoxidizing than the cis- and trans-ONOO−.The ΔG298K of oxidation of DMPO to DMPO•+ was found
to be 139.7 kcal mol−1;78 therefore, •NO3 is the only RNS inthis study that can spontaneously oxidize DMPO with anoverall ΔG298K,rxn = −12.6 kcal mol−1. Thermodynamics ofoxidation of DMPO by ONOO− is also unlikely due to thehighly endoergic free energy for this process of 84 kcal/mol ashad been previously been ruled out by other.64 Because of thehigh oxidation potential of DMPO (EDMPO•+/DMPO = 1.63 V)and by using [17O] labeling and EPR spin trapping, the
formation of DMPO•+ from HO• and SO4•− was discarded.60
Instead, nucleophilic addition to DMPO and its subsequentoxidation to give an EPR detectable species is the moreplausible mechanism for radical adduct formation in sulfite/horseradish peroxidase (HRP)/H2O2 system. RNS reactionswith nitrones may not be limited to simple electron transferreactions; therefore, nucleophilic addition reactions of RNS tonitrones will be explored in the subsequent sections.We performed additional calculations at the G3(MP2)//
B3LYP/6-31G* level of theory in aqueous phase. G3(MP2)values have been shown to give accurate gas-phase organicthermochemistries for small molecules.79 Using the G3(MP2)approach, more exoergic free energy of reduction was observedfor HO• as compared to that of •NO2 and is more consistentwith the experimental thermochemical data of E°(HO•
g/HO−
aq) = 1.98 V80 and E°(•NO2/NO2−aq) = 1.04 V.81
Nitrogen Dioxide Radical (•NO2). Figure 1 shows theoptimized geometries of •NO2 and the O- and N-centeredradical adducts, DMPO-NO2. NBO calculation of •NO2 showsnegative charges on the two O-atoms and a positive charge onthe N-atom with 44% of the spins residing on the N-atom and25% on each of the O-atoms. The N-centered radical adductformation was exoergic with ΔG298K,rxn = −1.4 kcal/mol, while
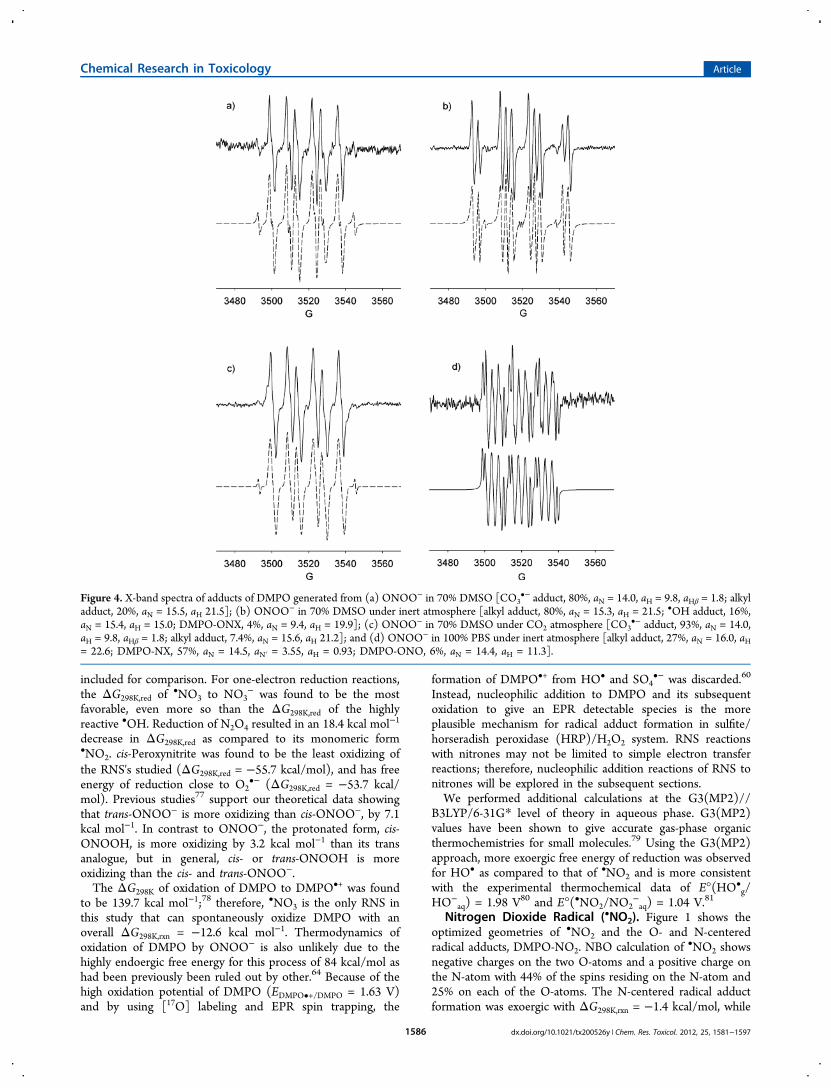
Figure 4. X-band spectra of adducts of DMPO generated from (a) ONOO− in 70% DMSO [CO3•− adduct, 80%, aN = 14.0, aH = 9.8, aHβ = 1.8; alkyl
adduct, 20%, aN = 15.5, aH 21.5]; (b) ONOO− in 70% DMSO under inert atmosphere [alkyl adduct, 80%, aN = 15.3, aH = 21.5; •OH adduct, 16%,aN = 15.4, aH = 15.0; DMPO-ONX, 4%, aN = 9.4, aH = 19.9]; (c) ONOO− in 70% DMSO under CO2 atmosphere [CO3
•− adduct, 93%, aN = 14.0,aH = 9.8, aHβ = 1.8; alkyl adduct, 7.4%, aN = 15.6, aH 21.2]; and (d) ONOO− in 100% PBS under inert atmosphere [alkyl adduct, 27%, aN = 16.0, aH= 22.6; DMPO-NX, 57%, aN = 14.5, aN′ = 3.55, aH = 0.93; DMPO-ONO, 6%, aN = 14.4, aH = 11.3].
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971586

the O-centered radical adduct is slightly endoergic withΔG298K,rxn = 1.2 kcal/mol. Unlike O2
•−, the more favoredaddition of an electropositive center (i.e., the N-atom) to thenitronyl-C indicates that the •NO2 addition to DMPO iselectrophilic in nature, which may be due to the high spindensity distribution on the N-atom. The small energy differencebetween the N-centered and the O-centered addition reactioncould also indicate that the latter mechanism can occur as willbe discussed below.Attempts to generate •NO2 by UV irradiation of NaNO2
with DMPO in PBS only yielded an acyclic adduct with aN =7.3, aH = 4.1, and aH = 4.1 (figure not shown). Spin trappingusing •NO/O2 solution in DMSO, however, gave anunidentified O-centered radical adduct, which will be calledDMPO-OX with aN = 13.8 and aH = 11.7 (see Figure 2a).Table 2 and Table S1 in the Supporting Information list thepredicted hfsc's for various •NO2 adducts (in the presence ofbulk dielectric effect of water as well as in the presence of twoexplicit water molecules) and shows that N-centered radicaladduct must exhibit a significant nitro-N hfsc, but experimentalevidence shows otherwise, indicating that the O-centeredradical adduct could be the major product in DMSO solution.In an attempt to further prove the formation of O-centeredDMPO adduct with NO2, the EPR spectrum was obtained from
mixing 17O-labeled O2 (28% O-17 atom) and •NO-saturatedDMSO solutions in the presence of DMPO. The solution onlyyielded the same spectrum as above with aN = 13.8 G and aH =11.8 G and did not yield hfsc originating from 17O due perhapsto the low concentration of N17O2 formed from the complexmechanism of its formation from •NO and O2 as well as thelow final yield of DMPO- N17O2. The nonobservance of the17O was exacerbated by its nuclear spin of I = 5/2 that candrastically reduce the amplitude of the spectrum by a factor of6. GC-MS analysis, however, showed a distinctive GC peak at∼4.4 min with a significantly intense base mass peak of 158.98m/z corresponding to the DMPO-NO2 adduct (see Figure S7ain the Supporting Information). Considering that the energydifference for the formation of N- and O-centered radicaladducts is only 2.6 kcal/mol, the formation of O-centeredradical adduct is not improbable. Moreover, the addition of theelectronegative center (i.e., the O atom) to the nitrone followsthat of the nucleophilic addition reaction of O2
•− to nitrones.On the basis of the thermodynamics of O-centered radicaladduct formation from NO2, which is only 2.6 kcal/moldifference as compared to the formation of the N-centeredadduct, the fact that the reaction of NO2 with DMPO yieldedan adduct with no additional hfsc due to N, and that the massspectral analysis is consistent with DMPO-NO2 formula, it isreasonable to assume that the adduct formed has a generalstructure of DMPO-ONO.Spin trapping using •NO/O2 in the presence of 5-carbamoyl-
5-methyl-pyrroline N-oxide (AMPO), 5-ethoxycarbonyl-5-methyl-pyrroline N-oxide (EMPO), or 5-(diethoxyphosphor-yl)-5-methyl-1-pyrroline N-oxide was also carried out (Figure2b−d). Similar to that observed for DMPO, significant amountsof nitrone-ONO adducts were evident. Previously,82 it has beenshown that olefins exhibited fast reactions with radicalsgenerated from •NO/O2 systems yielding •NO2 additionproduct via O-atom attack. Figure 1 shows the thermodynamicsof nitrone-NO2 formation, which generally shows slightlyhigher thermodynamic favorability for the formation of thetrans-isomers as compared to the corresponding cis-isomers, aswell as •NO2 addition via the O-atom rather than via the N-atom addition. Figure 2 shows the calculated hfsc's for the morepreferred nitrone-ONO adducts, and qualitative trends can bemade from the relative magnitudes of aN and aβ‑H, as well as thepresence of additional aN for the AMPO-ONO cis and transadducts as experimentally observed (Figure 2b).The formation of NO2 from NO and O2 is quite complex
and may form intermediate species that can react with nitrones.The formation of intermediate species such as N2O4 or NO3should yield nitrone-ONO and nitrone-ONO2 (to ultimatelygive DMPO-OH), respectively. Because the formation ofnitrone-OH adduct was not evident in any of the spectra inFigure 2, therefore, one cannot discount the formation ofnitrone-ONO from N2O4 since the free energy of N−Nhomolytic cleavage for N2O4 to give two molecules of NO2 isonly endoergic by 0.5 kcal/mol.
