Embed Size (px)
Citation preview

Reaction of Active Nitrogen with Tetrachloroethylene
WILLIAM ERNEST JONES AND MANIT RUJIMETHABHAS' Department of Chemistry, Dallrousie University, Halifax, Nova Scotio
Received January 2, 1973
The reaction of active nitrogen with tetrachloroethylene yielded cyanogen chloride, trichloroacetoni- trile, cyanogen, chlorine, and carbon tetrachloride. In addition, a yellow-brown polymer was found on the glass wall. A brief discussion of a possible mechanism is given.
La reaction de l'azote actif avec le titrachlorure d'ithylene conduit a un mClange de chlorure de cyano- gene, de trichloroacetonitrile, de cyanogene, de chlore et de tttrachlorure de carbone. De plus un poly- mere jaune-brun se retrouve sur la paroi du verre. Une breve discussion du mecanisme possible est prksentke. [Traduit par le journal]
Can. J . Chern. 51, 3680 (1973)
Introduction The reaction of active nitrogen with tetra-
chloroethylene (TCE) at room temperature gives a bright peach-colored flame. However, the flame boundary is not as well defined as it is in the reaction of active nitrogen with perfluoro- carbons. I t is therefore impossible to study the kinetics by the diffusion flame method as has been done with the perfluorocarbons (1). At a high reactant flow rate, the flame is quenched only a few centimeters from the reactant nozzle. As the flow rate of TCE decreases, the flame extends farther down stream from the reaction vessel. For example, at a very small TCE flow rate and in a well-darkened room, the flame emission can be seen to extend to the product trap. The gaseous products are: cyanogen chlo- ride (CNCI), trichloroacetonitrile (CCl,CN), cyanogen ((CN),), chlorine (Cl,), carbon tetra- chloride (CCI,), and unreacted TCE. A yellow- brown solid "polymer" forms as a deposit on the glass wall of the reaction vessel and on the reactant inlet.
Jennings and Linnett (2, 3) investigated the emission from the reaction flame of active nitrogen with TCE and found the CN violet and red systems and C-CI bands. However, no bands corresponding to the Cz swan bands or to the then unknown N-CI molecule were de- tected. Kiess and Broida (4) studied the reaction flame by using active nitrogen generated by electrodeless discharge and reported that the CN(0,O) violet emission was much more in-
'Holder of a Colombo Plan Scholarship.
tense than the CN(10,4) red emission. The authors could not indicate any specific mechan- ism leading to the CN emission.
Previous workers have not investigated the products of the reaction. In this paper, we report the determination of the reaction products.
Experimental Apparot~rs
The reaction was studied in a conventional fast flow system with a Pyrex reaction vessel of 40 cm length and 6 cm 0.d. Purified nitrogen from Canadian Liquid Air Corporation was further purified by passing it through (a) a liquid nitrogen trap packed with copper turnings, (b) a column of heated copper at 425 "C, and (c) another liquid nitrogen trap. The purified nitrogen passed through a capillary flow meter and was activated by a condensed electric discharge. The nitrogen atom concentration was obtained by the NO gas titration technique (5, 6) . The nitrogen atom flow rate was found to be 3.2 pmol s- ' at 1.75 Torr. The reaction products were studied by gas chromatography using a 10 ft column packed with chromosorb P coated with 3 0 z tricresyl phosphate. The separation was most effective when the column oven was subjected to a non-linear temperature program. After introduction of the sample, the column remained at room temperature for 7 min, and then was heated to 370 "K where it remained until the last compound ap- peared. The products were identified by comparison of retention times and the identification was checked by infrared and mass spectroscopy.
A reaction vessel 60 cm long and 2.5 crn in diameter was used to study the emission from the reaction. Both ends of this reaction vessel were equipped with quartz windows. The flame was monitored by viewing the reac- tion vessel end-on with a Heath EU-700 monochromator at a slit width of 350 p. Theemission bands were recorded from 2000-7500 A. For these reactions, the active nitrogen was generated by a 200 W microwave unit operating at 2450 M H z and at a power output of 75-80%. The reaction was studied at 1.2 Torr.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
TE
CH
NIS
CH
E U
NIV
ER
SIT
EIT
EIN
DH
OV
EN
on
11/1
5/14
For
pers
onal
use
onl
y.
