Embed Size (px)
Citation preview
immunoblot data was analyzed for pVHL suppression using imagequantification software. Branching morphogenesis assays were per-formed to assess phenotypic changes associated with the presence orabsence of pVHL. The in-vivo effect on tumor growth from VHLsuppression was assessed using stable anti-VHL siRNA cell lines(siRNA expression vector) and tumor xenograft assays (SCID/nudemice). Results: Real-time quantitative RT-PCR revealed statisti-cally significant suppression of VHL gene expression by RNAi(VHL2�VHL1). VHL1 siRNA resulted in pVHL levels comparable toscramble duplex (negative control). VHL2 siRNA transfection re-sulted in significant pVHL suppression (�90%) when compared toscramble siRNA. VHL siRNA transfected cells exhibited a branchingphenotype consistent with cells lacking pVHL. HeLa and 293 celllines were shown to be tumorigenic in both nude and SCID mice(SCID�nude). Anti-VHL siRNA cell lines showed significantlyhigher growth curves than the scramble siRNA control mouse tumorxenografts. Conclusions: The von Hippel Lindau tumor suppressorprotein (pVHL) can be successfully suppressed by RNA interference.This powerful tool will further assist in the understanding of theVHL-ubiquitin-mediated hypoxia induced factor (HIF) degradationpathway essential in renal cell carcinoma in the elderly.
QS97. CHEMOTHERAPY TREATED TUMOR CELLS UP-REGULATE TOLL LIKE RECEPTORS [TLR]. ReginaldAnunobi1, Steven C. Gribar1, Nicole Schapiro1, ChhinderSodhi2, David J. Hackam5, Micheal T. Lotze1, Herbert J. ZehIII1; 1University of Pittsburgh, Pittsburgh, PA; 2Children’sHospital of Pittsburgh., Pittsburgh, PA; 5Children’s Hospitalof Pittsburgh, Pittsburgh, PA
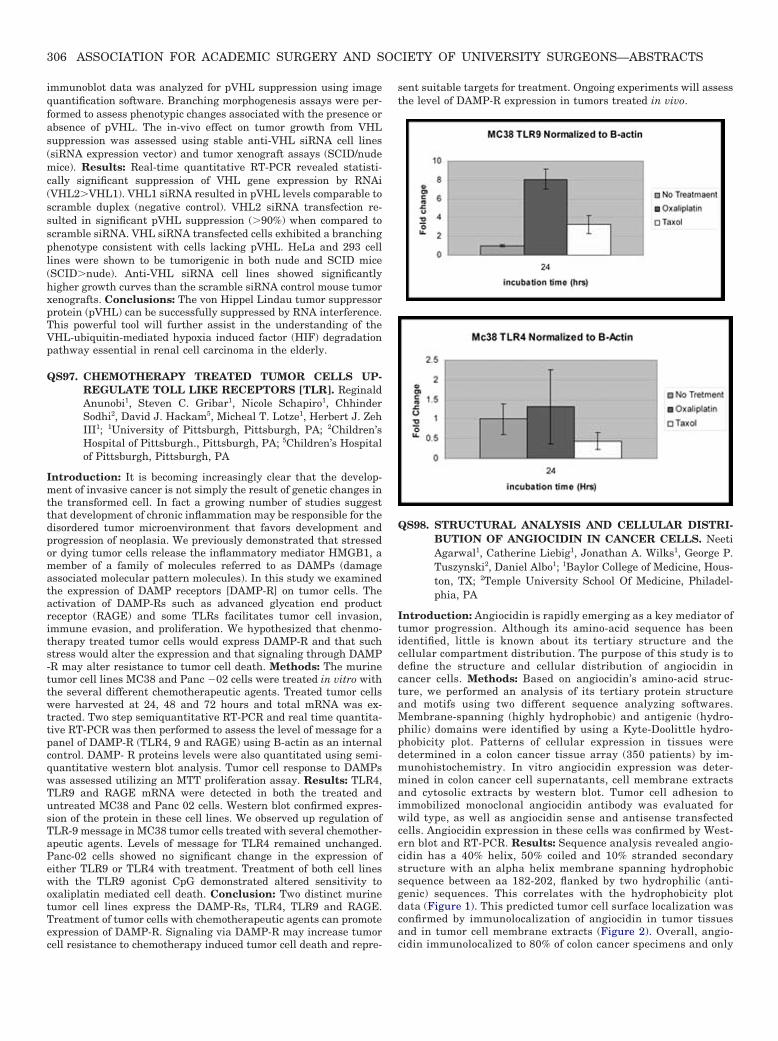
Introduction: It is becoming increasingly clear that the develop-ment of invasive cancer is not simply the result of genetic changes inthe transformed cell. In fact a growing number of studies suggestthat development of chronic inflammation may be responsible for thedisordered tumor microenvironment that favors development andprogression of neoplasia. We previously demonstrated that stressedor dying tumor cells release the inflammatory mediator HMGB1, amember of a family of molecules referred to as DAMPs (damageassociated molecular pattern molecules). In this study we examinedthe expression of DAMP receptors [DAMP-R] on tumor cells. Theactivation of DAMP-Rs such as advanced glycation end productreceptor (RAGE) and some TLRs facilitates tumor cell invasion,immune