Peroxynitrite (ONOO−) and Peroxynitrous Acid(ONOOH). The electronic property of ONOO− as shown inFigure 3 is characterized by high negative charge density on theterminal peroxy-O, O(3), for both cis and trans isomers(−0.68e and −0.72e, respectively). The charge density on thenitroso-O, O(1), is less negative with a charge of −0.21e. Thecis-ONOO− conformation is 4.2 kcal/mol more favored thanthe trans isomer. Scheme 3 shows the various modes ofaddition reactions of ONOO− to DMPO. Theoretical studies
Scheme 4. Proposed Reaction Mechanisms of ONOO− withDMPO with the Respective ΔG298K,rxn Calculated at thePCM/B3LYP/6-31+G**//B3LYP/6-31G* Level of Theory
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971587
show that nucleophilic addition of ONOO− to DMPO resultsin ring opening to form the nitroso-aldehyde and nitrite withΔG298K,rxn of −34.4 kcal/mol. The formation of a triplet adductfrom ONOO− and DMPO gave a highly endoergic product forthe O(1) addition [where O(1) corresponds to the doublybonded O] with ΔG298K,rxn = 54.1 kcal/mol (figure not shown).The addition of the terminal peroxyl-O, that is, O(3), toDMPO gave exoergic free energy of −9.0 kcal/mol yielding•NO2 and DMPO-O•− radicals, but the formation of DMPO-NO and O2
•− via addition through the N atom gave endoergicΔG298K,rxn of 26.2 kcal/mol. It has been previously shown thatthe reaction of ONOO− with DMPO in 17O-labeled wateryielded DMPO-OH spectra under low oxygen tensions as wellas a methyl radical adduct to DBNBS in the presence ofONOO− in DMSO.64 This experimental evidence supports thatnucleophilic addition of ONOO− to DMPO can result in theformation of DMPO-OH adduct from DMPO-O•− withslightly endoergic ΔG298K,rxn of 3.8 kcal/mol (Scheme 3). Theformation of doublet adducts from ONOO− via its addition to[DMPO]•+ is highly exoergic with the O(3) addition being themost favorable with ΔG298K,rxn = −26.3 kcal/mol.EPR spin trapping of ONOO− in the presence of DMPO was
initially performed in DMSO/H2O. Figure 4a shows that thespectrum obtained under normal atmospheric conditions wasmarkedly different than those obtained when the solution waspurged with argon (Figure 4b). EPR simulation of the resultingspectra under normal atmospheric conditions revealed hfsc'scorresponding to an unknown O-centered radical adduct,DMPO-OX (80%; aN = 14.0, aH = 9.8, and aHγ = 1.8) and a C-centered radical adduct, DMPO-R (20%; aN = 15.5 and aH =21.5). The nature of the DMPO-OX will be further discussed inthe succeeding section. Using the same spin trappingconditions with an argon purge yielded a different mixture ofradical adducts: a C-centered radical adduct DMPO-R (80%;
aN = 15.3 and aH = 21.5), DMPO-OH (16%; aN = 15.4 and aH= 15.0), and an adduct assigned to DMPO-ONX (4%; aN = 9.4and aH = 19.9). The DMPO-R adduct can be assigned to theformation of DMPO-CH3, perhaps due to the formation ofHO• from the O−O homolytic cleavage of ONOOH. The O−O homolytic cleavage of ONOOH to form HO• and •NO2 wascalculated to be slightly endoergic (ΔG298K,rxn = 2.2 kcal/mol)(Scheme 2), and while this process is likely to occur in solution,the formation of DMPO-OH, where the HO• originates fromONOOH, was previously confirmed using 17O-labeled water.64
The hfsc's observed for the formation of small amounts ofDMPO-ONX (aN = 9.4 and aH = 19.9) follow a qualitativetrend predicted for the formation of DMPO-OONO (Table 2)in which the aH is more improved (i.e., 14.2 G) in the presenceof explicit interaction of water molecules as compared to justconsidering the bulk dielectric effect of water. However,DMPO-OONO can only be formed via inverted spin trappingthrough ONOO− addition to [DMPO]•+. The question onhow [DMPO]•+ could be formed in solution can be accountedfor by the •NO3 oxidation of DMPO to DMPO•+. However,one could argue that HO• can further react with nitrateimpurity (as HNO3) to yield •NO3 (ΔG298K,rxn = −12.9 kcal/mol) or via H-atom abstraction from HOONO and isomer-ization of the •OONO formed to •NO3 with a total ΔG298K,rxn= −46.0 kcal/mol (see Scheme 2). Hence, •NO3 can oxidizeDMPO to DMPO•+ to form DMPO-OONO via inverted spintrapping with ONOO−. This oxidation process is exoergic withΔG298K,rxn of −12.6 kcal/mol, and subsequent addition ofONOO− to DMPO•+ gave highly favorable ΔG298K,rxn of −27.6kcal/mol (Figure 3) giving a net ΔG298K,rxn of −40.2 kcal/molfor the formation of an EPR-detectable DMPO-OONO. Theinitial endoergic process for the •NO3 oxidation of DMPO toDMPO•+ could explain the low yield (4%) of DMPO-OONOformed in solution. Also, the formation of DMPO-N(O)O2 can
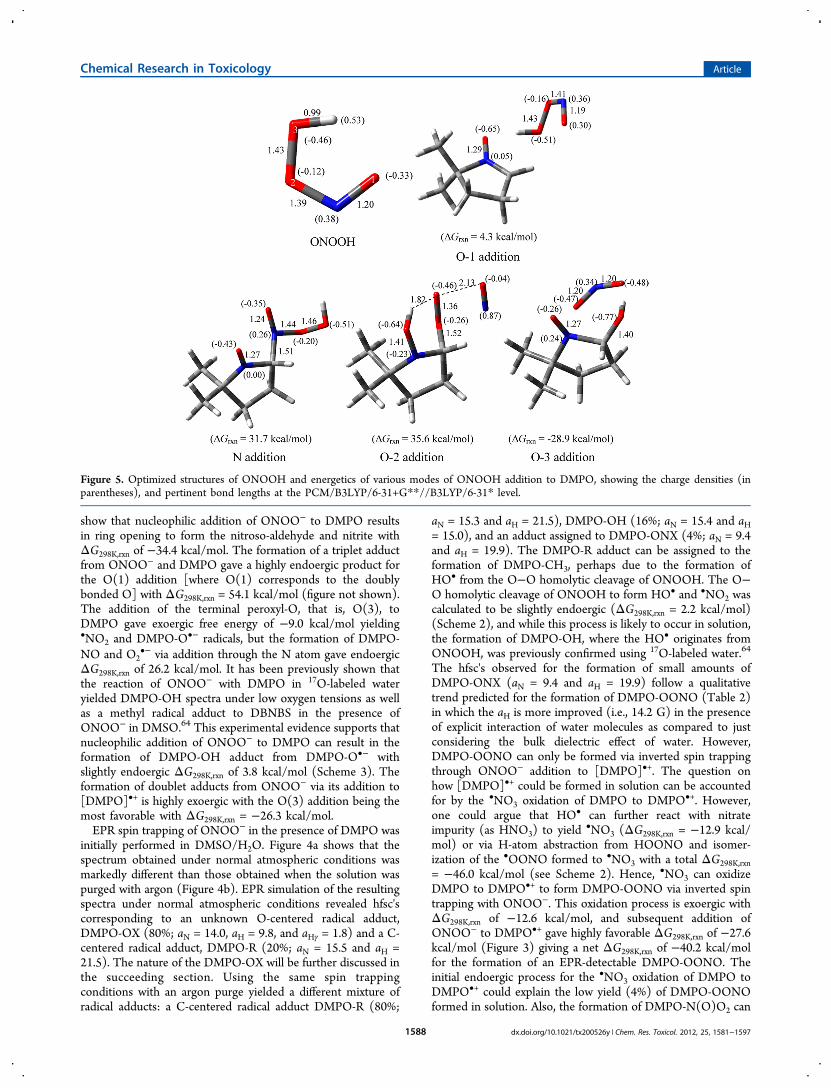
Figure 5. Optimized structures of ONOOH and energetics of various modes of ONOOH addition to DMPO, showing the charge densities (inparentheses), and pertinent bond lengths at the PCM/B3LYP/6-31+G**//B3LYP/6-31* level.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971588
be ruled out due to the lack of additional aN from the EPRspectrum and because the formation of DMPO-OONO is moreexoergic with ΔG298K,rxn = −40.2 kcal/mol than the formationof DMPO-N(O)O2 with ΔG298K,rxn = −0.5 kcal/mol (Scheme3).Under an inert atmosphere in aqueous solution (Figure 4d),
ONOO− yielded an N-centered radical adduct, DMPO-NX, asthe major product (∼57%) with aN = 14.5, aN′ = 3.55, and aH =0.93. The formation of a minor product, DMPO-ONO (6%),
was also observed with aN = 14.4 and aH = 11.3. The formationof the O- and N-centered adducts was further confirmed byGC-MS and showed distinctive peak at ∼6.2 min (not observedwith DMPO or ONOO− alone) with significantly intense basepeaks at 175.05 m/z and 159.02 m/z, corresponding toDMPO/ONOO− and DMPO-ONO adducts (Figure S8 in theSupporting Information). The time-of-flight mass spectrum alsoconfirmed the formation of a base peak at 175.14 m/z, whichwas not observed with ONOO− or DMPO alone (Figure S9 inthe Supporting Information). In addition to the formation ofO- and N-centered adducts, the same DMPO-R adduct wasalso observed as in DMSO, suggesting that the origin of theDMPO-R is independent of the solvent used. The only C-centered radical source would be from the nitrone itself viatrapping of another nitrone. As proposed above, •NO3 at high[ONOO−] may oxidize DMPO to DMPO•+, leading to theaddition of excess DMPO to DMPO•+ to form [DMPO−DMPO] •+ with ΔG298K,rxn = 0.5 kcal/mol. The predicted hfscvalues for the nitronyl-N and peroxynitrite-N (i.e., 11.0 and 5.8,respectively) of DMPO-N(O)OO gave reasonable agreementwith the experimental, while a significant discrepancy wasobserved between the predicted and the experimental hfsc foraH with 15.7 and 0.9, respectively (Table 2). Optimization ofDMPO-N(O)OO with two explicit water molecules wasproblematic, but we predict that the hfsc will not changesignificantly within ∼2 G as also observed for the other adductswith explicit water interaction. Therefore, the nature of DMPO-NX warrants further investigation.