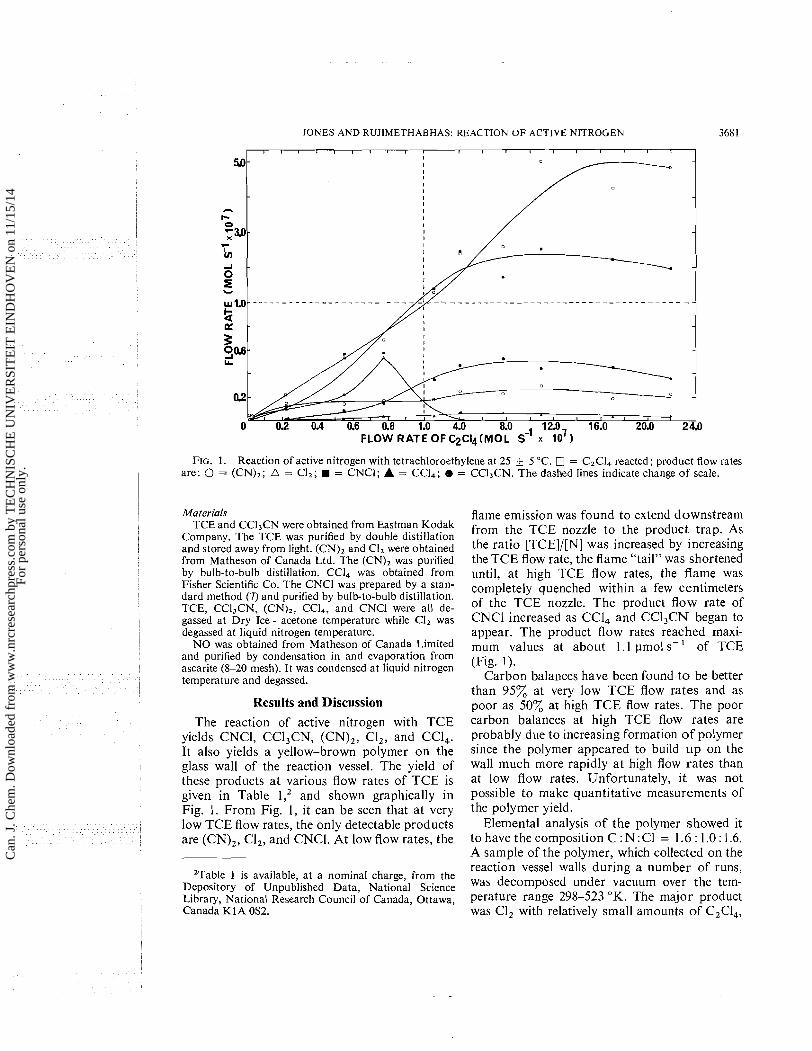
JONES AND RUJIMETHABHAS: REACTION OF ACTIVE NITROGEN 3681
0 02 0.4 0.6 0.8 1 . 4.0 8.0 12.0, 16.0 20.0 24.0 FLOW RATE OFC2CI4(MOL S-I x 10 )
FIG. 1. Reaction of active nitrogen with tetrachloroethylene at 25 ? 5 "C. = C2C14 reacted; product flow rates are: 0 = (CN),; A = CI,; W = CNCI; A = CCI4; 8 = CC1,CN. The dashed lines indicate change of scale.
Materials TCE and CC1,CN were obtained from Eastman Kodak
Company. The TCE was purified by double distillation and stored away from light. (CN), and CI, were obtained from Matheson of Canada Ltd. The (CN), was purified by bulb-to-bulb distillation. CCI, was obtained from Fisher Scientific Co. The CNCl was prepared by a stan- dard method (7) and purified by bulb-to-bulb distillation. TCE, CCI,CN, (CN),, CCI4, and CNCl were all de- gassed at Dry Ice - acetone temperature while C12 was degassed at liquid nitrogen temperature.
NO was obtained from Matheson of Canada Limited and purified by condensation in and evaporation from ascarite (8-20 mesh). It was condensed at liquid nitrogen temperature and degassed.
