Embed Size (px)
Citation preview

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 1/70
PAC ACCORD
Actions concertées dans les Cancers COloRectaux et Digestifs
PRODIGE 5 PROTOCOLE ACCORD 17/0707
PHASE II-III STUDY COMPARING RADIOCHEMOTHERAPY WITH THE FOLFOX
REGIMEN VERSUS RADIOCHEMOTHERAPY WITH 5FU-CISPLATIN (HERSKOVIC REGIMEN) IN FIRST LINE TREATMENT OF PATI ENTS WITH
INOPERABLE OESOPHAGEAL CANCER
Working version n°11 containing amendement 1, 2, 3, 4, 5, 6, 7 and 8 accepted by CPP 10/09/2009
COORDINATOR
Pr Thierry CONROY Centre Alexis Vautrin
Department of medical oncology 6, avenue de Bourgogne - 54511 Vandoeuvre les Nancy
Tel : 03.83.59.84.60 Fax : 03.83.59.85.50
e-mail : [email protected]
ASSOCIATED COORDINATOR
Pr. Laurent BEDENNE Hôpital du Bocage
2 Bd Maréchal de Lattre de Tassigny - 21079 DIJON Cedex Tél. : 03 80 29 37 50 Fax : 03 80 29 37 22
Email : [email protected]
SPONSOR
Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC)
101, rue de Tolbiac - 75654 PARIS CEDEX 13 - FRANCE Tel. +33.1.44.23.04.04 Fax: +33.1.44.23.55.69

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 2/70
STUDY PROTOCOL AGREEMENT FORM
ACCORD 17/0707
Prodige 5
PHASE II-III STUDY COMPARING RADIOCHEMOTHERAPY WITH THE FOLFOX REGIMEN VERSUS RADIOCHEMOTHERAPY WITH 5FU-CISPLATIN (HERSKOVIC
REGIMEN) IN FIRST LINE TREATMENT OF PATIENTS WITH I NOPERABLE OESOPHAGEAL CANCER
NAME AND FONCTION
ADDRESSES DATE (JJ-MM-AA) SIGNATURE
Directeur du BECT (FNCLCC)
Dr. Jean GENEVE
FNCLCC/BECT 101 rue de Tolbiac
75654 Paris cedex 13 Tel : 01.44.23.55.52 Fax : 01.44.23.55.69
E-mail : [email protected]
Project Manager (FNCLCC)
Christine MONTOTO-GRILLOT
FNCLCC/BECT 101 rue de Tolbiac
75654 Paris cedex 13 Tel : 01.44.23.55.67 Fax : 01.44.23.55.69
E-mail : [email protected]
Investigateur Coordonnateur
Pr Thierry CONROY
Centre Alexis Vautrin 6, avenue de Bourgogne
54511 Vandoeuvre les Nancy Tél. : 03.83.59.84.60 Fax : 03.83.59.85.50
E-mail : [email protected]
Data Manager
Sébastien LOUVEAU
Euraxi Pharma 10 rue Gutenberg
BP 80325 37303 Joué-lès-Tours Tél. : 02.47.74.30.47 Fax : 02.47.74.30.49
E-mail : [email protected]
Biostatistician
Sophie GOURGOU - BOURGADE
Centre Val-D’Aurelle Parc Euromédecine
34298 Montpellier cedex 5 tel :04.67.61.37.75 fax : 04.67.61.37.18
e-mail : [email protected]
PROTOCOLE WRITING COMITTEE
T. Conroy, A. Adenis, D. Azria, L. Bedenne, V. Boige, O. Bouché, M. Ducreux, P.L. Etienne, M. Giovannini, E. François, J. P Labat, F. Lorchel, V. Magnin, J.P Metges, P. Michel, F. Mornex, D. Peiffert, J.P. Pignon, T. Pignon, J.F. Seitz, F. Viret, M. Ychou, C. Montoto-Grillot.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 3/70
PROTOCOL SYNOPSIS
TITLE PHASE II-III STUDY COMPARING RADIOCHEMOTHERAPY WITH THE FOLFOX REGIMEN VERSUS RADIOCHEMOTHERAPY WITH 5FU-CISPLATIN (HERSKOVIC REGIMEN) IN FIRST LINE TREATME NT OF PATIENTS WITH INOPERABLE OESOPHAGEAL CANCER
INVESTIGATORS / TRIAL LOCATION
40 planned centers, France Coordinated by Prof. T. Conroy
STUDY OBJECTIVES PHASE II STUDY :
Primary
Secondary PHASE III STUDY :
Primary
Secondary
To assess the feasibility (completion of full treatment) in both arms. To assess endoscopic complete response rate in both arms. To assess the toxicity profile of each arm using NCI-CTC scale (version 3). To compare the progression-free survival (PFS) between 2 chemotherapy schedules delivered during concomitant radiotherapy: Folfox regimen versus Cisplatin/5-FU (Herskovic regimen). The PFS will be defined by the following events:
- tumor progression - metastasis diagnosis - esophageal second cancer - death from any cause
To compare overall survival, endoscopic complete response rate, incidence of grade 3-4 toxicities NCI-CTC and time to treatment failure between both regimens. To evaluate the quality of life using EORTC QLQ-C30 (version 3) and a validated disease- specific module EORTC QLQ-OES18.
STUDY DESIGN A multicenter randomized phase II trial followed by a phase III STUDY POPULATION Main Selection Criteria
Inclusion Criteria: Patients with: − Histologically proven adenocarcinoma, squamous cell or
adenosquamous carcinoma of the oesophagus ; − Inoperable oesophageal carcinoma (disease status :any T, N0 or N1,
M0 or M1a) or surgical contre-indication conditions ; − No prior treatment for oesophageal cancer (surgery, chemo- or
radiotherapy); − Peripheral neuropathy ≤ NCI-CTC grade 1; − Age ≥ 18 years; − ECOG Performance Status (PS) ≤ 2; − Sufficient (oral or with gastrostomy) calorific intake (> 1000
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 4/70
Kcal/m2/day); − Life expectancy ≥ 3 months ; − Adequate bone marrow reserve, normal renal and liver functions:
• Neutrophil count ≥ 1500/mm³ • Platelet count ≥ 100 000/mm³ • Hemoglobin ≥ 10 g/dl (after transfusion, if necessary) • Creatinine < 15mg/L • Total bilirubin level<1.5 x ULN • ALT/AST < 2.5 x ULN • Prothrombin time ≥ 60%;
− Laboratory values obtained the week preceding study entry; − Signed informed consent (prior to all study procedures); − Start of treatment within 28 days of inclusion.
Exclusion Criteria: − Metastatic disease except for third upper or cervical oesophagus tumor
with regional nodes, or third lower oesophagus tumor with celiac nodes (M1a);
− Multiple carcinomas of the oesophagus; − Small cell or undifferentiated carcinoma of the oesophagus; − Patients with cardia tumor (Siewert II) or gastric tumor extensive to the
oesophagus (Siewert III) are ineligible. − Complete dysphagia (grade 4 NCI-CTC), patient with exclusive
parenteral nutrition; − Weight loss within 3 months > 20% normal body weight; − Pregnant or breast-feeding woman; − Fertile patient not using adequate contraception; − Peripheral sensitive neuropathy with functional impairment; − Auditory disorders; − History of prior malignancies (other than cured non melanoma skin
cancer, cured cervical carcinoma in situ or stage I or II node negative head and neck cancer cured > 3 years ago);
− Prior cervical, thoracic and abdominal radiotherapy with field overlapping the proposed oesophageal radiotherapy field;
− Tracheo-oesophageal fistula or invasion of the tracheo-bronchial tree; − Previous myocardial infarction (inferior or equal to 6 months). Patients
with a previous myocardial infarction superior to 6 months, could be included only if: • no transient ischemia is shown by thallium myocardial scintigraphy
and • favourable advise for chemotherapy from a cardiologist is obtained;
− Other serious illness or medical conditions (such as symptomatic coronary disease, left ventricular failure or uncontrolled infection);
− Arterial disease stage II to IV according to the DE LERICHE and FONTAINE classification;
− Treatment with any other experimental drugs or participation in another clinical trial within 30 days of study screening;
− Concurrent treatment with any other anti-cancer therapy; − Concurrent treatment with phenytoine and Yellow fever vaccine; − Geographical, social or psychological circumstances preventing regular
follow-up.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 5/70
TOTAL EXPECTED NUMBER OF SUBJECTS
Phase II : to have 80 evaluable patients (40 in each arm), 88 patients (44 in each arm) will be included. Phase III : 169 other evaluable patients ; total of 266 patients, (133 par arm
including the phase II patients). EXPECTED NUMBER OF CENTRES
25 centers for Phase II, 40 centers for phase III.
STUDY DRUGS Formulations
Oxaliplatin : Powder for parenteral use, 100mg vials, dilution in 20 mls of water for injection or 5% glucose and reconstitution with 5% glucose solution to 250-500 ml.
Folinic acid : Commercially available formulation 5-FU : Commercially available formulation Cisplatin : Commercially available formulation
Administration route Intravenous. Dose regimen
Radiation (two arms) Rx > 6 MV, 2 to 4 beams, 50 Gy (at intersection of all fields), 2 Gy per fraction, 5 fractions per week, all fields used every day, maximum dose to spinal cord 40 Gy. In the case of lymph nodes radiation, total dose 50 Gy. Target volume: primary tumor (GTV : visible tumor ; PTV : expansion of 3 to 5cm of distal and proximal margins and lateral margins at mediastinal interface). If tumor of the upper 1/3 of the oesophagus, proximal margin must be adapted to the patient’s clinical situation. Patients with cervical primary tumor with positive supra-clavicular or cervical lymph nodes (defined as N1) are eligible. Patients with radiographic evidence of enlarged (superior or equal to 1,5 cm) celiac lymph nodes seen on CT scan or echography are ineligible. Patients with oesophageal tumor extensive to the cardia, classified Siewert I (center of the tumor lying > 1 cm – 5 cm above Gastro-Oesophageal Junction) are eligible. Patients with cardia tumor (Siewert II) or gastric tumor extensive to the oesophagus (Siewert III) are ineligible. The choice of technic (number and orientation of the beams, level-heading) will result from the analysis of the lungs DHV and CTV. Maximum dose to spinal cord will be 40 Gy. Gammagraph print or portal images will be done the first day of treatment for all the beams and at each beams change. They will be compared to the referential images. If these last don’t offer some anatomic locations, orthogonal prints will be realized.
Chemotherapy Arm A : Total treatment of six 2-weekly cycles of FOLFOX, the first 3 cycles starting on D1, D15 and D29 concomitant with 5 weeks’ radiotherapy. Oxaliplatin: 85 mg/m² as 2 hours infusion, in 250 to 500 ml of 5% glucose
solution on day 1 of each cycle with a separate infusion of folinic acid. Folinic acid : 200 mg/m² IV over 2 hours on day 1 of each cycle followed
by : 5-FU: 400 mg/m²/day IV bolus, on day 1 of each cycle then 5-FU: 1 600 mg/m² 46 h continuous IV infusion, over days 1 and 2 of
each cycle (approximately 800 mg/m² at day 1 and day 2)

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 6/70
Arm B : Two cycles of 5-FU / Cisplatin on week 1 and 5 of radiotherapy and two cycles of chemotherapy with 5-FU / Cisplatin on week 8 and 11 (one cycle each three weeks after the end of radiotherapy). Cisplatin : 75 mg/ m² continuous infusion (1 mg/minute) on day 1 of each
cycle followed by : 5-FU : 1000 mg/ m² per day continuous infusion from day 1 to day 4 of
each cycle.
EVALUATION CRITERIA
1° Phase II : In the two arms : - Percentage of patients having completed the full treatment. - Endoscopic complete response rate. - Toxicity profile (NCI-CTC). 2° Phase III : - Progression-free survival in each arm. - Overall Survival, complete response rate, time to treatment failure in
each arm. - Percentage of grade 3-4 toxicities in each arm.
STATISTICAL CONSIDERATIONS
General : Standard randomized phase II-III design Sample size: 97 patients were included in the phase II study and 169 supplementary patients will be included in the phase III (total of 266 patients: 133 in each arm).
DURATION OF STUDY PERIOD (per subject)
Arm A: 12 weeks or until disease progression, unacceptable toxicity, patient refusal to continue treatment, or treatment delay > 2 weeks. Arm B: 11 weeks or until disease progression, unacceptable toxicity, patient refusal to continue treatment, or treatment delay > 2 weeks.
STUDY DATES
Planned start date phase II : October 2004 Effective start date phase II : October 2004 Planned recruitment closure phase II : March 2006 Effective recruitment closure phase II : December 2005 Planned start date phase III : February 2008 Planned recruitment closure phase III : February 2010
Planned end date phase III: September 2011

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 7/70
TABLE OF CONTENTS
1. INTRODUCTION AND RATIONALE 10
1.1 Background 10 1.2 Information on study drugs 11
1.2.1 Oxaliplatin 11 1.2.2 5-FU 13 1.2.3 Folinic acid 14 1.2.4 Cisplatin 14
1.3 Study Rationale 14 2. STUDY OBJECTIVES 17 3. STUDY DESIGN 17
3.1 Type of Study 17 3.2 Expected Number of Patients 17 3.3 Method of Treatment Allocation 17 3.4 Duration of the Study Period for One patient 18
4. STUDY POPULATION 18 4.1 Inclusion Criteria 18 4.2 Exclusion Criteria 19
5. STUDY PROCEDURES 19 5.1 Treatments 19
5.1.1 Treatment administration 19 5.1.2 Dose modifications 23
5.2 Schedule of Visits and Observations 27 5.2.1 Pre-registration work up 27 5.2.2 Evaluations during treatment 28
5.2.3 Post treatment follow-up 29 5.3 Randomization procedures 30 5.4 Study Measurements 30
5.4.1 Completion of full treatment 30 5.4.2 Tumor Response 30 5.4.3 Time-related Parameters 32
6. SAFETY ASSESSMENTS 33 6.1 Concomitant Treatment Restrictions 33 6.2 Data Monitoring Committee 33
7. DRUG SUPPLIES 33 7.1 Study Drugs 33
7.1.1 Oxaliplatin 34 7.1.2 5-FU : Information can be found in the package insert. 35
7.1.3 Folinic acid : Information can be found in th e package insert. 35 7.2 Storage Conditions 35
7.3 Retrieval of treatments and/or destruction 35 8. TRial discontinuation criteria 35
9. Serious adverse event 36 10. COST AND EXCESS COSTS OF THE RESEARCH 38 11. QUALITY INSURANCE 39 12. OWNERSHIP AND CONFIDENTIALITY OF DATA 39 13. RULES FOR PUBLICATION 39 14. ETHICAL AND REGULATORY ASPECTS 40 15. COMMITTEE OF THE PROTECTION OF PERSONS 40

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 8/70
16. COMPETENT AUTHORITY 40 17. INFORMATION AND CONSENT OF PARTICIPANTS 41 18. SPONSOR'S RESPONSABILITIES 41 19. INVESTIGATOR'S RESPONSABILITIES 42 20. INTERRUPTION OF SUBJECT STUDY 42
20.1 Circumstance 42 20.2 Replacement of subjects 43
21. STATISTICAL CONSIDERATIONS 43 21.1 Parameters 43
21.1.1 Phase II 43 21.1.2 Phase III 43
21.2 Analysis Population 44 21.3 Statistical Methods 44
21.3 1. Demographics 44 21.3 2. Efficacy 44 21.3 3. Safety 44 21.3.4 Quality of life 45 21.3 5. Interim Analysis 45
21. 4. Sample Size Calculation 45
APPENDICES
Appendix 1 : ECOG Performance Status Scales/Scores 51 Appendix 2 : TNM classification of oesophageal carcinoma 52 Appendix 3 : US TNM echoendoscopic classification 53 Appendix 4 : De Leriche and Fontaine Classification 54 Appendix 5 : EORTC QLQ-C30 version 3 55 Appendix 6 : EORTC QLQ-OES18 57 Appendix 7 : Serious Adverse Event form 58 Appendix 8 : Notice d'information au patient et consentement éclairé 60 Appendix 9 : NCI-Common Toxicity Criteria version 3.0 68 Appendix 10 : Résumé des Caractéristiques du (des) produit(s) (RCP) 69 Appendix 11 : Siewert Classification 1998 70

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 9/70
LIST OF ABBREVIATIONS AND DEFINITION OF TERMS
AP Alkaline Phosphatase ASCO American Society of Clinical
Oncology AST Aspartate Amino-Transferase ALT Alanine Amino-Transferase BP Blood Pressure CDDP Cisplatin CEA Carcino-Embryonic Antigen CI Continuous Infusion CPP Comité pour la Protection des
Personnes CR Complete Response CRF Case Report Form CT Chemotherapy CV Cardiovascular d/D day DHV Dose-Volume Histogram DLT Dose Limiting Toxicity Dn Day n after treatment DNA Deoxyribonucleic Acid ECOG Eastern Cooperative Oncology
Group FA Folinic Acid FFCD Fédération Francophone de
Cancérologie Digestive FNCLCC Fédération Nationale des
Centres de Lutte Contre le Cancer
5-FU 5-Fluorouracil FOLFOX 5-FU, FA and Oxaliplatin
chemotherapy regimen γGT γ-Glutamyl Transferase GI Gastrointestinal GTV Growth Tumor Volume Gy Gray H Hour Hb Hemoglobin ICRU Intersection point of
radiotherapy beams IV Intravenous OXA Oxaliplatin LLN Lower Limit of Normal MeV Million electron Volts MMR Mismatch Repair MTD Maximum Tolerated Dose N Normal N°/n° number NCI National Cancer Institute NCI-CTC National Cancer Institute
Common Toxicity Criteria PD Progressive Disease PNN Polynuclear Neutrophil count PMH Primary Medical History PR Partial Response PS Performance Status
pt(s) Patient(s) PTV Primary Tumor Volume RNA Ribonucleic Acid RR Response Rate RTOG Radiation Therapy Oncology
Group SAE(s) Serious Adverse Event(s) SD Stable Disease Sd Standard deviation UICC Union Internationale Contre
le Cancer ULN Upper Limit of Normal vs Versus WBC White Blood Cells WHO World Health Organisation WNL Within Normal Limits

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 10/70
1. INTRODUCTION AND RATIONALE BACKGROUND Oesophageal carcinoma is more common in France than in any other European country (1). There are 4500 new cases every year, with a 94% mortality rate and a median survival of 9 months (2). Only 26% of tumors are operable, but even these patients have a perioperative mortality of 8% and three year survival of 25%. The incidence is 16 per 100 000 population. Survival of squamous cell oesophageal carcinoma improved significantly between 1984-86 and 1987-88 in Finistère, the benefit being attributed to increasing use of medical treatments either alone or in combination with surgery (3). These statistics clearly illustrate the fact that loco-regional treatment is not sufficient, and that systemic therapies need to be developed. Chemotherapy Many agents have been used over the years in single agent therapy or combination, but few have been tested in randomized trials (4). Response criteria have also been a discussion point, because symptomatic improvement can be due to a small tumor regression and primary tumors prove very difficult to measure at endoscopy, barium swallow and trans-oesophageal ultrasound. The most reliable objective response criteria appear to be those obtained with a combination of esophagoscopy, barium studies and CT-Scan. Treatment success in metastatic disease needs to be measured using RECIST response criteria, symptom-free survival, progression-free survival and overall survival (5-7). Fewer agents have been tested in oesophageal adenocarcinoma than in squamous cell carcinoma. Adenocarcinoma seems to respond less to chemotherapy, but this has never been proven in a randomized clinical trial. 7 single agents seem to show activity in oesophageal cancer, tested on small groups of patients (8-11) : mitomycin C (26% objective responses in historic trials), cisplatin (CDDP) (19%), vindesine (20%), bleomycin (15%), 5-fluorouracil (5-FU) (17-82%), vinorelbine (20%) and paclitaxel (28-34%)(4, 7, 12). The only randomized study comparing single agent therapy showed response rates of 5% for adriamycin, 12% for methotrexate and 15% for 5-FU. Various modalities of combination therapy using 5-FU, CDDP, bleomycin, vindesine and etoposide give response rates between 35% in metastatic disease and 50% in locally advanced disease. However, the results of these studies are difficult to interpret due to variable inclusion criteria. There are no large randomized trials that prove the superiority of one regimen over another. Standard combination therapy remains CDDP combined with a 5 day 5-FU continuous infusion. Patients with oesophageal cancer often present contraindications to this standard regimen: peripheral arterial disease, angina, previous vascular events, impaired renal function or impaired hearing. In a randomized phase II study in squamous cell oesophageal cancer, the EORTC found that this regimen was superior to CDDP alone, both in terms of response (36 vs 18%) and progression-free survival (9 vs 6 months), but there was no overall survival benefit mainly because of a significant number of toxic deaths in the CDDP-5-FU arm (13). Etoposide, irinotecan and paclitaxel appear to give similar results when combined with CDDP in phase II studies (14). Triplet combinations increase toxicity without improving outcome (8, 9). The addition of folinic acid to 5-FU/CDDP did not change outcome, but a 4-agent trial of 5-FU, FA, etoposide and CDDP gave encouraging results in a pre-operative setting (10).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 11/70
Radiotherapy Studies have shown that high-dose radiotherapy is capable of curing small oesophageal cancers (16-17), disproving the discouraging results of earlier, under-dosed, trials (18-20). 5 year survival has gone from 0-14% in early studies to 19-21% in the more recent trials. These results can be explained by better pre-treatment staging (CT), improved targeting, higher-energy sources and accelerated treatment reducing tumoral repopulation and sub-lethal lesion repair (16, 17, 21). Chemoradiotherapy In advanced oesophageal carcinoma, most exclusive chemoradiotherapy regimens tested in phase II included 5-FU and either CDDP or mitomycin with 40-60 Gy. Despite the absence of surgery, complete endoscopic responses ranged from 55-80% with a 29-40% two-year survival and a median of 11-22 months (29-35). Another study confirmed these results (36), and a third gave a non-significant 5-year survival difference of 16 vs 6% (27). A further study demonstrated a significantly improved disease-free survival with chemoradiotherapy (46) over radiotherapy alone. Eight studies comparing concomitant chemoradiotherapy to radiotherapy alone were published. The most recent trials used a combination of cisplatin and 5-FU. In the RTOG study 85-01 first published by Herskovic and coworkers (26,14), two courses of chemotherapy during 50 Gy radiation therapy followed by additional two courses of the same chemotherapy versus 64 Gy radiotherapy alone were investigated. No patient survived at three years in the radiotherapy group but 26 % of the patients in the concomitant chemoradiotherapy group were surviving at 5-years. A substantial reduction in local recurrences after chemoradiotherapy was demonstrated. However, this was at the expense of a higher risk of grade 3-4 toxicities : 20 % of the patients suffered from life-threatening toxicity and 40 % stopped chemotherapy before the completion of the treatment. Median survival was only 14 months in the chemoradiation arm. The Patterns of Care Study (22) confirmed these results and the superiority of chemoradiotherapy on radiotherapy alone, with 39% 2–year survival vs 20.6 % (p = 0.027) and lower 2-year local regional failure (30% vs 57.9 %; p = 0,0031). Two subsequent trials confirmed that the Herskovic regimen is now the standard chemoradiation regimen. The INT 0123 trial (23) compared high dose radiotherapy (64.8 Gy) versus standard dose (50.4 Gy) with 5FU and cisplatin in 218 patients. Toxic death rates were 10% (high dose) and 2% (standard dose) respectively, with no significant difference in median survival. The FNCLCC-FFCD 9305 trial (24) compared a split course radiotherapy to a standard fractionation radiotherapy (50 Gy in 25 fractions and 5 weeks). With a median follow-up of more than 6 years, the 5–year survival rate was significantly lower in the split course arm (10 vs 21% ; p = 0.047). Currently, patients with inoperable disease at diagnosis, contraindications or objections to surgery and metastatic disease with dysphagia are treated with combination chemoradiotherapy using 5-FU/ cisplatin (2 cycles during and 2 cycles after radiotherapy) and 50 Gy. Chemoradiotherapy is the treatment of choice for this group of patients (25), but it is by no means perfect. New combinations of drugs are required to improve the recurrence and metastasis rates, and to allow better dose-intensities. Synergistic combinations would be interesting in order to improve survival of this poor prognosis disease. INFORMATION ON STUDY DRUGS Oxaliplatin