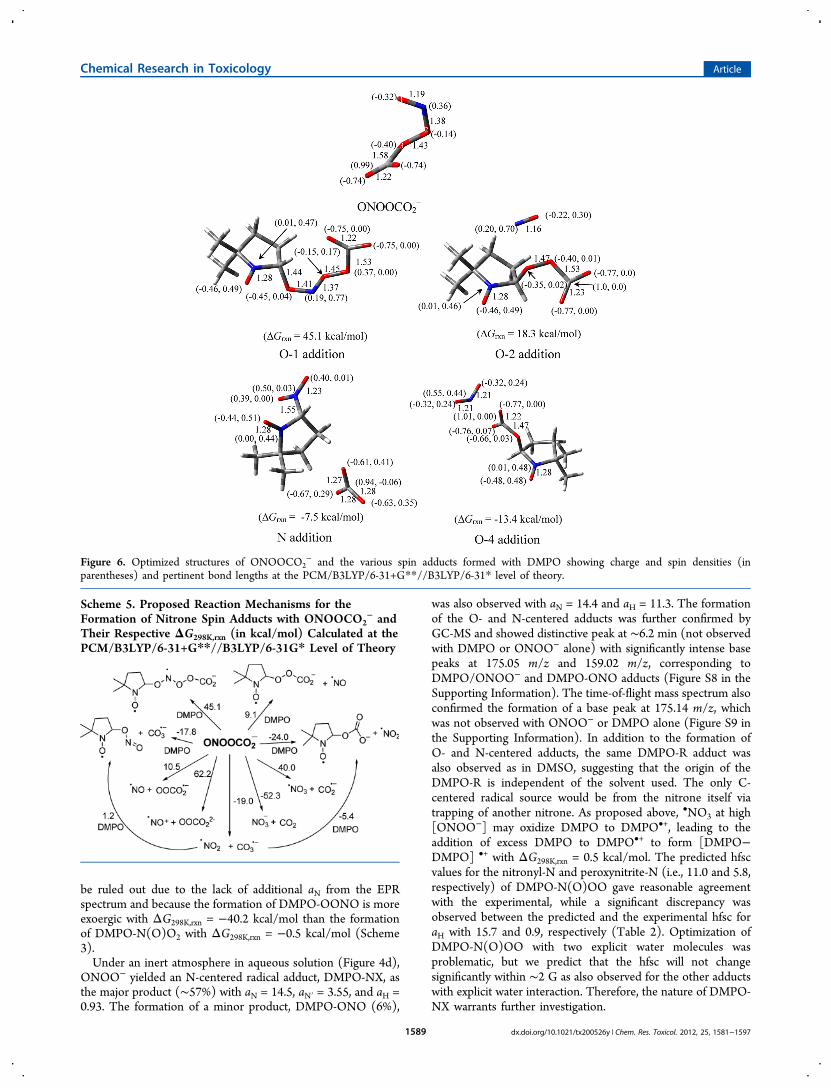
Figure 6. Optimized structures of ONOOCO2− and the various spin adducts formed with DMPO showing charge and spin densities (in
parentheses) and pertinent bond lengths at the PCM/B3LYP/6-31+G**//B3LYP/6-31* level of theory.
Scheme 5. Proposed Reaction Mechanisms for theFormation of Nitrone Spin Adducts with ONOOCO2
− andTheir Respective ΔG298K,rxn (in kcal/mol) Calculated at thePCM/B3LYP/6-31+G**//B3LYP/6-31G* Level of Theory
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971589

Scheme 4 shows the various modes of ONOOH addition toDMPO where two favorable pathways can be seen as follows:(1) the formation of singlet species, nitroso aldehyde andnitrous acid, via electrophilic addition of O(3) with highlyfavorable overall ΔG298K values of −45.3 kcal/mol and (2) theformation of doublet species, nitrogen dioxide and DMPO-OH,via O−O homolytic cleavage of ONOOH with also highlyfavorable ΔG298K,rxn of −35.9 kcal/mol. Figure 5 shows thevarious modes of ONOOH addition to DMPO with only theO(3) addition to form DMPO-OH and •NO2 being the mostfavorable pathway.Using the same spin trapping conditions employed for
DMPO (Figure 4b), the reactivity of ONOO− with EMPO,DEPMPO, and AMPO was also explored in an argon-purged70% DMSO solution. Shown in Figure S1 in the SupportingInformation, similar to DMPO, significant amounts of nitrone-R were formed from all nitrones, perhaps via formation of•CH3. While not observed for DEPMPO, nitrone-OX adductswere observed for EMPO and AMPO, which can be assigned tothe formation of DMPO-OONO most likely via inverted spintrapping. Figure 3 shows the thermodynamics of nitrone-OONO formation from [nitrone]•+ and its addition toONOO− giving exoergic free energies of ∼28 kcal/mol, with
the formation of the cis-isomer only slightly more exoergic by<1 kcal/mol as compared to the trans-isomer. The approxi-mated calculated hfsc's for the ONOO− adducts of DMPO,EMPO, DEPMPO, and AMPO are shown in Tables 2 (forDMPO) and S1 in the Supporting Information (for the rest ofthe nitrones) and generally shows higher aβ‑H than aN by asmuch as ∼4 G. This qualitative trend in the relative magnitudesof aβ‑H and aN follows the experimentally observed hfsc's withthe exception of EMPO, where the observed aβ‑H is lower thanaN.
