Embed Size (px)
Citation preview

Quenching kinetics of the acridine excited state by vinyl monomers inhomogeneous and micellar solution
Silmara F. Buchviser and Marcelo H. Gehlen*¤Instituto de de Carlos, Universidade de Paulo, Caixa Postal 780, 13560-970Qu•�mica Sa8 o Sa8 o Sa8 oCarlos [ SP, Brazil
The quenching of singlet and triplet electronic excited states of acridine cation by vinyl monomers is investigated in homogeneous(methanol), aqueous, and reverse micelles of sodium dodecyl sulfate (SDS). The Ñuorescence quenching of the dye by styrenederivatives in methanol as well as in aqueous SDS micelles is very efficient with quenching rate constants of the order of 1010M~1 s~1 and 5 ] 107 s~1, respectively. However, triplet quenching rate constants in methanol are much lower than the di†usioncontrolled limit, and no dye photoblenching is observed suggesting that a quenching process follows a reversible photoinducedcharge-transfer mechanism. The Ñuorescence quenching of the dye by styrene derivatives in reverse SDS micelles is a pseudo-Ðrst-order process with no micellar quenching transient. It is shown that placing the dye inside a reverse micelle substantially reducesthe quenching rate by styrene derivatives. The scope of this e†ect on photoinitiation efficiency for radical polymerization ofstyrene derivatives via a photoredox process of cationic dyes in reverse micelles is discussed. Association constants of vinylmonomers to SDS micelles, K, as well as partition coefficients in n-octanol/water have been also determined using the Ñuores-cence quenching method. K is correlated with the boiling points of the vinyl monomers.
Polymerization of vinyl monomers in microheterogeneoussystems like aqueous micelles, reverse micelles and micro-emulsions has been a topic of current interest in polymerscience.1h6 The host micellar environment usually constrainsthe molecular motion of the guest monomer, and the interplaybetween local arrangement and monomer reactivity may giverise to polymers with di†erent characteristics when comparedwith traditional polymer synthesis in homogeneous solution.Therefore, prior information about the degree of binding orassociation of the monomer to the micelle and the monomerintramicellar di†usion are important factors to understand thee†ects of the micellar structure in the polymerization kinetics.
Several studies of polymerization of vinyl monomers inmicroheterogeneous systems have been reported, but in mostof them the initiation step was based on thermal reaction ofclassical initiators.1h6 More recently, photoinitiators havebeen applied with great success, and the use of dyes in laser-initiated photopolymerization is a new Ðeld with many practi-cal applications.7h13
In radical photopolymerization of vinyl monomers in thehomogeneous phase, the photophysical quenching of dye initi-ator by the monomer, forming no active radicals, competeswith the photoredox step of radical formation and hencereduces substantially the photoinitiation rate.12,13
One of the strategies to avoid such an e†ect consists in aproper choice of a dye initiator/co-initiator (CoI) pair for agiven micelle/monomer system in such a way that a photo-initiation step occurs in a domain of lowest monomer concen-tration. In an intermediary step, the active radicals formedby a photoredox process should be efficiently trapped bythe domain of highest monomer concentration to initiatethe addition polymerization.
In micellar systems, three domains can be deÐned : thehydrophilic or aqueous domain ; the hydrophobic domain,formed by the surfactant alkyl chains ; and the micelle inter-face. The distribution of monomer, initiator and co-initiator
¤ E-mail : MARCELOG=IQSC.SC.USP.BR
between aqueous and micellar phase can be estimated frommeasurements of physicalÈchemical properties related to thepartition process of species.14 In the present contribution, theassociation constants K of vinyl monomers with SDS micellesare determined by a Ñuorescence quenching method (FQmethod).15 The K values or the equivalent association coeffi-cients of vinyl monomers to SDS micelles are compared withthose from the partition of the monomers in a water/n-octanolsystem. Protonated acridine with a decay time of ca.32.6^ 0.2 ns in SDS aqueous micelles and 24.6^ 0.2 ns inSDS reverse micelles is used as the Ñuorescence probe in theFQ method. The quenching rate constants of this cationic dyeby a series of monomers in homogeneous and micellar mediaare also determined from the analysis of time-resolved data.Quenching rate constants are used to indicate whether theprocess is chemical or di†usion controlled.
It will be seen that very efficient singlet and triplet quen-ching of the dye by styrene derivatives observed in homoge-neous solution as well as in SDS aqueous micelles is reducedby placing the dye in the water pool of a reverse micelle. Thescope of this e†ect on photoinitiation efficiency in reversemicelles will be discussed.
ExperimentalThe vinyl monomers, 3-vinylbenzyl chloride, VBC (Kodak) ;divinyl benzene, DVB (Fluka) ; vinyl n-butyl ether, VNBE(Polyscience) ; vinyl 2-pyrolidone, V2Prl (Aldrich) ; a-methylstyrene, MSty (Merck) ; styrene, Sty (EDN) ; vinyl acetate, VAc(3M) ; acrylamide, Aclmd (Merck) ; acrylonitrile, Acln(Aldrich) ; methyl methacrylate, MMA (Aldrich), were distilledat low pressure before use. Sodium dodecyl sulfate, SDS(Sigma), acridine hydrochloride (Carlo Erba), n-octanol, andcyclohexane were used as received. Milli-Q pure water wasused to prepare surfactant solutions. Working solutions wereprepared by adding, via a microsyringe, the appropriateamounts of concentrated stock solution of the dye in waterand monomer pure or in methanol to the surfactant solution,followed by stirring and sonication. In the studies of the quen-ching processes in homogeneous phase (methanol), the same
J. Chem. Soc., Faraday T rans., 1997, 93(6), 1133È1139 1133
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online / Journal Homepage / Table of Contents for this issue

kind of sample preparation was used. The pH of workingsolutions were kept below 3.0 by addition of a small amountof perchloric acid solution, and this procedure was used tohave only the protonated form of the acridine dye in groundand excited state and hence a monoexponential decay.16
Absorption measurements were performed on a HitachiU-2000 spectrophotometer. Corrected steady-state Ñuores-cence spectra were recorded on a CD-900 Edinburgh spectro-Ñuorimeter. Fluorescence decays were measured with asingle-photon timing technique using a CD-900 Edinburghspectrometer operating with a hydrogen-Ðlled nanosecondÑash lamp at 25È30 kHz pulse frequency range. All Ñuores-cence measurements were performed with samples thermalizedat 298 K, and in air equilibrated conditions with an excitationwavelength at 352 nm. Exponential and micellar quenchingmodels (vide infra) were used to analyze the decays by a non-linear least-squares iterative routine based on the Marquardtalgorithm. The software for data analysis was supplied byEdinburgh Instruments.