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 12/70
Oxaliplatin (oxa), a new third-generation cisplatin analog in the 1,2-diaminocyclohexane (DACH) family of platinum compounds, is active in several solid tumor types, including some cisplatin/carboplatin refractory diseases and is licensied in many countries for the treatment in first line of metastatic colorectal cancer (CRC). Mechanism of action Similarly to cisplatin, the main mechanism of action of oxaliplatin is mediated through the formation of DNA-adducts (38), but in spite of many similarities between the two drugs, there are important differences in their activity profiles(s). DACH platinum adducts are bulkier and more hydrophobic than cisplatin adducts (39,40). The mismatch repair protein complex may be prevented from binding to DACH-platinum DNA-adducts because of particular conformational DNA distortions in the region of the adducts (41). The induction of mismatch repair defects has been shown to correlate with acquired resistance to cisplatin while the sensitivity to oxaliplatin was maintained. Data concerning the role of DNA mismatch repair in platinum drug resistance indicate that oxaliplatin is likely to be more active in cisplatin-resistant cancer (42). Several laboratories have shown that the mismatch repair complexes recognize cisplatin diadducts, but not DACH-platinum diadducts in DNA (43,44). Finally, oxaliplatin induces primary and secondary DNA lesions that lead to apoptosis in human cancer cells. Preclinical activity Oxaliplatin has shown potent antiproliferative activity (as good or better than cisplatin) against mouse and human cervical carcinoma, non-small cell lung cancer, leukemia, colon, ovarian, breast, melanoma, bladder, glioma, and erythroleukemia cells lines (45,46). Tolerance of oxaliplatin The tolerability profile of oxaliplatin is as follows : The limiting toxicity of oxaliplatin is neurological. It involves a sensory peripheral neuropathy characterised by peripheral dysaesthesia and/or paraesthesia with or without cramps, often triggered by the cold (85 to 95% of patients). The duration of these symptoms, which usually regress between courses of treatment, increases with the number of treatment cycles. The onset of pain and /or a functional disorder and their duration are indications for dose adjustment, or even treatment discontinuation. This functional disorder, including difficulties in executing delicate movements, is a possible consequence of sensory impairment. The risk of occurrence of a functional disorder for a cumulative dose of approximately 800 mg/m² (i.e. 10 cycles) is 15% or less. The neurological signs and symptoms improve when treatment is discontinued in the majority of cases. Acute neuro-sensory manifestations have been reported. They start within hours of administration and often occurs on exposure to cold. They may present as transient paraesthesia, dysaesthesia and hypoaesthesia or as an acute syndrome of pharyngolaryngeal dysaesthesia. This acute syndrome of pharyngolaryngeal dysaesthesia (1-2% of patients) is characterized by subjective sensations of dysphagia or dyspnea, without any laryngospasm or bronchospasm (no stridor or wheezing); jaw spasm, abnormal tongue sensation, dysarthria and a feeling of chest pressure have also been observed. Such symptoms are rapidly reversible with or even in the absence of symptomatic treatment. Prolongation of the infusion time in subsequent cycle helps to reduce the incidence of this syndrome. Acute cold-induced dysesthesias : a frequent acute syndrome of dysesthesias of the hands, feet, or throat. This syndrome may be manifested as laryngo-pharyngeal dysesthesia when swallowing

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 13/70
cold food or drink. The intensity is generally mild to moderate and the symptoms are most often observed during or within a day or two after the oxaliplatin infusion, lasting for a few minutes to a few days, and are fully reversible. � Allergic Reactions : Uncommon (single agent) or common (in combination with 5-FU +/- folinic acid) anaphylactic reactions including bronchospasm, angioedema, hypotension and anaphylactic shock. Common allergic reactions such as skin rash (particularly urticaria), conjunctivitis, rhinitis have been observed. Typically, the reactions occured after several exposures to oxaliplatin. Frequent cases of fever without infection (immune type) or with infection (associated or no to neutropenia) have been reported. � Gastrointestinal disorders : As a single agent, oxaliplatin may cause anorexia, nausea, vomiting, diarrhoea and abdominal pain. In the majority of cases, these symptoms are not severe. Prophylaxis and/or treatment with potent antiemetic agents are indicated. In combination with 5-FU (with or without folinic acid), the frequency and severity of diarrhoea and mucositis is significantly increased compared with that observed with 5-FU alone. Stomatitis, mucositis, colitis including Clostridium difficile diarrhea has also been reported. Metabolic disorders including dehydration, ileus, intestinal obstruction, hypokaliemia, metabolic acidosis and renal impairment may be caused by severe diarrhea/emesis, particularly when combining oxaliplatin with 5-fluorouracil. Mild to moderate elevation of transaminases and alkaline phosphatases have been observed. � Haematological disorders : Anaemia, neutropenia and thrombocytopenia have been reported. The incidence of neutropenia and thrombocytopenia is greater when oxaliplatin is used in combination with 5-FU and folinic acid than that observed using a combination of 5-FU and folinic acid alone. � Other effects :
o Rare cases of acute interstitial lung disease and of pulmonary fibrosis have been reported.
o Rare cases of immuno-allergic thrombocytopenia and hemolytic anaemia. o Transient, reversible decreased vision within hours of receiving oxaliplatin, as well as
cases of optic neuritis has been rarely reported. o Rare cases of deafness have been reported. o Disturbance of renal function have been reported in approximately 3% of all patients
treated, with less than 1% of patients experiencing grade 3-4 abnormalities. o Moderate alopecia has been reported in 2% of patients treated. o Extravasation may result in local inflammation which may be severe and lead to
complication, especially when oxaliplatin is infused through a peripheral vein. o Dysarthria has been rarely reported.
5-FU 5-FU, is a pyrimidine analog, used in cancer chemotherapy since 1963 in France. It has been tested in a wide variety of bolus, short and long intravenous infusions for different indications in many single-agent and combination chemotherapies. The drug is metabolized to F d UMP (5 Fluoro-2'-deoxyuridinemonophosphate), which binds to thymidylate synthase, preventing DNA synthesis. The activity of 5-FU depends on peak

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 14/70
concentrations and on duration of exposure (47). Continuous infusion of 5-FU is very well tolerated. The limiting toxicity of 5-FU in continuous infusion is mucositis, hand-foot syndrome and diarrhea rather than hematological (48). The continuous infusion schedule has demonstrated efficacy in several tumor types (breast, head and neck, pancreas, gastric, rectal and colon cancer). Overall response rate in the published series of single agent 5-FU in patients with oesophageal cancer is between 18 and 82 % (4,7,10). 5-FU is included in most combination therapies for oesophageal cancer and forms part of the standard chemoradiotherapy for locally advanced disease. Folinic acid Folinic acid modulates the activity of 5-FU and thus improves outcome in both colorectal and gastric cancer (49-53). It has been added to 5-FU in several small studies of oesophageal cancer (54-58) with encouraging results, but large trials have yet to be performed. 5-FU and FA alone gave 19% OR rate in a small phase II study of oesophageal adenocarcinoma (4) and 45% combined with cisplatin and etoposide (69) with 31% two-year survival. Cisplatin Cis-dichlorodiammineplatine II (cisplatin) has been tested extensively against human tumor cell lines in vitro and in clinical trials and has proven to be one of the most active antineoplastic agents in clinical use, with a broad spectrum of antitumor activity, including esophageal cancer (Bleiberg EJC 1997). Cisplatin has the activity of a bifunctional alkylating agent and binds directly to DNA, inhibiting its synthesis by the formation of intrastrand cross-links. Although the nephrotoxicity of cisplatin was originally felt to be dose-limiting, renal toxicity has been preventable when adequate hydration is provided. Other toxicities include rare hypersensitivity, acute and delayed nausea and vomiting (needing agressive prophylactic antiemetics), hypomagnesemia, neurotoxicity and Raynaud’s phenomenon. Carboplatin, an less toxic analog of cisplatin, was demonstrated as poorly efficient in esophageal cancer (59). STUDY RATIONALE Oxaliplatin is a diaminocyclohexane platinum complex that is active in several solid tumor types, especially in some cisplatin/carboplatin refractory diseases, such as colorectal cancer, resistance being generally due to a loss of mismatch repair (MMR) function. Loss of MMR function, observed as genetic instability in microsatellite sequences, occurs in many types of sporadic cancers, including oesophageal carcinoma (28,65,66). Data concerning the role of DNA mismatch repair in platinum drug resistance indicate that oxaliplatin is likely to be more active in cisplatin-resistant cancer (42,67). Loss of MMR also causes resistance to other drugs, but not to 5-FU in some MMR-proficient and deficient cell lines (68). Oxaliplatin is licensed in many countries for the treatment of first line of metastatic colorectal cancer. A phase III European study of LV5FU2 +/- Oxaliplatin was conducted from August 1995 to July 1997. Four hundred twenty patients were randomized (210 in each arm) and received 5-FU 400 mg/m2 bolus + 5-FU 600 mg/m2 continuous infusion and FA 200 mg/m2 on day 1 and day 2

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 15/70
and Oxaliplatin 85 mg/m2 IV 2 hours on day 1, with cycles repeated every 2 weeks. The objective response rate was 22% for FU/LV versus 50% with the addition of oxaliplatin (p= 0.0001), with a median time to response in the oxaliplatin arm of 2.3 months. Time to progression was 6.2 vs 9.0 months (p= 0.0003) and overall survival 14.7 vs 16.2 months (p= 0.12) in the two arms respectively (71). Oxaliplatin demonstrates activity superior to that of cisplatin, having a broader spectrum of action than the parent compound (69-74). It has shown encouraging activity in tumors that respond to cisplatin or carboplatin: advanced ovarian cancer (75-77) and non small cell lung cancer (78). The combination of oxaliplatin and 5-FU is known to be active against several tumor types (79). The synergy of the two agents has been confirmed by clinical trials (69-74,80). Extension of this synergy to oesophageal cancer may be expected. Continuous 5-FU regimens appear to be more effective than bolus ones (71) and the oxaliplatin dose of 85 mg/m2 every 2 weeks is well-tolerated in combination with 5-FU with or without folinic acid. The 5-FU/oxaliplatin combination does not present the same cardiovascular or renal risks as 5-FU/cisplatin, as it does not require prehydration and has no renal toxicity. In vitro studies of squamous carcinoma cell lines (CAL 27) using FOLFOX and radiotherapy are currently underway. The safety profile of oxaliplatin combined with 5-FU, is now well established. Activity of oxaliplatin in esogastric carcinomas Gastric carcinoma and adenocarcinomas of the esopha go-gastric jonction Three trials assessed the activity of oxaliplatin combined to 5-FU and folinic acid. The first one, (60) included 57 patients with gastric adenocarcinoma (including 18 cardia). Patients received oxaliplatin 100 mg/ m2, folinic acid 400 mg/ m2, bolus 5-FU 400 mg/ m2 then 5-FU 3 g/ m2 as a continuous infusion over 46 hours every 14 days. Forty-nine were assessable for response. Two CR and 20 PR were observed, giving an overall RR of 44.9%. Median survival was 8.6 months. Another trial from the Chicago University (61) confirmed these data. Thirty-six patients with adenocarcinoma (n = 29), squamous cell carcinoma (n = 3) or poorly differentiated carcinoma (n = 3) of the esophagus or the cardia were included. They received a combination of oxaliplatin 85 mg/ m 2 day 1, folinic acid 500 mg/ m2 days 1 and 2, bolus 5-FU followed by 5-FU 600 mg/m2 22-hour continuous infusion days 1 and 2 every 2 weeks. Response rate in this heterogeneous population was 40% with a 8.6 month median survival. Another trial was reported during the 2002 ASCO (62). Forty-one patients with gastric cancer received a regimen consisting of Oxaliplatin 65 mg/ m2 days 1 and 8, 5-FU 2600 mg/ m2 plus folinic acid 300 mg/ m2 days 1 and 8 every 3 weeks. Out of 29 evaluable patients, the PR rate was 55.2 % and the median survival was not yet reached. The fourth trial was recently published (63). Twenty-six patients with advanced gastric cancer, whose disease progressed while receiving, or after discontinuing, chemotherapy with a 5-FU and platinum regimen, were enrolled. Treatment comprised oxaliplatin (85 mg/ m2 on day 1) as a 2-h infusion followed by bolus 5-FU (400 mg/ m2 on day 1), and 48-h infusion of 5-FU 2.4-3.0 g/ m2 concurrently with folinic acid 150 mg/ m2. Cycles were repeated at 2-weeks intervals. Of the 23 evaluable patients, there were six PR (response rate 26%). The median time to progression was 4.3 months and the median overall survival was 7.3 months. Another trial included patients with metastatic gastric cancer failing prior palliative first-line chemotherapy. The regimen consisted in a combination of raltitrexed (3 mg/ m2) and oxaliplatin (130 mg/ m2) every 3 weeks. One patient achieved a PR and 6 had stable disease, with a overall median survival of 4.5 months (64).
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 16/70
Oxaliplatin with radiation Some trials assessed the safety and the efficacy of oxaliplatin combined with radiation therapy, especially in rectal cancers. Two trials in patients with primary esophageal carcinoma are also available. One NCI-sponsored Phase I trial using oxaliplatin with protracted-infusion 5-FU plus radiotherapy for patients with esophageal cancer was recently published (81). Patients received oxaliplatin on days 1,15, and 29 with continuous 5-FU from days 8 to 42. Starting on day 8, patients received radiotherapy 36 to 39.6 Gy in 1.8 Gy fractions, then 5.4 Gy to 9 Gy in a smaller volume. Eligible patients could undergo surgery or begin a second cycle without radiotherapy. Six patients received oxaliplatin at 85 mg/m2 and 5-FU at 180 mg/m2/day. Then oxaliplatin was escalated to 100 mg/m2 with dose-limiting toxicities (DLT) in 2 out 3 patients and dose escalation was discontinued. However table 2 in the final report indicated 4 levels (2 dose levels for each drug) and no data is reported on the other levels. Forty patients were enrolled and 38 were treated, including 32 adenocarcinoma, 4 squamous cell carcinoma and 2 with mixed histology. At the end of cycle 1, 36 patients underwent endoscopy and a 81% complete response rate was observed. Of the 13 patients taken to surgery, 5 (38%) had no cancer in the resected specimen. Neurotoxicity was milder than expected and the combination appears less toxic than the Herskovic combination. The effect of this treatment on pulmonary function was also published (82). A Phase I study investigated the maximum tolerated dose (MTD) for an association of oxaliplatin when administered with folinic acid 200 mg/m2 followed by 5FU bolus and 22 h continuous infusion given on days 1 and 2, every 2 weeks with a 50 Gy radiation dose in patients with esophageal cancers. Patients also received 3 courses of Modified Folfox 4 (with 85 mg/m2 oxaliplatin) every 2 weeks after the end of the radiotherapy period. Five levels have been investigated, as indicated below. Limiting Toxicity and response rate :
Dose (mg/m2) Limiting toxicity Level
Number of evaluable patients Oxaliplatin 5FU bolus 5FU Cl Number of
patients Type of toxicity
Efficacy
Safety : 06 01 G3 asthenia and anorexia I
Efficacy : 05 50 300 400
01 G3 cutaneous reaction (Epithelite)
2 CR, 1 PR, 2 PD, 1 NE*
II 06 50 400 600 01 G3 anorexia 4 PR, 1 SD, 1PD
01 G4 febrile neutropenia after cycle 1
III 06 75 400 600 01 G3 asthenia and anorexia
1 CR, 4 PR, 1 SD, 1 NE*
IV 07 85 400 600 01 G3 anorexia 3 RP, 1 SD, 2 PD, 1 NE*,
V 06 100 400 600 03 G4 asthenia G3 diarrhea G3 asthenia
2 RP, 3 PD, 1 NE*
* NE = not evaluable

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 17/70
Dose-limiting Toxicities (DLT) occurred in 3 of 6 patients treated at level V, with a 100 mg/m2 dose. So 3 extra patients have been included at the level IV to confirm the recommended dose of 85 mg/m2 oxaliplatin every 2 weeks. Encouraging efficacy data have been observed, with a 61 % response rate in 28 evaluable patients. Given these results, it is necessary to assess in a randomized phase II / III study the tolerance and activity of chemoradiotherapy using Folfox 4 and Herskovic regimens in locally advanced oesophageal carcinomas.
2. STUDY OBJECTIVES Phase II : Primary Objectives : - To assess the feasibility (completion of full treatment) of combination chemotherapy containing
oxaliplatin, 5-fluorouracil and folinic acid (FOLFOX regimen) or 5-FU/Cisplatin with concomitant radiotherapy in first line treatment of inoperable advanced oesophageal cancer.
- To assess endoscopic complete response rate in both arms. Secondary Objective : To assess the toxicity profile of each arm using NCI-CTC version 2. Phase III : Primary Objective : To compare the progression-free survival between 2 chemotherapy schedules delivered during concomitant radiotherapy : Folfox regimen versus Cisplatin/5-FU (Herskovic regimen). Secondary Objectives : - To compare overall survival, endoscopic complete response rate, incidence of grade 3-4
toxicities NCI-CTC and time to treatment failure between both regimens. - To evaluate the quality of life using EORTC QLQ-C30 (version 3) and a validated disease-
specific module QLQ-OES18.
3. STUDY DESIGN 3.1 TYPE OF STUDY Multicenter randomised phase II trial followed by a phase III. 3.2 EXPECTED NUMBER OF PATIENTS 97 patients were included in the phase II study and 169 supplementary patients will be in the phase III (total of 266 evaluable patients :133 in each arm).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 18/70
3.3 METHOD OF TREATMENT ALLOCATION Eligible patients will be randomized according to a randomization by minimization, between the two arms of treatment with a stratification :
� by histological type (adenocarcinoma or adenosquamous versus squamous cell carcinoma),
� by pretreatment weight loss in the prior 6 months (grade 1 : < 10% versus grade 2 : ≥ 10%),
� by ECOG performance status (0 - 1 - 2), � and by centre.
The patient number and the patient initial are to be entered on each page of the Case Report Form. 3.4 DURATION OF THE STUDY PERIOD FOR ONE PATIENT Arm A : treatment will be given for a total of six 2-weekly cycles of FOLFOX, the first 3 cycles starting on D1, D15 and D29 concomitant with 5 weeks’ radiotherapy. Arm B : two cycles of 5-FU / Cisplatin on weeks 1 and 5 of radiotherapy and two cycles of chemotherapy with 5-FU / Cisplatin on weeks 8 and 11 (one cycle each three weeks after the end
of radiotherapy). Subjects will be considered to be on-study for the duration of their treatment and in the 30 days following treatment discontinuation. Treatment discontinuation is defined as the last day of study treatment (chemotherapy or radiotherapy). Subjects will continue on treatment until they have completed the study protocol, unless there is disease progression, unacceptable toxicity, patient refusal, or treatment delay > 2 weeks. All included patients will be followed up until recovery from residual toxicities/AEs, until progressive disease is observed, and until death occurs.
4. STUDY POPULATION 4.1 INCLUSION CRITERIA Patients with: − Histologically proven adenocarcinoma, squamous cell or adenosquamous carcinoma of the
oesophagus ; − Inoperable locally advanced oesophageal carcinoma (disease status : any T, N0 or N1,M0 or
M1a), or surgical contre-indication conditions ; − No prior treatment for oesophageal cancer (surgery, chemo- or radiotherapy); − Peripheral neuropathy ≤ NCI-CTC grade 1; − Age ≥ 18 years; − ECOG Performance Status (PS) ≤ 2; − Sufficient(oral or with gastrostomy) calorific intake (> 1000 Kcal/m2/day); − Life expectancy ≥ 3 months ;

