Embed Size (px)
Citation preview

of October 13, 2018.This information is current as
Brain Barrier−the BloodfParacellular Diapedesis in a Human Model o
Rapid Remodeling of Tight Junctions during
Richard M. Ransohoff and William A. MullerRyan C. Winger, Jennifer E. Koblinski, Takashi Kanda,
http://www.jimmunol.org/content/193/5/2427doi: 10.4049/jimmunol.1400700July 2014;
2014; 193:2427-2437; Prepublished online 25J Immunol
MaterialSupplementary
0.DCSupplementalhttp://www.jimmunol.org/content/suppl/2014/07/25/jimmunol.140070
Referenceshttp://www.jimmunol.org/content/193/5/2427.full#ref-list-1
, 23 of which you can access for free at: cites 68 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Rapid Remodeling of Tight Junctions during ParacellularDiapedesis in a Human Model of the Blood–Brain Barrier
Ryan C. Winger,* Jennifer E. Koblinski,*,1 Takashi Kanda,† Richard M. Ransohoff,‡ and
William A. Muller*
Leukocyte transendothelial migration (TEM; diapedesis) is a critical event in immune surveillance and inflammation. Most TEM
occurs at endothelial cell borders (paracellular). However, there is indirect evidence to suggest that, at the tight junctions of the
blood–brain barrier (BBB), leukocytes migrate directly through the endothelial cell body (transcellular). Why leukocytes migrate
through the endothelial cell body rather than the cell borders is unknown. To test the hypothesis that the tightness of endothelial
cell junctions influences the pathway of diapedesis, we developed an in vitro model of the BBB that possessed 10-fold higher
electrical resistance than standard culture conditions and strongly expressed the BBB tight junction proteins claudin-5 and
claudin-3. We found that paracellular TEM was still the predominant pathway (‡98%) and TEM was dependent on
PECAM-1 and CD99. We show that endothelial tight junctions expressing claudin-5 are dynamic and undergo rapid remodeling
during TEM. Membrane from the endothelial lateral border recycling compartment is mobilized to the exact site of tight junction
remodeling. This preserves the endothelial barrier by sealing the intercellular gaps with membrane and engaging the migrating
leukocyte with unligated adhesion molecules (PECAM-1 and CD99) as it crosses the cell border. These findings provide new
insights into leukocyte–endothelial interactions at the BBB and suggest that tight junctions are more dynamic than previously
appreciated. The Journal of Immunology, 2014, 193: 2427–2437.
The brain is under continuous immune surveillance bycirculating leukocytes, which patrol this specialized organto detect and eliminate potential infectious and damaging
agents (1–3). When left unabated, however, leukocyte recruitmentinto the brain is a prominent pathologic feature in many neuro-logic diseases, including cerebral infection (4, 5), stroke (6–9),trauma (10–12), multiple sclerosis (13, 14), and certain neurode-generative disorders (15). These disorders cause serious long-term neurologic deficits and significant morbidity and mortalityworldwide. Homeostasis within the CNS is largely maintained bya unique microvasculature called the blood–brain barrier (BBB)(16). The BBB is formed by highly specialized endothelial cells(ECs) that are interconnected by complex and continuous tightjunctions. These promote normal brain physiological function byrestricting the entry of ions, macromolecules, and noxious blood-borne agents (17). Astrocytes, which are in close apposition to thecerebral vasculature, are crucial inducers of the BBB phenotype
and help facilitate tight junction protein expression and mainte-nance through contact-dependent mechanisms and by releasingsoluble factors (18).Leukocytes have been speculated to take the path of least re-
sistance across the endothelium (19); therefore, the relativetightness of the endothelium may significantly influence thepathway of diapedesis (transendothelial migration [TEM]). Leu-kocytes have been reported to cross endothelium in vitro andin vivo using paracellular and transcellular pathways (20–22). Themechanisms underlying the leukocyte’s decision to migratethrough the EC body rather than the cell borders are unknown.Indirect evidence has suggested that, at the BBB, leukocytes mi-grate transcellularly through the EC body (23, 24). We hypothe-sized that the tightness of endothelial junctions and the ability ofleukocytes to breach them might dictate the pathway of TEM.ECs in vitro form monolayers of low resistance, and paracellularTEM predominates even under cytokine-activated conditions (25).However, if junctions are very tight, such as at the BBB, trans-cellular migration across the EC body at a thin point may be easierrather than having to disrupt and reform the complex three-dimensional interactions of the tight junctions.Whether leukocytes primarily cross the BBB paracellularly at
cell borders through tight junctions or directly through the EC bodytranscellularly to reach the brain remains unclear (26–28). Fur-thermore, if leukocytes migrate paracellularly, whether and howtight junctions are remodeled at the BBB is not known. This isa critical issue, as inflammation and BBB dysfunction are at theroot of most CNS pathologies (29, 30). Understanding the mo-lecular mechanisms governing the route of TEM in the CNS isfundamental to developing better reagents to modulate pathologicimmune responses or enhance host-protective mechanisms in neuro-inflammatory diseases. To test the hypothesis that transcellularmigration would be more common if EC junctions were tighter,we constructed a simplified and robust cell culture model of thehuman BBB by treating HUVECs with astrocyte-conditionedmedium (ACM) supplemented with agents that raise intracellular
*Department of Pathology, Feinberg School of Medicine, Northwestern University,Chicago, IL 60611; †Department of Neurology and Clinical Neuroscience,Yamaguchi University Graduate School of Medicine, Ube 755-8505, Japan; and‡Neuroinflammation Research Center, Department of Neurosciences, Lerner ResearchInstitute, Cleveland Clinic, Cleveland, OH 44195
1Current address: Department of Pathology, School of Medicine, Virginia Common-wealth University, Richmond, VA.
Received for publication March 18, 2014. Accepted for publication June 27, 2014.
This work was supported by National Institutes of Health Grants T32 AI7476-17(to R.C.W.) and R37 HL064774 and R01 HL046849 (to W.A.M.).
Address correspondence and reprint requests to Dr. William A. Muller, Departmentof Pathology, Feinberg School of Medicine, Northwestern University, 300 East Chi-cago Avenue, Chicago, IL 60611. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ACM, astrocyte-conditioned media; BBB, blood–brain barrier; CPT, 8-(4-chlorophenylthio); EC, endothelial cell; HSA, human serumalbumin; LBRC, lateral border recycling compartment; TEER, transendothelial elec-trical resistance; TEM, transendothelial migration; TR, targeted recycling; VE, vas-cular endothelial.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1400700
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

cAMP. This culture system was sufficient to induce BBB char-acteristics, significantly enhancing the barrier function to ions andsmall molecules and inducing the expression of BBB tight junc-tion proteins. We report that paracellular diapedesis is the preva-lent pathway used by monocytes and neutrophils at the BBBin vitro and that similar molecules and mechanisms regulate TEMunder these conditions as in low resistance endothelium. Moreimportantly, we report the novel finding that endothelial tight junc-tions are remarkably dynamic and undergo rapid remodeling to ac-commodate the passage of leukocytes across EC borders withoutperturbing the endothelial barrier. Junctional remodeling and barrierfunction preservation were facilitated by the recruitment of mem-brane from the lateral border recycling compartment (LBRC), a novelendothelial specific compartment containing PECAM-1 and CD99that we have previously shown to be essential for TEM (25, 31). Weconclude that rapid remodeling of tight junctions during paracellularTEM is a tightly regulated process, and endothelial tight junctions aremore dynamic than previously appreciated.
Materials and MethodsAll experimental protocols and procedures involving human subjects andmaterials were approved by the Institutional Review Board of NorthwesternUniversity Feinberg School of Medicine (Chicago, IL).
Reagents
The mAbs hec1 (anti–vascular endothelial [VE]-cadherin), hec7 (anti–PECAM-1), and hec2 (anti-CD99) were produced from hybridomas gen-erated in the laboratory, as previously described (32). P1.1 (nonblockinganti-PECAM mAb) was column purified on protein A–Sepharose (GEHealthcare) from ascites provided by P. J. Newman (Blood Center ofWisconsin, Milwaukee, WI) (33). Fab fragments of P1.1 were cut usingpapain (Thermo Fisher Scientific), followed by column purification on proteinA–Sepharose. Purity of Fab fragments was confirmed by SDS-PAGE. Un-labeled and Dylight 549–conjugated goat anti-mouse F(ab9)2 Abs used fortargeted recycling (TR) experiments of PECAM-1 were purchased fromJackson ImmunoResearch Laboratories. Abs for immunofluorescence wereconjugated to DyLight 488, DyLight 550, or Alexa Fluor 650, according tothe manufacturer’s protocol (Thermo Fisher Scientific). Alexa Fluor 488–conjugated mAbs against occludin and claudin-5 were purchased from LifeTechnologies (Invitrogen). Polyclonal Ab against claudin-3 was purchasedfrom Life Technologies (Invitrogen). The 8-(4-chlorophenylthio) (CPT)-cAMP was purchased from Sigma-Aldrich, and phosphodiesterase inhibitortype IV RO-20-1724 was purchased from Tocris Bioscience.
Cells
HUVECs were isolated from fresh human umbilical cords, as previouslydescribed (32). HUVECs for standard culture conditions were grown inM199 medium (Invitrogen) supplemented with 20% heat-inactivated nor-mal human serum and 100 U/ml penicillin-streptomycin at 37˚C in a hu-midified atmosphere of 5% CO2. HUVECs at passage two were culturedon thick hydrated collagen type 1 (Vitrogen; Cohesion Technologies) gelsset in 96-well plates coated with 5 mg/ml fibronectin. hCMEC/D3 brainECs were obtained as a gift from B. Weksler (Weill Cornell MedicalCollege). TY10 brain ECs generated by T. Kanda (Yamaguchi UniversityGraduate School of Medicine) were obtained as a gift from R. Ransohoff(Cleveland Clinic Lerner Research Institute). Primary human astrocyteswere purchased from ScienCell Research Laboratories. PBMCs wereisolated from healthy volunteers by density gradient centrifugation inFicoll-Paque (GE Healthcare), as previously described (34). Briefly, blooddrawn from healthy volunteers was immediately mixed with 10 mmol/Lfinal concentration of EDTA and an equal volume of HBSS (Mediatech,Herndon, VA) and then layered over Ficoll-Paque density gradient medium(GE Healthcare Biosciences AB, Uppsala, Sweden). After centrifugation,the upper plasma layer was collected into fresh tubes and the PBMCs at theinterface were collected into a separate tube, diluted with HBSS, andcentrifuged at 1500 rpm for 10 min. The cell pellet was resuspended in thespun plasma and centrifuged at 1200 rpm for 5 min. The resulting pelletwas washed two to three times with HBSS containing 0.1% human serumalbumin (HSA; Grifols Biologicals, Los Angeles, CA) via resuspensionand centrifugation. The final cell pellet was resuspended in M199 con-taining 0.1% HSA. Neutrophils were isolated and prepared from healthyvolunteers, as previously described (34).