Acridine triplet transients were monitored by the laserÑash-photolysis technique. A Nd-YAG laser (Spectron SL401) operating at 355 nm by frequency conversion was usedfor excitation. Triplet absorption transients were determinedwith an Applied Photophysics laser kinetic spectrometer.Transients were recorded and averaged on a HP 54504A digi-tizing oscilloscope before data treatment in a PC micro-computer.
Results and Discussion(i) Singlet and triplet quenching in homogeneous phase
The Ñuorescence decay of the protonated acridine in methanolis monoexponential with a lifetime of 27.1^ 0.2 ns at 298 K.This value is close to the lifetime of 29.7 ns of the dye in watersolution at pH 3 measured by Medeiros et al.16 The Ñuores-cence quenching of the dye by vinyl monomers was investi-gated by time-resolved and photostationary measurements. Itis well known that cationic dyes are good electron acceptorsand they can form ground-state charge-transfer complexeswith electron donors especially with aromatic compounds.17Thus, the quenching process of acridine by the styrene deriv-atives could have a static contribution, which combined witha dynamic quenching process could result in a non-linear andupper curvature of the relative Ñuorescence intensity withquencher concentration, mismatching the data of the ratio ofdecay times. However, for the systems studied, static quen-ching is ruled out because the plot of relative intensity withquencher concentration is linear and it Ðts very well the life-time ratios. Fig. 1 shows the linear SternÈVolmer plots of therelative Ñuorescence intensity and the ratio of lifetimes as afunction of quencher concentration for some of the systemsstudied.
Fig. 1 Fluorescence quenching of acridine by divinylbenzene (=,values ; values) ; a-methylstyrene values) and acryl-q0/q L, I0/I (|, I0/Iamide values)(…, I0/I
Triplet absorption transients of acridine in methanol wereobserved at 430 nm. The triplet lifetime calculated was 17.5 lsin absence of monomer. The second-order triplet quenchingrate constants were determined from the plot of the measuredpseudo-Ðrst-order rate constant as a function of monomerconcentration. The calculated values of the singlet and tripletquenching rate constants for the series of vinyl monomersstudied are given in Table 1. For some of the monomers, liter-ature values of the oxidation potential are available.¤ Theabsence of a more complete set of values precludes an analysisbased on the Rehm and Weller approach of photoinducedelectron transfer. Nevertheless, the decrease in quenching effi-ciency with an increase in the oxidation potentials of the vinylmonomer as observed, indicates that singlet and triplet quen-ching may occur via an exciplex mechanism and photoinducedcharge transfer. However, no appreciable photoblenching ofthe dye upon pulsed or constant light was observed. In astudy of the photoreduction of several di†erent dyes bystyrene, Timpe and Neuenfeld19 observed very weak photo-blenching quantum yields of acridine yellow and acridineorange in methanol (U\ 1.0] 10~4 and 2.6 ] 10~4,respectively) but working at very high styrene concentrationof 5 M. In methanol, styrene derivatives quench the singletexcited state of acridine with rate constants close to thedi†usion-controlled limit, but the quenching of the tripletexcited state is not so fast, which is a common trend in elec-
¤ Monomer oxidation potential volts in methanol : 1.44, 1.67,[email protected] and 2.00 for VNBE, V2Prl, MSty and Sty ;18a 2.15 and 3.64 forVAc and Acln.18b
Table 1 Singlet and triplet quenching rate constants of acridine by vinyl monomers in methanol and SDS aqueous micelles at 298 Ka
singlet (SDS)
kq/s~1 k~/s~1singlet (MeOH) triplet (MeOH)
kq/M~1 s~1 kq/M~1 s~1
VBC (1.47^ 0.07)] 1010 3.97] 107 \106 (9.48^ 0.23)] 108DVB (1.60^ 0.05)] 1010 5.09] 107 \106 (2.65^ 0.50)] 107MSty (1.13 ^ 0.03)] 1010 6.73] 107 1.58] 106 ÈSty (8.50 ^ 0.16)] 109 5.23] 107 2.10] 106 (5.76^ 1.03)] 105VNBE (1.36^ 0.04)] 1010 6.55] 107 7.75] 106 ÈV2Prl (1.22^ 0.04)] 1010 4.52] 107 2.57] 107 (1.58^ 0.18)] 107Aclmd (2.83^ 0.08)] 109VAc (1.56^ 0.15)] 108Acln (1.46^ 0.06)] 108MMA (9.24^ 1.22)] 106
a Acridine cation excited singlet lifetimes : 27.1^ 0.2 ns (MeOH), 32.7^ 0.2 ns (SDS micelles). is the exit rate constant of a monomer from ak~micelle (see text for details).