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 19/70
− Adequate bone marrow reserve, normal renal and liver functions: � Neutrophil count ≥ 1500/mm³ � Platelet count ≥ 100 000/mm³ � Hemoglobin ≥ 10 g/dl (after transfusion, if necessary) � Creatinine < 15mg/L � Prothrombin time ≥ 60%;
− Laboratory values obtained the week preceding study entry; − Signed informed consent (prior to all study procedures); − Start of treatment within 28 days of inclusion. 4.2 EXCLUSION CRITERIA − Metastatic disease except for third upper or cervical oesophagus tumor with regional nodes, or
third lower oesophagus tumor with celiac nodes (M1a); − Multiple carcinomas of the oesophagus; − Small cell or undifferentiated carcinoma of the oesophagus; − Patients with cardia tumor (Siewert II) or gastric tumor extensive to the oesophagus (Siewert III)
are ineligible. − Complete dysphagia (grade 4 NCI-CTC) patient with exclusive parenteral nutrition; − Weight loss within 3 months > 20% normal body weight; − Pregnant or breast-feeding woman; − Fertile patient not using adequate contraception; − Auditory disorders; − History of prior malignancies (other than cured non melanoma skin cancer, cured cervical
carcinoma in situ or stage I or II node negative head and neck cancer cured > 3 years ago); − Prior cervical, thoracic and abdominal radiotherapy with field overlapping the proposed
oesophageal radiotherapy field; − Tracheo-oesophageal fistula or invasion of the tracheo-bronchial tree; − Previous myocardial infarction (inferior or equal to 6 months). Patients with a previous
myocardial infarction superior to 6 months, could be included only if: � no transient ischemia is shown by thallium myocardial scintigraphy and � favourable advise for chemotherapy from a cardiologist is obtained ;
− Other serious illness or medical conditions (such as symptomatic coronary disease, left ventricular failure or uncontrolled infection);
− Arterial disease stage II to IV according to the DE LERICHE and FONTAINE classification; − Treatment with any other experimental drugs or participation in another clinical trial within 30
days of study screening; − Concurrent treatment with any other anti-cancer therapy; − Concurrent treatment with phenytoine or yellow fever vaccine. − Geographical, social or psychological circumstances preventing regular follow-up.
5. STUDY PROCEDURES

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 20/70
5.1 TREATMENTS Treatment administration Radiation Rx > 6 MV, 2 to 4 beams, 50 Gy (at intersection of all fields), 2 Gy per fraction, 5 fractions per week, all fields used every day, maximum dose to spinal cord 40 Gy. In the case of lymph nodes irradiation, total dose 50 Gy. Target volume: primary tumor (GTV : visible tumor ; PTV : expansion of 3 to 5 cm of distal and proximal margins and lateral margins at mediastinal interface). If tumor of the upper 1/3 of the oesophagus, proximal margin must be adapted to the patient’s clinical situation. Patients with cervical primary tumor with positive supra-clavicular or cervical lymph nodes (defined as N1) are eligible. Patients with radiographic evidence of enlarged (superior or equal to 1,5 cm) celiac lymph nodes seen on CT scan or echography are ineligible. Patients with oesophageal tumor extensive to the cardia, classified Siewert I (center of the tumor lying > 1 cm – 5 cm above Gastro-Oesophageal Junction) are eligible. Patients with cardia tumor (Siewert II) or gastric tumor extensive to the oesophagus (Siewert III) are ineligible. The choice of technic (number and orientation of the beams, level-heading) will result from the analysis of the lungs DHV and CTV. Maximum dose to spinal cord will be 40 Gy. Gammagraph print or portal images will be done the first day of treatment for all the beams and at each beams change. They will be compared to the referential images. If these last don’t offer some anatomic locations, orthogonal prints will be realized. Arm A : Total treatment of six 2-weekly cycles of FOLFOX, the first 3 cycles starting on D1, D15
and D29 concomitant with 5 weeks’radiotherapy. Arm B : Two cycles of 5-FU/Cisplatin on week 1 and 5 of radiotherapy and two cycles of
chemotherapy with 5-FU / Cisplatin on week 8 and 11 (one cycle each three weeks after the end of radiotherapy) [Herskovic regimen]
Gastrostomy or jejunostomy or nasogastric sound is recommended, if necessary before or during the treatment. Tumors of the upper 1/3 of the oesophagus : with the patient in dorsal decubitus position, arms at their sides, 3 equally weighted beams should be used, systematically including the subclavian lymph nodes, to a dose of 40 Gy at the ICRU point. The irradiated volume should then be reduced, by decreasing the size of the 3 fields, to add a boost of 10 Gy (ICRU point) at the tumor target and affected lymph nodes with a safety margin of 1 cm in all dimensions. Tumors of the middle 1/3 of the oesophagus : if the upper limit of the lesion exceeds the carena, the right and left retro-clavicular regions need to be included in the initial target volume. Tumors of the lower 1/3 of the oesophagus : the irradiated field will include the celiac lymph nodes.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 21/70
Dosimetry : Volumetric dosimetry will be performed on spinal cord and lung. Control gammagraphs print or portal images should be performed for each field at start and at each ballistic change (see section 5.4 for quality control measures). If possible, the following constraint will be respected : lungs dose (volume of the two lungs – PTV) receiving more than 20 Gy (V20) < 20 % of the total lung volume (V20 < 20 %). Target volume : Target volume includes the tumor and any suspect lymph nodes, with a 5 cm safety margin in the cranio-caudal axis above and below the tumor, and a 2 cm radial safety margin. If tumor of the upper 1/3 of the oesophagus, proximal margin must be adapted to the patient’s clinical situation. If the upper limit of tumor or lymph nodes exceeds the carena, the right and left retro-clavicular regions need to be included in the irradiated field. For lower 1/3 tumors, the irradiated field will include the celiac lymph nodes. After 40 Gy the irradiated volume will be reduced to the initial targets (tumor and nodes) with a 1 cm safety margin.
Chemotherapy :
Arm A (Modified Folfox regimen) : Oxaliplatin : 85 mg/m² as 2 hour infusion, in 250 to 500 ml of 5% glucose solution on days 1, 15,
29, 43, 57 and 71 (every 2 weeks) with a separate infusion of folinic acid Modified LV5FU scheme Folinic acid : 200 mg/m² IV over 2 hours on day 1 and of each cycle followed by 5-FU: 400 mg/m²/day 10 min IV bolus, on day 1 of each cycle followed by 5-FU: 1600 mg/m² 46 h continuous IV infusion, over days 1 and 2 of each cycle (approximately 800 mg/m² at day 1 and day 2). Modified FOLFOX regimen, every 2 weeks - 3 cycles during radiotherapy, 3 cycles after radiotherapy. RADIOTHERAPY:
↓↓↓↓Cycle 1 ↓↓↓↓Cycle 2 ↓↓↓↓Cycle 3 d1-2 d3-5 d8-d12 d15-16 d17-19 d22-d26 d29-30 d31-33 d36-40 CRT RT RT CRT RT RT CRT RT*
↓↓↓↓Cycle 4 ↓↓↓↓Cycle 5 ↓↓↓↓Cycle 6 Evaluation ↓↓↓↓
CT = chemotherapy RT = radiotherapy RT* = reduced field radiotherapy
d43-44 d57-58 d71-72
CT CT CT

