Embed Size (px)
Citation preview

CHIRALITY 6400404 (1994)
Racemisation of Drug Enantiomers by Benzylic Proton Abstraction at Physiological pH
CHRISTOPHER PEPPER, H. JOHN SMITH, KEVIN J. BARRELL, PAUL J. NICHOLLS, AND MICHAEL J.E. HEWLINS
Welsh School of Pharmacy (C. P., H. J.S., K . J . B. , P . J . N . ) and School of Chemistry and Applied Chemistry (M.J.E.H.), University of Wales College of Cardiff; Card18 U.K.
ABSTRACT The enantiomers of the aromatase inhibitors 3-(4-aminophenyl)-pyoli- dine-55-dione (WSP-3, 11), its N-pentyl derivative (III), and the antifungal econazole (IV), all possessing a benzylic proton at the chiral centre, are rapidly racemised in vitro in phosphate buffer (0.01 M) at pH 7.4 and 23°C with t,,, values of 7,6, and 5 h respectively. In vivo studies in rats show that (+)-econazole is racemised after intraperitoneal injection with t,,, = 1.24h. The enantiomers of the antifungal 1-[(benzofuran-2-yl)-4-chlorophenylmethyl] imidazole (V) were stable at pH 7.4, attributable to steric hindrance to carbanion formation in the racemisation step. o 1994 Wiey-Liss, Inc.
KEY WORDS: econazole, 3-(4-aminophenyl)pyrrolidine-2,5-diones, aromatase inhibitors, antifungals, drug registration
Several examples are known (see Discussion) of the lability at physiological pH of a C-H function at a chiral centre leading to racemisation or epimerisation of drug enantiomers or epimers, respectively. It is well known in organic chemistry that a benzylic proton, when attached to an electron with- drawing group, i.e., (I), can be readily abstracted by proton abstracting agents such as NaH, BuLi, LDA in nonaqueous media. We have found that for certain compounds of this type the benzylic proton at the chiral centre can be surprisingly labile at pH 7.4 in phosphate buffer leading to racemisation of enantiomeric forms. This followed from an initial observation that the aromatase inhibitor' (*)-WSP3 (11) underwent deu- teration within 1 day using D,O in 'H NMR studies at 20°C. We report here the rate of racemisation of the enantiomers of (11) at 20 and 37°C in phosphate buffer (0.01 M) at physiolog- ical pH (pH 7.4) as well as those of (IIIHV) to broaden the study.
MATERIALS AND METHODS Materials
D- and tartaric acid, D- and L-dibenzoyltartaric acid and sodium carbonate were obtained from Aldrich Chemical Com- pany, Poole, Dorset. Analar grade methanol, ethanol, methyl- ene chloride and diethyl ether were obtained from Fisons Chemicals, Loughborough, Leicestershire. Spectrophoto- metric grade methanol was purchased from Sigma Chemical Company Ltd., Poole, U.K. Acetonitrile, hexane and 2-pro- pan01 (HPLC grade) were obtained from Rathburn Chemicals Ltd. Walkburn, Scotland. Buffer salts (i.e., sodium dihydro- gen phosphate and disodium hydrogen phosphate) and econa- zole nitrate were supplied by Sigma. All optical rotation mea- surements were performed with a Bellingham and Stanley polarimeter using 1% methanolic solutions. All melting points 0 1994 Wiley-Liss, Inc.
were determined using an electrothermal melting point appa- ratus and are uncorrected.