Human BBB model culture conditions
Inducible HUVEC ACM BBB model. Astrocytes were grown on poly-D-lysine–coated tissue culture dishes and used until passage 10. ACM wascollected and treated, as previously described (35–37). Briefly, astrocytecultures were grown at 37˚C in a humidified atmosphere of 5% CO2 andwere fed with fresh astrocyte complete medium (ScienCell ResearchLaboratories) containing low concentration of FBS (2%). After ∼48 h,when confluent, the medium was collected, sterile filtered (0.2 mm),centrifuged, and diluted 1:1 with HUVEC media and either used imme-diately or stored at 280˚C. HUVECs used for the BBB model were cul-tured until confluence in ACM. Once confluent, ACM was then sup-plemented with 17.5 mM RO-20-1724 (Tocris Bioscience) and 250 mMCPT-cAMP (Sigma Aldrich) for 24 h. After 24 h, the monolayers wereextensively washed immediately prior to experimentation. In someexperiments, TNF was added to some monolayers to activate ECs. TNFtreatment was 20 ng/ml for 4 h, except for Supplemental Fig. 2, in whichtreatments are described in the figure legend.
hCMEC/D3 and TY10. hCMEC/D3 were cultured, as previously described(38), at 37˚C in a humidified atmosphere of 5% CO2. Briefly, hCMEC/D3were grown in endothelial basal medium-2 (Lonza) that was supplementedwith basic fibroblast growth factor (1 ng/ml), hydrocortisone (1.4 mM),ascorbic acid (5 mg/ml), chemically defined lipid concentrate (1/100 di-lution), 5% FBS, and 1% penicillin-streptomycin. TY10 (39) were grownin endothelial basal medium-2 containing 20% heat-inactivated FBS and0.6% antibiotic-antimycotic solution (Sigma-Aldrich) and supplementedwith EGM-2 SingleQuots (except for GA1000) at 33˚C. TY10 were grownat 33˚C for the growth phase and were then transferred to 37˚C for1–2 d when confluent to arrest growth and induce differentiation.
Characterization of barrier function
Endothelial barrier function was measured by two assays, as follows: 1)recording transendothelial electrical resistance (TEER) and 2) monitoringpermeability to FITC-conjugated dextran (20 kDa; Sigma-Aldrich). Briefly,the tightness of cell junctions was quantitated by measuring the ECjunctions’ ability to restrict paracellular flux of small ions via analysis ofTEER. ECs were plated on 3-mm Transwell filter inserts (Corning) thatwere precoated with collagen type I diluted 1:50 in 60% ethanol and thenoverlaid with 5 mg/ml fibronectin. For each filter, the electrical resistancewas measured using an electrical resistance system containing a current-passing and voltage-measuring electrode (World Precision Instruments,New Haven, CT). Resistances of blank filters were subtracted from thosefilters containing cells. Final resistance calculations were corrected forfilter surface area.
For permeability assays, ECs were cultured on hydrated fibrillar collagengels, as described above. Monolayers were washed with PBS, and 100 mg/ml20-kDa FITC-dextran was added to each well. Monolayers were then in-cubated at 37˚C in 5% CO2 for 1 h. After 1 h, monolayers were thenwashed extensively and the relative fluorescence intensity of FITC-dextranthat had passed across the monolayers into the collagen gels was measuredby a FilterMax F5 microplate reader (Molecular Devices, Sunnyvale, CA).Indicated results are the mean fluorescence intensity of at least three in-dependent experiments with replicates of six for each condition. In someexperiments measuring barrier function during TEM, monocytes wereadded (2 3 105 per well) at the same time as FITC-dextran and perme-ability was measured, as described.
TEM assay
Quantitative endpoint TEM assays were performed, as previously described(40). In brief, freshly isolated PBMC were resuspended in M199 mediumcontaining 0.1% HSA, added to monolayers at 23 105 cells/well, and thenwarmed for 1 h at 37˚C in a CO2 incubator for TEM to take place. Aspreviously published (34), only monocytes and not lymphocytes transmi-grate within this time frame. In experiments determining the dependenceof TEM on PECAM-1 or CD99, PBMCs (4 3 106 cell/ml) were mixed 1:1with either nonblocking control mAb (hec1; anti–VE-cadherin) or block-ing anti–PECAM-1 (hec7) or anti-CD99 (hec2) mAb to a final concen-tration of 20 mg/ml. Then 100 ml was added to the appropriate wells. Atthe end of the incubation, monolayers were washed twice with 1 mmol/LEDTA (Sigma-Aldrich) in HBSS, followed by two to three washes withPBS containing Ca2+ and Mg2+. The monolayers and leukocytes were thenfixed in freshly prepared 2.5% glutaraldehyde in 0.1 M sodium cacodylatebuffer (pH 7.4; Electron Microscopy Sciences). Before quantitative anal-ysis, the monolayers were stained with modified Wright-Giemsa (ProtocolHema3; Fisher Diagnostics, Middletown, VA) and mounted onto glassslides for visualization. Imaging was performed with a Zeiss Ultraphotmicroscope with Nomarski optics and a SPOT Insight Color CCD
2428 BBB TIGHT JUNCTION REMODELING DURING DIAPEDESIS
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

(Diagnostic Instruments, Sterling Heights, MI). TEM was analyzed bymanually counting at least 100 cells per collagen gel and noting theirposition relative to the endothelial monolayer (above or below, definingabove as any monocyte having most of the cell body on or above the focalplate of the EC nuclei). Cell adhesion was quantified as the mean of thenumber of leukocytes associated with each field.
Immunofluorescence microscopy
To visualize the distribution of adhesion molecules, adherens junctions, andtight junctions and their behavior during leukocyte TEM, leukocytes wereallowed to TEM for 10 min across BBB or standard control HUVECmonolayers. They were then rapidly washed and fixed in freshly prepared4% paraformaldehyde on ice, as previously described (25). Blocking buffer(PBS + 2% FBS, 2% FCS, and 2% species-specific normal serum) wasadded for 1 h at room temperature. Specific Abs for each adhesion mol-ecule and junctional protein were directly conjugated to Alexa Fluor dyesand incubated with monolayers at final concentration of 20 mg/ml for 1 h atroom temperature. In experiments visualizing tight junction proteins, cellswere permeabilized after fixation with 0.2% Triton X-100 (Sigma-Aldrich)for 10 min and incubated with Alexa Fluor 488–labeled claudin-5. Imagesand stacks of optical sections were collected with a spinning-disk confocalmicroscope using an Ultraview VoX imaging system (Ultraview, Waltham,MA) equipped with a Yokogawa CSU-1 spinning disk (Yokogawa Electric,Tokyo, Japan). Images were acquired through a 403 oil immersion ob-jective using Volocity software version 6.2.1 (Perkin Elmer, Waltham,MA) and subsequently processed and analyzed using ImageJ. The figuresshow representative optical sections through the region of interest orprojections of the whole stack using the maximum intensity method.
Quantification of transcellular and paracellular diapedesis
Monolayers were cultured, as described above, with or without TNF(20 ng/ml) activation for 4 h before each experiment. For some experiments,200 ng/ml CCL2 was added to the monolayers for 30 min and then washedextensively prior to experimentation, as previously described (41). A totalof 2 3 105 monocytes in cold M199 containing 0.1% HSA was added toeach well, and cells were allowed to settle for 15 min on ice at 4˚C.Monolayers were then warmed for 10 min at 37˚C in a CO2 incubator.Cells were rapidly washed with ice-cold PBS, fixed in ice-cold 4% para-formaldehyde, and then stained for VE-cadherin to label the cell junctionand CD18 to label monocytes. Fixed samples were examined by spinning-disk confocal microscopy. Image stacks were obtained by spinning diskconfocal microscopy and were analyzed for quantification of diapedesis.Monocytes crossing the endothelium at the cell junction with an inter-cellular gap in VE-cadherin were scored as paracellular, and those crossingthrough the EC body at a distance of 2 mm from the cell junction notassociated with gap in VE-cadherin were scored as transcellular. Totalmonocytes associated with the ECs in each field (i.e., monocytes on topand under endothelium plus those in the act of diapedesis) were countedfor each experiment. Percentage of TEM was calculated by dividing thetotal number of cells in the act of diapedesis by the total number ofmonocytes in each field (25).
TR experiments
TR experiments were performed, as previously described (25, 31), andperformed with resting or TNF-activated monolayers. Briefly, ECs platedon collagen gels were incubated with nonblocking Fab fragment P1.1 for1 h at 37˚C to label total PECAM-1. Monolayers were subsequentlywashed, chilled, and incubated with an excess of unlabeled goat anti-mouse F(ab9)2 IgG for 1 h on ice at 4˚C to saturate primary Ab boundto surface PECAM-1. Cells were then washed, and M199 containing AlexaFluor 594–conjugated goat anti-mouse F(ab9)2 IgG and 23 105 monocyteswas added to the endothelial monolayer. Cells were kept on ice for 15 minto allow for leukocytes to settle and then warmed at 37˚C for 10 min ina CO2 incubator. Monolayers were subsequently washed and fixed on icein freshly prepared 4% paraformaldehyde for 10 min at room temperature.Images were obtained and processed, and percentage of TR was quantified,as previously described (25, 31).
Quantification of adherens and tight junction gaps during TEM
Three-dimensional confocal imaging of leukocytes fixed in the act of TEMwas performed, as described above. Immunofluorescence staining was usedto identify changes in junctional staining distribution of adherens junctionVE-cadherin and tight junction claudin-5 with respect to monocytes stainedwith CD18. The number and location of monocytes extending pseudopodsin the act of TEM associated with or without disruption in staining of VE-cadherin and/or claudin-5 were quantitated. A monocyte in the act of TEM
was considered to be associated with an observed staining disruption if itwas located at the cell junction and the gap in VE-cadherin or claudin-5 wasat least 2 mm in width. In some experiments, transmigrating leukocytesassociated with gap in VE-cadherin or claudin-5 and recycled LBRC werequantified. Data were normalized to the total number of monocytes perfield.