Nitrosoperoxycarbonate (ONOOCO2−). The optimized
structure of ONOOCO2− was consistent with that of previous
theoretical studies,83 which show a 1.58 Å bond distancebetween the carboxylate-C and the peroxyl-O and a C−O−O−N dihedral angle of 82° (Figure 6). The ΔG298K,rxn of formationof ONOOCO2
− from trans-ONOO− is 0.5 kcal mol−183 and ismore favorable than its formation from cis-ONOO− withΔG298K,rxn = 2.8 kcal mol−1. However, the energy required forthe conformational change from cis-ONOO− to trans-ONOO−
was lowered upon complexation with CO2 (the free energies ofconformation change for free and CO2-complexed ONOO−
were determined to be 4.2 and 1.9 kcal mol−1, respectively).During its short half-life, the ONOOCO2
− complex can act as a
Scheme 6. Proposed Reaction Mechanisms for the Decomposition of SNAP in the Presence of DMPO and Their RespectiveΔG298K,rxn (in kcal/mol) Calculated at the PCM/B3LYP/6-31+G**//B3LYP/6-31G* Level of Theory
Figure 7. X-band spectra generated from visible light irradiation of DMPO (100 mM) and SNAP (65 mM) in aqueous solution: (a) under ambientatmosphere, HO• adducts: (major) aN = 15.2, aH = 14.8; (minor) aN = 15.8, aH = 15.2; (b) under Ar atmosphere, RS• adduct: aN = 14.6, aH = 16.0(lit. value for DMPO-SNAP: aN = 15.3, aH = 17.7).85
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971590
strong oxidant and, as previously discussed, can undergohomolytic cleavage to form CO3
•− and •NO2 species(ΔG298K,rxn = −19.0 kcal mol−1) or rearrange to form NO3
−
and CO2 (ΔG298K,rxn = −52.3 kcal mol−1) (Scheme 5). Thesecalculations are consistent with previous models in which theyield of radical products was 30%. Thus, in neutral pH tomoderately basic solution, ONOO− is energetically able to existin equilibrium with ONOOCO2
− where it can act as a strongoxidizing agent and spontaneously form radical species. Atneutral pH, ONOO− can undergo competing mechanisms forradical formation such as protonation to form ONOOH orONOOCO2
− formation. As the pH is lowered, the protonationof ONOO− can dominate, forming ONOOH.We further investigated the nature of the DMPO-OX EPR
signal in Figure 4a. Under normal atmospheric conditions, theobserved formation of DMPO-OX adduct could originate fromDMPO-OCO2
−. This hypothesis was confirmed by bubblingpure CO2 through argon-purged DMSO before ONOO− spintrapping (Figure 4c). The spectra obtained from the CO2
bubbled solution yielded one radical adduct at low ONOO−
concentration, which is identified as DMPO-OCO2− based on
our previously reported hfsc's values of aN = 14.32, aH = 10.68,and aβ‑H = 1.37 for DMPO-OCO2
−.84 Figure S2 shows thatonly when [ONOO−] ≫ [CO2] the DMPO-R adduct isformed due to the formation of •CH3 from HO• via O−Ohomolytic cleavage of ONOOH as discussed above, while at[ONOO−] ≪ [CO2], the DMPO-OCO2 adduct is morepredominant. Figure 6 shows the optimized geometries ofDMPO-ONOOCO2
− arising from the various modes ofONOOCO2
− addition reaction to DMPO. Results suggestthat N- and carboxylate-O(4) addition to DMPO gave the mostexoergic ΔG298K,rxn with −7.5 and −13.4 kcal/mol, respectively.Because of the highly negative charge density of thecarboxylate-O(4)'s ONOOCO2
− (−0.74e), its addition toDMPO is therefore nucleophilic in nature and is the mostfavorable to give DMPO-OCO2
− and •NO2, further confirmingthe EPR results observed in the presence of CO2 and ONOO−.GC-MS analysis of the sample showed evidence of DMPO-OCO2 adduct formation where the chromatogram showed aunique peak at RT 3.1 min with a base mass peak 174.91 m/zcorresponding to [DMPO-OCO2H + H]+ (Figure S10 in theSupporting Information).
Figure 8. Optimized structures of •NO3,•NH2,
•N3, and their respective adducts with DMPO showing the free energies of reaction, charge and spindensities (in parentheses), and pertinent bond lengths at the PCM/B3LYP/6-31+G**//B3LYP/6-31* level of theory.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971591

In DMSO, the possibility of forming the decompositionproducts, CH3SO(NO2) + •CH3 and CH3SO(CO3)
− +•CH3, due to oxidation by •NO2 and CO3
•−, respectively, wasalso considered. However, results show endoergic free energiesof 40.5 and 22.3 kcal mol−1, for reactions of •NO2 or CO3
•−
with DMSO, respectively, as compared to the exoergic •OHreaction to DMSO to form CH3SO(OH) + •CH3 withΔG298K,rxn = −2.3 kcal/mol indicate that •CH3 formation is lesslikely to occur from •NO2 and CO3
•− oxidation of DMSO.Furthermore, the formation of N2O4 from •NO2 withΔG298K,rxn = −0.5 kcal/mol can compete with •NO2 oxidationof DMSO.Spin Trapping Studies Using SNAP and 3-Morpholi-
nosydnonimine (SIN-1). Scheme 6 shows the reaction of
SNAP with DMPO. Homolytic cleavage of the S−N bond ofSNAP into •NO and thiol radical (RS•) was determined to beendoergic, while the ionic cleavage of SNAP into NO+ and thiylanion (RS−) was calculated to be highly unfavorable withΔG298K,rxn = 14.5 kcal/mol and ΔG298K,rxn = 50.5 kcal/mol,respectively, suggesting that the only relevant mechanism ofSNAP decomposition is the release of •NO and RS•. While it isenergetically favorable for the free RS• to form an adduct withDMPO (ΔG298K,rxn = −1.1 kcal/mol), it was calculated that theformation of a disulfide from two RS• molecules wassignificantly more favorable with ΔG298K,rxn = −33.6 kcal/mol.EPR spin trapping from SNAP decomposition was
performed. Solutions of irradiated SNAP in water aloneunder inert atmosphere and air did not give any quartet EPRsignal. The EPR spectra of DMPO in the presence of SNAPunder normal atmosphere were determined to be HO• adducts(Figure 7a) with slightly different g values (i.e., major species:aN = 15.2 and aH = 14.8; and minor species: aN = 15.8 and aH =15.2), indicating that the presence of O2 can lead to theformation of DMPO-OH via perhaps the initial reaction of O2with the NO to form an oxidizing species. However, under Aratmosphere, the formation of an RS• adduct was observed(Figure 7b) with aN = 14.6 and aH = 16.0 close to the reportedhfsc values for DMPO-SNAP of aN = 15.3 and aH = 17.7 andDMPO-SG of aN = 15.2 and aH = 16.3.85 GC-MS analysis ofthe irradiated DMPO/SNAP solution gave a significantlyintense GC peak at 6.45 min as compared to DMPO andSNAP alone with a base mass peak of 302.94 m/z (see FigureS11 in the Supporting Information).The formation of S-centered radical adducts from SNAP was
further confirmed by spin trapping using AMPO, EMPO, andDEPMPO, showing hfsc's consistent to that of the formation ofS-centered radical (see Figure S3 in the SupportingInformation). The signal intensity increased significantlywhen the experiment was repeated under inert atmosphere(Figure 7b), which suggests that •NO2 formation underatmospheric conditions can annihilate the signal, probably viaits direct reaction to the DMPO-SR adduct. It has beenpreviously shown that as •NO concentration increases in thepresence of O2, and the rate of •NO2 formation increases, suchthat at 1.0 mM •NO, the half-life of •NO due to •NO2formation is 0.56 s.19 Calculations suggest that the formationof a DMPO-NO adduct is thermodynamically unfavorable, withΔG298K,rxn = 14.0 kcal/mol,86 while the formation of a DMPO-NO2 adduct is favorable, with ΔG298K,rxn = −1.4−1.2 kcal/mol.While DMPO may display thermodynamically favorabletrapping reactions with •NO2, the radical characteristic of•NO2 can be “hidden” via its complexation to form N2O4,which can compete with its addition to the nitrone; hence, no•NO2 adduct was observed (see Scheme 6).As expected, the unfavorable reactivity of •NO to DMPO
only yielded O2•− adduct using SIN-1 as shown in Figure S4 in
the Supporting Information. While the SIN-1 decompositionmechanism was proposed87 to include the formation of a C-centered radical, only DMPO-OOH was observed in this study.
