Embed Size (px)
Citation preview

Protein Structure
Disclaimer: All information and images were taken from outside sources and the author claims no legal ownership of any material.Sources for images are linked on each slide and the information from each slide is derived from the 4th edition of the
biochemistry text written by Voet, Voet, and Pratt.

Overview• Secondary Structure
• Geometry of peptide bonds• Alpha helices • Beta Sheets• Fibrous proteins • Non-repetitive Protein Structure
• Tertiary Structure • X-ray crystallography • Nuclear Magnetic Resonance (NMR)• Side Chain Polarity • Types and IMF’s• Protein Databases
• Quaternary Structure
• Protein Stability
• Protein Folding Source: http://chemistry.csulb.edu/research-biological.html

Protein Structure Overview
• There are four levels of structure
• Primary is the sequence of amino acids
• Secondary is the manner in which the primary sequence is arranged in 3-D space
• Tertiary is the 3-D arrangement of the secondary structures
• Quaternary is the combination of multiple individual polypeptides.
Source: https://www.boundless.com/biology/textbooks/boundless-biology-textbook/biological-macromolecules-3/proteins-56/protein-structure-304-11437/
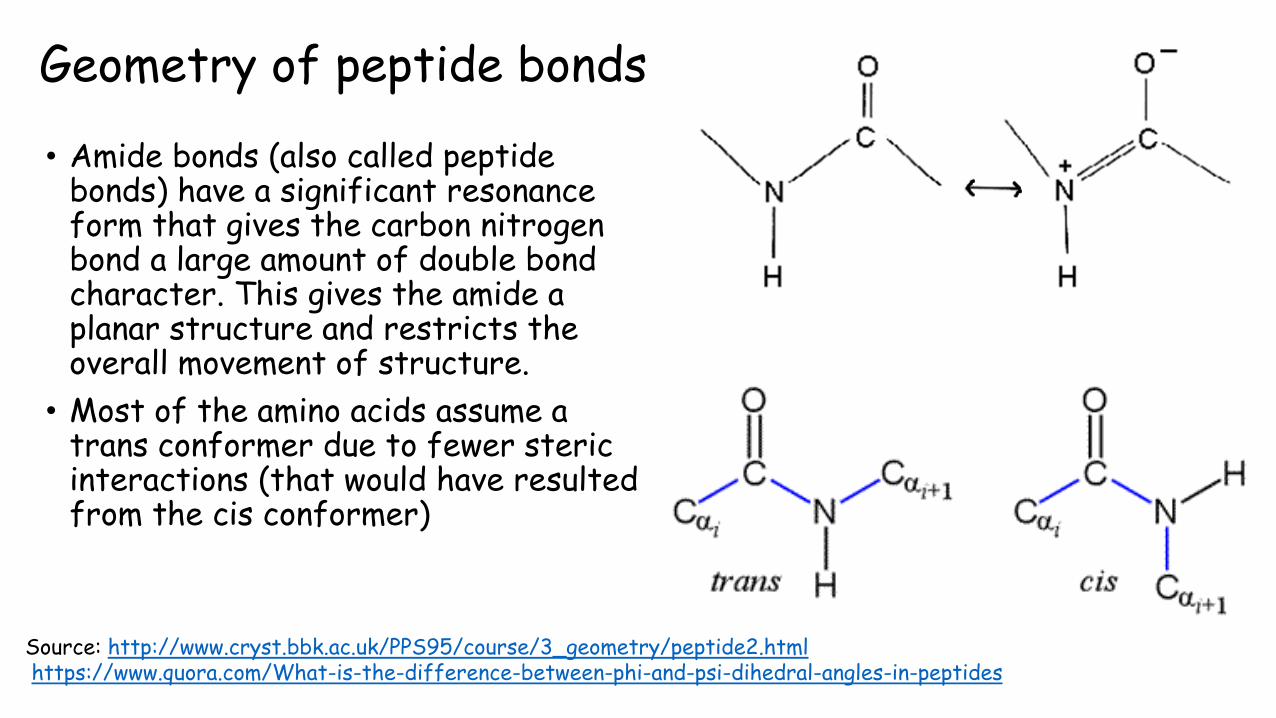
Geometry of peptide bonds
• Amide bonds (also called peptide bonds) have a significant resonance form that gives the carbon nitrogen bond a large amount of double bond character. This gives the amide a planar structure and restricts the overall movement of structure.
• Most of the amino acids assume a trans conformer due to fewer steric interactions (that would have resulted from the cis conformer)
Source: http://www.cryst.bbk.ac.uk/PPS95/course/3_geometry/peptide2.htmlhttps://www.quora.com/What-is-the-difference-between-phi-and-psi-dihedral-angles-in-peptides

Torsional angles of the peptide bond
• The backbone or main chain of the protein refers to the atoms that participate in the peptide bonds and the atoms that connect them.
• The backbone can be drawn as a linked sequence of rigid planar peptide groups. The conformation of the backbone can be described using torsional angles.
• The angle between the carbonyl carbon and the alpha carbon can be described by the psi value (Ψ)
• The angle between the alpha carbon and the nitrogen of the next amide bond can be described by the phi value (Φ)
• The angel between the carbonyl carbon and the nitrogen can be described by the omega value (ω)
• Images showing the location of psi and phi can be seen on the next slide.

