Embed Size (px)
Citation preview

Quantum-Mechanical Investigations of Unstable Intermediates Relevant to the Mechanism of
Chemical Carcinogenesis by N- A1 k y lni trosamines
COLIN THOMSON, DAVID PROVAN, AND SUSAN CLARK Department of Chemistry, Uniuersity of St. Andrews, St. Andrews. Scotland
Abstract
A6 inifio LCAO-MO-SCF calculations on the transformation of nitroxylamide to hydroxydiimide, and of monomethylnitrosamine to the corresponding diazohydroxide are reported. A large barrier was obtained in both cases, and this was lowered by -1 0% when the molecules were allowed to in- teract with a single water molecule. I t was concluded that it is more likely that the diazohydroxide arises from the a-hydroxy derivative rather than from the monomethylnitrosamine. The relevance of these investigations to the mechanism of carcinogenesis by N-nitrosamines is discussed.
Introduction
N-Nitrosamines have been extensively studied with respect to both their carcinogenic and mutagenetic effects during the last twenty or so years,* fol- lowing the initial observation of Magee and Barnes [2] that dimethylnitrosamine (DMN) produced a high incidence of hepatomas when fed to rats, and this field has been extensively reviewed by Magee and Barnes [3] and others [5-71.
More recent evidence has led to the suggestion that N-nitrosamines are im- portant as environmental carcinogens [8,9], and they may well be implicated in various types of human cancer [ 101. There is thus an urgent need to under- stand their mode of action in in uiuo situations, and much research has been carried out during the last few years with this object in mind. Despite much progress, there remain a number of unresolved questions concerning the mechanism of carcinogenesis by these compounds.
It was established by Magee [ 1 1,3], and Dutton and Heath [ 121 that DMN requires metabolic activation in order to cause tumours, and a number of elegant studies have shown that this activation is carried out by the microsomal enzyme system known as the mixed function oxidases which are located primarily, but not exclusively, in the liver [ 13, 141. As a result of this activation the nitrosamines break down and give rise to intermediates which are able to alkylate nucleic acids and proteins [3, 14, 151 and possibly other cellular macromolecules. However, the exact toxicological consequences of alkylation are by no means clear, par- ticularly since the extent of alkylation of nucleic acid bases in various organs of a given test organism with several different nitroso compounds did not cor- relate with the predominant sites of tumour localization [ 16, 171.
Despite this uncertainty, it is clearly important that the metabolism of the
* Heidelberger [ I ] contains a comprehensive bibliography on chemical carcinogenesis.
International Journal of Quantum Chemistry: Quantum Biology Symposium 4.205-21 5 (1977) Q 1977 by John Wiley & Sons, Inc. 205
206 THOMSON, PROVAN, AND CLARK
C H’ 3 1
CH&N + OH-
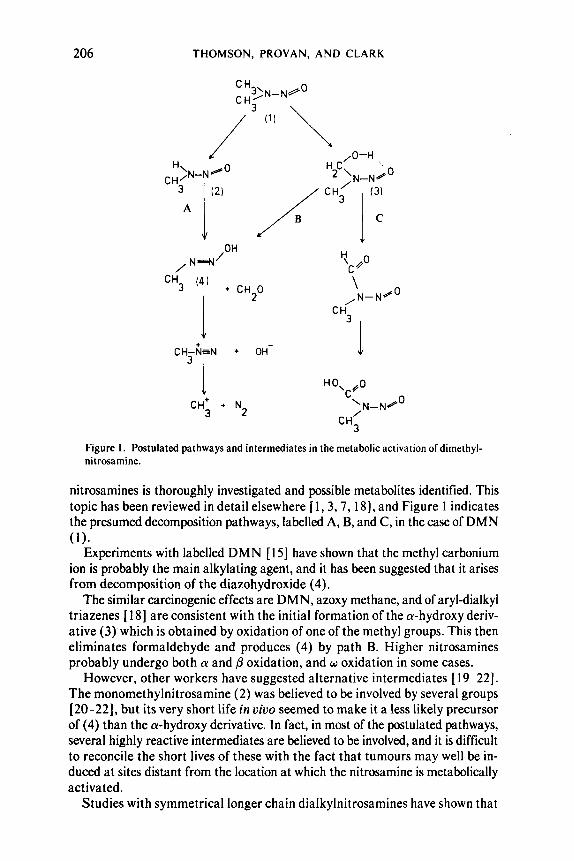
Figure I . Postulated pathways and intermediates in the metabolic activation ofdimethyl- nitrosamine.
nitrosamines is thoroughly investigated and possible metabolites identified. This topic has been reviewed in detail elsewhere [ 1 ,3 ,7 , 181, and Figure 1 indicates the presumed decomposition pathways, labelled A, B, and C, in the case of DMN ( 1 ) .
