Embed Size (px)
Citation preview

J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
Ava i l ab l e on l i ne a t www.sc i enced i r ec t . com
ScienceDirectwww.e l sev i e r . com/ loca te / j p ro t
Proteomic analysis of protein methylation in the
yeast Saccharomyces cerevisiaeKeyun Wanga,b,1, Yongjin J. Zhoua,1, Hongwei Liua,1, Kai Chenga,b, Jiawei Maoa,b,Fangjun Wanga, Wujun Liua, Mingliang Yea, Zongbao K. Zhaoa,⁎, Hanfa Zoua,⁎aDivision of Biotechnology, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, ChinabUniversity of Chinese Academy of Sciences, Beijing 100049, China
A R T I C L E I N F O
Abbreviations: SAM, S-adenosyl-L-methionography tandem mass spectrometry; FDR, f⁎ Corresponding authors at: Division of Biotechel.: +86 411 84379066, +86 411 84379610 ; faE-mail addresses: [email protected] (Z.K
1 These authors contributed equally to this
http://dx.doi.org/10.1016/j.jprot.2014.07.0321874-3919/© 2014 Elsevier B.V. All rights rese
A B S T R A C T
Article history:Received 24 January 2014Accepted 20 July 2014
Protein methylation catalyzed by SAM-dependent methyltransferase represents a majorPTM involved in many important biological processes. Because methylation can occur onnitrogen, oxygen and sulfur centers and multiple methylation states exist on the nitrogencenters, methylproteome remains poorly documented. Here we present the methylation byisotope labeled SAM (MILS) strategy for a highly-confident analysis of the methylproteomeof the yeast Saccharomyces cerevisiae based on the online multidimensional μHPLC/MS/MStechnology. We identified 43 methylated proteins, containing 68 methylation eventsassociated with 64 methylation sites. More than 90% of these methylation events werepreviously unannotated in Uniprot database. Our results indicated, 1) over 2.6% of identifiedS. cerevisiae proteins are methylated, 2) the amino acid residue preference of proteinmethylation follows the order Lys ≫ Arg > Asp > Asn ≈ Gln ≈ His > Glu > Cys, and 3) themethylation state on nitrogen center is largely exclusive. As our dataset covers varioustypes of methylation centers, it provides rich information about yeast methylproteome andshould significantly contribute to the field of protein methylation.
Biological significanceIn this paper, we presented the methylation by isotope labeled SAM (MILS) strategy for ahighly-confident analysis of the methylproteome of the yeast S. cerevisiae and collected acomprehensive list of proteins methylated on a set of distinct residues (K, R, N, E, D, Q, H, C).Our study provided useful information about the amino acid residue preference andmethylation state distributions on nitrogen centers of protein methylation in S. cerevisiae.
© 2014 Elsevier B.V. All rights reserved.
Keywords:Saccharomyces cerevisiaeMethylationProteomicsLC/MS/MS
1. Introduction
Protein methylation catalyzed by SAM-dependent methyltrans-
ine; PTM, post-translational modification; HPLC-MS/MS, high performance liquid chroma-alse discovery rate.nology, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.x: +86 411 84379211, +86 411 84379620.. Zhao), [email protected] (H. Zou).
t
T
work.
rved.
ferase represents a major PTM [1] involved in many importantbiological processes [2–4]. Protein methylation can occur onnitrogen, oxygen and sulfur centers and multiple methylationstates, namely, mono-, di- and trimethylation, can occur on

227J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
nitrogen center [5]. The dynamic nature of protein methylationfurther promotes efforts to understand the importance andfunction of this PTM [3,6]. Irrespective of copious publications inthis field, studies have been extensively focused on theN-methylation on Lys or Arg residue [2–4,7–13]. Because theintroduction of methyl groups within a protein usually has littleeffect in terms of analytical properties [2], challenges areremarkable for a global analysis of the methylated protein.Therefore, a comprehensive survey of methylproteome ofSaccharomyces cerevisiae remains elusive [14–17]. Several funda-mental questions in the field remain unanswered. For example,to what extent a proteome is methylated? Are there anypreferences among different methylation centers or amino acidresidues? In the case of methylation on nitrogen center, what isthe distribution profile for differentmethylation states? Here wepresent the methylproteome of the yeast S. cerevisiae and themethylation by isotope labeled SAM (MILS) strategy dedicated forthe analysis of the SAM-auxotrophic yeast [18] proteome basedon the online multidimensional μHPLC/MS/MS technology. Ourdataset addressed most of those questions and provided richinformation for further research on protein methylation.
Several strategies have been applied to identify proteinmethylation. Traditional [methyl-3H] labeling in combinationwith fluorography is laborious and it is difficult to locate themethylation sites [19]. The data processing tool FindMod wasused to analyze partial yeast proteome and 83 methylationevents on Lys or Arg were found [17]. The approach combiningantibody-enrichment of Arg-methylated proteins and MSanalysis identified 200 putatively methylated proteins, yetexact methylation sites were unavailable [14]. In recent years,antibody enrichment was widely utilized to the global identifi-cation ofmethylation sites on Lys orArg residue [8–11]. It shouldbe noted that the use of pan-antibody significantly advancedthe detection of protein methylome, however, the enrichmentefficiency remains to be improved. The strategy based on stableisotope labeling by amino acids in cell culture was alsoemployed to quest protein methylation, where the cell cultureis supplemented with [13CD3]methionine, the biological precur-sor of labeled methyl donor, [13CD3]SAM [8–10,15]. In thismethod, the confidence of identified methylation sites wasenhanced by the presence of MS ion pairs. However, a fewissues are obvious. First, [13CD3]methionine per se can also beincorporated into the proteome, which would complicate theMS data analysis. Moreover, SAM-dependent transmethylationactivities co-produce S-adenosylhomocysteine (SAH), whichcan be hydrolyzed to homocysteine. Methionine synthase, anessential housekeeping enzyme for sulfur and cofactor metab-olism, can methylate homocysteine to produce unlabeledmethionine depending on methyltetrahydrofolate [20]. Suchan inevitable cellular process can dilute the labeled methyldonor, leading to complication in data analysis and high falsenegative results.
