Embed Size (px)
Citation preview

Rate Theories of elementary reaction

2 Transition state theory (TST) for bimolecular reactions
Theory of Absolute reaction Rates
Theory of activated complex theory

A + B-C A-B + C During reaction, massive changes of form are occurring, energies are being redistributed among bonds: old bonds are being ripped apart and new bonds formed.
H + H–H H∙∙∙∙∙∙∙∙∙ H∙∙∙∙∙∙H
H∙∙∙∙∙∙H∙∙∙∙∙∙H (activated state)
H∙∙∙∙∙∙H∙∙∙∙∙∙∙∙∙∙∙∙H H–H + H
This process can be generalized as:
A + B-C [A B C] A-B + C
Activated complex Transition state

Whether or not the energy change of the reaction can be used to explain the reaction on the basis of thermodynamics?
The transition state theory (TST), attempting to explain
reaction rates on the basis of thermodynamics, was devel
oped by H. Eyring and M. Polanyi during 1930-1935. TS
T treated the reaction rate from a quantum mechanical vi
ewpoint involves the consideration of intramolecular forc
es and intermolecular forces at the same time.

According to TST, before undergoing reaction, reactant
molecules must form an activated complex which is in
thermodynamic equilibrium with the molecules of the
reactants. The activated complexes, the energy of which
is higher than both reactants and products, is treated as
an ordinary molecule except that it has transient
existence and decomposes at a definite rate to form the
product.
Basic consideration

2.1 Potential energy surfaces
According to the quantum mechanics, the nature of the chemical interaction (chemical bond) is a potential energy which is the function of interatomic distance (r):
( )V V r
The function can be obtained by solving Schrödinger equation for a fixed nuclear configuration, i.e., Born-Oppenheimer approximation.
The other way is to use empirical equation. The empirical equation usually used for system of two atoms is the Morse equation:

)]}(exp[2)](2{exp[)( 00 rrarraDrV e
where De is the depth of the wall of potential, or the dissociati
on energy of the bond. r0 is the equilibrium interatomic distanc
e, a is a parameter with the unit of cm-1 which can be determined from spectroscopy.
Morse equation:
When r = r0, Vr (r = r0) = -De
r, Vr (r) = 0
When r > r0, interatomic attraction exists,
r < r0, interatomic repulsion appears.
The equilibrium distance r0 is the bond length.
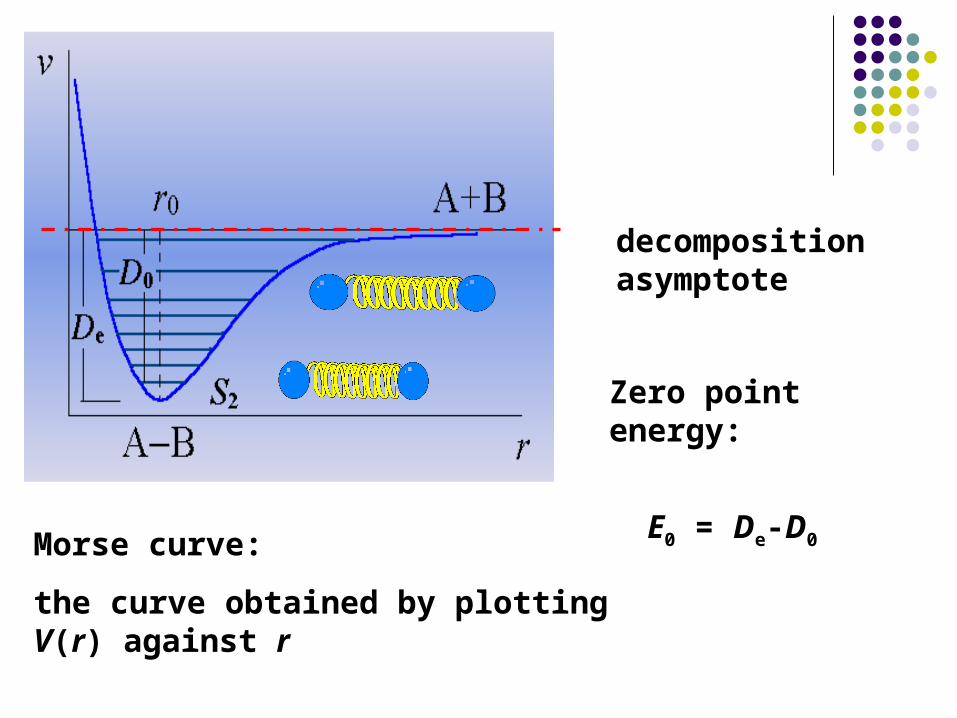
Morse curve:
the curve obtained by plotting V(r) against r
Zero point energy:
E0 = De-D0
decomposition asymptote
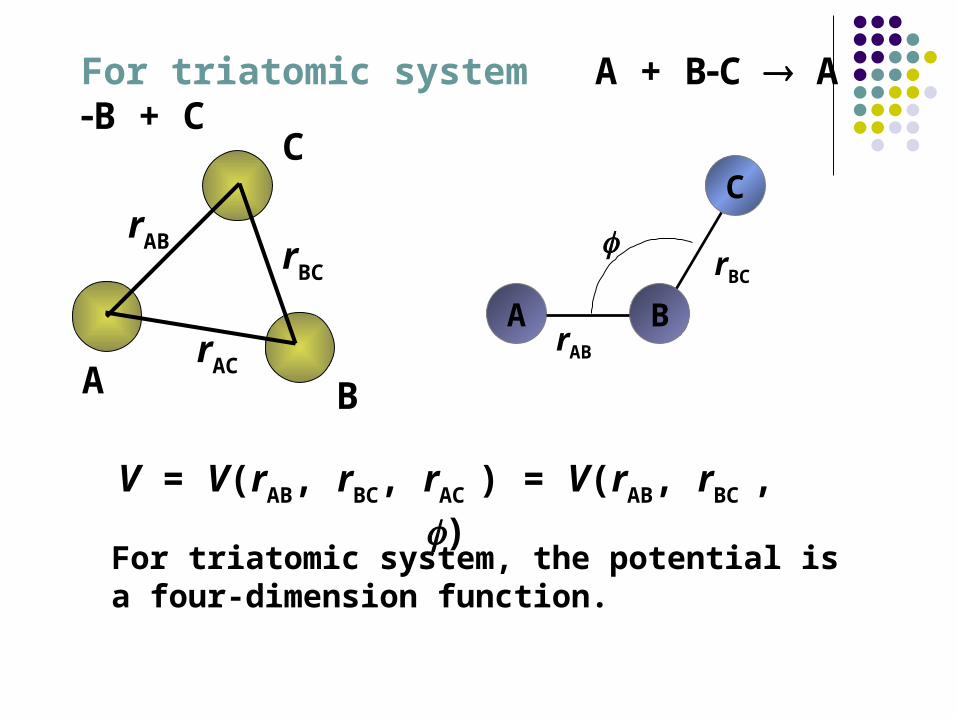
For triatomic system A + BC AB + C
V = V(rAB, rBC, rAC ) = V(rAB, rBC , )
A B
C
rAB rBC
rAC
For triatomic system, the potential is a four-dimension function.
A B
C
rBC
rAB