Experiments with labelled DMN [ 151 have shown that the methyl carbonium ion is probably the main alkylating agent, and it has been suggested that it arises from decomposition of the diazohydroxide (4).
The similar carcinogenic effects are DMN, azoxy methane, and of aryl-dialkyl triazenes [ 181 are consistent with the initial formation of the a-hydroxy deriv- ative (3) which is obtained by oxidation of one of the methyl groups. This then eliminates formaldehyde and produces (4) by path B. Higher nitrosamines probably undergo both a and @ oxidation, and w oxidation in some cases.
However, other workers have suggested alternative intermediates [ 19-22]. The monomethylnitrosamine (2) was believed to be involved by several groups [20-221, but its very short life in uiuo seemed to make it a less likely precursor of (4) than the a-hydroxy derivative. In fact, in most of the postulated pathways, several highly reactive intermediates are believed to be involved, and it is difficult to reconcile the short lives of these with the fact that tumours may well be in- duced at sites distant from the location at which the nitrosamine is metabolically activated.
Studies with symmetrical longer chain dialkylnitrosamines have shown that

CHEMICAL CARCINOGENESIS BY N-ALKYLNITROSAMINES 207
I11
IV
V
TABLE 1. Relative energieso of the isomers of hydroxydiimide computed with STO-3G and ST04-31G basis sets and standard geometries [36,37].
0.31 - 0.26 2.48 1.22
11.16 17.21
B a s i s S e t I I Isomer I 3c I
Values in aldehydic and carboxylic nitrosamino compounds are among the products [23] (path C) and Schoental [ 191 suggested that the alkylnitrosamine aldehydes may well be the proximate carcinogens. The presence of the carbonyl group would enable the molecule to effect cross linkages with cellular macromolecules.
Fahmy and coworkers [24-281 have provided further evidence for this suggestion in examining the mutagenic action of various nitrosamines and the recently synthesised N-a-acetoxymethyl-N-methylnitrosamine [29, 301. The latter was found to be more mutagenic and carcinogenic than DMN, and is easily hydrolyzed to the hydroxy derivative.
In view of the difficulty of studying the postulated intermediates experi- mentally, we have begun detailed quantum-mechanical investigations of various possible intermediate compounds that may arise in the decomposition of N - nitrosamines, and hopefully these investigations should shed some light on the reaction mechanism. It should also be pointed out that the alkylation of cellular macromolecules could also involve free radical intermediates [3 11 during the decomposition of the nitrosamines, and it is likely that theoretical studies of possible radical intermediates will also give more information on this possibili- ty.
It is important at the outset to be clear as to both the value of quantum the- oretical investigations in studying biologically interesting molecules and reac- tions, and also the limitations of the method which may well affect the conclu- sions that are presented. The use of MO theory in carcinogenesis research has been reviewed recently by Scribner [32], and we will not repeat the arguments here. Suffice it to say that with a quantum-mechanical calculation we can cal- culate the relative stabilities of different possible intermediates and their elec- tronic charge distribution, and investigate theoretically the reaction pathways between the intermediates, together with the energy barriers. It is thus likely that useful information which has a bearing on the relative reactivity of different intermediates will be forthcoming from such calculations. This paper and sub- sequent papers in this series attempt to provide such information. In addition, information on the physicochemical properties of these molecules is also obtained which can be compared with experimental data and so confirm the usefulness of the calculations in a predictive sense.