In this study, we designed the MILS strategy to analyze theyeast methylproteome by taking the advantage that yeast cantake up sulfonium compounds including SAM from the culturemedium. When [CD3]SAM was used, the SAM-auxotrophicyeast cells generated [CD3]-modifications by protein methyl-transferases. Because the biosynthetic route of methionine toSAMwas blocked, the formation of fresh, regular SAM from themethylation co-product SAH upon [CD3]SAM consumption
was not possible. This prevented regular methylation events.Moreover, we showed that the conversion of [CD3]SAM into[CD3]methionine was negligible, which prevented the incor-poration of [CD3]methionine into protein biosynthesis.Therefore, the MILS strategy had higher labeling specificityover the conventional use of [CD3]methionine, which couldreduce false negative results and increase the confidence ofour dataset. A total of 43methylated proteins were identified,containing 68 methylation events associated with 64 meth-ylation sites. More than 90% of these methylation eventswere previously unannotated in the Uniprot database or inthe Pang's study [17].
2. Materials and methods
2.1. SAM and [CD3]SAM synthesis
The AdoMet synthetase geneMetKwas PCR amplified from theEscherichea coli genome with the primer pair MetK-F/MetK-R.MetK and expression vector pET28a (Invitrogen) were digestedwithNde I and EcoR I and ligated, resulting in pMetK, which wastransformed into E. coli Bl21 (DE3). The procedure for prepara-tion of SAM synthase was modified from a previous study [21].Briefly, Bl21(DE3) (pMetK) was cultivated at 30 °C and 200 rpmin 500 mL Luria–Bertani (LB) rich medium with 50 μg/mLkanamycin sulfate up to an optical density at 600 nm (OD600)of 0.4–0.6. Then, 0.2 mM isopropyl β-D-1-thiogalactopyranosidewas added to induce the recombinant protein expression. After12 h, the cells were collected by centrifugation (2000 g, 5 min),resuspended in 50 mL of 100 mM Tris–HCl buffer (pH 8.0)containing 1 mM EDTA, and treated with 50 μg/mL lysozymeat 30 °C for 30 min. After adding phenylmethylsulfonyl fluorideto a final concentration of 0.1 mM, the cells were lysed byultrasonication in an ice bath and the lysatewas centrifuged for20 min at 12,000 g in a Hitachi 46 rotor to remove the cell debris.After dialysis against Tris–KCl buffer which contains 50 mMTris–HCl, 50 mM KCl and 1 mM EDTA (pH 8.0), the crudeAdoMet synthase was either used immediately or stored at −20 °C with 20% glycerol until needed.
Enzymatic preparation of SAM and [CD3]SAM was per-formed in 90 mL of 100 mM Tris–HCl buffer (pH 8.0) in thepresence of 50 mM KC1, 26 mM MgC12, 1 mM EDTA, 20% (v/v)acetonitrile, 13 mM ATP, 10 mM methionine or [CD3]methio-nine, and 10 mL of crude AdoMet synthase. The reaction wasincubated at 30 °C for 12 h, and the progress of the reactionwas analyzed by thin layer chromatography. Acetonitrile wasremoved under reduced pressure. The aqueous phase wascentrifuged, and the supernatant was collected, concentrated,re-dissolved in 40 mL of 1.0 M HCl, and centrifuged again toremove residual protein. The supernatant was concentratedand the residue was re-dissolved in 30 mL of H2O, and passedthrough an IRC86 cation resin column (formate form, elute:H2O, 0.02 N HCl to 0.04 N HCl). The absorbance of the elutewas monitored at 254 nm, and SAM or [CD3]SAM fractionswere collected, and lyophilized to give a white solid (150 mg,95%). 1H-NMR spectra of synthetic SAM and [CD3]SAM wereshown in Fig. S1.

228 J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
2.2. Strains and culture conditions
The SAM auxotroph yeast strain S. cerevisiae (ΔSAM1, ΔSAM2)was constructed on the SAM1 disruption strain S. cerevisiaeYDR502C (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; ΔSAM2)background, which was obtained from the American TypeCulture Collection. Firstly, the SAM1 disruption cassette withlong homologous regions was constructed by restriction-free(RF) cloning strategy. Then the linear SAM1 disruption cassettewas prepared with a PCR reaction using a primer pair SAM1-F/SAM1-R and transformed into S. cerevisiae YDR502C for SAM1disruption with homologous recombination as described previ-ously [22]. The potential colony was verified by PCR using theirgenomic DNA samples as templates, P5/P7 and P6/P8 as primerpairs for SAM1disruptant analysis, as well as phenotypecharacterization by spotting on SC + SAM or SC − SAM plate.