In 1930, Eyring and Polanyi make = 180 o, i.e., collinear c
ollision and the potential energy surface can be plotted in a
three dimensions / coordination system.
V = V(rAB, rBC)
Eyring et al. calculated the energy of the triatomic system:
HA + HBHC HAHB+ HC
using the method proposed by London.
A B CrBCrAB
= 180 o
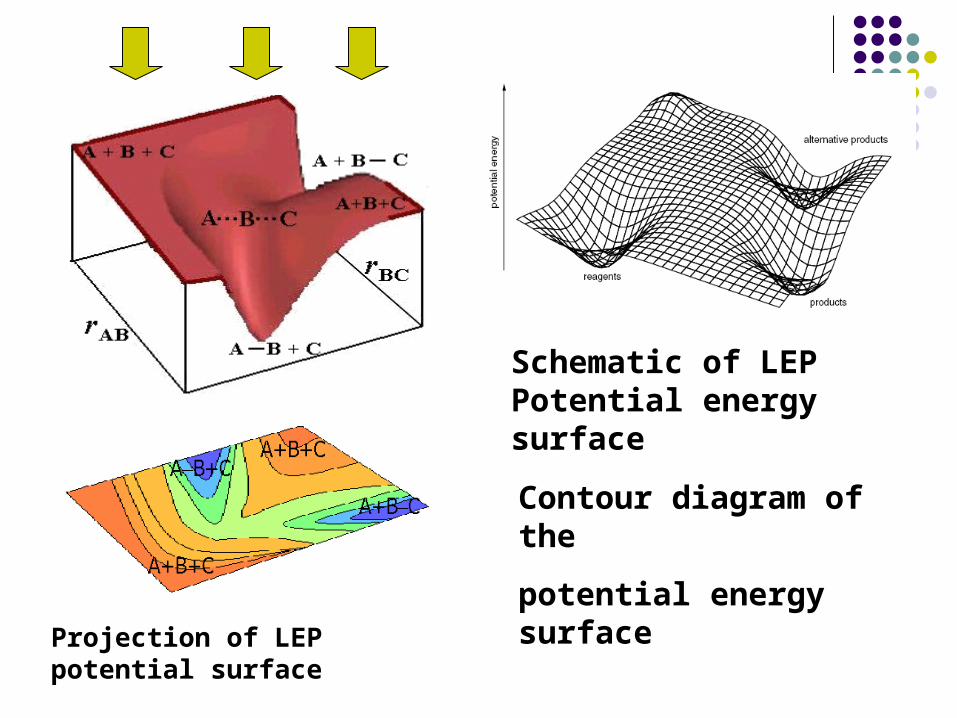
Schematic of LEP Potential energy surface
Contour diagram of the
potential energy surface