evasion, and proliferation. We hypothesized that chenmo-therapy treated tumor cells would express DAMP-R and that suchstress would alter the expression and that signaling through DAMP-R may alter resistance to tumor cell death. Methods: The murinetumor cell lines MC38 and Panc �02 cells were treated in vitro withthe several different chemotherapeutic agents. Treated tumor cellswere harvested at 24, 48 and 72 hours and total mRNA was ex-tracted. Two step semiquantitative RT-PCR and real time quantita-tive RT-PCR was then performed to assess the level of message for apanel of DAMP-R (TLR4, 9 and RAGE) using B-actin as an internalcontrol. DAMP- R proteins levels were also quantitated using semi-quantitative western blot analysis. Tumor cell response to DAMPswas assessed utilizing an MTT proliferation assay. Results: TLR4,TLR9 and RAGE mRNA were detected in both the treated anduntreated MC38 and Panc 02 cells. Western blot confirmed expres-sion of the protein in these cell lines. We observed up regulation ofTLR-9 message in MC38 tumor cells treated with several chemother-apeutic agents. Levels of message for TLR4 remained unchanged.Panc-02 cells showed no significant change in the expression ofeither TLR9 or TLR4 with treatment. Treatment of both cell lineswith the TLR9 agonist CpG demonstrated altered sensitivity tooxaliplatin mediated cell death. Conclusion: Two distinct murinetumor cell lines express the DAMP-Rs, TLR4, TLR9 and RAGE.Treatment of tumor cells with chemotherapeutic agents can promoteexpression of DAMP-R. Signaling via DAMP-R may increase tumorcell resistance to chemotherapy induced tumor cell death and repre-
sent suitable targets for treatment. Ongoing experiments will assessthe level of DAMP-R expression in tumors treated in vivo.
QS98. STRUCTURAL ANALYSIS AND CELLULAR DISTRI-BUTION OF ANGIOCIDIN IN CANCER CELLS. NeetiAgarwal1, Catherine Liebig1, Jonathan A. Wilks1, George P.Tuszynski2, Daniel Albo1; 1Baylor College of Medicine, Hous-ton, TX; 2Temple University School Of Medicine, Philadel-phia, PA
Introduction: Angiocidin is rapidly emerging as a key mediator oftumor progression. Although its amino-acid sequence has beenidentified, little is known about its tertiary structure and thecellular compartment distribution. The purpose of this study is todefine the structure and cellular distribution of angiocidin incancer cells. Methods: Based on angiocidin’s amino-acid struc-ture, we performed an analysis of its tertiary protein structureand