Embed Size (px)
Citation preview

science/technology
PROTEIN STRUCTURE BY MASS SPEC MS is carving out a complementary role in more complicated structural analysis
Celia M. Henry C&EN Washington
M ass spectrometry is undisput-edly a valuable tool for the identification of proteins. For exam
ple, mass spectrometry of fragments from proteolytic digests can be used to search databases and identify proteins. In addition, mass spectrometry can be used to sequence unknown proteins.
Thus, the idea of using mass spec to determine a protein's primary structure is not new. But what about using MS methods for more complicated structural analysis, such as secondary and tertiary structure? The technique might not be an obvious choice, but a small and growing group of scientists is using mass spectrometry to help determine three-dimensional structure of proteins.
At the conference "Characterization of Protein Conformation by Mass Spectrometry," held in late October in Pacific Grove, Calif., scientists described their research in using mass spectrometry for structural analysis of proteins. The conference was the 16th Asilomar Conference on Mass Spectrometry, sponsored by the American Society for Mass Spectrometry, which is based in Santa Fe, N.M.
David E. Wemmer, a professor of biophysical chemistry at the University of California, Berkeley, and a structural biologist at Lawrence Berkeley National Laboratory, helped set the stage for how mass spectrometry can contribute to the field of protein structural analysis. The usual techniques for structural analysis of proteins are X-ray crystallography and nuclear magnetic resonance spectroscopy. Despite the power of these techniques, they also have their drawbacks.
"Crystallography is superb for determining the structures of well-defined parts of proteins that crystallize," Wemmer told C&EN. "A large fraction of proteins do crystallize, and a large fraction
of the amino acids in proteins that do crystallize are well ordered. But you need crystals, and you only see the well-ordered parts" of proteins.
Sometimes the view generated with crystallography is incomplete. For example, in multidomain proteins the different domains are often linked by flexi-
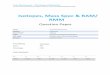
The IS distance constraints generated by chemical cross-links (dashed yellow lines) are superimposed on the average NMR structure of bovine fibroblast growth factor, FGF-2. The 14 lysines of FGF-2 are shown In red.
ble chains, which either don't crystallize or crystallize in a way that tells nothing about the interaction between the domains, Wemmer said. Or, a protein could require multiple states for its function, but only one of those states crystallizes. "In a very crude sense, what you are getting are snapshots. It's like being in a dark room where things are moving around. You flash a flashbulb, take a photograph, and see where everything is, but it's frozen."
NMR's Achilles' heel is its sensitivity. The low sensitivity of the technique means that a higher concentration of material is needed. "Not all proteins or nucleic acids of interest are water soluble at a high enough level to do the analysis by NMR," Wemmer noted. In addi
tion, NMR does not work well for high-molecular-weight proteins.
Wemmer sees a role for mass spectrometry in protein structure determination. "There are advantages of mass spec, but none of them translate conventionally into anything that approaches three-dimensional structure," Wemmer told C&EN. "You have to create a situation where in solution something happens to the protein that reflects its structure. Then you analyze it with mass spec. If you know the rules for what affects the mass spec result, you can infer structural implications from that mass measurement."
Irwin D. (Tack) Kuntz Jr., professor of pharmaceutical chemistry and director of the Molecular Design Institute at the University of California, San Francisco, de
scribed one such method. He and his collaborators have used chemical cross-linking in combination with mass spectrometry to identify the "fold family" of proteins [Proc. Natl Acad. Sci. USA, 97, 5802 (2000)]. Kuntz said there are approximately 1,500 known fold families, which are a way of classifying protein structure.
Bradford W. Gibson, a mass spec-trometrist who is professor and director of chemistry at Buck Institute, No-vato, Calif., as well as an adjunct professor of pharmaceutical chemistry at UCSF and one of Kuntz's collaborators, spoke with C&EN about the technique. The researchers use homo- and heterobifunctional cross-linkers that react with specific amino acid side chains. Two residues will be cross-linked if they are within the distance dictated by the length of the cross-linker. After cross-linking, the protein is digested, and the frag
ments are analyzed by MS. The chemical cross-linking provides
structural information, albeit at low resolution. The data reveal that two particular residues were close enough in the 3-D structure that they could be cross-linked. The real breakthrough came when the team realized that these "fuzzy" distance constraints could be used to evaluate computational models, Gibson said.
