Embed Size (px)
Citation preview

Quality(assessment(and(quality(control(of(NGS(data(Blast(
Finding(SSR(
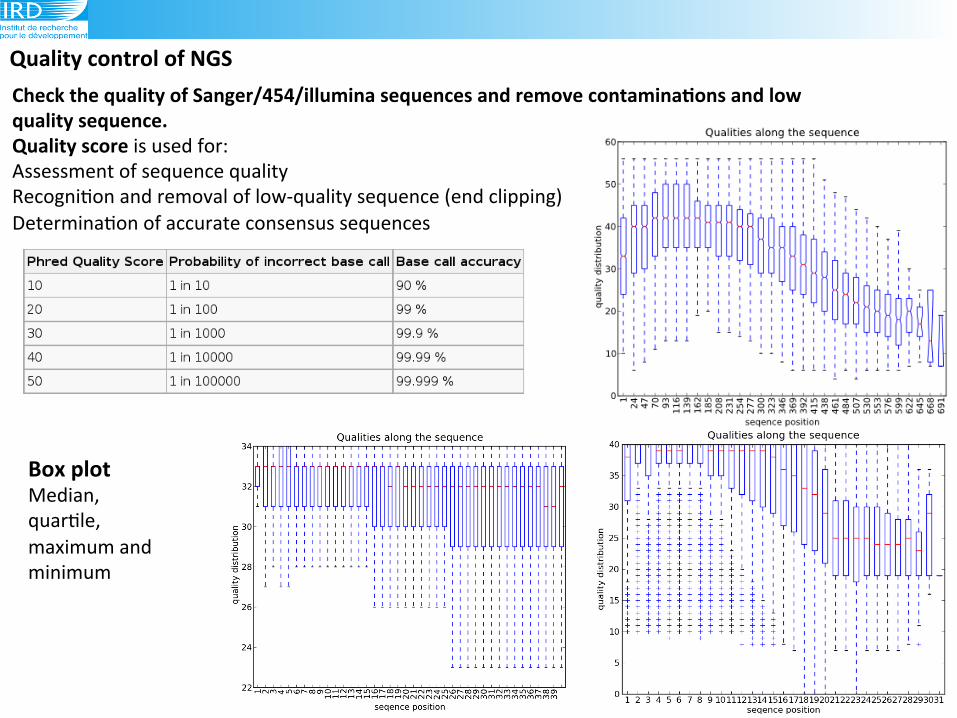
Check(the(quality(of(Sanger/454/illumina(sequences(and(remove(contaminaAons(and(low(quality(sequence.(!Quality(score(is(used!for:!Assessment!of!sequence!quality!
Recogni6on!and!removal!of!low9quality!sequence!(end!clipping)!
Determina6on!of!accurate!consensus!sequences((
Quality(control(of(NGS(
Box(plot(Median,!
quar6le,!
maximum!and!
minimum!
!
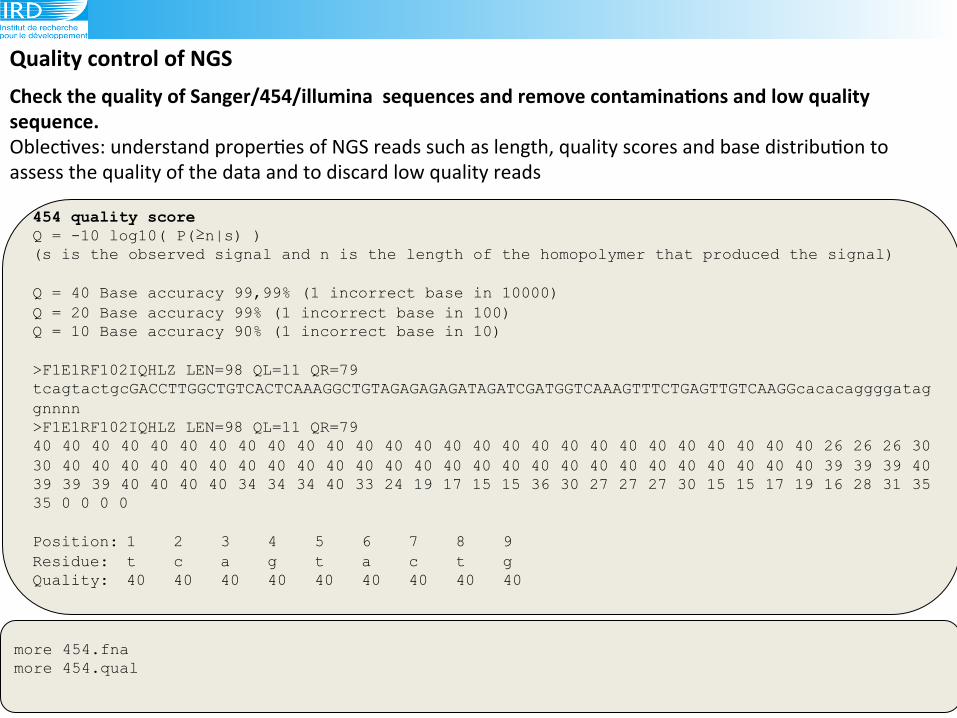
454 quality score Q = -10 log10( P(≥n|s) ) (s is the observed signal and n is the length of the homopolymer that produced the signal) Q = 40 Base accuracy 99,99% (1 incorrect base in 10000) Q = 20 Base accuracy 99% (1 incorrect base in 100) Q = 10 Base accuracy 90% (1 incorrect base in 10) >F1E1RF102IQHLZ LEN=98 QL=11 QR=79 tcagtactgcGACCTTGGCTGTCACTCAAAGGCTGTAGAGAGAGATAGATCGATGGTCAAAGTTTCTGAGTTGTCAAGGcacacaggggataggnnnn >F1E1RF102IQHLZ LEN=98 QL=11 QR=79 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 26 26 26 30 30 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 39 39 39 40 40 40 40 34 34 34 40 33 24 19 17 15 15 36 30 27 27 27 30 15 15 17 19 16 28 31 35 35 0 0 0 0 Position: 1 2 3 4 5 6 7 8 9 Residue: t c a g t a c t g Quality: 40 40 40 40 40 40 40 40 40
Check(the(quality(of(Sanger/454/illumina((sequences(and(remove(contaminaAons(and(low(quality(sequence.(Oblec6ves:!understand!proper6es!of!NGS!reads!such!as!length,!quality!scores!and!base!distribu6on!to!
assess!the!quality!of!the!data!and!to!discard!low!quality!reads!!!
(
more 454.fna more 454.qual
Quality(control(of(NGS(
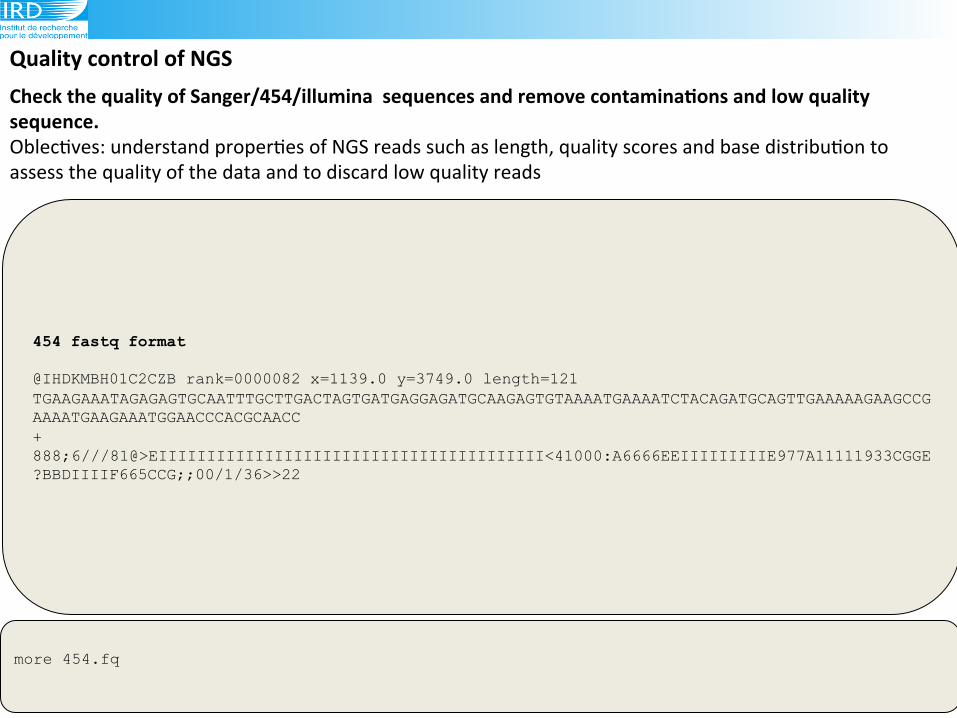
454 fastq format @IHDKMBH01C2CZB rank=0000082 x=1139.0 y=3749.0 length=121 TGAAGAAATAGAGAGTGCAATTTGCTTGACTAGTGATGAGGAGATGCAAGAGTGTAAAATGAAAATCTACAGATGCAGTTGAAAAAGAAGCCGAAAATGAAGAAATGGAACCCACGCAACC + 888;6///81@>EIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII<41000:A6666EEIIIIIIIIIE977A11111933CGGE?BBDIIIIF665CCG;;00/1/36>>22
Check(the(quality(of(Sanger/454/illumina((sequences(and(remove(contaminaAons(and(low(quality(sequence.(Oblec6ves:!understand!proper6es!of!NGS!reads!such!as!length,!quality!scores!and!base!distribu6on!to!
assess!the!quality!of!the!data!and!to!discard!low!quality!reads!!!