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 22/70
Arm B (Herskovic regimen) :
Two cycles of chemotherapy with 5-FU / Cisplatin on weeks 1(day 1) and 5 (day 29) of radiotherapy and two cycles each three weeks after the end of radiotherapy on weeks 8 (day 50) and 11 (day 71). Hydration before Cisplatin administration : During 03 hours : 1 liter of NaCL Isotonic + 22, 5 ml of KCL 10% + 9 ml of MgCL2 (adapted to the blood ionogramme). During 03 other hours : 1 liter of NaCL Isotonic + 22, 5 ml of KCL 10% + 9 ml of MgCL2. Investigators could use this proposed scheme or another according to the local habits. If diuresis is inferior to 2 liters, administer 100 ml of Mannitol 20%. Cisplatin : 75 mg/m 2 continuous infusion (1 mg/minute) in 250 ml of sodium chloride solution on
day 1of each cycle (or in case of hydration delay on day 2) followed by: Hydration : During 03 hours : 1 liter of NaCL Isotonic + 22, 5 ml of KCL 10% + 9 ml of MgCL2. During 03 other hours : 1 liter of NaCL Isotonic + 22, 5 ml of KCL 10% + 9 ml of MgCL2. 5-FU : 1000 mg/m 2 per day continuous infusion from day 1 to day 4 of each cycle.
RADIOTHERAPY:
↓↓↓↓Cycle 1 ↓↓↓↓Cycle 2 d1-4 d5 d8-d12 d15-19 d22-d26 d29-32 d33 d34-49 CRT RT RT RT RT CRT RT*
↓↓↓↓Cycle 3 ↓↓↓↓Cycle 4 Evaluation ↓↓↓↓
CT = chemotherapy RT = radiotherapy RT* = reduced field radiotherapy Notice the number of vomiting and precise the diuresis curve each three hours. Excessives loss of water and electrolytes will be compensated according to the ionogramme (with calcemia and magnesemia) results, done the day of Cisplatin administration and the following day. Vomiting will be prevented using antiemetics: corticosteroids, anti-5HT3. FOLFOX should continue every 2 weeks after the end of radiotherapy for 3 cycles or until disease progression, unacceptable toxicity, patient refusal to continue treatment, or treatment delay > 2 weeks. 5-FU/Cisplatin should continue for two cycles each three weeks after the end of radiotherapy or until disease progression, unacceptable toxicity, patient refusal to continue treatment, or treatment delay > 2 weeks.
d50-53 d71-74
CT CT
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 23/70
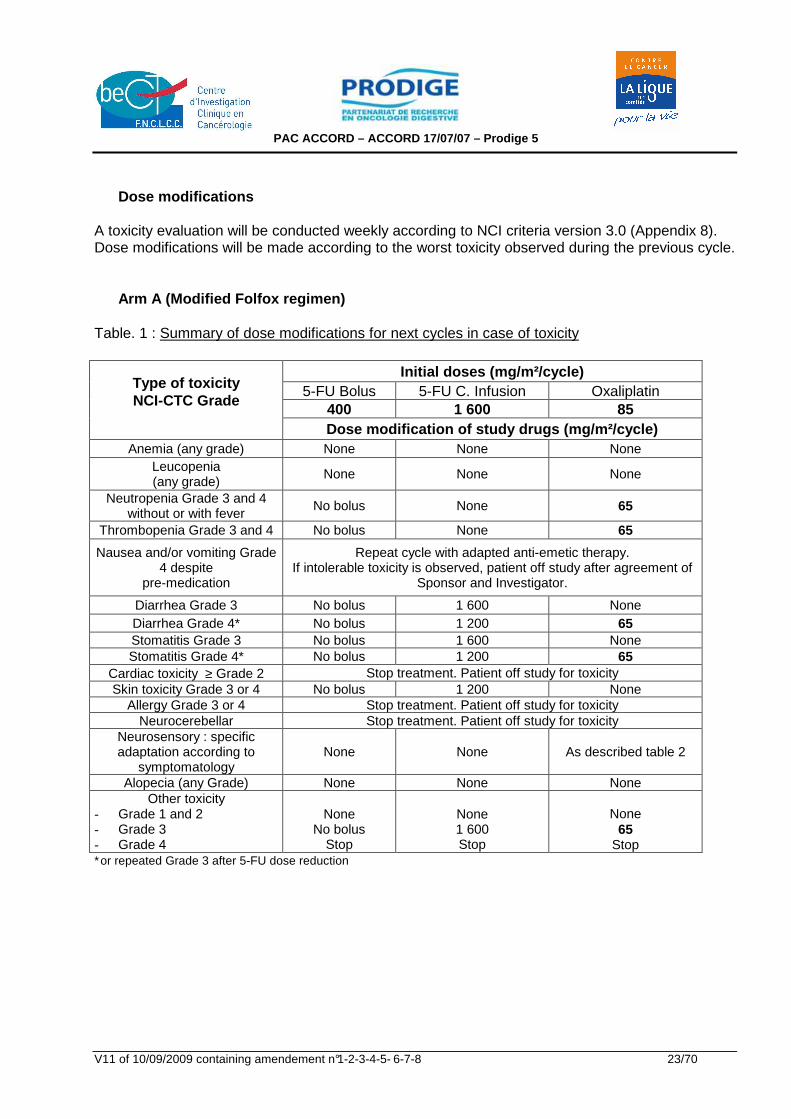
Dose modifications A toxicity evaluation will be conducted weekly according to NCI criteria version 3.0 (Appendix 8). Dose modifications will be made according to the worst toxicity observed during the previous cycle. Arm A (Modified Folfox regimen) Table. 1 : Summary of dose modifications for next cycles in case of toxicity
Initial doses (mg/m²/cycle) 5-FU Bolus 5-FU C. Infusion Oxaliplatin
400 1 600 85
Type of toxicity NCI-CTC Grade
Dose modification of study drugs (mg/m²/cycle)
Anemia (any grade) None None None Leucopenia (any grade)
None None None
Neutropenia Grade 3 and 4 without or with fever
No bolus None 65
Thrombopenia Grade 3 and 4 No bolus None 65
Nausea and/or vomiting Grade 4 despite
pre-medication
Repeat cycle with adapted anti-emetic therapy. If intolerable toxicity is observed, patient off study after agreement of
Sponsor and Investigator.
Diarrhea Grade 3 No bolus 1 600 None Diarrhea Grade 4* No bolus 1 200 65 Stomatitis Grade 3 No bolus 1 600 None Stomatitis Grade 4* No bolus 1 200 65
Cardiac toxicity ≥ Grade 2 Stop treatment. Patient off study for toxicity Skin toxicity Grade 3 or 4 No bolus 1 200 None
Allergy Grade 3 or 4 Stop treatment. Patient off study for toxicity Neurocerebellar Stop treatment. Patient off study for toxicity
Neurosensory : specific adaptation according to
symptomatology None None As described table 2
Alopecia (any Grade) None None None Other toxicity
- Grade 1 and 2 - Grade 3 - Grade 4
None
No bolus Stop
None 1 600 Stop
None
65 Stop
∗or repeated Grade 3 after 5-FU dose reduction
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 24/70
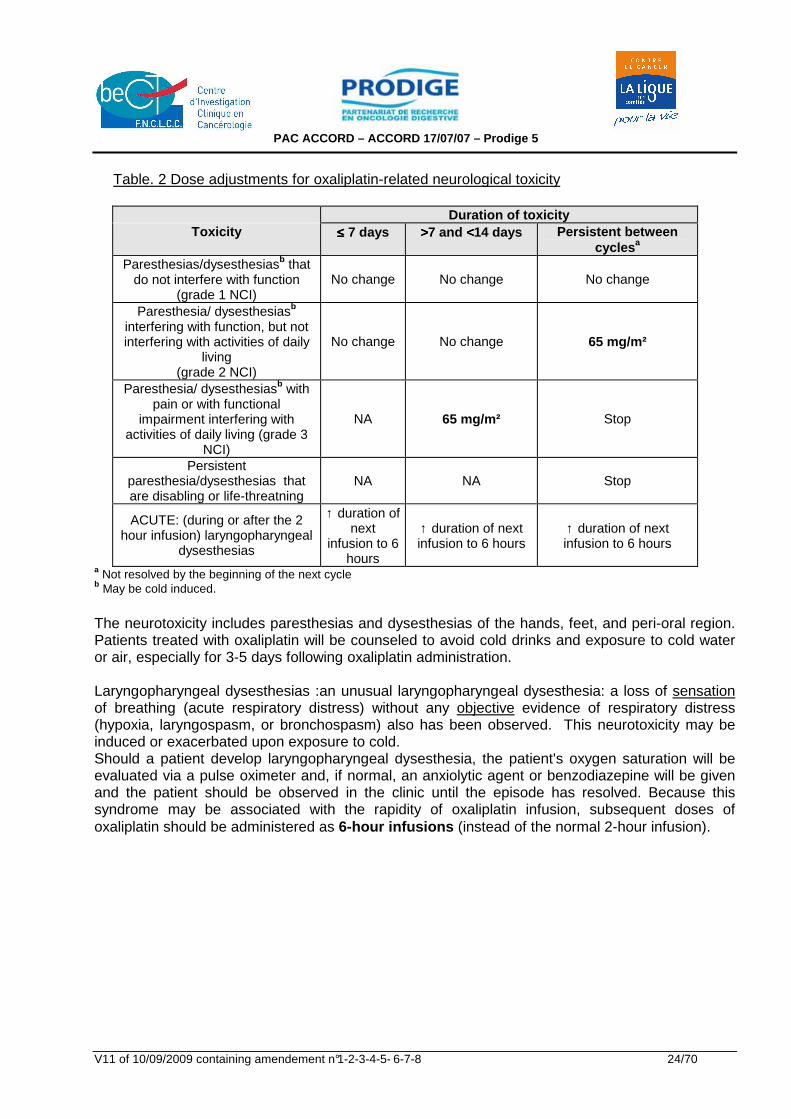
Table. 2 Dose adjustments for oxaliplatin-related neurological toxicity
Duration of toxicity
Toxicity ≤≤≤≤ 7 days >>>>7 and <<<<14 days Persistent between cycles a
Paresthesias/dysesthesiasb that do not interfere with function
(grade 1 NCI) No change No change No change
Paresthesia/ dysesthesiasb interfering with function, but not interfering with activities of daily
living (grade 2 NCI)
No change No change 65 mg/m²
Paresthesia/ dysesthesiasb with pain or with functional
impairment interfering with activities of daily living (grade 3
NCI)
NA 65 mg/m² Stop
Persistent paresthesia/dysesthesias that are disabling or life-threatning
NA NA Stop
ACUTE: (during or after the 2 hour infusion) laryngopharyngeal
dysesthesias
↑ duration of next
infusion to 6 hours
↑ duration of next infusion to 6 hours
↑ duration of next infusion to 6 hours
a Not resolved by the beginning of the next cycle b May be cold induced.
The neurotoxicity includes paresthesias and dysesthesias of the hands, feet, and peri-oral region. Patients treated with oxaliplatin will be counseled to avoid cold drinks and exposure to cold water or air, especially for 3-5 days following oxaliplatin administration. Laryngopharyngeal dysesthesias :an unusual laryngopharyngeal dysesthesia: a loss of sensation of breathing (acute respiratory distress) without any objective evidence of respiratory distress (hypoxia, laryngospasm, or bronchospasm) also has been observed. This neurotoxicity may be induced or exacerbated upon exposure to cold. Should a patient develop laryngopharyngeal dysesthesia, the patient's oxygen saturation will be evaluated via a pulse oximeter and, if normal, an anxiolytic agent or benzodiazepine will be given and the patient should be observed in the clinic until the episode has resolved. Because this syndrome may be associated with the rapidity of oxaliplatin infusion, subsequent doses of oxaliplatin should be administered as 6-hour infusions (instead of the normal 2-hour infusion).
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 25/70
Pulmonary fibrosis : In the case of unexplained respiratory symptoms such as non-productive cough, dyspnea, crackles or radiological pulmonary infiltrates, oxaliplatin should be discontinued until further investigations exclude interstitial pulmonary fibrosis.
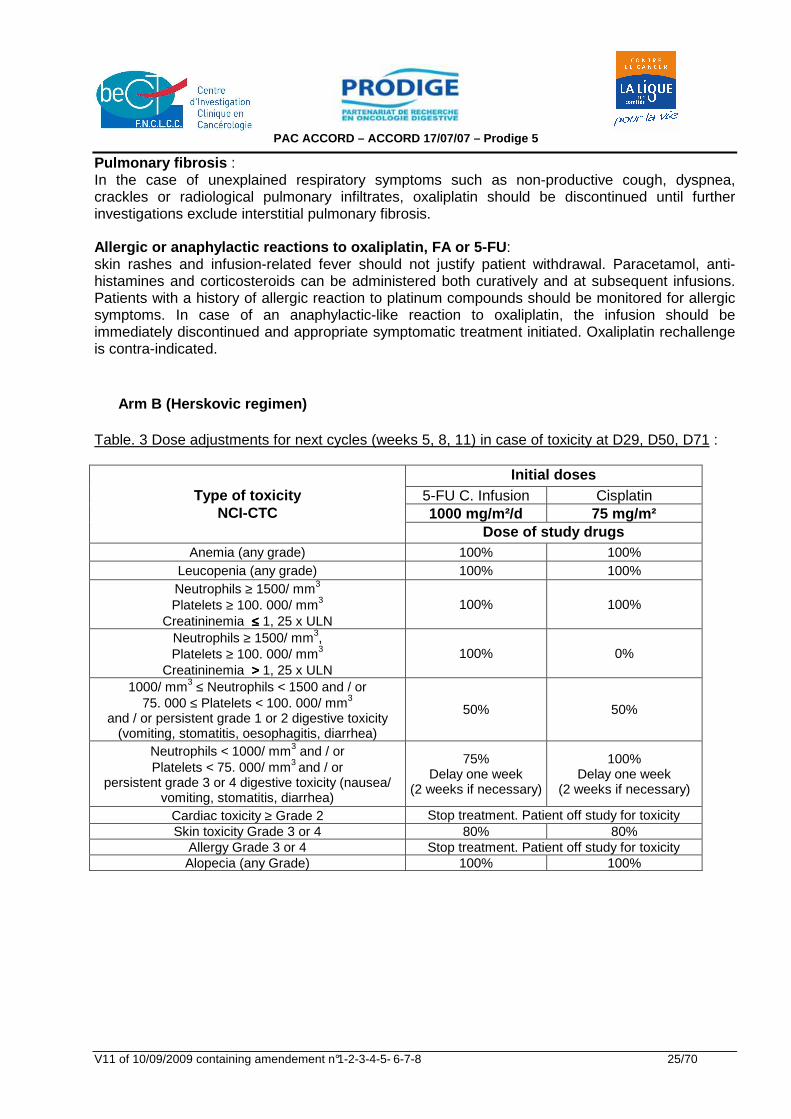
Allergic or anaphylactic reactions to oxaliplatin, FA or 5-FU : skin rashes and infusion-related fever should not justify patient withdrawal. Paracetamol, anti-histamines and corticosteroids can be administered both curatively and at subsequent infusions. Patients with a history of allergic reaction to platinum compounds should be monitored for allergic symptoms. In case of an anaphylactic-like reaction to oxaliplatin, the infusion should be immediately discontinued and appropriate symptomatic treatment initiated. Oxaliplatin rechallenge is contra-indicated. Arm B (Herskovic regimen) Table. 3 Dose adjustments for next cycles (weeks 5, 8, 11) in case of toxicity at D29, D50, D71 :
Initial doses 5-FU C. Infusion Cisplatin 1000 mg/m²/d 75 mg/m²
Type of toxicity NCI-CTC
Dose of study drugs Anemia (any grade) 100% 100%
Leucopenia (any grade) 100% 100% Neutrophils ≥ 1500/ mm3 Platelets ≥ 100. 000/ mm3
Creatininemia ≤≤≤≤ 1, 25 x ULN 100% 100%
Neutrophils ≥ 1500/ mm3, Platelets ≥ 100. 000/ mm3
Creatininemia >>>> 1, 25 x ULN 100% 0%
1000/ mm3 ≤ Neutrophils < 1500 and / or 75. 000 ≤ Platelets < 100. 000/ mm3
and / or persistent grade 1 or 2 digestive toxicity (vomiting, stomatitis, oesophagitis, diarrhea)
50% 50%
Neutrophils < 1000/ mm3 and / or Platelets < 75. 000/ mm3 and / or
persistent grade 3 or 4 digestive toxicity (nausea/ vomiting, stomatitis, diarrhea)
75% Delay one week
(2 weeks if necessary)
100% Delay one week
(2 weeks if necessary)
Cardiac toxicity ≥ Grade 2 Stop treatment. Patient off study for toxicity Skin toxicity Grade 3 or 4 80% 80%
Allergy Grade 3 or 4 Stop treatment. Patient off study for toxicity Alopecia (any Grade) 100% 100%
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 26/70
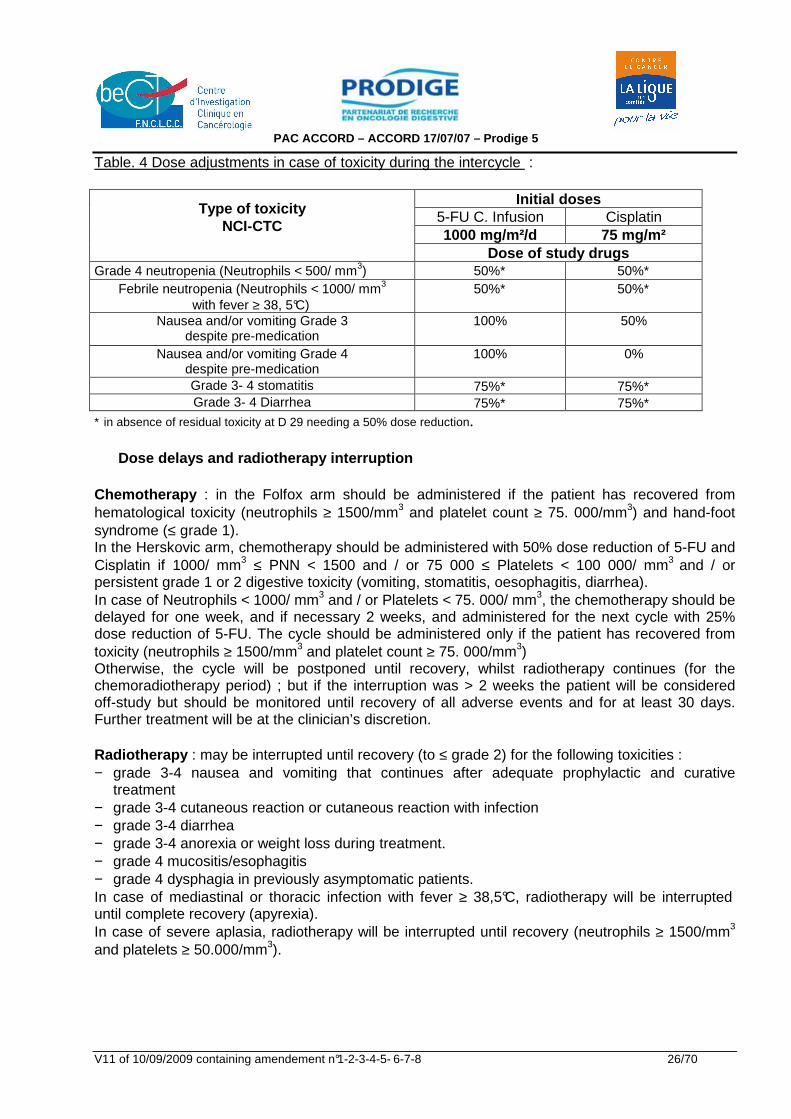
Table. 4 Dose adjustments in case of toxicity during the intercycle :
Initial doses 5-FU C. Infusion Cisplatin 1000 mg/m²/d 75 mg/m²
Type of toxicity NCI-CTC
Dose of study drugs
Grade 4 neutropenia (Neutrophils < 500/ mm3) 50%∗ 50%∗ Febrile neutropenia (Neutrophils < 1000/ mm3
with fever ≥ 38, 5°C) 50%∗ 50%∗
Nausea and/or vomiting Grade 3 despite pre-medication
100% 50%
Nausea and/or vomiting Grade 4 despite pre-medication
100% 0%
Grade 3- 4 stomatitis 75%∗ 75%∗ Grade 3- 4 Diarrhea 75%∗ 75%∗
∗ in absence of residual toxicity at D 29 needing a 50% dose reduction. Dose delays and radiotherapy interruption Chemotherapy : in the Folfox arm should be administered if the patient has recovered from hematological toxicity (neutrophils ≥ 1500/mm3 and platelet count ≥ 75. 000/mm3) and hand-foot syndrome (≤ grade 1). In the Herskovic arm, chemotherapy should be administered with 50% dose reduction of 5-FU and Cisplatin if 1000/ mm3 ≤ PNN < 1500 and / or 75 000 ≤ Platelets < 100 000/ mm3 and / or persistent grade 1 or 2 digestive toxicity (vomiting, stomatitis, oesophagitis, diarrhea). In case of Neutrophils < 1000/ mm3 and / or Platelets < 75. 000/ mm3, the chemotherapy should be delayed for one week, and if necessary 2 weeks, and administered for the next cycle with 25% dose reduction of 5-FU. The cycle should be administered only if the patient has recovered from toxicity (neutrophils ≥ 1500/mm3 and platelet count ≥ 75. 000/mm3) Otherwise, the cycle will be postponed until recovery, whilst radiotherapy continues (for the chemoradiotherapy period) ; but if the interruption was > 2 weeks the patient will be considered off-study but should be monitored until recovery of all adverse events and for at least 30 days. Further treatment will be at the clinician’s discretion. Radiotherapy : may be interrupted until recovery (to ≤ grade 2) for the following toxicities : − grade 3-4 nausea and vomiting that continues after adequate prophylactic and curative
treatment − grade 3-4 cutaneous reaction or cutaneous reaction with infection − grade 3-4 diarrhea − grade 3-4 anorexia or weight loss during treatment. − grade 4 mucositis/esophagitis − grade 4 dysphagia in previously asymptomatic patients. In case of mediastinal or thoracic infection with fever ≥ 38,5°C, radiotherapy will be interrupted until complete recovery (apyrexia). In case of severe aplasia, radiotherapy will be interrupted until recovery (neutrophils ≥ 1500/mm3 and platelets ≥ 50.000/mm3).
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 27/70
Radiotherapy will be resumed on recovery, but if the interruption was > 2 weeks the patient will be considered off-study but should be monitored until recovery of all adverse events and for at least 30 days. Further treatment will be at the clinician’s discretion. SCHEDULE OF VISITS AND OBSERVATIONS Pre-registration work up
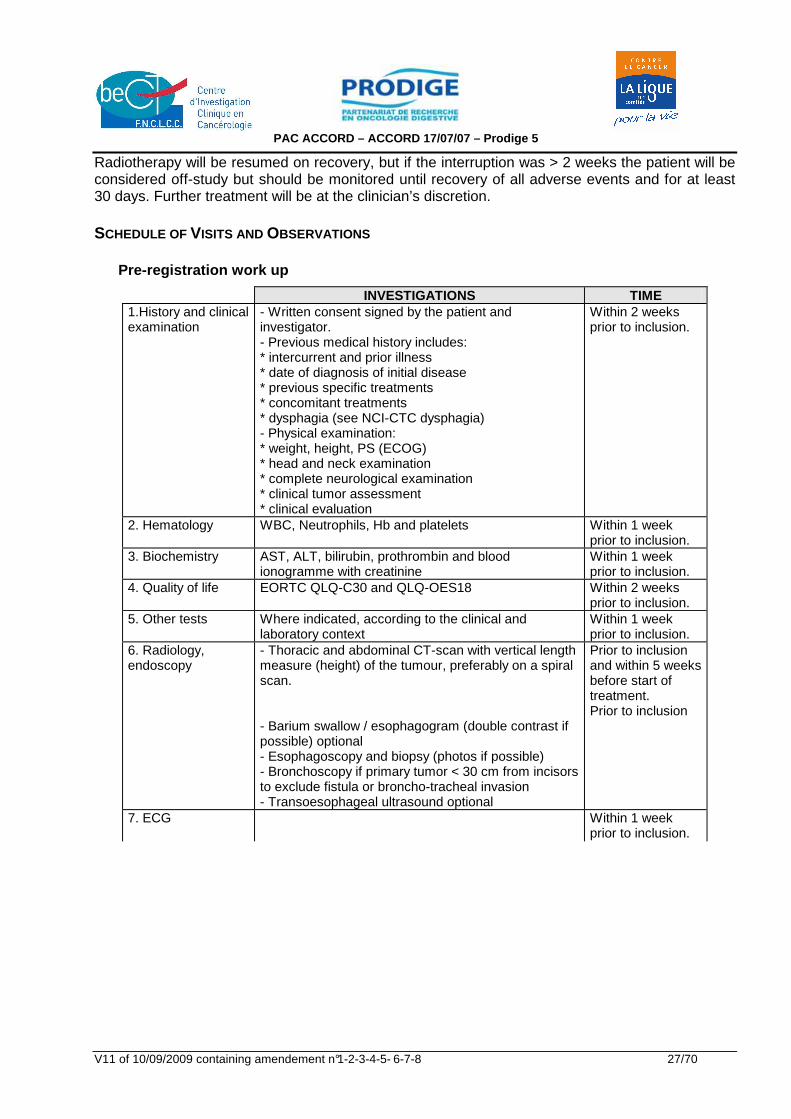
INVESTIGATIONS TIME 1.History and clinical examination
- Written consent signed by the patient and investigator. - Previous medical history includes: * intercurrent and prior illness * date of diagnosis of initial disease * previous specific treatments * concomitant treatments * dysphagia (see NCI-CTC dysphagia) - Physical examination: * weight, height, PS (ECOG) * head and neck examination * complete neurological examination * clinical tumor assessment * clinical evaluation
Within 2 weeks prior to inclusion.
2. Hematology WBC, Neutrophils, Hb and platelets Within 1 week prior to inclusion.
3. Biochemistry AST, ALT, bilirubin, prothrombin and blood ionogramme with creatinine
Within 1 week prior to inclusion.
4. Quality of life EORTC QLQ-C30 and QLQ-OES18 Within 2 weeks prior to inclusion.
5. Other tests Where indicated, according to the clinical and laboratory context
Within 1 week prior to inclusion.
6. Radiology, endoscopy
- Thoracic and abdominal CT-scan with vertical length measure (height) of the tumour, preferably on a spiral scan. - Barium swallow / esophagogram (double contrast if possible) optional - Esophagoscopy and biopsy (photos if possible) - Bronchoscopy if primary tumor < 30 cm from incisors to exclude fistula or broncho-tracheal invasion - Transoesophageal ultrasound optional
Prior to inclusion and within 5 weeks before start of treatment. Prior to inclusion
7. ECG Within 1 week prior to inclusion.
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 28/70
Evaluations during treatment : Folfox arm
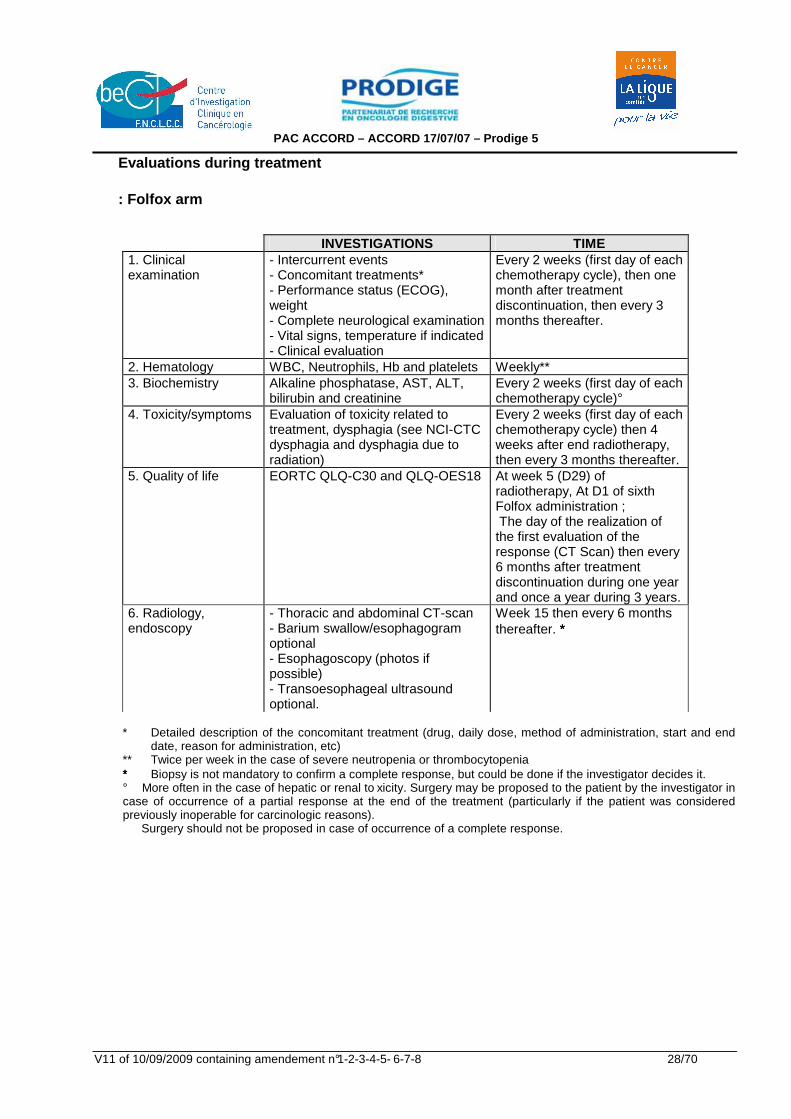
INVESTIGATIONS TIME 1. Clinical examination
- Intercurrent events - Concomitant treatments* - Performance status (ECOG), weight - Complete neurological examination - Vital signs, temperature if indicated - Clinical evaluation
Every 2 weeks (first day of each chemotherapy cycle), then one month after treatment discontinuation, then every 3 months thereafter.
2. Hematology WBC, Neutrophils, Hb and platelets Weekly** 3. Biochemistry Alkaline phosphatase, AST, ALT,
bilirubin and creatinine Every 2 weeks (first day of each chemotherapy cycle)°
4. Toxicity/symptoms Evaluation of toxicity related to treatment, dysphagia (see NCI-CTC dysphagia and dysphagia due to radiation)
Every 2 weeks (first day of each chemotherapy cycle) then 4 weeks after end radiotherapy, then every 3 months thereafter.
5. Quality of life EORTC QLQ-C30 and QLQ-OES18 At week 5 (D29) of radiotherapy, At D1 of sixth Folfox administration ; The day of the realization of the first evaluation of the response (CT Scan) then every 6 months after treatment discontinuation during one year and once a year during 3 years.
6. Radiology, endoscopy
- Thoracic and abdominal CT-scan - Barium swallow/esophagogram optional - Esophagoscopy (photos if possible) - Transoesophageal ultrasound optional.
Week 15 then every 6 months thereafter. ∗∗∗
* Detailed description of the concomitant treatment (drug, daily dose, method of administration, start and end
date, reason for administration, etc) ** Twice per week in the case of severe neutropenia or thrombocytopenia ∗∗∗ Biopsy is not mandatory to confirm a complete response, but could be done if the investigator decides it. ° More often in the case of hepatic or renal to xicity. Surgery may be proposed to the patient by the investigator in case of occurrence of a partial response at the end of the treatment (particularly if the patient was considered previously inoperable for carcinologic reasons). Surgery should not be proposed in case of occurrence of a complete response.
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 29/70
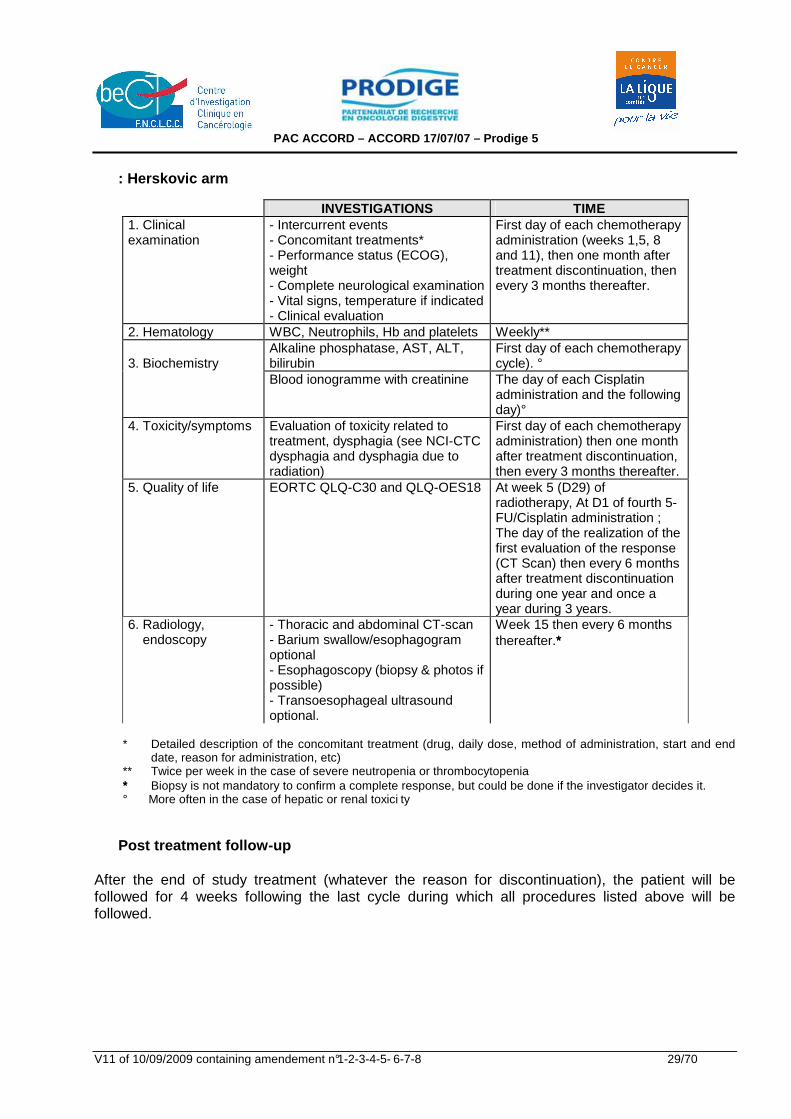
: Herskovic arm
INVESTIGATIONS TIME 1. Clinical examination
- Intercurrent events - Concomitant treatments* - Performance status (ECOG), weight - Complete neurological examination - Vital signs, temperature if indicated - Clinical evaluation
First day of each chemotherapy administration (weeks 1,5, 8 and 11), then one month after treatment discontinuation, then every 3 months thereafter.
2. Hematology WBC, Neutrophils, Hb and platelets Weekly** Alkaline phosphatase, AST, ALT, bilirubin
First day of each chemotherapy cycle). °
3. Biochemistry
Blood ionogramme with creatinine The day of each Cisplatin administration and the following day)°
4. Toxicity/symptoms Evaluation of toxicity related to treatment, dysphagia (see NCI-CTC dysphagia and dysphagia due to radiation)
First day of each chemotherapy administration) then one month after treatment discontinuation, then every 3 months thereafter.
5. Quality of life EORTC QLQ-C30 and QLQ-OES18 At week 5 (D29) of radiotherapy, At D1 of fourth 5-FU/Cisplatin administration ; The day of the realization of the first evaluation of the response (CT Scan) then every 6 months after treatment discontinuation during one year and once a year during 3 years.
6. Radiology, endoscopy
- Thoracic and abdominal CT-scan - Barium swallow/esophagogram optional - Esophagoscopy (biopsy & photos if possible) - Transoesophageal ultrasound optional.
Week 15 then every 6 months thereafter.∗∗∗
* Detailed description of the concomitant treatment (drug, daily dose, method of administration, start and end
date, reason for administration, etc) ** Twice per week in the case of severe neutropenia or thrombocytopenia ∗∗∗ Biopsy is not mandatory to confirm a complete response, but could be done if the investigator decides it. ° More often in the case of hepatic or renal toxici ty
Post treatment follow-up After the end of study treatment (whatever the reason for discontinuation), the patient will be followed for 4 weeks following the last cycle during which all procedures listed above will be followed.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 30/70
From the date of discontinuation for a period of 30 days, all SAEs will be reported. In the case of toxicity persisting after the end of treatment, the patient will be followed until full recovery or stabilisation. A tumor evaluation should be performed during the week 15 and then every 6 months until progression. If, on week 15 any doubt exist about endoscopic complete response evaluation (ulceration or stenosis), a new endoscopy for control will be necessary within 2 months. Evaluation of dysphagia should be performed at the same intervals according to NCI common toxicity criteria (see appendix 8). As precised in paragraph 5.2.2, EORTC QLQ-C30 and QLQ-OES18 must be collected every six months after treatment discontinuation during one year and once a year during 3 years. If feasible, post-treatment follow-up must be pursued until patient's death. RANDOMIZATION PROCEDURES After signing the consent form and validating the results of the baseline inclusion examination, eligible patients will be randomised by the trial randomization centre. The investigator must fax the completed and signed randomization form to the management centre of the company EURAXI Pharma, Biometrics Department. In return, the data management Centre will fax the confirmation of randomization by specifying the treatment arm and patient number. The contact details of the randomization centre are:
Management of Randomization and Data-Management
Sébastien LOUVEAU EURAXI PHARMA
Department of Biometrics from Monday to Friday between 9 am and 4 pm
Fax: 02.47.74.30.82 Phone: 02.47.74.30.47 Treatment must begin within 28 days after randomization. STUDY MEASUREMENTS Completion of full treatment A patient included in the Folfox arm is considered as having completed the treatment if he has received three cycles of radiochemotherapy and three cycles of chemotherapy alone. A patient included in the Herskovic arm is considered as having completed the treatment if he has received two cycles of radiochemotherapy and two cycles of chemotherapy alone. Tumor Response