Synthetic dextrose (SD) medium consisted of 2% (w/v)glucose, 0.67% (w/v) yeast nitrogen base (Difco, Detroit, USA)with (NH4)2SO4 and without amino acids. SD complete was SDmedium with required essential nutrients (20 mg/L uracil,20 mg/L histidine, 20 mg/L methionine and 100 mg/L leucine).SD complete lacking uracil and supplemented with 60 μM SAM(SD-Ura + SAM) was used for selecting SAM1 disruptants. YPDwhich consisted of 1% (w/v) yeast extract (Oxoid, Basingstoke,UK), 2% (w/v) peptone (Difco) and 2% (w/v) dextrosewas used togrow yeast strain S. cerevisiae YDR502C. YPD + SAM (60 μM) andYPD + [CD3]SAM (60 μM)were used to grow S. cerevisiae (ΔSAM1,ΔSAM2). S. cerevisiae (ΔSAM1, ΔSAM2) was inoculated into100 mL of medium to an OD600 of 0.02 and cultivated for 24 h.
2.3. Protein extraction and digestion
After a 24 h cultivation, the ‘heavy’ labeled cell and the ‘light’labeled cell were separately harvested by centrifugation,washed four times with cold PBS buffer and lysed in amodified RIPA buffer (pH 7.4) containing 150 mM NaCl,50 mM Tris–HCl, 1% (w/v) Nonidet P-40 (NP-40), 0.25% (w/v)sodium dexoxycholate, 1 mM EDTA and protease cocktailinhibitors using glass beads (0.4–0.6 mm) vortexing (celldisrupter Fastprep®-24 from MP Biomedicals, Inc.) on ice.Lysates were centrifuged at 13,000 g, mixed 1:1 (w/w, Brad-ford assay) and digested by trypsin in the same procedure asreported previously [23].
2.4. MS analysis
HPLC system (Thermo, San Jose, CA) consisted of a degasser andaquaternary SurveyorMSpumpwas coupled to a LTQ-OrbitrapXLmass spectrometer (Thermo, San Jose, CA) equipped with ananospray source and a six-port/two-position vale. Online multi-dimensional μHPLC/MS/MSanalysiswas performed to analyze thetryptic digestions according to a reported method [23] with minormodifications. Generally, 200 μg tryptic digestions were loadedonto thephosphatemonolithSCX trap column,whichwasdirectlyconnected to the reversed phase (RP) separation column. Thepeptides were gradually eluted from SCX segment into the RPseparation column using a serial stepwise elution with saltconcentration of 100, 200, 250, 300, 350, 400, 450, 500, 600 and1000 mMNH4OAcgeneratedbybuffersA (H2O: formicacid, 100:0.1,v/v) and C (1.0 M NH4OAc at pH 2.7), and then a 90 min RP linear
separation gradient elution from 95% buffer A to 35% buffer B(acetonitrile:formic acid, 100:0.1, v/v)witha flowrateof 300 nL/minwas used to separate the peptides retained in the RP separationcolumn. Once the RP gradient separation was started, the dataacquisition of MS began immediately. The samples were triplicateanalyzed in parallel.
The mass spectrometer was set as follows: ion transfercapillary 200 °C, spray voltage 1.8 kV, full MS range 400–2000,and full mass spectra was acquired in the Orbitrap at aresolution of 60,000 at m/z 400. One full MS scan was followedby 10 MS2 scans; the dynamic exclusion function was set asfollows: repeat count 2, repeat duration 30 s, and exclusionduration 60 s.
2.5. Data analysis
MS/MS spectra were converted into Mascot-compatible peaklists using DTA Supercharge (v2.0a7) [24] then searched againstthe yeast database (5885 entries) (ftp://genomeftp.Stanford.edu/yeast/datadownload/sequence/genomic_sequence/orf_protein/orf_trans.fasta.gz) using Mascot Version 2.3 (Matrix Science).To evaluate the false discovery rate (FDR), reversed se-quences were appended to the database. Carbamidomethylcysteine was set as a fixed modification of +57.0215. BecauseMascot allows a maximum of nine variable modifications tobe searched concurrently, the variable modifications insearch of methylation in different amino acids were set in 6groups as follows: 1) in light methyl Lys and Arg searches:mono-methyl-Lys, mono-methyl-Arg, di-methyl-Arg, di-methyl-Lys, tri-methyl-Lys; 2) in heavy methyl Arg and Lyssearches: mono-methyl (CD3)-Arg, mono-methyl (CD3)-Lys,di-methyl (CD3)-Arg, di-methyl (CD3)-Lys, tri-methyl (CD3)-Lys;3) in methyl Cys, Asp and Glu searches: mono-methyl-Cys,mono-methyl-Asp,mono-methyl-Glu,mono-methyl (CD3)-Cys,mono-methyl (CD3)-Asp, mono-methyl (CD3)-Glu; 4) in methylHis, Asn and Gln searches: mono-methyl-His, mono-methyl-Asn, mono-methyl-Gln, mono-methyl (CD3)-His,mono-methyl (CD3)-Asn, mono-methyl (CD3)-Gln; 5) in methylSer, Thr and Tyr searches: mono-methyl-Ser, mono-methyl-Thr,mono-methyl-Tyr, mono-methyl (CD3)-Ser, mono-methyl(CD3)-Thr, mono-methyl (CD3)-Tyr; and 6) in methyl Trp search:mono-methyl-Trp, mono-methyl (CD3)-Trp. The detailed massshifts per methylated modification are shown in Table S1.Oxidized methionine as variable modification of +15.9949 wasincluded in all the four groups. When testing the possibility ofconverting SAM of heavy form into methionine of heavy form,(CD3)-Met was set as variable modification. Peptides weresearched using fully tryptic cleavage constraints and up to twomissed cleavages sites were allowed for tryptic digestion. Themass tolerances were 50 ppm for parent masses and 0.8 Da forfragmentmasses. It should be noted that nomore than 5 variablemodifications were allowed for a single peptide and theminimalpeptide length allowed is 6. The identification of a proteinrequires at least one unique corresponding peptide. Methylatedpeptides with Mascot score ≥ 20 (P ≤ 0.05) were saved for furthermethylated sites' assignment. Methylated peptides without H:Lpairs of approximately the same abundance were removed fromthe identification list. In the manual verification process, onlymethylated peptides with at least 3 sequential b ions or y ionswere saved; while for those peptides with low signal/noise, the
229J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
number of sequential b ions or y ions was 5. Methylation siteswere initially determined depending on database searching, andfurther verified by manual inspection to remove peptides thatwere not properly assigned. The surface accessibility of identifiedmethylation sites was analyzed using NetSurfP [25] by uploadingsequences of methylated proteins.