motifs using two different sequence analyzing softwares.Membrane-spanning (highly hydrophobic) and antigenic (hydro-philic) domains were identified by using a Kyte-Doolittle hydro-phobicity plot. Patterns of cellular expression in tissues weredetermined in a colon cancer tissue array (350 patients) by im-munohistochemistry. In vitro angiocidin expression was deter-mined in colon cancer cell supernatants, cell membrane extractsand cytosolic extracts by western blot. Tumor cell adhesion toimmobilized monoclonal angiocidin antibody was evaluated forwild type, as well as angiocidin sense and antisense transfectedcells. Angiocidin expression in these cells was confirmed by West-ern blot and RT-PCR. Results: Sequence analysis revealed angio-cidin has a 40% helix, 50% coiled and 10% stranded secondarystructure with an alpha helix membrane spanning hydrophobicsequence between aa 182-202, flanked by two hydrophilic (anti-genic) sequences. This correlates with the hydrophobicity plotdata (Figure 1). This predicted tumor cell surface localization wasconfirmed by immunolocalization of angiocidin in tumor tissuesand in tumor cell membrane extracts (Figure 2). Overall, angio-cidin immunolocalized to 80% of colon cancer specimens and only
306 ASSOCIATION FOR ACADEMIC SURGERY AND SOCIETY OF UNIVERSITY SURGEONS—ABSTRACTS
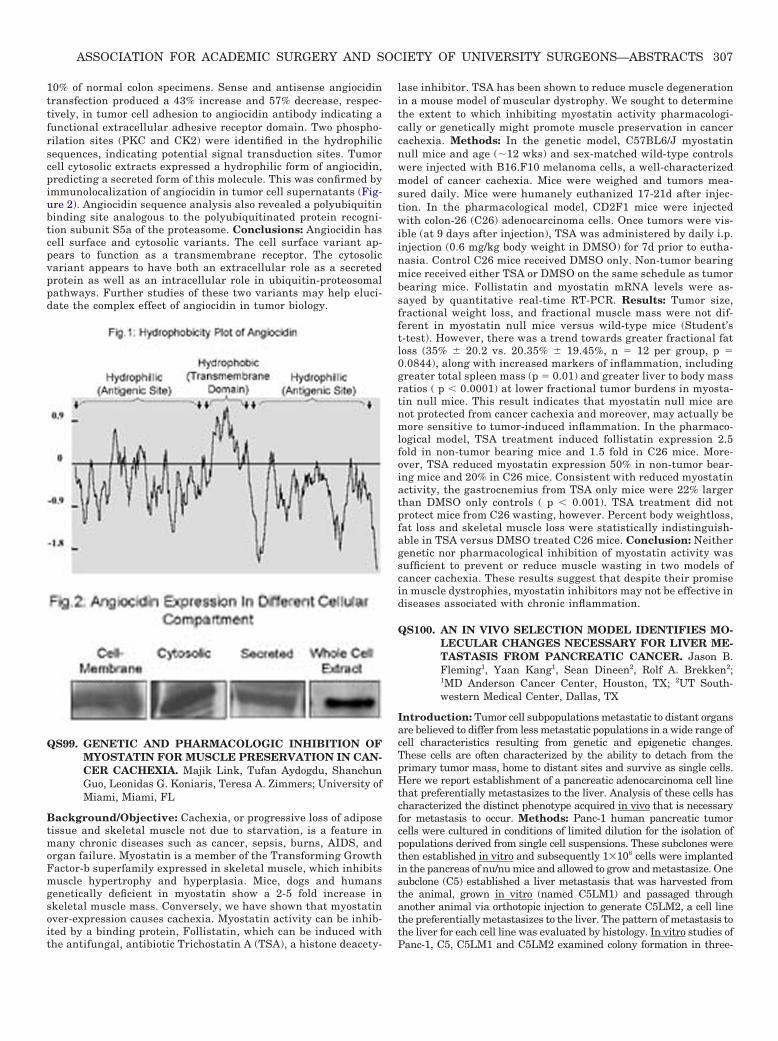
10% of normal colon specimens. Sense and antisense angiocidintransfection produced a 43% increase and 57% decrease, respec-tively, in tumor cell adhesion to angiocidin antibody indicating afunctional extracellular adhesive receptor domain. Two phospho-rilation sites (PKC and CK2) were identified in the hydrophilicsequences, indicating potential signal transduction sites. Tumorcell cytosolic extracts expressed a hydrophilic form of angiocidin,predicting a secreted form of this molecule. This was confirmed byimmunolocalization of angiocidin in tumor cell supernatants (Fig-ure 2). Angiocidin sequence analysis also revealed a polyubiquitinbinding site analogous to the polyubiquitinated protein recogni-tion subunit S5a of the proteasome. Conclusions: Angiocidin hascell surface and cytosolic variants. The cell surface variant ap-pears to function as a transmembrane receptor. The cytosolicvariant appears to have both an extracellular role as a secretedprotein as well as an intracellular role in ubiquitin-proteosomalpathways. Further studies of these two variants may help eluci-date the complex effect of angiocidin in tumor biology.