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 31/70
Patient Evaluability Folfox arm : patients must have received a minimum of 3 cycles of chemotherapy and 50 Gy radiotherapy to be considered evaluable for response unless early disease progression occurs. Herskovic arm : patients must have received a minimum of 2 cycles of chemotherapy (week 1 and 5) and 50 Gy radiotherapy to be considered evaluable for response unless early disease progression occurs. Patients on therapy for at least this period will have their response classified according to the definitions set out below. Tumor assessment for all lesions must have been performed at baseline, week 15 and every 6 months on follow-up visits for patients off study for other reason than PD. It is essential to examine all target lesions at every evaluation. For inclusion, endoscopy, oesophagogram and CT-Scan have to be performed (if possible spiral CT-Scan should be preferred). The reference method is the CT scan . Oesophageal tumor must be evaluated according to the vertical length of the tumor (height on CT scan or Barium swallow), maximal dimension of the oesophageal tumor in the transverse plane on CT-Scan and the maximal thickness of the oesophageal wall on CT scan. If measurement of the tumor height is not feasible on CT scan, tumor height should be measured on oesophagogram (or endoscopy if oesophagogram is not available). Endoscopic complete response is defined by IDMC members as follow:
- All endoscopic reports before and after treatment must be available - Disappearance of the tumor lesion and no stenosis - No ulceration (slough) - No budding - No appearance of any lesion on endoscopy (or echo-endoscopy) - No local progression on CT-Scan
If, on week 15, any doubt exists about complete response evaluation (ulceration or stenosis), a new endoscopy for control will be necessary within 2 months. At inclusion, impassable tumors during the endoscopy will be classified as ycT3 or ycT4 if the CT scan shows an extension to the adjacent organs. At each tumor evaluation, dysphagia must be evaluat ed using NCI common toxicity criteria (appendix 9). Disease Measurability : RECIST Criteria Complete response (CR) : disappearance of all target lesions associated with the disappearance of all non target lesions and the normalization of tumor marker level. The persistence of one or more non-target lesion and/or maintenance of tumor marker level above the normal limits will lead to the evaluation of the response as partial. CR is confirmed if determined by 2 observations no less than 4 weeks apart. Partial response (PR) : at least a 30% decrease of in the sum of the longest diameter of the target lesions, taking as reference the baseline sum longest diameter or a CR associated with abnormal tumor marker level and/or persistence of non target lesions as described above.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 32/70
Progressive disease (PD) : at least 20% increase in the sum of the longest diameter of target lesions, taking as reference the smallest sum longest diameter recorded since the treatment started or the appearance of one or more new lesions. Stable disease (SD) : neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease, taking as reference the smallest sum longest diameter since the treatment started. Development of brain metastasis : The development of brain metastasis will be considered as a sign of progression, even if the disease is responding outside the brain. However, the investigator may decide to continue the study drug until progression outside the brain concomitantly with the most appropriate treatment of the brain metastasis (e.g. radiotherapy). Time-related Parameters Progression-free survival Will start from the day of randomization until the documentation of tumor progression, metastasis diagnosis, esophageal second cancer or death form any cause. Time to treatment failure Will start from the day of randomization until the documentation of progression, treatment discontinuation for : toxicity, patient’s refusal, patient lost of view or death. Overall Survival: Survival is measured from the day of randomization to death, last contact or cutoff. Quality of life Quality of life will be evaluated using EORTC QLQ-C30 and a validated disease-specific module EORTC QLQ-OES18 (see sections 5.2.1 and 5.2.2). The investigators must explain to the patients the interest in answering the questionnaire and identifie precisely for each of them the dates of collect of these questionnaires (see paragraphs 5.2.1 to 5.2.3). An identified person must be in charge of the colle ct of the questionnaires in each centre and a second person must be designed in case of abs ence of the first one. The first questionnaires must be given and explained to the patient before the inclusion. The patient must complete it before the randomization. During the treatment, the questionnaires must be redacted by the patient in the centre (day 29 of radiotherapy and the day of the administration of the sixth cycle of Folfox or fourth cycle of 5-FU/Cisplatin).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 33/70
After the end of the treatment, the questionnaires must be completed by the patient in the centre at the moment of the visit control each 6 months during the first year and once a year the next 3 years. If a patient refuses to complet a questionnaire, the reason must be précised on the questionnaire by the person in charge of the collect. Clinical research associate of the sponsor will send the collected questionnaires to the data-management service (a copy being conserved in the patient’s file).
6. SAFETY ASSESSMENTS All patients having received at least one dose of chemotherapy and one fraction of radiotherapy will be considered as evaluable for safety. Safety parameters will include: description of toxic deaths, premature withdrawals from treatment for toxicity, description of Adverse Events, Serious Adverse Events (SAE), and evaluation of toxicity, using NCI-CTC version 3. 6.1 CONCOMITANT TREATMENT RESTRICTIONS No concomitant anti-cancer therapy, no high-dose long-term corticosteroids, no Calcium Gluconate/ Magnesium Sulfate infusions, no hematopoietic growth factors and no oesophageal dilatation are allowed during the trial. All concomitant medications must be thoroughly documented in the source data. Refrigerated headwear should be avoided during/after oxaliplatin infusion. Appropriate antiemetic treatment should be prescribed, both preventatively and curatively for each chemotherapy administration. 6.2 DATA MONITORING COMMMITTEE : An independent Data Monitoring Committee (DMC) will be set up to review trial enrolment, safety and efficacy of the administered treatments and dosimetric documents. The DMC, including 2 medical oncologists, 2 radiotherapists and a statistician not participating to the trial will convene at least every 6 months to review safety (toxicities, treatment exposure, SAEs including the rate of deaths on study i.e. within 30 days of last chemotherapy administration) and other issues related to the appropriate conduct of the trial. Statistical listings, tables and analysis will be performed by an external statistician who will provide the results to the committee. The DMC will participate, at the end of the phase II, to the discussion about the continuation of the trial as a phase III randomized study (see paragraph 9, ‘statistical considerations’).
7. DRUG SUPPLIES STUDY DRUGS

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 34/70
Oxaliplatin and 5-FU/FA will be administered in accordance with standard procedures for these cytotoxic agents at the institution. Products will be prepared according to Good Chemotherapy Practises and the procedures described below. Oxaliplatin, 5-FU and folinic acid will be provided by the Sponsor. The other products will be taken from the usual pharmacy stock. � This product will be labelled in accordance with article R.5123 of the Code of Public Health and
the Recommendations of Appendix 1 of European Good Manufacturing Practises. � This product will be distributed to the different pharmacies of the healthcare centres according
to Good Distribution Practices (GDP). Traceability of all products used within the scope of this clinical study, whether they are provided by the sponsor or the hospital pharmacy, m ust be guaranteed throughout the duration of the study. 7.1.1 Oxaliplatin
Active Ingredient Chemical name : Trans-/-diaminocyclohexane oxalatoplatinum cis-
[oxalato(trans-/-1,2-diaminocyclohexane) platinum(II)] International non proprietary name : Oxaliplatin Empirical formula : C8H14N2O4Pt Relative molecular weight : 397.3 Physical properties : White to off white cake or powder Slightly soluble in water Very slightly soluble in methanol Insoluble in ethanol and acetone The pH of an aqueous solution of 2 mg/ml is between
4.8 and 5.7 Finished Product Presentation and composition Oxaliplatin is presented in the form of a white to off-white cake or powder contained in clear glass vials sealed with a rubber stopper and an aluminium seal with a flip-off cover. 100 mg vials Oxaliplatin 100 mg Lactose monohydrate 900 mg Nominal volume of vial 50 ml Reconstitution The freeze-dried powder is reconstituted by adding 20 ml of water for injection or 5 % glucose solution and then by diluting in an infusion solution of 250 to 500 ml of 5% glucose solution.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 35/70
Dispose of any reconstituted solution that shows evidence of precipitation. Always use the recommended solvants. Never administer undiluted solution. RECONSTITUTION OR FINAL DILUTION MUST NEVER BE PERFORMED WIT H SODIUM CHLORIDE SOLUTION . Incompatibilities Do not combine with alkaline medications or media which cause oxaliplatin to degrade. Do not administer simultaneously by the same line. Flush line after oxaliplatin administration. Do not use preparation or administration needles or intravenous sets containing aluminium components. There is a risk of degrading oxaliplatin. Precautions All cancer chemotherapeutic agents should be handled with utmost care during preparation and administration. They should not be prepared by pregnant or breast-feeding women or by persons allergic to that agent. To avoid any form of physical contact with the drug by the health care provider, gown, gloves and masks should be worn when appropriate. As a parenteral agent, oxaliplatin should be prepared in a vertical-flow biologic safety cabinet. All equipment used to prepare and administer the drug should be destroyed according to standard hospital procedures for disposal of cytotoxic waste. Refer to hospital guidelines for any additional precautions that may apply. 7.1.2 5-FU : Information can be found in the packag e insert. 7.1.3 Folinic acid : Information can be found in th e package insert.
7.2 STORAGE CONDITIONS
Oxaliplatin : - Freeze-dried powder : May be stored for three years at 25°C ; excursions permitted to 15°C to 30°C, not exceeding 30°C. - Reconstituted solution : In 5% dextrose solution or water for injection and in the original vial, the solution should be diluted immediately. - Solution for infusion : After dilution in 250 to 500 ml of 5% dextrose solution, the shelf-life is 24 hours at 2°C to 8°C.
8. TRIAL DISCONTINUATION CRITERIA The trial may be suspended or stopped by the sponsor after discussion with the coordinator at the request of the Competent Authority and/or Committee for the Protection of Persons (CPP) for the following reasons:
- Unexpected incidence and/or severity of toxicity, - Insufficient recruitment of patients, - Poor quality of collected data.
9. SERIOUS ADVERSE EVENT

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 36/70
GENERALE DEFENITION Any event is considered to be a serious adverse eve nt (SAE) when it:
• Results in death, • Is life-threatening, • Requires patient hospitalization or prolongs existing hospitalization, • Causes a permanent disability or a serious temporary incapacity, • Causes a congenital malformation, foetal malformation or an abortion, • Is medically significant.
The terms disability and incapacity correspond to any temporary or permanent, clinical ly
significant physical or mental handicap with an imp act on the patient’s physical activity
and/or quality of life.
A medically significant event is any clinical event or laboratory result which the investigator considers to be serious and which does not correspond to the above-defined criteria of seriousness. Such an event may represent a risk to the patient and require medical intervention in order to prevent an outcome corresponding to one of the above criteria of seriousness (for example: overdosage, a second cancer, pregnancy and new facts may be considered to be medically significant). The following adverse events are not considered to be serious adverse events (SAE):
• Hospitalisation < 24 hours, • Elective hospitalisation scheduled before the start of the trial and/or planned in the protocol
(biopsy, chemotherapy). DEFINITION OF AN EXPECTED SERIOUS ADVERSE EVENT (E-SAE) An ESAE is an event already mentioned in the most recent version of the investigator's brochure or in the Summary of Product Characteristics (SPC) for medicinal products that have already been granted marketing authorisation (MA). This definition also applies to the trial medicinal product when it is administered off-label for a same population. DEFINITION OF AN UNEXPECTED SERIOUS ADVERSE EVENT (U-SAE) An USAE is an event not mentioned or with a different nature, severity or outcome from that described in the investigator's brochure or in the Summary of Product Characteristics (SPC) for medicinal products that have already been granted marketing authorisation (MA). CRITERIA OF SEVERITY The criterion of severity should not be confused wi th the criterion of seriousness which is used to define the need for expedited reporting. The severity of events will be assessed according to version 3.0 of the CTC-AE classification (cf. appendix 7). The severity of adverse events not listed in this classification should be assessed using the following terms:

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 37/70
Mild (grade 1) : does not affect the patient’s usual daily activity Moderate (grade 2) : disturbs the patient’s usual daily activity Severe (grade 3) : prevents the patient’s usual daily activity Very Severe (grade 4): Requires life-support/is life-threatening Death (grade 5) RECOMMENDED PROCEDURE IN THE EVENT OF AN SAE The investigator must inform the Therapeutic and Clinical Studies Office drug safety monitor (PV-BECT) of all expected (ESAE) and unexpected (USAE) Serious Adver se Events , whether or not these are caused by the research, occurring during the study or in the 30 days following the last dose of complete treatment (pre-operative + surgery + post-operative). All delayed Serious Adverse Events (occurring after this 30 day period) which may reasonably be considered to be related to the study treatment(s) or research must be reported with no time limit. SAE must be reported by sending a fax, within 48 working hours of their observation to BECT PV.
Bureau d’Etudes Cliniques et Thérapeutiques (BECT) Pharmacovigilance
Tél. : 01 44 23 04 16 – Fax : 01 44 23 55 70 Courriel : [email protected]
using the SAE report form (cf appendix 7). The investigator should note for each event:
• As clear a description as possible according to medical terminology, • Its severity, • Time of onset and termination of the event, • Measures taken and the need or not for corrective treatment, • If the trial treatment was discontinued, • Its outcome, in the event of a non-fatal event, the clinical course must be followed up
until cure or return to previous condition or stabilisation of any sequelae, • The causal relation between this event and the trial treatment or a constraint related to
the research (period without treatment, additional examinations requested for the research etc.),
• The causal relation with the trial medication(s), the disease treated or any other concomitant disease or treatment. The investigator must also enclose with the serious adverse event report, whenever possible:
• A copy of the hospital discharge report or prolongation of hospital stay report, • A copy of the autopsy report, • A copy of all the results of the additional tests performed, including the relevant
negative results together with normal laboratory values, • Any other document that the investigator considers to be useful and relevant.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 38/70
All these documents must be anonymized. Complementary information may be requested (by fax, telephone or during a visit) by the monitor. In the event of an unexpected SAE which may reasonably be related to one of the study treatments, the investigator will be asked to provide pharmacovigilance with additional information. Nevertheless, any expected event with a different severity, outcome or incidence will be considered to be unexpected by pharmacovigilance. FOLLOW-UP OF SAE The investigator is responsible for providing appropriate medical follow-up to patients until resolution or stabilisation of the effect or until the death of the patient. This may sometimes involve continuation of follow-up after the patient has left the trial. The investigator should send this complementary information to BECT PV using an SAE report form (by ticking the box “Follow-up n° X” to specif y that this concerns follow-up and is not an initial report) within 48 hours of obtaining it. The final follow-up form should also be sent after resolution or stabilisation of the SAE. The investigator should keep the documents about a suspected adverse effect in order to be able to complete the previous information whenever necessary. He must answer requests for additional information from BECT PV in order to document the initial case report. Toxic Deaths A toxic death is defined as any death to which toxicity is thought to have made a contribution. It should be treated as an SAE.
10. COST AND EXCESS COSTS OF THE RESEARCH Any additional expenses stipulated in article R.112-1 of the Code of Public Health are subject to an agreement negotiated between the FNCLCC and the representative of the hospital, taking into account the financial means available to the FNCLCC for its public promotion activity. The FNCLCC however, ensures the organisation of the trial and provides the following items: protocol, case report file, investigator's dossier; and oxaliplatin, acid folinic and 5-FU in the FOLFOX arm supply of the study medications. The equipment, treatments, or services provided by other partners, must be specified in the clinical trial agreement.
11. QUALITY ASSURANCE In order to guarantee the authenticity and credibility of the data in accordance with GCP, the sponsor must set up a quality assurance system comprising:

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 39/70
- Trial management according to the Federation’s procedures, - Quality control of investigator site data by the monitor, whose role is to check the agreement and consistency of data in the CRF by comparison with the source document,
- Possible audit of investigator centres.
12. OWNERSHIP AND CONFIDENTIALITY OF DATA The investigator undertakes, for himself and all the persons involved in following up the conduct of the trial, to guarantee the confidentiality of all information provided by the French National Federation of Cancer Centres (FNCLCC) until the publication of the trial results. This confidentiality agreement does not apply to information that the investigator may have to give to patients within the scope of their participation in the trial or to information that has already been published. The investigator undertakes not to publish, disclose or use, either directly or indirectly in any way, the scientific or technical information related to the trial. However, pursuant to article R 5121-13 of the Code of Public Health, the centre and the investigator may give information about the trial to the following:
- Minister of Health, - Public health inspectors, - Public health inspecting pharmacists, - The General Director and Inspectors from Afssaps.
No written or oral comment about the trial may be made without the agreement of the sponsor and the coordinator; all the information made available or obtained during the conduct of the trial belongs, as of right, to the French National Federation of Cancer Centres which may use it as it so wishes.
13. RULES FOR PUBLICATION Publishing PRODIGE trials in good-quality medical journals within a short period of time is one essential objective to achieve therapeutical progress. Publications must be carried out under the direct supervision of the PRODIGE Coordinating Committee (PCC) that decides for: - the time for publishing the trial's preliminary and definitive results. - the writing committee composition (at most 5 members). All information resulting from the trial are deemed confidential, at least until the sponsor, the trial's principal investigator and statistician have proceeded to an appropriate analysis and control. The Coordinating Committee (PCC) may delegate its functions to the trial's principal investigator. In any cases, the PCC validates the choices and insures that the time-delays are respected. A text

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 40/70
submitted by the writing committee is considered as accepted if the PCC has not answered within 1 month after its submission. The Writing Committee comprises: - The investigator (or both investigators when they are two) who has written the initial project.
He/she will be first author unless decided otherwise. - The statistician(s) who have analyzed the data. - The most important contributors. - Possibly, one expert who has brought essential contribution to the data analysis (biologist,
anatomopathologist, …)
• The first author commits to submit an article for publication within a short period of time fixed by the PCC. The time delay should not exceed one year after the trial has ended. In case the first author has difficulty complying with the writing, the PCC will designate a new writer who will subsequently become first author. Recourse to a medical writer can be appropriately used and writing workshops organized for the first author in collaboration with the statistician. Before any publication is submitted, a comprehensive list of inclusions and investigators per center will be kept at the disposal of all the investigators participating in the trial.
• The publication authors are listed in the following order, according to the amount of work and the number of patients included:
- The first author. - The members of the Writing Committee (see definition above). - A limited number of investigators (1 per center) listed in order of their participation, in principle
only one investigator per center, excepted special centers for which 2 investigators may be selected by the Steering Committee. This rule may be modulated to allow some small- and/or medium-size centers that have included a large number of patients to be included in the list of authors. The PCC will validate this process to insure that nobody is penalized.
- The maximum number of authors will be defined as the limit fixed by the medical journals. - Whatever the number of included patients is, there will be at least one author representing
one of the two partners: FFCD or FNCLCC. - In case of a second publication or a substudy, the list of authors may differ from the initial
article and reflects the medical expertise involved in the research work. For instance, in the RCT trials, an article dedicated to radiotherapy may be signed by the co-investigator radiotherapists of the center that have included patients. The last author of this derived publication (having possibly equally contributed) is the first author of the initial article.
- The Prodige partnership must be cited in the title or after the list of authors. In case of a cooperative trial, the first association that is quoted is the one that has initiated the trial and the others are mentioned, in the order of their participation, under the condition that they have included at least 5 % of the patients.
- Beside exception, in all the trials sponsored or managed by FFCD, a member of the INSERM Unit U866, if he/she is not already first author, will be last author in order to insure that his/her work is accounted for by the INSERM. In that case, the author before the last one will be signaled as 'having equally contributed', whenever it is applicable with respect to the

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 41/70
concerned medical journals. - As a general rule, the statistician will be listed with the authors beyond the third position.
He/she may be the first or second author of a specific publication (derived article or substudy). All participants who are not listed with the authors are cited at the end of the article. The Data Manager will be also cited and he/she may be included in the list of authors if it is deemed justified by the PCC. The various partners are thanked. The authors and the sponsor will receive a manuscript for internal review before it is submitted to a journal. They commit to respond within a period of 15 working days (30 days during the summer period). Beyond this delay their opinion and critics will not be taken into account.
• Oral communication based on the trial's results: With both the PCC and Steering Committee agreements, an investigator may, in his/her name, present all or part of the results orally. As a general rule, the list of authors is the same as for written articles, but the order of appearance of the authors may sometimes differ between articles and oral communications and vary according to congresses. In some cases (multidisciplinary studies, or pathological, biological, echo-endoscopic studies carried out in parallel with a Prodige trial) other authors may be selected in function of their contribution. The PRODIGE partnership, and possibly other associations, must be cited.
14. ETHICAL AND REGULATORY ASPECTS The clinical trial must be conducted in accordance with the following texts: - Ethical principles of the current version of the Helsinki declaration, - Good Clinical Practices of the International Conference of Harmonization (ICH–E6, 17/07/96), - European Directive (2001/20/EC) on the conduct of clinical trials, - The Huriet law (n° 88-1138) of December 20, 1988 concerning the Protection of Persons
participating in Biomedical Research and modified by the Public Health law (n° 2004-806) of August 9, 2004,
- The Data processing and Liberties law n° 78-17 of January 6, 1978 modified by law n° 2004-801 of August 6, 2004 concerning the protection of persons with regard to the processing of personal data,
- Bioethics law n° 2004-800 of August 6, 2004.
15. COMMITTEE FOR THE PROTECTION OF PERSONS Before conducting biomedical research on human subjects, the sponsor must submit the project for review to the competent Committee for the Protection of Persons of the site where the co-ordinating investigator practises his activity. The application for an ethics opinion on the biomedical research project is sent to the committee by the sponsor. Applications for substantial amendments to the preliminary drafts are also sent by the sponsor to the committee for an approbation.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 42/70
16. COMPETENT AUTHORITY Before conducting or requesting the conduct of biomedical research the sponsor of this research must send the competent authority an authorisation application. The timeframe for examination of the authorisation application, except for cell therapy products with a timeframe of 90 days and gene therapy products with a timeframe is 120 days, cannot exceed 60 days from the date of receipt of the complete dossier. The competent authority must inform the sponsor if it has justified objections to the implementation of the research within 30 days from the date of receipt of the complete dossier, and inform the committee for the protection of persons concerned.
17. INFORMATION AND CONSENT OF PARTICIPANTS Before conducting biomedical research on human subjects, their informed consent must be collected in writing after they have been informed, by the investigator during a visit and after a sufficient period of reflection. The information intended for participants in the trial must include all the items defined in the law of public health of August 9, 2004 and must be written in a simple way, in a language comprehensible to the patient (cf appendix 8). After reading the information sheet, the participant must initial all the pages and the original must be placed in the investigator file and a copy given to the participant in the research. The consent form must be dated and signed personally by the participant in the research and the investigator (the original form is archived by the investigator and a copy is given to the participant). The information sheet and informed consent forms (appendix 8) must be grouped together on the same document in order to ensure that all the information is given to the participant in the research. In the case of trials performed in order to conduct genomic or proteomic analyses, the information form must specify the type of research that will be carried out and the patient must be able to accept or refuse the keeping of his biological samples for the purpose of scientific research.
18. SPONSOR’S RESPONSIBILITIES In accordance with the Huriet law, the sponsor is responsible for: - Taking out a civil liability insurance policy to cover any harm or injury caused by this research
on subjects taking part (art. L.1121-7), - Paying the competent Regional Health and Social Affairs Management (DRASS) the fixed
charge for consulting the Independent Ethics Committee (CCPPRB) (Order of 7/5/91 and 13/12/01),
- Sending the French Agency for the Safety of Health Products (AFSSAPS), the initial declaration of intent describing the essential data of the research accompanied by the IEC opinion (art. L.1123-8),
- Informing the Directors (art. L.1123-10, R.5124) and Pharmacists (art. L.5125-19, R.5124-1) of the hospitals concerned,