Projection of LEP potential surface
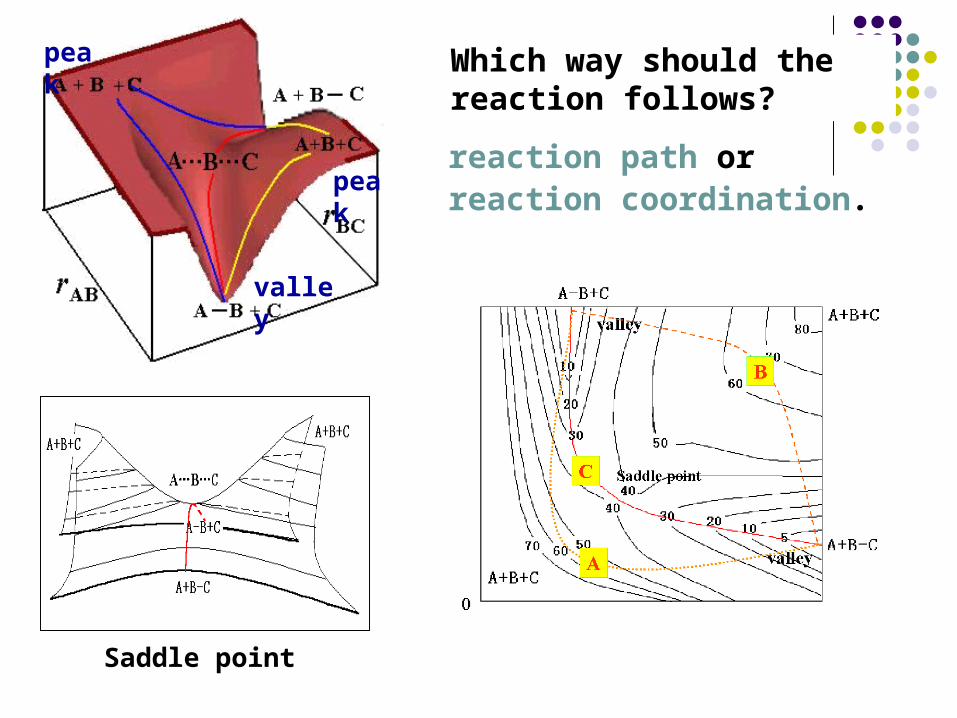
Which way should the reaction follows?
Saddle point
valley
peak
peak
reaction path or reaction coordination.
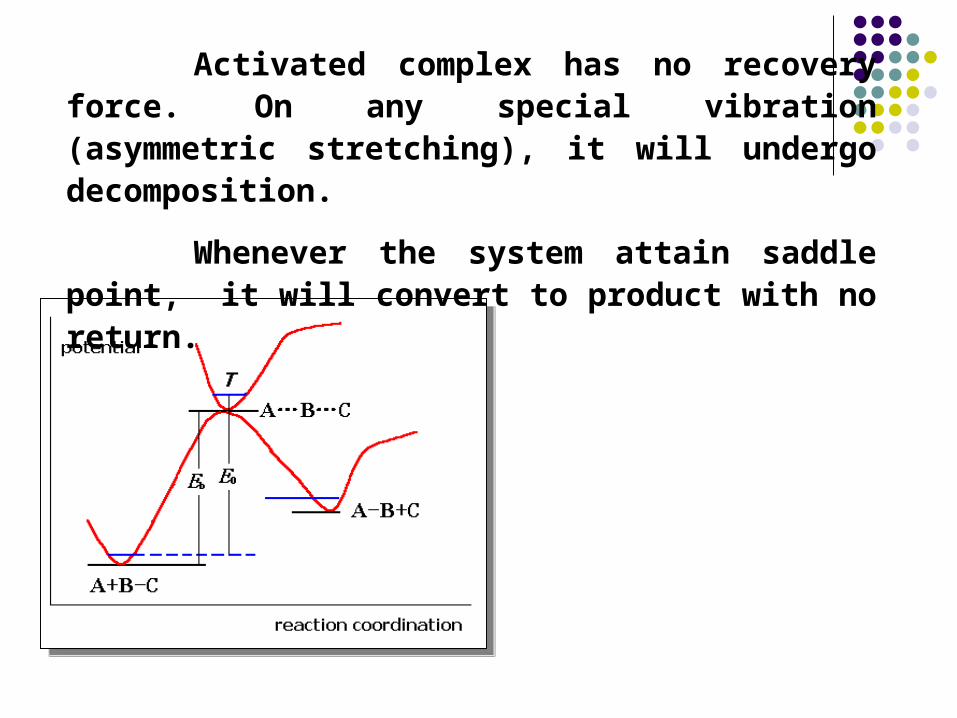
Activated complex has no recovery force. On any special vibration (asymmetric stretching), it will undergo decomposition.
Whenever the system attain saddle point, it will convert to product with no return.
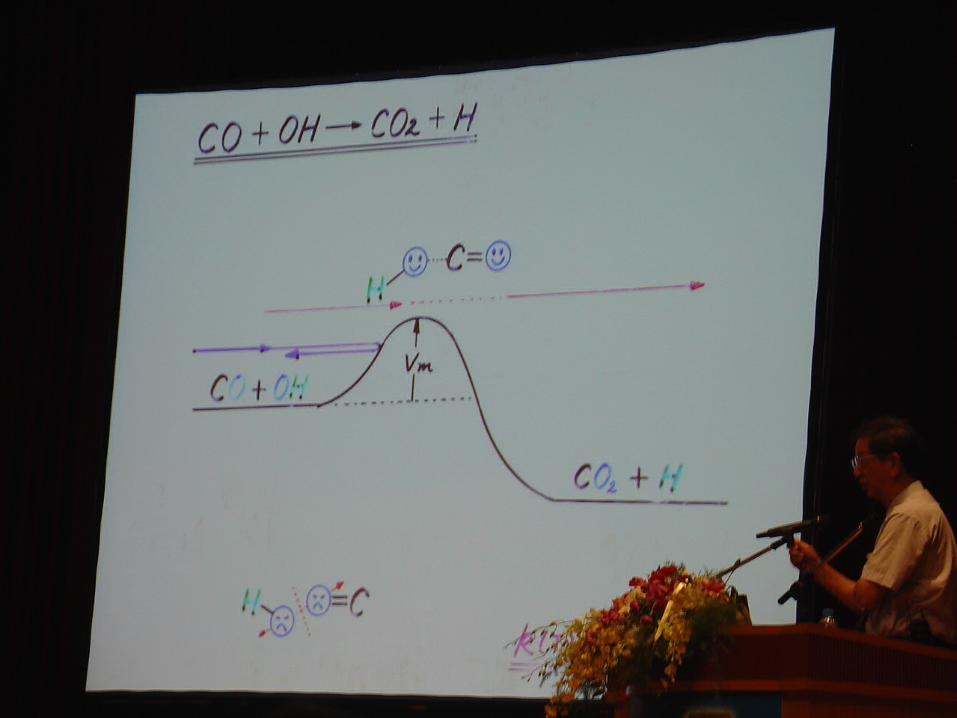