Source: http://www.cryst.bbk.ac.uk/PPS95/course/9_quaternary/3_geometry/torsion.htmlhttp://www.bioinf.org.uk/teaching/bbk/molstruc/practical2/peptide.htmlhttps://en.wikipedia.org/wiki/Dihedral_angle

Ramachandran Plot• Plot that shows the allowed
conformations of polypeptides.
• There are only a few regions of the plot that are sterically allowed conformations.
• The major exceptions are: proline, which is restricted due to the ring and glycine, whose sidechain is significantly smaller than any other amino acid and so the polypeptide can assume otherwise forbidden conformations.
Source: http://sandwalk.blogspot.com/2008/04/ramachandron-plots.html
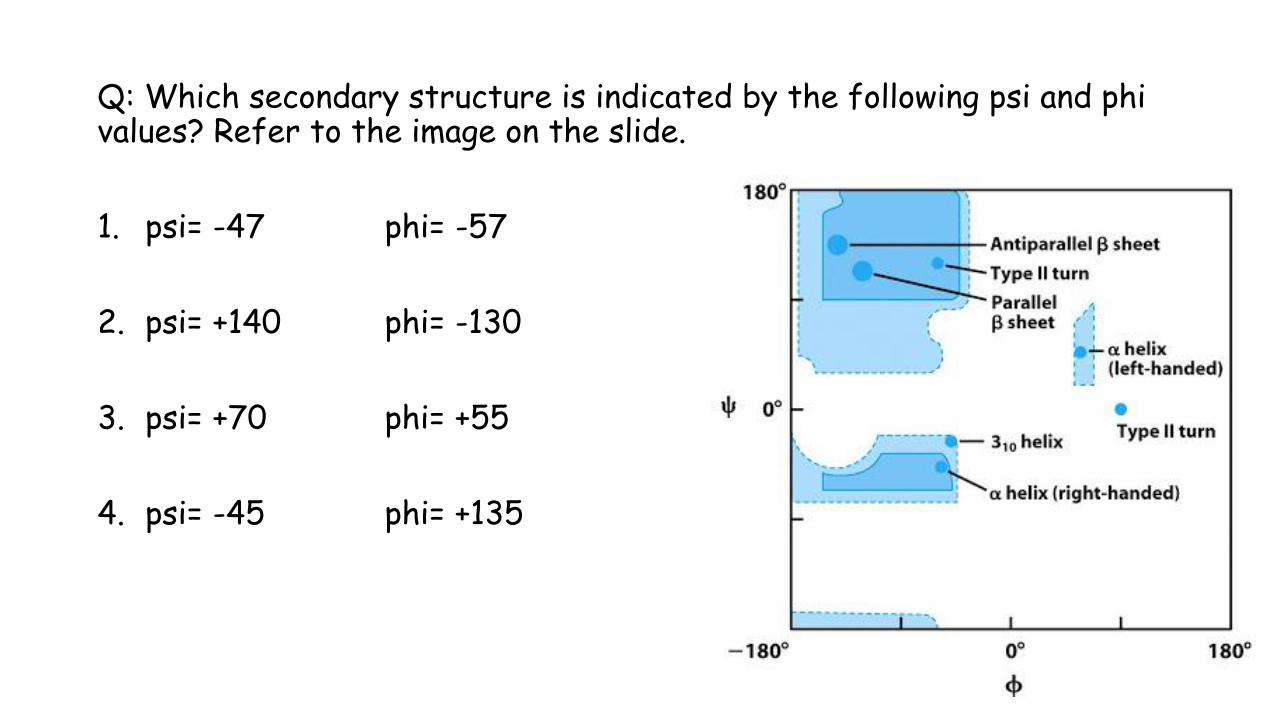
Q: Which secondary structure is indicated by the following psi and phi values? Refer to the image on the slide.
1. psi= -47 phi= -57
2. psi= +140 phi= -130
3. psi= +70 phi= +55
4. psi= -45 phi= +135

