Embed Size (px)
Citation preview

E L S E V I E R Biochimica et Biophysica Acta 1249 (1995) 15-22
BB Biochi~ic~a et Biophysica A~ta
Purification and characterization of phosphoenolpyruvate carboxykinase from Trypanosoma brucei
Marjory Hunt, Peter Ki3hler *
l~stitute of Parasitology, Universi~' of Z~rich, Winterthurerstr. 266a, 8057 ZUrich, Switzerland
Received 18 October 1993; accepted 11 November 1994
Abstract
ATP-dependent phosphcenolpyruvate carboxykinase (EC 4.1.1.49; PEPCK, ATP) was purified from glycosomes of cultured procyclic Trypanosoma brucei to electrophoretic homogeneity. The purified enzyme exhibited a mean specific activity of 83 units mg- i , as measured in the carboxylation direction at 30 ° C. A similar activity was obtained for the decarboxylation reaction. The enzyme was shown to be a homodimer in solution with a subunit molecular mass of 59 kDa. Amino acid sequence analysis suggested that the PEPCK (ATP) is identical to the tl2.¢panosomal protein p60, the sequence of which was previously predicted from the corresponding nucleotide sequence by other investigators. The basic nature of the enzyme was indicated by a high isoelectric point (pH 8.9). The enzyme was found to be strictly dependent on adenosine nucleotides for activity, as well as on the presence of Mn ~+. Mg 2+ was found to be ineffective as activator of the trypanosomal enzyme, but a combination of subsaturating ( < 300 ~M) concentrations of Mn 2+ and high concentrations of Mg 2+ caused a synergistic effect on the carboxylation activity, indicating a dual cation requirement. Mn 2+ is necessary to activate the enzyme and 1tin 2+ or Mg 2+ most likely forms the cation-nucleotide complex as the active form of the substrate. Relatively high (5 mM) levels of ATP were required to produce a significant inhibition of the carboxylation reaction. Quinolinic acid, a structural analogue of oxaloacetate, completely inhibited the decarboxylation reaction at a 1 mM concentration. The apparent Michaelis constants of the enzyme were 490 /~M for PEP, 37/~M for oxaloacetate, 40 /~M for ADP, 10.3 /~M for ATP, 970 /~M for Mn 2+ and 26 mM for HCOy. Endogenous substrate concentrations were found to be 327 nmol PEP, 1486 nmol ADP, 4200 nmol ATP and 11.5 nmol Mn 2+ (ml cell volume)- i. Our kinetic data suggest that under physiological conditions PEPCK (ATP) in T. brucei is bidirectional and that its activity is regulated primarily by mass action. The physiological relevance of the enzyme in procyclic T. brucei is discussed.
Keywords: Phosphoenolpyruvate carboxykinase (ATP); (Topanosoma brucei)
1. Introduction
Trypanosoma brucei, the causative agent of sleeping sickness in man and related diseases in animals, undergoes a series of differentiation steps during its complex develop- mental cycle which are associated with remarkable alter- ations in ultrastructure and metabolism [1-7]. As blood- stream form T. brucei differentiate into procyclic insect forms, they switch from a primarily glycolytical utilization of glucose to oxidatwe pathways mediating substrate
Abbreviations: PEP, phcsphoenolpyruvate; PEPCK, phospho- enolpyruvate carboxykinase; PSG, phosphate-buffered saline containing glucose; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel elec- trophoresis; TCA, tricarboxylic acid; TED buffer, 25 mM Tris-HC1, 5 mM EDTA, 1 mM DTr and 1 mM NaN 3, pH 7.8.
* Corresponding author. Fax: +41 1 3630478.
0167-4838/95/$09.50 © 1995 Elsevier Science B.V. All rights reserved SSDI 0 1 6 7 - 4 8 3 8 ( 9 5 ) 0 0 0 6 1 - 5
catabolism to CO 2 and organic acids. During this process, the activity and distribution of many enzymes change dramatically, including an increase in the activity of phos- phoenolpyruvate carboxykinase (PEPCK) up to levels ten times higher than those observed in bloodstream forms [8,9]. Apart from this stage specificity, trypanosomal PEPCK is unique in that it is localized in microbody-l ike organelles, the glycosomes, and its activity is dependent on ATP. It has been suggested that PEPCK (ATP) (EC 4.1.1.49) within the glycosomal compartment of procyclic T. brucei constitutes part of a pathway involved in the reoxidation of glycolytically produced NADH and in ATP synthesis [10,11]. In the tissues of higher animals, the reaction catalyzed by PEPCK is the first committed step in the process of gluconeogenesis, from pyruvate or other low molecular mass carbon precursors. This enzyme (EC 4.1.1.32) requires GTP (or ITP) as nucleoside triphosphate

16 M. Hunt, P. Ki~hler / Biochimica et Biophysica Acre 1249 (1995) 15-22
and is located in the cytosol and mitochondria of animal cells [12-14].
Besides in kinetoplastid flagellates [ 15,16], the presence of ATP-dependent PEPCKs have been reported in bacteria [17], yeast [18] and plants [19]. The enzyme has been partially purified from T. brucei procyclic forms in analyt- ical scale, but the properties of this preparation were not described [20]. Recently, Parsons and Smith [21] have reported that the sequence of the gene coding for the T. brucei glycosomal protein, p60, previously described by Kueng et al. [22], is homologous to the PEPCK (ATP) genes from yeast and bacteria.
In order to study the physiological role of PEPCK (ATP) in insect form T. brucei, we have purified the enzyme from these stages of the parasite and characterized its major properties. We have also demonstrated by N- terminal amino acid sequence analysis of the purified protein that the trypanosomal PEPCK (ATP) is identical to the glycosomal protein, p60, previously described in the same organism.