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 43/70
- Making available all the necessary information to conduct the research to the investigators (art. R.5122),
- Informing AFSSAPS about any Serious Adverse Event that may be related to the research and about any premature discontinuation of the study as soon as they are known (art. L.1123-8).
The sponsor must also draft a declaration on the data processing of personal medical data, to the Advisory board and French Data Protection Agency (CNIL), in accordance with the conditions of application of the simplified procedure (Law n°94-5 48 dated 1/07/94 completing Law n°78-17 dated 6/01/78). The sponsor must ensure the archiving of essential study documents under conditions ensuring their safekeeping for the minimal duration specified by GCP, which is 15 years after the end of research. The sponsor of the clinical trial is the person or entity taking the initiative to conduct biomedical research on human subjects, ensuring management and checking that it is adequately funded. The sponsor must be established in the European Community or if not, have a legal representative in a member state.
19. INVESTIGATOR’S RESPONSIBILITIES The principal investigator of each hospital involved undertakes to conduct the clinical trial in accordance with the protocol approved by the CPP and competent authority. The investigator must make no amendment to the protocol without the written authorisation of the sponsor and without the CCPPRB and the competent authority giving their authorisation for the proposed amendments The principal investigator is responsible for: - Providing the sponsor with his/her curriculum vitae and those of the Co-investigators, - Identifying the members of the team participating in the trial and defining their responsibilities, - Starting patient recruitment after authorisation from the sponsor, Each investigator is responsible for: - Collecting the informed consent form which has been personally dated and signed by the
participant in the research before any trial-specific screening procedure, - Regularly filling in the case report files (CRF) for each patient included in the trial and allowing
the Clinical Research Associate (CRA), appointed by the sponsor, direct access to the source documents in order to validate the data in the CRF,
- Dating, correcting and signing corrections of the CRF for each patient included in the trial, - Accepting regular visits by the monitor and where necessary by the auditors appointed by the
sponsor or inspectors from the regulatory authorities. All the trial documents (protocol, consent forms, CRFs, investigator’s dossier, etc…), and all the original documents (laboratory results, X-rays, study visit reports, clinical examination reports, etc) must be considered to be confidential and kept in a safe place. The Principal Investigator must keep the data as well as a patient identification list for a minimum period of 15 years after the end of the study.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 44/70
20. INTERRUPTION OF SUBJECT STUDY PERIOD CIRCUMSTANCES Subjects will discontinue treatment in the event of :
• Completion of treatment (as described in section 5.4.1); • Disease progression (or death due to progressive disease) : the date and evidence for
disease progression will be documented in the medical record; • Adverse event, treatment limiting toxicity, inter-current medical problem; • Voluntary withdrawal, investigator’s decision; • Interruption of treatment > 2 weeks.
The reason and date of discontinuation are to be documented in the CRF. The investigator should complete all end of treatment procedures when a patient withdraws from treatment. REPLACEMENT OF SUBJECTS Ineligible or or non-evaluable patients will not be replaced ; the sample size is increased of 10% (4 patients in more in each arm) to include 80 evaluable patients in phase II. A new calculation of the phase III population will be done at the end of the phase II.
21. STATISTICAL CONSIDERATIONS : At the end of the first part of the trial (randomised phase II) on 88 patients (44 in each arm), the decision to continue the study will be taken by the coordinator, the sponsor and the statistician of the study, with the help of the Data Monitoring Committee. Three conditions should be fulfilled to decide to continue the trial :
- Good inclusion rates (88 patients in less than 18 months). - Completion of full treatment over 60 % in the experimental arm. - Endoscopic complete response rate of Folfox arm equal or superior to the endoscopic
response rate of 5FU/Cisplatin arm.
If it is decided to continue the study as a phase III randomised study, the main objective will be to increase the progression-free survival. With a global alpha risk of 5 % and 90% power, a period of the inclusion of 48 months (28 months phase II and 20 months for the continuation) and a minimum follow-up of 12 months, the inclusion of 169 supplementary patients (total of 266 patients; 133 in each arm) will be necessary to demonstrate an improvement of 20 % in progression-free survival at three years (from 30 % in the Herskovic arm to 50 % in the Folfox arm, based on phase II results). The corresponding number of events is around 144. However, a new calculation of the number of patients can be done after the interim analysis to adjust the sample size (this calculation could confirm the sample size or increase it). The analysis of results will be done on the “intent to treat” basis (all patients randomised will be included in the final analysis) and per-protocol (eligible and evaluable patients).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 45/70
PARAMETERS : PHASE II : Primary Endpoints : percentage of patients having completed the full treatment in the two arms ; percentage of endoscopic complete response in the two arms. Secondary Endpoints : description of safety profile of each arm using NCI-CTC scale. Characterisation of toxicity, SAEs, and treatment withdrawals. PHASE III : Primary Endpoint : comparison of the progression-free survival between the two arms. Secondary Endpoints : comparison of overall survival, complete response rate, incidence of grade 3-4 toxicities and time to treatment failure between both arms. Evaluation of the quality of life using EORTC QLQ-C30 (version 3) and a validated disease- specific module QLQ-OES 18. ANALYSIS POPULATION Safety: All subjects having received at least one dose of chemotherapy or one fraction of radiotherapy will be considered evaluable for safety. Response rate will be analysed in the per-protocol population all eligible, evaluable patients. i.e., patients having received at least : 3 cycles and 50 Gy in the Folfox arm, 2 cycles of chemotherapy (week 1 and 5) and 50 Gy in the Herskovic arm, or having progressed before the first evaluation. An intent-to treat analysis (on all randomized patients) will be performed for all the endpoints. For the analysis performed on eligible, evaluable patients, patients categorized as early death, toxic death and progressive disease will be considered as treatment failure. Response will be evaluated in all patients. STATISTICAL METHODS DEMOGRAPHICS : Descriptive statistics (mean, median, standard deviation and 95% confidence interval, range of value) will be used for this study. EFFICACY : Response rate will be reported with the 95 % confidence interval.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 46/70
Rate of progression-free survival, time to treatment failure, overall survival and the corresponding curves will be computed using the Kaplan-Meier method. The rates will be reported with their 95% confidence interval. Efficacy parameters will also be subjected to further appropriate analysis, considering correlation with factors or probable prognostic value such as patient PS at entry, histology type (adenocarcinoma / squamous cell carcinoma / adenosquamous), location of the tumor, weight loss at inclusion (> ou ≤ 10%), length of the tumor (≤ ou > 5 cm). Response parameters will undergo univariate analysis using the pearson X2 test and multivariate analysis via logistic regression. Univariate analysis of time parameters will be carried out using the log-rank test and the Cox test for multivariate analysis. SAFETY : Descriptive statistics will be employed to characterise the toxicity, toxic death, SAE and toxicity-related treatment discontinuation profiles.Toxicity will be analyzed per cycle and per patient ; the rate of grade 3-4 toxicities will be compared using chi2 test. QUALITY OF LIFE : The analysis will consist in the comparison of the different scores of “Global health status/Qol” (primary objective) and dysphagia, asthenia, pain and physical functions (secondary objectives). INTERIM ANALYSIS : One interim analysis is planned during the phase III after the occurrence of 72 of the 144 planned events. For this analysis, comparative analysis on efficacy (progression-free survival, response rate, survival) and safety will be performed. The results will be presented to the Data Monitoring Committee. The results of this analysis will be given to the principal investigator only if the Data Monitoring Committee proposes to stop the trial. The Data Monitoring Committee will propose to the sponsor and the coordinator to stop accrual of new patients and to publish preliminary results earlier than anticipated only if after interim analysis of the trial one of the following conditions is met: - The results show clearly that experimental arm is either beneficial (increase of progression-free survival, p<0.0031) or harmful (p>0,84, decrease of survival and/or major toxicity according to IDMC appreciation’s). - All the data from this or other ongoing trials are convincing enough to influence the practice of most physicians. SAMPLE SIZE CALCULATION : Standard randomized phase II/ III design.88 patients will be included in the phase II study (44 in each arm) and 266 in the phase III (133 in each arm).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 47/70
REFERENCES : 1. La Vecchia C, Negri E, Levi F et al. Cancer mortality in Europe : effects of age, cohort of birth and period of death. Eur J Cancer 1998; 34: 118-41 2. Ménégoz F, Black RJ, Arveux P et al. Cancer incidence and mortality in France in 1975-1995. Eur J Cancer 1997; 6: 442-66 3. Calament G, Cauvin JM, Robaszkiewicz M et al. Traitement et survie du cancer épidermoïde de l’oesophage dans le département du Finistère entre 1984 et 1988 (716 cas). Gastroenterol Clin Biol 1993; 17: 9-16 4. Kok TC.Chemotherapy in oesophageal cancer : a review. Cancer Treat Rev 1997; 23: 65-85 5. Agha FP, Gennis MA, Orringer MB et al. Evaluation of response to preoperative chemotherapy in oesophageal and gastric cardia cancer using biphasic esophagograms and surgical-pathological correlation. Am J Clin Oncol 1986 ; 9 :227-32 6. Conroy T. Measurement of the primary oesophageal tumor. In EORTC GI tract cancer cooperative group Manual on diagnosis and treatment of GI tract cancer. Wils J, Bleiberg H eds, Brentwood Multiscience Publishing Co. 1993, 28-30 7. François E, Olivier JM, Rougier P et al. Standards, options et recommandations pour la prise en charge des patients atteints de cancer de l’oesophage. In Standards, options et recommandations pour la pratique clinique, FNCLCC ed. CD ROM, John Libbey Eurotext 1998 8. Conroy T, Etienne PL, Adenis A et al. Phase II trial of vinorelbine in metastatic squamous cell oesophageal carcinoma. J Clin Oncol 1996 ;14 :164-70 9. Ilson DH, Ajani JA, Bhalla K et al. Phase II trial of paclitaxel, fluorouracil and cisplatin in patients with advanced carcinoma of the oesophagus. J Clin Oncol 1998 ; 16 :1826-34 10. Stahl M, Wilke H, K. Walz et al. Randomised phase III trial in locally advanced squamous cell carcinoma (SCC) of the esophagus : chemoradiation with and without surgery. Proc Am Soc Clin Oncol 2003 ; 22, 250 : abstract 10001. 11. Nygaard K, Hagen S, Hansen HS et al. Pre-operative radiotherapy prolongs survival in operable oesophageal carcinoma : a randomized multicenter study of pre-operative radiotherapy and chemotherapy. The second Scandinavian trial in oesophageal cancer. World J Surg 1992 ; 16 :1104-10 12. Ajani JA, Ilson DH, Daugherty K et al. Activity of taxol in patients with squamous cell carcinoma and adenocarcinoma of the oesophagus. J Natl Cancer Inst 1994 ;86 :1086-91 13. Bleiberg H, Conroy T, Paillot B et al. Randomized phase II study of cisplatin and 5-FU vs cisplatin alone in advanced squamous cell oesophageal cancer Eur J Cancer 1997; 33:1216-20 14. Ezdinli EZ, Gelver R, Desai DV et al. Chemotherapy of advanced oesophageal carcinoma : Eastern Cooperative Oncology Group Experience. Cancer 1980 ; 2149-53 15. Pearson JG. The present status and future potential of radiotherapy in the management of ooesophageal cancer. Cancer 1977 ;39 :882-6 16. Sykes AJ, Burt PA, Slevin NJ et al. Radical radiotherapy for carcinoma of the ooesophagus : an effective alternative to surgery. Radiother Oncol 1998 ;48 :15-21 17. Girinsky T, Auperin A, Marsiglia H et al. Accelerated fractionation in oesophageal cancers : a multivariate analysis on 88 patients. Int J Radiat Oncol Biol Phys 1997 ;38 :1013-8 18. Newaishy GA, Read GA, Duncan W, Kerr GR. Results of radical radiotherapy of squamous cell carcinoma of the oesophagus. Clin Radiol 1982 ;33 :347-352 19. Slevin NJ, Stout R. Carcinoma of the oesophagus : a review of 108 cases treated by radical radiation therapy. Clin Radiol 1989 ;40 :200-3 20. Harrison LN, Fogel TD, Picone JR et al. Radiation therapy for carcinoma of the oesophagus. J Surg Oncol 1998 ; 37 :40-3 21. De-Ren S. Ten year follow-up of oesophageal cancer treated by radical radiation therapy : analysis of 869 patients. Int J Radiat Oncol Biol Phys 1989 ;16 :329-34 22. Coia LR, Minsky BD, Berkey BA, John MJ, Haller D, Landry J, Pisansky TM, Willett CG, Hoffman JP, Owen JB, Hanks GE. Outcome of patients receiving radiation radiation for cancer of the esophagus: results of the 1992-1994 Patterns of Care Study. J Clin. Oncol. 2000;18:455-62 23. Minsky BD, Pajak TF, Ginsberg RJ, Pisansky TM, Martenson J, Komaki R, Okawara G, Rosenthal SA, Kelsen DP; int 0123 (Radiation Therapy Oncology Group 94-05) phase III trial of combined modality therapy for esophageal cancer: high-dose versus standard-dose radiation therapy.J Clin Oncol. 2002;20: 1167-74

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 48/70
24. J.F Seitz, J. Jacob, D.Lapalus, J.L Raoul, J.Y. Douillard, O. Bouché, T. Conroy, M. Ychou, A. Adenis, P. Haegele, Y. Becouarn, M. Giovannini, G. Macquart-Moulin, C. Langlois, M. Henry-Amar. Etude nationale randomisée (FNCLCC-FFCD 9305) de radio-chimiothérapie dans les cancers épidermoîdes de l’œsophage : résultats à 5 ans et analyse des facteurs pronostiques. « Journées Francophones de Pathologie Digestive », Paris 29 mars-2 avril 2003. Gastroenterol. Clin. Biol. 2003;27, Hors série I, A54. 25. Wong RK, Malthaner RA, Zuraw L, Rumble RB; Cancer Care Ontario Practice Guidelines Initiative Gastrointestinal Cancer Disease Site Group. Combined modality radiotherapy and chemotherapy in nonsurgical management of localized carcinoma of the esophagus: a practice guideline. Int. J. Rad. Oncol. Biol. Phys 2003;55 (4) 930-42 26. Al-Sarraf M, Martz K, Herskovic A et al. Progress report of combined chemoradiotherapy versus radiotherapy alone in patients with oesophageal cancer : an intergroup study. J Clin Oncol 1997 ;15 :277-84 27. Araujo CMM, Souhami L, Gil RA et al. A randomized trial comparing radiation therapy versus concomitant radiation therapy and chemotherapy in carcinoma of the thoracic oesophagus . Cancer 1991 ;67 :2258-61 28. Forastière AA, Orringer MB, Perez-Tamayo C et al. Preoperative chemoradiation followed by transhiatal esophagectomy for carcinoma of the oesophagus : final report. J Clin Oncol 1993 ;11 :1118-23 29. Coia LR, Engstrom PF, Paul A. Nonsurgical management of oesophageal cancer : a report of a study of combined radiotherapy and chemotherapy. J Clin Oncol 1987 ;5 :1783-90 30. Coia LR, Engstrom PF, Paul A et al. Long-term results of infusional 5-FU, mitomycin C and radiation as primary management of oesophageal carcinoma. Int J Radiat Oncol Biol Phys 1991 ;20 :29-36 31. Keane TJ, Harwood AR, Elhakim T et al. Radical radiation therapy with 5-FU infusion and mitomycin C for oesophageal squamous carcinoma. Radiother Oncol 1985 ;4 :205-10 32. John MJ, Flam MS, Ager Mowry PA et al. Radiotherapy alone and chemoradiation for nonmetastatic oesophageal carcinoma. A critical review of chemoradiation. Cancer 1989 ;63 :2397-403 33. Leichman L, Herskovic A, Leichman CG et al. Nonoperative therapy for squamous cell cancer of the oesophagus. J Clin Oncol 1987 ;5 :365-70 34. Seitz JF, Giovanni M, Padaut-Cesana J et al. Inoperable nonmetastatic squamous cell carcinoma of the oesophagus managed by concomitant chemotherapy (5-FU and cisplatin) and radiation therapy. Cancer 1990 ;66 :214-9 35. Seitz JF, Milan C, Dumas F et al. Radiochimiothérapie concomitante concentrée dans les cancers épidermoïdes de l’oesophage : résultats d’un essai de phase II multicentrique de la FFCD chez 119 patients non opérables. Gastroenterol Clin Biol 1995 ;19 :A21 36. Kolaric K, Zupanc D, Zivkovic M. Radiation alone vs radiochemotherapy (DDP + 5-FU) in locoregionally advanced oesophageal cancer. Interim report of a prospective randomized study. Proc Am Soc Clin Oncol 1992 ;11 :175a 37. Roussel A, Haegelé P, Paillot B et al. Results of the EORTC-GTCCG phase III trial of irradiation vs irradiation and CDDP in operable oesophageal cancer. Proc Am Soc Clin Oncol 1994 ; 13 :199a 38. Knox RJ, Folellos F, Lydall D, et al. Mechanisms of cytotoxicity of anticancer platinum evidence that cis-diamine-dichloroplatinum (II) and cis-diamine (1,1-cyclobutaneol, carboxylato) platinum (II) differ only in the kinetics of their interaction with DNA. Cancer Res 1986;46: 1972-9 39. Woynarowski JM, Chapman WG, Napier C et al. Sequence and Region-Specificity of Oxaliplatin Adducts in Naked and Cellular DNA. Mol Pharm 1998;54: 770-7 40. Page JD, Hussain I, Sancar A, et al. Effect of the diaminocyclothexane carrier ligand on platinum adduct formation, repair, and lethality. Biochemistry 1990;29: 1016-24 41. Scheeff ED, Howell SB. Computer modeling of the primary cisplatin and oxaliplatin DNA adducts and relevance to mismatch repair recognition. Proc Am Assoc Cancer Res 1998;39: 1082a 42. Fink D, Nebel S, Norris PS, et al. Enrichment for DNA mismatch repair-deficient cells during treatment with cisplatin. Int J Cancer 1998;77: 741-6 43. Aebi S, Kurdihaidar B, Gordon R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res 1996;53: 3087-90 44. Mu D, Tursun M, Duckett DR, et al. Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair system. Mol Cel Biol 1997;17: 760-9 45. Rixe O, Ortuzar W, Alvarez M, et al. Cisplatin and Carboplatin : spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute. Anticancer drug screen panel biochem pharmacol 1996;52: 1855-6 46. Pendyala L, Creaven PJ. In vitro cytotoxicity, protein binding, red blood cell partitioning, and biotransformation of oxaliplatin. Cancer Res. 1993;53: 5970-6 47. Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet 1989;16: 215

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 49/70
48. Dorr RT, Von Hoff D. Drug Neurographs. Appleton, Lange, editors. In: Cancer Chemotherapy Handbook 2nd ed. Connecticut: 1994. 5-Fluorouracil, p. 500-15 49. Rubin J,Gallagher JG, Schroeder G et al. Phase II trials of 5-fluorouracil and leucovorin in patients with metastatic gastric or pancreatic carcinoma.Cancer 1996 Nov1;78(9):1888-91 50. Louvet C, de Gramont A, Demuyinck B et al. High dose folinic acid, 5-FU bolus and continuous infusion in poor prognosis patients with advanced measurable gastric cancer. Ann Oncol 1991 ;2 :229-230 51. Johnson PWM, Thompson PJ, Seymour MT et al. A less toxic regimen of 5-FU and high dose folinic acid for advanced gastrointestinal adenocarcinoma.Br JCancer 1991 ;64 :603-5 52. Machover D, Goldschmid E, Chollet P et al. Treatment of advanced colorectal and gastric adenocarcinoma with 5-FU and high dose folinic acid. J Clin Oncol 1986 ;4 :685-95 53. Machover D. A comprehensive review of 5-fluorouracil and leucovorin in patients with metastatic colorectal carcinoma. Cancer 1997;80(7):1179-87 54. Roca E, et al. Intensive chemoradiotherapy for organ preservation in patients with resectable and non-resectable ooesophageal cancer. Eur J Cancer 1996;32A(3):429-32 55. Seitz JF, et al. 5-fluorouracil, high dose folinic acid and mitomycin C in the treatment of advanced digestive cancers. Bull Cancer 1994;81(2):134-7 56. Stahl M, et al. 5-Fluorouracil, folinic acid, etoposide and cisplatin chemotherapy for locally advanced or metastatic carcinoma of the ooesophagus. Eur J Cancer 1994; 30A(3): 325-8 57. Highley MS, et al. High-dose folinic acid with 5-fluorouracil bolus and continuous infusion in the treatment of advanced gastric and ooesophageal adenocarcinoma. Br J Cancer 1993;67(2):407-8 58. Trudeau M, et al. A phase I study of escalating interferon alpha-2a combined with 5-fluorouracil and leucovorin in patients with gastrointestinal malignancies. Acta Oncol 1993; 32(5):537-9 59. Lovett D, Kelsen D, Eisenberger M, Houston C. A Phase II trial of carboplatin and vinblastine in the treatment of advanced squamous cell carcinoma of the esophagus. Cancer 1991; 67: 354-6. 60. Sternberg C, Kelsen D, Dukemen M, Leichman L, Heelan R. Carboplatin: a new platinum analog in the treatment of epidermoid carcinoma of the esophagus. Cancer Treat Rep. 1985; 69 (11): 1305-7. 61. Louvet C, Andre T, Tigaud JM, Gamelin E, Douillard JY, Brunet R, Francois E, Jacob JH, Levoir D, Taamma A, Rougier P, Cvitkovic E, De Gramont A. Phase II study of oxaliplatin, fluorouracil and folinic acid in locally advanced or metastatic gastric cancer. J Clin Oncol; 20 (23) : 4543-8. 62. Phase II study of oxaliplatin (OX), fluorouracil (FU) and leucovorin (LV) in metastatic carcinoma of the esophagus/gastric cardia. Mauer AM et al Proc Am . Soc. Clin. Oncol 2002; 21, 139a, abstract 554. 63. A phase II study of oxaliplatin and weekly 24-hour infusion of high dose 5-fluorouracil and leucovorin (HDFL) in the first-line treatment of inoperable locally advanced or recurrent/metastatic gastric cancers. Chao Y et al Proc. Am. Soc. Clin. Oncol 2002; 21, abstract 651. 64. Kim DY, Kim JH, Lee SH, Kim TY, Heo DS, Bang YJ, Kim NK. Phase II study of oxaliplatin, fluorouracil and leucovorin in previously platinum-treated patients with advanced gastric cancer. Ann Oncol. 2003; 14 (3): 383-7. 65. Schmid KE, Kornek GV, Schull B, Raderer M, Lenauer A, Depisch D, Lang F, Scheithauer W. Second-line treatment of advanced gastric cancer with oxaliplatin plus raltitrexed. Onkologie. 2003; 26 (3): 255-8. 66. Dolan K, et al. Allelotype analysis of ooesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer 1998;78(7):950-7 67. Meltzer SJ. The molecular biology of oesophageal carcinoma. Recent Results Cancer Res 1996;142:1-8 68. Fink D, Nebel S, Aebi S, et al. The Role of DNA Mismatch Repair in Platinum Drug Resistance. Cancer Res. 1996;56: 4881-6 69. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58: 5248-57 70. Giacchetti S, Zidani R, Perpoint B, et al. Phase III trial of 5-fluorouracil, folinic acid, with or without oxaliplatin in previously untreated patients with metastatic colorectal cancer. Proc Am Soc Clin Oncol 1997;16: 805a 71. De Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first line treatment in advanced colorectal cancer.J Clin Oncol 2000, 16, pp 2938-2947 72. André T, Bensmaïne MA, Louvet C, et al. Multicenter phase II study of bimonthly high-dose Leucovorin, 5-Fluorouracil infusion and Oxaliplatin for metastatic colorectal cancer resistant to the same leucovoron regimen (Folfox 3/ 4). Journal of Clinical Oncology, 1999.17.(11).3560-3568 73. Becouarn Y, Ychou M, Ducreux M, et al. Phase II Trial of Oxaliplatin as First Line Chemotherapy in Metastatic Colorectal Cancer Patients. J Clin Oncol 1998;16: 2739-44 74. Gamelin E, Le Bouil A, Boisdron-Celle M, et al. Cumulative Pharmacokinetic Study of Oxaliplatin , Administered Every Three Weeks, combined with 5-Fluorouracil in Colorectal Cancer Patients. Clin Cancer Res 1997;3: 891-9

