Embed Size (px)
Citation preview

RD
PE*†‡
R
vpiaTlnplvndtwPddsulPtPadA
p
BYE
ikT
Biochemical and Biophysical Research Communications 255, 502–507 (1999)
Article ID bbrc.1999.0234, available online at http://www.idealibrary.com on
0CA
al and Rho-Dependent Activation of Phospholipasein v-Raf-Transformed Cells
aul Frankel,* Miguel Ramos,* Judith Flom,* Sergei Bychenok,* Troy Joseph,*ugen Kerkhoff,† Ulf R. Rapp,† Larry A. Feig,‡ and David A. Foster*,1
Department of Biological Sciences, Hunter College of the City University of New York, New York, New York 10021;Institut fur Medizinische Strahlenkunde und Zellforschung, University of Wurzburg, Wurzburg, Germany; andDepartment of Biochemistry, Tufts University School of Medicine, Boston, Massachusetts 02111
eceived January 14, 1999
to most, if not all mitogenic signals, there is an increaseivtPsvtwta(mPso
rtraicRatdavivria
M
Dp
Phospholipase D (PLD) activity is commonly ele-ated in response to mitogenic signals. We reportedreviously that although the transformed phenotype
nduced by v-Src was dependent upon Raf-1, the PLDctivity induced by v-Src was independent of Raf-1.his observation suggested to us that Raf would not
ikely be an activator of PLD. However, upon exami-ation of PLD activity in v-Raf-transformed cells, sur-risingly, we found that PLD activity is elevated to
evels that were even higher than that observed in-Src-transformed cells. To characterize the mecha-ism of v-Raf-induced PLD activity, we examined theependence of v-Raf-induced PLD activity upon pro-ein kinase C (PKC) the small GTPases Ral and Rho,hich have all been implicated in the activation ofLD. The v-Raf-induced PLD activity was inhibited byominant negative mutants for both Ral and Rho. Theependence upon Ral was particularly surprisingince Ral is a downstream target of Ras, which is anpstream activator of Raf. Depleting cells of PKC by
ong term phorbol ester treatment actually increasedLD activity in v-Raf-transformed cells, indicating
hat v-Raf-induced PLD activity is not dependent onKC. These data describe a novel mechanism for PLDctivation by v-Raf that is independent of PKC, butependent upon both Ral and Rho GTPases. © 1999
cademic Press
There is a strong correlation between the activation ofhospholipase D (PLD) and mitogenesis (1). In response
1 To whom correspondence should be addressed at Department ofiological Sciences, Hunter College of the City University of Nework, 695 Park Ave., New York, NY 10021. Fax: (212) 772-5227.-mail: [email protected] used: DG, diacylglycerol; 4-OH-T, 4-hydroxytamox-
fen; PA, phosphatidic acid; PBt, phosphatidylbutanol; PKC, proteininase C; PLD, Phospholipase D; TLC, thin layer chromatography;PA, 12-O-tetradecanoyl phorbol 13-acetate.
502006-291X/99 $30.00opyright © 1999 by Academic Pressll rights of reproduction in any form reserved.
n PLD activity. PLD activity is elevated in response to aariety of mitogens including platelet-derived growth fac-or (2) epidermal growth factor (3, 4), and insulin (5, 6).LD activity is also elevated in cells transformed by aeveral transforming oncogenes including v-Src (7, 8),-Ras (9–12), and v-Fps (13). The activation of PLD ac-ivity results in the production of phosphatidic acid (PA),hich has been implicated as a lipid second messenger
hat stimulates a variety of signaling molecules (14–16)nd in the formation of vesicles for membrane trafficking17–19). A role for PLD and PA in the transduction ofitogenic signals is not clear; but the observation thatLD is elevated in response to most if not all mitogenictimuli suggests that PLD may be an integral componentf mitogenesis.We demonstrated previously that although Raf is
equired for induction of the transformed phenotype byhe oncogenic tyrosine kinase v-Src (20), Raf was notequired for the activation of PLD by v-Src (10, 20). Thectivation of PLD by v-Src is mediated by Ras (10) andts downstream target Ral guanine nucleotide ex-hange factor, which activates the Ras-family GTPasealA (21). Thus, the v-Src induction of PLD is medi-ted by a Ras signaling pathway that is independent ofhe well characterized Ras/Raf pathway (22). Theseata led us to the prejudice that v-Raf would not likelyctivate PLD activity. However, the recent report that-Raf-induced transformation was inhibited by a dom-nant negative RalA mutant (23) led us to further in-estigate the affect of v-Raf on PLD activity. In thiseport, we present evidence indicating that PLD activ-ty is activated in response to v-Raf via a novel mech-nism involving both Ral and Rho GTPases.