2. Materials and methods
2.1. Organisms and chemicals
Procyclic T. brucei brucei used in these experiments derived from cultures of bloodstream form trypanosomes, stock STIB 348 TBAAB (Swiss Tropical Institute, Basel). The organisms were cultured at 27 ° C in SDM-79 medium supplemented with 10% heat-inactivated fetal bovine serum and 10 /xg m1-1 gentamycin, in the presence of 3 mM citrate, as described previously [9]. Chemicals, reagents and materials used in this work were of the highest quality commercially available.
2.2. Purification o f P E P C K (ATP)
All purification steps described below were performed at 0 - 4 ° C, unless otherwise stated. Pellets of cultured procyclic trypanosomes of approximately 25 g wet wt. were washed three times by successive cycles of resuspen- sion in an ice-cold phosphate buffered saline containing glucose (PSG) (60 mM sodium phosphate, 44 mM NaCI, 60 mM glucose, pH 7.4) and disrupted by grinding with 1.5 volumes of dry silicon carbide. The extent of cell breakage was monitored microscopically. When the major- ity of the cells (approximately 80%) were disintegrated, the paste was resuspended in TED buffer (25 mM Tris-HCl, 5 mM EDTA, 1 mM DTT and 1 mM NAN3, pH 7.8) containing 250 mM sucrose to a protein concentration of approx. 5 mg ml 1. After a first centrifugation for 5 min at 100 × g, the supernatant was centrifuged for 10 min at 1000 × g. The glycosome enriched pellet was obtained by centrifugation of the 1000 × g supernatant for 30 min at 10 000 X g and diluted in TED buffer containing 250 mM
sucrose to 5 mg protein ml- 1 prior to storage at - 80 ° C. After thawing, about 50 mg of glycosomal protein were resuspended in modified TED buffer (TED buffer supple- mented with 1 /zM leupeptin, pH 7.0) containing 1 M (NH4)2SO 4, 0.1% Triton X-100 and 0.5 M NaCI to give a final protein concentration of 1 mg ml- ~. The mixture was left to equilibrate on ice for 30 min and then centrifuged for 10 rain at 14000 X g. The supematant was filtered on gauze (Rhena, Switzerland) and applied to a 22 × 1.6 cm Phenyl-Sepharose CL-4B column (Pharmacia LKB), equi- librated in modified TED buffer containing 1 M (NH4)2SO 4. This chromatographic step was carried out at 18 ° C. Sample application and washing with the equilibra- tion buffer were performed at a flow rate of 25 ml h -1. PEPCK (ATP) was eluted overnight at a flow rate of 11.5 ml h -1 with a 0-70% linear gradient of 3.2 M 2- methoxyethanol, 0.85 M ethanol and I% Brij-35 (w /v ) in modified TED buffer. The gradient was established in modified TED buffer containing 1 M (NH4)2SO 4 as the initial concentration, which decreased during the elution process to 0.3 M. Fractions (5 ml aliquots) containing PEPCK (ATP) activity were pooled, concentrated and desalted by ultrafiltration and washing with modified TED buffer in Macrosep centrifugal concentrators (Filtron, MD, USA). The concentrated protein solution was then chro- matographed on a 10.2 × 1.0 cm Cibacron Blue 3GA column (Type 3000-CL, insolubilized on cross-linked 4% beaded agarose, Sigma), equilibrated in modified TED buffer containing 50 mM NaC1. After sample application, the column was washed with the latter buffer and subse- quently with modified TED buffer containing 50 mM NaC1 and 10 mM glycerol 3-phosphate, at a flow rate of 15.6 ml h -1. PEPCK (ATP) was eluted overnight with a 0.05-0.65 M linear gradient of NaC1 in modified TED buffer. Flow rate and fraction size were 7.8 ml h - 1 and 2.5 ml, respectively. Pooled fractions from the Cibacron chro- matography were concentrated to 0.5 ml as described above and further purified by gel filtration on an 85 × 1.7 cm Bio-Gel P-60 column (Bio-Rad), equilibrated with modified TED buffer (pH 7.0) containing 0.4 M (NH4)2SO 4, 15% methoxy-ethanol, 3% ethanol and 0.6% Brij 35. PEPCK (ATP) was eluted overnight with the same buffer at a flow rate of 2.8 ml h- 1. Fractions of 5 ml were collected and stored at - 8 0 ° C.
2.3. Enzyme assays
PEPCK (ATP) activity was determined spectrophoto- metrically by following the decrease in absorbance at 340 nm at 30°C in the presence of an appropriate amount of enzyme. The carboxylation of phosphoenolpyruvate (PEP) to oxaloacetate was measured in a 1.0-ml reaction mixture containing 100 mM Mops, 150 mM NaHCO3, 1 mM DTT, 10 mM NaF, 3 mM MnCI2, 5 mM PEP, 0.15 mM NADH and 6 units of malate dehydrogenase, pH 6.9. The reaction was started by the addition of 2 mM ADP. The decarboxy-

M. Hunt, P. K6hler / Biochimica et Biophysica Acta 1249 (1995) 15-22 17
lation of oxaloacetate w~Ls measured in a 1.0-ml reaction mixture containing 100 mM Mops-KOH, 1 mM DTT, 1 mM MnC12, 1 mM ATP, 0.2 mM oxaloacetate, 0.15 mM NADH, 0.6 units pyruvate kinase, 1.3 units lactate dehydrogenase, pH 7.5. The reaction was initiated by the addition of 1 mM ATP. The enzyme-catalyzed rates re- ported here have been corrected for the spontaneous decar- boxylation of oxaloacetate in the absence of PEPCK (ATP). The decarboxylation of oxaloacetate to pyruvate was deter- mined in a 1.0-ml reaction mixture containing 100 mM Mops, 1 mM DTT, 1 mM MnC12, 0.15 mM NADH, 1.3 units of lactate dehydrogenase, pH 7.5. The reaction was initiated by the addition of 0.2 mM oxaloacetate. Apparent Michaelis constants (K m) were determined from Lineweaver-Burk double reciprocal plots of variation of enzyme activity with sub:~trate concentration. These values were calculated using total concentrations of substrates and divalent metal ion. Attempts have not been made to deter- mine the concentrations of free Mn 2+ present in the complex assay system. For all measurements of the en- zyme's carboxylation reaction, prior to the initiation of the reaction the pH of the assay mixtures was set to 8.0 with NaOH followed by the adLdition of solid NaHCO 3 and final setting of the pH to 6.9 with HC1. Hexokinase and phos- phoglucose isomerase, which were used as glycosomal marker enzymes throughout the glycosome purification procedure, were assayed according to Bergmeyer [23]. Effectors and inhibitors of PEPCK (ATP) were added to the reaction mixture before initiation of the assay, at the concentrations indicated in Table 3. One enzyme unit corresponds to 1 /zmol of substrate transformed by PEPCK (ATP) per min at 30 ° C.