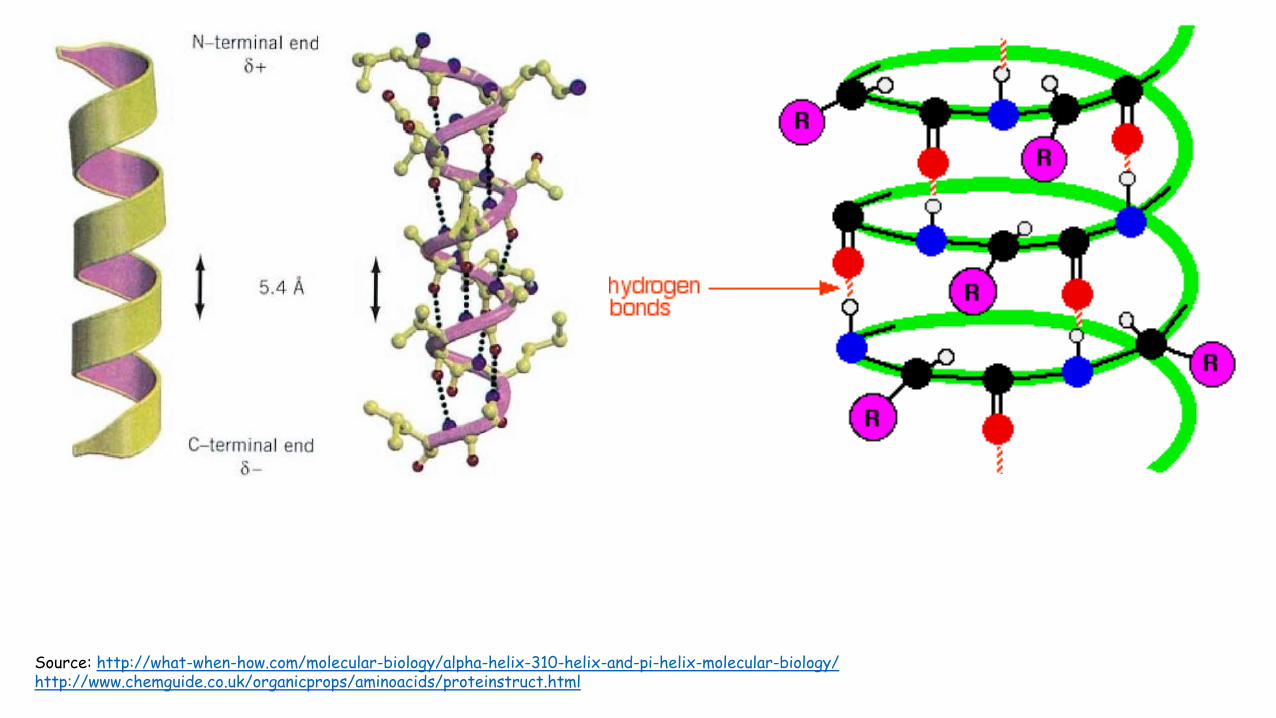
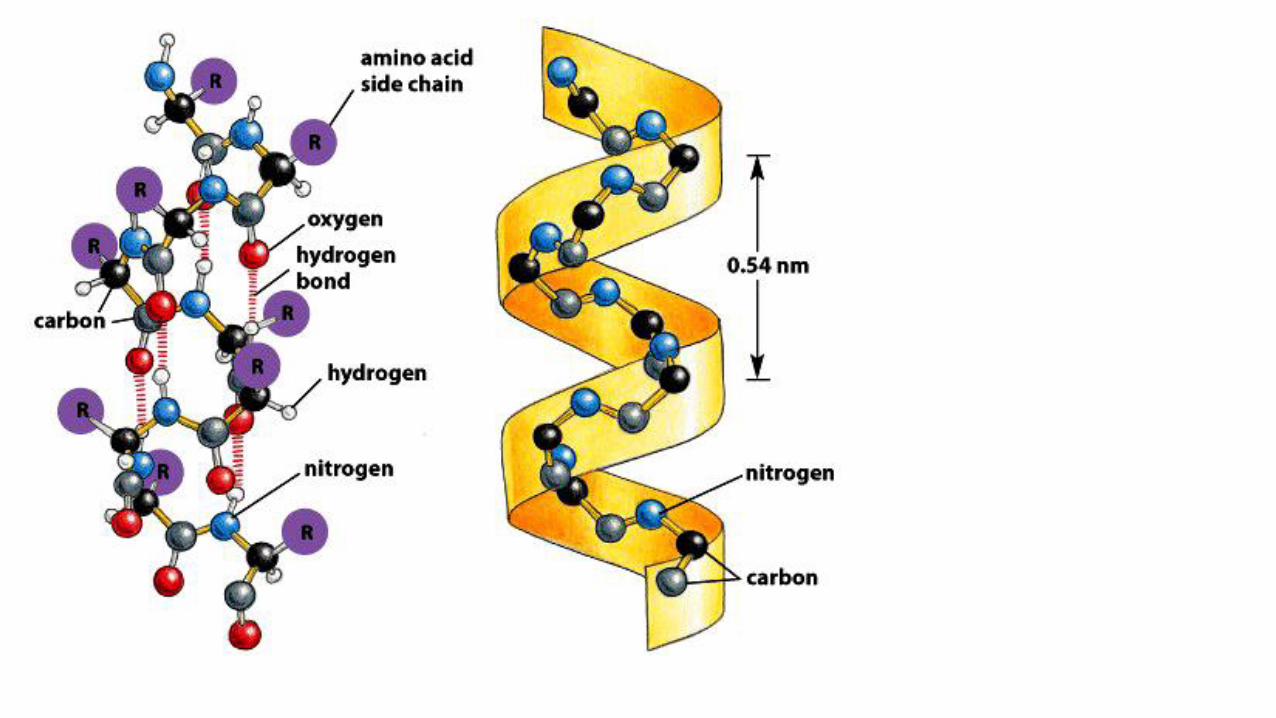
Alpha helices
• Structure that has both favorable hydrogen bond patterns and psi and phi values that fall within the fully allowed regions of the Ramachandran plot.
• Discovered by Linus Pauling in 1951
• The helix is right handed and has 3.6 residues per turn. Its pitch (distance risen per each turn) is 5.4 Å.
• The alpha helix backbone’s carbonyls bond with the hydrogen of the amide linkages. The carbonyl bonds to the hydrogen of the amide that is 4 residues after it. These hydrogen bonds are 2.8 Å apart.
• The side chains point outward and downward from the helix thereby avoiding contact with the backbone and with each other.
Source: http://what-when-how.com/molecular-biology/alpha-helix-310-helix-and-pi-helix-molecular-biology/http://www.chemguide.co.uk/organicprops/aminoacids/proteinstruct.html

Q: Answer each of the following in regards to alpha helices
1. How many residues are needed to make an alpha helix that is 20 Å long?
2. Assuming average amino acid weight of 110 Daltons, what is the contour length of an alpha helix that weighs 1.4 KiloDaltons
3. If the average cell membrane is 7-10 nm in thickness, how many amino acids are required to form an alpha helix that will span the entire membrane. (give answer in the form of a range and in units of Å)

Q: Answer each of the following in regards to alpha helices.
1. What would happen to the helix if the hydrogen bonds occurred between a carbonyl and every 6th residue instead of every 4th?
2. What factor can be changed to drastically alter the solubility of alpha helices in a given solvent?
3. How many amino acids are in the helix shown below?