1134 J. Chem. Soc., Faraday T rans., 1997, V ol. 93
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online
tron transfer reactions where usually the excited singlet stateshows a higher reactivity than the triplet state.20
(ii) Kinetics of micellar Ñuorescence quenching21
The Ñuorescence quenching of acridine by vinyl monomers inaqueous and reverse micellar phase of SDS is now studied bymeans of time-resolved and photostationary measurements.Fluorescence transients are analyzed with micellar quenchingmodels in order to determine the intramicellar quenching rateconstant, and the monomer exit rate constant from akq ,micelle, Relative Ñuorescence quenching data is used tok~ .estimate association constants of vinyl monomers to aqueousSDS micelles using the EncinasÈLissi method.15
Several theoretical and experimental studies of Ñuorescencequenching in micellar systems have been reported in the lastfew years.21 Depending on the time range of the Ñuorescenceevent, which is dictated by the probeÏs lifetime, several di†er-ent models can be used to explain the decay kinetics. In thepresent studies, intermicellar mobility of the probe isneglected. The Ñuorescence decay of acridine in aqueous andreverse micelles of SDS is monoexponential with lifetimes ofabout 33 and 25 ns, respectively. This cationic dye binds verystrongly to aqueous SDS micelles due to electrostatic andhydrophobic factors. When the dye is dispersed in a reversemicellar solution, its very low solubility in apolar media con-Ðnes the probe to the water pool of the reverse micelle. Atmoderate micellar concentration, the frequency of inter-micellar exchange of the probe via micellar collisions islow,22,23 and therefore the micellar residence time of the acri-dine probe is much higher than its excited singlet lifetime. Insuch situations, the probe can be considered as an immobilespecies trapped in the same micelle during its excited-state life-time.
Aqueous micelles. Among the di†erent monomers used asquenchers, three kinds of behaviour related to the quencherintermicellar mobility during the Ñuorescence decay of acri-dine in SDS aqueous solution were identiÐed. In the case ofhydrophobic and low water soluble monomers like VBC andDVB, Ñuorescence decay analysis reveals that these quenchersare immobile species during the Ñuorescence event. Such aconclusion is supported by the independence of the factorA2with quencher concentration which means that the decay rateat long times is of the order of the self-decay rate constantk0 ;of the acridine in the absence of added quencher [vide eqn.(1)]. Fig. 2 shows the decay proÐles of acridine in SDSaqueous micelles at di†erent concentrations of DVB. Decays
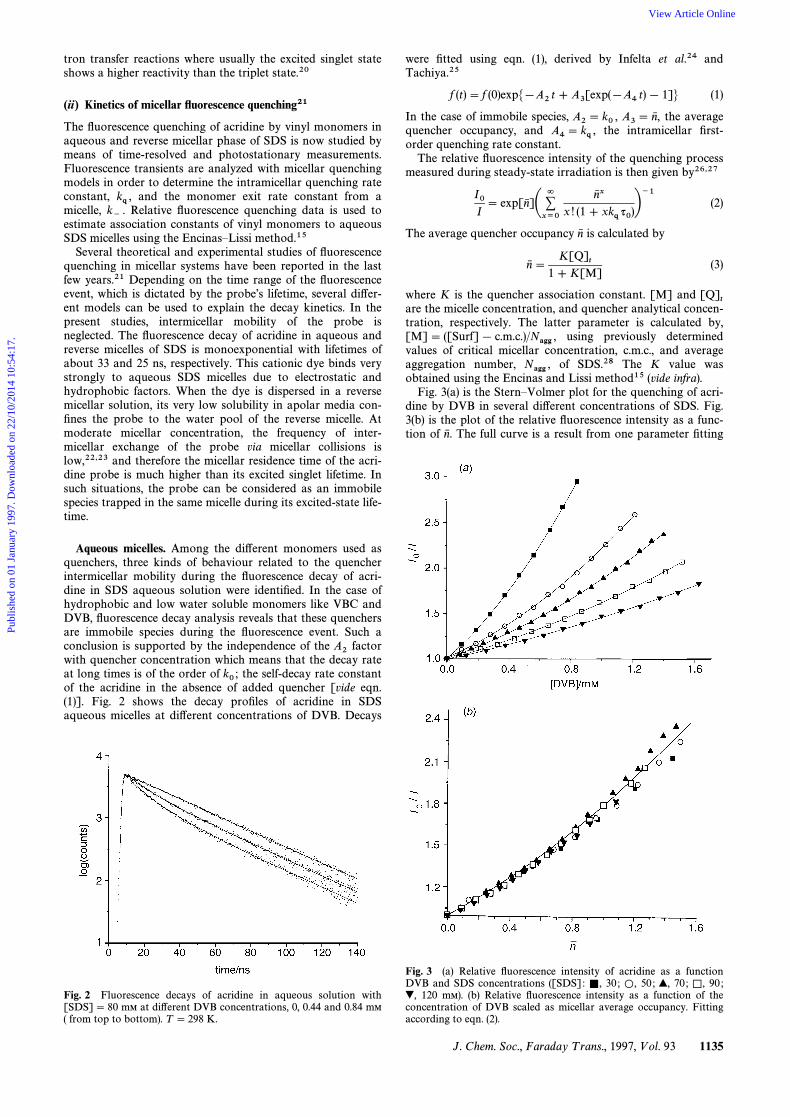
Fig. 2 Fluorescence decays of acridine in aqueous solution with[SDS]\ 80 mM at di†erent DVB concentrations, 0, 0.44 and 0.84 mM
( from top to bottom). T \ 298 K.
were Ðtted using eqn. (1), derived by Infelta et al.24 andTachiya.25
f (t) \ f (0)expM[A2 t ] A3[exp([A4 t)[ 1]N (1)
In the case of immobile species, the averageA2 \ k0 , A3\ n6 ,quencher occupancy, and the intramicellar Ðrst-A4 \ kq ,order quenching rate constant.
The relative Ñuorescence intensity of the quenching processmeasured during steady-state irradiation is then given by26,27
I0I
\ exp[n6 ]A
;x/0
= n6 xx ! (1] xkq q0)
B~1(2)
The average quencher occupancy is calculated byn6
n6 \K[Q]
t1 ] K[M]
(3)
where K is the quencher association constant. [M] and [Q]tare the micelle concentration, and quencher analytical concen-
tration, respectively. The latter parameter is calculated by,using previously determined[M]\ ([Surf][ c.m.c.)/Nagg ,
values of critical micellar concentration, c.m.c., and averageaggregation number, of SDS.28 The K value wasNagg ,obtained using the Encinas and Lissi method15 (vide infra).