2.4. Molecular mass and p H optimum determinations
NaHCO 3. The buffers used were a mixture of 25 mM each of sodium acetate, Mes, Hepes and glycine (pH 4.9-8.1), or a mixture of Mes and Mops, 60 mM each (pH 5.0-8.4), or 200 mM Mops (pH 6.5-7.5). For the decarboxylation direction, the assay conditions were the same as described under 'enzyme assays', except that 200 mM Hepes (pH 5.5-8.5) or a mixture of 600 mM each of Mes and Mops (pH 5.5-8.5) was used. Under the various pH conditions substrate and cofactor concentrations were always saturat- ing.
2.5. SDS-PAGE, isoelectric focusing and protein quantita- tion
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out according to the method of Laemmli [25] under reducing and denaturing conditions. Gels composed of a 4% stacking and a 12% separating gel of 0.75 mm thickness were employed. Protein bands were visualized by staining with Coomassie brilliant blue R-250. Isoelec- tric focusing was performed at 5 ° C on polyacrylamide gels of 0.5 mm thickness, containing Pharmalyte 8.0-10.5 (Pharmacia LKB) on an isoelectric focusing apparatus (Pharmacia LKB). The electrophoresis buffers employed were 0.25 M Hepes (anode) and 1 M NaOH (cathode). The p l of the PEPCK (ATP) was determined graphically by plotting the migration of standard proteins from a high p I calibration kit (Pharmacia) against their pls. All solutions were degassed and a container with solid NaOH was placed in the electrophoresis chamber, which was made air-tight, to protect the pH gradient from atmospheric CO 2. Protein determinations were performed using the Coomassie brilliant blue G-250 based assay from Bio-Rad, with bovine serum albumin as standard.
The molecular mass of PEPCK (ATP) was estimated by using a 91 × 1.6 cm Sephadex G-200 column, equilibrated with 25 mM Tris-HCl, 5 mM EDTA, 1 mM NaN 3, 100 mM NaC1, pH 7.0. The enzyme was eluted from the column with the same buffer at a flow rate of 4.8 ml h- i. The column was calibrated with standard proteins of known molecular mass. The size of the PEPCK (ATP) was calcu- lated following the recoramendations of the manufacturer (Pharmacia LKB). The molecular mass markers used were dextran blue 2000 (2000 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa) and ribonuclease A (12,.7 kDa). The pH optimum of PEPCK (ATP) was detemained according to the procedure used by Krakow and W~ng [24] for the estimation of the pH optimum of trypanosomal glycerol kinase. For the carboxylation reaction the assay conditions were the same as described above, except that the 100 mM Mops buffer was replaced by other buffer components, and the pH of the reaction mixture was adjusted to the desired values with NaOH or HC1, following the addition of 20 mM
2.6. Amino acid sequence analysis
N-terminal amino acid sequence analysis was per- formed using a pulsed-liquid-phase sequencer, equipped with an on-line PTH-amino acid analyzer (Applied Biosys- tems, Model 477A and 120A, respectively). Prior to analy- sis, samples of purified PEPCK (ATP) were concentrated and washed several times with 30% isopropanol in extra pure deionized water (Merck) using a Macrosep centrifu- gal concentrator. To avoid N-terminal blocking of the protein, all reagents were of the purest quality available. Sequencing of PEPCK (ATP) was performed three times.
2.7. Determination o f metabolites and divalent metal ions
Procyclic trypanosomes were grown in vitro as de- scribed above. The cells were pelleted and washed three times with PSG. For the measurement of metabolite con- centrations, the final pellet was resuspended in 15% per- chloric acid (2 ml per g wet wt. of pellet) and left to stand on ice for 30 min. The mixture was centrifuged 15 min at

18 M. Hunt, P. KShler / Biochimica et Biophysica Acta 1249 (1995) 15-22
10 000 X g and the supernatant further treated with a small volume of 15% perchloric acid and then processed as described above. The pH of the final supernatant was adjusted to 7.0 with 1 M KOH. Concentrations of sub- strates and nucleotides were determined according to Bergmeyer [23]. For the determination of manganese and cobalt, the washed trypanosome pellet was resuspended in about 10 volumes of 50% ( v / v ) HNO 3. The mixture was heated to 80°C under constant stirring until the volume was reduced to a few ml. The residual solution was diluted with extra pure deionized water (Merck, Switzerland), filtrated through filter paper, and the pH adjusted to 7.0 with 1 M NaOH. Solid Chelex-100 (Bio-Rad) was added to a concentration of approximately 50 mg ml-1 and the mixture was left to stand for 1 b at room temperature on a revolving mixer. Following centrifugation for a few sec- onds at 14000 × g the supernatant was removed and the metal ions were eluted off of the chelating resin by stirring in 1.5 ml 1 N HC1 for 5 min. The suspension was centrifuged as above and the cation concentrations were determined in the acidic supernatant by atomic absorption spectrometry (Instrumentation Laboratory, Milano, Italy), using an air-acetylene flame. Cobalt and manganese were measured at 240.7 and 279.5 nm, respectively.