(
more 454.fq
Quality(control(of(NGS(

Illumina fastq format @DE18INS655:127:C6DV3ANXX:3:1101:1407:2122 1:N:0:NCAGTG NTGCACTTGACACTCACCGAATGATTCAAGTAGCAAGTTCAAATGATGGTTTAGGCGTCTCCCGTGAGATCCTCTGGAAGGTCCTCCTGAGCGCCTGAAC + #<<BBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBFBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
Check(the(quality(of(Sanger/454/illumina((sequences(and(remove(contaminaAons(and(low(quality(sequence.(Oblec6ves:!understand!proper6es!of!NGS!reads!such!as!length,!quality!scores!and!base!distribu6on!to!
assess!the!quality!of!the!data!and!to!discard!low!quality!reads!!!
(
File 1_OA22_ACAGTG_L003_R1_001.fastq contains 310693256 lines. Extract first 5 Millions sequences. head –n 20000000 1_OA22_ACAGTG_L003_R1_001.fastq > 5M.1_OA22_ACAGTG_L003_R1_001.fastq
Quality(control(of(NGS(

Check(the(quality(of(Sanger/454/illumina(sequences(and(remove(contaminaAons(and(low(quality(sequence.(!(Use!of!FASTX9Toolkit:!hLp://hannonlab.cshl.edu/fastx_toolkit/!!FASTA!&!FASTQ!files!
Use!of!FASTQC!hLp://www.bioinforma6cs.babraham.ac.uk/projects/fastqc/!FASTQ!files!!
Use!of!prinseq!!hLp://prinseq.sourceforge.net!FASTA!&!FASTQ!files!
Use!of!cutadapt!hLps://cutadapt.readthedocs.org/en/stable/!!!
Quality(control(of(NGS(
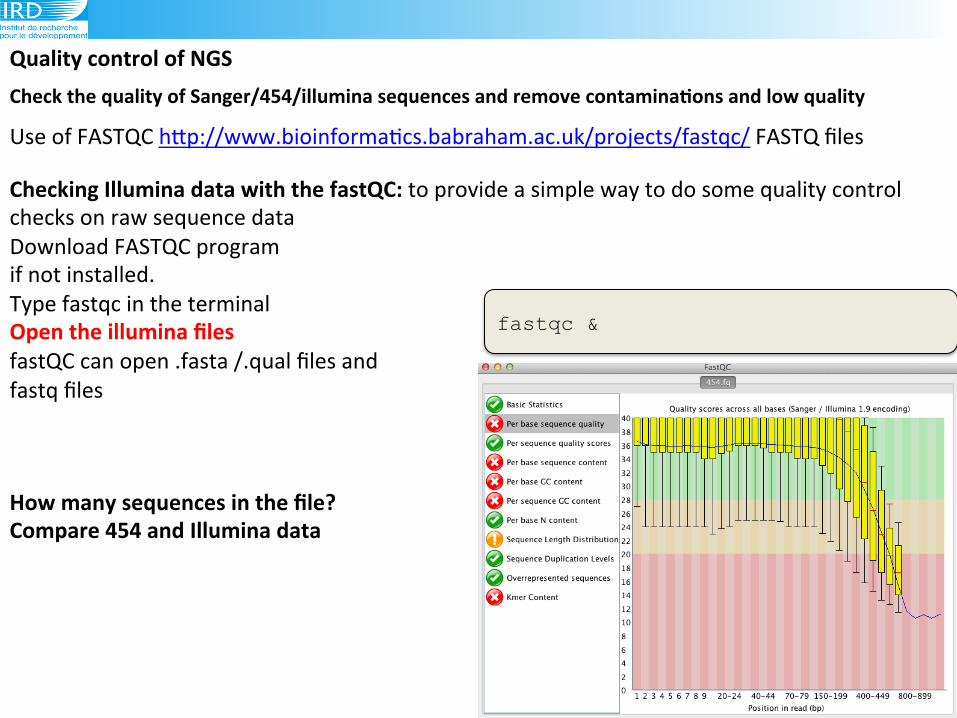
Check(the(quality(of(Sanger/454/illumina(sequences(and(remove(contaminaAons(and(low(quality((!(Use!of!FASTQC!hLp://www.bioinforma6cs.babraham.ac.uk/projects/fastqc/!FASTQ!files!
!!
Quality(control(of(NGS(
Checking(Illumina(data(with(the(fastQC:(to!provide!a!simple!way!to!do!some!quality!control!
checks!on!raw!sequence!data!(Download!FASTQC!program!!
if!not!installed.!
Type!fastqc!in!the!terminal!
Open(the(illumina(files(fastQC!can!open!.fasta!/.qual!files!and!!
fastq!files!!!
fastqc &
How(many(sequences(in(the(file?(Compare(454(and(Illumina(data!
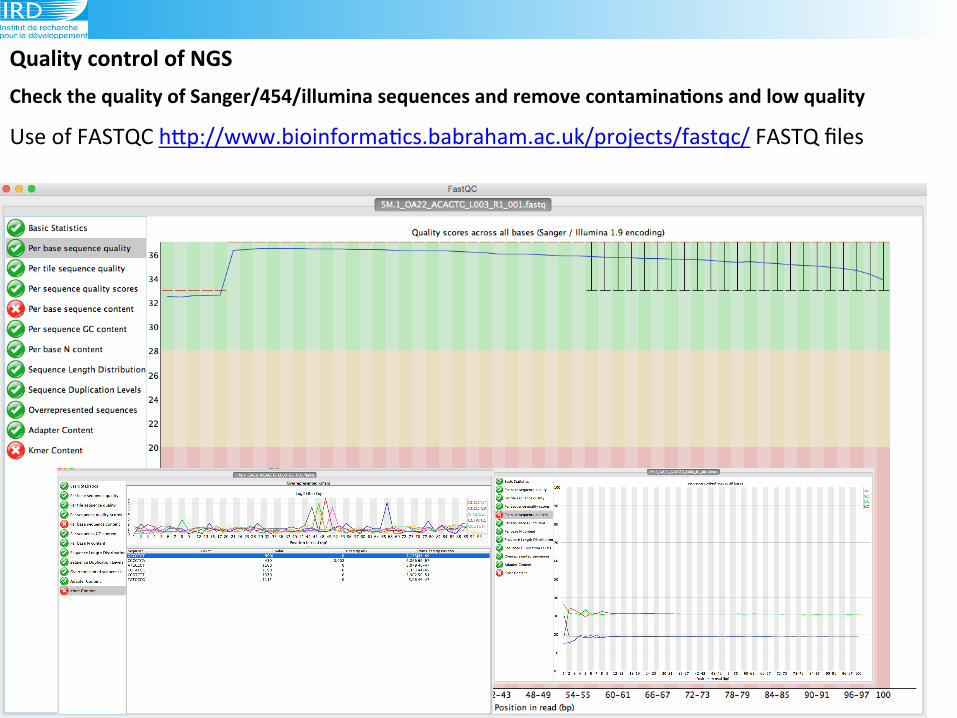
Check(the(quality(of(Sanger/454/illumina(sequences(and(remove(contaminaAons(and(low(quality((!(Use!of!FASTQC!hLp://www.bioinforma6cs.babraham.ac.uk/projects/fastqc/!FASTQ!files!