Fig. 3(a) is the SternÈVolmer plot for the quenching of acri-dine by DVB in several di†erent concentrations of SDS. Fig.3(b) is the plot of the relative Ñuorescence intensity as a func-tion of The full curve is a result from one parameter Ðttingn6 .
Fig. 3 (a) Relative Ñuorescence intensity of acridine as a functionDVB and SDS concentrations ([SDS] : 30 ; 50 ; 70 ; 90 ;=, L, >, K,
120 mM). (b) Relative Ñuorescence intensity as a function of the@,concentration of DVB scaled as micellar average occupancy. Fittingaccording to eqn. (2).
J. Chem. Soc., Faraday T rans., 1997, V ol. 93 1135
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online
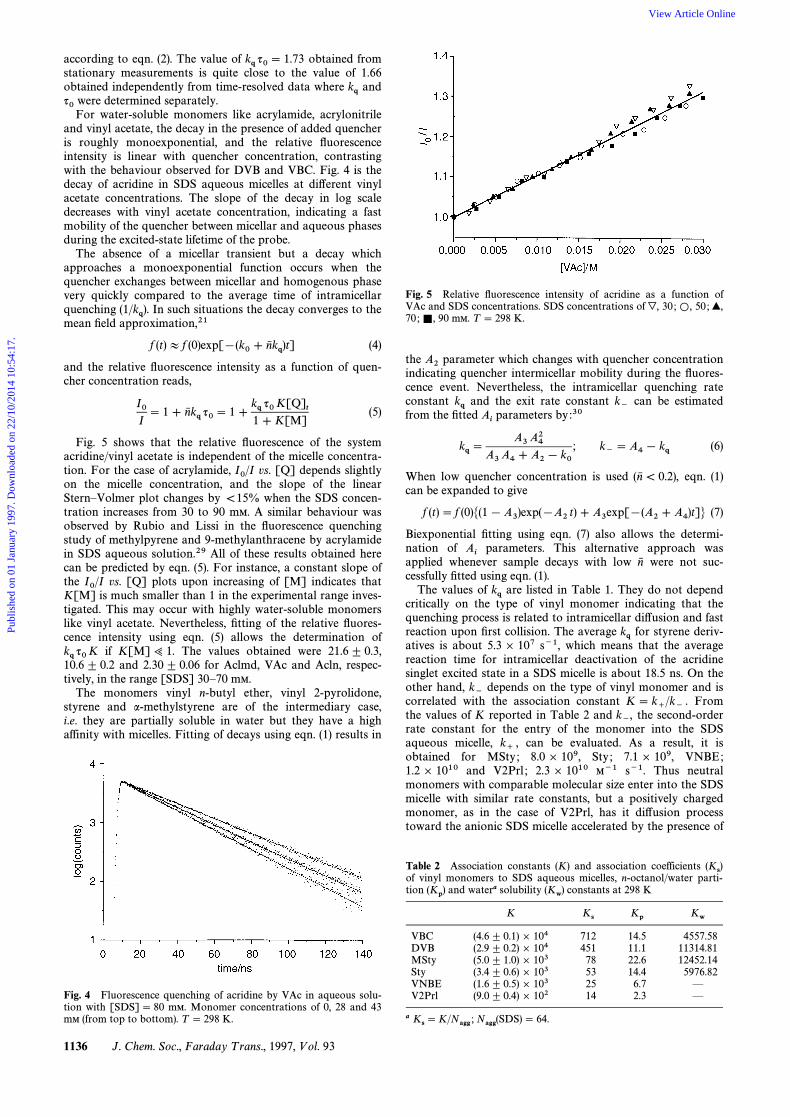
according to eqn. (2). The value of obtained fromkq q0\ 1.73stationary measurements is quite close to the value of 1.66obtained independently from time-resolved data where andkqwere determined separately.q0For water-soluble monomers like acrylamide, acrylonitrileand vinyl acetate, the decay in the presence of added quencheris roughly monoexponential, and the relative Ñuorescenceintensity is linear with quencher concentration, contrastingwith the behaviour observed for DVB and VBC. Fig. 4 is thedecay of acridine in SDS aqueous micelles at di†erent vinylacetate concentrations. The slope of the decay in log scaledecreases with vinyl acetate concentration, indicating a fastmobility of the quencher between micellar and aqueous phasesduring the excited-state lifetime of the probe.
The absence of a micellar transient but a decay whichapproaches a monoexponential function occurs when thequencher exchanges between micellar and homogenous phasevery quickly compared to the average time of intramicellarquenching In such situations the decay converges to the(1/kq).mean Ðeld approximation,21
f (t) B f (0)exp[[(k0 ] n6 kq)t] (4)
and the relative Ñuorescence intensity as a function of quen-cher concentration reads,
I0I
\ 1 ] n6 kq q0\ 1 ]kq q0K[Q]
t1 ] K[M]
(5)
Fig. 5 shows that the relative Ñuorescence of the systemacridine/vinyl acetate is independent of the micelle concentra-tion. For the case of acrylamide, vs. [Q] depends slightlyI0/Ion the micelle concentration, and the slope of the linearSternÈVolmer plot changes by \15% when the SDS concen-tration increases from 30 to 90 mM. A similar behaviour wasobserved by Rubio and Lissi in the Ñuorescence quenchingstudy of methylpyrene and 9-methylanthracene by acrylamidein SDS aqueous solution.29 All of these results obtained herecan be predicted by eqn. (5). For instance, a constant slope ofthe vs. [Q] plots upon increasing of [M] indicates thatI0/IK[M] is much smaller than 1 in the experimental range inves-tigated. This may occur with highly water-soluble monomerslike vinyl acetate. Nevertheless, Ðtting of the relative Ñuores-cence intensity using eqn. (5) allows the determination of
if K[M]> 1. The values obtained were 21.6^ 0.3,kq q0K10.6^ 0.2 and 2.30 ^ 0.06 for Aclmd, VAc and Acln, respec-tively, in the range [SDS] 30È70 mM.