3. Results and discussion
3.1. Stable isotope-labeling by SAM in culture to label proteinmethylation sites in vivo
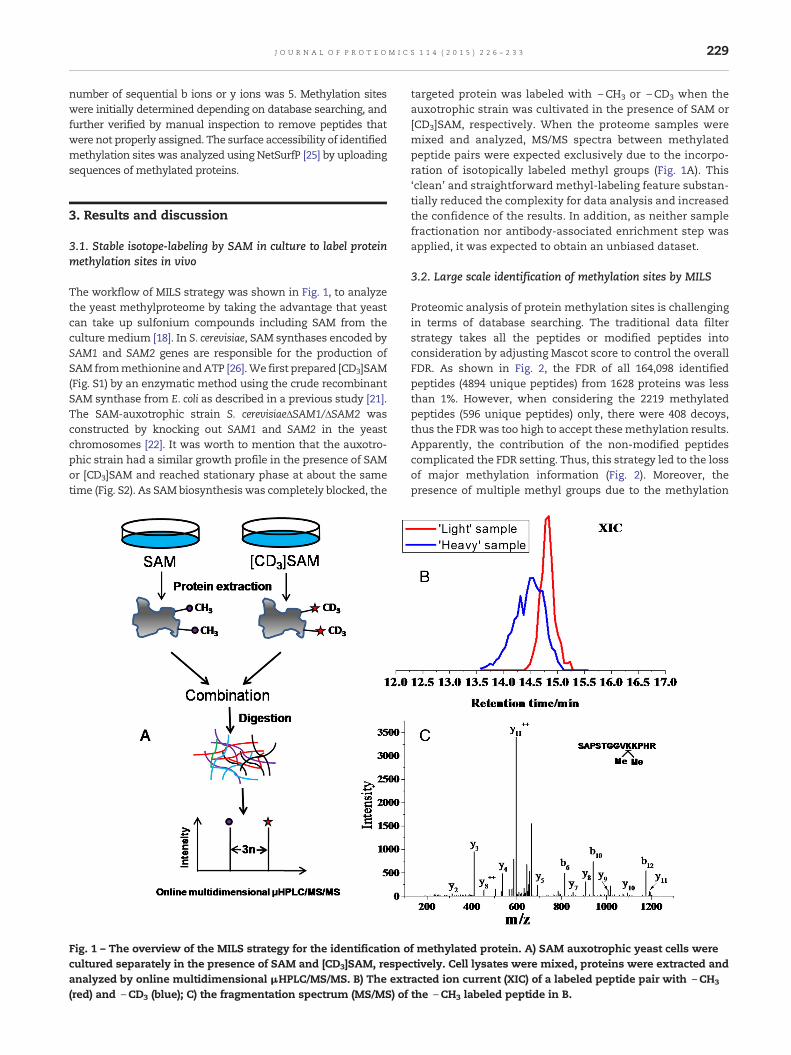
The workflow of MILS strategy was shown in Fig. 1, to analyzethe yeast methylproteome by taking the advantage that yeastcan take up sulfonium compounds including SAM from theculture medium [18]. In S. cerevisiae, SAM synthases encoded bySAM1 and SAM2 genes are responsible for the production ofSAM frommethionine andATP [26].We first prepared [CD3]SAM(Fig. S1) by an enzymatic method using the crude recombinantSAM synthase from E. coli as described in a previous study [21].The SAM-auxotrophic strain S. cerevisiaeΔSAM1/ΔSAM2 wasconstructed by knocking out SAM1 and SAM2 in the yeastchromosomes [22]. It was worth to mention that the auxotro-phic strain had a similar growth profile in the presence of SAMor [CD3]SAM and reached stationary phase at about the sametime (Fig. S2). As SAMbiosynthesis was completely blocked, the
Fig. 1 – The overview of the MILS strategy for the identification ocultured separately in the presence of SAM and [CD3]SAM, respeanalyzed by online multidimensional μHPLC/MS/MS. B) The extr(red) and \CD3 (blue); C) the fragmentation spectrum (MS/MS) of
targeted protein was labeled with \CH3 or \CD3 when theauxotrophic strain was cultivated in the presence of SAM or[CD3]SAM, respectively. When the proteome samples weremixed and analyzed, MS/MS spectra between methylatedpeptide pairs were expected exclusively due to the incorpo-ration of isotopically labeled methyl groups (Fig. 1A). This‘clean’ and straightforward methyl-labeling feature substan-tially reduced the complexity for data analysis and increasedthe confidence of the results. In addition, as neither samplefractionation nor antibody-associated enrichment step wasapplied, it was expected to obtain an unbiased dataset.