Beta Sheets• Polypeptide chains next to each other form hydrogen bonds, rather than
within the same chain.
• There are two types: antiparallel and parallel. • Antiparallel: the chains run in opposite directions • Parallel: chains run in the same direction
• The ideal formation of hydrogen bonds gives the sheet a slightly pleated appearance instead of being perfect flat.
• Side chains of the polypeptide extend to opposite sides of the sheet with a two residue repeat that has a distance of 7 Å.
• Beta sheets usually exhibit a slight right handed twist due to a compromise between optimizing the conformation and keeping the hydrogen bonds.
• The favored structure is antiparallel over parallel (see hydrogen bonding on next slide to justify)
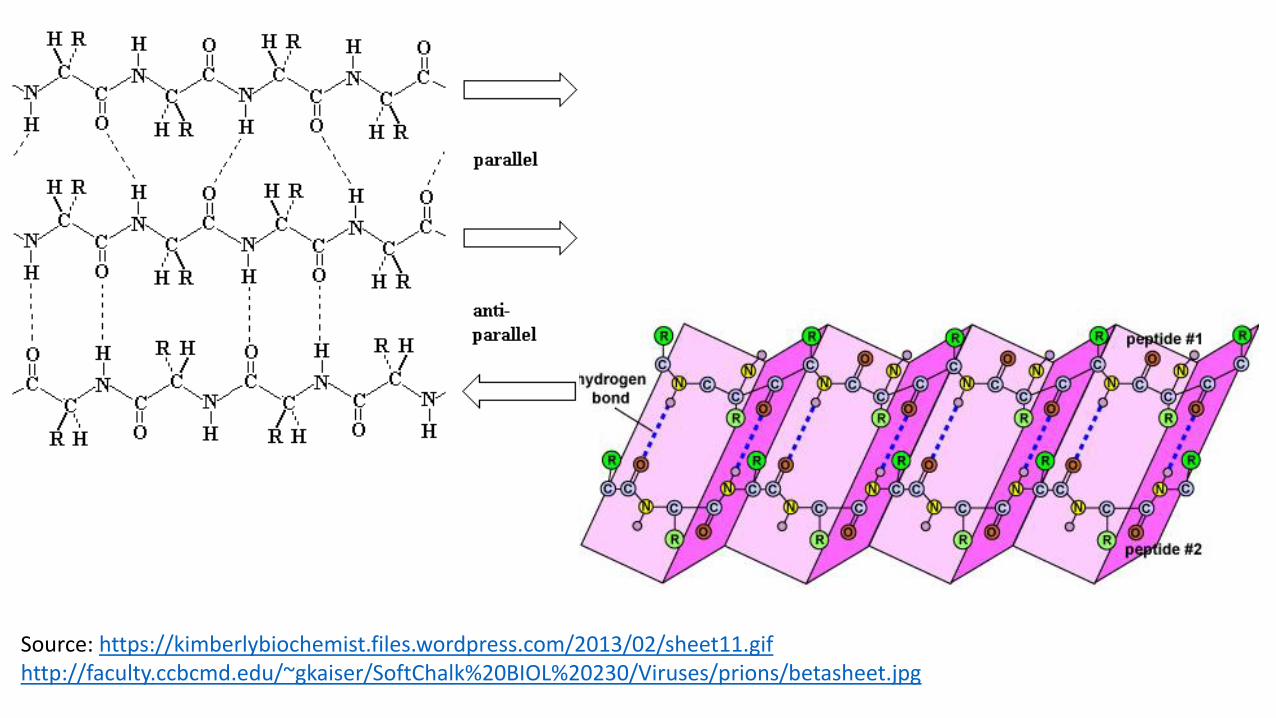
Source: https://kimberlybiochemist.files.wordpress.com/2013/02/sheet11.gifhttp://faculty.ccbcmd.edu/~gkaiser/SoftChalk%20BIOL%20230/Viruses/prions/betasheet.jpg


Q: Answer the following in regards to beta sheets
1. What causes beta sheets to have a pleated appearance instead of being flat?
2. Why do beta sheets have a slight twist to them?
3. How long is a beta sheet that has 14 residues?
4. How many residues are in a beta sheet that is 28 Å long?
5. How long is a beta sheet that weighs 1.6 kiloDaltons, assume the average amino acid weighs 110 Daltons.