!!
Quality(control(of(NGS(
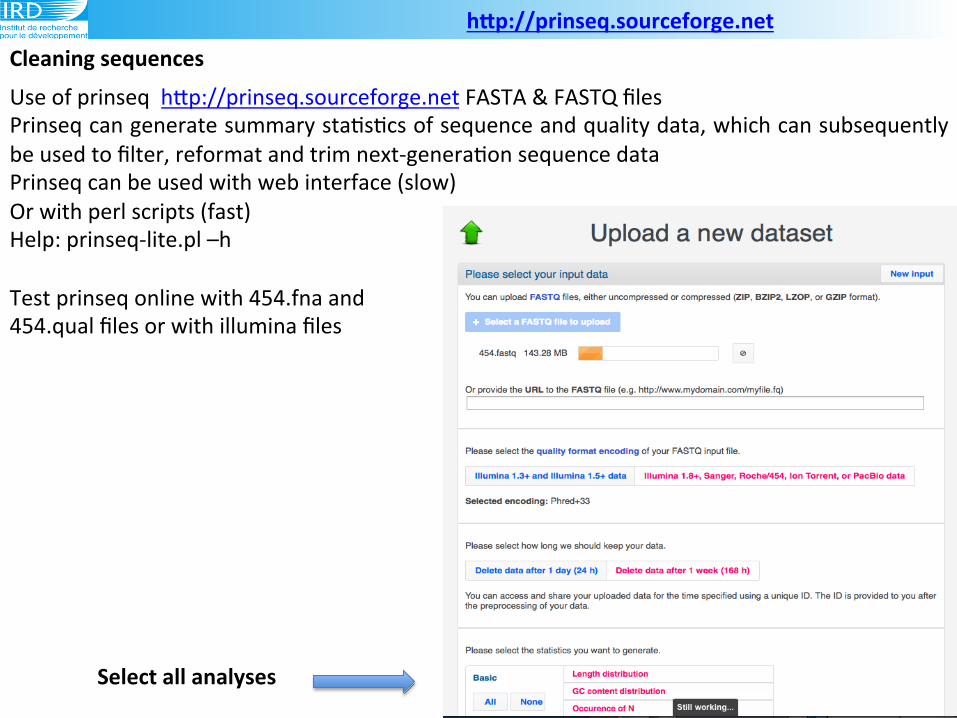
Cleaning(sequences(hLp://prinseq.sourceforge.net((
Use!of!prinseq!!hLp://prinseq.sourceforge.net!FASTA!&!FASTQ!files!
Prinseq!can!generate!summary!sta6s6cs!of!sequence!and!quality!data,!which!can!subsequently!
be!used!to!filter,!reformat!and!trim!next9genera6on!sequence!data!
Prinseq!can!be!used!with!web!interface!(slow)!
Or!with!perl!scripts!(fast)!
Help:!prinseq9lite.pl!–h!
!
Test!prinseq!online!with!454.fna!and!!
454.qual!files!or!with!illumina!files!
Select(all(analyses(
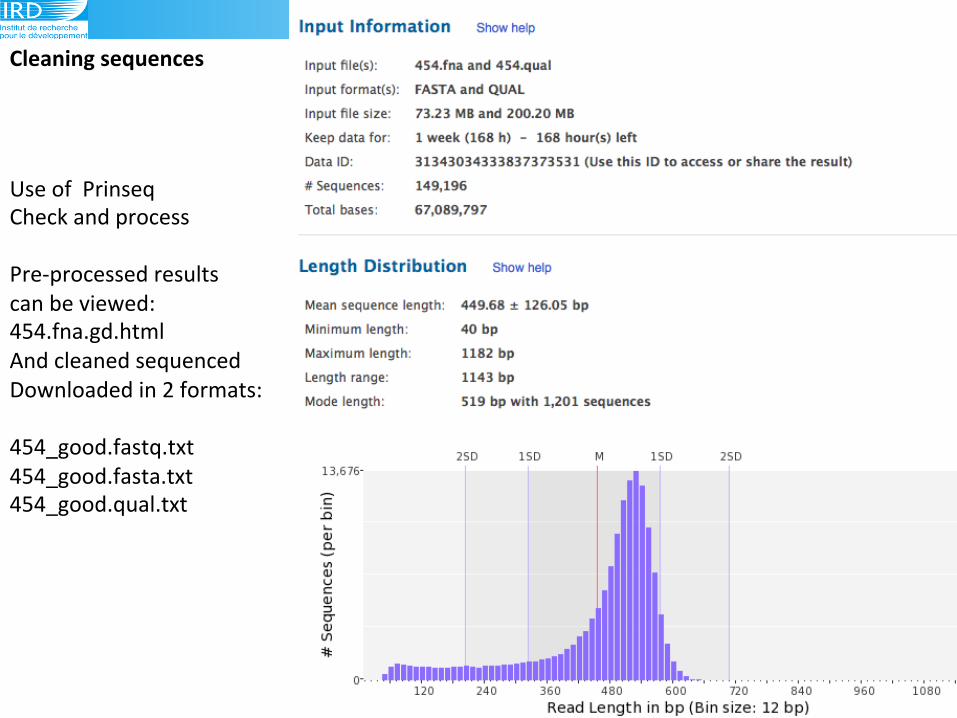
hLp://prinseq.sourceforge.net((
Use!of!!Prinseq!
Check!and!process!
!
Pre9processed!results!!
can!be!viewed:!
454.fna.gd.html!
And!cleaned!sequenced!
Downloaded!in!2!formats:!
!
454_good.fastq.txt!
454_good.fasta.txt!
454_good.qual.txt!
!
!
Cleaning(sequences(
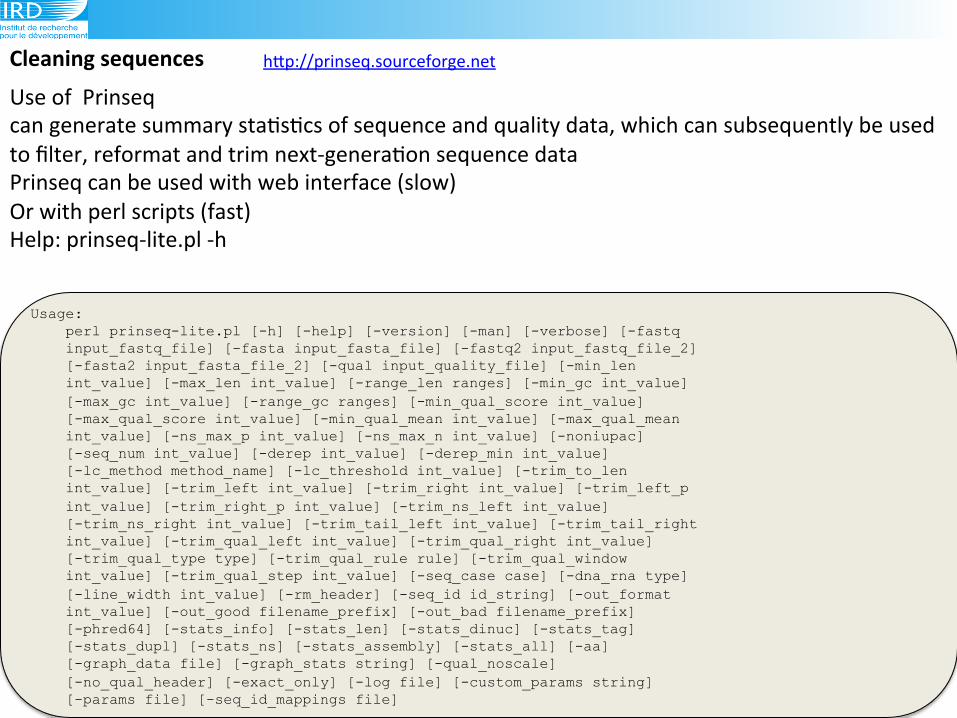
Cleaning(sequences( hLp://prinseq.sourceforge.net!
!Use!of!!Prinseq!
can!generate!summary!sta6s6cs!of!sequence!and!quality!data,!which!can!subsequently!be!used!
to!filter,!reformat!and!trim!next9genera6on!sequence!data!
Prinseq!can!be!used!with!web!interface!(slow)!
Or!with!perl!scripts!(fast)!
Help:!prinseq9lite.pl!9h!!