3. Results
3.1. Pur i f i ca t ion
PEPCK (ATP) from cultured procyclic T. bruce i was purified to electrophoretic homogeneity (Fig. 1). In the representative example of the purification procedure (Ta- ble 1), the enzyme was purified 122-fold, with a specific activity of 81.8 units m g - ~ and an overall recovery of 7%. Based on the average of four purifications, the enzyme possesses a specific activity of 83 units mg-1 and consti- tutes 5 - 1 0 % of the glycosomal protein fraction. The data presented in Fig. 1 and Table 1 show that Cibacron Blue affinity chromatography results in a remarkable decrease of contaminating glycosomal proteins and a substantial purification of PEPCK (ATP). However, it was noticed that the type of elution pattern of this chromatography step varied among different purifications. In some preparations, as that shown in Fig. 1, only one minor contaminant band
1 2 3 4
PEPCK 97 66
43
31
22
14
Fig. 1. Analysis of the purification of PEPCK (ATP) from T. brucei on SDS-PAGE. Lanes: 1, hydrophobic interaction chromatography.; 2, Cibacron 3GA chromatography; 3, gel filtration chromatography; 4, molecular mass markers (kDa). Glycosomal fraction is not shown.
was detectable, whereas in others two to four additional bands were visible on the gel resulting in correspondingly lower specific activities of the enzyme following purifica- tion by Cibracron Blue chromatography. Pure PEPCK (ATP) was relatively stable at 4 ° C, with no notable loss of activity within 24 h. Storage of the enzyme in the buffer system used for the gel filtration chromatography step did not result in a significant change in activity when main- tained at - 80 ° C over a period of three months.
3.2. P h y s i c a l and c h e m i c a l p roper t i e s
The molecular mass of the enzyme was determined by SDS-PAGE under denaturing conditions and by gel filtra- tion chromatography on Sephadex G-200. On SDS-poly- acrylamide gels the subunit molecular mass of PEPCK (ATP) was 59 _ 1.3 kDa (n = 4), while under nondenatur- ing conditions, gel filtration chromatography resulted in an estimated native molecular mass of 115 + 3 kDa (n = 3), indicating that in solution this enzyme is a homodimer. The isoelectric point ( p I ) of the enzyme was found to be relatively high (8.86 +__ 0.01; n = 3), an observation which is in accordance with the basic nature of most of the glycosomal proteins so far described. In Fig. 2, the align-
Table 1 Purification of PEPCK (ATP) from T. brucei ~
Purification stage Total protein Total activity Specific activity Purification Yield (mg) (units) (units/mg protein) (%)
Crude homogenate 600 404 0.67 1.0 100 Glycosomes 39 350 9.1 13.6 87 Phenyl-Sepharose 23 273 11.9 17.8 68 Cibacron Blue 3GA 1.5 114 76.0 113 28 Bio-Gel P60 0.33 27 81.8 122 7
a Result of a representative purification procedure. One unit is defined as 1 /xmol of PEP transformed to oxaloacetate per min at 30 ° C.

M. Hunt, P. K6hler / Biochimica et Biophysica Acta 1249 (1995) 15-22 19
H2N-GIn-Pro-IIe-Ile-His-Lys-Asn-Leu-Thr--PEPCK(ATP) w C O O H
H2N-Met-Ala-Pro-lle-Ile-His-Lys-Asn-Leu-Thr p60 COOH
Fig. 2. Comparison of the N-terminal amino acid sequence of PEPCK
(ATP) with that of the glycosomal protein p60 from cultured procyclic T. brucei. The sequence of the PEPCK (ATP) was determined by amino
acid sequencing analysis as described in Section 2. The sequence of the
p60 was predicted from the cor~:esponding DNA sequence [21].
ment of the sequence of the first nine amino acids obtained from our purified PEPCK (ATP) protein and the corre- sponding amino acid sequence predicted from the DNA sequence, coding for the glycosomal protein p60 [22] shows an almost perfect :match. The absence of the initial methionine in our sequence is not unduly surprising, as this N-terminus is known to be frequently cleaved post- translationally. The observed glutamine vs alanine mis- match is most probably due to intraspecific biochemical variation between the different trypanosome strains used in the two studies.
3.3. pH optimum
The trypanosomal PEPCK (ATP) showed a broad pH optimum in the decarboxylation direction, displaying max- imum activity around pH 7.3. In the carboxylation direc- tion, the pH optimum of 1:he enzyme was 6.9.
3.4. Kinetic studies
The kinetic parameters of purified PEPCK (ATP) were determined for both reaction directions using an enzyme preparation obtained from one of the purification proce- dures. Under the standard assay conditions employed, a preparation of the purified enzyme catalyzed the decar- boxylation of 78 + 3.5 /J,mol of oxaloacetate min ~(mg protein)-l at 30 ° C. A siimilar catalytic activity was ob- served for the opposite reaction direction, in which 69 ___ 3.0 /xmol of phosphoenolpymvate were converted to oxaloac- etate min - l (mg enzyme) -1 at 30°C. The apparent Michaelis constants of the enzyme for its substrates are
summarized in Table 2. Comparison of these data with the endogenous metabolite and ion concentrations indicates that adenosine nucleotides are propably available in satu- rating amounts (Table 2). However, endogenous PEP lev- els were found to be in the range of the K m for this substrate and oxaloacetate levels are expected to be lower than the apparent K m value of the enzyme calculated for this compounds, indicating that the activity of the enzyme is regulated by substrate availability. The failure to detect oxaloacetate within the trypanosome cell is probably due to both the low endogenous concentration and the gradual loss of this compound due to spontaneous or metal-cata- lyzed decarboxylation prior to analysis. The amounts of Mn 2÷ detected within procyclic T. brucei (11.5 nmol ml l) were far below those required for optimal PEPCK (ATP) activation. However, these low endogenous Mn 2÷ levels were found to be close to those sufficient to produce a slight activation of the enzyme when millimolar concen- trations of Mg 2+ were simultaneously present in the assay system (Table 4).