Statistical analysis
All experiments with quantification were performed independently at leastthree times with a minimum of three replicates for each sample within eachexperiment. For the TEM assay, the values for the replicates were averagedtogether within each experiment. The average and SD of these averages areshown in the figures. Group differences were tested with one-way ANOVA(or two-way ANOVA when appropriate), followed by the Tukey multiplecomparison post hoc test to calculate p values and determine that the meansare statistically and significantly different from each other. Differencesbetween two groups were analyzed with nonparametric Mann–WhitneyU test. Statistical analyses were done using GraphPad Prism software(GraphPad, San Diego, CA).
ResultsEstablishment of a cell culture model of the human BBB
To investigate the relationship between the tightness of EC junc-tions and the route of diapedesis in vitro, we sought to simulate theBBB by generating monolayers of tight endothelium with BBBfeatures. Brain ECs, irrespective of their species, lose many of theirBBB properties in vitro (42, 43). Pharmacologically increasinglevels of intracellular cAMP in combination with ACM enhancesendothelial barrier function in vitro and induces expression ofBBB markers in brain ECs and noncerebral peripheral vascularECs (35, 36, 44). Therefore, we attempted to establish a humanBBB model by culturing HUVECs in ACM supplemented with thecAMP analog CPT-cAMP and phosphodiesterase inhibitor RO-20-1724, known agents that raise intracellular cAMP (35, 36).HUVECs grown under standard culture conditions in which theydo not express organized tight junctions (45, 46) were used asa control for all experiments.We first performed functional studies to assay the barrier
function of our in vitro model by measuring TEER and per-meability to 20-kDa FITC-dextran. As demonstrated in Fig. 1A,HUVECs grown under BBB culture conditions displayed elec-trical resistances up to 250 ohm-cm2, nearly 10 times greater thanstandard culture controls (p , 0.0001). In fact, this TEER is ashigh as has been reported for human ECs of any kind in vitro (47–49). In addition, we used two stable human BBB brain EC lines,TY10 and hCMEC/D3, grown in special medium that is reportedto allow them to maintain tight junction characteristics and BBBproperties (38, 39). Although these cell lines formed junctions thathad higher electrical resistance than conventional ECs (Fig. 1Aand hCMEC/D3, data not shown), neither were as high as ourHUVEC-ACM model. Permeability studies supported our findingswith TEER (Fig. 1B). Our BBB culture system was 10 timesmore restrictive to permeability of 20-kDa FITC-dextran acrossits junctions than standard HUVEC controls. In addition,our HUVEC-ACM model behaves physiologically and can bedisrupted by sufficient stimulation with TNF, a proinflammatorycytokine and inducer of vascular permeability that is commonlyassociated with neuroinflammation (Supplemental Fig. 1).We next examined the induction of the BBB phenotype by
determining the expression and localization of BBB-associatedtight junction proteins occludin, claudin-5, and claudin-3. In ad-dition, we examined endothelial adhesion and signaling moleculesknown to be involved in TEM that are also expressed at brain ECjunctions, including PECAM-1, CD99, and VE-cadherin (50). Asdepicted in Fig. 1C, our BBB model exhibits continuous stainingof PECAM-1, CD99, and VE-cadherin at EC borders similar to
The Journal of Immunology 2429
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
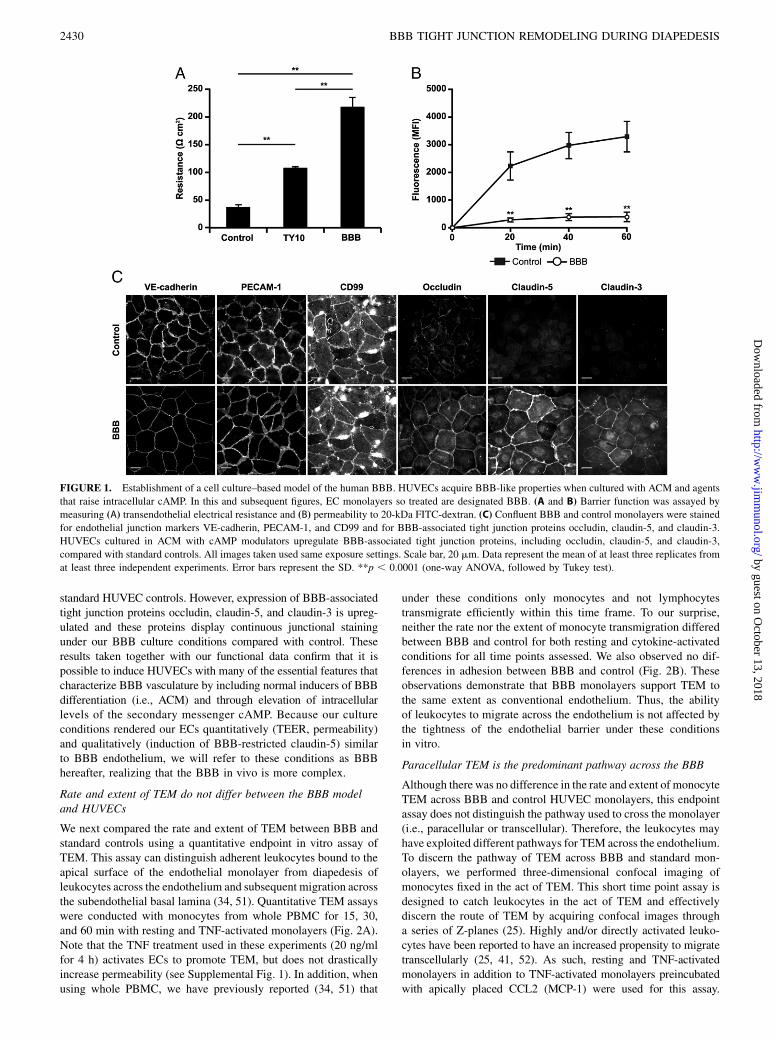
standard HUVEC controls. However, expression of BBB-associatedtight junction proteins occludin, claudin-5, and claudin-3 is upreg-ulated and these proteins display continuous junctional stainingunder our BBB culture conditions compared with control. Theseresults taken together with our functional data confirm that it ispossible to induce HUVECs with many of the essential features thatcharacterize BBB vasculature by including normal inducers of BBBdifferentiation (i.e., ACM) and through elevation of intracellularlevels of the secondary messenger cAMP. Because our cultureconditions rendered our ECs quantitatively (TEER, permeability)and qualitatively (induction of BBB-restricted claudin-5) similarto BBB endothelium, we will refer to these conditions as BBBhereafter, realizing that the BBB in vivo is more complex.
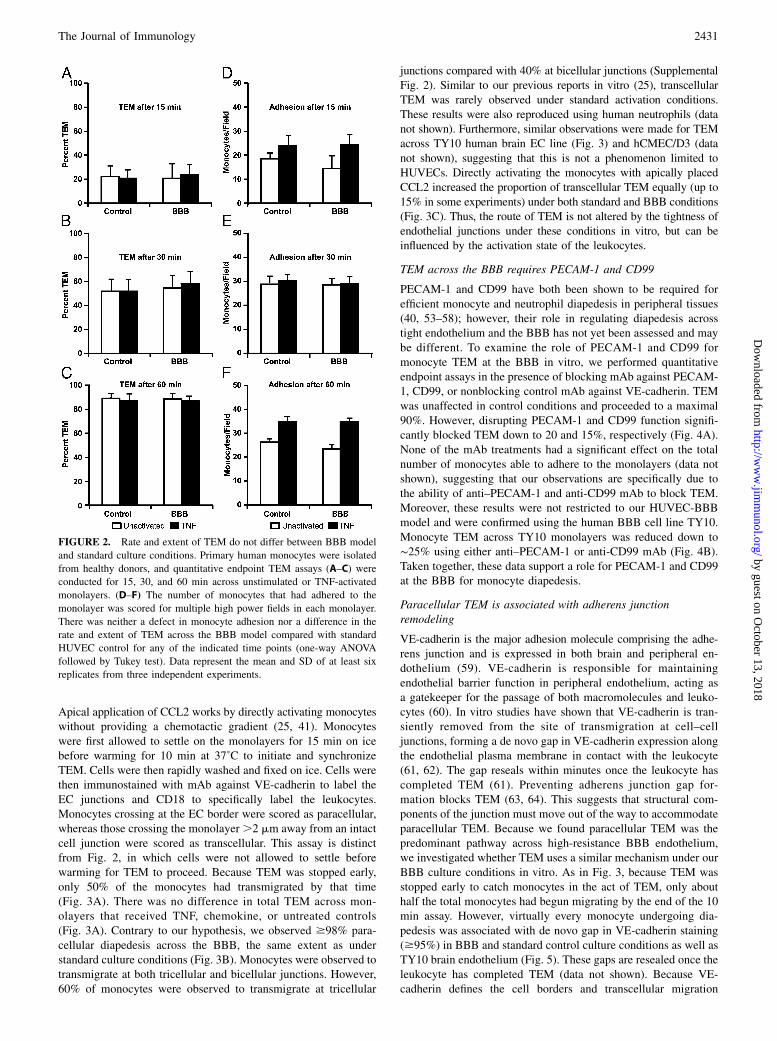
Rate and extent of TEM do not differ between the BBB modeland HUVECs
We next compared the rate and extent of TEM between BBB andstandard controls using a quantitative endpoint in vitro assay ofTEM. This assay can distinguish adherent leukocytes bound to theapical surface of the endothelial monolayer from diapedesis ofleukocytes across the endothelium and subsequent migration acrossthe subendothelial basal lamina (34, 51). Quantitative TEM assayswere conducted with monocytes from whole PBMC for 15, 30,and 60 min with resting and TNF-activated monolayers (Fig. 2A).Note that the TNF treatment used in these experiments (20 ng/mlfor 4 h) activates ECs to promote TEM, but does not drasticallyincrease permeability (see Supplemental Fig. 1). In addition, whenusing whole PBMC, we have previously reported (34, 51) that
under these conditions only monocytes and not lymphocytestransmigrate efficiently within this time frame. To our surprise,neither the rate nor the extent of monocyte transmigration differedbetween BBB and control for both resting and cytokine-activatedconditions for all time points assessed. We also observed no dif-ferences in adhesion between BBB and control (Fig. 2B). Theseobservations demonstrate that BBB monolayers support TEM tothe same extent as conventional endothelium. Thus, the abilityof leukocytes to migrate across the endothelium is not affected bythe tightness of the endothelial barrier under these conditionsin vitro.