Usage: perl prinseq-lite.pl [-h] [-help] [-version] [-man] [-verbose] [-fastq input_fastq_file] [-fasta input_fasta_file] [-fastq2 input_fastq_file_2] [-fasta2 input_fasta_file_2] [-qual input_quality_file] [-min_len int_value] [-max_len int_value] [-range_len ranges] [-min_gc int_value] [-max_gc int_value] [-range_gc ranges] [-min_qual_score int_value] [-max_qual_score int_value] [-min_qual_mean int_value] [-max_qual_mean int_value] [-ns_max_p int_value] [-ns_max_n int_value] [-noniupac] [-seq_num int_value] [-derep int_value] [-derep_min int_value] [-lc_method method_name] [-lc_threshold int_value] [-trim_to_len int_value] [-trim_left int_value] [-trim_right int_value] [-trim_left_p int_value] [-trim_right_p int_value] [-trim_ns_left int_value] [-trim_ns_right int_value] [-trim_tail_left int_value] [-trim_tail_right int_value] [-trim_qual_left int_value] [-trim_qual_right int_value] [-trim_qual_type type] [-trim_qual_rule rule] [-trim_qual_window int_value] [-trim_qual_step int_value] [-seq_case case] [-dna_rna type] [-line_width int_value] [-rm_header] [-seq_id id_string] [-out_format int_value] [-out_good filename_prefix] [-out_bad filename_prefix] [-phred64] [-stats_info] [-stats_len] [-stats_dinuc] [-stats_tag] [-stats_dupl] [-stats_ns] [-stats_assembly] [-stats_all] [-aa] [-graph_data file] [-graph_stats string] [-qual_noscale] [-no_qual_header] [-exact_only] [-log file] [-custom_params string] [-params file] [-seq_id_mappings file]

prinseq-lite.pl -stats_all –fasta 454.fna –qual 454.qual > stats
Cleaning(sequences(
Use!of!!Prinseq!
can!generate!summary!sta6s6cs!of!sequence!and!quality!data,!which!can!subsequently!be!used!
to!filter,!reformat!and!trim!next9genera6on!sequence!data!
Prinseq!can!be!used!with!web!interface!(slow)!
Or!with!perl!scripts!(fast)!
Help:!prinseq9lite.pl!9h!!
We!use!.fasta!and!.qual!files!or!.fastq!
1.!To!get!sta6s6cs!
Note!the!number!of!bases,!reads!and!min/maximum!length!of!reads!

Cleaning(sequences(
prinseq-lite.pl –fasta 454.fna –qual 454.qual –out_format 3 –min_len 100 –trim_qual_left 20 –trim_qual_right 20 -trim_qual_window 1
Use!prinseq!with!the!following!parameters:!
Minimum!size!of!reads!100!bp!
minimum!qual!20!
minimum!mean!qual!20!
(
Chek(the(output(file(with(fastQC(How(many(sequences(passed(the(cleaning(step?(
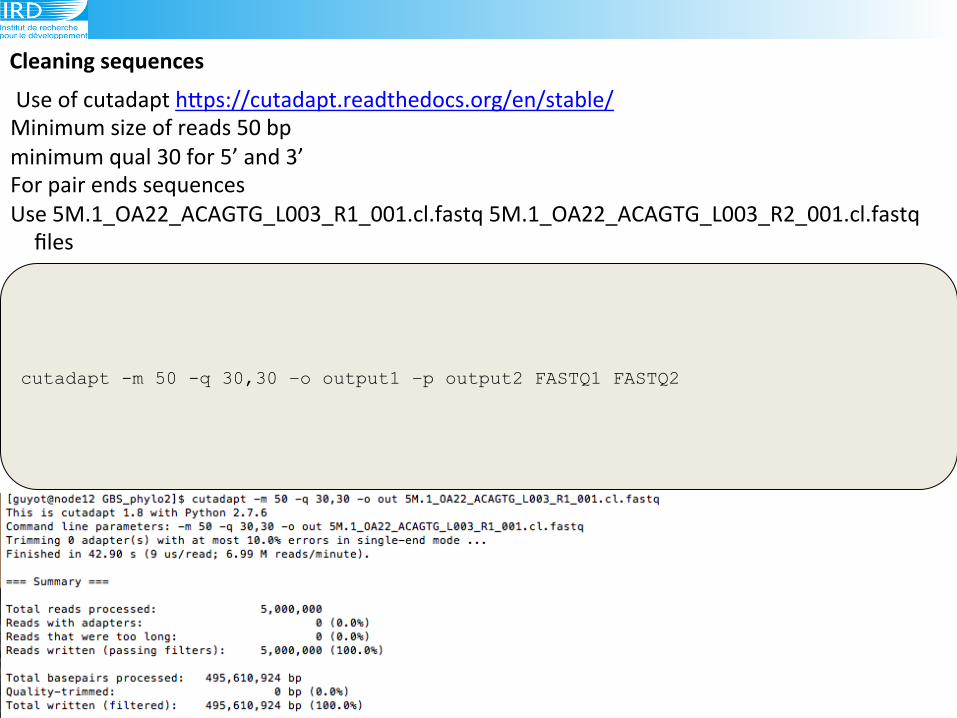
Cleaning(sequences(
cutadapt -m 50 -q 30,30 –o output1 –p output2 FASTQ1 FASTQ2
!Use!of!cutadapt!hLps://cutadapt.readthedocs.org/en/stable/!!!
Minimum!size!of!reads!50!bp!
minimum!qual!30!for!5’!and!3’!
For!pair!ends!sequences!
Use!5M.1_OA22_ACAGTG_L003_R1_001.cl.fastq!5M.1_OA22_ACAGTG_L003_R2_001.cl.fastq!
files!!
(
Sequence(queries(against(database:(BLAST(and(FASTA(
PairRwise(sequences(comparisons!
Dynamic(programming(is(Ame(consuming,(need(of(faster(algorithm((!BLAST,(a(local(alignment((Lipman,(Karlin,(Altschul,(1990)(The!most!popular!local!alignment.!Blast2!gapped!alignments.!!!!

Sequence(queries(against(database:(BLAST(and(FASTA(Dynamic(programming(is(Ame(consuming,(need(of(faster(algorithm((!BLAST,(a(local(alignment((Lipman,(Karlin,(Altschul,(1990)(The!most!popular!local!alignment.!Blast2!gapped!alignments.!!!!
PairRwise(sequences(comparisons!

Sequence(queries(against(database:(BLAST(and(FASTA(Dynamic(programming(is(Ame(consuming,(need(of(faster(algorithm((!
Expecta6on!value!
(E9value):!number!
of!solu6ons!
expected!by!chance!
Score!depends!of!
the!subs6tu6on!
matrix,!gaps!(open!
and!length)!
BLAST,(a(local(alignment((Lipman,(Karlin,(Altschul,(1990)(The!most!popular!local!alignment.!Blast2!gapped!alignments.!!!!
PairRwise(sequences(comparisons!
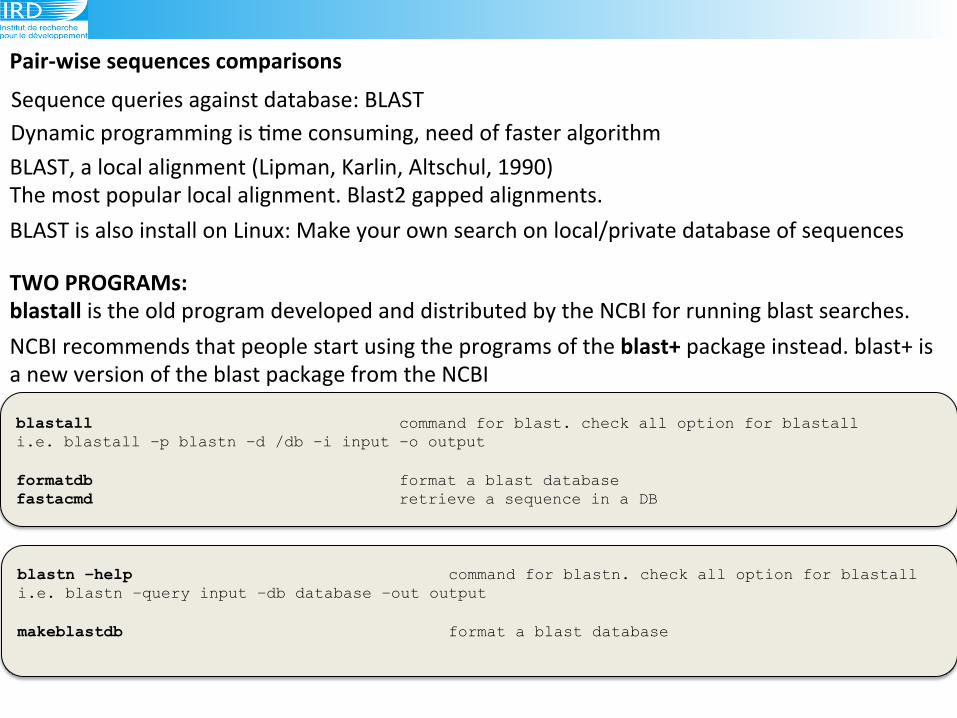
Sequence!queries!against!database:!BLAST!