Q: In the crystal structure shown: identify the beta sheets and alpha helices. Estimate the length of each helix and how many amino acids are in each helix. Could you use an alpha helix to estimate the number of amino acids in the beta sheets? If so, how would you do this?
Source: http://csgid.org/deposits/view/1513
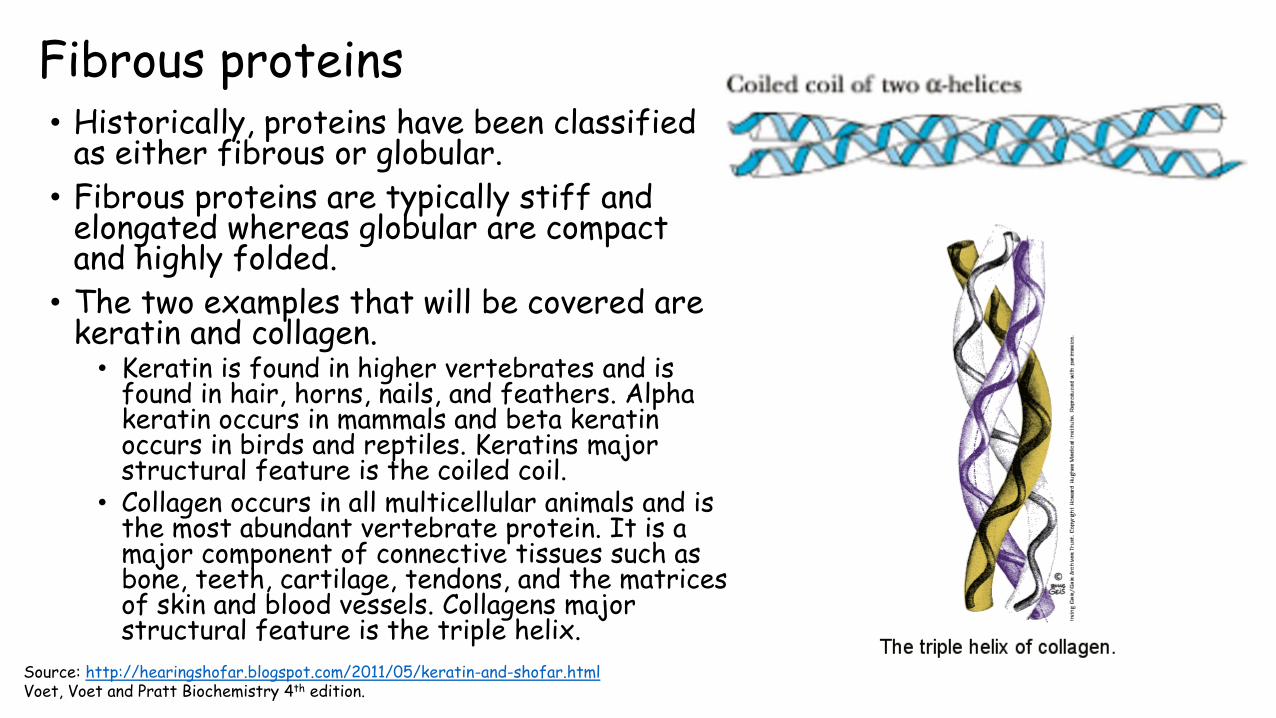
Fibrous proteins • Historically, proteins have been classified
as either fibrous or globular.
• Fibrous proteins are typically stiff and elongated whereas globular are compact and highly folded.
• The two examples that will be covered are keratin and collagen.• Keratin is found in higher vertebrates and is
found in hair, horns, nails, and feathers. Alpha keratin occurs in mammals and beta keratin occurs in birds and reptiles. Keratins major structural feature is the coiled coil.
• Collagen occurs in all multicellular animals and is the most abundant vertebrate protein. It is a major component of connective tissues such as bone, teeth, cartilage, tendons, and the matrices of skin and blood vessels. Collagens major structural feature is the triple helix.
Source: http://hearingshofar.blogspot.com/2011/05/keratin-and-shofar.htmlVoet, Voet and Pratt Biochemistry 4th edition.

Keratin • Keratin is similar to an alpha helix except that
it has a spacing of 5.1 Å instead of the 5.4 Åexpected for an alpha helix. The difference between the two “is the result of two alpha keratin polypeptides, each of which forms an alpha helix, twisting around each other to form a left handed coil.”
• The two keratin polypeptides being wrapped around each other is called a coiled coil.
• The coiled coil is a result of the primary sequence of the polypeptide. The central segment has a 7 amino acid pseudorepeat 1-2-3-4-5-6-7 with nonpolar residues predominating at positions 1 and 4. This results in a hydrophobic strip along one side of the helix that can then interact favorably with the hydrophobic strip of another polypeptide.
Source: http://slideplayer.com/slide/6117668/
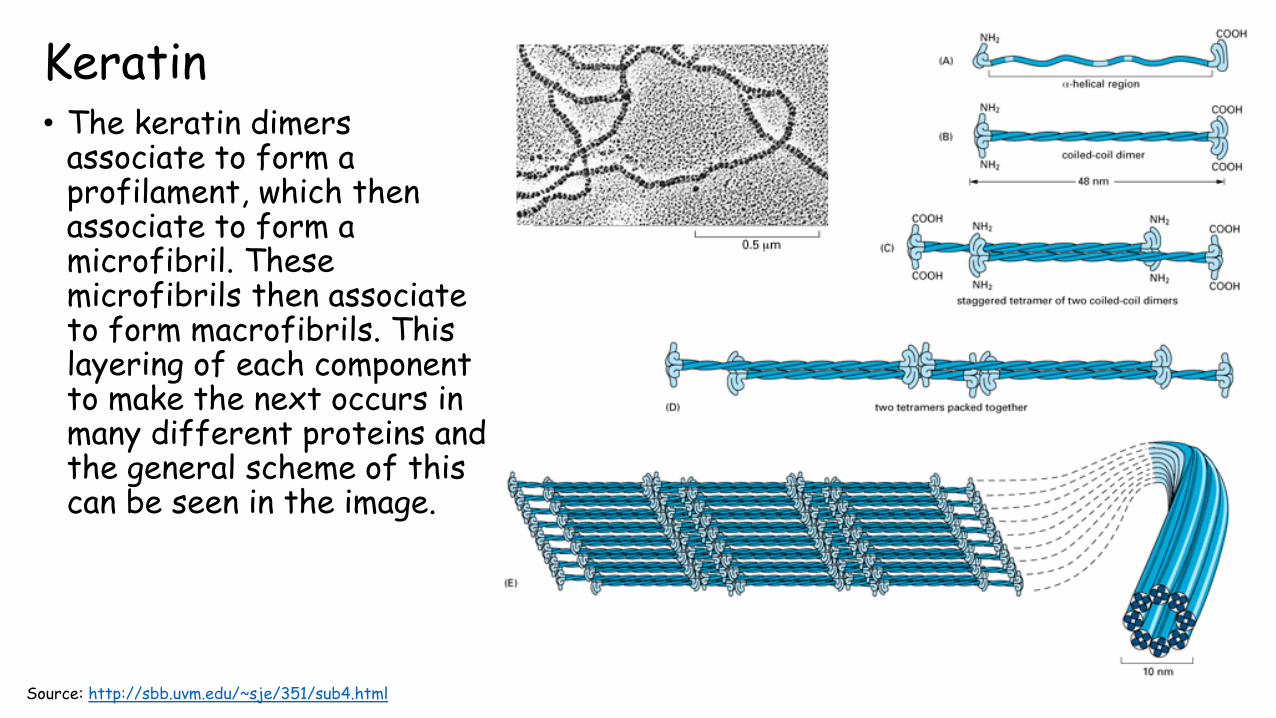
Keratin• The keratin dimers
associate to form a profilament, which then associate to form a microfibril. These microfibrils then associate to form macrofibrils. This layering of each component to make the next occurs in many different proteins and the general scheme of this can be seen in the image.
Source: http://sbb.uvm.edu/~sje/351/sub4.html