3.2. Large scale identification of methylation sites by MILS
Proteomic analysis of protein methylation sites is challengingin terms of database searching. The traditional data filterstrategy takes all the peptides or modified peptides intoconsideration by adjusting Mascot score to control the overallFDR. As shown in Fig. 2, the FDR of all 164,098 identifiedpeptides (4894 unique peptides) from 1628 proteins was lessthan 1%. However, when considering the 2219 methylatedpeptides (596 unique peptides) only, there were 408 decoys,thus the FDRwas too high to accept thesemethylation results.Apparently, the contribution of the non-modified peptidescomplicated the FDR setting. Thus, this strategy led to the lossof major methylation information (Fig. 2). Moreover, thepresence of multiple methyl groups due to the methylation
f methylated protein. A) SAM auxotrophic yeast cells werectively. Cell lysates were mixed, proteins were extracted andacted ion current (XIC) of a labeled peptide pair with \CH3
the \CH3 labeled peptide in B.

Fig. 2 – Filtering strategies to find high confidence modified peptides with methylated sites.
230 J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
on multiple sites or on a nitrogen center with different statescould lead to a large number of false positives due to theincreasing number of possible peptide matches in theexpanded searching space [15]. The MILS strategy overcamethese problems by determining the mass differences of twoindependent fragmentation spectra appearing in pairs due tothe presence of isotopically labeled methyl groups (Fig. 1B).Hence, mono-, di- and tri-methylated peptide pairs could bedifferentiated by 3.0188, 6.0376 and 9.0565 Da, respectively(Table S1). Although mono-acetylation and tri-methylation ofpeptide fragment share a very close mass, these two eventscan be easily distinguished by the absence and presence of a9 Da ion pair, respectively.
To rule out the possibility of converting [CD3]SAM into[CD3]methionine, we searched replicate 1 with methionine asa variable modification, in the final dataset of 41,873 peptides(5168 unique peptides) with score > 20, only 57 peptides wereidentified with heavy labeled methionine. If score > 36 wasrequired, no peptide was observed, which means in ourexperiment, the conversion of [CD3]SAM into [CD3]methioninewas negligible, and this is accordance with our databasesearching parameters that did not consider the presence ofany [CD3]methionine. On the other hand, it is also suggestedthat the vast majority of MS pairs with 3 × n Daltonincrements should be originated from protein methylationwith [CD3]SAM.
In proteomics study, it is important to identify moremodified sites without sacrificing confidence. However, theincrease of confidence always fails the identification of sometrue modification sites, i.e. high false negative. In thisexperiment, the light form and heavy form of true methyla-tion sites should coexist in similar abundance. Thus, it isacceptable to use this standard to filter the datasets to ensurethe confidence of the identified modification sites. There areindeed some known methylation sites that were rejected byfurther filtering due to bad fragmentation or absence of peakpair, the further filtering is still necessary to ensure thereliability of high throughput study.
Weperformeddatabase searching for all chemically possibleproteinmethylation except thosewhich occurred on residues of
protein C-terminus and N-terminus. Thus, database searchingwas set to include N-methylation on Arg, Asn, Gln, His, Lys andTrp; O-methylation on Asp, Glu, Ser, Thr and Tyr; as well asS-methylation on Cys (Fig. 3). We identified 43 methylatedproteins, containing 68 methylation events associated with 64methylation sites (Fig. 3, Table S5). Itwas found that Lyswas themostmethylated residue andArg followed.Moreover, a numberof N-methylation events also occurred on Asn (4), Gln (4) andHis (4). O-methylation on carboxyl group was difficult to beascertained, as a recent exhaustive screening of the yeastproteome only picked up 2 peptides bearing a methylation onAsp and 1 peptide on Glu [16]. However, our results identified 6peptides containing methylation on Asp and 3 peptidescontaining methylation on Glu. We also identified that 1peptide contained S-methylation on Cys. However, no proteinmethylation was found on Ser, Thr, Trp and Tyr. Because therewere no selective enrichment procedures, the presence ofmethylation information should be with no bias. Overall, theamino acid residue preference of protein methylation followedthis sequence, Lys ≫ Arg > Asp > Asn ≈ Gln ≈ His > Glu > Cys.It was noticed that arginine methylation was more abundantthan lysine methylation inmammal cell lines [8]. However, ourresults indicated that lysine methylation was more abundant.This trend was in consistent with data shown a previous yeastmethylproteome study which identified 40 lysine-methylatedproteins and 31 arginine-methylated proteins [17]. In theUniprot database, there were 15 lysine-methylated proteinsand 7 arginine-methylated proteins. Therefore, yeast may havedifferent residue preferences over mammal cells in terms ofprotein methylation. Taken together, this dataset coveredmajor possibilities in terms of protein methylation on differentcenters andwith different states, representing a comprehensivemethylproteome of the S. cerevisiae. We also noticed that thenewly introduced MS instrument could help to reach nearlycomprehensive coverage of the yeast proteome [27]. With thedevelopment of MS technology, proteomics approach usingadvanced MS system and MILS strategy can enable a morein-depth understanding of the methylproteome of S. cerevisiaein the near future.