Results and Discussion The reaction of active nitrogen with TCE
yields CNCl, CCl,CN, (CN),, CI,, and CCl,. It also yields a yellow-brown polymer on the glass wall of the reaction vessel. The yield of these products a t various flow rates of TCE is given in Table and shown graphically in Fig. 1. From Fig. 1, it can be seen that at very low TCE flow rates, the only detectable products are (CN),, CI,, and CNCl. At low flow rates, the
'Table 1 is available, at a nominal charge, from the Depository of Unpublished Data, National Science Library, National Research Council of Canada, Ottawa, Canada K I A 0S2.
flame emission was found to extend downstream from the TCE nozzle to the product trap. As the ratio [TCE]/[N] was increased by increasing the TCE flow rate, the flame "tail" was shortened until, at high TCE flow rates, the flame was completely quenched within a few centimeters of the TCE nozzle. The product flow rate of CNCl increased as CCl, and CC1,CN began to appear. The product flow rates reached maxi- mum values at about 1.1 pmol s-' of TCE (Fig. 1).
Carbon balances have been found t o be better than 95% at very low TCE flow rates and as poor as 50% at high TCE flow rates. The poor carbon balances at high TCE flow rates are probably due to increasing formation of polymer since the polymer appeared to build u p on the wall much more rapidly at high flow rates than at low flow rates. Unfortunately, it was not possible to make quantitative measurements of the polymer yield.
Elemental analysis of the polymer showed it to have the composition C : N : C1 = 1.6 : 1.0 : I .6. A sample of the polymer, which collected on the reaction vessel walls during a number of runs, was decomposed under vacuum over the tem- perature range 298-523 OK. The major product was C1, with relatively small amounts of C2C14,
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
TE
CH
NIS
CH
E U
NIV
ER
SIT
EIT
EIN
DH
OV
EN
on
11/1
5/14
For
pers
onal
use
onl
y.

3682 CAN. J . CHEM. VOL. 51, 1973
CCl,, CNC1, and N, also being formed. After several hours of decomposition, the residue had turned very dark in color.
Up to a TCE flow rate of 0.4 pmol s-', approximately 50% of the chlorine originally present could be accounted for. Above this flow rate, the amount of chlorine which could be accounted for by the products decreased drastically. The polymer contained a certain amount of chlorine. However, this amount was not sufficient to account for the chlorine loss at high flow rates. It was noted that, even at very low temperatures (80 "C), small amounts of C1, evolved from the polymer. It is possible that C1, was physically adsorbed by the polymer. If physical adsorption did not occur, the reported elemental analysis for chlorine must be low. It was also possible that C1, was adsorbed by stopcock grease present in the system. This was found to occur in the reaction of active nitrogen with COCl, (8).
The reaction flame consisted of an intense emission composed of the CN red (A211-X2C+) and CN violet (B2C+-X2C+) systems (9). Since the work of Jennings and Linett (2,3), a spectrum of the NCl molecule has been observed in the region 61 5-703 nm (10). Although this region was studied carefully, no spectrum of the NCl radical was observed. However, as noted by Colin and Jones (lo), the NCl spectrum is extremely weak and difficult to observe against the CN emission background.
When the flow rate of TCE was adjusted properly, the flame emission near the nozzle was very intense while, farther down stream, the emission appeared to oscillate with an extremely bright pink-white flame. Such an oscillatory phenomenon suggested that the flame involved chain reactions (1 1). This flame oscillation was clearly visible in the system in which the active nitrogen was generated by the microwave discharge. However, it was barely observed in the system where the con- densed electric discharge was used to activate the nitrogen. This was probably due to the oscillation of the discharge itself.
Because of the formation of polymer and the resulting significant loss of carbon and chlorine, it was difficult to determine the kinetics and, with the present information, it is only possible to discuss several general reactions which are important in the overall mechanism.
The primary step may involve attack by N(,S) as in reaction 1 or energy transfer from the excited molecule N2(A3C,+) as in reaction 2.
[I] N('S) + C2C14 -> [C2C14N]co,,,plc, -> products
Both reactions 1 and 2 would yield chlorocarbon radicals and C1 while reaction 1 would also yield CNCI.
The chlorocarbon radicals would subsequently react with nitrogen atoms as in the following reactions :
[3] RCI + N -> CNCl + R'
[41 RCI + N -> CNCl + R" + CI
Reactions 3 and 4 as well as reaction 1 would account for the major product CNCl.
In addition to the chlorocarbon radicals formed in reactions 1 and 2, the radical C,Cl, would be formed by adding chlorine atoms to C2C14 as in reaction 5.
This reaction is known to occur readily (12-14). Cyanogen results from the recombination of
CN radicals formed when CNCl is excited by N2(A3C,+) as in reactions 6 and 7.
[61 N2(A3 C,+) + CNCl -> N2(X1 Z,+) + CNCl*
171 CNCl* -> CN + C1
These reactions have been proposed by Bayes (15) from studies of the reaction of active nitrogen with CNCI.