ATERIALS AND METHODS
Cells and cell culture conditions. All cell lines were maintained inulbecco’s modified Eagle medium (DMEM, Life Technologies) sup-lemented with 10% calf serum except for cells expressing the BxB-

Evaeevg(pucftacnay(as4ad
EecStf
6[0
tttta0rtSs
ppht(p(RtuQ
R
3aIn
thhicittisgplwotapoc4isgt2tMikmmdabivo
peatbcPritl
cP(ae
Vol. 255, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
RTM Raf-1 which were maintained with phenol red-free DMEM.-Raf-transformed NIH 3T3 cells were generated by amplification oftransformed focus induced by transfection of the p3611-MSV-v-Raf
xpression plasmid (24) into NIH 3T3 cells. The generation of cellsxpressing the BxB-ERTM Raf-1 was described previously (25).-Raf-transformed cells overexpressing the S28N Ral A mutant wereenerated by transfection of the p-Zip-RalA (S28N) expression vector23) followed by selection of G418 resistant colonies which were thenooled. The dominant negative RhoC T19N mutant was constructedsing overlap PCR as described previously (26). v-Raf-transformedells overexpressing the T19N RhoC mutant was generated by trans-ection of the pZip-RhoC (T19N) expression vector followed by selec-ion of G418 resistant colonies. Empty vector control cell lines werelso established for both v-Raf-transformed and parental NIH 3T3ells. Transfection was performed using Lipofectamine (Life Tech-ologies) according to the vendors instructions. RalA, RhoC, Raf-1,nd Phospho-ERK protein levels were verified by Western blot anal-sis using commercially-obtained antibodies as described previously10,21). Cell cultures were made quiescent by growing to confluencend then replacing with fresh media containing 0.5% newborn calferum overnight (;16hr). For induction of BxB-ERTM Raf-1 with-hydroxytamoxifen (4-OH-T), a solution of 1 mg/ml in ethanol wasdded to the medium to a final concentration of 200 nM (5,000 foldilution).
Materials. [3H]-Myristate (NET-830), was obtained from Newngland Nuclear. Phosphatidylbutanol (PBt) , PA, and diacylglyc-rol (DG) standards were obtained from Avanti Polar Lipids. Pre-oated silica 60A thin layer chromatography (TLC) plates were fromcientific Products. Antibody for RalA was obtained from Transduc-ion Laboratories. Antibodies for Raf-1, RhoC and Phospho-Erk wererom Santa Cruz Biotechnology.
Prelabeling of phospholipids. Unless otherwise indicated, cells in0 mm culture dishes were prelabeled for 4 to 6 h with 3 mmCi of
3H]-Myristate in 2 ml of Dulbecco’s modified Eagle media containing.5% newborn calf serum.
Extraction of lipids. Extraction of lipids was performed accordingo procedures described by Song et al. (7, 27) with minor modifica-ions. Cells were washed with isotonic tris-saline buffer and rapidlyreated with 0.5 ml of MeOH:6N HCl (50:2). Lipids were extracted byhe addition of 0.5 ml of CHCl3. Phase separation was obtained bydding 155 mml of 1M NaCl. The organic phase was reextracted with.12 ml of 1M NaCl, 0.35 ml of H2O and 155 mml of MeOH andecovered. An aliquot (10 mml) of the organic phase was removed andotal cpm incorporated into cellular lipid/sample was determined.amples were normalized for total cpm, dried under N2 and redis-olved in CHCl3:MeOH (9:1).
Characterization of phospholipid metabolites by TLC. Extracts ofhospholipid metabolites were characterized by TLC as describedreviously (27). The following solvent systems were used: For DG,exane:diethylether:MeOH: glacial acetic acid (90:20:3:2); for PBt,he organic phase of ethylacetate:trimethylpentane:acetic acid:H20100:50:20:100). Lipid standards were visualized by treating TLClates with iodine vapor. TLC plates were sprayed with EN3HANCEDupont) and exposed to Kodak XAR-5 film at 270°C for 2-3 days.elative levels of PLD activity was then determined by measuring
he intensity of the corresponding PBt band in the autoradiogramsing a Molecular Dynamics scanning densotometer and Image-uant software.