3.5. Substrate and metal ion specificities
In both reaction directions no activity was noted for T. brucei PEPCK (ATP) when the adenosine nucleotide was replaced by the corresponding guanosine or inosine nu- cleotide substrates (Table 3). In both reaction directions, the enzyme was strictly dependent on the presence of Mn 2÷ for activity. In the presence of millimolar Mg 2÷ concentrations, but absence of Mn 2÷, only residual activity levels were noted that may be caused by contaminant levels of Mn 2÷ in the system. In the carboxylation direc- tion, the divalent cation requirement was studied in more detail. As shown in Table 4, enzyme activity was strongly dependent on Mn 2÷ with an apparent K m for Mn 2÷ of 0.97 mM (calculated as total cation concentration) and maximal velocities at 3 mM levels. There was very little observed activation of enzymatic activity with Mg 2+ over a concentration range of 1-10 mM. The low levels of activity ( < 3% compared to Mn 2+ at 3 mM)) observed
Table 2
Michael is constants for substrates and activator of PEPCK (ATP) and contents of substrates and activator in cultured procyclic T. brucei
Substrates and manganese K m ( / zM) a Endogenous substrate and manganese contents (nmol per ml cell volume) b
PEP 490 ± 100 327 + 24
Oxaloacetate 37 + 18 not detectable
ADP 40 _+ 11 1486 ___ 232 ATP 10.3 + 0.2 4200 + 30
HCO 3 25.7 d + 7.4 c not determined Mn 2+ 970 11.5 d
Enzyme activity measurements and calculation of apparent K m values were carried out as described in Section 2. Values represent means of three determinations + S.D.
a Apparent K m values were calculated using total concentration of substrate or activator. b Calculated on the basis that the volume of a trypanosome cell is approximately 30 fl (3 - 10 I° cells equals 1 ml). c Expressed in mM.
d Values represent means of twc, determinations.
20 M. Hunt, P. KiJhler / Biochimica et Biophysica A cta 1249 (1995) 15-22
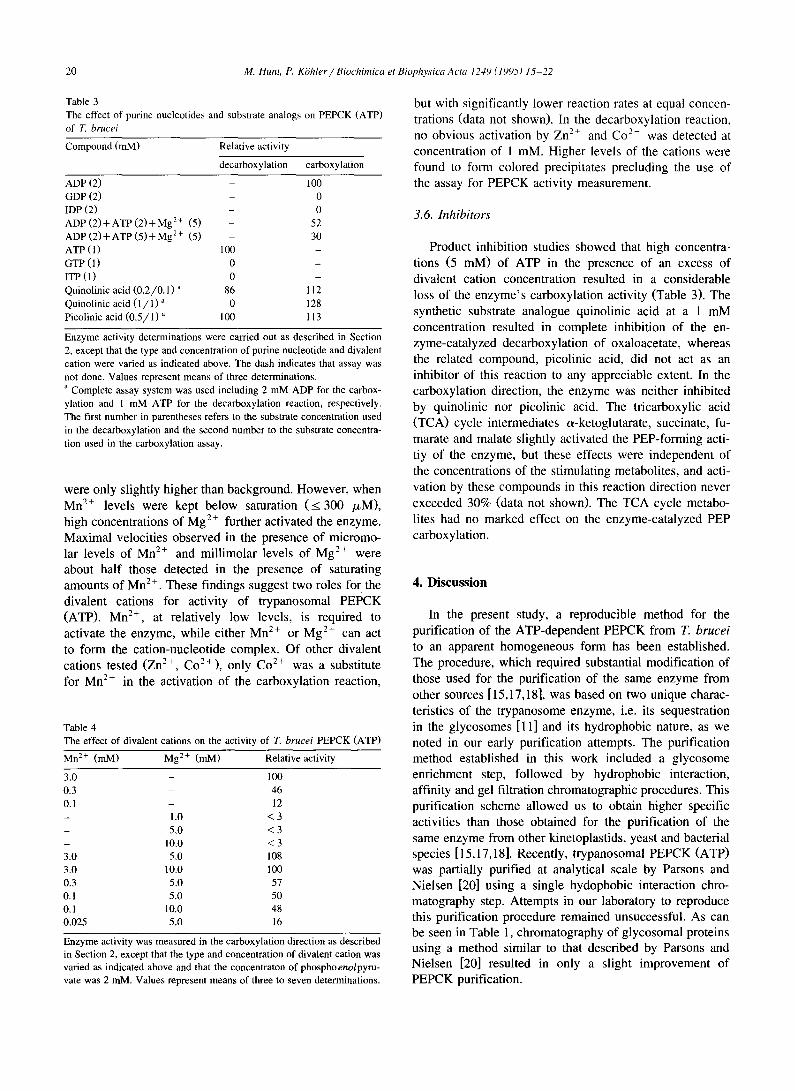
Table 3
The effect of purine nucleotides and substrate analogs on PEPCK (ATP) of T. brucei
Compound (mM) Relative activity
decarboxylation carboxylation
ADP (2) - 100 GDP (2) - 0
IDP (2) - 0 ADP ( 2 ) + A T P ( 2 ) + M g 2+ (5) - 52
ADP ( 2 ) + A T P ( 5 ) + M g 2+ (5) - 30
ATP (1) 100 -
GTP (1) 0 - ITP ( l ) 0 -
Quinolinic acid (0 .2 /0 .1 ) a 86 112
Quinolinic acid ( 1 / 1 ) a 0 128
Picolinic acid (0 .5 /1 ) a 100 113
Enzyme activity determinations were carried out as described in Section 2, except that the type and concentration of purine nucleotide and divalent
cation were varied as indicated above. The dash indicates that assay was
not done. Values represent means of three determinations. a Complete assay system was used including 2 mM ADP for the carbox-
ylation and 1 mM ATP for the decarboxylation reaction, respectively. The first number in parentheses refers to the substrate concentration used
in the decarboxylation and the second number to the substrate concentra-
tion used in the carboxylation assay.