Paracellular TEM is the predominant pathway across the BBB
Although there was no difference in the rate and extent of monocyteTEM across BBB and control HUVEC monolayers, this endpointassay does not distinguish the pathway used to cross the monolayer(i.e., paracellular or transcellular). Therefore, the leukocytes mayhave exploited different pathways for TEM across the endothelium.To discern the pathway of TEM across BBB and standard mon-olayers, we performed three-dimensional confocal imaging ofmonocytes fixed in the act of TEM. This short time point assay isdesigned to catch leukocytes in the act of TEM and effectivelydiscern the route of TEM by acquiring confocal images througha series of Z-planes (25). Highly and/or directly activated leuko-cytes have been reported to have an increased propensity to migratetranscellularly (25, 41, 52). As such, resting and TNF-activatedmonolayers in addition to TNF-activated monolayers preincubatedwith apically placed CCL2 (MCP-1) were used for this assay.
FIGURE 1. Establishment of a cell culture–based model of the human BBB. HUVECs acquire BBB-like properties when cultured with ACM and agents
that raise intracellular cAMP. In this and subsequent figures, EC monolayers so treated are designated BBB. (A and B) Barrier function was assayed by
measuring (A) transendothelial electrical resistance and (B) permeability to 20-kDa FITC-dextran. (C) Confluent BBB and control monolayers were stained
for endothelial junction markers VE-cadherin, PECAM-1, and CD99 and for BBB-associated tight junction proteins occludin, claudin-5, and claudin-3.
HUVECs cultured in ACM with cAMP modulators upregulate BBB-associated tight junction proteins, including occludin, claudin-5, and claudin-3,
compared with standard controls. All images taken used same exposure settings. Scale bar, 20 mm. Data represent the mean of at least three replicates from
at least three independent experiments. Error bars represent the SD. **p , 0.0001 (one-way ANOVA, followed by Tukey test).
2430 BBB TIGHT JUNCTION REMODELING DURING DIAPEDESIS
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
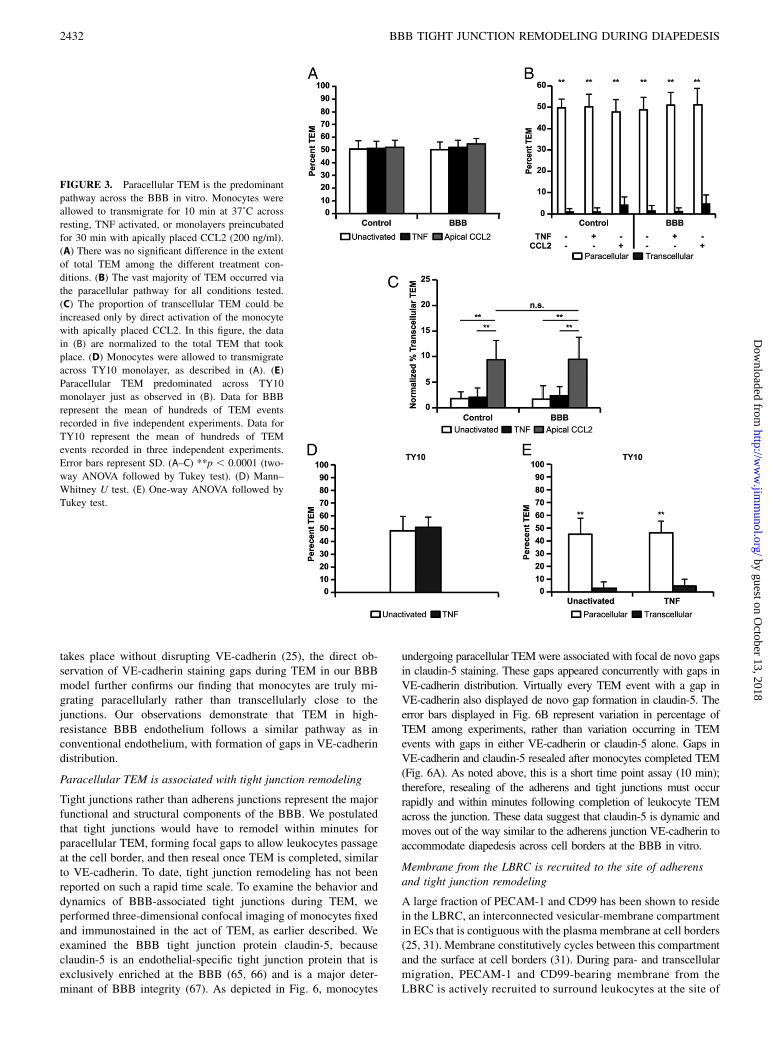
Apical application of CCL2 works by directly activating monocyteswithout providing a chemotactic gradient (25, 41). Monocyteswere first allowed to settle on the monolayers for 15 min on icebefore warming for 10 min at 37˚C to initiate and synchronizeTEM. Cells were then rapidly washed and fixed on ice. Cells werethen immunostained with mAb against VE-cadherin to label theEC junctions and CD18 to specifically label the leukocytes.Monocytes crossing at the EC border were scored as paracellular,whereas those crossing the monolayer.2 mm away from an intactcell junction were scored as transcellular. This assay is distinctfrom Fig. 2, in which cells were not allowed to settle beforewarming for TEM to proceed. Because TEM was stopped early,only 50% of the monocytes had transmigrated by that time(Fig. 3A). There was no difference in total TEM across mon-olayers that received TNF, chemokine, or untreated controls(Fig. 3A). Contrary to our hypothesis, we observed $98% para-cellular diapedesis across the BBB, the same extent as understandard culture conditions (Fig. 3B). Monocytes were observed totransmigrate at both tricellular and bicellular junctions. However,60% of monocytes were observed to transmigrate at tricellular
junctions compared with 40% at bicellular junctions (SupplementalFig. 2). Similar to our previous reports in vitro (25), transcellularTEM was rarely observed under standard activation conditions.These results were also reproduced using human neutrophils (datanot shown). Furthermore, similar observations were made for TEMacross TY10 human brain EC line (Fig. 3) and hCMEC/D3 (datanot shown), suggesting that this is not a phenomenon limited toHUVECs. Directly activating the monocytes with apically placedCCL2 increased the proportion of transcellular TEM equally (up to15% in some experiments) under both standard and BBB conditions(Fig. 3C). Thus, the route of TEM is not altered by the tightness ofendothelial junctions under these conditions in vitro, but can beinfluenced by the activation state of the leukocytes.
TEM across the BBB requires PECAM-1 and CD99
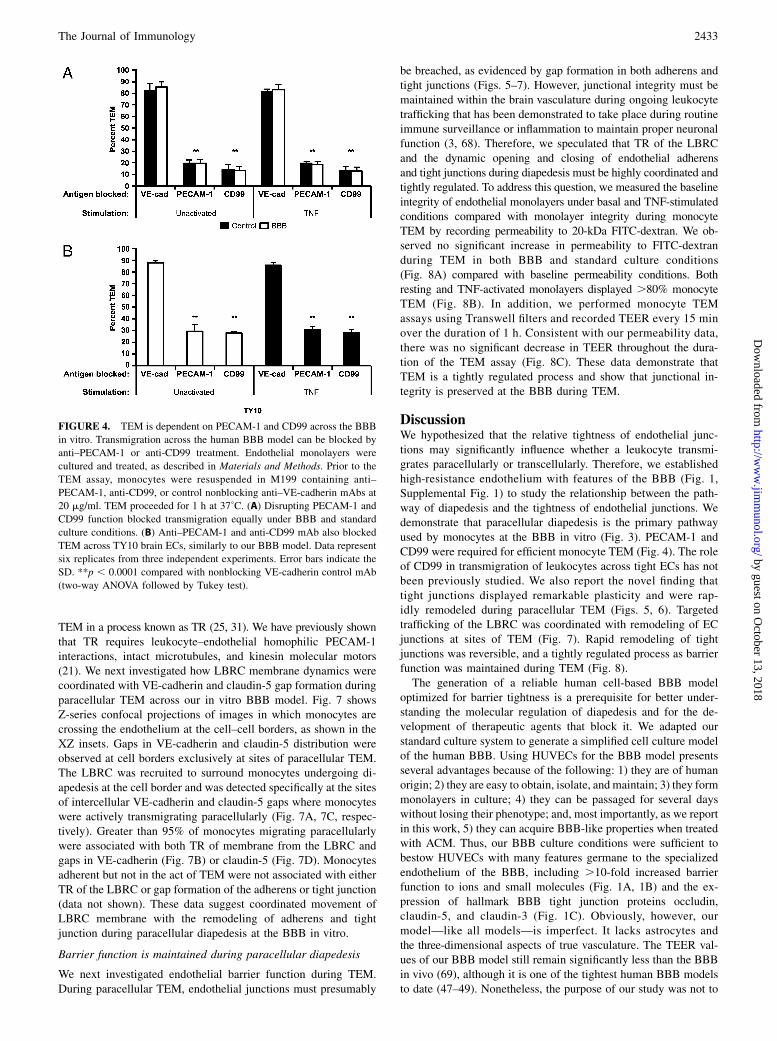
PECAM-1 and CD99 have both been shown to be required forefficient monocyte and neutrophil diapedesis in peripheral tissues(40, 53–58); however, their role in regulating diapedesis acrosstight endothelium and the BBB has not yet been assessed and maybe different. To examine the role of PECAM-1 and CD99 formonocyte TEM at the BBB in vitro, we performed quantitativeendpoint assays in the presence of blocking mAb against PECAM-1, CD99, or nonblocking control mAb against VE-cadherin. TEMwas unaffected in control conditions and proceeded to a maximal90%. However, disrupting PECAM-1 and CD99 function signifi-cantly blocked TEM down to 20 and 15%, respectively (Fig. 4A).None of the mAb treatments had a significant effect on the totalnumber of monocytes able to adhere to the monolayers (data notshown), suggesting that our observations are specifically due tothe ability of anti–PECAM-1 and anti-CD99 mAb to block TEM.Moreover, these results were not restricted to our HUVEC-BBBmodel and were confirmed using the human BBB cell line TY10.Monocyte TEM across TY10 monolayers was reduced down to∼25% using either anti–PECAM-1 or anti-CD99 mAb (Fig. 4B).Taken together, these data support a role for PECAM-1 and CD99at the BBB for monocyte diapedesis.