(+ )- and (-)-3-(4-Aminophenyl)pyrrolidine-2,5-di- one (WSP-3; 11) A mixture of (*)-(II)' (2.98 g, 0.015 mol) was stirred with a 2:l molar equivalent of (+)-tartaric acid (1.176 g, 0.0075 moll and methanol (84 ml). This mixture was heated until dissolution was complete and then allowed to cool to room temperature. The precipitate, a yellow solid, was filtered off and recrystallised six times from methanol to yield the (+)-WSP-3 tartrate salt (1.09 g). The recrystallisation steps were monitored using polarimetry, i.e., a 1% methan- olic solution of the salt was measured for its optical rotation after each recrystallisation until a steady state optical rotation was achieved. An identical procedure was employed using (-)-tartaric acid to yield the (-)-WSP-3 tartrate salt (1.03 g) after six recrystallisations. Aqueous solutions of the respec- tive salts were than added to equimolar amounts of aqueous sodium carbonate to yield the (+)- and (-)-free bases. (+I- form: m.p. 172-4"C, [ ( w ] , ~ ~ +131.2". (-)-form: m.p. 172-
( + 1- and (- )-1-Pentyl-3-(4-aminophenyl)pyrroli- dine-2,5-dione (WSP-3; 111) (2)-(IIIj2 (3.0 g, 0.012 mol) was reacted with (+)-tartaric acid as previously described for (II), and the (+)-tartrate salt (1.96 g) was precipitated on cooling as a fine white crystalline solid. The (-)-tartrate salt was recovered from the mother liquors on addition of (-)- tartaric acid and yielded 1.95 g of the (-)-tartrate salt. Six recrystallisations gave a steady state optical rotation for both
174"C, [ c x ] " ~ ~ - 130.9".
Received for publication December 9, 1993; accepted February 18, 1994. Address reprint requests to Dr. H. J. Smith, Welsh School of Pharmacy, U. W.C.C., Cardiff CF13XF. U.K.
RACEMISATION OF ENANTIOMERS AT pH 7.4 401
2 / p H Y
X
( I ) : X and Y I electron withdrawing groups
SH O Y
S c h e m e 1. Formulae of the aromatase inhibitors 3- (4-aminophenyl)-pyrrolidine-2, 5-done (WSP-3.11). its N-pentyl derivative (111). the antifungal econazole (IV). and the antifungal 1- [(benzofuran-Z-yl)-4-chlorophenylmethyl] imidazole (V).
salts, sodium carbonate was then used to liberate the free bases. (+)-form: m.p. 102-103"C, [a]25u + 161.2". (-)-form: m.p. 102-103"C, -162.0".
(+)- and (-)-Econazole (IV). (?I-Econazole nitrate (6 g, 0.013 mol) was reacted with excess sodium hydroxide to yield the free base which was fdtered off and dned. The free base (4 g; 9 x lo-" mol) was then reacted with tartaric acid using the method described for (111). The (+)-tartrate salt (2.4 g) was precipitated as a fine white solid, the (-)-tartrate salt yielded 2.1 g from the mother liquors. The free bases were recovered by the method previously described using aqueous sodium carbonate. (+)-form: m.p. 85.9-87.3"C, [a]",, + 102.4". (-)-form: m.p. 85.9-87.3"C, [a125D - 103".
(+)- and (-)-l-[(Benzofuran-2-yl)-4-chlorophenyl- methyl] imidazole (V). (&)-(V)3 (1.69 g, 5.5 mmol) in dry ether was mixed with a solution of (+)-dibenzoyltartaric acid monohydrate (2.1 g, 5.5 mmol) in dry ether (50 ml). The precipitate (2.1 g) was removed and recrystallised six times from ethanol. Use of (-)-dibenzoyltartaric acid gave the (-1- salt. The bases were released in the manner previously de- scribed. Enantiomeric purity was monitored by HPLC using hexane: 2-propano1:diethylamine (80: 200.2) on a Chiralpak AD (amylose coated silica gel) column. (+)-salt form: m.p. 128.4-129. TC, [ O L ] ~ ' ~ ) +71.84". (-)-form: m.p. 130-
Liquid Chromatography A Milton-Roy LC system was used consisting of a Model
3000 Constametric pump, a Rheodyne injection unit, and a Model 3100 variable wavelength spectromonitor. A Model
130.5"C, [ C X ] ~ ' ~ ) -71.61".