were only slightly higher than background. However, when Mn 2÷ levels were kept below saturation ( < 300 /zM), high concentrations of Mg 2+ further activated the enzyme. Maximal velocities observed in the presence of micromo- lar levels of Mn 2÷ and millimolar levels of Mg 2÷ were about half those detected in the presence of saturating amounts of Mn 2÷. These findings suggest two roles for the divalent cations for activity of trypanosomal PEPCK (ATP). Mn 2+, at relatively low levels, is required to activate the enzyme, while either Mn 2÷ or Mg 2+ can act to form the cation-nucleotide complex. Of other divalent cations tested (Zn 2+, C02+), only Co 2÷ was a substitute for Mn 2+ in the activation of the carboxylation reaction,
Table 4 The effect of divalent cations on the activity of T. brucei PEPCK (ATP)
Mn 2+ (mM) Mg 2+ (mM) Relative activity
3.0 - 100
0.3 - 46
0.1 - 12 - 1 .0 < 3
- 5 . 0 < 3
- 1 0 . 0 < 3
3.0 5,0 108 3.0 10,0 100
0.3 5,0 57 0.1 5 . 0 50 0.1 10.0 48 0.025 5,0 16
Enzyme activity was measured in the carboxylation direction as described in Section 2, except that the type and concentration of divalent cation was varied as indicated above and that the concentraton of phosphoenolpyru- vate was 2 mM. Values represent means of three to seven determinations.
but with significantly lower reaction rates at equal concen- trations (data not shown). In the decarboxylation reaction, no obvious activation by Zn 2+ and Co 2+ was detected at concentration of 1 mM. Higher levels of the cations were found to form colored precipitates precluding the use of the assay for PEPCK activity measurement.
3.6. Inhibitors
Product inhibition studies showed that high concentra- tions (5 raM) of ATP in the presence of an excess of divalent cation concentration resulted in a considerable loss of the enzyme's carboxylation activity (Table 3). The synthetic substrate analogue quinolinic acid at a 1 mM concentration resulted in complete inhibition of the en- zyme-catalyzed decarboxylation of oxaloacetate, whereas the related compound, picolinic acid, did not act as an inhibitor of this reaction to any appreciable extent. In the carboxylation direction, the enzyme was neither inhibited by quinolinic nor picolinic acid. The tricarboxylic acid (TCA) cycle intermediates c~-ketoglutarate, succinate, fu- marate and malate slightly activated the PEP-forming acti- tiy of the enzyme, but these effects were independent of the concentrations of the stimulating metabolites, and acti- vation by these compounds in this reaction direction never exceeded 30% (data not shown). The TCA cycle metabo- lites had no marked effect on the enzyme-catalyzed PEP carboxylation.
4. Discussion
In the present study, a reproducible method for the purification of the ATP-dependent PEPCK from T. brucei to an apparent homogeneous form has been established. The procedure, which required substantial modification of those used for the purification of the same enzyme from other sources [15,17,18], was based on two unique charac- teristics of the trypanosome enzyme, i.e. its sequestration in the glycosomes [11] and its hydrophobic nature, as we noted in our early purification attempts. The purification method established in this work included a glycosome enrichment step, followed by hydrophobic interaction, affinity and gel filtration chromatographic procedures. This purification scheme allowed us to obtain higher specific activities than those obtained for the purification of the same enzyme from other kinetoplastids, yeast and bacterial species [ 15,17,18]. Recently, trypanosomal PEPCK (ATP) was partially purified at analytical scale by Parsons and Nielsen [20] using a single hydophobic interaction chro- matography step. Attempts in our laboratory to reproduce this purification procedure remained unsuccessful. As can be seen in Table 1, chromatography of glycosomal proteins using a method similar to that described by Parsons and Nielsen [20] resulted in only a slight improvement of PEPCK purification.

M. Hunt, P. Krhler / Biochimica et Biophysica Acta 1249 (1995) 15-22 21
PEPCK (ATP) from T. brucei resembles the homolo- gous enzyme from the related kinetoplastid T. cruzi in that it is composed of two identical polypeptide chains [15]. In contrast, ATP-dependent PEPCKs from Saccharomyces cerevisiae and Escherichia coli were reported to be te- trameric and monomeric, respectively [ 17,18]. The subunit size of PEPCK (ATP), as estimated by SDS-PAGE, varies among different species, with 42 kDa for T. cruzi [15], 54 kDa for E. coli [17], 59 kDa for T. brucei (this work) and 64 kDa for the yeast enzyme [18]. The subunit molecular mass of 59 kDa observed by SDS-PAGE for the T. brucei enzyme is in agreement with the 60 kDa value reported by Parsons and Nielsen [20] and the 52.5 kDa molecular mass predicted from the gene ,;equence coding for the glycoso- mal protein, p60. This protein was previously described by Kueng et al. [22] as a novel glycosomal protein with membrane binding capacities, expressed in procyclic forms of T. brucei. However, it was subsequently shown that the sequence of the cloned gene is 50% homologous to the gene coding for S. cere~isiae PEPCK (ATP) [21]. The N-terminal amino acid sequence of the purified PEPCK (ATP) obtained here confirms this observation and demon- strates that the P60 glycosomal protein is identical to PEPCK (ATP). The p l observed for the purified PEPCK (ATP) is distinctly higher than that measured for its cyto- plasmic homologue in other species. This unique structural feature is shared by mo,;t glycosomal enzymes and was suggested to be related to the mechanism of import of these proteins into the glycosomes [20,26,27].