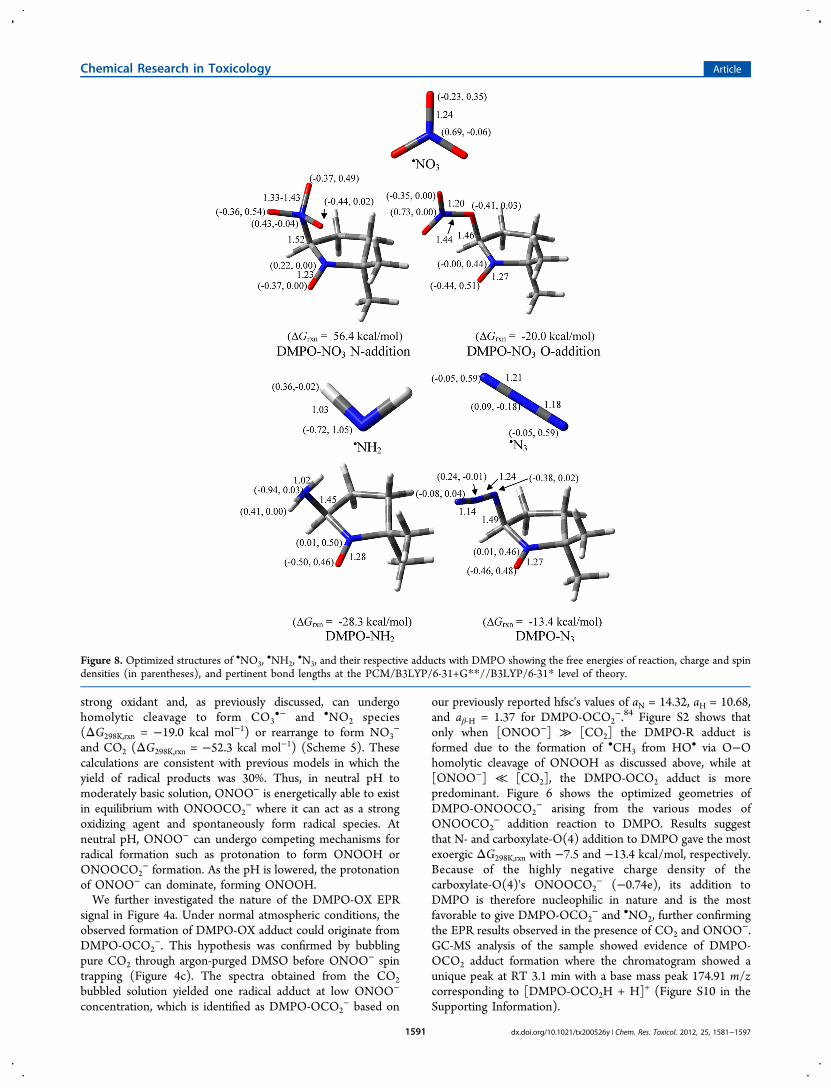
Nitrogen Trioxide (•NO3), Amino (•NH2), Azidyl (•N3),
and Nitroxyl (HNO). Figure 8 shows the various radicaladducts formed from •NO3,
•NH2, and•N3
−. The formation ofO-centered radical adduct, DMPO-ONO2, is more favorablethan the N-centered radical adduct, DMPO-NO3, withΔG298K,rxn of −20.0 and 56.4 kcal/mol, respectively. Becauseof the higher reduction potential of NO3 E°(
•NO3/NO3−aq) =
1.9−2.6 V88 as compared to the oxidation potential of DMPO
Scheme 7. Proposed Decomposition Mechanisms forDMPO-NO3 and Their Respective ΔG298K,rxn (in kcal/mol)Calculated at the PCM/B3LYP/6-31+G**//B3LYP/6-31G*Level of Theory
Figure 9. X-band spectra in PBS of DMPO adducts of (a) •N3: aN =14.75, aβ‑H = 14.18, aN′ = 3.4 (lit. value for DMPO-N3: aN = 14.8, aH =14.2 and aN‑azide = 3.1);51 and (b) •NH2: aN = 15.75, aβ‑H = 19.30, aN′= 1.62 (lit. value for DMPO-NH2: aN = 15.9, aH = 19.3, and aN‑amino =1.60).93
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971592
E°(DMPO/DMPO•+) of E° = 1.6 V89 (consistent with the freeenergies of reduction shown in Table 1), it is expected that themechanism of DMPO-NO3 formation should proceed viainverted spin trapping where DMPO is oxidized to DMPO•+
•NO3 and subsequent addition of NO3− to DMPO•+. The high
negative charge on the O atoms of NO3− favored the
nucleophilic addition of the radical to the nitrone. The spindensity distribution on the nitro-N in DMPO-ONO2 isnegligible. EPR spin trapping of •NO3 only gave a spectrumcorresponding to DMPO-OH. Although the formation ofDMPO-ONO2 was already exoergic, its hydrolysis to DMPO-OH and HNO3 is also favorable with ΔG298K,rxn = −4.1 kcal/mol (Scheme 7)significantly more favorable than the highlyendoergic formation of DMPO-OH and HOONO withΔG298K,rxn = 28.0 kcal/mol. Scheme 7 further shows thatDMPO-OH and HNO3 are more likely to be formed via O−Nhomolytic cleavage of DMPO-ONO2 to form DMPO-O• and•NO2 with ΔG298K,rxn = 19.1 kcal/mol as compared to the O−Nheterolytic cleavages, resulting in the formation of DMPO-O+
and NO2− or DMPO-O− and NO2
+ with ΔG298K,rxn values of41.1 and 67.5 kcal/mol, respectively.Because only the DMPO-OH adduct was observed during
irradiation of DMPO in the presence of HNO3 and H2O2, theeffect of HNO3 concentration on DMPO-OH formation wasfurther investigated. It is expected that the DMPO-OHconcentration will be dependent on the HNO3 concentrationat a constant H2O2 concentration. Results show that lowerDMPO-OH is formed at higher HNO3 concentration, whichindicates that HNO3 can be competing with DMPO forreaction with HO• (see Figure S5 in the SupportingInformation). Moreover, DMPO-OH formation was evidenteven at a large excess of HNO3 over H2O2 (i.e., 800 mMHNO3: 140 mM H2O2), which may also indicate •NO3
addition to DMPO; however, the use of HN17O3 would have
been more straightforward for the elucidation of themechanism of DMPO-OH formation. Nevertheless, thecalculated thermochemistries and formation of DMPO-OHadduct at large excess of HNO3 suggest origination of DMPO-OH from DMPO-NO3.Formations of DMPO-NH2 and DMPO-N3
− gave exoergicΔG298K,rxn values of −28.3 and −13.4 kcal/mol, respectively,with the most electronegative N-atoms adding to the nitrone.Spin densities of the N-atom directly bound to the nitronerange from 2 to 3%, which is enough to impart additional aNdue to the N-atom as shown in Figure 9. The formation ofDMPO-NH2 was previously reported by Kirino, et al.90 (aN =15.9, aH = 19.0, and aN‑amino = 1.7) as well as by Chignell et al.91
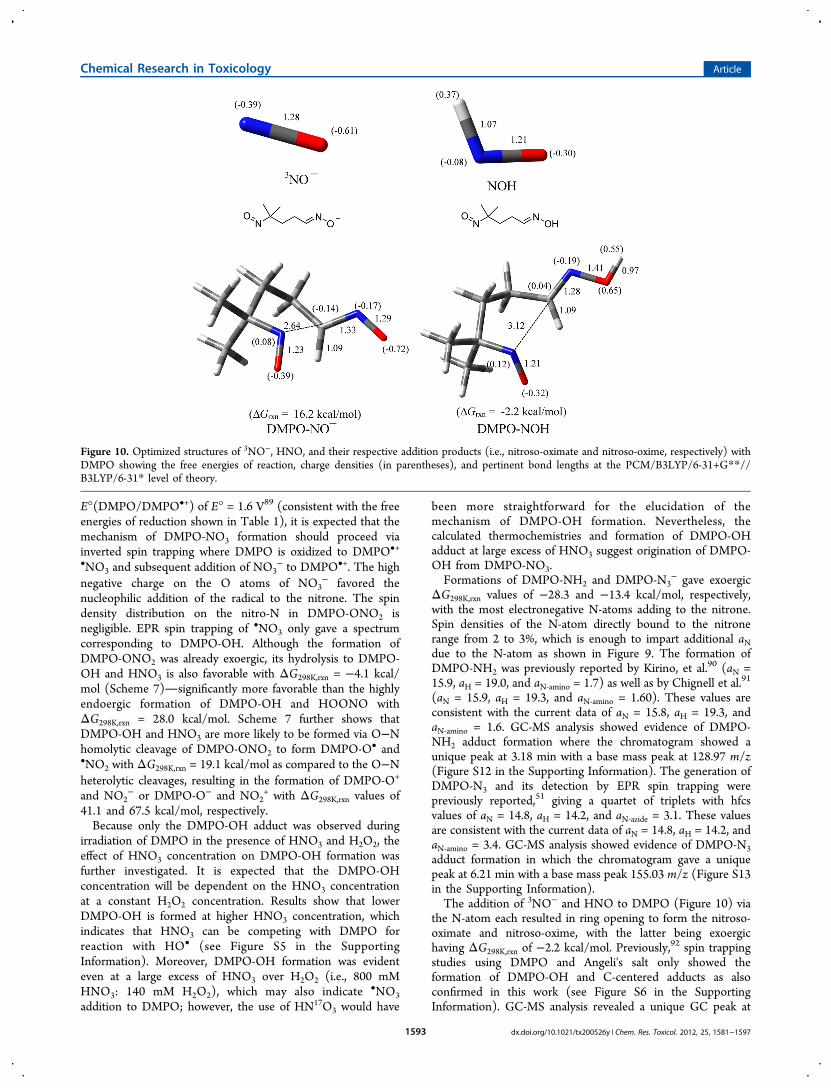
(aN = 15.9, aH = 19.3, and aN‑amino = 1.60). These values areconsistent with the current data of aN = 15.8, aH = 19.3, andaN‑amino = 1.6. GC-MS analysis showed evidence of DMPO-NH2 adduct formation where the chromatogram showed aunique peak at 3.18 min with a base mass peak at 128.97 m/z(Figure S12 in the Supporting Information). The generation ofDMPO-N3 and its detection by EPR spin trapping werepreviously reported,51 giving a quartet of triplets with hfcsvalues of aN = 14.8, aH = 14.2, and aN‑azide = 3.1. These valuesare consistent with the current data of aN = 14.8, aH = 14.2, andaN‑amino = 3.4. GC-MS analysis showed evidence of DMPO-N3adduct formation in which the chromatogram gave a uniquepeak at 6.21 min with a base mass peak 155.03 m/z (Figure S13in the Supporting Information).The addition of 3NO− and HNO to DMPO (Figure 10) via
the N-atom each resulted in ring opening to form the nitroso-oximate and nitroso-oxime, with the latter being exoergichaving ΔG298K,rxn of −2.2 kcal/mol. Previously,92 spin trappingstudies using DMPO and Angeli's salt only showed theformation of DMPO-OH and C-centered adducts as alsoconfirmed in this work (see Figure S6 in the SupportingInformation). GC-MS analysis revealed a unique GC peak at
Figure 10. Optimized structures of 3NO−, HNO, and their respective addition products (i.e., nitroso-oximate and nitroso-oxime, respectively) withDMPO showing the free energies of reaction, charge densities (in parentheses), and pertinent bond lengths at the PCM/B3LYP/6-31+G**//B3LYP/6-31* level of theory.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971593

3.21 min with a base mass peak 113.08 m/z (Figure S14 in theSupporting Information), corresponding to an ene-aldoxime,which can be formed from the elimination of the NO from theC-nitroso moiety of the originally formed nitroso-oxime.