CL-4100 computing integrator was used to process the data. (*)-(IIHIV) were separated on an a'-acid glycoprotein
column (Chromtech AB; 100 x 4.0 mm) and a pre-column (10 x 4.0 mm) both packed with identical material with a mean particle size of 5 pm. A heated column jacket was supplied by Jones Chromatography (Hengoed, Mid Glamor- gan) to standardise temperature. Injection on the column was achieved using a Hamilton syringe (50 p1) into a Rheodyne 20 p1 loop.
(k)-(V) was separated on an amylose coated silica gel (Chi- ralpak AD; 4.6 x 250 mm) column using a precolumn (4.6 x 50mm).
Isocratic chromatographic conditions used were as follows:
Mobile phase: (II), (1111, and (IV): 0.01 M phosphate buffer pH 7.4 containing for (II), 5% 2-propanol; (111) 12% 2-propanol; (IV) 10% acetonitrile; (V): hexane:2-pro- panokdiethylamine (8020:0.2)
Flow: 0.5 ml min-' UV detection: 250 nm (V:288 nm) Temperature: 23°C Injection volume: 20 pl Pressure: 260 psi (V:70 psi)
Racemisation Studies Stock solutions (50 nM) of the pure enantiomers of (IIMV)
were prepared in phosphate buffer (0.01 M) pH 7.4 and ali- quots (20 pi) injected onto the respective HPLC column at
402 PEPPER ET AL.
B
x- A c
I B !
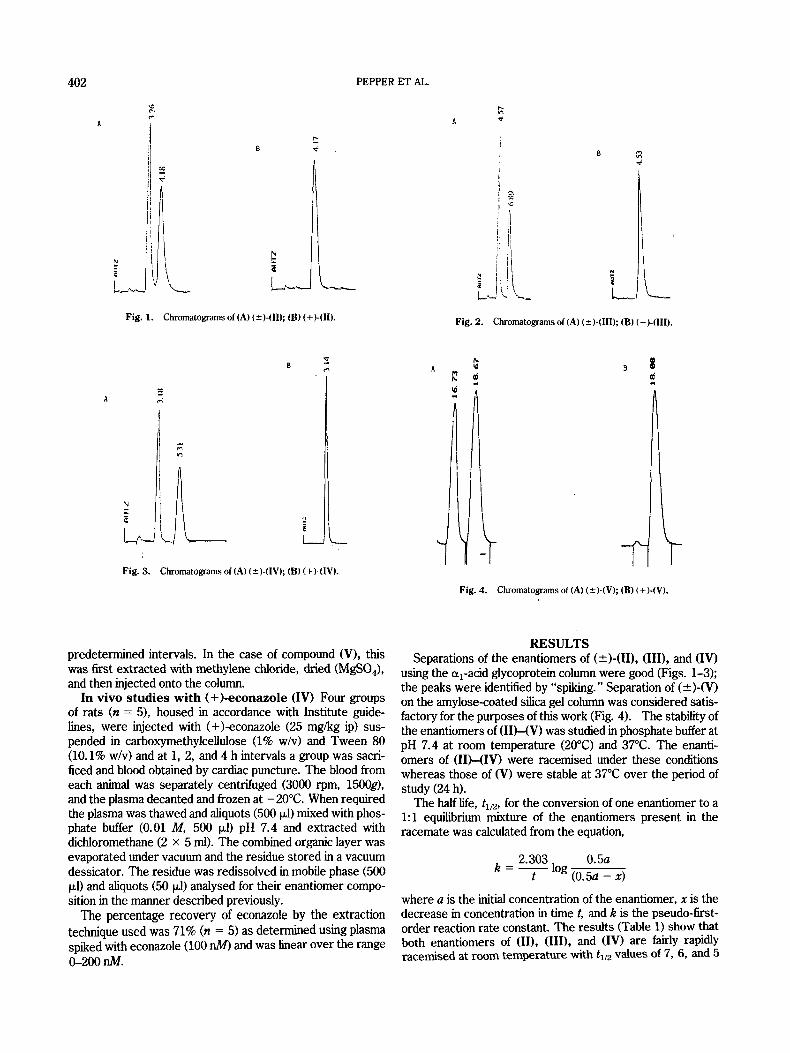
Fig. 2. Chromatograms of (A) (*)-(III); (B) (+)-(III).
i- Fig. 3. Chromatograms of (A) (?)-(IVk (8) (+)-(IV).