The monomers vinyl n-butyl ether, vinyl 2-pyrolidone,styrene and a-methylstyrene are of the intermediary case,i.e. they are partially soluble in water but they have a highaffinity with micelles. Fitting of decays using eqn. (1) results in
Fig. 4 Fluorescence quenching of acridine by VAc in aqueous solu-tion with [SDS]\ 80 mM. Monomer concentrations of 0, 28 and 43mM (from top to bottom). T \ 298 K.
Fig. 5 Relative Ñuorescence intensity of acridine as a function ofVAc and SDS concentrations. SDS concentrations of 30 ; 50 ;È, L, >,70 ; 90 mM. T \ 298 K.=,
the parameter which changes with quencher concentrationA2indicating quencher intermicellar mobility during the Ñuores-cence event. Nevertheless, the intramicellar quenching rateconstant and the exit rate constant can be estimatedkq k~from the Ðtted parameters by :30A
i
kq \A3 A42
A3 A4 ] A2 [ k0; k~\ A4 [ kq (6)
When low quencher concentration is used eqn. (1)(n6 \ 0.2),can be expanded to give
f (t) \ f (0)M(1 [ A3)exp([A2 t) ] A3exp[[(A2] A4)t]N (7)
Biexponential Ðtting using eqn. (7) also allows the determi-nation of parameters. This alternative approach wasA
iapplied whenever sample decays with low were not suc-n6cessfully Ðtted using eqn. (1).
The values of are listed in Table 1. They do not dependkqcritically on the type of vinyl monomer indicating that thequenching process is related to intramicellar di†usion and fastreaction upon Ðrst collision. The average for styrene deriv-kqatives is about 5.3] 107 s~1, which means that the averagereaction time for intramicellar deactivation of the acridinesinglet excited state in a SDS micelle is about 18.5 ns. On theother hand, depends on the type of vinyl monomer and isk~correlated with the association constant FromK \ k
`/k~ .
the values of K reported in Table 2 and the second-orderk~,rate constant for the entry of the monomer into the SDSaqueous micelle, can be evaluated. As a result, it isk
`,
obtained for MSty ; 8.0] 109, Sty ; 7.1] 109, VNBE;1.2] 1010 and V2Prl ; 2.3 ] 1010 M~1 s~1. Thus neutralmonomers with comparable molecular size enter into the SDSmicelle with similar rate constants, but a positively chargedmonomer, as in the case of V2Prl, has it di†usion processtoward the anionic SDS micelle accelerated by the presence of
Table 2 Association constants (K) and association coefficients (Ks)of vinyl monomers to SDS aqueous micelles, n-octanol/water parti-tion and watera solubility constants at 298 K(Kp) (Kw)
K Ks Kp KwVBC (4.6^ 0.1)] 104 712 14.5 4557.58DVB (2.9^ 0.2)] 104 451 11.1 11314.81MSty (5.0 ^ 1.0)] 103 78 22.6 12452.14Sty (3.4 ^ 0.6)] 103 53 14.4 5976.82VNBE (1.6^ 0.5)] 103 25 6.7 ÈV2Prl (9.0^ 0.4)] 102 14 2.3 È
a Ks\K/Nagg ; Nagg(SDS)\ 64.
1136 J. Chem. Soc., Faraday T rans., 1997, V ol. 93
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online

the attractive potential and its entry rate constant becomeslarger than of neutral species.
The association constants of the vinyl monomers (VBC, Sty,MSty, DVB) to SDS aqueous micelles were determined usingthe method of Encinas and Lissi.15 In order to evaluate thehydrophobic contribution as well as the speciÐc site e†ect onthe binding process of monomers to SDS micelles, partitionconstants in the water/n-octanol system and water solubilityof the monomer were determined. To obtain such parameters,partition and solubility experiments were carried out at 298K, and the amounts of monomer in water and in n-octanolwere determined by measuring the Ñuorescence quenchingratio when aliquots of the phases were added to 3 ml ofI0/Iacridine methanol solution. In this procedure, the previouslydetermined SternÈVolmer equation of each monomer wasused as a calibration curve. Table 2 reports the association,partition and solubility constants for the vinyl monomersstudied. The solubility data shows no clear correlationbetween SDS/water and n-octanol/water partition constants.This fact indicates that micellar solubilization is not entirelygoverned by hydrophobic contribution but some speciÐc sitee†ect may be present in some cases. On the other hand, theratio of solubility data of methylstyrene and styrene gives afactor about 1.5 for and which may be ascribed to aKs Kp ,similar free energy change in the partition between water andapolar phase upon addition of a methyl group, and in thiscase absence of any additional speciÐc site e†ect on the micel-lar solubilization. The association constants determined forstyrene derivatives are within the range found for arenes. Forbenzene, toluene and p-xylene in SDS micellar solution (0.05M) the values of K are (1.6, 5.3, 16)] 103 M~1, respectively.31
Following the work of Almgren et al.31 association con-stants are correlated with the boiling points of the vinylmonomers. The empirical correlation observed (see Fig. 6)may be used as a guide in estimating other association con-stants which have not been measured. However, a moreprecise determination of association constants of organicsolutes to aqueous micelles can be obtained by using a linearsolvation free-energy relationship analysis as described recent-ly by Quina et al.32
Reverse micelles. SDS surfactant does not form a stablereverse system unless a cosurfactant is added. In the presentstudies, the reverse micellar solution of SDS in cyclohexanewas formed with 0.1 M of surfactant, 0.25 M of hexanol as co-surfactant, and the water to surfactant molar ratio was keptapproximately at 10. The average aggregation number, Nagg ,of the reverse SDS micelles was determined from time-resolved Ñuorescence quenching using acridine dye as probe
Fig. 6 Association constants of vinyl monomers to SDS micelles asa function of the boiling point
and Cu2` as quencher. The value of found in the com-Naggposition used was 430 ^ 30.The Ñuorescence decay of the acridine cation in SDS reverse
micellar solution is monoexponential with a lifetime of 24.6 ns.The quenching of acridine by styrene derivatives in reversemicelles follows a pseudo Ðrst-order process and no micellartype of transient is observed. Decay proÐles of acridine in SDSreverse micelles at di†erent MSty concentrations are shown inFig. 7. The linear SternÈVolmer plots in the case of Sty andMSty in SDS reverse micellar solutions are presented in Fig.8.