We have chosen initially to investigate the prototype nitrosamine, nitroxyl amide, and its transformation to the diazohydroxide (hydroxydiimide):
HzNNO - HN=NOH (1)

208 THOMSON. PROVAN, A N D CLARK
‘ A ( 4 Figure 2. Variable R and 13 defining the position of the migrating H atom in processes A(a) and A(b) (see text).
and the corresponding transformation of monomethylnitrosamine to 1 - methyl-2-hydroxydiimide:
In addition to studying the isolated molecule transformation, we have also investigated the effect of solvation on the process, and present our preliminary conclusions below. Subsequent papers will deal with the electronic structure of the hydroxy and aldehyde derivatives of DMN and also various other interme- diates. Of course, the prototype is an atypical nitrosamine in that it contains no CH3 group and hence cannot be hydroxylated in the same way as the other ni- trosamines. Nevertheless, as we shall show, the reaction path for the transfor- mation to the diazohydroxide is similar to that in the monomethyl derivative, and as the calculations on the protype system are less expensive, we have initially studied this system more thoroughly.
Method of Calculation
Although semiempirical methods such as CND0/2, INDO, MIND0/3, etc., have been widely used in studying biologically interesting molecules,* we felt at the outset that it was preferable to use ab initio SCF methods in view of the success of these and the ready availability of efficient programmes for treating molecules of the size we are considering here (containing up to 12 atoms). The good agreement between the SCF results and those obtained with the PCILO method [34] have also led us to use the latter, which is computationally much cheaper, in geometry optimization studies on parts of the potential energy sur- face, particularly for the hydrated species.
The majority of calculations were carried out using the Gaussian 70 pro- gramme with the STO-~G or s ~ o - 4 G minimal basis set [35], although some calculations were carried out with the 4-3 1G extended basis set. Most of the calculations were carried out on the IBM 360/44 at St. Andrews, using modified versions of the distributed programmes, but a few were carried out on the IBM 370/165 at Cambridge. Energies are tabulated in atomic units (hartrees) but we shall also use kilocalories in discussions of relative energies and barrier heights. Distances are given in angstroms.
* For extensive references, see [33].
CHEMICAL CARCINOGENESIS BY N-ALKYLNITROSAMINES 209
Results A. Nitrosyl Amide-Hydroxydiimide System
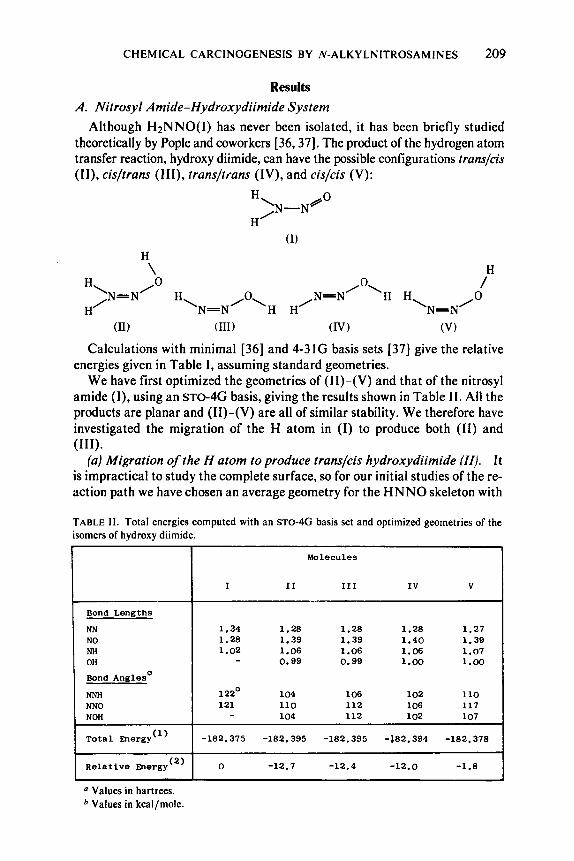
Although H*NNO(I) has never been isolated, it has been briefly studied theoretically by Pople and coworkers [36,37]. The product of the hydrogen atom transfer reaction, hydroxy diimide, can have the possible configurations trans/cis (II), cisltrans (111), trans/l/ans (IV), and cislcis (V):
H \ H
(11) ( 111 ) (Iv) (V)
Calculations with minimal [36] and 4-31G basis sets [37] give the relative energies given in Table I, assuming standard geometries.