Fig. 4. Chromatograms of (A) (?)-(Vk (B) (+)-(V).
predetermined intervals. In the case of compound (V), this was first extracted with methylene chloride, dried (MgSO,), and then injected onto the column.
In vivo studies with (+I-econazole (IV) Four groups of rats (n = 51, housed in accordance with Institute guide- lines, were injected with (+)-econazole (25 mgkg ip) sus- pended in carboxymethylcellulose (1% w/v) and Tween 80 (10.1% w/v) and at 1, 2, and 4 h intervals a group was sacri- ficed and blood obtained by cardiac puncture. The blood from each animal was separately centrifuged (3000 rpm, 1500g), and the plasma decanted and frozen at -20°C. When required the plasma was thawed and aliquots (500 1.1) mixed with phos- phate buffer (0.01 M, 500 1.1) pH 7.4 and extracted with dichloromethane (2 x 5 ml). The combined organic layer was evaporated under vacuum and the residue stored in a vacuum dessicator. The residue was redissolved in mobile phase (500 1.1) and aliquots (50 1.1) analysed for their enantiomer compo- sition in the manner described previously.
The percentage recovery of econazole by the extraction technique used was 71% (n = 5) as determined using plasma spiked with econazole (100 nM) and was linear Over the range 0-200 nM.
RESULTS Separations of the enantiomers of (*)-(II), (III), and (IV)
using the a,-acid glycoprotein column were good (Figs. 1-3); the peaks were identified by “spiking.” Separation of (*)-(V) on the amylose-coated silica gel column was considered satis- factory for the purposes of this work (Fig. 4). The stability of the enantiomers of ( IIHV) was studied in phosphate buffer at pH 7.4 at room temperature (20°C) and 37°C. The enanti- omers of (IIHIV) were racemised under these conditions whereas those of (V) were stable at 37°C over the period of study (24 h).
The half life, tllz, for the conversion of one enantiomer to a 1:l equilibrium mixture of the enantiomers present in the racemate was calculated from the equation,
2.303 0 . 5 ~ k = - t log ( 0 . 5 ~ - x )
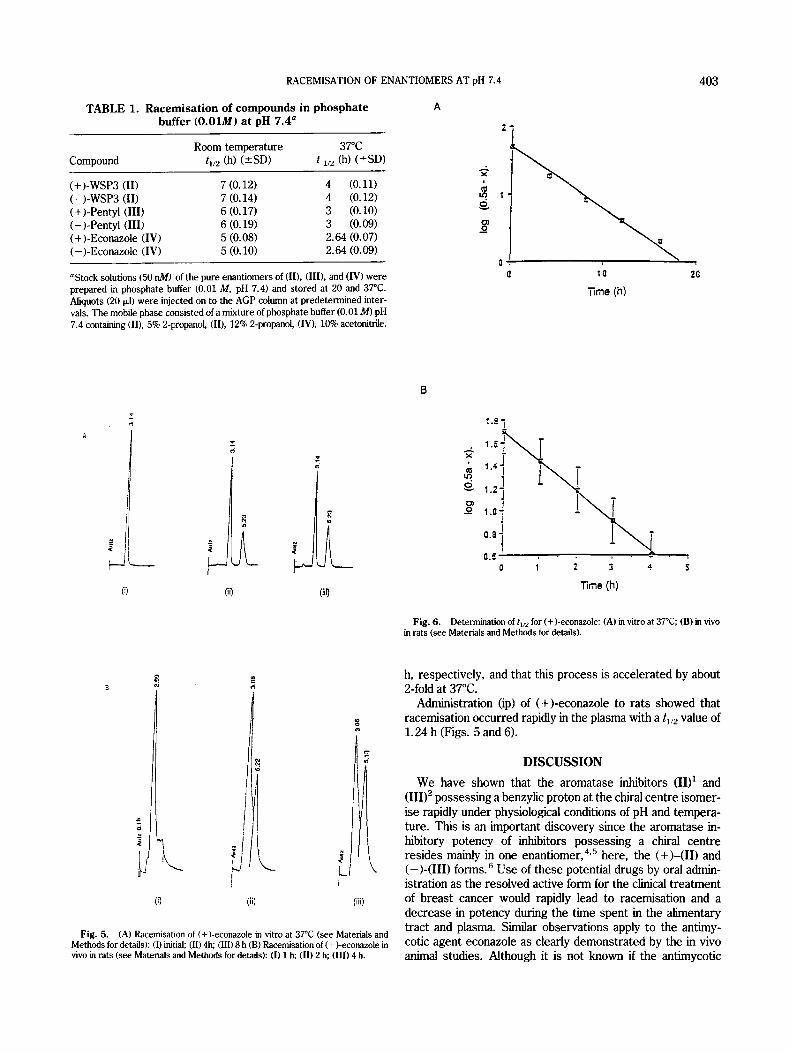
where a is the initial concentration of the enantiomer, x is the decrease in concentration in time t, and k is the pseudo-fkst- order reaction rate constant. The results (Table 1) show that both enantiomers of (II), (III), and (IV) are fairly rapidy racemised at room temperature with tllz values of 7, 6, and 5
RACEMISATION OF ENANTIOMERS AT pH 7.4
I
403
1 1
TABLE 1. Racemisation of compounds in phosphate A buffer (0.01M) at pH 7.4"