Considering the low water solubility of the styrene deriv-atives, the intramicellar quenching process should involve anencounter of excited probe and quencher on the surface regionof the reverse micelle delimited by the surfactant stabilizedwater nanodroplet. In such cases, the quenching rate may be afunction of the surface concentration of the quencher, [Q]s ,being written as
d[P*]
dt\ kqs[Q]s[P*] (8)
where is the bimolecular quenching rate constant on thekqsmicellar surface.
Fig. 7 Fluorescence decay proÐles of acridine in SDS reverse micel-lar solution in the absence and presence of a-methylstyrene. Concen-trations of 0, 8 and 17 mM (from top to bottom). Exponential Ðttingaccording to eqn. (9).
Fig. 8 SternÈVolmer plot of the Ñuorescence quenching of acridineby styrene (Sty) and a-methylstyrene (MSty) in reverse micellar solu-tions of SDS. and values) ; and values).= … (I0/I | K (q0/q
J. Chem. Soc., Faraday T rans., 1997, V ol. 93 1137
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online
Eqn. (8) holds if the two-dimensional di†usion transient onthe surface is very short lived and exchange of quencherbetween micellar surface and homogeneous phase is fast. Con-sidering a photophysical deactivation of no more than oneexcited probe per micelle, the radial distribution of monomerfrom the micelle surface to bulk remains practically at equi-librium, then is related to the average concentration in[Q]ssolution, [Q], by a Boltzmann factor whereexp[[V0/kT ], V0is the potential energy of the quencher on the micellar surface(potential is considered zero in solution). Introducing this rela-tion into eqn. (8), followed by integration results in an expo-nential decay given by
f (t) \ f (0)exp[[(k0 ] kqr[Q])t] (9)
where is the observed quenching ratekqr\ kqs exp([V0/kT )constant.
Table 3 shows the values of for styrene derivatives. Thekqrvalues found are within the range of (4È9) ] 108 M~1 s~1,which are 10È30 times lower than the quenching rate con-stants reported in methanol. This clearly shows that placingthe dye photoinitiator inside a reverse micelle reduces sub-stantially the quenching rate by the hydrophobic vinyl mono-mers.
(iii) Photoinitiation efficiency
A complete description of the photoinitiation efficiency woulddepend on the kinetic scheme for excited state dye (D*) deacti-vation (singlet and/or triplet) and radical formation pathwaysin the presence of a co-initiator and monomer (m). If activeradicals are formed from a singlet exciplex, the mechanismwould be :
D ] hl ÈÈÈÕ D* (10)
k0D* ÈÈÈÕ D (11)
kq1D* ] CoI ÈÈÈÕ [DÉ É ÉCoI]* ÈÈÈÕ D ] CoI (12)B
active radicals
kq2D* ] m ÈÈÈÕ D ] m (13)
From the above mechanism [eqn. (10)È(13)], the radicalquantum yield, will be given by eqn. (14) :Ur ,
Ur \bkq1q0[CoI]
1 ] kq1q0[CoI]] kq2 q0[m](14)
where b stands for the fraction of excited complex that decom-poses to active radicals. and are the singlet lifetime,q0 , kq1 kq2and quenching rate constants by co-initiator and monomer,respectively.
If the singlet excited state of the dye initiator is short lived,a photoredox reaction may proceed through the triplet excitedstate and the radical quantum yield will be given approx-imately by the product of eqn. (14) and the intersystem cross-
Table 3 Second-order quenching rate constants of the acridinesinglet excited-state by vinyl monomers in SDSÈhexanol reversemicelles in cyclohexane at 298 Ka
kqr/10~8 M~1 s~1
DVB 5.63^ 0.20VBC 4.77^ 0.20MSty 9.31 ^ 0.30Sty 5.2 ^ 0.10VNBE 3.38^ 0.20
a Acridine singlet excited-state lifetime ns.qs \ 24.60^ 0.2
ing quantum yield in the absence of co-initiator andmonomer. In this situation, the kinetic parameters of eqn. (14)are related to the dye-triplet photochemistry. In several photo-initiating systems, a dye triplet is the reactive species whichgenerates radicals from a redox process with a co-initiator.33
Eqn. (14) applies to an homogeneous solution. In a reversemicellar solution where the dye initiator and co-initiator areconÐned to the water domain but the hydrophobic monomerstands apart in the apolar media, the radical quantum yield,considering monodisperse micellar conÐnements, single dyeoccupancy but multiple co-initiator occupancy, and absenceof exchange of reactant among micelles, may be evaluated by :
Urm \ b@ ;x
xkq1m q0 Px
1 ] xkq1m q0 ] kqr q0[m](15)
where is the probability of Ðnding a micelle with x co-Pxinitiator molecules, which in the ideal solubilization follows a
Poisson distribution with average co-initiator occupancygiven by (for a co-initiator insoluble in thex6 \ [CoI]/[M]apolar phase). In eqn. (15), the reaction rate, like in the case ofintramicellar Ñuorescence quenching, is assumed to be pro-portional to the occupancy number x. b@ has the samemeaning as b, but now including the e†ect of radical recombi-nation in the micelle cage. and are the intramicellarkq1m kqrquenching rate constant by the co-initiator, and the second-order quenching rate constant by the monomer, respectively.