PairRwise(sequences(comparisons!
Dynamic!programming!is!6me!consuming,!need!of!faster!algorithm!!!
blastall command for blast. check all option for blastall i.e. blastall –p blastn –d /db –i input –o output formatdb format a blast database fastacmd retrieve a sequence in a DB
BLAST!is!also!install!on!Linux:!Make!your!own!search!on!local/private!database!of!sequences!
!
BLAST,!a!local!alignment!(Lipman,!Karlin,!Altschul,!1990)!
The!most!popular!local!alignment.!Blast2!gapped!alignments.!!!!
TWO(PROGRAMs:(blastall!is!the!old!program!developed!and!distributed!by!the!NCBI!for!running!blast!searches.!
NCBI!recommends!that!people!start!using!the!programs!of!the!blast+!package!instead.!blast+!is!a!new!version!of!the!blast!package!from!the!NCBI!
blastn –help command for blastn. check all option for blastall i.e. blastn –query input –db database –out output makeblastdb format a blast database !
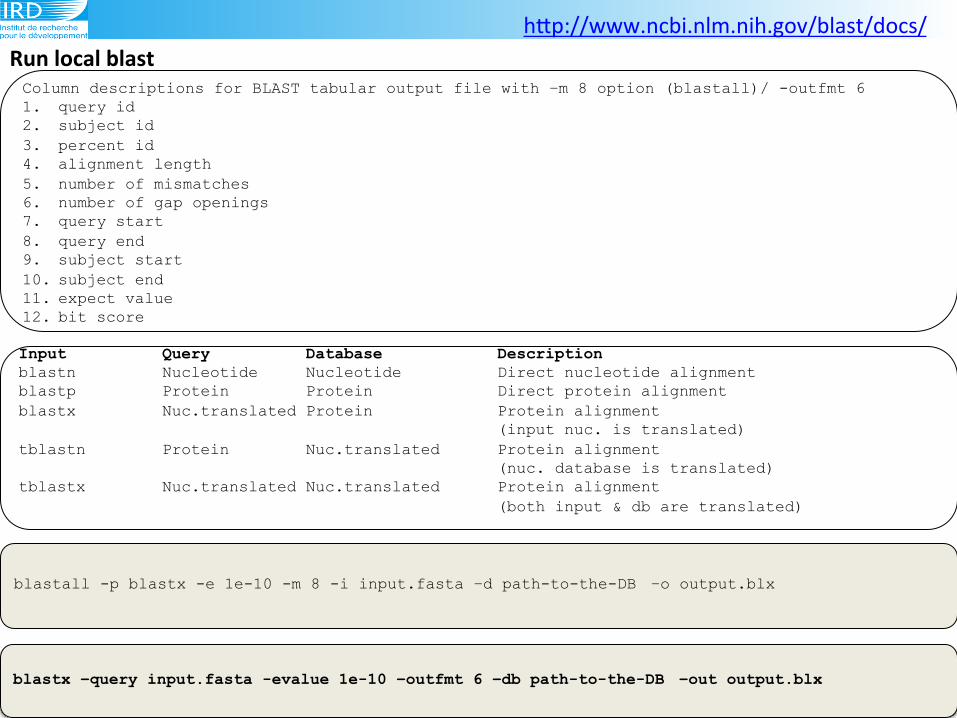
Run(local(blast(
blastall -p blastx -e 1e-10 -m 8 -i input.fasta –d path-to-the-DB –o output.blx
Column descriptions for BLAST tabular output file with –m 8 option (blastall)/ -outfmt 6 1. query id 2. subject id 3. percent id 4. alignment length 5. number of mismatches 6. number of gap openings 7. query start 8. query end 9. subject start 10. subject end 11. expect value 12. bit score
Input Query Database Description blastn Nucleotide Nucleotide Direct nucleotide alignment blastp Protein Protein Direct protein alignment blastx Nuc.translated Protein Protein alignment
(input nuc. is translated) tblastn Protein Nuc.translated Protein alignment
(nuc. database is translated) tblastx Nuc.translated Nuc.translated Protein alignment
(both input & db are translated)
hLp://www.ncbi.nlm.nih.gov/blast/docs/!
!
blastx –query input.fasta -evalue 1e-10 –outfmt 6 –db path-to-the-DB –out output.blx
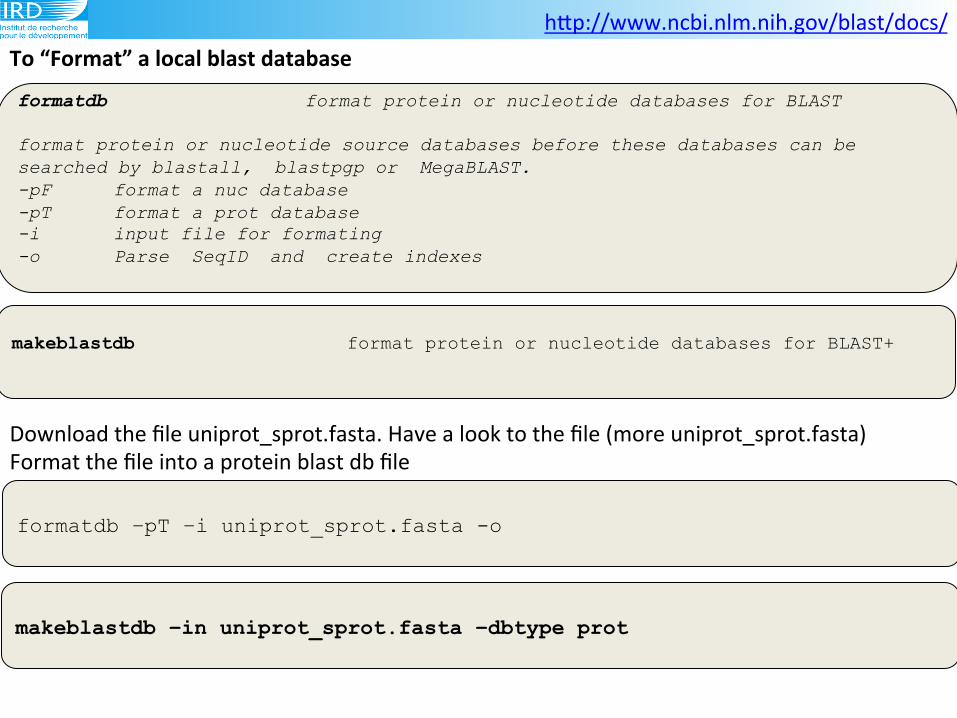
To(“Format”(a(local(blast(database(
formatdb –pT –i uniprot_sprot.fasta -o
formatdb format protein or nucleotide databases for BLAST format protein or nucleotide source databases before these databases can be searched by blastall, blastpgp or MegaBLAST. -pF format a nuc database -pT format a prot database -i input file for formating -o Parse SeqID and create indexes
makeblastdb format protein or nucleotide databases for BLAST+
hLp://www.ncbi.nlm.nih.gov/blast/docs/!
!
Download!the!file!uniprot_sprot.fasta.!Have!a!look!to!the!file!(more!uniprot_sprot.fasta)!