ESULTS
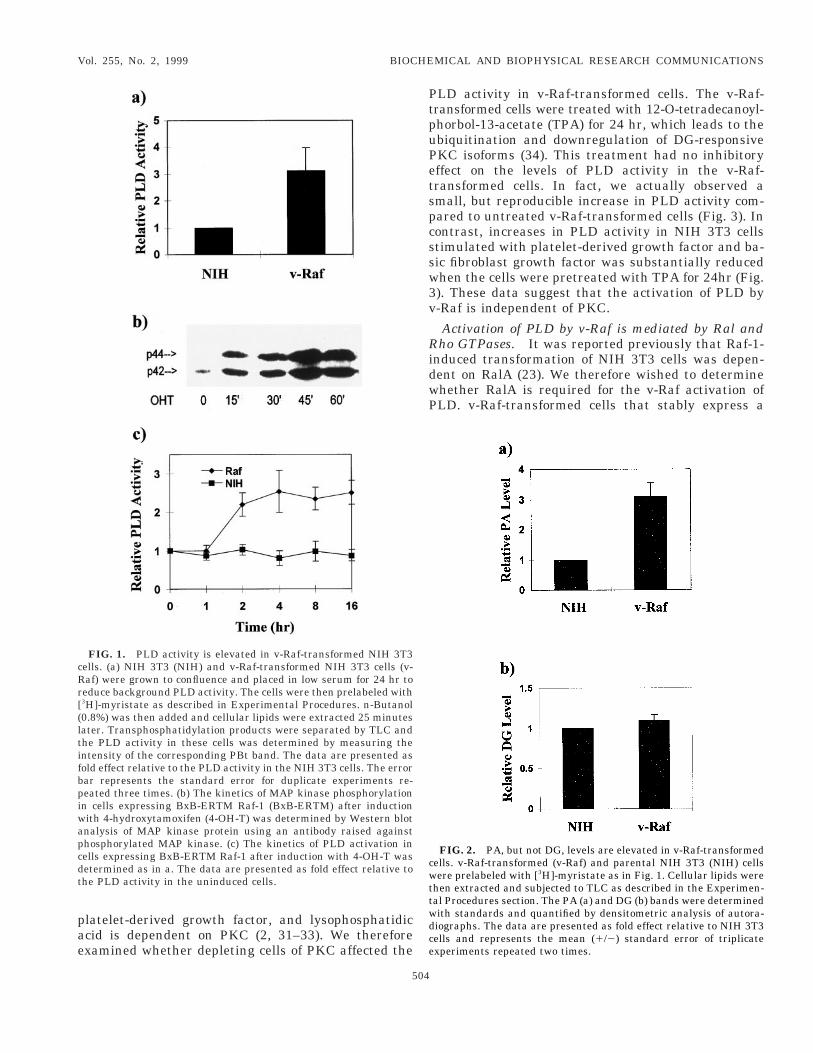
PLD activity is elevated in v-Raf-transformed NIHT3 cells. A unique property of mammalian PLD is itsbility to catalyze a transphosphatidylation reaction.n this reaction short chain primary alcohols act asucleophilic acceptors in place of H2O. This results in
503
he formation of a biologically stable phosphatidylalco-ol instead of PA. Formation of phosphatidylalcoholsas been used widely as an assay for PLD activity. To
nvestigate the PLD activity in v-Raf-transformedells, cells were prelabeled [3H]-myristate, which isncorporated exclusively into phosphatidylcholine (27),he substrate for PLD and then examined theransphopshatidylation of phosphatidylcholine to PBtn the presence of exogenously-provided n-butanol. Ashown in Fig. 1a, the v-Raf-transformed cells displayedreater than 3 fold elevated PLD activity than thearental NIH 3T3 cells. To determine if the elevatedevels of PLD activity in the v-Raf-transformed cellsas an acute response activated Raf or due to a sec-ndary effect of transformation, we looked at condi-ional activation of Raf-1 in NIH 3T3 cells expressingn inducible form of an oncogenic activated c-Raf-1rotein (BxB-ERTM) (25). This Raf protein is a fusionf an activated Raf-1 with the estrogen receptor thatonfers responsiveness to the estrogen analogue-hydroxytamoxifen (25). NIH 3T3 cells stably express-ng the BxB-ERTM Raf-1 were established as de-cribed in Experimental Procedures. We first investi-ated the ability to induce MAP kinase phosphoryla-ion which is an early response to activation of Raf (28,9). The cells expressing BxB-ERTM c-Raf-1 werereated with 4-OH-T and the phosphorylation state ofAP kinase was examined at various time points after
nduction. As observed previously (25) increased MAPinase phosphorylation could be detected within 15in after induction and reached maximal levels by 45in (Fig. 1b). We next examined the ability of 4-hy-
roxytamoxifen to induce PLD activity in these cells;nd, as shown in Fig. 1c, increased PLD activity coulde detected within two hr after treatment. These datandicate that the activation of PLD is a response to-Raf kinase activity and not an indirect consequencef the transformed phenotype induced by v-Raf.It is not yet clear what role PLD-generated PA might
lay in the transduction of intracellular signals, how-ver PA can be metabolically converted to the PKCgonist DG by phosphatidate phosphohydrolase. Weherefore examined the relative levels of PA and DG inoth the v-Ras-transformed and parental NIH 3T3ells. As shown in Fig. 2a, there was about 3 fold moreA in the v-Raf-transformed cells relative to the pa-ental NIH 3T3 cells; whereas there was no differencen DG levels between the two cell lines (Fig. 2b). Thus,he activation of PLD by v-Raf results in increasedevels of PA.