Room temperature 37°C Compound t i /% (h) (5SD) t 112 (h) (*SD)
(+)-WSP3 (11) 7 (0.12) 4 (0.11) (-)-WSPS (11) 7 (0.14) 4 (0.12) (+)-Pentyl (111) 6 (0.17) 3 (0.10) (-)-Pentyl (111) 6 (0.19) 3 (0.09) (+)-Econazole (IV) 5 (0.08) 2.64 (0.07) (-)-Econazole (11') 5 (0.10) 2.64 (0.09)
"Stock solutions (50 nM) of the pure enantiomers of (11). (111). and (IV) were prepared in phosphate buffer (0.01 M, pH 7.4) and stored at 20 and 37°C. Aliquots (20 (*I) were injected on to the AGP column at predetermined inter- vals. The mobile phase consisted of a mixture of phosphate buffer (0.01 M) pH 7.4 containing (11). 5% 2-propano1, (II), 12% 2-propanol, (IV), 10% acetonitrile.
(ii) (iii)
c c CI
r l 11 ( /-
(4 (iij (iii)
Fig. 5. (A) Racemisation of (+)-econazole in vitro at 37°C (see Materials and Methods for details): (I) initial; (11) 4h; (111) 8 h (B) Racemisation of (+)-econazole in vivo in rats (see Materials and Methods for details): (I) 1 h; (11) 2 h; (111) 4 h.
0 a 1 0 20
Eme (h)
Time (h)
Fig. 6. Determination oft,,, for (+)-econazole: (A) in vitro at 37°C; (B) in viva in rats (see Materials and Methods for details).
h, respectively, and that this process is accelerated by about 2-fold at 37°C.
Administration (ip) of (+)-econazole to rats showed that racemisation occurred rapidly in the plasma with a t,,, value of 1.24 h (Figs. 5 and 6) .
DISCUSSION We have shown that the aromatase inhibitors (11)' and
(IIU2 possessing a benzylic proton at the chiral centre isomer- ise rapidly under physiological conditions of pH and tempera- ture. This is an important discovery since the aromatase in- hibitory potency of inhibitors possessing a chiral centre resides mainly in one e n a n t i ~ m e r , ~ ' ~ here, the (+)-(11) and (-)-(III) forms6 Use of these potential drugs by oral admin- istration as the resolved active form for the clinical treatment of breast cancer would rapidly lead to racemisation and a decrease in potency during the time spent in the alimentary tract and plasma. Similar observations apply to the antimy- cotic agent econazole as clearly demonstrated by the in vivo animal studies. Although it is not known if the antimycotic

404 PEPPER ET AL.
activity lies in one isomer we have found that aromatase inhibitory potency lies mainly in the (+)-form.6 It is to be expected that structurally related analogues of econazole, e. g., miconazole, isoconazole, tioconazole, and sulconazole, would similarly show enantiomer racemisation under these conditions.