■ CONCLUSIONS
EPR and theoretical studies of the reaction between DMPOand •NO2 suggest the formation of an O-centered radicaladduct, DMPO-ONO. The formation of an O-centered radicaladduct from •NO2 was also demonstrated using AMPO,EMPO, and DEPMPO. The addition reactions of DMPO withONOO−/ONOOH primarily result in ring opening to form thenitroso-aldehyde and nitrite/nitrous acid. The formation ofDMPO-OH, as detected by EPR, from the O−O homolyticcleavage of ONOOH was also thermodynamically favorable.Decomposition of ONOOCO2
− in the presence of DMPOyielded CO3
•− adduct at a high CO2 concentration. However,alkyl adduct formation in an all-PBS system suggests invertedspin trapping of DMPO by DMPO•+, which is likely generatedfrom the highly oxidizing •NO3. The EPR-detectable nitronyladduct as DMPO-OONO, whose formation can also berationalized by inverted spin trapping, was shown to be alsoformed using AMPO and EMPO as well. EPR studies of theNO-donor SNAP in the presence of DMPO show S-centeredradical adduct formation under an inert atmosphere. Thereaction of SIN-1 with DMPO only yielded O2
•−, and while•NO3 addition to DMPO initially formed DMPO-ONO2, itssubsequent hydrolysis yielded DMPO-OH and HNO3 as theformer detected by EPR, consistent with the calculated exoergicfree energy. Additions of •N3 and
•NH2 to DMPO yielded N-centered radicals with significant hyperfine contributions fromthe N-atoms of the radical moieties. Reaction of HNO toDMPO only yielded a ring-opening product to give nitroso-oxime and DMPO-OH. This study shows a myriad of RNSreaction with nitrones and, through the use of spin traps, canprovide insights into the mechanisms of RNS-mediatedreactions in chemical and biological systems. Moreover, thisstudy can lead to the development of better probe designs forRNS detection and to the understanding the antioxidativeproperties of nitrones.
■ ASSOCIATED CONTENT
*S Supporting InformationAdditional EPR spectra, tables, thermodynamic data of all RNS,ROS, nitrones, and their respective spin adducts, and completeref 67. This material is available free of charge via the Internetat http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*Tel: 614-292-8215. Fax: 614-699-0999. E-mail: [email protected].
FundingThis publication was made possible by Grant RO1 HL81248from the NIH National Heart, Lung, and Blood Institute. Thiswork was supported by an allocation of computing time fromthe Ohio Supercomputer Center.
NotesThe authors declare no competing financial interest.
■ ABBREVIATIONS
EPR, electron paramagnetic resonance; ROS, reactive oxygenspecies; RNS, reactive nitrogen species; SNAP, S-nitroso-N-acetyl-D,L-penicillamine; TEMPONE-H, 1-hydroxy-2,2,6,6-tet-ramethyl-4-oxo-piperidine; HRP, horseradish peroxidase;DMPO, 5,5-dimethyl-pyrroline N-oxide; AMPO, 5-carbamoyl-5-methyl-pyrroline N-oxide; EMPO, 5-ethoxycarbonyl-5-meth-yl-pyrroline N-oxide; DEPMPO, 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide
■ REFERENCES(1) Bossy-Wetzel, E., Schwarzenbacher, R., and Lipton, S. A. (2004)Molecular pathways to neurodegeneration. Nat. Med. 10 (Suppl.), S2−S9.(2) Escobales, N., and Crespo, M. J. (2005) Oxidative-nitrosativestress in hypertension. Curr. Vasc. Pharmacol. 3, 231−246.(3) Kaneki, M., Shimizu, N., Yamada, D., and Chang, K. (2007)Nitrosative stress and pathogenesis of insulin resistance. Antioxid.Redox Signaling 9, 319−329.(4) Llorens, S., and Nava, E. (2003) Cardiovascular diseases and thenitric oxide pathway. Curr. Vasc. Pharmacol. 1, 335−346.(5) Sugiura, H., and Ichinose, M. (2011) Nitrative stress ininflammatory lung diseases. Nitric Oxide 25, 138−144.(6) Szabo, C. (2009) Role of nitrosative stress in the pathogenesis ofdiabetic vascular dysfunction. Br. J. Pharmacol. 156, 713−727.(7) Thomas, D. D., Ridnour, L. A., Isenberg, J. S., Flores-Santana, W.,Switzer, C. H., Donzelli, S., Hussain, P., Vecoli, C., Paolocci, N., Ambs,S., Colton, C. A., Harris, C. C., Roberts, D. D., and Wink, D. A. (2008)The chemical biology of nitric oxide: Implications in cellular signaling.Free Radical Biol. Med. 45, 18−31.(8) Ferguson, L. R. (2010) Chronic inflammation and mutagenesis.Mutat. Res. 690, 3−11.(9) Bartesaghi, S., Valez, V., Trujillo, M., Peluffo, G., Romero, N.,Zhang, H., Kalyanaraman, B., and Radi, R. (2006) Mechanistic studiesof peroxynitrite-mediated tyrosine nitration in membranes using thehydrophobic probe N-tert-BOC-L-tyrosine tert-butyl ester. Biochemistry45, 6813−6825.(10) Kalyanaraman, B. (2004) Nitrated lipids: A class of cell-signalingmolecules. Proc. Natl. Acad. Sci. U.S.A. 101, 11527−11528.(11) Lancaster, J. R., Jr. (2008) Protein cysteine thiol nitrosation:Maker or marker of reactive nitrogen species-induced non-erythroidcellular signaling? Nitric Oxide 19, 68−72.(12) Nuriel, T., Hansler, A., and Gross, S. S. (2011) Proteinnitrotryptophan: Formation, significance and identification. J.Proteomics,.(13) Ridnour, L. A., Thomas, D. D., Mancardi, D., Espey, M. G.,Miranda, K. M., Paolocci, N., Feelisch, M., Fukuto, J., and Wink, D. A.(2004) The chemistry of nitrosative stress induced by nitric oxide andreactive nitrogen oxide species. Putting perspective on stressfulbiological situations. Biol. Chem. 385, 1−10.(14) Shahani, N., and Sawa, A. (2011) Nitric oxide signaling andnitrosative stress in neurons: Role for S-nitrosylation. Antioxid. RedoxSignaling 14, 1493−1504.(15) Tiago, T., Palma, P. S., Gutierrez-Merino, C., and Aureliano, M.(2010) Peroxynitrite-mediated oxidative modifications of myosin andimplications on structure and function. Free Radical Res. 44, 1317−1327.(16) Rosen, G. M., Tsai, P., and Pou, S. (2002) Mechanism of free-radical generation by nitric oxide synthase. Chem. Rev. 102, 1191−1200.(17) McAninly, J., Williams, D. L. H., Askew, S. C., Butler, A. R., andRussell, C. (1993) Metal ion catalysis in nitrosothiol (RSNO)decomposition. Chem. Commun. 23, 1758−1759.(18) Angelo, M., Singel, D. J., and Stamler, J. S. (2006) An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizesnitrite as a substrate. Proc. Natl. Acad. Sci. U.S.A. 103, 8366−8371.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971594

(19) Beckman, J. S., and Koppenol, W. H. (1996) Nitric oxide,superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J.Physiol. 271, C1424−C1437.(20) Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., andFreeman, B. A. (1990) Apparent hydroxyl radical production byperoxynitrite: Implications for endothelial injury from nitric oxide andsuperoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620−1624.(21) Czapski, G., and Goldstein, S. (1995) The role of the reactionsof ·NO with superoxide and oxygen in biological systems: A kineticapproach. Free Radical Biol. Med. 19, 785−794.(22) Alvarez, B., Demicheli, V., Duran, R., Trujillo, M., Cervenansky,C., Freeman, B. A., and Radi, R. (2004) Inactivation of human Cu,Znsuperoxide dismutase by peroxynitrite and formation of histidinylradical. Free Radical Biol. Med. 37, 813−822.(23) Ischiropoulos, H., Zhu, L., Chen, J., Tsai, M., Martin, J. C.,Smith, C. D., and Beckman, J. S. (1992) Peroxynitrite-mediatedtyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem.Biophys. 298, 431−437.(24) Pearce, L. L., Martinez-Bosch, S., Manzano, E. L., Winnica, D.E., Epperly, M. W., and Peterson, J. (2009) The resistance of electron-transport chain Fe-S clusters to oxidative damage during the reactionof peroxynitrite with mitochondrial complex II and rat-heartpericardium. Nitric Oxide 20, 135−142.(25) Rubbo, H., Denicola, A., and Radi, R. (1994) Peroxynitriteinactivates thiol-containing enzymes of Trypanosoma cruzi energeticmetabolism and inhibits cell respiration. Arch. Biochem. Biophys. 