A unique property of PEPCK (ATP) from kinetoplastids is their strict dependency on Mn 2+ for activity (this study and [15]), whereas the yeast and E. coli enzymes are also active, although to a lesse:r extent, in the presence of Mg 2+ and other divalent cations [17,28]. The concentrations of Mn 2÷ required for activation of the trypanosome enzyme were relatively high as compared to the trace amounts of this metal ion demonstrated in procyclic trypanosomes. A possible explanation for this discrepancy is provided by the studies with mixed divalent metal ion (Mn 2÷ and Mg 2÷) assays (Table 4). From these experiments it ap- pears likely that two independent functions for the two metal ions exist in the cztalysis by trypanosomal PEPCK (ATP). Mn 2÷ but not l~[g 2+ binds to and activates the enzyme while either Mn z'÷ or Mg 2÷ may serve to form the cation-nucleotide substrate. The concentrations of diva- lent metal ions in the glycosome of procyclic T. brucei have not been determined. However, the amount of total cellular Mn 2+ in these cells was estimated at about 10/zM and the total cellular Mg 2+ is likely to be in the lower millimolar range. This, together with our experimental data suggests that the divalent metal ion concentrations avail- able in the glycosome cculd approximate values that are required for PEPCK (ATP) activation. Similar observa- tions were previously made with the homologous enzyme in T. cruzi that was found to depend solely on Mn 2÷ for activity, while Mg 2÷ was suggested to form the cation-
nucleotide substrate in vivo [ 15]. Kinetic studies of GTP- depending PEPCKs (EC 4.1.1.32) from a few other sources have also been reported to require a dual function for cations in catalysis. For example, the enzymes from chicken liver mitochondria [29] and Ascaris muscle tissue [30] are activated by Mn 2+, but either Mn 2+ or Mg 2+ can serve to form the divalent cation-nucleotide complex. On the other hand, several other GTP-dependent PEPCKs were reported to be activated by either Mg 2÷ or Mn 2+ or even by other divalent metal ions, such as Co 2+ or Cd 2+ (for refs. see [29]). Further kinetic analyses are needed to elucidate the precise roles of the two cations in T. brucei PEPCK (ATP) catalysis.
The nucleotide specificity of PEPCK (ATP) and the inhibitory effect of ATP on the carboxylation reaction observed here are in agreement with previous reports. Urbina [15] showed that PEPCK (ATP) from T. cruzi was not active in the decarboxylation direction with GTP or ITP as nucleotide, and that ATP, GTP and ITP inhibited the carboxylation reaction. In the present study we could not demonstrate a significant inhibition of PEPCK (ATP) activity by picolinic acid, although the structurally related compound 3-mercaptopicolinic acid was reported to pro- duce a strong inhibitory effect on the homologous enzyme of T. cruzi [31]. However, a 1 mM concentration of quinolinic acid, another known substrate analogue inhibitor of PEPCKs, blocked completely the T. brucei PEPCK (ATP) reaction, even though only in the direction of oxaloacetate decarboxylation. Similar differences in the inhibition of PEPCK between both reaction directions have previously been observed with other substrate analogues [32], and may be determined by the reaction mechanism, the substrate affinities of the enzyme and the specificities of the assay conditions used to measure the enzymatic reaction.
The kinetic constants for the substrates of both reaction directions of the T. brucei PEPCK (ATP) were also found to be similar to those of the homologous enzyme in T. cruzi [15]. An exception is the K m for bicarbonate which was found to be 7-fold greater for the T. brucei enzyme and thus more similar to the values reported for the GTP-dependent PEPCKs from higher animal sources. This striking kinetic difference between the two homologous enzymes may indicate differences in their physiological function between both systems. Compared to the levels of PEP and adenine nucleotides detected in the trypanosome cell, the K m values for these compounds are either similar or much lower. The K m value for oxaloacetate is also low and in view of the expected high pCO 2 in the natural environment of the procyclic trypanosome, the mid-gut of the tsetse fly, the K m estimated for bicarbonate may also be considered as relatively low.
Bloodstream form T. brucei depend entirely on glycoly- sis for energy generation and excrete pyruvate as the major end product [9]. Upon transformation to procyclic forms, the composition and activities of some of the glycosomal

22 M. Hunt, P. KShler / Biochimica et Biophysica Acta 1249 (1995) 15 22
enzymes change dramatically. For example, the specific activities of PEPCK (ATP) and malate dehydrogenase increase 10- and 100-fold respectively, whereas hexoki- nase and pyruvate kinase drop to levels 15- to 20-fold below those present in bloodstream forms [8,9]. Simultane- ously, trypanosomes aquire a fully functional TCA cycle and a cytochrome-mediated electron transport chain, which allows them to oxidize carbohydrates more efficiently than bloodstream forms, as indicated by the formation of CO 2 as the predominant end product. In this pathway, PEPCK (ATP) is likely to play an important role, in that it catalyzes the conversion of glycolytically formed PEP into oxaioacetate and thus link glycolysis with the TCA cycle. However, procyclic T. brucei are known to grow in vitro solely on an amino acid diet [1,33] and in the mid-gut of the tsetse fly these organisms are assumed to live primarily on these substrates, especially proline, rather than on carbohydrates [34]. This suggests the functional activity of both a gluconeogenic pathway, in particular under condi- tions where the supplies of exogenous carbohydrate are scarce or absent, and oxidative routes to fulfil the energy requirement of the parasite. Within this complex metabolic network, PEPCK (ATP) very likely plays a central regula- tory role. Since its reaction is freely reversible this enzyme can perform either a gluconeogenic or catabolic reaction. The kinetic data of the present work support the assump- tion that the direction of catalysis of this enzyme in vivo is determined by several factors, i.e. the phosphate potential, the concentrations of oxaloacetate, PEP and bicarbonate and possibly also by small changes in the Mn 2+ concentra- tion within the glycosomal compartment of the try- panosome cell.