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 50/70
75. Levi F, Zidani R, Misset JL, for the International Organisation for Cancer chemotherapy. Randomized multiCentertrial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. Lancet 1997 ; 350 : 681-86 76. Chollet P, Bensmaïne MA, Brienza S, et al. Single Agent Activity of Oxaliplatin in Heavily Pretreated Advanced Epithelial Ovarian Cancer. Ann.Oncol. 1996;7: 1065-70 77. Misset JL, Chollet P, Vennin P, et al. Multicentric phase II-III trial of oxaliplatin versus cisplatin both in association with cyclophosphamide in the treatment of advanced ovarian cancer: toxicity and efficacy results. Proc Am Soc Clin Oncol 1997;16: 1266a. 78. V. Dieras, P. Bougnoux, T. Petit, et al. Multicentic phase II study of oxaliplatin as single agent in cisplatin/carboplatin + or – taxane-pretreated ovarian cancer patients. Annals of oncology 13: 258-266, 2002. 79. Monnet I, Brienza S, Hugret F, et al. Phase II study of Oxaliplatin in poor prognosis non small cell lung cancer (NSCLC). Eur J Cancer 1998;34: 1124-7. 80. Raymond E, Buquet-Fagot C, Djelloul S, et al. Antitumor activity of oxaliplatin in combination with 5-fluorouracil and the thymidilate synthase inhibitor AG337 in human colon, breast and ovarian cancers. Anti-Cancer Drugs 1997;8: 876-85. 81. The meta-analysis group in cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol 1998;16: 301-308. 82. Khushulani NI, Leichman CG, Proulx G, Nava H, Bodnar L, Klippenstein D, Litwin A, Smith J. Nava E, Pendyala L, Smith P, Greco W, Berdzik J, Douglass H, Leichman L. Oxaliplatin in combination with protracted-infusion Fluorouracil and radiation : report of a clinical trialfor patients with esophageal cancer. J. Clin. Oncol. 2002; 20: 2844-50. 83. Gergel TJ, Leichman L, Nava HR, Blumenson LE, Loewen GM, Gibbs JE, Khushalani NI, Leichman CG, Bodnar LM, Douglass HO, Smith JL, Kuettel MR, Proulx GM. Effect of concurrent radiation therapy and chemotherapy on pulmonary function in patients with esophageal cancer: dose-volume histogram analysis. Cancer J. 2002; 8(6) 451-60

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 51/70
APPENDICES
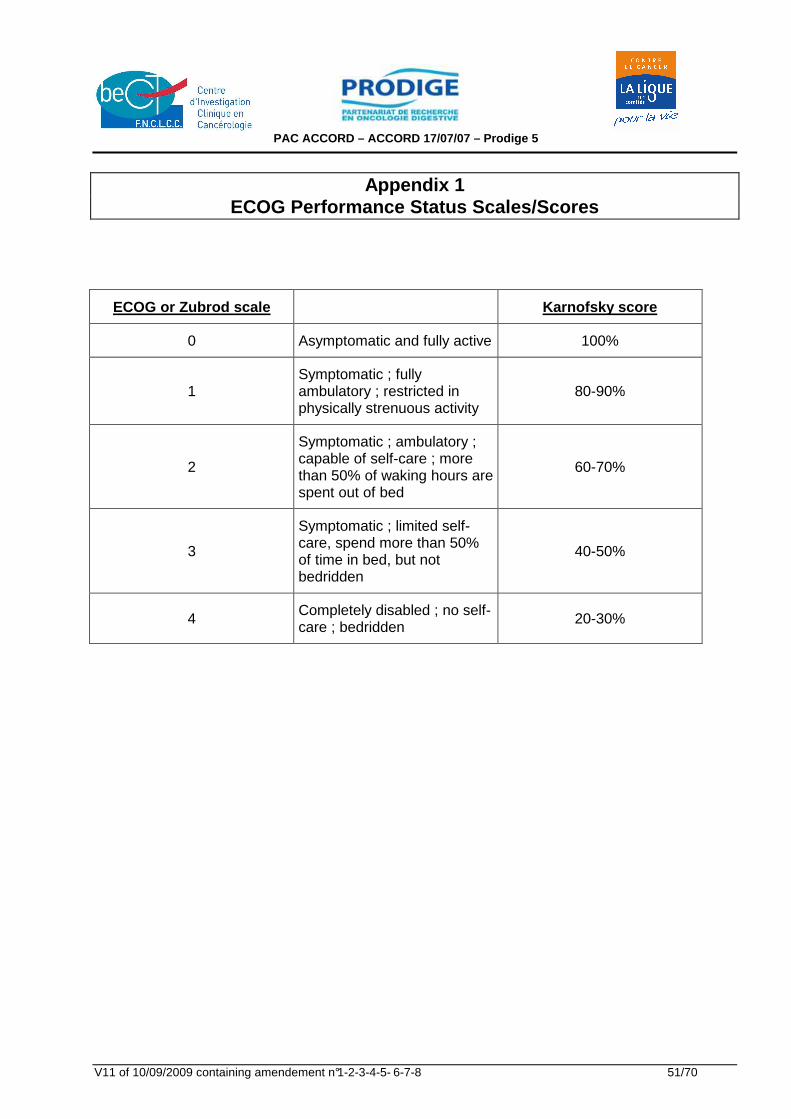
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V11 of 10/09/2009 containing amendement n°1-2-3-4-5- 6-7-8 51/70
Appendix 1 ECOG Performance Status Scales/Scores
ECOG or Zubrod scale Karnofsky score
0 Asymptomatic and fully active 100%
1 Symptomatic ; fully ambulatory ; restricted in physically strenuous activity
80-90%
2
Symptomatic ; ambulatory ; capable of self-care ; more than 50% of waking hours are spent out of bed
60-70%
3
Symptomatic ; limited self-care, spend more than 50% of time in bed, but not bedridden
40-50%
4 Completely disabled ; no self-care ; bedridden 20-30%

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 52/70
Appendix 2 TNM classification for oesophageal carcinoma (ICD-O C15)
T1 T2 T3 T4 N1 M1
M1a M1b
M1a M1b
M1b
Lamina propria, submucosa Muscularis propria Adventitia Adjacent structures Regional* Distant metastasis: Lower thoracic oesophagus Celiac nodes Other distant metastasis Upper thoracic oesophagus Cervical nodes Other distant metastasis Mid-thoracic oesophagus Distant metastasis including non-regional lymph nodes
* regional lymph nodes: cervical oesophagus - cervical nodes including supraclavicular nodes. intrathoracic oesophagus - mediastinal and perigastric nodes, excluding celiac nodes. Rules for classification Physical examination, imaging and/or surgical exploration should be used to classify patients by N and M. Primary tumor should also be assessed by endoscopy, including bronchoscopy. Anatomical subsites Cervical oesophagus Between lower border of cricoid cartilage and thoracic inlet (suprasternal notch),
18 cm from incisors
Upper thoracic oesophagus Between thoracic inlet and tracheal bifurcation, 24 cm from incisors
Mid-thoracic oesophagus Proximal half of oesophagus between tracheal bifurcation and esophagogastric junction, lower border 32 cm from incisors
Lower thoracic oesophagus Distal half of oesophagus between tracheal bifurcation and esophagogastric junction, lower border 40 cm from incisors

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 53/70
APPENDIX 3 : US TNM ECHOENDOSCOPIC CLASSIFICATION (TIO TL and al, Gastroenterology 1989 ; 96 : 1478-86)
T1 : Tumor invading mucosea and submucosea
T2 : Tumor invading muscularis but not exceeding it
T3 : Tumor invading adventitia (or serous)
T4 : Tumor invading adjacent structures
N0 : No lymph node invasion
N1 : invaded peritumoral lymph nodes : round, with the same echogenicity than tumor
N2 : invaded distant lymph nodes (5 cm above or below the tumor inferior or superior pole).

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 54/70
APPENDIX 4 : De Leriche and Fontaine Classification for arteritis
Stage I : no clinical symptom
Stage II : clinical symptoms during the effort
Stage III : clinical symptoms during the rest
Stage IV : necrosis.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 55/70
Appendix 5 : EORTC QLQ-C30 (version 3 )
Nous nous intéressons à vous et à votre santé. Répondez vous-même à toutes les questions en entourant le chiffre qui correspond le mieux à votre situation. Il n'y a pas de "bonne" ou de "mauvaise" réponse. Ces informations sont strictement confidentielles.
Merci de préciser: Vos initiales : |__|__|__|
Date de naissance (jour/mois/année): |__׀__׀ __׀__| |__׀__׀__| |__׀__|
La date d`aujourd`hui (jour/mois/année): 31 |__׀__׀ __׀__| |__׀__׀__| |__׀__|
Pas du tout
Un peu
Assez Beaucoup
1. Avez-vous des difficultés à faire certains efforts physiques pénibles comme porter un sac à provision chargé ou une valise ?
1
2
3
4
2. Avez-vous des difficultés à faire une longue promenade ?
1 2 3 4
3. Avez-vous des difficultés à faire un petit tour dehors ?
1 2 3 4
4. Etes-vous obligé(e) de rester au lit ou dans un fauteuil pendant la journée ?
1 2 3 4
5. Avez-vous besoin d'aide pour manger, vous habiller, faire votre toilette ou aller aux toilettes ?
1 2 3 4
Au cours de la semaine passée : Pas du
tout Un peu
Assez Beaucoup
6. Avez-vous été gêné(e) pour faire votre travail ou vos activités de tous les jours ?
1 2 3 4
7. Avez-vous été gêné(e) dans vos activités de loisirs ?
1 2 3 4
8. Avez-vous eu le souffle court ? 1 2 3 4
9. Avez-vous ressenti de la douleur ? 1 2 3 4
10.Avez-vous eu besoin de repos ? 1 2 3 4
11.Avez-vous eu des difficultés pour dormir ? 1 2 3 4
12.Vous êtes-vous senti(e) faible ? 1 2 3 4
13.Avez-vous smanqué d'appétit ? 1 2 3 4
14.Avez-vous eu des nausées (mal au coeur) ? 1 2 3 4
15.Avez-vous vomi ? 1 2 3 4
Passez à la page suivante S.V.P
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 56/70
Au cours de la semaine passée : Pas du
tout Un peu
Assez Beaucoup
16.Avez-vous été constipé(e) ? 1 2 3 4
17.Avez-vous eu de la diarrhée ? 1 2 3 4
18.Etiez-vous fatigué(e) ? 1 2 3 4
19.Des douleurs ont-elles perturbé vos activités quotidiennes ?
1 2 3 4
20. Avez-vous eu des difficultés à vous concentrer sur certaines choses, par exemple, pour lire le journal ou regarder la télévision ?
1 2 3 4
21.Vous êtes-vous senti(e) tendu(e) ? 1 2 3 4
22.Vous êtes-vous fait du souci ? 1 2 3 4
23.Vous êtes-vous senti(e) irritable ? 1 2 3 4
24.Vous êtes-vous senti(e) déprimé(e) ? 1 2 3 4
25. Avez-vous eu des difficultés pour vous souvenir de certaines choses ?
1 2 3 4
26. Votre état physique ou votre traitement médical vous ont-ils gêné(e) dans votre vie familiale ?
1 2 3 4
27. Votre état physique ou votre traitement médical vous ont-ils gêné(e) dans vos activités sociales (par exemple, sortir avec des amis, aller au cinéma...) ?
1
2
3
4
28. Votre état physique ou votre traitement médical vous ont-ils causé des problèmes financiers ?
1 2 3 4
Pour les questions suivantes, veuillez répondre en entourant le chiffre entre 1 et 7 qui s'applique le mieux à votre situation : 29. Comment évalueriez-vous votre état de santé au cours de la semaine passée ? 1 2 3 4 5 6 7 Très mauvais Excellent 30. Comment évalueriez-vous l'ensemble de votre qualité de vie au cours de la semaine passée ? 1 2 3 4 5 6 7 Très mauvais Excellent © Copyright 1995 EORTC Study Group on Quality of Life. Tous droits réservés Version 3.0.
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 57/70
Appendix 6 : EORTC QLQ-OES18 Les patients rapportent parfois les symptômes ou problèmes suivants.
Pourriez-vous indiquer, s’il vous plait, si, durant la semaine passée, vous avez été affecté(e) par l’un de ces symptômes ou problèmes
Entourez, s’il vous plait, le chiffre qui correspond le mieux à votre situation.
Au cours de la semaine passée : Pas du
tout Un peu
Assez Beaucoup
31. Pouviez-vous manger des aliments solides ? 1 2 3 4
32. Pouviez-vous manger des aliments mixés ou mous ?
1 2 3 4
33. Pouviez-vous boire des liquides ? 1 2 3 4
34. Avez-vous eu du mal à avaler votre salive ? 1 2 3 4
35. Vous-êtes vous étranglé(e) en avalant ? 1 2 3 4
36. Avez-vous eu du mal à apprécier vos repas ? 1 2 3 4
37. Vous êtes-vous senti(e) rassasié(e) trop vite ? 1 2 3 4
38. Avez-vous eu du mal à manger ? 1 2 3 4
39. Avez-vous été gêné(e) de manger devant d’autres personnes ?
1 2 3 4
40. Avez-vous eu la bouche sèche ? 1 2 3 4
41. Avez-vous eu des problèmes de goût ? 1 2 3 4
42. Avez-vous eu du mal à tousser ? 1 2 3 4
43. Avez-vous eu du mal à parler ? 1 2 3 4
44. Avez-vous eu des acidités ou des brûlures d’estomac ?
1 2 3 4
45. Avez-vous été gêné(e) par des renvois d’acide ou de bile dans la bouche ?
1 2 3 4
46. Avez-vous eu des douleurs en mangeant ? 1 2 3 4
47. Avez-vous eu mal dans la poitrine ? 1 2 3 4
48. Avez-vous eu des douleurs dans le ventre ? 1 2 3 4 © QLQ-C30-OES24 Copyright 1999 EORTC Study Group on Quality of Life. Tous droits réservés (phase III module)
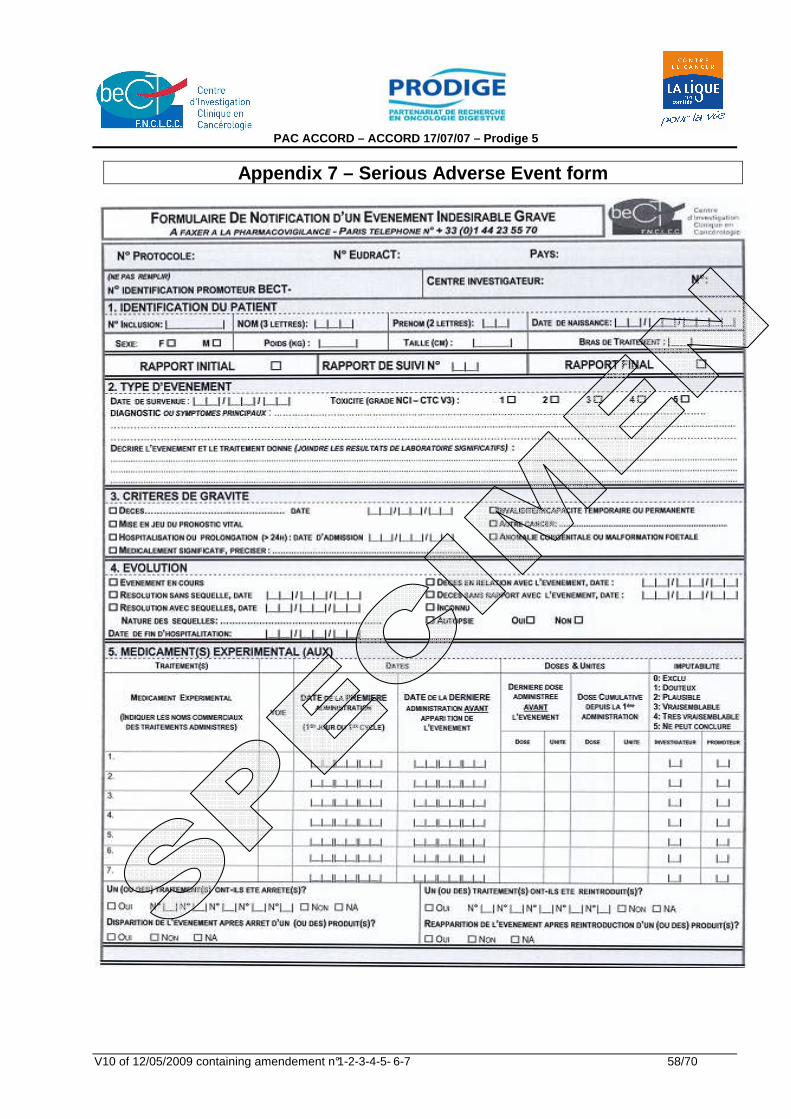
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 58/70
Appendix 7 – Serious Adverse Event form
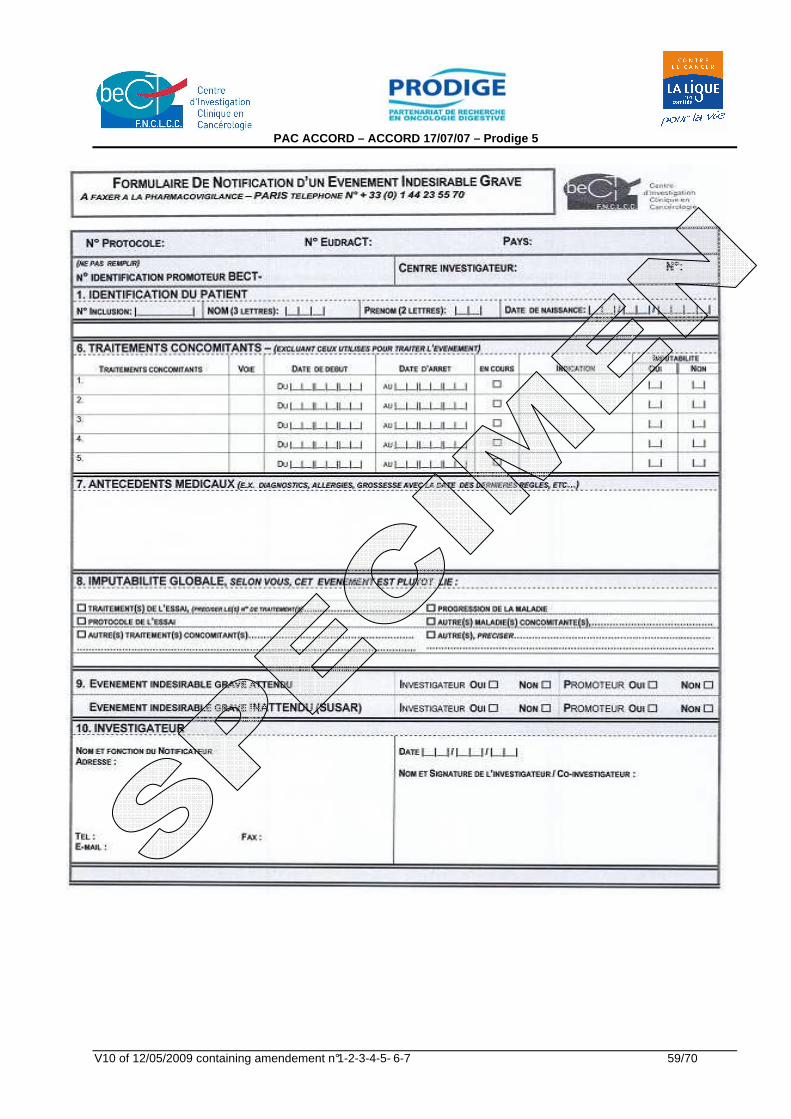
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 59/70