We have first optimized the geometries of (11)-(V) and that of the nitrosyl amide (I), using an sTo-4G basis, giving the results shown in Table 11. All the products are planar and (11)-(V) are all of similar stability. We therefore have investigated the migration of the H atom in (I) to produce both (11) and
(a) Migration of the H atom to produce translcis hydroxydiimide (II). It is impractical to study the complete surface, so for our initial studies of the re- action path we have chosen an average geometry for the HNNO skeleton with
(111).
TABLE I I . Total energies computed with an STO-4G basis set and optimized geometries of the isomers of hydroxy diimide.
-.--- Bond Lengths
NN NO NH OH
Bond Angleso
"H NNO NOH
Total mergy ' l )
Relative m e r g y ( 2 )
Values in hartrees. Values in kcal/mole.
Molecules
I I1 I11 IV V
1.34 1 .28 1 .28 1.28 1 .27 1 .28 1.39 1 .39 1 . 4 0 1 .39 1 .02 1 .06 1 .06 1 .06 1 .07
0 . 9 9 0 .99 1 . 0 0 1 . 0 0 -
122O 104 106 102 110 121 110 112 106 117
104 112 102 107 -
-182.375 -182.395 -182.395 -182.394 -182.378
0 -12.7 -12.4 -12 .0 -1.8
210 THOMSON, PROVAN, AND CLARK
I -l82.380
1 L
0 e' 90 180 90 0
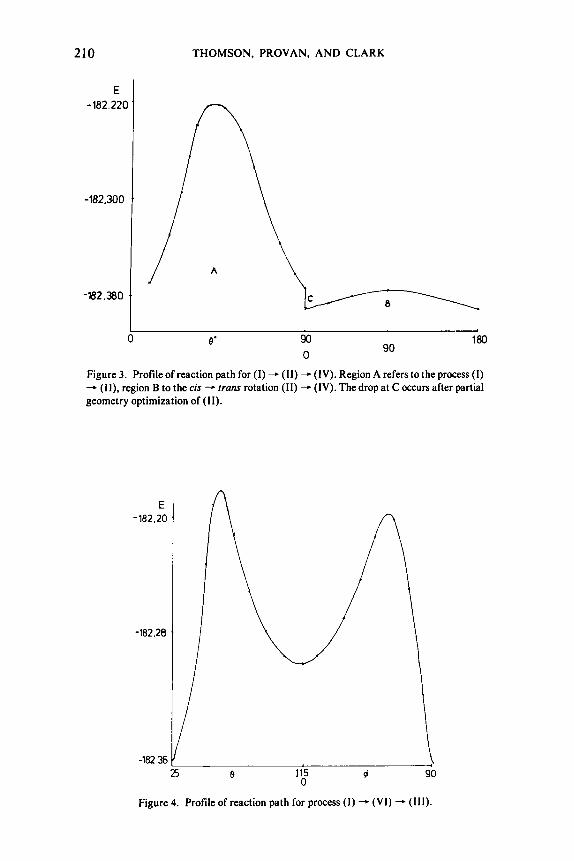
Figure 3. Profile of reaction path for (1) - (11) - (IV). Region A refers to the process ( I ) - (11). region B to the cis - trans rotation (11) - (IV). The drop at C occurs after partial geometry optimization of (11).
E -182.20
-182.28 .
-182 36 a e 115 d 90
0
Figure 4. Profile of reaction path for process ( I ) - (VI) + ( I l l ) .

CHEMICAL CARCINOGENESIS BY N-ALKYLNITROSAMINES 21 1
TABLE 1 1 1 . Total energies of methylnitrosamine and 2-methyl-l-hydroxydiimide, and optimized geometries.