Paracellular TEM is associated with adherens junctionremodeling
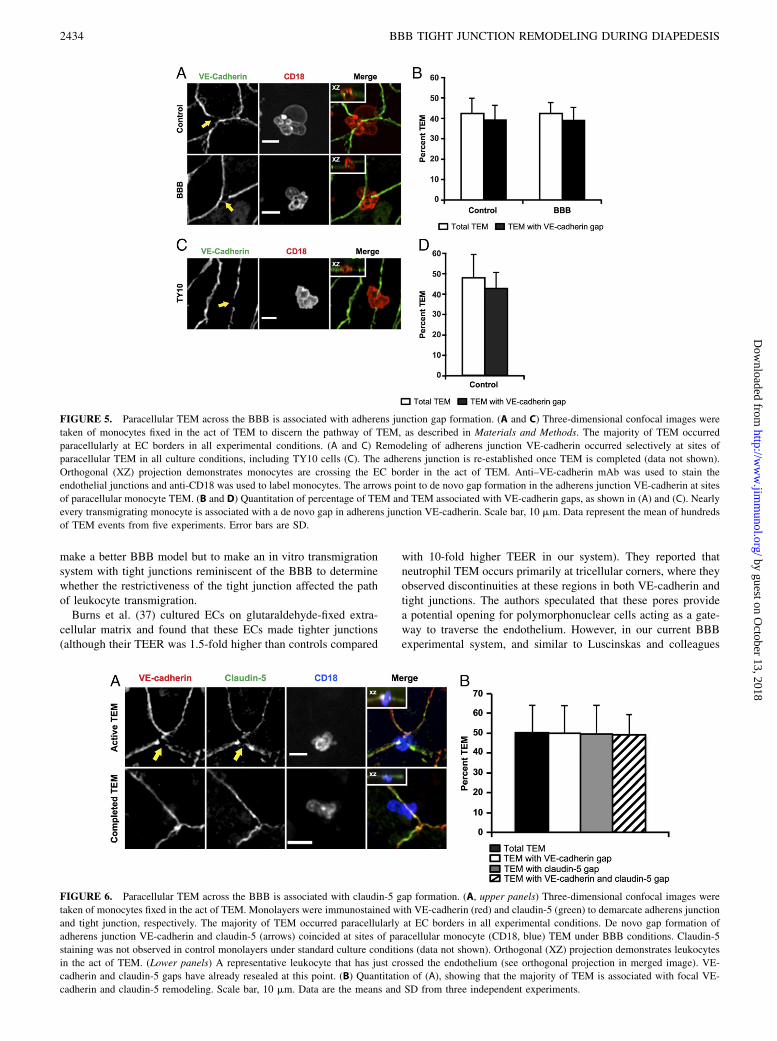
VE-cadherin is the major adhesion molecule comprising the adhe-rens junction and is expressed in both brain and peripheral en-dothelium (59). VE-cadherin is responsible for maintainingendothelial barrier function in peripheral endothelium, acting asa gatekeeper for the passage of both macromolecules and leuko-cytes (60). In vitro studies have shown that VE-cadherin is tran-siently removed from the site of transmigration at cell–celljunctions, forming a de novo gap in VE-cadherin expression alongthe endothelial plasma membrane in contact with the leukocyte(61, 62). The gap reseals within minutes once the leukocyte hascompleted TEM (61). Preventing adherens junction gap for-mation blocks TEM (63, 64). This suggests that structural com-ponents of the junction must move out of the way to accommodateparacellular TEM. Because we found paracellular TEM was thepredominant pathway across high-resistance BBB endothelium,we investigated whether TEM uses a similar mechanism under ourBBB culture conditions in vitro. As in Fig. 3, because TEM wasstopped early to catch monocytes in the act of TEM, only abouthalf the total monocytes had begun migrating by the end of the 10min assay. However, virtually every monocyte undergoing dia-pedesis was associated with de novo gap in VE-cadherin staining($95%) in BBB and standard control culture conditions as well asTY10 brain endothelium (Fig. 5). These gaps are resealed once theleukocyte has completed TEM (data not shown). Because VE-cadherin defines the cell borders and transcellular migration
FIGURE 2. Rate and extent of TEM do not differ between BBB model
and standard culture conditions. Primary human monocytes were isolated
from healthy donors, and quantitative endpoint TEM assays (A–C) were
conducted for 15, 30, and 60 min across unstimulated or TNF-activated
monolayers. (D–F) The number of monocytes that had adhered to the
monolayer was scored for multiple high power fields in each monolayer.
There was neither a defect in monocyte adhesion nor a difference in the
rate and extent of TEM across the BBB model compared with standard
HUVEC control for any of the indicated time points (one-way ANOVA
followed by Tukey test). Data represent the mean and SD of at least six
replicates from three independent experiments.
The Journal of Immunology 2431
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
takes place without disrupting VE-cadherin (25), the direct ob-servation of VE-cadherin staining gaps during TEM in our BBBmodel further confirms our finding that monocytes are truly mi-grating paracellularly rather than transcellularly close to thejunctions. Our observations demonstrate that TEM in high-resistance BBB endothelium follows a similar pathway as inconventional endothelium, with formation of gaps in VE-cadherindistribution.
Paracellular TEM is associated with tight junction remodeling
Tight junctions rather than adherens junctions represent the majorfunctional and structural components of the BBB. We postulatedthat tight junctions would have to remodel within minutes forparacellular TEM, forming focal gaps to allow leukocytes passageat the cell border, and then reseal once TEM is completed, similarto VE-cadherin. To date, tight junction remodeling has not beenreported on such a rapid time scale. To examine the behavior anddynamics of BBB-associated tight junctions during TEM, weperformed three-dimensional confocal imaging of monocytes fixedand immunostained in the act of TEM, as earlier described. Weexamined the BBB tight junction protein claudin-5, becauseclaudin-5 is an endothelial-specific tight junction protein that isexclusively enriched at the BBB (65, 66) and is a major deter-minant of BBB integrity (67). As depicted in Fig. 6, monocytes
undergoing paracellular TEMwere associated with focal de novo gapsin claudin-5 staining. These gaps appeared concurrently with gaps inVE-cadherin distribution. Virtually every TEM event with a gap inVE-cadherin also displayed de novo gap formation in claudin-5. Theerror bars displayed in Fig. 6B represent variation in percentage ofTEM among experiments, rather than variation occurring in TEMevents with gaps in either VE-cadherin or claudin-5 alone. Gaps inVE-cadherin and claudin-5 resealed after monocytes completed TEM(Fig. 6A). As noted above, this is a short time point assay (10 min);therefore, resealing of the adherens and tight junctions must occurrapidly and within minutes following completion of leukocyte TEMacross the junction. These data suggest that claudin-5 is dynamic andmoves out of the way similar to the adherens junction VE-cadherin toaccommodate diapedesis across cell borders at the BBB in vitro.
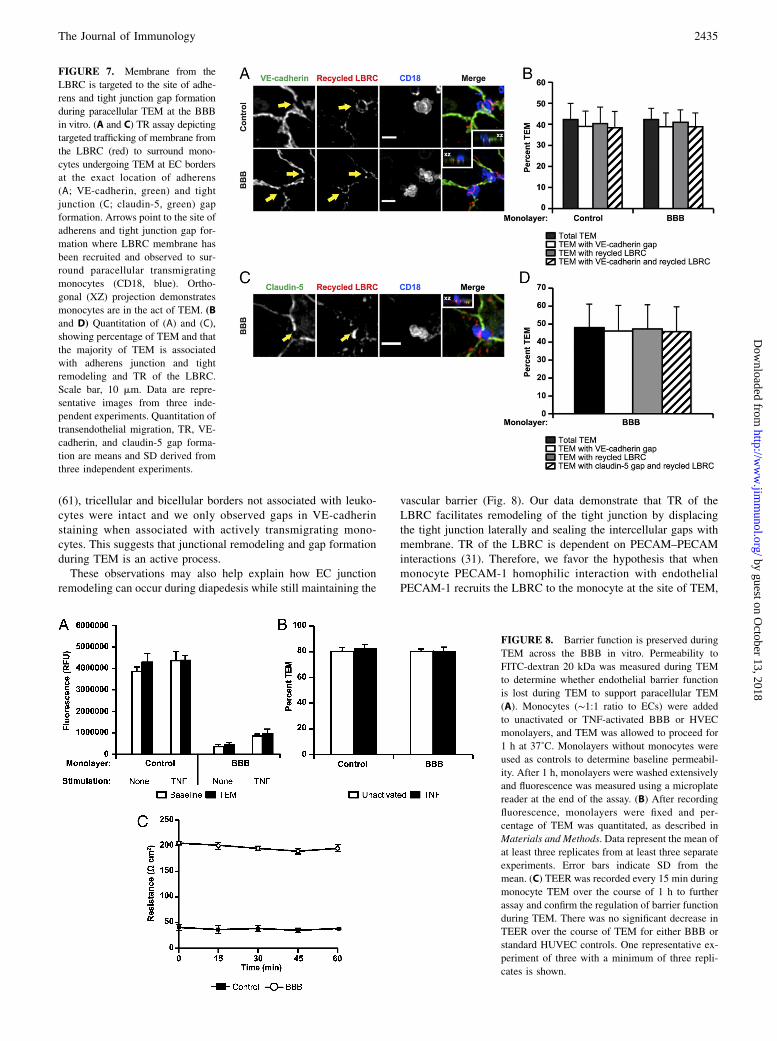
Membrane from the LBRC is recruited to the site of adherensand tight junction remodeling
A large fraction of PECAM-1 and CD99 has been shown to residein the LBRC, an interconnected vesicular-membrane compartmentin ECs that is contiguous with the plasma membrane at cell borders(25, 31). Membrane constitutively cycles between this compartmentand the surface at cell borders (31). During para- and transcellularmigration, PECAM-1 and CD99-bearing membrane from theLBRC is actively recruited to surround leukocytes at the site of
FIGURE 3. Paracellular TEM is the predominant
pathway across the BBB in vitro. Monocytes were
allowed to transmigrate for 10 min at 37˚C across
resting, TNF activated, or monolayers preincubated
for 30 min with apically placed CCL2 (200 ng/ml).