v-Raf induced PLD activity is insensitive to depletingells of PKC. It has been reported that activation ofLD by either v-Src or v-Ras is independent of PKC
27, 30). However, several groups have shown thatctivation of PLD by other mitogenic stimuli includingpidermal growth factor, fibroblast growth factor,
pae
PtpuPetspcssw3v
RidwP
cwttwdce
cRr[(ltifbpiwapcdt
Vol. 255, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
latelet-derived growth factor, and lysophosphatidiccid is dependent on PKC (2, 31–33). We thereforexamined whether depleting cells of PKC affected the
FIG. 1. PLD activity is elevated in v-Raf-transformed NIH 3T3ells. (a) NIH 3T3 (NIH) and v-Raf-transformed NIH 3T3 cells (v-af) were grown to confluence and placed in low serum for 24 hr toeduce background PLD activity. The cells were then prelabeled with3H]-myristate as described in Experimental Procedures. n-Butanol0.8%) was then added and cellular lipids were extracted 25 minutesater. Transphosphatidylation products were separated by TLC andhe PLD activity in these cells was determined by measuring thentensity of the corresponding PBt band. The data are presented asold effect relative to the PLD activity in the NIH 3T3 cells. The errorar represents the standard error for duplicate experiments re-eated three times. (b) The kinetics of MAP kinase phosphorylationn cells expressing BxB-ERTM Raf-1 (BxB-ERTM) after inductionith 4-hydroxytamoxifen (4-OH-T) was determined by Western blotnalysis of MAP kinase protein using an antibody raised againsthosphorylated MAP kinase. (c) The kinetics of PLD activation inells expressing BxB-ERTM Raf-1 after induction with 4-OH-T wasetermined as in a. The data are presented as fold effect relative tohe PLD activity in the uninduced cells.
504
LD activity in v-Raf-transformed cells. The v-Raf-ransformed cells were treated with 12-O-tetradecanoyl-horbol-13-acetate (TPA) for 24 hr, which leads to thebiquitination and downregulation of DG-responsiveKC isoforms (34). This treatment had no inhibitoryffect on the levels of PLD activity in the v-Raf-ransformed cells. In fact, we actually observed amall, but reproducible increase in PLD activity com-ared to untreated v-Raf-transformed cells (Fig. 3). Inontrast, increases in PLD activity in NIH 3T3 cellstimulated with platelet-derived growth factor and ba-ic fibroblast growth factor was substantially reducedhen the cells were pretreated with TPA for 24hr (Fig.). These data suggest that the activation of PLD by-Raf is independent of PKC.
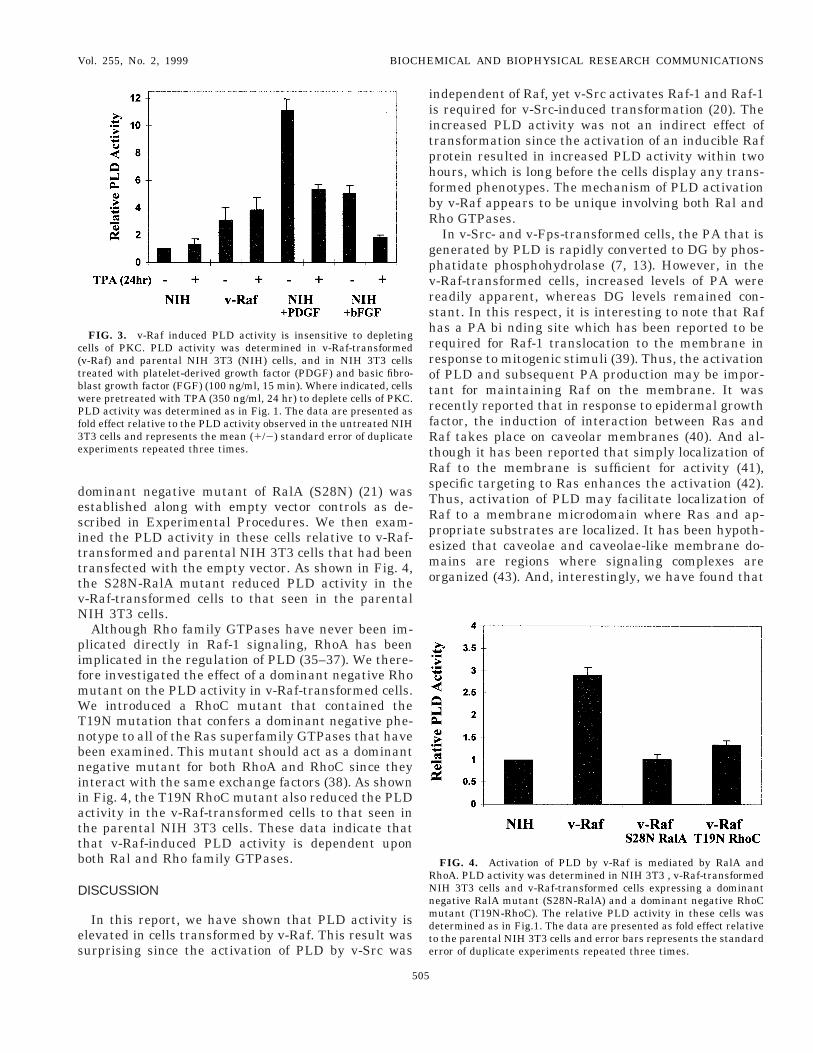
Activation of PLD by v-Raf is mediated by Ral andho GTPases. It was reported previously that Raf-1-
nduced transformation of NIH 3T3 cells was depen-ent on RalA (23). We therefore wished to determinehether RalA is required for the v-Raf activation ofLD. v-Raf-transformed cells that stably express a