Acknowledgements
This study was supported by grants of the Swiss Na- tional Science Foundation (31-8880.86 and 31-29380.90). The authors wish to thank Dr. D.L. Pountney for atomic absorption analysis and Dr. P. Hunziker and Mr. N.E. Birchler for N-terminal amino acid sequence analysis.
References
[1] Brown, R.C., Evans, D.A. and Vickerman, K. (1973) Int. J. Para- sitol. 3, 691-704.
[2] Ghiotto, V., Brun, R., Jenni, L. and Hecker, H. (1979) Exp. Para- sitol. 48, 447-456.
[3] Overath, P., Czichos, J. and Haas, C. (1986) Eur. J. Biochem. 160, 175-182.
[4] Czichos, J., Nonnengaesser, C. and Overath, P. (1986) Exp. Para- sitol. 62, 283-291.
[5] Fairlamb, A.H. and Opperdoes, F.R. (1986) Carbohydrate Metabolism in Cultured Cells (Morgan, M.J., ed.), pp. 183-224, Plenum Press, New York.
[6] Giffin, B.F., McCann, P.P., Bitonti, A.J. and Bacchi, C.J. (1986) J. Protozool. 33, 238-243.
[7] KShler, P. (1989) Parasitology in Focus (Mehlhorn, H., ed.), pp. 412-453, Springer-Verlag, Heidelberg.
[8] Hart, D.T., Misset, O., Edwards, S.W. and Opperdoes, F.R. (1984) Mol. Biochem. Parasitol. 12, 25-35.
[9] Durieux, P., Schtitz, P., Brun, R. and K~Shler, P. (1991) Mol. Biochem. Parasitol. 45, 19-28.
[10] Opperdoes, F.R. and Cottem, D. (1982) FEBS Lett. 143, 60-64. [11] Broman, K., Knupfer, A.L., Ropars, M. and Deshusses, J. (1983)
Mol. Biocbem. Parasitol. 8, 79-87. [ 12] Chang, H.C. and Lane, M.D. (1966)J. Biol. Chem. 241,2413-2420. [13] Sato, A., Suzuki, T. and Kochi, H. (1986) J. Biochem. 100, 671-678. [14] Johnson, W.V., Kemp, J.R. and Anderson, P.M. (1990) Arch.
Biochem. Biophys. 280, 376-382. [15] Urbina, J.A. (1987) Arch. Biochem. Biophys. 258, 186-195. [16] Mottram, J. and Coombs, G.H. (1985) Biochim. Biophys. Acta 827,
310-319. [17] Goldie, A.H. and Sanwal, B.D. (1980) J. Biol. Chem. 255, 1399-
1405. [18] Tortora, P., Hanozet, G.M. and Guerritore, A. (1985) Anal. Biochem.
144, 179-185. [19] Utter, M.F. and Kolenbrander, H.M. (1972) The Enzymes (Boyer,
P.D., ed.) Vol. 6, pp. 136-154, Academic Press, New York. [20] Parsons, M. and Nielsen, B. (1990) Exp. Parasitol. 70, 276-285. [21] Parsons, M. and Smith, J.M. (1989) Nucleic Acids Res. 17, 6411. [22] Kueng, V., Schlaeppi, K., Schneider, A. and Seebeck, T. (1989) J.
Biol. Chem. 264, 5203-5209. [23] Bergmeyer, H.U. (1974) Methods of Enzymatic Analysis, 2nd edn.,
Academic Press, New York. [24] Krakow, J.L. and Wang, C.C. (1990) Mol. Biochem. Parasitol. 43,
17-26. [25] Laemmli, U.K. (1970) Nature 227, 680-685. [26] Borst, P. (1986)Biochim. Biophys. Acta 866, 179-203. [27] Michels, P.A.M. (1989) Exp. Parasitol. 69, 310-315. [28] Medina, V., Pontarollo, R., Glaeske, D., Tabel, H. and Goldie, H.
(1990) J. Bacteriol. 172, 7151-7156. [29] Lee, H.M., Hebda, C.A. and Nowak, T.J. (1981) Biol. Chem. 256,
12793-12801. [30] Rohrer, S.P., Saz, H.J. and Nowak, T. (1986) J. Biol. Chem. 261,
13049-13055. [31] Urbina, J.A., Osorno, C.E. and Rojas, A. (1990) Arch. Biochem.
Biophys. 282, 91-99. [32] Ash, D.E., Emig, F.A., Chowdhury, S.A., Satoh, Y. and Schramm,
V.L. (1990) J. Biol. Chem. 265, 7377-7384. [33] Evans, D.A. and Brown, R.C. (1972) J. Protozool. 19, 686-690. [34] Cross, G.A.M., Klein, R.A. and Linstead, D.J. (1975) Parasitology
71,311-326.





