Bond Lengths R Nn NC NN NO cn on Bond Angles'
"C "0 Non
I Mol ecu 1 e
V I I VIII
1.02 1.47 1.39 1.24 1.12 -
119 120 107.5
- 1.49 1.28 1.38 1.12 1.01
106 120 107.5
-~ -220.962 -220.970
0 -5.2
Values in hartrees. Values in kcal/mole.
nI\ R(NN)= 1.31A,R(NO)= I .33A,R(NH)= 1 .02A,HNN=NNO= 120°, and define the motion of the H atom in terms of the distances R and 8 in Figure 2. The amide corresponds to 8 = 26'. R = 2.01 A, and the translcis product (11) to 8 = 90°, R = 1.90 A. 8 was varied between 26' and 90°, and R was optimized for each value of 8. The results of the calculations are depicted graphically in Figure 3.-
Since the energies of (11) and ( IV) are similar, the conversion of (I) - (IV) by the out-of-plane rotation about the NO bond in (11) was also investigated, keeping all bond lengths and angles the same as in the HNNOH skeleton, except for NOH which varied from 107.5" in (11) to 100.6' in (IV). The energy changes in this transformation are also depicted in Figure 2. Partial geometry optimi- zation for the product (11) gives the drop in energy at 8 = 90' compared to the value of E using average bond lengths. It is clear that there is a considerable barrier of -91 kcal/mole-' to the process (I) - (11), but the barrier to the in- terconversion of (11) and (IV) is low (1 1 kcal/mole).
(b) Migration of the H atom to produce cisltrans hydroxydiimide (III). It is apparent that this process could take place via an intermediate (VI):
(W) and the transformation was studied as in (a). A similar coordinate system was used as in (a) (Fig. 2), and the intermediates (VI) and (111) correspond to 8 = 114', R = 1.06 A and 8 = 21 5" , R = 1.778 A, respectively. The optimized ge- ometry of the intermediate has NH = l .OO A, NH = l .06 A, NN = l .3 l A, NO

212 THOMSON, PROVAN, AND CLARK
A A A
= 1.33 A, HNN = 97O, NNO = 127O, and NNHM = 114'. Figure 4 depicts the energy changes during this transformation, where it is clear that a stable intermediate is formed. However, the barrier found ( 1 12 kcal/mole) is even higher than in transformation (a), and therefore it was decided to investigate the transformation (11) - (11) for the monomethylnitrosamine.
B. Monomethylnitrosamine - Methyl Hydroxydiimide
H \
,N=N /o CH,
(W) (VIII)
The method of investigation was analogous to that used in Section A(a). Geometry optimizations for reactant and product were initially carried out (Table III), and an average geometry selected for the migration. Pathways analogous to A(a) and A(b) [namely, B(a) and B(b)] were studied. The results were very similar to those for the prototype shown in Figure 1, and we do not reproduce them here. The barrier was calculated to be -92 kcal/mole for the first step and -1 1 kcal/mole for the rotation. Thus it is clear that the intro- duction of the CH3 group makes very little difference to the barrier, and in view of the similarity to A(a) it was decided that the analogous study of B(b) would not be fruitful.
These calculations of course are entirely hypothetical insofar as they refer to the isolated molecules. It was considered possible that with molecules with a substantial charge asymmetry as in these cases, solvation might well lower the barrier, and many fruitful studies of the solvation of organic molecules have recently been carried out by the Pullmans and coworkers [38]. We therefore report the results of the first stage of a study of the effect of solvation on the process, in which we have introduced a single H20 molecule during the trans- formation.
C. The Effect of a Water Molecule on the Migration of the H Atom
We have investigated the interaction of a single water molecule with the species in the transformation A(a). This process was selected as it gave the lowest barrier, and sihce the migration of the H atom in B(a) gave a similar barrier,
Figure 5. Position of the water molecule with respect to ( I ) and the variables R. R'. and B for the migration of the H atom and HzO molecule.