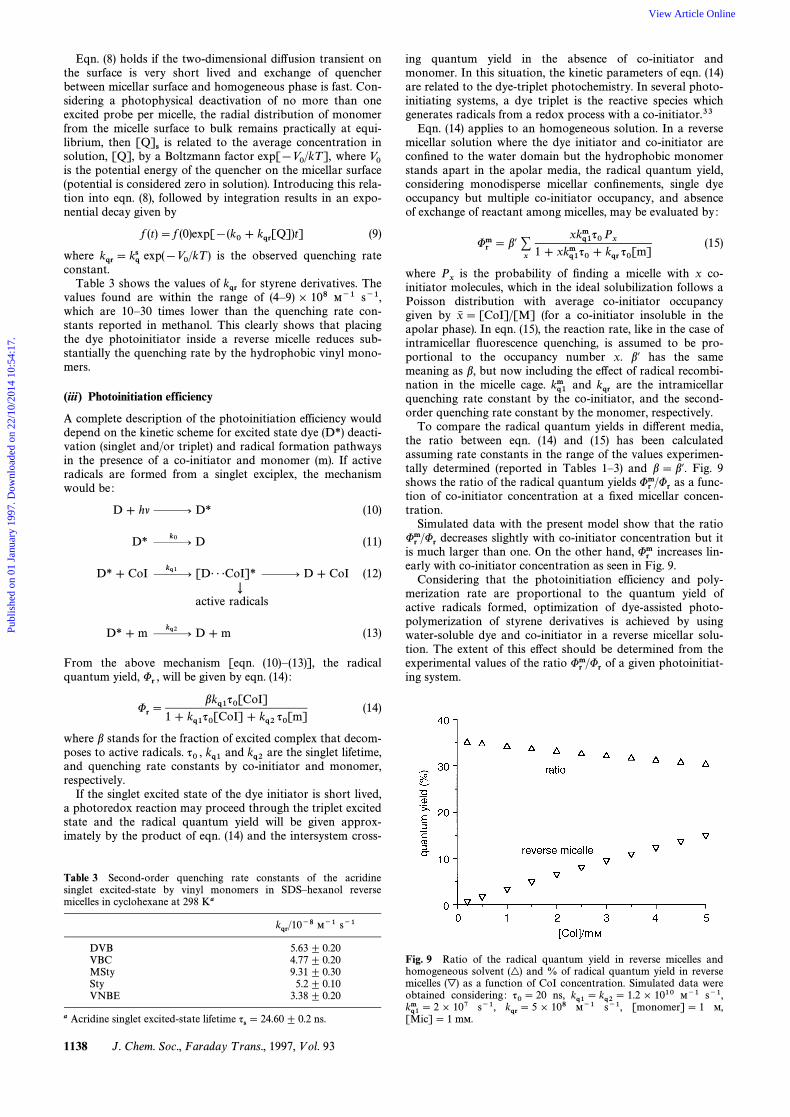
To compare the radical quantum yields in di†erent media,the ratio between eqn. (14) and (15) has been calculatedassuming rate constants in the range of the values experimen-tally determined (reported in Tables 1È3) and b \ b@. Fig. 9shows the ratio of the radical quantum yields as a func-Urm/Urtion of co-initiator concentration at a Ðxed micellar concen-tration.
Simulated data with the present model show that the ratiodecreases slightly with co-initiator concentration but itUrm/Uris much larger than one. On the other hand, increases lin-Urmearly with co-initiator concentration as seen in Fig. 9.
Considering that the photoinitiation efficiency and poly-merization rate are proportional to the quantum yield ofactive radicals formed, optimization of dye-assisted photo-polymerization of styrene derivatives is achieved by usingwater-soluble dye and co-initiator in a reverse micellar solu-tion. The extent of this e†ect should be determined from theexperimental values of the ratio of a given photoinitiat-Urm/Uring system.
Fig. 9 Ratio of the radical quantum yield in reverse micelles andhomogeneous solvent and % of radical quantum yield in reverse(|)micelles as a function of CoI concentration. Simulated data were(È)obtained considering : ns, M~1 s~1,q0\ 20 kq1 \ kq2 \ 1.2] 1010
s~1, M~1 s~1, [monomer]\ 1 M,kq1m \ 2 ] 107 kqr \ 5 ] 108[Mic]\ 1 mM.
1138 J. Chem. Soc., Faraday T rans., 1997, V ol. 93
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online

Conclusions and general remarksIn this paper, the excited-state quenching of acridine by vinylmonomer in homogeneous (methanol) and in micro-heterogeneous (SDS micelles) systems is investigated withattention to the e†ect of the excited-state dye deactivation asan important problem in photoinitiation of addition poly-merization.
Association constants of vinyl monomers to SDS aqueousmicelles have been determined by using the Ñuorescence quen-ching method and for styrene derivatives they are within therange of 103È104 M~1. The high affinities of the cationic dyeand monomers to SDS aqueous micelles as well as the veryefficient Ñuorescence quenching of the excited dye by vinylmonomers given no active radicals predicts a poor photoini-tiation quantum yield in addition photopolymerization inaqueous micelle or o/w emulsion of anionic surfactants. Fluo-rescence quenching is also very high in a homogeneous phaselike methanol which makes it difficult for the use of commonsolvents for dye-assisted photopolymerization of styrenederivatives.
On the other hand, in a reverse micellar solution the quen-ching process is substantially reduced when the dye photoini-tiator is conÐned to the water domain and the hydrophobicmonomer is solubilized apart in the apolar media. The use ofa water-soluble co-initiator in a photoredox reaction with awater-soluble dye in a reverse micelle may improve the effi-ciency of the photoinitiation step by reducing the competitivequenching by the monomer.
Co-initiators like arylsulÐnates are water soluble and theirphotoredox processes with cationic dyes generate active rad-icals with no liquid charge34,35 which should migrate to theapolar media to initiate the addition polymerization of vinylhydrophobic monomers. Preliminary results show that addi-tion photopolymerization of styrene using acridine dyes/arylsulÐnates in reverse micelles of AOT in hexane occurswith much higher conversion than when the same process iscarried out in an homogenous solvent.36
support by CNPq and FAPESP (Brazil) is gratefullyFinancialacknowledged. S.F.B. thanks CAPES and FAPESP for agraduate fellowship.