308,96−102.(26) Yang, D., Wang, H.-L., Sun, Z.-N., Chung, N.-W., and Shen, J.-G. (2006) A highly selective fluorescent probe for the detection andimaging of peroxynitrite in living cells. J. Am. Chem. Soc. 128, 6004−6005.(27) Sun, Z.-N., Wang, H.-L., Liu, F.-Q., Chen, Y., Tam, P. K. H., andYang, D. (2009) BODIPY-based fluorescent probe for peroxynitritedetection and imaging in living cells. Org. Lett. 11, 1887−1890.(28) Lancaster, J. R., Jr. (2010) The use of diaminofluorescein fornitric oxide detection: Conceptual and methodological distinctionbetween NO and nitrosation. Free Radical Biol. Med. 49, 1145.(29) Cortes, J. S., Granados, S. G., Ordaz, A. A., Jimenez, J. A. L.,Griveau, S., and Bedioui, F. (2007) Electropolymerized manganesetetraaminophthalocyanine thin films onto platinum ultramicroelec-trode for the electrochemical detection of peroxynitrite in solution.Electroanalysis 19, 61−64.(30) Lorch, S., Lightfoot, R., Ohshima, H., Virag, L., Chen, Q.,Hertkorn, C., Weiss, M., Souza, J., Ischiropoulos, H., Yermilov, V.,Pignatelli, B., Masuda, M., and Szabo, C. (2002) Detection ofperoxynitrite-induced protein and DNA modifications. Methods Mol.Biol. 196, 247−275.(31) Ikeda, K., Yukihiro Hiraoka, B., Iwai, H., Matsumoto, T., Mineki,R., Taka, H., Takamori, K., Ogawa, H., and Yamakura, F. (2006)Detection of 6-nitrotryptophan in proteins by Western blot analysisand its application for peroxynitrite-treated PC12 cells. Nitric Oxide 16,18−28.(32) Kalyanaraman, B., Karoui, H., Singh, R. J., and Felix, C. C.(1996) Detection of thiyl radical adducts formed during hydroxylradical- and peroxynitrite-mediated oxidation of thiols-a highresolution ESR spin-trapping study at Q-band (35 GHz). Anal.Biochem. 241, 75−81.(33) Goldstein, S., Czapski, G., Lind, J., and Merenyi, G. (2000)Tyrosine nitration by simultaneous generation of ·NO and O2 underphysiological conditions. J. Biol. Chem. 275, 3031−3036.(34) Tsai, P., Porasuphatana, S., Pou, S., and Rosen, G. M. (2000)Investigations into the spin trapping of nitric oxide and superoxide:models to explore free radical generation by nitric oxide synthase. J.Chem. Soc., Perkin Trans. 2, 983−988.(35) Sikora, A., Zielonka, J., Lopez, M., Joseph, J., and Kalyanaraman,B. (2009) Direct oxidation of boronates by peroxynitrite: Mechanismand implications in fluorescence imaging of peroxynitrite. Free RadicalBiol. Med. 47, 1401−1407.
(36) Goldstein, S., Lind, J., and Merenyi, G. (2005) Chemistry ofperoxynitrites as compared to peroxynitrates. Chem. Rev. 105, 2457−2470.(37) Merenyi, G. b., Lind, J., Czapski, G., and Goldstein, S. (2003)Direct determination of the Gibbs' energy of formation ofperoxynitrous acid. Inorg. Chem. 42, 3796−3800.(38) Goldstein, S., and Rabani, J. (2007) Mechanism of nitriteformation by nitrate photolysis in aqueous solutions: The role ofperoxynitrite, nitrogen dioxide, and hydroxyl radical. J. Am. Chem. Soc.129, 10597−10601.(39) Merenyi, G., Lind, J., Goldstein, S., and Czapski, G. (1999)Mechanism and thermochemistry of peroxynitrite decomposition inwater. J. Phys. Chem. 103, 5685−5691.(40) Bonini, M. G., Radi, R., Ferrer-Sueta, G., Ferreira, A. M. D. C.,and Augusto, O. (1999) Direct EPR detection of the carbonate radicalanion produced from peroxynitrite and carbon dioxide. J. Biol. Chem.274, 10802−10806.(41) Meli, R., Nauser, T., Latal, P., and Koppenol, W. H. (2002)Reaction of peroxynitrite with carbon dioxide: intermediates anddetermination of the yield of CO3
.‑ and NO2. J. Biol. Inorg. Chem. 7,31−36.(42) Goldstein, S., and Czapski, G. (1999) Viscosity effects on thereaction of peroxynitrite with CO2: Evidence for radical formation in asolvent cage. J. Am. Chem. Soc. 121, 2444−2447.(43) Massari, J., Tokikawa, R., Zanolli, L., Tavares, M. F. M.,Assuncao, N. A., and Bechara, E. J. H. (2010) Acetyl radicalproduction by the methylglyoxal-peroxynitrite system: A possibleroute for L-lysine acetylation. Chem. Res. Toxicol. 23, 1762−1770.(44) Royer, L. O., Knudsen, F. S., De Oliveira, M. A., Tavares, M. F.M., and Bechara, E. J. H. (2004) Peroxynitrite-initiated oxidation ofacetoacetate and 2-methylacetoacetate esters by oxygen: Potentialsources of reactive intermediates in keto acidoses. Chem. Res. Toxicol.17, 1725−1732.(45) Imaram, W., Gersch, C., Kim, K. M., Johnson, R. J., Henderson,G. N., and Angerhofer, A. (2010) Radicals in the reaction betweenperoxynitrite and uric acid identified by electron spin resonancespectroscopy and liquid chromatography mass spectrometry. FreeRadical Biol. Med. 49, 275−281.(46) Imaram, W., Johnson, R. J., and Angerhofer, A. (2009) ESR spintrapping of the reaction between urate and peroxynitrite: Thehydrogen adduct. Appl. Magn. Reson. 37, 463−472.(47) Olson, L. P., Bartberger, M. D., and Houk, K. N. (2003)Peroxynitrate and peroxynitrite: A complete basis set investigation ofsimilarities and differences between these NOx species. J. Am. Chem.Soc. 125, 3999−4006.(48) Shiri, M., Zolfigol, M. A., Kruger, H. G., and Tanbakouchian, Z.(2010) Advances in the application of N2O4/NO2 in organic reactions.Tetrahedron 66, 9077−9106.(49) Batinic-Haberle, I., Spasojevic, I., Stevens, R. D., Hambright, P.,Neta, P., Okado-Matsumoto, A., and Fridovich, I. (2004) New class ofpotent catalysts of O2·- dismutation. Mn(III) ortho-methoxyethylpyr-idyl- and di-ortho-methoxyethylimidazolylporphyrins. Dalton Trans.,1696−1702.(50) Bowler, M. W., Montgomery, M. G., Leslie, A. G. W., andWalker, J. E. (2006) How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. U.S.A. 103, 8646−8649.(51) Kalyanaraman, B., Janzen, E. G., and Mason, R. P. (1985) Spintrapping of the azidyl radical in azide/catalase/hydrogen peroxide andvarious azide/peroxidase/hydrogen peroxide peroxidizing systems. J.Biol. Chem. 260, 4003−4006.(52) Partridge, R. S., Monroe, S. M., Parks, J. K., Johnson, K., Parker,W. D., Jr., Eaton, G. R., and Eaton, S. S. (1994) Spin trapping of azidyland hydroxyl radicals in azide-inhibited rat brain submitochondrialparticles. Arch. Biochem. Biophys. 310, 210−217.(53) Amadelli, R., Maldotti, A., Bartocci, C., and Carassiti, V. (1989)ESR spin-trapping investigation of azide oxidation on cadmium sulfideand zinc oxide suspension. J. Phys. Chem. 93, 6448−6453.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971595

(54) Clarke, K., Edge, R., Johnson, V., Land, E. J., Navaratnam, S.,and Truscott, T. G. (2008) Direct observation of NH2 reactions withoxygen, amino acids, and melanins. J. Phys. Chem. A 112, 1234−1237.(55) Kemp-Harper, B. K. (2011) Nitroxyl (HNO): A novel redoxsignaling molecule. Antioxid. Redox Signaling 14, 1609−1613.(56) Dikalov, S., Skatchkov, M., Fink, B., and Bassenger, E. (1997)Quantification of superoxide radicals and peroxynitrite in vascular cellsusing oxidation of sterically hindered hydroxylamines and electronspin resonance. Nitric Oxide 1, 423−431.(57) Rosen, G. M., and Rauckman, l. J. (1977) Formation andreduction of a nitroxide radical by liver microsomes. Biochem.Pharmacol. 26, 675−678.(58) Finkelstein, E., Rosen, G. M., and Rauckman, E. J. (1980) Spintrapping. Kinetics of the reaction of superoxide and hydroxyl radicalswith nitrones. J. Am. Chem. Soc. 102, 4994−4999.(59) Rosen, G. M., Britigan, B. E., Halpern, H. J., and Pou, S. (1999)Free Radicals: Biology and Detection by Spin Trapping, OxfordUniversity Press, New York, NY.(60) Ranguelova, K., and Mason, R. P. (2011) The fidelity of spintrapping with DMPO in biological systems. Magn. Reson. Chem. 49,152−158.