Q: Assuming each sequence repeats, which of them could possibly be found in a coiled coil? Explain each case.
1. WRHITSE
2. HYTLVQN
3. IVELACN
4. lRTTLLV

Collagen • A single collagen fiber consists of three polypeptide chains.
• Collagen has a distinct amino acid composition: about 30% is glycine and another 15-30% is proline. Some amino acid derivatives are also found including: 4-hydroxylprolyl (Hyp), 3-hydroxylprolyl, and 5-hydroxylysyl (Hyl)
• The nonstandard residues are formed after collagen polypeptides are synthesized. The sequence of a typical collagen polypeptide consist of the repeating triplet sequence Gly-X-Y where X is often proline and Y is often Hyp. Hyl sometimes appears at the Y position as well.
• The proline resides cannot form an alpha helix and instead “the collagen polypeptide assumes a left-handed helical conformation with about three residues per turn. Three parallel chains wind around each other with a gentle, right handed, ropelike twist to form the triple helical structure of a collagen molecule.”
• Every third residue passes through the center of the helix, which is so crowded that only glycine can fit.

Collagen• The three polypeptide chains are staggered so that Gly, X and Y residues
occur at every level along the triple helix.
• The peptide groups are oriented so that the hydrogen of the amide bond of the glycine reside makes a strong hydrogen bond with the carbonyl oxygen of the X residue on the neighboring chain.
• The entire protein gets its rigidity from the proline and proline derivatives. (Proline is a relatively bulky and inflexible residue)
• Collagen is covalently cross linked. These cross links are not disulfide bonds but instead are formed between Lysine and Histidine residues.
• The amount of cross linking in collagen increase with the age of the organism.

Non-repetitive Protein Structure • Majority of proteins are globular and contain several types of regular
secondary structures.
• “non-repetitive structures are no less ordered than are helices or beta sheets; they are simply irregular and hence more difficult to describe”
• Random coil: totally disordered and rapidly fluctuating conformations taken on by a denatured protein.
• Denatured proteins are fully unfolded and native proteins are folded proteins.
• Beta bulges are created from an “extra” residue that doesn’t hydrogen bond to neighbors like it should.
• Alpha helices can be destabilized by sequential repeats of residues with bulky side chains
• Proline can create a kink in alpha helices or beta sheets.
• Helix capping is when a residue such as Asn or Gln folds back to form hydrogen bonds with one of the four terminal residues of an alpha helix

Q: Draw each of the following amino acids: 4-Hydroxylprolyl, 3-Hydroxylprolyl and 5-hydroxylysyl.
Q: Why can’t collagen strands be broken down by BME?
Q: Briefly explain how collagen strands are cross-linked (see page 141 of Voet, Voet, and Pratt textbook 4th edition)
Q: Which of the following sequences is most likely to be found in an alpha helix? Least likely? Why?
AEMFHAVQ
TSPPMLIQ
AVCYYYGL

Tertiary Structure • The determination of atomic positions is done with X-ray crystallography and
NMR spectroscopy.
• Tertiary structure consists of secondary elements that combine to form motifs and domains.
• The polarity of side chains can heavily dictate the overall shape of the protein.
• Motifs are super-secondary structures.
• Structure is more conserved than sequence.
• There are a multitude of databases that store protein structural information and is called structural bioinformatics.

X-ray Crystallography• Technique that directly images
molecules.
• According to optical principles: the uncertainty in locating an object is approximately equal to the wavelength of radiation used to observe it.
• Covalent bond distances are about 1.5 Å and the wavelength of visible light is 4000 Å whereas X-rays are about 1.5 Å. So x-rays are needed to study molecules.
• A crystal of the compound of interest is exposed to x-rays and the diffraction pattern gives information regarding its structure. Since x-rays interact almost exclusively with electrons, the patterns are really electron densities.
“Precession diagram of a lysozyme crystal. One can easily distinguish a four-fold symmetry axis perpendicular to the diagram. According to the relationships between direct and reciprocal lattices, if the axes of the unit cell are large (as in this case), the separation between reciprocal points is small”
See source
Source: http://www.xtal.iqfr.csic.es/Cristalografia/parte_06-en.html

X-ray Crystallography• Proteins differ from smaller molecules because they are often highly
hydrated and are typically 40-60% water by volume in crystal form. • Because of the large amount of hydration, protein crystals are often
jellylike. • These crystals have a limit of resolution that ranges from 1.5-3 Å.
Source: http://www.nature.com/nprot/journal/v10/n9/full/nprot.2015.069.html

The question arises if the structure of a protein in crystal is the same as in solution. Some evidence has been presented to justify that the structures are the same.
1. The protein is essentially in solution because of the large hydration factor.
2. Other techniques have given identical information. NMR structures are determined in solution and this data correlates well with x-ray crystallography data.
3. Many enzymes are active in their crystal state. Since enzymes require a very specific conformation to work the crystalline enzymes must have structures nearly identical to those in solution.
X-ray Crystallography

Nuclear Magnetic Resonance (NMR)
• NMR is based on the principle that atomic nuclei resonates in an applied magnetic field in a way that is sensitive to its electronic environment and its interactions with nearby nuclei.
• Since even small proteins have 100’s of protons, the 2-D spectrum is used to help determine which protons are close to each other in 3-D space.
• The size of the molecule currently limits if NMR can be used. The current limit is approximately 100 kD but this may eventually increase.
• Protein structures are often simplified to help reduce clutter in the image. This is done by using ribbons to represent alpha helices and beta sheets and to use thin lines tracing the alpha carbons through space.
Source: http://nmr.umn.edu/adv_protein.html

Q: Why is it important to have multiple techniques to determine protein structure?
Q: What are some of the limitations of x-ray crystallography?
Q: What are some of the limitations of NMR?
Q: Why are images of proteins often simplified?