References1 M. S. El-Aasser, in An Introduction to Polymer Colloids, ed. F.
Candau and R. H. Ottewill, Kluwer Academic Publishers, Dor-drecht, 1990, p. 1.
2 F. Candau, in An Introduction to Polymer Colloids, ed. F. Candauand R. H. Ottewill, Kluwer Academic Publishers, Dordrecht,1990, p. 73.
3 Q. Wang, S. Fu and T. Yu, Prog. Polym. Sci., 1994, 19, 703.4 A. Jayakrisman and D. O. Shah, J. Polym. Sci., Polym. L ett. Ed.,
1984, 22, 31.5 V. Vaskova� , V. Juranicova� and J. Barton, Makromol. Chem.,
1990, 191, 717 ; V. Vaskova� , V. Juranicova� and J. Barton, Makro-mol. Chem., 1991, 192, 989 ; V. Vaskova� , V. Juranicova� and J.Barton, Makromol. Chem., 1991, 192, 1339.
6 V. Vaskova� , Zuzanz Hlouskova, J. Barton and V. Juranicova� ,Makromol. Chem., 1992, 193, 717.
7 H. F. Gruber, Prog. Polym. Sci., 1992, 17, 953.8 Ping-Lin Kuo, N. J. Turro, Chi-Ming Tseng, M. S. El-Aasser and
J. W. Vanderho†, Macromolecules, 1987, 20, 1216.9 D. Cochin, F. Candau and R. Zana, Macromolecules, 1993, 26,
5755.10 D. Cochin, R. Zana and F. Candau, Macromolecules, 1993, 26,
5765.11 C. M. Previtali, S. G. Bertolotti, M. G. Neumann, I. A. Pastre,
A. M. Rufs and M. V. Encinas, Macromolecules, 1994, 27, 7454.12 J. P. Fouassier, P. Jacques, D. J. Lougnot and T. Pilot, Polym.
Photochem., 1984, 5, 57.13 P. K. Seugupta and S. K. Modak, Makromol. Chem., 1985, 186,
1593.14 L. Sepulveda, E. A. Lissi and F. H. Quina, Adv. Colloid Interface
Sci., 1986, 25, 1.15 M. V. Encinas and E. A. Lissi, Chem. Phys. L ett., 1982, 91, 55.16 G. M. M. Medeiros, M. F. Leita8 o and S. M. B. Costa, J. Photo-
chem. Photobiol. A: Chem., 1993, 72, 225.17 M. G. Neumann, F. Gessner, A. R. Souza and C. C. Schmitt, J.
Photochem. Photobiol. A: Chem., 1988, 45, 355.18 (a) H. Siegermann, in Oxidation and Reduction Half-wave Poten-
tials of Organic Compounds, T echnique of Electroorganic SynthesisPart II, Techniques of Chemistry, ed. N. L. Weinberg, JohnWiley & Sons, 1975, vol. V, p. 667 ; (b) R. A. Caldwell, D. Creed,D. C. De Marco, L. A. Melton, H. Ohta and P. M. Wine, J. Am.Chem. Soc., 1980, 102, 2369.
19 H. J. Timpe and S. Neuenfeld, J. Chem. Soc., Faraday T rans.,1992, 88, 2329.
20 E. Volgelmann, W. Rauscher, R. Traber and H. E. A. Kramer, Z.Phys. Chem. Neue Folge, 1981, 124, 13.
21 M. H. Gehlen and F. C. De Schryver, Chem. Rev., 1993, 93, 199.22 P. D. I. Fletcher, A. M. Howe and B. H. Robinson, J. Chem. Soc.,
Faraday T rans. 1, 1987, 83, 985.23 P. D. I. Fletcher and D. I. Horsup, J. Chem. Soc., Faraday T rans.
1992, 88, 855.24 P. P. Infelta, M. Gra� tzel and J. K. Thomas, J. Phys. Chem., 1974,
78, 190.25 M. Tachiya, Chem. Phys. L ett., 1975, 33, 289.26 A. Henglein and R. Scheerer, Ber. Bunsen-Ges. Phys. Chem., 1978,
82, 1007.27 P. P. Infelta, Chem. Phys. L ett., 1978, 61, 88.28 M. H. Gehlen and F. C. De Schryver J. Phys. Chem., 1993, 97,
11242.29 M. A. Rubio and E. A. Lissi, J. Photochem. Photobiol. A: Chem.,
1993, 71, 175.30 A. Malliaris, J. Lang and R. Zana, J. Chem. Soc., Faraday T rans.
1, 1986, 82, 109.31 M. Almgren, F. Grieser and J. K. Thomas, J. Am. Chem. Soc.,
1979, 101, 279.32 F. H. Quina, E. O. Alonso and J. P. S. Farah, J. Phys. Chem.,
1995, 99, 11708.33 H. J. Timpe, S. Jockusch and K. Ko� rner, in Radiation Curing in
Polymer Science and T echnology, ed. J. P. Fouassier and J. F.Rabek, Elsevier, London, 1993, vol. 2, p. 575.
34 J. D. Margerum, A. M. Lackner, M. J. Little and C. T. Petrusis,J. Phys. Chem., 1971, 75, 3066.
35 D. F. Eaton, in Adv. Photochem., 1986, 13, 427.36 S. F. Buchvieser and M. H. Gehlen, Proc. Eighth Inter. Am.
Photochem. Soc. Conf., Foz do 1996, p. 6.IguacÓ u,
Paper 6/05032H; Received 18th July, 1996
J. Chem. Soc., Faraday T rans., 1997, V ol. 93 1139
Publ
ishe
d on
01
Janu
ary
1997
. Dow
nloa
ded
on 2
2/10
/201
4 10
:54:
17.
View Article Online





