Format!the!file!into!a!protein!blast!db!file!!
makeblastdb –in uniprot_sprot.fasta –dbtype prot
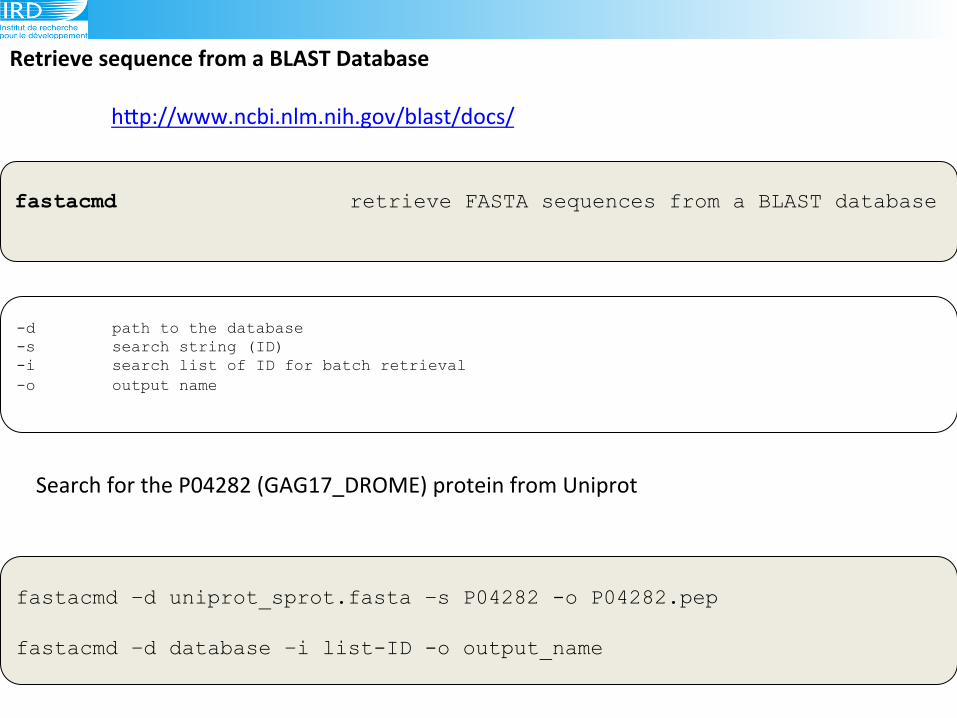
Retrieve(sequence(from(a(BLAST(Database(
fastacmd –d uniprot_sprot.fasta –s P04282 -o P04282.pep fastacmd –d database –i list-ID -o output_name
fastacmd retrieve FASTA sequences from a BLAST database
-d path to the database -s search string (ID) -i search list of ID for batch retrieval -o output name
hLp://www.ncbi.nlm.nih.gov/blast/docs/!
!
Search!for!the!P04282!(GAG17_DROME)!protein!from!Uniprot!
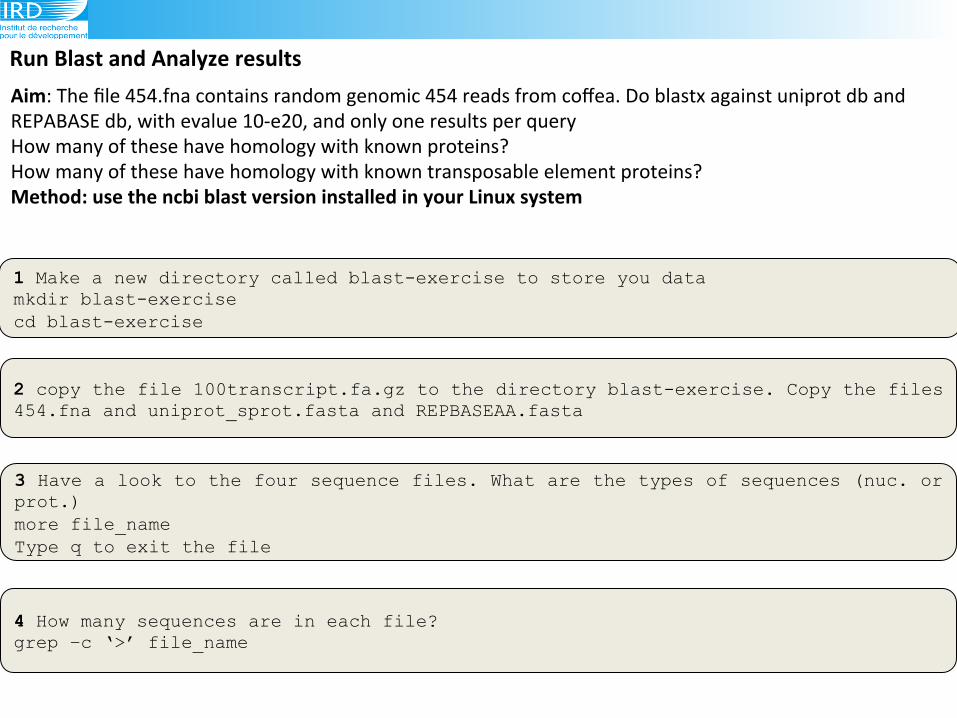
Run(Blast(and(Analyze(results(
1 Make a new directory called blast-exercise to store you data mkdir blast-exercise cd blast-exercise
Aim:!The!file!454.fna!contains!random!genomic!454!reads!from!coffea.!Do!blastx!against!uniprot!db!and!
REPABASE!db,!with!evalue!109e20,!and!only!one!results!per!query!!!
How!many!of!these!have!homology!with!known!proteins?!
How!many!of!these!have!homology!with!known!transposable!element!proteins?(Method:(use(the(ncbi(blast(version(installed(in(your(Linux(system(!!!
2 copy the file 100transcript.fa.gz to the directory blast-exercise. Copy the files 454.fna and uniprot_sprot.fasta and REPBASEAA.fasta
3 Have a look to the four sequence files. What are the types of sequences (nuc. or prot.) more file_name Type q to exit the file
4 How many sequences are in each file? grep –c ‘>’ file_name
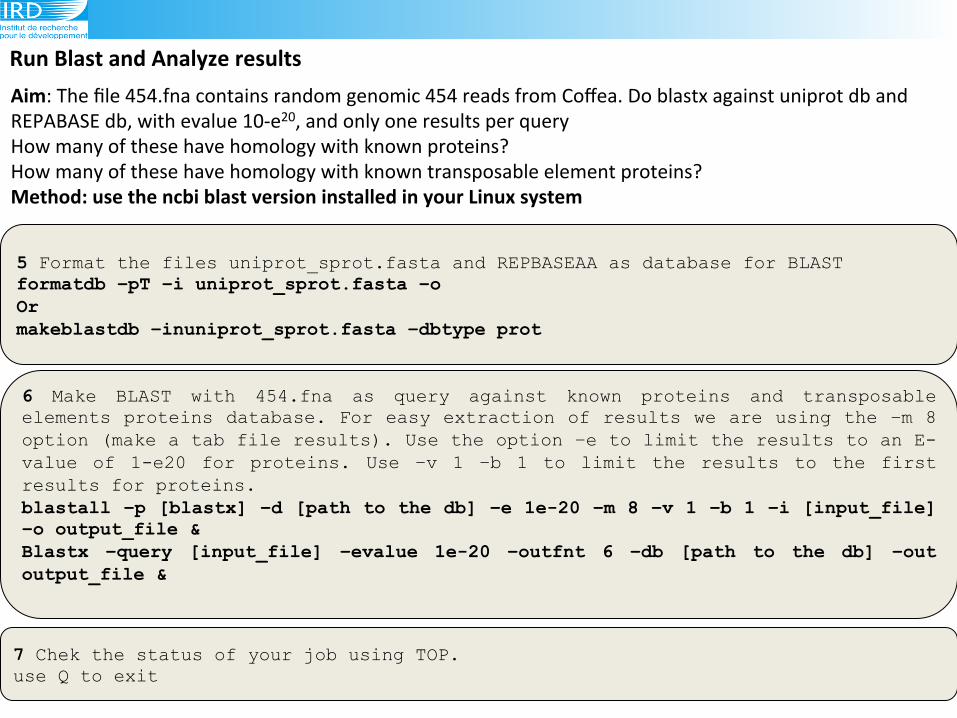
Run(Blast(and(Analyze(results(
6 Make BLAST with 454.fna as query against known proteins and transposable elements proteins database. For easy extraction of results we are using the –m 8 option (make a tab file results). Use the option –e to limit the results to an E-value of 1-e20 for proteins. Use –v 1 –b 1 to limit the results to the first results for proteins. blastall –p [blastx] –d [path to the db] –e 1e-20 –m 8 –v 1 –b 1 –i [input_file] –o output_file & Blastx –query [input_file] –evalue 1e-20 –outfnt 6 –db [path to the db] –out output_file &
5 Format the files uniprot_sprot.fasta and REPBASEAA as database for BLAST formatdb –pT –i uniprot_sprot.fasta –o Or makeblastdb –inuniprot_sprot.fasta –dbtype prot
7 Chek the status of your job using TOP. use Q to exit
Aim:!The!file!454.fna!contains!random!genomic!454!reads!from!Coffea.!Do!blastx!against!uniprot!db!and!