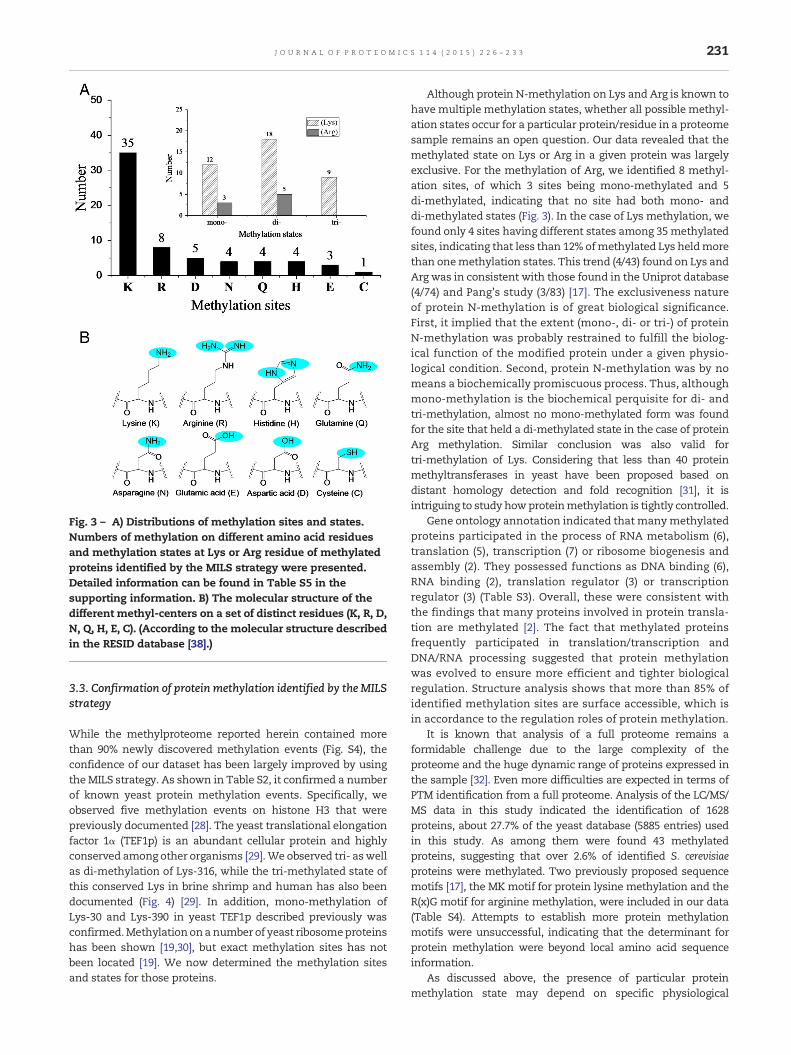
Fig. 3 – A) Distributions of methylation sites and states.Numbers of methylation on different amino acid residuesand methylation states at Lys or Arg residue of methylatedproteins identified by the MILS strategy were presented.Detailed information can be found in Table S5 in thesupporting information. B) The molecular structure of thedifferent methyl-centers on a set of distinct residues (K, R, D,N, Q, H, E, C). (According to the molecular structure describedin the RESID database [38].)
231J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
3.3. Confirmation of protein methylation identified by the MILSstrategy
While the methylproteome reported herein contained morethan 90% newly discovered methylation events (Fig. S4), theconfidence of our dataset has been largely improved by usingtheMILS strategy. As shown in Table S2, it confirmed a numberof known yeast protein methylation events. Specifically, weobserved five methylation events on histone H3 that werepreviously documented [28]. The yeast translational elongationfactor 1α (TEF1p) is an abundant cellular protein and highlyconserved among other organisms [29].We observed tri- aswellas di-methylation of Lys-316, while the tri-methylated state ofthis conserved Lys in brine shrimp and human has also beendocumented (Fig. 4) [29]. In addition, mono-methylation ofLys-30 and Lys-390 in yeast TEF1p described previously wasconfirmed.Methylationonanumber of yeast ribosomeproteinshas been shown [19,30], but exact methylation sites has notbeen located [19]. We now determined the methylation sitesand states for those proteins.
Although protein N-methylation on Lys and Arg is known tohavemultiple methylation states, whether all possible methyl-ation states occur for a particular protein/residue in a proteomesample remains an open question. Our data revealed that themethylated state on Lys or Arg in a given protein was largelyexclusive. For the methylation of Arg, we identified 8 methyl-ation sites, of which 3 sites being mono-methylated and 5di-methylated, indicating that no site had both mono- anddi-methylated states (Fig. 3). In the case of Lys methylation, wefound only 4 sites having different states among 35methylatedsites, indicating that less than 12% ofmethylated Lys heldmorethan onemethylation states. This trend (4/43) found on Lys andArg was in consistent with those found in the Uniprot database(4/74) and Pang's study (3/83) [17]. The exclusiveness natureof protein N-methylation is of great biological significance.First, it implied that the extent (mono-, di- or tri-) of proteinN-methylation was probably restrained to fulfill the biolog-ical function of the modified protein under a given physio-logical condition. Second, protein N-methylation was by nomeans a biochemically promiscuous process. Thus, althoughmono-methylation is the biochemical perquisite for di- andtri-methylation, almost no mono-methylated form was foundfor the site that held a di-methylated state in the case of proteinArg methylation. Similar conclusion was also valid fortri-methylation of Lys. Considering that less than 40 proteinmethyltransferases in yeast have been proposed based ondistant homology detection and fold recognition [31], it isintriguing to study howproteinmethylation is tightly controlled.