(A) There was no significant difference in the extent
of total TEM among the different treatment con-
ditions. (B) The vast majority of TEM occurred via
the paracellular pathway for all conditions tested.
(C) The proportion of transcellular TEM could be
increased only by direct activation of the monocyte
with apically placed CCL2. In this figure, the data
in (B) are normalized to the total TEM that took
place. (D) Monocytes were allowed to transmigrate
across TY10 monolayer, as described in (A). (E)
Paracellular TEM predominated across TY10
monolayer just as observed in (B). Data for BBB
represent the mean of hundreds of TEM events
recorded in five independent experiments. Data for
TY10 represent the mean of hundreds of TEM
events recorded in three independent experiments.
Error bars represent SD. (A–C) **p , 0.0001 (two-
way ANOVA followed by Tukey test). (D) Mann–
Whitney U test. (E) One-way ANOVA followed by
Tukey test.
2432 BBB TIGHT JUNCTION REMODELING DURING DIAPEDESIS
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
TEM in a process known as TR (25, 31). We have previously shownthat TR requires leukocyte–endothelial homophilic PECAM-1interactions, intact microtubules, and kinesin molecular motors(21). We next investigated how LBRC membrane dynamics werecoordinated with VE-cadherin and claudin-5 gap formation duringparacellular TEM across our in vitro BBB model. Fig. 7 showsZ-series confocal projections of images in which monocytes arecrossing the endothelium at the cell–cell borders, as shown in theXZ insets. Gaps in VE-cadherin and claudin-5 distribution wereobserved at cell borders exclusively at sites of paracellular TEM.The LBRC was recruited to surround monocytes undergoing di-apedesis at the cell border and was detected specifically at the sitesof intercellular VE-cadherin and claudin-5 gaps where monocyteswere actively transmigrating paracellularly (Fig. 7A, 7C, respec-tively). Greater than 95% of monocytes migrating paracellularlywere associated with both TR of membrane from the LBRC andgaps in VE-cadherin (Fig. 7B) or claudin-5 (Fig. 7D). Monocytesadherent but not in the act of TEM were not associated with eitherTR of the LBRC or gap formation of the adherens or tight junction(data not shown). These data suggest coordinated movement ofLBRC membrane with the remodeling of adherens and tightjunction during paracellular diapedesis at the BBB in vitro.
Barrier function is maintained during paracellular diapedesis
We next investigated endothelial barrier function during TEM.During paracellular TEM, endothelial junctions must presumably
be breached, as evidenced by gap formation in both adherens andtight junctions (Figs. 5–7). However, junctional integrity must bemaintained within the brain vasculature during ongoing leukocytetrafficking that has been demonstrated to take place during routineimmune surveillance or inflammation to maintain proper neuronalfunction (3, 68). Therefore, we speculated that TR of the LBRCand the dynamic opening and closing of endothelial adherensand tight junctions during diapedesis must be highly coordinated andtightly regulated. To address this question, we measured the baselineintegrity of endothelial monolayers under basal and TNF-stimulatedconditions compared with monolayer integrity during monocyteTEM by recording permeability to 20-kDa FITC-dextran. We ob-served no significant increase in permeability to FITC-dextranduring TEM in both BBB and standard culture conditions(Fig. 8A) compared with baseline permeability conditions. Bothresting and TNF-activated monolayers displayed .80% monocyteTEM (Fig. 8B). In addition, we performed monocyte TEMassays using Transwell filters and recorded TEER every 15 minover the duration of 1 h. Consistent with our permeability data,there was no significant decrease in TEER throughout the dura-tion of the TEM assay (Fig. 8C). These data demonstrate thatTEM is a tightly regulated process and show that junctional in-tegrity is preserved at the BBB during TEM.
DiscussionWe hypothesized that the relative tightness of endothelial junc-tions may significantly influence whether a leukocyte transmi-grates paracellularly or transcellularly. Therefore, we establishedhigh-resistance endothelium with features of the BBB (Fig. 1,Supplemental Fig. 1) to study the relationship between the path-way of diapedesis and the tightness of endothelial junctions. Wedemonstrate that paracellular diapedesis is the primary pathwayused by monocytes at the BBB in vitro (Fig. 3). PECAM-1 andCD99 were required for efficient monocyte TEM (Fig. 4). The roleof CD99 in transmigration of leukocytes across tight ECs has notbeen previously studied. We also report the novel finding thattight junctions displayed remarkable plasticity and were rap-idly remodeled during paracellular TEM (Figs. 5, 6). Targetedtrafficking of the LBRC was coordinated with remodeling of ECjunctions at sites of TEM (Fig. 7). Rapid remodeling of tightjunctions was reversible, and a tightly regulated process as barrierfunction was maintained during TEM (Fig. 8).The generation of a reliable human cell-based BBB model
optimized for barrier tightness is a prerequisite for better under-standing the molecular regulation of diapedesis and for the de-velopment of therapeutic agents that block it. We adapted ourstandard culture system to generate a simplified cell culture modelof the human BBB. Using HUVECs for the BBB model presentsseveral advantages because of the following: 1) they are of humanorigin; 2) they are easy to obtain, isolate, and maintain; 3) they formmonolayers in culture; 4) they can be passaged for several dayswithout losing their phenotype; and, most importantly, as we reportin this work, 5) they can acquire BBB-like properties when treatedwith ACM. Thus, our BBB culture conditions were sufficient tobestow HUVECs with many features germane to the specializedendothelium of the BBB, including .10-fold increased barrierfunction to ions and small molecules (Fig. 1A, 1B) and the ex-pression of hallmark BBB tight junction proteins occludin,claudin-5, and claudin-3 (Fig. 1C). Obviously, however, ourmodel—like all models—is imperfect. It lacks astrocytes andthe three-dimensional aspects of true vasculature. The TEER val-ues of our BBB model still remain significantly less than the BBBin vivo (69), although it is one of the tightest human BBB modelsto date (47–49). Nonetheless, the purpose of our study was not to
FIGURE 4. TEM is dependent on PECAM-1 and CD99 across the BBB
in vitro. Transmigration across the human BBB model can be blocked by
anti–PECAM-1 or anti-CD99 treatment. Endothelial monolayers were
cultured and treated, as described in Materials and Methods. Prior to the
TEM assay, monocytes were resuspended in M199 containing anti–
PECAM-1, anti-CD99, or control nonblocking anti–VE-cadherin mAbs at
20 mg/ml. TEM proceeded for 1 h at 37˚C. (A) Disrupting PECAM-1 and
CD99 function blocked transmigration equally under BBB and standard
culture conditions. (B) Anti–PECAM-1 and anti-CD99 mAb also blocked
TEM across TY10 brain ECs, similarly to our BBB model. Data represent
six replicates from three independent experiments. Error bars indicate the
SD. **p , 0.0001 compared with nonblocking VE-cadherin control mAb
(two-way ANOVA followed by Tukey test).
The Journal of Immunology 2433
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
make a better BBB model but to make an in vitro transmigrationsystem with tight junctions reminiscent of the BBB to determinewhether the restrictiveness of the tight junction affected the pathof leukocyte transmigration.Burns et al. (37) cultured ECs on glutaraldehyde-fixed extra-
cellular matrix and found that these ECs made tighter junctions(although their TEER was 1.5-fold higher than controls compared
with 10-fold higher TEER in our system). They reported thatneutrophil TEM occurs primarily at tricellular corners, where theyobserved discontinuities at these regions in both VE-cadherin andtight junctions. The authors speculated that these pores providea potential opening for polymorphonuclear cells acting as a gate-way to traverse the endothelium. However, in our current BBBexperimental system, and similar to Luscinskas and colleagues
FIGURE 5. Paracellular TEM across the BBB is associated with adherens junction gap formation. (A and C) Three-dimensional confocal images were
taken of monocytes fixed in the act of TEM to discern the pathway of TEM, as described in Materials and Methods. The majority of TEM occurred
paracellularly at EC borders in all experimental conditions. (A and C) Remodeling of adherens junction VE-cadherin occurred selectively at sites of
paracellular TEM in all culture conditions, including TY10 cells (C). The adherens junction is re-established once TEM is completed (data not shown).
Orthogonal (XZ) projection demonstrates monocytes are crossing the EC border in the act of TEM. Anti–VE-cadherin mAb was used to stain the
endothelial junctions and anti-CD18 was used to label monocytes. The arrows point to de novo gap formation in the adherens junction VE-cadherin at sites
of paracellular monocyte TEM. (B and D) Quantitation of percentage of TEM and TEM associated with VE-cadherin gaps, as shown in (A) and (C). Nearly
every transmigrating monocyte is associated with a de novo gap in adherens junction VE-cadherin. Scale bar, 10 mm. Data represent the mean of hundreds
of TEM events from five experiments. Error bars are SD.
FIGURE 6. Paracellular TEM across the BBB is associated with claudin-5 gap formation. (A, upper panels) Three-dimensional confocal images were
taken of monocytes fixed in the act of TEM. Monolayers were immunostained with VE-cadherin (red) and claudin-5 (green) to demarcate adherens junction
and tight junction, respectively. The majority of TEM occurred paracellularly at EC borders in all experimental conditions. De novo gap formation of
adherens junction VE-cadherin and claudin-5 (arrows) coincided at sites of paracellular monocyte (CD18, blue) TEM under BBB conditions. Claudin-5
staining was not observed in control monolayers under standard culture conditions (data not shown). Orthogonal (XZ) projection demonstrates leukocytes
in the act of TEM. (Lower panels) A representative leukocyte that has just crossed the endothelium (see orthogonal projection in merged image). VE-
cadherin and claudin-5 gaps have already resealed at this point. (B) Quantitation of (A), showing that the majority of TEM is associated with focal VE-
cadherin and claudin-5 remodeling. Scale bar, 10 mm. Data are the means and SD from three independent experiments.