QS99. GENETIC AND PHARMACOLOGIC INHIBITION OFMYOSTATIN FOR MUSCLE PRESERVATION IN CAN-CER CACHEXIA. Majik Link, Tufan Aydogdu, ShanchunGuo, Leonidas G. Koniaris, Teresa A. Zimmers; University ofMiami, Miami, FL
Background/Objective: Cachexia, or progressive loss of adiposetissue and skeletal muscle not due to starvation, is a feature inmany chronic diseases such as cancer, sepsis, burns, AIDS, andorgan failure. Myostatin is a member of the Transforming GrowthFactor-b superfamily expressed in skeletal muscle, which inhibitsmuscle hypertrophy and hyperplasia. Mice, dogs and humansgenetically deficient in myostatin show a 2-5 fold increase inskeletal muscle mass. Conversely, we have shown that myostatinover-expression causes cachexia. Myostatin activity can be inhib-ited by a binding protein, Follistatin, which can be induced withthe antifungal, antibiotic Trichostatin A (TSA), a histone deacety-
lase inhibitor. TSA has been shown to reduce muscle degenerationin a mouse model of muscular dystrophy. We sought to determinethe extent to which inhibiting myostatin activity pharmacologi-cally or genetically might promote muscle preservation in cancercachexia. Methods: In the genetic model, C57BL6/J myostatinnull mice and age (�12 wks) and sex-matched wild-type controlswere injected with B16.F10 melanoma cells, a well-characterizedmodel of cancer cachexia. Mice were weighed and tumors mea-sured daily. Mice were humanely euthanized 17-21d after injec-tion. In the pharmacological model, CD2F1 mice were injectedwith colon-26 (C26) adenocarcinoma cells. Once tumors were vis-ible (at 9 days after injection), TSA was administered by daily i.p.injection (0.6 mg/kg body weight in DMSO) for 7d prior to eutha-nasia. Control C26 mice received DMSO only. Non-tumor bearingmice received either TSA or DMSO on the same schedule as tumorbearing mice. Follistatin and myostatin mRNA levels were as-sayed by quantitative real-time RT-PCR. Results: Tumor size,fractional weight loss, and fractional muscle mass were not dif-ferent in myostatin null mice versus wild-type mice (Student’st-test). However, there was a trend towards greater fractional fatloss (35% � 20.2 vs. 20.35% � 19.45%, n � 12 per group, p �0.0844), along with increased markers of inflammation, includinggreater total spleen mass (p � 0.01) and greater liver to body massratios ( p � 0.0001) at lower fractional tumor burdens in myosta-tin null mice. This result indicates that myostatin null mice arenot protected from cancer cachexia and moreover, may actually bemore sensitive to tumor-induced inflammation. In the pharmaco-logical model, TSA treatment induced follistatin expression 2.5fold in non-tumor bearing mice and 1.5 fold in C26 mice. More-over, TSA reduced myostatin expression 50% in non-tumor bear-ing mice and 20% in C26 mice. Consistent with reduced myostatinactivity, the gastrocnemius from TSA only mice were 22% largerthan DMSO only controls ( p � 0.001). TSA treatment did notprotect mice from C26 wasting, however. Percent body weightloss,fat loss and skeletal muscle loss were statistically indistinguish-able in TSA versus DMSO treated C26 mice. Conclusion: Neithergenetic nor pharmacological inhibition of myostatin activity wassufficient to prevent or reduce muscle wasting in two models ofcancer cachexia. These results suggest that despite their promisein muscle dystrophies, myostatin inhibitors may not be effective indiseases associated with chronic inflammation.
QS100. AN IN VIVO SELECTION MODEL IDENTIFIES MO-LECULAR CHANGES NECESSARY FOR LIVER ME-TASTASIS FROM PANCREATIC CANCER. Jason B.Fleming1, Yaan Kang1, Sean Dineen2, Rolf A. Brekken2;1MD Anderson Cancer Center, Houston, TX; 2UT South-western Medical Center, Dallas, TX
Introduction: Tumor cell subpopulations metastatic to distant organsare believed to differ from less metastatic populations in a wide range ofcell characteristics resulting from genetic and epigenetic changes.These cells are often characterized by the ability to detach from theprimary tumor mass, home to distant sites and survive as single cells.Here we report establishment of a pancreatic adenocarcinoma cell linethat preferentially metastasizes to the liver. Analysis of these cells hascharacterized the distinct phenotype acquired in vivo that is necessaryfor metastasis to occur. Methods: Panc-1 human pancreatic tumorcells were cultured in conditions of limited dilution for the isolation ofpopulations derived from single cell suspensions. These subclones werethen established in vitro and subsequently 1�106 cells were implantedin the pancreas of nu/nu mice and allowed to grow and metastasize. Onesubclone (C5) established a liver metastasis that was harvested fromthe animal, grown in vitro (named C5LM1) and passaged throughanother animal via orthotopic injection to generate C5LM2, a cell linethe preferentially metastasizes to the liver. The pattern of metastasis tothe liver for each cell line was evaluated by histology. In vitro studies ofPanc-1, C5, C5LM1 and C5LM2 examined colony formation in three-
307ASSOCIATION FOR ACADEMIC SURGERY AND SOCIETY OF UNIVERSITY SURGEONS—ABSTRACTS





