REPABASE!db,!with!evalue!109e20,!and!only!one!results!per!query!!!
How!many!of!these!have!homology!with!known!proteins?!
How!many!of!these!have!homology!with!known!transposable!element!proteins?(Method:(use(the(ncbi(blast(version(installed(in(your(Linux(system(!!!
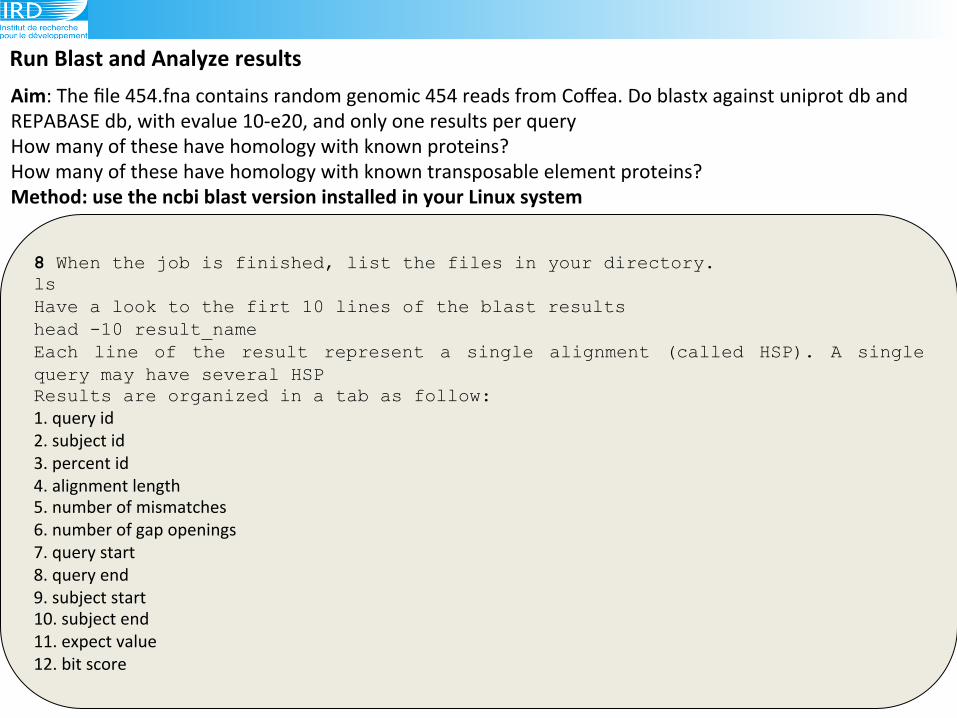
Run(Blast(and(Analyze(results(
8 When the job is finished, list the files in your directory. ls Have a look to the firt 10 lines of the blast results head -10 result_name Each line of the result represent a single alignment (called HSP). A single query may have several HSP Results are organized in a tab as follow: 1.!query!id!
2.!subject!id!!
3.!percent!id!
4.!alignment!length!
5.!number!of!mismatches!!
6.!number!of!gap!openings!!
7.!query!start!
8.!query!end!
9.!subject!start!!
10.!subject!end!!
11.!expect!value!!
12.!bit!score
Aim:!The!file!454.fna!contains!random!genomic!454!reads!from!Coffea.!Do!blastx!against!uniprot!db!and!
REPABASE!db,!with!evalue!109e20,!and!only!one!results!per!query!!!
How!many!of!these!have!homology!with!known!proteins?!
How!many!of!these!have!homology!with!known!transposable!element!proteins?(Method:(use(the(ncbi(blast(version(installed(in(your(Linux(system(!!!
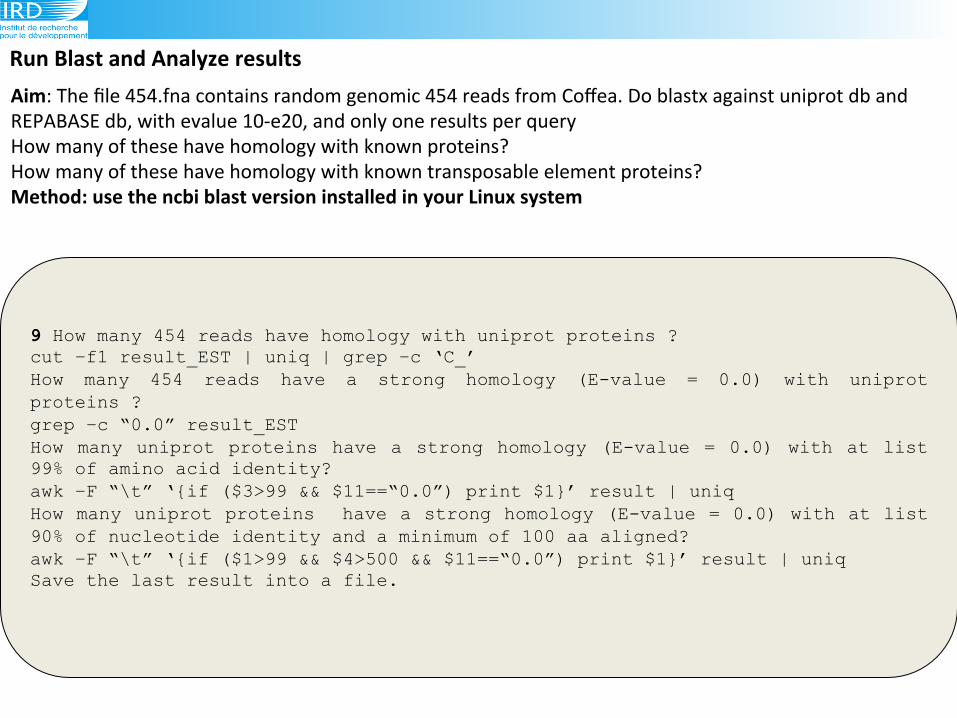
Run(Blast(and(Analyze(results(
9 How many 454 reads have homology with uniprot proteins ? cut –f1 result_EST | uniq | grep –c ‘C_’ How many 454 reads have a strong homology (E-value = 0.0) with uniprot proteins ? grep –c “0.0” result_EST How many uniprot proteins have a strong homology (E-value = 0.0) with at list 99% of amino acid identity? awk –F “\t” ‘{if ($3>99 && $11==“0.0”) print $1}’ result | uniq How many uniprot proteins have a strong homology (E-value = 0.0) with at list 90% of nucleotide identity and a minimum of 100 aa aligned? awk –F “\t” ‘{if ($1>99 && $4>500 && $11==“0.0”) print $1}’ result | uniq Save the last result into a file.!
Aim:!The!file!454.fna!contains!random!genomic!454!reads!from!Coffea.!Do!blastx!against!uniprot!db!and!
REPABASE!db,!with!evalue!109e20,!and!only!one!results!per!query!!!
How!many!of!these!have!homology!with!known!proteins?!
How!many!of!these!have!homology!with!known!transposable!element!proteins?(Method:(use(the(ncbi(blast(version(installed(in(your(Linux(system(!!!

Run(Blast(and(Analyze(results(
10 Retrieve the uniprot proteins with a strong homology (E-value = 0.0) with at list 90% of amino-acid identity and a minimum of 100 aa aligned. awk –F “\t” ‘{if ($1>99 && $4>500 && $11==“0.0”) print $2}’ result list-accession-EST Save the result into a file Use the fastacmd command to retrieve the sequences fastacmd –d database –i input_list-accession-EST Save the sequence into a file (> file_name)
Aim:!The!file!100transcript.fa!contains!100!assembled!coffea&transcripts!(coffea&pseudozanguebarie)!How!many!of!these!have!homology!with!EST!from!coffea&canephora?!How!many!of!these!have!homology!with!known!proteins?!
How!many!of!these!have!homology!with!known!transposable!element!proteins?(Method:(use(the(ncbi(blast(version(installed(in(you(Linux(system(!!!