Gene ontology annotation indicated that manymethylatedproteins participated in the process of RNA metabolism (6),translation (5), transcription (7) or ribosome biogenesis andassembly (2). They possessed functions as DNA binding (6),RNA binding (2), translation regulator (3) or transcriptionregulator (3) (Table S3). Overall, these were consistent withthe findings that many proteins involved in protein transla-tion are methylated [2]. The fact that methylated proteinsfrequently participated in translation/transcription andDNA/RNA processing suggested that protein methylationwas evolved to ensure more efficient and tighter biologicalregulation. Structure analysis shows that more than 85% ofidentified methylation sites are surface accessible, which isin accordance to the regulation roles of protein methylation.
It is known that analysis of a full proteome remains aformidable challenge due to the large complexity of theproteome and the huge dynamic range of proteins expressed inthe sample [32]. Even more difficulties are expected in terms ofPTM identification from a full proteome. Analysis of the LC/MS/MS data in this study indicated the identification of 1628proteins, about 27.7% of the yeast database (5885 entries) usedin this study. As among them were found 43 methylatedproteins, suggesting that over 2.6% of identified S. cerevisiaeproteins were methylated. Two previously proposed sequencemotifs [17], the MKmotif for protein lysine methylation and theR(x)G motif for arginine methylation, were included in our data(Table S4). Attempts to establish more protein methylationmotifs were unsuccessful, indicating that the determinant forprotein methylation were beyond local amino acid sequenceinformation.
As discussed above, the presence of particular proteinmethylation state may depend on specific physiological

Fig. 4 – Cross-species comparison of lysine methylation in translational elongation factor 1α. Methylated K residues areindicated with arrows. The symbol ‘&’ stands for mono-methylation, ‘#’ for di-methylation and ‘$’ for tri-methylation.
232 J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
condition. Thus, besides the limitation of the LC/MS/MSworkflow, the culture conditions and genetic background ofthe yeast strain may all contribute to the profile of our dataset,which might miss the identification of some known proteinmethylation events [28,30–37]. As the dataset described hereincovers various types of methylation centers, it provides richinformation about yeast methylproteome and should signifi-cantly contribute to the field of protein methylation as well asrelated biological sciences. In addition, the MILS strategy mayalso be useful to explore themethylation ofDNA/RNAandsmallmetabolites in yeast.
Supplementary data to this article can be found online athttp://dx.doi.org/10.1016/j.jprot.2014.07.032.
Competing interests
The authors declare they have no competing interests.Accession information. All described nanoLC-MS/MS
data can be downloaded from ProteomeXchange with theProteomeXchange accession: PXD000606.
Transparency document
The Transparency document associated with this article canbe found in the online version.
Acknowledgment
Thiswork was supported by the China State Key Basic ResearchProgram Grant (2013CB911202, 2012CB910101, 2012CB910604),the Creative Research Group Project of NSFC (21321064), theNational Natural Science Foundation of China (21235006,81361128015), and Analytical Method Innovation Program ofMOST (2012IM030900) to HZ.
R E F E R E N C E S
[1] Walsh CT, Garneau-Tsodikova S, Gatto GT. Proteinposttranslational modifications: the chemistry of proteomediversifications. Angew Chem Int Ed 2005;44:7342–72.
[2] Polevoda B, Sherman F. Methylation of proteins involved intranslation. Mol Microbiol 2007;65:590–606.
[3] Huang J, Berger SL. The emerging field of dynamic lysinemethylation of non-histone proteins. Curr Opin Genet Dev2008;18:152–8.
[4] Teyssier C, Le RM, Sentis S, Jalaguier S, et al. Protein argininemethylation in estrogen signaling and estrogen-relatedcancers. Trends Endocrinol Metab 2010;21:181–9.
[5] Grillo MA, Colombatto S. S-adenosylmethionine and proteinmethylation. Amino Acids 2005;28:357–62.
[6] Klose RJ, Yi Z. Regulation of histone methylation bydemethylimination and demethylation. Nat Rev Mol Cell Biol2007;8:307–18.
[7] Bedford MT, Richard S. Arginine methylation: an emergingregulator of protein function. Mol Cell 2005;18:263–72.
[8] Bremang M, Cuomo A, Agresta AM, et al. Massspectrometry-based identification and characterisation oflysine and arginine methylation in the human proteome. MolBiosyst 2013;9:2231–47.
[9] Uhlmann T, Geoghegan VL, Thomas B, et al. A method forlarge-scale identification of protein arginine methylation.Mol Cell Proteomics 2012;11:1489–99.
[10] Cao XJ, Arnaudo AM, Garcia BA. Large-scale globalidentification of protein lysine methylation in vivo.Epigenetics 2013;8:477–85.
[11] Guo A, Gu H, Zhou J, et al. Immunoaffinity enrichment andmass spectrometry analysis of protein methylation. Mol CellProteomics 2014;13:372–87.
[12] Clarke SG. Protein methylation at the surface and burieddeep: thinking outside the histone box. Trends Biochem Sci2013;38:243–52.
[13] Afjehi-Sadat L, Garcia BA. Comprehending dynamic proteinmethylation with mass spectrometry. Curr Opin Chem Biol2013;17:12–9.
[14] Boisvert FM, Cote J, Boulanger MC, Richard S. A proteomicanalysis of arginine-methylated protein complexes. Mol CellProteomics 2003;2:1319–30.