FIG. 2. PA, but not DG, levels are elevated in v-Raf-transformedells. v-Raf-transformed (v-Raf) and parental NIH 3T3 (NIH) cellsere prelabeled with [3H]-myristate as in Fig. 1. Cellular lipids were
hen extracted and subjected to TLC as described in the Experimen-al Procedures section. The PA (a) and DG (b) bands were determinedith standards and quantified by densitometric analysis of autora-iographs. The data are presented as fold effect relative to NIH 3T3ells and represents the mean (1/2) standard error of triplicatexperiments repeated two times.
desitttvN
pifmWTnbniiattb
D
es
iiitphfbR
gpvrshrrotrfRtRsTRpemo
RNnmdte
c(tbwPf3e
Vol. 255, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
ominant negative mutant of RalA (S28N) (21) wasstablished along with empty vector controls as de-cribed in Experimental Procedures. We then exam-ned the PLD activity in these cells relative to v-Raf-ransformed and parental NIH 3T3 cells that had beenransfected with the empty vector. As shown in Fig. 4,he S28N-RalA mutant reduced PLD activity in the-Raf-transformed cells to that seen in the parentalIH 3T3 cells.Although Rho family GTPases have never been im-
licated directly in Raf-1 signaling, RhoA has beenmplicated in the regulation of PLD (35–37). We there-ore investigated the effect of a dominant negative Rhoutant on the PLD activity in v-Raf-transformed cells.e introduced a RhoC mutant that contained the
19N mutation that confers a dominant negative phe-otype to all of the Ras superfamily GTPases that haveeen examined. This mutant should act as a dominantegative mutant for both RhoA and RhoC since they
nteract with the same exchange factors (38). As shownn Fig. 4, the T19N RhoC mutant also reduced the PLDctivity in the v-Raf-transformed cells to that seen inhe parental NIH 3T3 cells. These data indicate thathat v-Raf-induced PLD activity is dependent uponoth Ral and Rho family GTPases.
ISCUSSION
In this report, we have shown that PLD activity islevated in cells transformed by v-Raf. This result wasurprising since the activation of PLD by v-Src was
FIG. 3. v-Raf induced PLD activity is insensitive to depletingells of PKC. PLD activity was determined in v-Raf-transformedv-Raf) and parental NIH 3T3 (NIH) cells, and in NIH 3T3 cellsreated with platelet-derived growth factor (PDGF) and basic fibro-last growth factor (FGF) (100 ng/ml, 15 min). Where indicated, cellsere pretreated with TPA (350 ng/ml, 24 hr) to deplete cells of PKC.LD activity was determined as in Fig. 1. The data are presented as
old effect relative to the PLD activity observed in the untreated NIHT3 cells and represents the mean (1/2) standard error of duplicatexperiments repeated three times.