2434 BBB TIGHT JUNCTION REMODELING DURING DIAPEDESIS
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
(61), tricellular and bicellular borders not associated with leuko-cytes were intact and we only observed gaps in VE-cadherinstaining when associated with actively transmigrating mono-cytes. This suggests that junctional remodeling and gap formationduring TEM is an active process.These observations may also help explain how EC junction
remodeling can occur during diapedesis while still maintaining the
vascular barrier (Fig. 8). Our data demonstrate that TR of theLBRC facilitates remodeling of the tight junction by displacingthe tight junction laterally and sealing the intercellular gaps withmembrane. TR of the LBRC is dependent on PECAM–PECAMinteractions (31). Therefore, we favor the hypothesis that whenmonocyte PECAM-1 homophilic interaction with endothelialPECAM-1 recruits the LBRC to the monocyte at the site of TEM,
FIGURE 7. Membrane from the
LBRC is targeted to the site of adhe-
rens and tight junction gap formation
during paracellular TEM at the BBB
in vitro. (A and C) TR assay depicting
targeted trafficking of membrane from
the LBRC (red) to surround mono-
cytes undergoing TEM at EC borders
at the exact location of adherens
(A; VE-cadherin, green) and tight
junction (C; claudin-5, green) gap
formation. Arrows point to the site of
adherens and tight junction gap for-
mation where LBRC membrane has
been recruited and observed to sur-
round paracellular transmigrating
monocytes (CD18, blue). Ortho-
gonal (XZ) projection demonstrates
monocytes are in the act of TEM. (B
and D) Quantitation of (A) and (C),
showing percentage of TEM and that
the majority of TEM is associated
with adherens junction and tight
remodeling and TR of the LBRC.
Scale bar, 10 mm. Data are repre-
sentative images from three inde-
pendent experiments. Quantitation of
transendothelial migration, TR, VE-
cadherin, and claudin-5 gap forma-
tion are means and SD derived from
three independent experiments.
FIGURE 8. Barrier function is preserved during
TEM across the BBB in vitro. Permeability to
FITC-dextran 20 kDa was measured during TEM
to determine whether endothelial barrier function
is lost during TEM to support paracellular TEM
(A). Monocytes (∼1:1 ratio to ECs) were added
to unactivated or TNF-activated BBB or HVEC
monolayers, and TEM was allowed to proceed for
1 h at 37˚C. Monolayers without monocytes were
used as controls to determine baseline permeabil-
ity. After 1 h, monolayers were washed extensively
and fluorescence was measured using a microplate
reader at the end of the assay. (B) After recording
fluorescence, monolayers were fixed and per-
centage of TEM was quantitated, as described in
Materials and Methods. Data represent the mean of
at least three replicates from at least three separate
experiments. Error bars indicate SD from the
mean. (C) TEER was recorded every 15 min during
monocyte TEM over the course of 1 h to further
assay and confirm the regulation of barrier function
during TEM. There was no significant decrease in
TEER over the course of TEM for either BBB or
standard HUVEC controls. One representative ex-
periment of three with a minimum of three repli-
cates is shown.
The Journal of Immunology 2435
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

this membrane then facilitates the remodeling of the junctionlocally by displacing the tight junction laterally. This is consistentwith Luscinskas and colleagues (61), who have demonstrated thatVE-cadherin is pushed laterally to accommodate paracellularTEM. VE-cadherin expression at the site where TEM occurred isthen quickly re-established within minutes when TEM is com-pleted. The additional membrane provided by the LBRC can sealthe de novo gaps while tightly engaging the leukocyte withhomophilic adhesion molecules such as PECAM-1 and CD99 as itpasses between endothelial junctions. This would prevent plasmaleakage and thereby maintain the endothelial barrier. We speculatethat because both adherens junction proteins and tight junctionsare attached to actin filaments, and these junctions are intermixedin ECs (70–72), similar mechanisms allow for tight junctions andadherens junctions to undergo lateral movement in the plane of themembrane.In conclusion, we report that, within the limits tested, the relative
tightness of BBB EC junctions does not influence the pathway ofTEM in vitro. Leukocyte TEM at the BBB in vitro is a complexprocess that requires complex coordination of diverse signalingevents, including PECAM-1, CD99, and TR of the LBRC. Theseevents all converge to facilitate the remodeling of adherens andtight junctions to allow for the passage of the migrating leukocytesat cell borders. The LBRC serves as a novel EC mechanism topromote diapedesis and maintain vascular integrity. The degree ofcrosstalk between adherens and tight junctions is undoubtedly morecomplex than previously appreciated. Future studies geared towardunderstanding the interconnection between these pathways willbring us closer to novel and selective therapies that can regulateinflammation.
AcknowledgmentsWe thank Clifford D. Carpenter for excellent technical assistance,
Dr. Babette Weksler for providing the hCMEC/D3 cell line, Dr. Deyu Feng
for use of the microplate reader, Dr. Yukio Takeshita for technical assistance
with the TY10 cell line, and Dr. David Sullivan, Dr. Birgit Obermeier, Evan
Weber, and Richard Watson for thoughtful and helpful review of the
manuscript.
DisclosuresThe authors have no financial conflicts of interest.
References1. Ousman, S. S., and P. Kubes. 2012. Immune surveillance in the central nervous
system. Nat. Neurosci. 15: 1096–1101.2. Hickey, W. F. 2001. Basic principles of immunological surveillance of the
normal central nervous system. Glia 36: 118–124.3. Hickey, W. F. 1999. Leukocyte traffic in the central nervous system: the par-
ticipants and their roles. Semin. Immunol. 11: 125–137.4. Koedel, U., M. Klein, and H. W. Pfister. 2010. New understandings on the
pathophysiology of bacterial meningitis. Curr. Opin. Infect. Dis. 23: 217–223.5. Roberts, T. K., C. M. Buckner, and J. W. Berman. 2010. Leukocyte transmi-
gration across the blood-brain barrier: perspectives on neuroAIDS. Front. Biosci.(Landmark Ed.) 15: 478–536.
6. Jin, R., G. Yang, and G. Li. 2010. Inflammatory mechanisms in ischemic stroke:role of inflammatory cells. J. Leukoc. Biol. 87: 779–789.
7. Wang, Q., X. N. Tang, and M. A. Yenari. 2007. The inflammatory response instroke. J. Neuroimmunol. 184: 53–68.
8. Kochanek, P. M., and J. M. Hallenbeck. 1992. Polymorphonuclear leukocytesand monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke.Stroke 23: 1367–1379.
9. Yilmaz, G., and D. N. Granger. 2008. Cell adhesion molecules and ischemicstroke. Neurol. Res. 30: 783–793.
10. Shechter, R., O. Miller, G. Yovel, N. Rosenzweig, A. London, J. Ruckh,K. W. Kim, E. Klein, V. Kalchenko, P. Bendel, et al. 2013. Recruitment ofbeneficial M2 macrophages to injured spinal cord is orchestrated by remote brainchoroid plexus. Immunity 38: 555–569.
11. Schwarzmaier, S. M., R. Zimmermann, N. B. McGarry, R. Trabold, S. W. Kim,and N. Plesnila. 2013. In vivo temporal and spatial profile of leukocyte adhesionand migration after experimental traumatic brain injury in mice. J. Neuro-inflammation 10: 32.
12. Feuerstein, G. Z., X. Wang, and F. C. Barone. 1997. Inflammatory gene ex-pression in cerebral ischemia and trauma: potential new therapeutic targets. Ann.N. Y. Acad. Sci. 825: 179–193.
13. Greenwood, J., S. J. Heasman, J. I. Alvarez, A. Prat, R. Lyck, and B. Engelhardt.2011. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier:a prerequisite for successful immune cell entry to the brain. Neuropathol. Appl.Neurobiol. 37: 24–39.
14. Awad, A. M., and O. St€uve. 2010. Immunopathogenesis of multiple sclerosis:new insights and therapeutic implications. Continuum (Minneap. Minn.) 16:166–180.
15. Amor, S., F. Puentes, D. Baker, and P. van der Valk. 2010. Inflammation inneurodegenerative diseases. Immunology 129: 154–169.
16. Daneman, R. 2012. The blood-brain barrier in health and disease. Ann. Neurol.72: 648–672.
17. Obermeier, B., R. Daneman, and R. M. Ransohoff. 2013. Development, main-tenance and disruption of the blood-brain barrier. Nat. Med. 19: 1584–1596.
18. Abbott, N. J., L. Ronnback, and E. Hansson. 2006. Astrocyte-endothelialinteractions at the blood-brain barrier. Nat. Rev. Neurosci. 7: 41–53.
19. Lossinsky, A. S., and R. R. Shivers. 2004. Structural pathways for macromo-lecular and cellular transport across the blood-brain barrier during inflammatoryconditions: review. Histol. Histopathol. 19: 535–564.
20. Muller, W. A. 2001. Migration of leukocytes across endothelial junctions: someconcepts and controversies. Microcirculation 8: 181–193.
21. Muller, W. A. 2011. Mechanisms of leukocyte transendothelial migration. Annu.Rev. Pathol. 6: 323–344.
22. Carman, C. V., and T. A. Springer. 2008. Trans-cellular migration: cell-cellcontacts get intimate. Curr. Opin. Cell Biol. 20: 533–540.
23. Wolburg, H., K. Wolburg-Buchholz, and B. Engelhardt. 2005. Diapedesis ofmononuclear cells across cerebral venules during experimental autoimmuneencephalomyelitis leaves tight junctions intact. Acta Neuropathol. 109: 181–190.
24. von Wedel-Parlow, M., S. Schrot, J. Lemmen, L. Treeratanapiboon, J. Wegener,and H. J. Galla. 2011. Neutrophils cross the BBB primarily on transcellularpathways: an in vitro study. Brain Res. 1367: 62–76.
25. Mamdouh, Z., A. Mikhailov, and W. A. Muller. 2009. Transcellular migration ofleukocytes is mediated by the endothelial lateral border recycling compartment.J. Exp. Med. 206: 2795–2808.
26. Raine, C. S., B. Cannella, A. M. Duijvestijn, and A. H. Cross. 1990. Homingto central nervous system vasculature by antigen-specific lymphocytes. II.Lymphocyte/endothelial cell adhesion during the initial stages of autoimmunedemyelination. Lab. Invest. 63: 476–489.