[15] Ong SE, Mittler G, Mann M. Identifying and quantifying invivo methylation sites by heavy methyl SILAC. Nat Methods2004;1:119–26.
[16] Sprung R, Chen Y, Zhang K, Cheng D, et al. Identification andvalidation of eukaryotic aspartate and glutamate methylationin proteins. J Proteome Res 2008;7:1001–6.
[17] Pang CNJ, Gasteiger E, Wilkins MR. Identification ofarginine- and lysine-methylation in the proteome of

233J O U R N A L O F P R O T E O M I C S 1 1 4 ( 2 0 1 5 ) 2 2 6 – 2 3 3
Saccharomyces cerevisiae and its functional implications. BMCGenomics 2010;11:92–107.
[18] Rouillon A, Surdin-Kerjan Y, Thomas DJ. Transport ofsulfonium compounds. Characterization of theS-adenosylmethionine and S-methylmethionine permeasesfrom the yeast Saccharomyces cerevisiae. J Biol Chem 1999;274:28096–105.
[19] Lhoest J, Lobet Y, Costers E, Colson C. Methylated proteinsand amino-acid in the ribosomes of Saccharomyces-cerevisiae.Eur J Biochem 1984;141:585–90.
[20] Finkelstein JD. Methionine metabolism in mammals. NutrBiochem 1990;1:228–37.
[21] Park J, Tai JZ, Roessner CA, Scott AI. Enzymatic synthesisof S-adenosyl-L-methionine on the preparative scale. BioorgMed Chem 1996;4:2179–85.
[22] Zhou YJ, Yang F, Zhang SF, Tan HD, Zhao ZK. Efficient genedisruption in Saccharomyces cerevisiae using marker cassetteswith long homologous arms prepared by the restriction-freecloning strategy. World J Microbiol Biotechnol 2011;27:2999–3003.
[23] Wang FJ, Dong J, Jiang XG, Ye ML, Zou HF. Capillary trapcolumnwith strong cation-exchangemonolith for automatedshotgun proteome analysis. Anal Chem 2007;79:6599–606.
[24] Mortensen P, Gouw JW, Olsen JV, Ong SE, et al. MSQuant,an open source platform for mass spectrometry-basedquantitative proteomics. J Proteome Res 2010;9:393–403.
[25] Petersen B, Petersen TN, Andersen P, et al. A generic methodfor assignment of reliability scores applied to solventaccessibility predictions. BMC Struct Biol 2009;9:51.
[26] Thomas D, Surdin-Kerjan Y. The synthesis of the twoS-adenosyl-methionine synthetases is differently regulatedin Saccharomyces cerevisiae. Mol Gen Genet 1991;226:224–32.
[27] Nagaraj N, Kulak NA, Cox J, et al. System-wide perturbationanalysis with nearly complete coverage of the yeastproteome by single-shot ultra HPLC runs on a bench topOrbitrap. Mol Cell Proteomics 2012;11 [M111. 013722].
[28] Garcia BA, Hake SB, Diaz RL, Kauer M, et al. Organismaldifferences in post-translational modifications in histones H3and H4. J Biol Chem 2007;282:7641–55.
[29] Cavallius J, ZollW, ChakraburttyK,MerrickWC. Characterizationof yeast EF-1-alpha-nonconservation of posttranslationalmodifications. Biochim Biophys Acta 1993;1163:75–80.
[30] Webb KJ, Laganowsky A, Whitelegge JP, Clarke SG.Identification of two SET domain proteins required formethylation of lysine residues in yeast ribosomal proteinRpl42ab. J Biol Chem 2008;283:35561–8.
[31] Wlodarski T, Kutner J, Towpik J, Knizewski L, et al.Comprehensive structural and substrate specificityclassification of the Saccharomyces cerevisiaemethyltransferome. PLoS One 2001;6:e23168.
[32] Domon B, Aebersold R. Mass spectrometry and proteinanalysis. Science 2006;312:212–7.
[33] Ng HH, Feng Q, Wang H, Erdjument-Bromage H, et al. Lysinemethylation within the globular domain of histone H3 byDot1 is important for telomeric silencing and Sir proteinassociation. Genes Dev 2002;16:1518–27.
[34] Heurgue-Hamard V, Champ S, Mora L, Merkulova-Rainon T,et al. The glutamine residue of the conserved GGQ motif inSaccharomyces cerevisiae release factor eRF1 ismethylated by theproduct of the YDR140w gene. J Biol Chem 2005;280:2439–45.
[35] Polevoda B, Span L, Sherman F. The yeast translation releasefactors Mrf1p and Sup45p (eRF1) are methylated, respectively,by the methyltransferases Mtq1p and Mtq2p. J Biol Chem2006;281:2562–71.
[36] Webb KJ, Zurita-Lopez CI, Al-Hadid Q, Laganowsky A, et al. Anovel 3-methylhistidine modification of yeast ribosomalprotein Rpl3 is dependent upon the YIL110Wmethyltransferase. J Biol Chem 2010;285:37598–606.
[37] Webb KJ, Al-Hadid Q, Zurita-Lopez CI, Young BD, et al. Theribosomal L1 protuberance in yeast is methylated on a lysineresidue catalyzed by a seven-beta-strand methyltransferase.J Biol Chem 2001;286:18405–13.
[38] Garavelli JS. The RESID database of protein modifications as aresource and annotation tool. Proteomics 2004;4:1527–33.





