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 60/70
NOTE D'INFORMATION (1) DESTINEE AUX PERSONNES PARTICIPANT AU PROTOCOLE DE RECHERCHE BIOMEDICALE
(1) Toutes les pages de cette note d'information doiven t être paraphées par le participant à la recherche et l'investigateur .
ACCORD 17/0707 – Prodige 5
ETUDE DE PHASE II / III COMPARANT LA RADIOCHIMIOTHE RAPIE AVEC LE SCHEMA FOLFOX A LA
RADIOCHIMIOTHERAPIE PAR 5FU/CISPLATINE (SCHEMA HERS KOVIC) EN TRAITEMENT DE PREMIERE LIGNE DES PATIENTS PRESENTANT UNE TUMEUR D E L’OESOPHAGE INOPERABLE
Promoteur : Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), 101 rue de Tolbiac, 75654 PARIS CEDEX 13 Investigateur Coordonnateur : Pr. Thierry CONROY, Département d’oncologie médicale, Centre Alexis Vautrin, 6 avenue de Bourgogne, 54511 Vandoeuvre-lès-Nancy cedex
Document constitué en application de la loi du 20 décembre 1988 relative à la protection des personnes participant à la recherche biomédicale (Loi Huriet) et ses amendements ainsi qu'à la Loi du 6 janvier 1978 relative à l'Informatique, aux
Fichiers et aux Libertés et ses amendements. Ce document ne peut faire l’objet d’aucun traitement informatique.
****
NOTICE D'INFORMATION
Avant de participer à cette étude de recherche biomédicale, il est important que vous lisiez ce document fournissant les informations relatives au déroulement de cette étude. Vous pourrez donner votre consentement après un délai de réflexion. En participant à cette étude, vous contribuez à un effort de recherche qui pourra aider d’autres personnes.
Appendix 8 : Notice d’information et de consentemen t éclairé

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 61/70
A. BUT DE L’ETUDE
Le traitement de votre tumeur de l’œsophage nécessitera des perfusions (chimiothérapie), des rayons (radiothérapie), et les 2 associés.
Nous vous proposons de participer à un projet de recherche conduit par le Docteur ....................... en collaboration avec la Fédération Nationale de Centres de Lutte Contre le Cancer (FNCLCC), portant sur un médicament de chimiothérapie, l’oxaliplatine, en association avec le 5-fluorouracile (5-FU), l’acide folinique (association connue sous le nom FOLFOX) et la radiothérapie. Le but de cette étude est de montrer que le FOLFOX associé à la radiothérapie est mieux toléré et aussi efficace que le traitement de référence, l’association 5-FU/Cisplatine avec la radiothérapie.
Deux cent soixante six (266) patients seront traités dans différents centres français.
Les sels de platine (dont le cisplatine) en perfusion ont une efficacité connue dans les tumeurs de l’œsophage, en association avec le 5-FU et la radiothérapie.
L’oxaliplatine est un nouveau sel de platine, qui pourrait être efficace dans votre cas. Il est utilisé dans le traitement d’autres tumeurs avec de bons résultats. Le 5-FU est un médicament actif dans votre maladie et connu depuis de nombreuses années. L’acide folinique est une vitamine qui augmente l’action du 5-FU; il a déjà été utilisé dans des cas comme le vôtre. La tolérance de ces 3 produits de chimiothérapie en association a été étudiée chez des dizaines de milliers de patients et est donc bien connue.
La radiothérapie (rayons) est un traitement des tumeurs de l’œsophage dont l’efficacité est prouvée. Cette efficacité est augmentée par une chimiothérapie administrée en même temps.
B. DEROULEMENT DE L’ETUDE
� VOTRE TRAITEMENT :
Le choix du traitement (association de la radiothérapie au FOLFOX ou au 5-FU/cisplatine) sera établi par un tirage au sort centralisé dans l’ensemble de la France. • Si vous devez recevoir l’association Folfox + Radiothérapie :
Le traitement durera 12 semaines. Vous aurez une séance de rayons quelques minutes par jour, 5 jours par semaine pendant 5 semaines. Toutes les 2 semaines, vous recevrez également de la chimiothérapie sur 2 jours: le premier jour de chaque cycle de chimiothérapie vous recevrez l’oxaliplatine en perfusion intraveineuse pendant 2 heures, et l’acide folinique, aussi en perfusion de 2 heures. Ensuite, le 5-FU sera administré en perfusion très courte, suivi par une perfusion continue de 46 heures, par voie intraveineuse à l’aide d’une petite pompe portable. Vous continuerez de recevoir de la chimiothérapie deux jours toutes les 2 semaines à trois reprises, après la fin des rayons. Néanmoins le traitement sera interrompu en cas de mauvaise tolérance ou de refus de votre part de le poursuivre. • Si vous devez recevoir l’association 5-FU/cisplatine + Radiothérapie :
Le traitement durera 11 semaines. Vous aurez une séance de rayons quelques minutes par jour, 5 jours par semaine pendant 5 semaines. Vous recevrez également quatre cycles de chimiothérapie sur 4 jours: le premier jour de chaque cycle de chimiothérapie vous recevrez du cisplatine en perfusion intraveineuse puis une perfusion continue de 5-FU d’environ 96 heures du premier au quatrième jour de chaque cycle. Le deuxième cycle vous sera administré à la cinquième semaine, le troisième cycle à la huitième semaine et le quatrième et dernier cycle à la onzième semaine. Les doses de chaque médicament seront adaptées en fonction de votre tolérance au traitement. Votre médecin vous préviendra de toute modification.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 62/70
� SUIVI DU TRAITEMENT ET QUALITE DE VIE :
Les médecins responsables de votre chimiothérapie ou de vos rayons vous examineront régulièrement avant l’administration de chaque cycle de chimiothérapie. A l’occasion de chacune de ces consultations, ils prescriront les examens habituels nécessaires à la surveillance de votre état de santé et de votre traitement. D’autre part, des prises de sang seront réalisées chaque semaine tandis que les radiographies, scanners et endoscopies, eux, seront réalisés avant et 15 semaines après le début de votre traitement pour évaluer l’efficacité du traitement. Par la suite, votre médecin suivra l’évolution de votre état de santé tous les 3 mois. Deux questionnaires de qualité de vie vous seront remis; nous vous demandons de bien vouloir répondre soigneusement aux questions qui vous sont posées et de remettre régulièrement ces deux questionnaires à l’équipe en charge de votre radio-chimiothérapie. • Si vous êtes traité(e) par l’association Folfox-Radiothérapie : nous vous prions de les remplir avant le début de votre traitement puis au premier jour de la cinquième semaine de traitement, au premier jour du sixième cycle, lors de la première évaluation de l’efficacité de votre traitement puis tous les 6 mois pendant une année et une fois par an pendant trois ans. • Si vous êtes traité(e) par l’association 5-FU/cisplatine-Radiothérapie : nous vous serions reconnaissant de les remplir avant le début de votre traitement puis au premier jour de la cinquième semaine de traitement, au premier jour du quatrième cycle, lors de la première évaluation de l’efficacité de votre traitement puis tous les 6 mois pendant une année et une fois par an pendant trois ans.
L’analyse de ces questionnaires est indispensable en vue de l’évaluation de votre qualité de vie en rapport avec votre traitement et votre état de santé; aussi, nous vous demandons de ne pas oublier de les remplir.
C. EFFETS INDESIRABLES POTENTIELS DU TRAITEMENT
Comme vous le savez, toute chimiothérapie ou radiothérapie peut entraîner des effets indésirables, appelés « effets secondaires ». L’oxaliplatine est déjà commercialisé en France pour d’autres tumeurs. Des effets secondaires ont pu être constatés chez certains patients. Tous ces effets secondaires sont le plus souvent réversibles. • Les effets indésirables les plus courants sont des nausées, vomissements ou diarrhée (qui peuvent
provoquer dans les cas sévères une altération de la fonction rénale et des complications intestinales sévères), des aphtes, une perte de l’appétit ainsi qu’une modification de la sensibilité des extrémités qui peut être provoquée ou aggravée par le froid. Si vous constatez une sensation anormale de fourmillements, des douleurs au niveau des mains, des pieds ou une gêne pour réaliser certains mouvements, avertissez immédiatement votre médecin. De même, des sensations de crampes ou des troubles de la sensibilité de la région buccale ou de la gorge doivent être signalés à votre médecin.
• D’autres effets indésirables peuvent être dus à une diminution des cellules sanguines. Il peut s’agir d’une anémie (diminution des globules rouges), de saignements anormaux ou d’ecchymoses (baisse des plaquettes), d’infections parfois sévères (diminution des globules blancs).
• Une surveillance de la numération de la formule sanguine est donc nécessaire pendant le traitement par l’oxaliplatine.
• Très fréquemment, une fièvre peut survenir lors du traitement; la signaler à votre médecin. • On retrouve fréquemment des réactions allergiques lors de l’injection : éruption cutanée, conjonctivite,
rhinite, sensation de malaise, baisse de la tension artérielle, difficultés pour respirer.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 63/70
• Rarement : on constate une diminution de la vue (qui doit être signalée immédiatement à votre médecin), une diminution de l’audition, des troubles de la parole, une insuffisance rénale, l’apparition de symptômes respiratoires ou une perte des cheveux.
• Un contact du produit de perfusion au niveau de la peau peut provoquer des rougeurs légères.
Le 5-FU peut entraîner les effets suivants: • Des aphtes (petites ulcérations) dans la bouche qui sont prévenus par des bains de bouche
systématiques; • Des diarrhées; • Une rougeur avec douleur des mains et des pieds et plus rarement la peau qui pèle; • Des larmoiements, voire des conjonctivites; • Des troubles de l’équilibre; • Des douleurs au niveau de la poitrine; • Une réaction allergique.
Lors d’un traitement par l'oxaliplatine et le 5-FU, des élévations des paramètres hépatiques lors des examens sanguins de contrôle sont communément reportées. Ces élévations sont généralement de faible sévérité et n'entraînent aucun symptôme que vous pouvez ressentir. Dans quelques cas, des symptômes cliniques peuvent être associés avec ces élévations des paramètres hépatiques et consister en une jaunisse, ascite (accumulation de liquide dans l'abdomen), une augmentation de volume du foie et/ou de la rate. Ces anomalies du foie et les symptômes cliniques peuvent être liés à votre maladie ou, dans de rares cas, refléter un effet direct du traitement sur le tissu hépatique, qui pourrait inclure des modifications des vaisseaux sanguins dans le foie (maladie veino-occlusive). Pendant la durée de l'étude, la fonction hépatique sera contrôlée de façon régulière par des examens sanguins et, lorsque nécessaire (si un dysfonctionnement hépatique ou des symptômes cliniques non reliés à votre maladie) surviennent, des investigations complémentaires vous seront proposées. L’acide folinique peut accentuer les effets secondaires du 5-FU.
La combinaison 5-FU-oxaliplatine et acide folinique (Folfox) peut entraîner la survenue des effets suivants: • Des diarrhées, souvent modérées et bien contrôlées par un traitement anti-diarrhéique oral; • Des aphtes dans la bouche, plus souvent qu’avec le 5-FU ou l’oxaliplatine seuls; • Une diminution un peu plus importante du nombre des globules blancs pour laquelle les précautions à
prendre ont été détaillées plus haut; • Une chute modérée des cheveux est rapportée chez moins de 2% des patients : lorsqu’elle survient, elle
est le plus souvent réversible.
L’addition des rayons à une chimiothérapie (Folfox ou 5-FU/cisplatine) peut entraîner la survenue des effets suivants : • Des rougeurs de la peau; • Une perte d’appétit; • Des nausées et vomissements; • Des difficultés pour avaler; • Une fatigue générale; • Eventuellement d’autres effets secondaires que nous ne pouvons pas prévoir entièrement car
l’association de la radiothérapie avec l’oxaliplatine est un nouveau traitement.
Tous ces effets secondaires, dont nous tenons à vous donner le descriptif détaillé, sont le plus souvent modérés et ne nécessitent pas l’arrêt du traitement. Ils ne justifient pas l’interruption systématique du traitement. Il faut les signaler à votre médecin qui vous proposera l’attitude la plus adaptée à votre situation. Les effets secondaires n’influencent en aucun cas l’efficacité du traitement.

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 64/70
Le cisplatine peut entraîner les effets suivants : • Une élévation du taux d’urée ou de la créatininémie, mais votre médecin vous administrera des
perfusions préventives à base de perfusions de sérum salé; • Des bourdonnements d’oreilles et une baisse de l’audition; • Une baisse du nombre de globules blancs et/ou de plaquettes ou de globules rouges; • Des nausées et vomissements mais un traitement préventif sera systématiquement prescrit par votre
médecin, juste avant l’administration du traitement; • Une élévation du taux d’acide urique; • Des fourmillements dans les pieds et les mains, le plus souvent après des traitements prolongés (4 à 7
mois); • Des réactions allergiques peuvent survenir dans les minutes qui suivent l’administration du cisplatine; • Plus rarement, ont été observés des troubles cardiaques et une perte d’appétit.
Il est obligatoire d’utiliser une contraception pendant l’étude et les six mois suivants.
D. PROTECTION DES PATIENTS
Le médecin qui vous propose cette étude vous laissera le temps de réfléchir. Vous êtes libre de participer ou non à cette étude. Vous pouvez vous réserver le droit à tout moment d’interrompre votre participation sans que cela ne porte préjudice aux soins qui vous seront prodigués et sans avoir à en donner les raisons. Ce protocole de recherche a été soumis au Comité Consultatif de Protection des Personnes se prêtant à la Recherche Biomédicale de Lorraine qui a émis un avis favorable à sa réalisation le 27 Mai 2004. Pour participer à cette étude, vous devez être bénéficiaire d’un régime de sécurité sociale en tant qu’assuré ou ayant-droit. Vous ne serez inclus dans l’étude que si vous signez et paraphez ce consentement qui vous est remis. Vous ne pourrez participer en même temps a une autre recherche biomédicale. Vous pouvez ensuite vous retirer à tout moment de l’essai sans justification, sans conséquence sur la suite de votre traitement ni sur la qualité des soins qui vous seront fournis et sans conséquence sur la relation avec votre médecin ; vous pourrez être suivi par la même équipe médicale. Le promoteur de cet essai qui en assure la gestion et la responsabilité est la Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) située au 101, rue de Tolbiac, 75654 Paris Cedex 13 – France. Elle en assure également la prise en charge globale. La FNCLCC a pris toutes les dispositions prévues par la loi sur les Recherches Biomédicales (anciennement Loi Huriet- décret d’application 2006-477 du 26 avril 2006 modifiant le titre II du livre 1 du Code de la Santé Publique) relative à la protection des personnes se prêtant à des recherches biomédicales. La FNCLCC devant assumer l’indemnisation des éventuelles conséquences dommageables de la recherche biomédicale pour la personne qui s’y prête, a souscrit une assurance de recherches biomédicales, conformément à la législation en vigueur (n° de contrat 906812007009 Protocole ACCORD 17 / 0707), auprès de la Société Gerling France (111-113 rue de Longchamp, 75016 Paris – Tél. 01 44 05 56 00) Lorsque la responsabilité du promoteur n’est pas engagée, les participants peuvent être indemnisés auprès de l’ONIAM, (Office National d’Indemnisation des Accidents Médicaux, 36, Avenue du Général de Gaulle, 93175 BAGNOLET Cedex , N° Vert : 0800 779 887). Votre dossier médical restera confidentiel et ne pourra être consulté que sous la responsabilité du médecin s’occupant de votre traitement ainsi que par les autorités de santé et par des personnes dûment mandatées par le promoteur de l’essai et soumises au secret professionnel.
Dans le cadre de la recherche biomédicale à laquelle il vous est proposé de participer, un traitement automatisé et anonymisé de vos données personnelles va être mis en œuvre pour permettre d’analyser les

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 65/70
résultats de la recherche au regard de l’objectif de cette dernière qui vous a été présenté. A cette fin, les données médicales vous concernant et les données relatives seront transmises au Promoteur de la recherche ou aux personnes ou sociétés agissant pour son compte, en France ou à l’étranger. Ces données seront identifiées par un numéro de code et/ou vos initiales. Ces données pourront également, dans des conditions assurant leur confidentialité, être transmises aux autorités de santé françaises ou étrangères. Conformément aux dispositions de loi relative à l’informatique et aux libertés (loi n° 78-17 du 6 janv ier 1978 modifiée par la loi n° 2004-801 du 6 août 2004) vou s disposez d’un droit d’accès, de rectification et d’opposition relatif au traitement de vos données personnelles. Ces droits s’exercent auprès du médecin en charge de la recherche qui seul connaît votre identité. Vous pouvez également accéder directement ou par l’intermédiaire d’un médecin de votre choix à l’ensemble de vos données médicales en application des dispositions de l’article L 1111-7 du Code de la Santé Publique. Les informations concernant votre identité seront tenues confidentielles par votre médecin. De plus, vous serez informé, à votre demande auprès du médecin qui vous a pris en charge, des résultats globaux de l’essai par l’investigateur. Enfin, il est également important de vous signaler que vous pouvez avoir accès à des informations sur l’essai en consultant le site Internet de la Fédération (http://www.fnclcc.fr/)
E. A QUI DEVEZ-VOUS VOUS ADRESSER EN CAS DE QUESTIO NS OU DE PROBLEMES ?
En cas de problèmes, d’événements indésirables en cours d’essai ou de questions, vous pouvez-vous adresser aux personnes suivantes :
Vos contacts dans l’étude (titre, nom, prénom, adre sse et téléphone) :
……………………………………………………………………………………………………………………………
……………………………………………………………………………………………………………………………
Coordonnées du médecin référent du patient
……………………………………………………………………………………………………………………………
……………………………………………………………………………………………………………………………
Votre médecin traitant sera tenu informé de votre suivi dans cet essai par votre médecin investigateur

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 66/70
FORMULAIRE DU RECUEIL DE CONSENTEMENT
ACCORD 17/0707 – Prodige 5
ETUDE DE PHASE II / III COMPARANT LA RADIOCHIMIOTHE RAPIE AVEC LE SCHEMA FOLFOX A LA RADIOCHIMIOTHERAPIE PAR 5FU/CISPLATINE (SCHEMA H ERSKOVIC) EN TRAITEMENT DE PREMIERE LIGNE DES PATIENTS PRESENTANT UNE TUMEUR D E L’OESOPHAGE INOPERABLE
Je soussigné(e) : Nom :………………………………………….Prénom :…………………………………………………. Adresse :……………………………………………………………………………………………………
CONSENS EXPRESSEMENT A PARTICIPER A CETTE RECHERCHE DANS LES CONDITIONS QUI M’ONT ETE PRECISEES DANS LA FICHE D’ INFORMATION.
J'ai été bien informé(e) des objectifs de cette étude et de ses conditions de réalisation, ainsi que des risques qu’elle pourrait comporter. J’ai eu réponse à toutes mes questions concernant l’étude. Je sais que mon médecin doit me communiquer, à tout moment pendant et après ma participation à l'étude, toute nouvelle information concernant l'étude ou tout dommage éventuel qui y serait lié. J’ai donné librement mon consentement pour participer à cette étude et je sais que j'ai le droit de me retirer à tout moment de cette étude, sans préjudice d'aucune sorte. J'accepte que, pour le besoin de l’étude, le promoteur et/ou les Autorités Sanitaires puissent accéder directement aux documents médicaux originaux pour vérifier les procédures cliniques et/ou les données sans violation de la confidentialité. J’accepte que les données médicales enregistrées à l’occasion de cette recherche, puissent faire l’objet d’un traitement informatisé par le promoteur ou pour son compte. J’ai bien noté que le droit d’accès prévu par la loi « Informatique et Liberté » (article 40 et suivants) s’exerce à tout moment auprès du médecin de l’étude. Je pourrais exercer mon droit de rectification auprès de ce même médecin. J’ai reçu une copie du présent document et j’ai été informé(e) qu’une copie sera également conservée par le médecin qui me propose de participer à cette étude, dans des conditions garantissant la confidentialité, et j’y consens. Mon consentement ne décharge pas de leurs responsabilités les organisateurs de la recherche. Je conserve tous mes droits garantis par la loi. Je déclare avoir répondu à toutes les questions qui m'ont été posées à propos de mes antécédents médicaux et je m'engage à suivre toutes les consignes et instructions qui me seront données par l'équipe médicale et qui sont détaillées dans la notice d'information.
• ______________________________________________ I__I__I I__I__I I200__I ______________________________ Nom du patient ou de son représentant légal Date Signature • ______________________________________________ I__I__I I__I__I I200__I ______________________________ Nom du médecin investigateur ou du médecin qui le représente (co-investigateur)
Date Signature

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 67/70
Se référer à l’échelle d’évaluation de la toxicité CTCAE qui est jointe séparément et que l’on peut télécharger sur le site du NCI
http://ctep.cancer.gov/
Common Terminology Criteria for Adverse Events v3.0 (CTCAE)
(Publish Date December 12, 2003)
CTCAE v3.0, a new version of the CTEP, NCI CTC v2.0 , includes Adverse Events applicable to all oncology clinical trials regardless of chronicity o r modality.
Appendix 9 – NCI-Common Toxicity Criteria version 3 .0
PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 68/70
Les RCP des produits (à préciser) sont joints sépar ément.
Se référer aux dernières versions en vigueur sur le site des agences réglementaires Afssaps, EMEA ou sur le site du VIDAL :
http://agmed.sante.gouv.fr/ http://www.emea.eu.int/ http://www.vidalpro.net/
Appendix10 – Résumé des Caractéristiques du (des) P roduit (s)

PAC ACCORD – ACCORD 17/07/07 – Prodige 5
V10 of 12/05/2009 containing amendement n°1-2-3-4-5- 6-7 69/70
Appendix 11 – Siewert Classification 1998 : tumour center lying
Type I : > 1 cm – 5 cm above GEJ Usually Barrett’s adenocarcinomas Type II : 1 cm above to 2 cm below GEJ = true carcinoma of the cardia Type III : tumour center 2 to 5 cm below GEJ = subcardia, proximal gastric center
From Siewert JR, Stein HJ, Br J Surg 1998; 85 : 1457-9








![[PPT]TUMOR TRAKTUS UROGENITAL - FK UWKS 2012 C | … · Web viewTUMOR TRAKTUS UROGENITAL I. Tumor Ginjal A. Tumor Grawitz B. Tumor Wilms II. Tumor Urotel III. Tumor Testis IV. Karsinoma](https://img.pdfslide.us/doc/110x75/5ade93b87f8b9ad66b8bb718/ppttumor-traktus-urogenital-fk-uwks-2012-c-viewtumor-traktus-urogenital.jpg)




