2.2 Kinetic treatment of the rate constant of TST
For reaction:
The rate of the reaction depends on two factors:
1) the concentration of the activated complex (c)
2) the rate at which the activated complex dissociates into products()
ABr c
AB
A B
cK
c c
According to equilibrium assumption
A Br K c c Kk

According to statistical thermodynamics, K can be
expressed using the molecular partition function.
0AB
A B A B A B
K expEc q f
c c q q f f RT
E0 is the difference between the zero point energy of
activated complex and reactants. q is the partition function, f
is the partition function without E0 stem and volume stem.
For activated complex with three atoms, f can be written a
s a product of partition function for three translational, thre
e rotational, and five vibrational degrees of freedom.
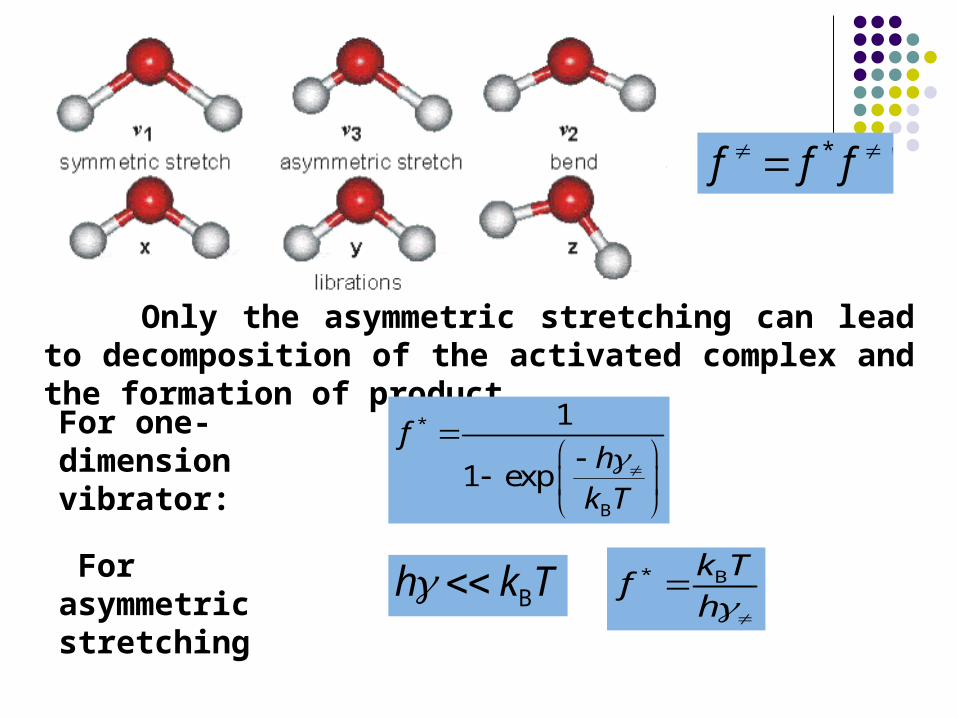
Only the asymmetric stretching can lead to decomposition of the activated complex and the formation of product.
'* fff
For one-dimension vibrator:
*
B
1
1 exp
fhk T
For asymmetric stretching Bh k T * Bk Tf
h