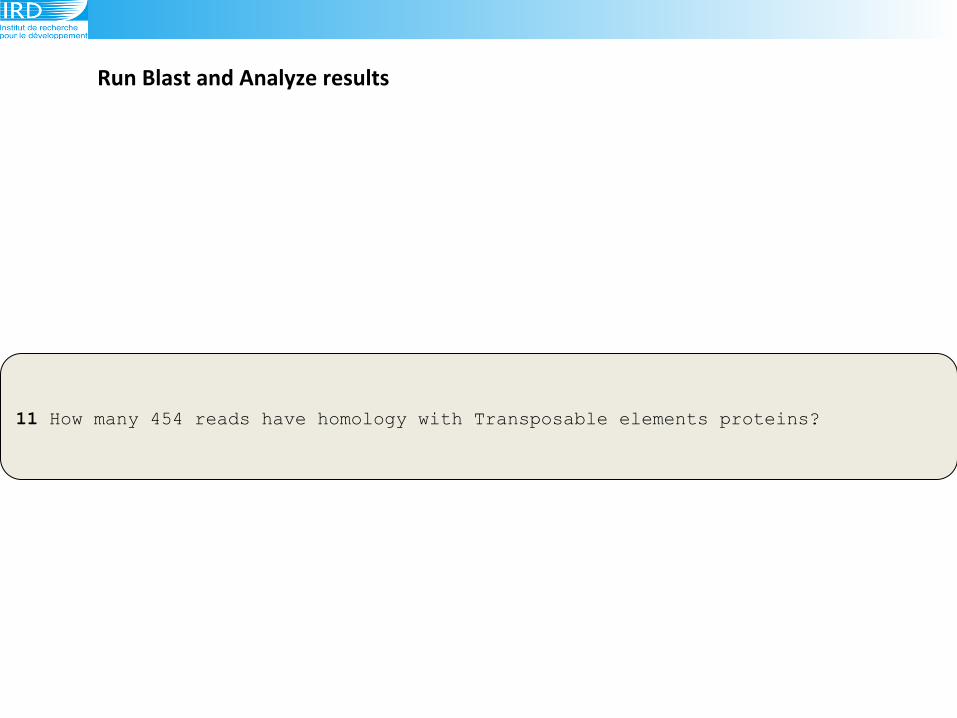
Run(Blast(and(Analyze(results(
11 How many 454 reads have homology with Transposable elements proteins?
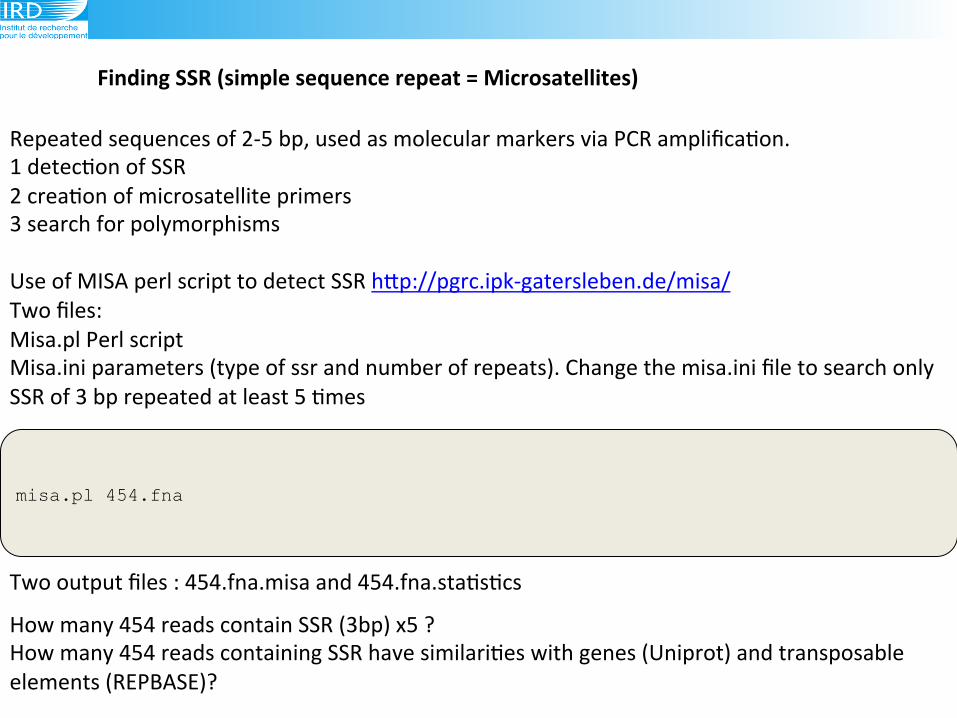
Finding(SSR((simple(sequence(repeat(=(Microsatellites)(
misa.pl 454.fna
Repeated!sequences!of!295!bp,!used!as!molecular!markers!via!PCR!amplifica6on.!
1!detec6on!of!SSR!
2!crea6on!of!microsatellite!primers!
3!search!for!polymorphisms!
!
Use!of!MISA!perl!script!to!detect!SSR!hLp://pgrc.ipk9gatersleben.de/misa/!!
Two!files:!
Misa.pl!Perl!script!
Misa.ini!parameters!(type!of!ssr!and!number!of!repeats).!Change!the!misa.ini!file!to!search!only!
SSR!of!3!bp!repeated!at!least!5!6mes!
Two!output!files!:!454.fna.misa!and!454.fna.sta6s6cs!
How!many!454!reads!contain!SSR!(3bp)!x5!?!
How!many!454!reads!containing!SSR!have!similari6es!with!genes!(Uniprot)!and!transposable!
elements!(REPBASE)?!

Unix(Shell(script(
If(you(want(to(do(a(lot(of(runs(of(the(same(program(using(slightly(different(input,(you(can(create(mulAple(input(files(and(then(write(a(script!((Allow(to(automate(your(work((Different(languages((bash,(perl,(python)((Script(must(be(executable(using(the(chmod(command((To(run(the((new(script,(tell(to(the(operaAng(system(the(path(to(the(program:(./myprogr.sh or /home/user/Desktop/myprog.sh
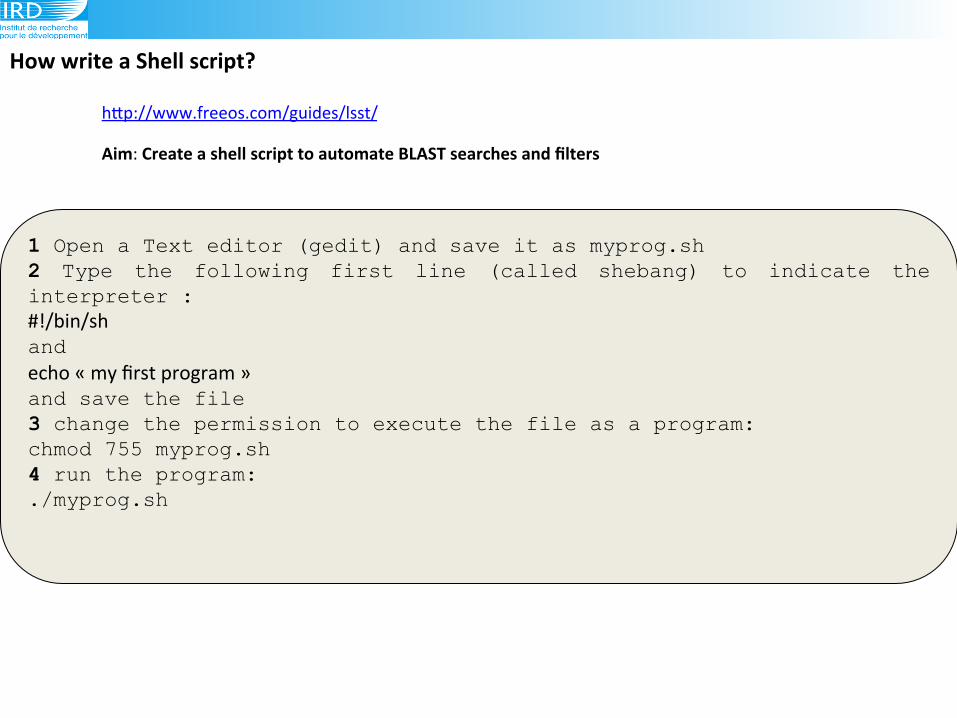
How(write(a(Shell(script?(
1 Open a Text editor (gedit) and save it as myprog.sh 2 Type the following first line (called shebang) to indicate the interpreter : #!/bin/sh!
and echo!«!my!first!program!»!
and save the file 3 change the permission to execute the file as a program: chmod 755 myprog.sh 4 run the program: ./myprog.sh
Aim:!Create(a(shell(script(to(automate(BLAST(searches(and(filters((
hLp://www.freeos.com/guides/lsst/!
!




