505
ndependent of Raf, yet v-Src activates Raf-1 and Raf-1s required for v-Src-induced transformation (20). Thencreased PLD activity was not an indirect effect ofransformation since the activation of an inducible Rafrotein resulted in increased PLD activity within twoours, which is long before the cells display any trans-ormed phenotypes. The mechanism of PLD activationy v-Raf appears to be unique involving both Ral andho GTPases.In v-Src- and v-Fps-transformed cells, the PA that is
enerated by PLD is rapidly converted to DG by phos-hatidate phosphohydrolase (7, 13). However, in the-Raf-transformed cells, increased levels of PA wereeadily apparent, whereas DG levels remained con-tant. In this respect, it is interesting to note that Rafas a PA bi nding site which has been reported to beequired for Raf-1 translocation to the membrane inesponse to mitogenic stimuli (39). Thus, the activationf PLD and subsequent PA production may be impor-ant for maintaining Raf on the membrane. It wasecently reported that in response to epidermal growthactor, the induction of interaction between Ras andaf takes place on caveolar membranes (40). And al-
hough it has been reported that simply localization ofaf to the membrane is sufficient for activity (41),pecific targeting to Ras enhances the activation (42).hus, activation of PLD may facilitate localization ofaf to a membrane microdomain where Ras and ap-ropriate substrates are localized. It has been hypoth-sized that caveolae and caveolae-like membrane do-ains are regions where signaling complexes are
rganized (43). And, interestingly, we have found that
FIG. 4. Activation of PLD by v-Raf is mediated by RalA andhoA. PLD activity was determined in NIH 3T3 , v-Raf-transformedIH 3T3 cells and v-Raf-transformed cells expressing a dominantegative RalA mutant (S28N-RalA) and a dominant negative RhoCutant (T19N-RhoC). The relative PLD activity in these cells was
etermined as in Fig.1. The data are presented as fold effect relativeo the parental NIH 3T3 cells and error bars represents the standardrror of duplicate experiments repeated three times.

ct
avbRmAtaceoilrddiisaushwmttasivtarmtmsPe
ici(cgipmme
A
sAorIN
R
1
1
1
1
111
1
1
1
2
2
2
2
2
2
Vol. 255, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
aveolar membranes are highly enriched for PLD ac-ivity and RalA (Our unpublished data).
The PLD activity induced by v-Raf was inhibited bydominant negative mutant of RalA. We reported pre-
iously that the activation of PLD by v-Src and v-Ras islocked by the dominant negative RalA mutant (21).alA interacts directly with PLD1 (44) and apparentlyediates interaction with the PLD1 activator proteinrf (45). This RalA/Arf/PLD complex constitutes a dis-
inct downstream signaling pathway from Ras medi-ted by interaction between Ras and the RalA ex-hange factor Ral-GDS (Ral-guanine nucleotidexchange factor), which is a direct downstream targetf the Ras effector domain (46). Although v-Raf-nduced transformation was dependent upon RalA, aink between Raf-1 and the RalA-PLD1 pathway is noteadily apparent since Raf is generally considered aownstream target of Ras. Since it is believed that theominant negative RalA mutant functions by interact-ng with and rendering inactive Ral-GDS, the sensitiv-ty of v-Raf-induced PLD activity to this RalA mutantuggests a role for Ral-GDS. How Ral-GDS might bectivated by Raf, is unclear, but it suggests that simplepstream and downstream models based on genetictudies may not sufficiently explain what is actuallyappening. The involvement of a Rho family GTPaseas indicated by a sensitivity to a dominant negativeutant of RhoC. The Rho family GTPases are reported
o be involved in the regulation of the actin cytoskele-on and specifically in the formation of focal adhesionsnd actin stress fibers (47–49). RhoA has also beenhown to be involved in the regulation of PLD bothn-vitro and in-vivo (35–37, 50, 51). Whether the in-olvement of Rho in the regulation of the actin cy-oskeleton and the formation of focal adhesions andctin stress fibers has any thing to do with its apparentole in regulating PLD activity remains to be deter-ined. However, PLD has been reported to mediate
he formation of actin stress fibers (52). Since transfor-ation results in dramatic changes in cytoskeletal
tructure, it is possible that the Rho requirement forLD activation by Raf involves regulation of cytoskel-tal structure.Recently several groups have reported that very high
ntensity Raf signaling, rather than stimulating cellycle progression, induces expression of the cell cyclenhibitor p21 Waf1/Cip1 and causes cell cycle arrest53–55). Consistent with this observation, it was re-ently suggested that activated Rho functions syner-istically with Ras and Raf to transform cells by block-ng p21 Waf1/Cip1 expression (56) indicating areviously unsuspected role for Rho in Ras and Rafitogenic signals. Thus, it is also possible that Rho-ediated PLD activation may inhibit p21 Waf1/Cip1
xpression and facilitate cell cycle progression.
506
CKNOWLEDGMENTS
We thank Alan Hall for the RhoC gene. This investigation wasupported by National Institutes of Health Grant CA46677 andmerican Cancer Society Grant BE-243 to DAF; National Institutesf Health Grant GM47707 and an American Cancer Society facultyesearch award (to L.A.F.); and a Research Centers in Minoritynstitutions (RCMI) award from the Division of Research Resources,ational Institutes of Health (RR 03037) to Hunter College.