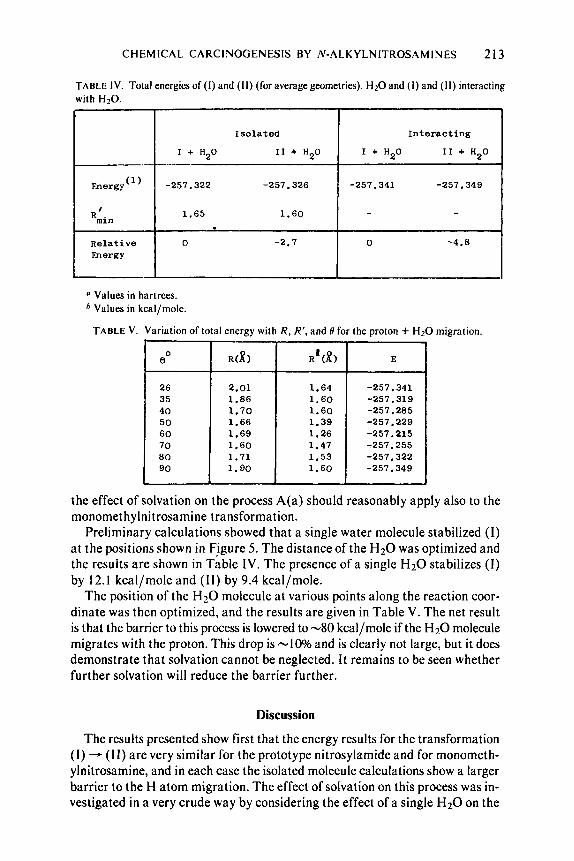
CHEMICAL CARCINOGENESIS BY N-ALKYLNITROSAMINES 213
.
Energy (' )
I
Rmin
Relative Energy
TABLE IV. Total energies of ( I ) and (11) (for average geometries), H20 and ( I ) and (11) interacting with H,O.
Isolated Interacting
I + H20 I1 + H20 I + H20 I 1 + H20 .---
-257.322 -257.326 -257.341 -257.349
1.65 1.60 -
0 -2.7 0 -4.8
-
T A B L E V. Variation of total energy with R, R', and 0 fc
I 26 2.01 1.64 35 1.86 1.60 40 1.70 1.60 50 1.66 1.39 60 1.69 1.26 70 1.60 1.47 80 1.71 1.53 90 1.90 1.60
r the proton + H 2 0 migration.
-257.341 -257.319 -257.285 -257.229 -257.215 -257.255 -257.322 -257.349
the effect of solvation on the process A(a) should reasonably apply also to the monomethylnitrosamine transformation.
Preliminary calculations showed that a single water molecule stabilized (1) at the positions shown i n Figure 5. The distance of the H20 was optimized and thc results are shown in Table IV. The presence of a single H2O stabilizes ( I ) by 12. I kcal/mole and (11) by 9.4 kcal/mole.
The position of the H20 molecule at various points along the reaction coor- dinate was then optimized, and the results are given in Table V. The net result is that the barrier to this process is lowered to-80 kcal/mole if the H20 molecule migrates with the proton. This drop is -10% and is clearly not large, but it does demonstrate that solvation cannot be neglected. It remains to be seen whether further solvation will reduce the barrier further.
Discussion
The results presented show first that the energy results for the transformation (I) - ( I I ) are very similar for the prototype nitrosylamide and for monometh- ylnitrosamine, and in each case the isolated molecule calculations show a larger barrier to the H atom migration. The effect of solvation on this process was in- vestigated in a very crude way by considering the effect of a single H 2 0 on the

214 THOMSON, PROVAN. AND CLARK
barrier height. In both the nitrosylamide case and that of monomethylnitrosa- mine [39] the effect was similar, with a decrease in the barrier by -10%. Further solvation might well be expected to lower the barrier further, but it is difficult to envision that this effect would be large enough to make this transformation a likely route to the diazohydroxide, and it seems more likely that this would occur via a concerted reaction involving the a-hydroxy derivative. Calculations are in progress on this system and will be reported at a later date. We have also recently included several water molecules in the calculations of the migration [39] and find little difference in the barrier height. Our tentative conclusion therefore is that the diazohydroxide is unlikely to arise from this kind of process and that the pathway via the hydroxy (or possibly aldehyde) derivative is probably preferred.
Acknowledgment The authors are indebted to the staff of the Computing Laboratory, University
of St. Andrews, for their help in carrying out these calculations, and C. T. wishes to thank Professor Lowdin and the organizers of the Symposium for the invi- tation to present this work at Sanibel. This work was supported by the National Foundation for Cancer Research.