Several potent aromatase inhibitors have been described5v7 which possess a dibenzylic proton at the chiral centre; the benzofuran (W3 is representative of this type. The enanti- omers of (V) were stable in phosphate buffer pH 7.4 for 24 h. The stability of this type of structure may be attributed to the crowded nature of the benzylic proton which may either pre- vent exchange, due to sterically hindering approach of the base, or impose constraints on incipient carbanion formation due to lack of coplanarity.
Reports on the racemisation of enantiomers possessing a C-H at the chiral centre are few (see review by Testa and Trager' and Testa e t al.'). Amfepramone undergoes deute- rium exchange" in buffered D20 at pH 7.4 and 37°C with tIlz = 15 h and its monodeethylated derivative has tllz = 21 h whereas thalidomide is racemised" in phosphate buffer pH 7.4 at 37°C with tll2 = 2.5 h. Two analogues of thalidomide, 2-phthalimidinoglutarimide and 2-phthalimidoadipie, are rapidly racemised, 12,13 the former with tl,z = ca. 3 h. l2 The benzylic C-H at the chiral centre of arylpropionic acids is known to be labile in plasma due to enzymic inver~ion'~ and in oxazepam the presence of a conjugated benzylic C-H at the chiral centre leads to racemi~ationl~ (tl12 = 14 min) in the pH 6-8 range temperature due to ring opening and closure of the 2-hydroxyketone function. Examples of racemisation of a CH at the chiral centre in an epimer of drugs possessing two or more chiral centres is also known. Pilocarpine (2S:3R) is epimerised at the C (2)-chiral centre with a calculated tlIz = 36 days at pH 7.4 and 35"C.16 The ACE inhibitor RS-10085 (S: S: S) was slowly epimeri~ed'~ (S: S: R) at pH > 7 and 40°C. 16-hido and 16-choro derivatives of estrone-3-methyl ether were extensively epimerised in vivo, 18p19 an inversion shown to occur nonenzymatically.
Testa e t al., ' using the above examples, rationalised CH- lability at a chiral centre with the effects of attached acid- strengthening and acid-weakening groups (RR'RC-H) in a broad manner. Our observaions by being confined to a ben- zylic proton at the chiral centre amplify the above examples and further support the broad structural generalisations made by Testa e t al. Our observations suggest the possibility that other established drugs possessing a benzylic type C-H at the chiral centre may on examination of their enantiomers be found to exhibit a similar lability in near neutral solution.
Should the observations made here have a wider application then a general awareness of benzylic type C-H, lability at the chiral centre in enantiomers is essential to limit tissue and animal experimentation programmes and the associated de- velopment costs of chiral synthesis, since these could be better oriented towards the racemate at an early stage prior to submission for drug licensing. Furthermore, worldwide drug registration authorities, now moving towards licensing the racemic form of a drug only if it has been previously shown that the less active enantiomer possesses little undesirable side effects,20.21 could be faced in the future with formulating
a requirement for a minimum cutoff lability t,, value for an enantiomer presented for registration.
LITERATURE CITED 1.
2.
3.
4.
5.