(61) Makino, K., Hagiwara, T., Hagi, A., Nishi, M., and Murakami, A.(1990) Cautionary note for DMPO spin trapping in the presence ofiron ion. Biochem. Biophys. Res. Commun. 172, 1073−1080.(62) Eberson, L. (1994) Inverted spin trapping. Part III. Furtherstudies on the chemical and photochemical oxidation of spin traps inthe presence of nucleophiles. J. Chem. Soc., Perkin Trans. 2, 171−176.(63) Astolfi, P., Greci, L., and Panagiotaki, M. (2005) Spin trappingof nitrogen dioxide and of radicals generated from nitrous acid. FreeRadical Res. 39, 137−144.(64) Gatti, R. M., Alvarez, B., Vasquez-Vivar, J., Radi, R., andAugusto, O. (1998) Formation of spin trap adducts during thedecomposition of peroxynitrite. Arch. Biochem. Biophys. 349, 36−46.(65) Durand, G., Prosak, R. A., Han, Y., Ortial, S., Rockenbauer, A.,Pucci, B., and Villamena, F. A. (2009) Spin trapping andcytoprotective properties of fluorinated amphiphilic carrier conjugatesof cyclic versus linear nitrones. Chem. Res. Toxicol. 22, 1570−1581.(66) Labanowski, J. W., and Andzelm, J. (1991) Density FunctionalMethods in Chemistry, Springer, New York, NY.(67) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.;Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.;Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.;Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.;Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao,O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.;Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.;Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman,J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.;Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.;Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.;Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen,W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, revisionD.01; Gaussian, Inc.: Wallingford, CT, 2003.(68) Scott, A. P., and Radom, L. (1996) Harmonic vibrationalfrequencies: An evaluation of Hartree-Fock, Moeller-Plesset, quadraticconfiguration interaction, density functional theory, and semiempiricalscale factors. J. Phys. Chem. 100, 16502−16513.(69) Cossi, M., and Barone, V. (1998) Analytical second derivativesof the free energy in solution by polarizable continuum models. J.Chem. Phys. 109, 6246−6254.(70) Cossi, M., Barone, V., Cammi, R., and Tomasi, J. (1996) Abinitio study of solvated molecules: a new implementation of thepolarizable continuum model. Chem. Phys. Lett. 255, 327−335.
(71) Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988)Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899−926.(72) Baboul, A. G., Curtiss, L. A., and Redfern, P. C. (1999)Gaussian-3 theory using density functional geometries and zero-pointenergies. J. Chem. Phys. 110, 7650−7657.(73) Izgorodina, E. I., Brittain, D. R., Hodgson, J. L., Krenske, E. H.,Lin, C. Y., Namazian, M., and Coote, M. L. (2007) Shouldcontemporary density functional theory methods be used to studythe thermodynamics of radical reactions? J. Phys. Chem. A 111, 10754−10768.(74) Rockenbauer, A., and Korecz, L. (1996) Automatic computersimulations of ESR spectra. Appl. Magn. Reson. 10, 29−43.(75) Robinson, K. M., and Beckman, J. S. (2005) Synthesis ofperoxynitrite from nitrite and hydrogen peroxide. Methods Enzymol.396, 207−214.(76) Pace, M. D., and Carmichael, A. J. (1997) Quantitative EPR spintrapping. 1. Nitrogen dioxide radicals and nitrite ions from energeticmaterials in alkaline aqueous solution. J. Phys. Chem. A 101, 1848−1853.(77) Crow, J. P., Spruell, C., Chen, J., Gunn, C., Ischiropoulos, H.,Tsai, M., Smith, C. D., Radi, R., Koppenol, W. H., and Beckman, J. S.(1994) On the pH-dependent yield of hydroxyl radical products fromperoxynitrite. Free Radical Biol. Med. 16, 331−338.(78) Villamena, F. A. (2010) Superoxide radical anion adduct of 5,5-dimethyl-1-pyrroline N-oxide. 6: Redox properties. J. Phys. Chem. A114, 1153−1160.(79) Henry, D. J., Parkinson, C. J., and Radom, L. (2002) Anassessment of the performance of high-level theoretical procedures inthe computation of the heats of formation of small open-shellmolecules. J. Phys. Chem. A 106, 7927−7936.(80) Koppenol, W. H., and Liebman, J. F. (1984) The oxidizingnature of the hydroxyl radical. A comparison with the ferryl ion(FeO2+). J. Phys. Chem. A 88, 99−101.(81) Stanbury, D. M. (1989) Reduction potentials involvinginorganic free radicals in aqueous solution. Adv. Inorg. Chem. 33,69−138.(82) Rockenbauer, A., Gyor, M., and Tudos, F. (1986) Spin trappingreactions with nitric oxides IV. Reactions with olefins. Tetrahedron Lett.27, 3425−3428.(83) Houk, K. N., Condroski, K. R., and Pryor, W. A. (1996) Radicaland concerted mechanisms in oxidations of amines, sulfides, andalkenes by peroxynitrite, peroxynitrous acid, and the peroxynitrite-CO2
adduct: Density functional theory transition structures and energetics.J. Am. Chem. Soc. 118, 13002−13006.(84) Villamena, F. A., Locigno, E. J., Rockenbauer, A., Hadad, C. M.,and Zweier, J. L. (2007) Theoretical and experimental studies of thespin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline N-oxide(DMPO). 2. Carbonate radical anion. J. Phys. Chem. 111, 384−391.(85) Singh, R. J., Hogg, N., Joseph, J., and Kalyanaraman, B. (1996)Mechanism of nitric oxide release from S-nitrosothiols. J. Biol. Chem.271, 18596−18603.(86) Villamena, F. A., Hadad, C. M., and Zweier, J. L. (2005)Comparative DFT study of the spin trapping of methyl, mercapto,hydroperoxy, superoxide, and nitric oxide radicals by varioussubstituted cyclic nitrones. J. Phys. Chem. A 109, 1662−1674.(87) Rojas Wahl, R. U. (2004) Decomposition mechanism of 3-N-morpholinosydnonimine (SIN-1)-a density functional study onintrinsic structures and reactivities. J. Mol. Model. 10, 121−129.(88) Dutton, A. S., Fukuto, J. M., and Houk, K. N. (2005)Theoretical reduction potentials for nitrogen oxides from CBS-QB3energetics and (C)PCM solvation calculations. Inorg. Chem. 44, 4024−4028.(89) McIntire, G. L., Blount, H. N., Stronks, H. J., Shetty, R. V., andJanren, E. G. (1980) Spin trapping in electrochemistry. 2. Aqueous andnonaqueous electrochemical characterizations of spin traps. J. Phys.Chem. A 84, 916−921.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971596

(90) Kirino, Y., Ohkuma, T. a., and Kwan, T. (1981) Spin trappingwith 5,5-dimethylpyrroline-N-oxide in aqueous solution. Chem. Pharm.Bull. 29, 29−34.(91) Chignell, C. F., Kalyanaraman, B., Sik, R. H., and Mason, R. P.(1981) Spectroscopic studies of cutaneous photosensitizing agents. II.Spin trapping of photolysis products from sulfanilamide and 4-aminobenzoic acid using 5,5-dimethyl-1-pyrroline-1-oxide. Photochem.Photobiol. 34, 147−156.(92) Stoyanovsky, D. A., Clancy, R., and Cederbaum, A. I. (1999)Decomposition of sodium trioxodinitrate (Angeli’s Salt) to hydroxylradical: An ESR spin-trapping study. J. Am. Chem. Soc. 121, 5093−5094.(93) Chignell, C. F., Kalyanaraman, B., Mason, R. P., and Sik, R. H.(1980) Spectroscopic studies of cutaneous photosensitizing agents. I.Spin trapping of photolysis products from sulfanilamide, 4-amino-benzoic acid, and related compounds. Photochem. Photobiol. 32, 563−571.
Chemical Research in Toxicology Article
dx.doi.org/10.1021/tx200526y | Chem. Res. Toxicol. 2012, 25, 1581−15971597







![arXiv:1901.04857v1 [nucl-th] 14 Jan 2019 · Our reaction rates di er from previous results by 2.9% near 1.0 GK. Our reactivities Our reactivities are smaller than previous results,](https://img.pdfslide.us/doc/110x75/5d55040688c9938f688b6e50/arxiv190104857v1-nucl-th-14-jan-2019-our-reaction-rates-di-er-from-previous.jpg)





