Side Chain Polarity
• In globular proteins, side chains are distributed based on their polarity.
• For instance, nonpolar residues are often found in the interior of globular proteins away from the aqueous environment.
• Charged residues are most likely to be found on the surface of a protein in order to form hydrogen bonds with the aqueous solvent. This is because placing an ion inside an essentially anhydrous environment is energetically unfavorable.
• Polar but uncharged residues are found on both the surface and interior of the protein. When in the interior, these residues are often hydrogen bonded to other groups.
• Water is often excluded from the inside of proteins and when it is found inside it often acts to bridge two residues that need to hydrogen bond.

Q: cell membranes have a nonpolar, hydrophobic interior with aqueous environments on each side of the double membrane. Given the following hypothetical polypeptide sequence, which portions would you predict would be inside the membrane and which should be outside the membrane?
AVLILAAVTREDCINQWFPILEDCSYTEEDLILILAVWFPFPHDMED
Q: if the same sequence in the question above was a globular protein, which portions would likely be on the interior and which portions would likely be on the surface? Explain.

Types of Tertiary Structure
• βαβ motif: very common form. An α helix connects two parallel β sheets.
• β hairpin: antiparallel β strands connected by relatively tight reverse turns.
• αα motif: two successive antiparallel α helices stack against each other with their axes inclined. Similar to keratin structure expect the helices are antiparallel instead of parallel.
• Greek key motif: a β hairpin is folded over to form a 4 stranded antiparallel β sheet.
• Many proteins can be classified based on the amount of α helices or βsheets they contain. If a protein is mostly α helices its classified as an αprotein and if its mostly β sheets its classified as a β protein and if its an even mix of the two its an α/β protein.

Q: classify each of the motifs below using the terminology presented on the previous slide. (Hint: there are two examples of each)
Source: all images on this slide were taken from Google Images

Protein Databases• PDB: (Protein Data Bank) contains nearly 80,000 macromolecular structures
and information associated with the structure. A variety of viewing platforms can be used to view the structures compiled in the database.
• Computational tools allow the classification and comparison of protein strucutes. Some of these are outlined below• CATH: Class Architecture Topology and Homologous super family- Categorizes proteins
based on the four characteristics listed. • CE: Combinatorial Extension- finds all proteins in the PDB that can be structurally aligned
with the query structure. • Pfam: Protein Families- database of nearly 11,000 of multiple sequence alignments of
protein domains. Used to view occurrence of protein domains across species. • SCOP: Structural Classification Of Proteins- classifies based on class, fold, superfamily,
family, protein and species. • VAST: Vector Alignment Search Tool- gives a list of proteins that structurally resemble
the query protein.

Quaternary Structure
• Quaternary structure is the association of multiple polypeptides to form an active protein. Not all proteins have a quaternary structure, it a protein is only composed of one strand of amino acids than it won’t have a quaternary structure.
• Larger proteins are often composed of multiple subunits because its easier to repair one subunit instead of repairing the entire protein.
• Proteins with more than one subunit are called oligomers. Identical subunits are called protomers.
• Protein subunits usually associate non-covalently through hydrophobic interactions or ionic interactions. Sometimes protein subunits may be held together by disulfide bonds.
• Proteins can have rotational symmetry but not inversion or mirror symmetry.

Protein Stability • Native proteins are only marginally stable under physiological conditions and
the energy required to denature a protein is approximately 0.4 kJ per molper amino acid. So a 100 amino acid protein is only about 40 kJ per mol more stable in its native state than in its denatured state.
• Protein structures are governed primarily by hydrophobic effects.
• The hydrophobic effect causes nonpolar substances to minimize their contacts with water. The aggregation of nonpolar side chains in the center of a globular protein is an entropically driven process.
• The combined hydrophobic and hydrophilic tendencies of individual amino acid residues in proteins can be expressed as hydropathies. The greater the hydropathy the more likely it is to be on the interior of a protein and vice versa.
• Overall contribution of hydrogen bonds to protein stability is negligible since these can formed in either the native or denatured state.
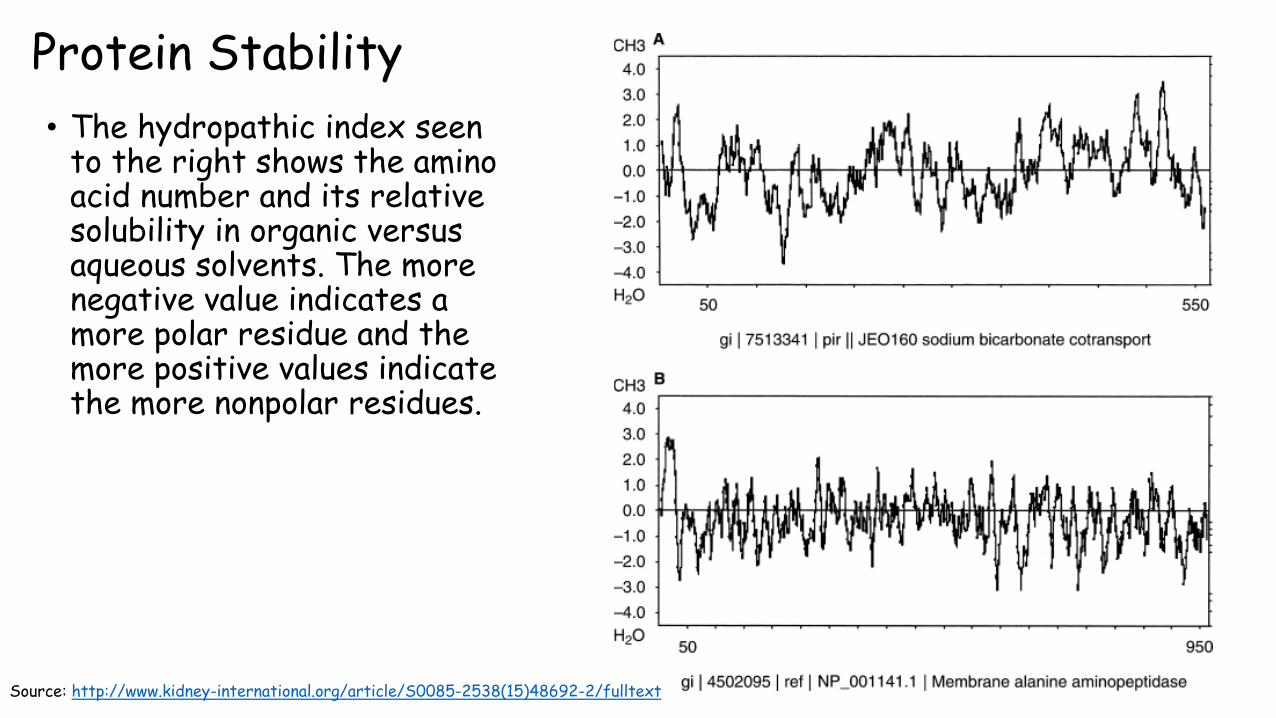
Protein Stability
• The hydropathic index seen to the right shows the amino acid number and its relative solubility in organic versus aqueous solvents. The more negative value indicates a more polar residue and the more positive values indicate the more nonpolar residues.
Source: http://www.kidney-international.org/article/S0085-2538(15)48692-2/fulltext