Bibliography [ I ] C. Heidelberger, Ann. Rev. Biochem. 44,79 (1975). (21 P. N. Magee and J. M. Barnes, Brit. J. Cancer 10. 114 (1956). (31 P. N. Magee and J. M. Barnes, Adv. Cancer Res. 10, 163 (1967). (41 H . Druckey, R. Preussmann, S. Ivankovic, and D. Schmahl. Z. Krebsforsch. 69, 103
[5] P. N. Magee and P. F. Swann, Brit. Med. Bull. 25,240 (1969). [6] Topics in Chemical Carcinogenesis. W. Nakahara, S. Takaya, T. Sugimura, and S. Odashirna,
[7] J. H. Weissburger and G. M. Williams, in Cancer: A Comprehensiw Treatise. vol I , F. F.
[8] H. Druckey, D. Steinhoff, H. Beuthner, H. Schneider, and P. Klarner, Arzneirn. Forsch. 13,
[9] N-Nitroso Compounds in the Enuironment. P. Bogovski, E. A. Walker, and W. Davis, Eds.
(1967).
Eds. (Univ. of Tokyo Press, Tokyo, 1972).
Becker, Ed. (Plenum, New York, 1975). p. 185.
320 (1963).
(Int. Agency for Research on Cancer Lyon, France, 1972). [ 101 W. Liijinsky and S. S. Epstein, Nature 225.21 (1970). [ I I ] P. N. Magee, Biochem. J. 64,676 (1956). [ 121 A. H. Dutton and D. F. Heath, Nature 178,644 (1956). [ 131 E. C. Miller and J. A. Miller, in Molecular Biology ofCancer. H. Busch. Ed. (Academic Press,
(141 P. N. Magee and E. Farber, Biochem. J. 83,114 (1972). [ 151 W. Liijinsky, J . Loo, and A. E. Ross. Nature 218, 1 I74 (1968). [ 161 P. F. Swann and P. N. Magee, Biochem. J. 110.39 (1968). [I71 P. F. Swann and P. N. Magee, Biochem. J. 125,841 (1971). [ 181 H. Druckey, Xenobiotica 3,271 (1973). [ 191 R. Schoental, Brit. J. Cancer 28,436 (1973). [20] D. F. Heath, Biochem. J. 85.72 (1962). (211 P. N. Magee and R. Schoental, Brit. Med. Bull. 20, 102 (1964). (221 S. Grilli and G. Prodi, Gann 66,473 (1975). [23] L. Blattmann and H. Preussmann, Z. Krebsforsch. 79.3 (1973).
New York, 1974).

CHEMICAL CARCINOGENESIS BY N-ALKYLNITROSAMINES 2 15
[24] 0. G. Fahmy and M. J . Fahmy, Chem.-Biol. Interactions 10, 141 (1975). [25] 0. G. Fahmy and M. J . Fahmy, Chem.-Biol. Interactions 11,395 (1975). [26] 0. G. Fahmy, M. J . Fahmy, and M. Wiessler, Biochem. Pharm. 24, 1145 (1975). [27] 0. G. Fahmy and M. J. Fahmy, Chem.-Biol. Interactions 14.21 (1976). [28] 0. G. Fahmy and M. J. Fahmy, Cancer Res. 35,3780 (1975). [29] P. P. Roller, D. R. Shimp, and L. K. Keefer, Tetrahedron Lett., 2065 (1975). [30] M. Weissler. Angew. Chem., Int. Ed. 13,743 (1974). (311 L. N. Ferguson, Chem. Soc. Rev. 4,289 (1975). [32] J . D. Scribner, J. Nat. Cancer Inst. 55, 1035 (1975). [33] Quantum Mechanics of Molecular Conformations. B. Pullman, Ed. (Wiley, New York,
[34] S. Diner, J. P. Malrieu, F. Jordan, and M. Gilbert, Theor. Chim. Acta 15, 100 (1968). [35] Gaussian 70. Quantum Chemistry Program Exchange, Program 236, Indiana University,
[36] L. Radom, W. J. Hehre, and J. A. Pople, J . Chem. Soc. A, 2299 (1971). [37] L. Radom, W. J. Hehre, and J. A. Pople. J. Amer. Chem. SOC. 93,289 (1971). 1381 A. Pullman and B. Pullman, Quart. Rev. Biophys. 7, 505 (1975). (391 C. Thomson and S. Clark, unpublished results.
Received February 1, 1977
1976).
Bloomington, In.