EFERENCES
1. Boarder, M. R. (1994) TIPS 15, 57–62.2. Plevin, R., Cook, S. G., Palmer, S., and Wakelam, M. J. O. (1991)
Biochem. J. 279, 559–565.3. Kaszkin, M., Seidler, L., Kast, R., and Kinzel, V. (1992) Biochem.
J. 287, 51–57.4. Song, J., Jiang, Y.-J., and Foster, D. A. (1994) Cell Growth Differ.
5, 79–85.5. Karnam, P., Standaert, M. L., Galloway, L., and Farese, R. V.
J. Biol. Chem. 272, 6136–6140.6. Shome, K., Vasudevon, C., and Romero, G. (1997) Curr. Biol. 7,
387–396.7. Song, J., Pfeffer, L. M., and Foster, D. A. (1991) Mol. Cell. Biol.
11, 4903–4908.8. Wyke A. W., Cook S. J., MacNulty E. E., and Wakelam M. J.
(1992) Cell Signal. 4, 267–274.9. Teegarden D., Taparowsky E. J., and Kent, C. (1990). J. Biol.
Chem. 265, 6042–6047.0. Jiang, H., Lu, Z., Luo, J. Q., Wolfman, A., and Foster, D. A.
(1995) J. Biol. Chem. 270, 6006–6009.1. Carnero, A., Cuadrado, A., del Peso, L., and Lacal, J. C. (1994)
Oncogene 9, 1387–1395.2. Martin A., Duffy P. A., Liossis C., Gomez-Munoz A., O’Brien L.,
Stone J. C., and Brindley D. N. (1997). Oncogene 14, 1571–1580.3. Jiang, Y.-W., Song, J., Zang, Q. and Foster, D. A. (1994) Biochem.
Biophys. Res. Commun. 203,1195–1203.4. Exton, J. H. (1994) Biochim. Biophys. Acta. 1212, 26–42.5. Exton, J. H. (1997) Physiol. Rev. 77, 303–320.6. Singer, W. D., Brown, H. A., and Sternweis, P. C. (1997) Annu.
Rev. Biochem. 66, 475–509.7. Ktistakis, N. T., Brown, H. A., Waters, M. G., Sternweis, P. C.,
and Roth, M. G. (1996) J. Cell Biol. 134, 295–306.8. Chen, Y.-G., Siddhanta, A., Austin, C. D., Hammond, S. M.,
Sung, T.-C., Frohman, M. A., Morris, A. J., and Shields, D. (1997)J. Cell. Biol. 138, 495–504.
9. Roth, M. G., and Sternweis, P. C. (1997) Curr. Opin. Cell Biol. 9,519–526.
0. Qureshi, S. A., Joseph, C. K., Gupta, R., Hendrickson, M., Song,J., Bruder, J., Rapp, U. R., and Foster, D. A. (1993) Biochem.Biophys. Res. Commun. 192, 969–975.
1. Jiang, H., Luo, J.-Q., Urano, T., Frankel, P., Lu, Z., Foster, D. A.,and Feig, L. (1995) Nature 378, 409–412.
2. Avruch, J., Zhang, X. F., Kyriakis, J. M. (1994) Trends Biochem.Sci. 19, 279–283.
3. Urano, T., Emkey, R., and Feig, L. A. (1996) EMBO J. 16,810–816.
4. Rapp, U. R., Goldsborough, M. D., Mark, G. E., Bonner, T. I.,Groffen, J., Reynolds, F. H. and Stephenson, J. R. (1983) Proc.Natl. Acad. Sci. USA 80, 4218–4222.
5. Kerkhoff, E., and Rapp, U. R. (1997) Mol. Cell. Biol. 17, 2576–2586.

2
22
2
3
3
3
3
3
3
3
3
33
4
4
4
4
4
4
4
4445
5
5
5
5
5
5
Vol. 255, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
6. Farnsworth, C. L., Freshney, N. W., Rosen, L. B., Ghosh, A.,Greenberg, M. E., and Feig, L. A. (1995) Nature 376, 524–527.
7. Song, J., and Foster, D. A. (1993) Biochem. J. 294, 711–717.8. Kyriakis, J. M., App, H., Zhang, X. F., Banerjee, P., Brautigan,
D. L., Rapp, U. R., and Avruch, J. (1992) Nature 358, 417–421.9. Howe, L. R., Leevers, S. J., Gomez, N., Nakielny, S., Cohen, P.,
and Marshall, C. J. (1992) Cell 71, 335–342.0. del Paso, L., Hernandez, R., Esteve, P., and Lacal, J. C. (1996)