6. 7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
Daly, M.J., Jones, G.W., Nicholls, P.J., Smith, H.J., Rowlands, M.G., Bunnett, M.A. Synthesis and biochemical evaluation of analogues of ami- noglutethimide based on phenylpyrrolidine-2.5-dione. J. Med. Chem. 2 9 520-523, 1986. Whomsley, R., Smith H.J., Nicholls, P.J., Nazareth, W., Ahmadi, M. Some 1-, and 3-substituted 3-(4'-aminophenyl) pyrrolidine-2,5diones as selective inhibitors of aromatase. J. Enz. Inhibit. 6317-330, 1993. PesteUi, V., Giannotti, D.. Giollitti, A,, Fanto, N., Riviera, L., Bellotti, M. G. New benzofuran-imidazoles as antimycotic agents. Chemioterapia
Laughton, C.A., McKenna, R., Neidle, S., Jarman. M., McCague, R., Rowlands, M.G. Crystallographic and molecular modelling studies on 3-ethyl-3-(4-pyridyl)piperidine-2,6-dione and its butyl analogue. inbibitors of mammalian aromatase. Comparison with natural substrates: Prediction of enantioselectivity for N-alkyl derivatives. J. Med. Chem. 33267% 2679, 1990. Vanden Bossche, H., Willemsens, G., Roels, I., Bellens, D., Moereels, H., Coene, M., Jeune, L.L., Lauwers, W., Janseen, P.A.J. R76713 and enantiomers: Selective, nonsteroidal inhibitors of the cytochrome P450- dependent oestrogen synthesis. Biochem. Pharmacol. 40(8): 1707-1718, 1990. Pepper, C., Smith, H.J., Nicholls, P.J. Unpublished work. Hirsch, K.S., Jones, C.D., Lmdstrom, T.D., Stamm, N.B., Sutton, G.P., Taylor, H.M., Weaver, D.E. Discovery and development of a novel class of nonsteroidal aromatase inhibitors. Steroids 50201-217, 1987. Testa, B., Trager, W. F. Racemates versus enantiomers in drug develop- ment: Dogmatism or pragmatism. Chirality 2(3): 129-133, 1990. Testa, B., Carrupt, P.-A., Gal, J. The so-called "interconversion" of stereoisomeric drugs: an attempt at clarification. Chirality 5 105-111, 1993. Testa, B. Some chemical and stereochemical aspects of diethylpropan metabolism in man. Acta Pharm. Suec. 10441454, 1973. Knoche, B., Blaschke, G. Racemisation of thalidomide in uzfro. 4th Inter- national Symposium on Chiral Disctimination, 19-22 September 1993, Montreal, Canada. Abstract 144. Schmahl, H.J., Heger. W., Nau, H. The enantiomers of the teratogenic thalidomide analogue EM 12. Toxicol. Lett., 4523-33, 1989. Eger, K., Jalalian, M.. Verspohl, E.J., Lupke, N.P. Synthesis, central nervous system activity and teratogenicity of a homothalidomide. Arz- neim.-Forsch. (Drug Res.) 4 0 107?-1075, 1990. Kaye, B. Chiral drug metabolism: A perspective. Biochem. Soc. Trans. 1945e-459, 1991. Yang, S.K., Lu, X-L. Racemisation Kinetics of enantiomeric oxazepams and stereoselective hydrolysis of enantiomeric oxazepam 3-acetates in rat liver microsomes and brain homogenate. J. Pharm. Sci. 78789-795, 1989. Nunes, M.A.. Brochmann-Hanssen, E. Hydrolysis and epimerization ki- netics of pilocarpine in aqueous solution. J. Pharm. Sci. 63:716-721, 1974. Gu, L., Strickley, R. G. Diketopiperazine formation, hydrolysis, and epimerization of the new dipeptide angiotensin-converting enzyme inhibi- tor RS-10085. Pharm. Res. 4:392-397, 1987. Schumann, W. Zur Biotransformation von s s 267 (16a-Azido-3-meth- oxyestra-1,3,5(10)-trien-17-one). Pharmazie 4332%332, 1988. Nambara, T., Nokubo, M. Epimerization of halogen during in vivo trans- formation of l6a-chlorestrone methyl ether. Chem. Pharm. Bull. 21: 2812-2813, 1973. Anon. Rules Governing Medifinal Products in the European Community, Vol. I1 1989. European Economic Community, Brussels. De Camp, W.H. The FDA perspective on the development of stereoiso- mers. Chirality 12-6, 1989.
6 (4):269-271; 272-276 1987.





