The CN radicals, formed as in reaction 7, may also attack CNCl to form (CN), and C1. They might also attack CCl, to yield the second major product of the reaction CC1,CN as in the following reaction :
The product CCl,, which is formed in rela- tively smaller amounts, probably results from the addition of C1 atoms to CCl,.
Despite the fact that no spectra of NC1 were observed in the emission flame, it is impossible to eliminate it as an intermediate species. NCl would be expected to form as in the following reaction:
This reaction has been proposed by Phillips and
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
TE
CH
NIS
CH
E U
NIV
ER
SIT
EIT
EIN
DH
OV
EN
on
11/1
5/14
For
pers
onal
use
onl
y.

JONES AND RUJIMETHABHAS: REACTION OF ACTIVE NITROGEN 3683
co-workers (16, 17) in their study of the reaction of active nitrogen with Cl,. If NCl formed, it would react rapidly (18) with N(4S) as in reaction 10.
[lo] NCI + N(4S) -> NZ + CI
Thus, the formation of NCl and its subsequent reaction with N(4S) could explain the significant loss of N atoms during the active nitrogen-TCE reaction. A maximum 23% of the original con- centration of N atoms appears in the products (Table I).
M.R. wishes to acknowledge his scholarship from the Canadian International Development Agency. We also acknowledge financial assistance from the National Research Council of Canada.
1. M. RUJIMETHABHAS and W. E. JONES. Can. J. Chem. 50, 346 (1972).
2. K. R. JENNINGS and J. W. LINNETT. Nature, 180, 1272 (1957).
I 3. K. R. JENNINGS and J. W. LINNETT. Trans. Fara- day Soc. 56, 1737 (1960).
I 4. N. H. KIESS and H. P. BROIDA. Proc. 7th Symp. 1 (Int.) Combust. 1958. (Pub. 1959). p. 207.
5. (a) A. A. WESTENBERG and N. DE HAAS. J.
Chem. Phys. 40, 3087 (1964); ( b ) L. ELIAS. J. Chem. Phys. 42, 4311 (1965).
6. J. T. HERRON. J. Phys Chem. 69, 2736 (1965). 7. G. BRAUER. ( E d i t o r ) . Handbook of preparative
inorganic chemistry. Vol. 1. 2nd ed. Academic Press. New York, N.Y. 1963. Method I. p. 662.
8. R. R. BAKER and C. A. WINKLER. Can. J. Chem. 49, 3846 (1971).
9. R. W. B. PEARSE and A. G. GAYDON. The iden- tification of molecular spectra. 3rd ed., Chap- man and Hall, London. 1963.
10. R. COLIN and W. E. JONES. Can. J. Phys. 45, 301 (1967).
11. D. A. FRANK-KAMENET~KII. Diffusion and heat transfer in chemical kinetics. Plenum Press, New York. 1969.
12. J. A. FRANKLIN, G. HUYBRECHTS, and C. CILLIEN. Trans. Faraday Soc. 65, 2094 (1969).
13. J. KNOX and K. D. WAUGH. Trans. Faraday Soc. 65, 1585 (1969).
14. D. D. DAVIS, W. BRAUN, and A. M. BASS. Inter. J. Chem. Kinet. 2, 101 (1970).
15. K. D. BAYES. Can. J. Chem. 39, 1074 (1961). 16. K. S. RAXWORTHY and L. F. PHILLIPS. Can. J.
Chem. 42, 2928 (1964). 17. M. R. GRIGOR and L. F. PHILLIPS. Proc. 11th
Symp. (Int.) Combust. 1966. (Pub. 1967). p. 1171.
18. C. G. FREEMAN and L. F. PHILLIPS. J. Phys. Chem. 72, 3028 (1968).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
TE
CH
NIS
CH
E U
NIV
ER
SIT
EIT
EIN
DH
OV
EN
on
11/1
5/14
For
pers
onal
use
onl
y.


















![TETRACHLOROETHYLENE (PCE) · Tetrachloroethylene (PCE) ... bKd = [(Koc) × (fraction organic matter)], a site and soil zone specific parameter eestimated parameter value iii](https://img.pdfslide.us/doc/110x75/5ae720597f8b9acc268e36af/tetrachloroethylene-pce-pce-bkd-koc-fraction-organic-matter-a-site.jpg)