J. Cell. Biochem. 61, 599–608.1. Yeo, E. J., and Exton, J. H. (1995) J. Biol. Chem. 270, 3980–
3988.2. Yeo, E. J., Kazlauskas, A., and Exton, J. H. (1994) J. Biol. Chem.
269, 27823–27826.3. van der Bend, R. L., de Widt, J., van Corven, E. J., Moolenaar,
W. H., and van Blitterswijk, W. J. (1992) Biochem. J. 285,235–240.
4. Lu, Z., Liu, D., Hornia, A., Devonish, W., Pagano, M., and Foster,D. A. (1998). Mol. Cell. Biol. 18, 839–845.
5. Bowman, E. P., D. J. Uhlinger and J. D. Lambeth. (1993) J. Biol.Chem. 268, 21509–21512.
6. Malcolm, K. C., Ross, A. H., Qiu, R. G., Symons, M., and Exton,J. H. (1994) J. Biol. Chem. 269, 25951–25954.
7. Hammond, S. M., Jenco, J. M., Nakashima, S., Cadwallader, K.,Gu, Q., Cook, S., Nozawa, Y., Prestwich, G. D., Frohman, M. A.,and Morris, A. J. (1997) J. Biol. Chem. 272, 3860–3868.
8. Feig, L. A. (1994) Curr. Opin. Cell Biol. 6, 204–211.9. Ghosh, S., Strum, J. C., Sciorra, V. A., Daniel, L., and Bell, R. M.
(1996) J. Biol. Chem. 271, 8472–8480.0. Mineo, C., James, G. L., Smart, E. J., and Anderson, R. G. W.
(1996) J. Biol. Chem. 271, 11930–11935.1. Stokoe, D., Macdonald, S. G., Cadwallader, K., Symons, M.,
Hancock, J. F. (1994) Science 264, 1463–1467.
507
2. Mineo, C., Anderson, R. G., White, M. A. (1997) J. Biol. Chem.272, 10345–10348.
3. Okamoto, T., Schlegel, A., Sherer, P. E., and Lisanti, M. P. (1998)J. Biol. Chem. 273, 5419–6422.
4. Luo, J.-Q., Liu, X., Hammond, S. M., Colley, W. C., Feig, L. A,Frohman, M. A. Morris, A. J., and Foster, D. A. (1997) Biochem.Biophys. Res. Commun. 235, 854–859.
5. Luo, J.-Q., Liu, X., Frankel, P., Rotunda, T., Ramos, M., Flom, J.,Jiang, H., Feig, L. A., Morris, A, Kahn, R. A., and Foster, D. A.(1998). Proc. Natl. Acad. Sci. USA 95, 3632–3637.
6. Cantor, S. B., Urano, T., and Feig, L. A. (1995) Mol. Cell. Biol. 15,4578–4584.
7. Ridley, A. J., and Hall, A. (1992) Cell 70, 389–399.8. Hotchin, N. A., and Hall, A. (1996) Cancer Surv. 27, 311–322.9. Tapon, N., and Hall, A. (1997) Curr. Opin. Cell Biol. 9, 86–92.0. Schmidt, M., Rumenapp, U., Bienek, C., Keller, J., von Eichel-
Streiber, C., and Jakobs, K. H. (1996) J. Biol. Chem. 271, 2422–2426.
1. Hess, J. A., Ross, A. H., Qiu, R. G., Symons, M., and Exton, J. H.(1997) J. Biol. Chem. 272, 1615–1620.
2. Cross, M. J., Roberts, S., Ridley, A. J., Hodgkin, M. N., Stewart,S., Claesson-Welsh, L., and Wakelam, M. J. O. (1996) Curr. Biol.6, 588–597.
3. Kerkhoff, E., and Rapp, U. R. (1998) Cancer Res. 58, 1636–1640.
4. Sewing, A., Wiseman, B., Lloyd, A. C., and Land, H. (1997) Mol.Cell. Biol. 17, 5588–5597.
5. Woods, D., Parry, D., Cherwinski, H., Bosch, E., Lees, E., andMcMahon, M. (1997) Mol. Cell. Biol. 17, 5598–5611.
6. Olson, M. F., Paterson, H. F., and Marshall, C. J. (1998) Nature394, 295–299.







![Phospholipase pPLAIIIα Increases Germination Rate and ......Phospholipase pPLAIIIa Increases Germination Rate and Resistance to Turnip Crinkle Virus when Overexpressed1[OPEN] Jin](https://img.pdfslide.us/doc/110x75/60c23bedb7cd7e20713772ef/phospholipase-pplaiii-increases-germination-rate-and-phospholipase-pplaiiia.jpg)
















![[RAF CAREERS] What Careers are there in the RAF?](https://img.pdfslide.us/doc/110x75/55cf8f58bb61ebe4598b4842/raf-careers-what-careers-are-there-in-the-raf.jpg)