B 'k T
f fh
0B
A B
'exp
Ek T fk K
h f f RT
0B
A B
'exp
Ek T fk
h f f RT
statistical expression for the rate constant of TST
For general elementary reaction
0B 'exp
i
Ek T fk
h f RT
0B 'exp
i
Ek T fk
h f RT
In which f’ can be obtained from partition equation and E0 can be obtained from potential surface. Therefore, k of
TST can be theoretically calculated. Absolute rate theory

For example:
For elementary equation:
H2+ F HH F H + HF
Theoretical:
k = 1.17 1011 exp(-790/T)
Experimental:
k = 2 1011 exp(-800/T)

2.3 Thermodynamic treatment of TST For nonideal systems, the intermolecular interacti
on makes the partition function complex. For these c
ases, the kinetic treatment becomes impossible. In 19
33, LaMer tried to treat TST thermodynamically.
Bk Tk Kh
0B
A B
'exp
Ek T fk
h f f RT
0
A B
'exp
EfK
f f RT
lnG RT K y y
G H T S y y y
Standard molar entropy of activation, standard molar enthalpy of activation

B expk T G
kh RT
y
expG
KRT
yy
B exp expk T S H
h R RT
y y
G H T S y y y
The thermodynamic expression of the rate of TST is different from Arrhenius equation
Bk Tk Kh
lnG RT K y y
Bk Tk Kh
Bln ln lnk T
k Kh
dT
Kd
TdT
kd
ln1ln
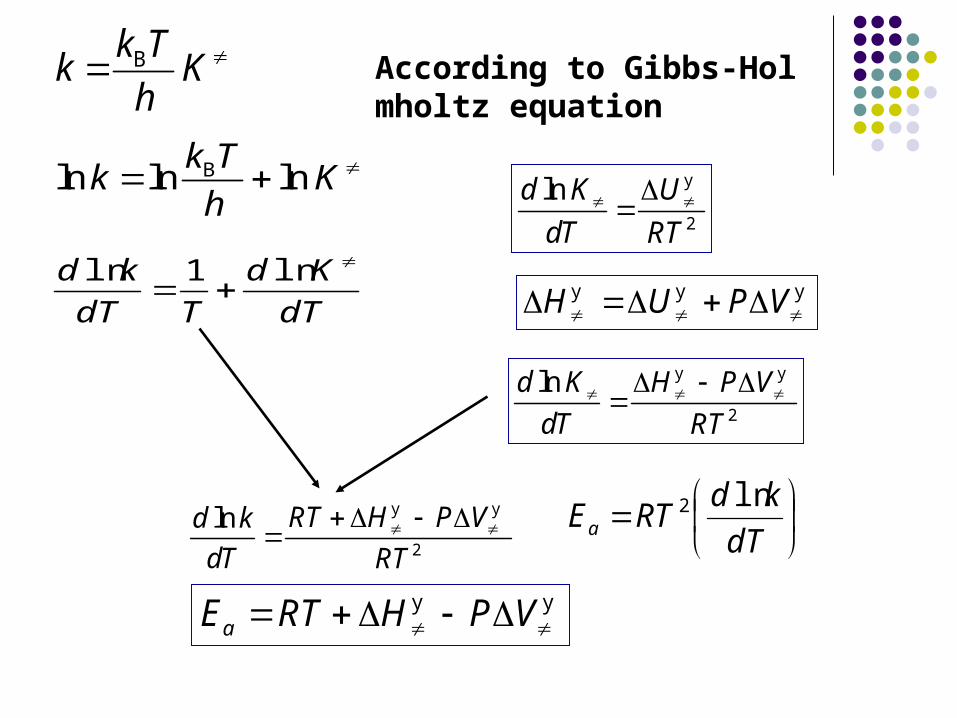
According to Gibbs-Holmholtz equation
2
lnd K U
dT RT
y
H U P V y y y
2
lnd K H P V
dT RT
y y
2
ln RT H P Vd k
dT RT
y y
dT
kdRTEa
ln2
aE RT H P V y y