27. Cross, A. H., and C. S. Raine. 1991. Central nervous system endothelial cell-polymorphonuclear cell interactions during autoimmune demyelination. Am. J.Pathol. 139: 1401–1409.
28. Lampert, P. 1967. Electron microscopic studies on ordinary and hyperacuteexperimental allergic encephalomyelitis. Acta Neuropathol. 9: 99–126.
29. Alvarez, J. I., R. Cayrol, and A. Prat. 2011. Disruption of central nervous systembarriers in multiple sclerosis. Biochim. Biophys. Acta 1812: 252–264.
30. Hawkins, B. T., and T. P. Davis. 2005. The blood-brain barrier/neurovascular unitin health and disease. Pharmacol. Rev. 57: 173–185.
31. Mamdouh, Z., X. Chen, L. M. Pierini, F. R. Maxfield, and W. A. Muller. 2003.Targeted recycling of PECAM from endothelial surface-connected compart-ments during diapedesis. Nature 421: 748–753.
32. Muller, W. A., C. M. Ratti, S. L. McDonnell, and Z. A. Cohn. 1989. A humanendothelial cell-restricted, externally disposed plasmalemmal protein enriched inintercellular junctions. J. Exp. Med. 170: 399–414.
33. Liao, F., H. K. Huynh, A. Eiroa, T. Greene, E. Polizzi, and W. A. Muller. 1995.Migration of monocytes across endothelium and passage through extracellularmatrix involve separate molecular domains of PECAM-1. J. Exp. Med. 182:1337–1343.
34. Muller, W. A., and S. A. Weigl. 1992. Monocyte-selective transendothelial mi-gration: dissection of the binding and transmigration phases by an in vitro assay.J. Exp. Med. 176: 819–828.
35. Gaillard, P. J., L. H. Voorwinden, J. L. Nielsen, A. Ivanov, R. Atsumi,H. Engman, C. Ringbom, A. G. de Boer, and D. D. Breimer. 2001. Establishmentand functional characterization of an in vitro model of the blood-brain barrier,comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur. J.Pharm. Sci. 12: 215-222.
36. Rubin, L. L., D. E. Hall, S. Porter, K. Barbu, C. Cannon, H. C. Horner,M. Janatpour, C. W. Liaw, K. Manning, J. Morales, et al. 1991. A cell culturemodel of the blood-brain barrier. J. Cell Biol. 115: 1725–1735.
37. Burns, A. R., D. C. Walker, E. S. Brown, L. T. Thurmon, R. A. Bowden,C. R. Keese, S. I. Simon, M. L. Entman, and C. W. Smith. 1997. Neutrophiltransendothelial migration is independent of tight junctions and occurs prefer-entially at tricellular corners. J. Immunol. 159: 2893–2903.
38. Weksler, B. B., E. A. Subileau, N. Perriere, P. Charneau, K. Holloway,M. Leveque, H. Tricoire-Leignel, A. Nicotra, S. Bourdoulous, P. Turowski, et al.2005. Blood-brain barrier-specific properties of a human adult brain endothelialcell line. FASEB J. 19: 1872–1874.
39. Sano, Y., Y. Kashiwamura, M. Abe, L.-H. Dieu, J. Huwyler, F. Shimizu,H. Haruki, T. Maeda, K. Saito, A. Tasaki, and T. Kanda. 2013. Stable humanbrain microvascular endothelial cell line retaining its barrier-specific natureindependent of the passage number. Clin. Exp. Neuroimmunol. 4: 92–103.
40. Muller, W. A., S. A. Weigl, X. Deng, and D. M. Phillips. 1993. PECAM-1 isrequired for transendothelial migration of leukocytes. J. Exp. Med. 178: 449–460.
41. Carman, C. V., and T. A. Springer. 2004. A transmigratory cup in leukocytediapedesis both through individual vascular endothelial cells and between them.J. Cell Biol. 167: 377–388.
2436 BBB TIGHT JUNCTION REMODELING DURING DIAPEDESIS
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

42. Rubin, L. L. 1991. The blood-brain barrier in and out of cell culture. Curr. Opin.Neurobiol. 1: 360–363.
43. Calabria, A. R., and E. V. Shusta. 2008. A genomic comparison of in vivo andin vitro brain microvascular endothelial cells. J. Cereb. Blood Flow Metab. 28:135-148.
44. Prat, A., K. Biernacki, K. Wosik, and J. P. Antel. 2001. Glial cell influence on thehuman blood-brain barrier. Glia 36: 145–155.
45. Tio, S., M. Deenen, and E. Marani. 1990. Astrocyte-mediated induction of al-kaline phosphatase activity in human umbilical cord vein endothelium: anin vitro model. Eur. J. Morphol. 28: 289–300.
46. Elgjo, R. F., T. Henriksen, and S. A. Evensen. 1975. Ultrastructural identificationof umbilical cord vein endothelium in situ and in culture. Cell Tissue Res. 162:49–59.
47. Eigenmann, D. E., G. Xue, K. S. Kim, A. V. Moses, M. Hamburger, andM. Oufir. 2013. Comparative study of four immortalized human brain capillaryendothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimizationof culture conditions, for an in vitro blood-brain barrier model for drug per-meability studies. Fluids Barriers CNS 10: 33.
48. Reichel, A., D. J. Begley, and N. J. Abbott. 2003. An overview of in vitrotechniques for blood-brain barrier studies. Methods Mol. Med. 89: 307–324.
49. Forster, C., M. Burek, I. A. Romero, B. Weksler, P. O. Couraud, andD. Drenckhahn. 2008. Differential effects of hydrocortisone and TNFalpha ontight junction proteins in an in vitro model of the human blood-brain barrier.J. Physiol. 586: 1937–1949.
50. Coisne, C., and B. Engelhardt. 2011. Tight junctions in brain barriers duringcentral nervous system inflammation. Antioxid. Redox Signal. 15: 1285–1303.
51. Muller, W. A., and F. W. Luscinskas. 2008. Assays of transendothelial migrationin vitro. Methods Enzymol. 443: 155–176.
52. Feng, D., J. A. Nagy, K. Pyne, H. F. Dvorak, and A. M. Dvorak. 1998. Neu-trophils emigrate from venules by a transendothelial cell pathway in response toFMLP. J. Exp. Med. 187: 903–915.
53. Schenkel, A. R., T. W. Chew, and W. A. Muller. 2004. Platelet endothelial celladhesion molecule deficiency or blockade significantly reduces leukocyte emi-gration in a majority of mouse strains. J. Immunol. 173: 6403–6408.
54. Muller, W. A. 1995. The role of PECAM-1 (CD31) in leukocyte emigration:studies in vitro and in vivo. J. Leukoc. Biol. 57: 523–528.
55. Bogen, S., J. Pak, M. Garifallou, X. Deng, and W. A. Muller. 1994. Monoclonalantibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J. Exp.Med. 179: 1059–1064.
56. Dufour, E. M., A. Deroche, Y. Bae, and W. A. Muller. 2008. CD99 is essentialfor leukocyte diapedesis in vivo. Cell Commun. Adhes. 15: 351–363.
57. Lou, O., P. Alcaide, F. W. Luscinskas, and W. A. Muller. 2007. CD99 is a key me-diator of the transendothelial migration of neutrophils. J. Immunol. 178: 1136–1143.
58. Schenkel, A. R., Z. Mamdouh, X. Chen, R. M. Liebman, and W. A. Muller.2002. CD99 plays a major role in the migration of monocytes through endo-thelial junctions. Nat. Immunol. 3: 143–150.
59. Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecularorganization and role in vascular homeostasis. Physiol. Rev. 84: 869–901.
60. Dejana, E., F. Orsenigo, and M. G. Lampugnani. 2008. The role of adherensjunctions and VE-cadherin in the control of vascular permeability. J. Cell Sci.121: 2115–2122.
61. Shaw, S. K., P. S. Bamba, B. N. Perkins, and F. W. Luscinskas. 2001. Real-timeimaging of vascular endothelial-cadherin during leukocyte transmigration acrossendothelium. J. Immunol. 167: 2323–2330.
62. Allport, J. R., W. A. Muller, and F. W. Luscinskas. 2000. Monocytes inducereversible focal changes in vascular endothelial cadherin complex duringtransendothelial migration under flow. J. Cell Biol. 148: 203–216.
63. Alcaide, P., G. Newton, S. Auerbach, S. Sehrawat, T. N. Mayadas, D. E. Golan,P. Yacono, P. Vincent, A. Kowalczyk, and F. W. Luscinskas. 2008. p120-Cateninregulates leukocyte transmigration through an effect on VE-cadherin phos-phorylation. Blood 112: 2770–2779.
64. Allingham, M. J., J. D. van Buul, and K. Burridge. 2007. ICAM-1-mediated, Src-and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation isrequired for leukocyte transendothelial migration. J. Immunol. 179: 4053–4064.
65. Morita, K., M. Furuse, K. Fujimoto, and S. Tsukita. 1999. Claudin multigenefamily encoding four-transmembrane domain protein components of tightjunction strands. Proc. Natl. Acad. Sci. USA 96: 511–516.
66. Morita, K., H. Sasaki, M. Furuse, and S. Tsukita. 1999. Endothelial claudin:claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. CellBiol. 147: 185–194.
67. Nitta, T., M. Hata, S. Gotoh, Y. Seo, H. Sasaki, N. Hashimoto, M. Furuse, andS. Tsukita. 2003. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 161: 653–660.
68. Cramer, E. B. 1992. The Ability of Leukocytes to Cross the Tight Junctions. CRCPress, Boca Raton, FL.
69. Butt, A. M. 1995. Effect of inflammatory agents on electrical resistance acrossthe blood-brain barrier in pial microvessels of anaesthetized rats. Brain Res. 696:145–150.
70. Vorbrodt, A. W., and D. H. Dobrogowska. 2004. Molecular anatomy of inter-endothelial junctions in human blood-brain barrier microvessels. Folia Histo-chem. Cytobiol. 42: 67–75.
71. Dejana, E. 2004. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol.Cell Biol. 5: 261–270.
72. Schulze, C., and J. A. Firth. 1993. Immunohistochemical localization of adhe-rens junction components in blood-brain barrier microvessels of the rat. J. CellSci. 104: 773–782.
The Journal of Immunology 2437
by guest on October 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from