• While hydrogen bonds don’t contribute that much to overall stability they do slightly control tertiary structure because while a few conformations may be possible in regards to hydrophobic interactions, only one of these may allow the formation of hydrogen bonds.
• Ions pairs have little contribution to native protein stability because the free energy usually fails to compensate for the loss of entropy of the side chains.
• Disulfide bonds function to lock in a certain conformation and aren’t necessarily essential stabilizing forces. Proteins with disulfide bonds in the cytoplasm are rare because the cytoplasm is a reducing environment.
Protein Stability

Protein Stability: Zinc Fingers
• Zinc fingers are often found as nucleic acid binding proteins and are structures that contain 25-60 residues arranged around one or two Zinc ions that are tetrahedrallycoordinated by the side chains of Cys, His, and occasionally Asp or Glu.
• These proteins are too small to be stable without the zinc ion
• Zinc is the ideal metal for this because its filled d electron shell permits it to interact strongly with a variety of ligands. Zinc also only has one oxidation state and so it cant undergo oxidation-reduction reactions.
Source: https://en.wikipedia.org/wiki/Zinc_fingerhttps://en.wikibooks.org/wiki/Structural_Biochemistry/Zinc_fingers

• Proteins can be denatured by a number of methods, each with its pros and cons. • Heating: causes conformation sensitive properties to change abruptly over a narrow
range. These properties include optical rotation, viscosity and UV absorption. The unfolding is called melting and when heated this occurs cooperatively (simultaneously) across the entire polypeptide.
• Changing pH: changes charge distribution and hydrogen bonding• Detergents: associate with nonpolar residues and reduce the interactions between
residues that contribute to stability of the native conformation. • Chaotropic agents: include compounds like guanidinium ion and urea. In the range of 5-10
M these compounds increase the solubility of nonpolar compounds in water. The mechanism of action is not well understood.
• In 1957 Christian Anfinsen showed that ribonuclease A (RNase A) could be denatured and then renatured, resulting in a protein with almost 100% enzymatic activity. This indicated that proteins renature spontaneously.
Protein Stability: Denaturing and Renaturing

Protein Renaturation • Christian Anfinsen’s experiment with Ribonuclease A.
• Conclusions: proteins must renature spontaneously and proteins primary structure dictates its tertiary structure.
Source: https://kimberlybiochemist.wordpress.com/tag/anfinsen-experiment/https://biochem1362.wordpress.com/2013/03/23/anfinsen-experiment/

Protein Folding• Assume a protein randomly folds until it reaches the correct conformation.
• Assume that an n residue protein’s 2n torsion angles have three stable conformations. Then there are 10n conformations available to the protein (large underestimate due to ignoring side chains).
• If the protein could explore a new conformation every 10-13 s (a fraction of a second, a very small number) than the time required for a protein to explore all possible conformations is 10n/1013.
• For a 100 residue protein that is 1087 seconds which is longer than the age of the universe (4.3x1017 s).
• In fact protein folding is not random and follows a directed pathway. Proteins fold in less than a few seconds to their native conformation.

Protein Folding
Source: https://en.wikipedia.org/wiki/Folding_funnelhttps://www.learner.org/courses/physics/visual/visual.html?shortname=funnel

References
1. Voet, D. Voet, J.G. Pratt, C.W. Fundamentals of Biochemistry: Life at the Molecular Level. 4th edition. Wiley and Sons. 2013.















![Determining protein structure by tyrosine bioconjugation · INTRODUCTION Protein structure is driven by the interactions of the 20 amino acids with solvent and other amino acids[1]](https://img.pdfslide.us/doc/110x75/5f2af327b5a59d74a66e7b0a/determining-protein-structure-by-tyrosine-bioconjugation-introduction-protein-structure.jpg)