aE RT H yFor liquid reaction: PV = 0
aE H nRT y
For gaseous reaction: (1 )P V nRT n RT y
n is the number of reactant molecules
exp expn aB Ek T Sk e
h RT RT
y
exp expn aB Ek T Sk e
h RT RT
y
exp expBk T S Hk
h R RT
y y
thermodynamic expression of the rate of TST.
aE RT H P V y y

B exp expn aEk T Sk e
h RT RT
yB exp expn aEk T S
k eh RT RT
y
RT
EAk aexp
B expnk T SA e
h RT
yB expnk T S
A eh RT
y
RT
EPZk a
SCT exp'
B'k T
Zh
expS
PRT
y
is a general constant with unit of s-1 of the magnitude of 1013.
The pre-exponential factor depends on the standard entropy of activation and related to the structure of activated complex.
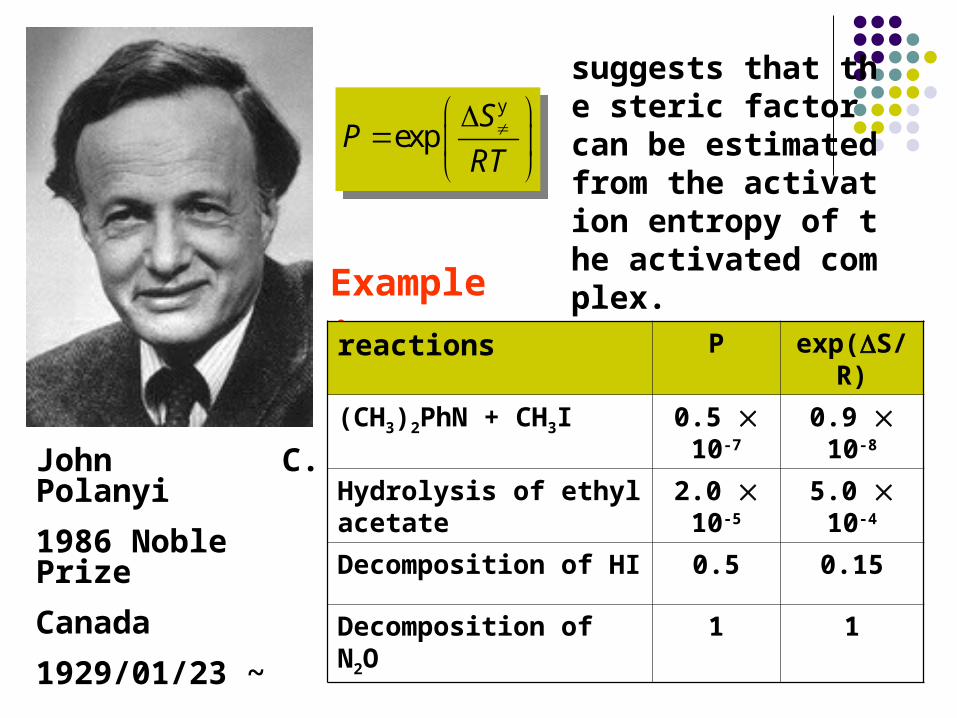
Example:
reactions P exp(S/R)
(CH3)2PhN + CH3I 0.5 10-7 0.9 10-8
Hydrolysis of ethyl acetate 2.0 10-5 5.0 10-4
Decomposition of HI 0.5 0.15
Decomposition of N2O 1 1
suggests that the steric factor can be estimated from the activation entropy of the activated complex.
expS
PRT
y
expS
PRT
y
John C. Polanyi
1986 Noble Prize
Canada
1929/01/23 ~





