The researchers studied the bovine fibroblast growth factor, FGF-2, the structure of which has been determined by both crystallography and NMR. They generated a set of computational models by finding in a protein database the 20 best structural models based on the sequence of FGF-2. The correct fold family, as experimentally determined,
2 2 NOVEMBER 27,2000 C&EN

appeared in that list, but not at the " top. "When we used homology modeling by itself, the correct fold family was number five or six on the list," Gibson said. "When we re-rated the list based on experimental distance constraints taken from chemical cross-linking, the correct fold family moved right to the top of the list."
To avoid aberrant distance constraints, the stoichiometry of the reaction is kept low enough so there is only one cross-link per individual protein molecule. 'That's one of the analytical challenges," Gibson said. "If you keep the stoichiometry low, you're always looking for the needle in the haystack."
To improve the precision of the distance constraints, Gibson noted that the group is planning to vary the length of the spacer on the cross-linkers. The group also is contemplating using cross-linkers that have a residue-specific functional group at one end and a photoreactive group at the other. Such a cross-linker would lose specificity, but would increase the ability to probe tertiary structure because it wouldn't be based on the distribution of particular amino acids.
Although not yet borne out in practice, the combination of mass spectrometry and chemical cross-linking has the potential to be a high-throughput method of determining a low-resolution 3-D protein structure. Kuntz and his collaborators hope to use the method in drug discovery. Kuntz said that cross-linking could help prioritize ongoing projects, where early information—even if limited—would be helpful.
Physical chemists are using MS to study proteins in the gas phase. Martin F. Jarrold, a physical chemistry professor at Northwestern University, is one of those studying intrinsic gas-phase structure. He is often asked, "Why on Earth would you look at peptides in die gas phase?"
Jarrold told C&EN that his stock answer to that question is that biology happens in many different environments— in membranes as well as in aqueous solutions. "My argument has always been that the best place to start understanding a complex system like a protein is to take away all the solvent, take away everything, and understand it as an isolated system," he said.
Evan R. Williams, a chemistry professor at UC Berkeley, agrees. "One of the advantages of gas-phase measurements is that you can get rid of the solvent en-
Mass spectrometry and hydrogen-deuterium exchange revealed regions of the chap-erone GroEL that unfold quickly (red) and slowly (blue). Shown is the structure of one of the subunits of GroEL, illustrating the apical, intermediate, and equatorial domains. Reprinted from Biochemistry [39, 4250 (2000)1.
tirely. You can find out what is intrinsic to the molecule and what properties are a function of the solvent."
Jarrold has been using a combination of ion mobility, which provides a measure of molecular conformation, and mass spectrometry to determine the propensity of amino acids to form helices in the gas phase. The ordering of the amino acids is different in solution than in the gas phase.
'The hydrophobic interactions between the side chain and the solvent probably play quite a big role in determining the helix propensities in aqueous solutions," he said. "But the helix propensity in aqueous solution is really only part of the story. What about the helix propensities in membranes? Much less is known about that."
Jarrold said his group's measurements more closely match what happens in membranes. That's not surprising, he told C&EN, when the dielectric constants are considered—1 in the gas phase, about 2 in a membrane, and close to 80 in solution. "The gas phase is probably a pretty good model for what's happening in a membrane," he noted.
In the studies of helix propensities, Jarrold started with alanine and glycine. Alanine forms helices more readily than other amino acids in solution, whereas
• glycine has a low helix propensity. However, in the gas phase, Jarrold discovered that peptides needed to be designed to be helical.
This was accomplished by acety-lating the N-terminus and adding a single lysine residue to the peptides. With the acetylated N-terminus, the lysine side chain is the most basic site, so it is the site that is protonat-ed. By changing the position of the lysine residue, the peptide could be switched from a helix to a random globule.
Then they turned to valine. "We were expecting valine not to be helical," Jarrold says. "The surprise to us was that valine was actually way better than alanine at making helices in the gas phase. In solution, it's not thought of as a helix maker at all." According to Jarrold, the group has enough information to be able to say that the helix propensity scales differ in the gas phase and in the solution phase.
Gas-phase techniques also are being used to study how proteins interact with metal ions. Michael T. Bowers, a professor of chemistry at
the University of California, Santa Barbara, told C&EN that a crucial question hasn't been answered: Why do metals associate with proteins at all?
"Why don't they just stay in the solution?" Bowers asked. "They'd much rather be surrounded by water, which is a much better ion charge stabilization medium than the protein. We know they associate with the protein. The question is, why do they do it and what governs that?"
When a protein and metal ion interact, they form one of two structures. In one structure, the protein can wrap around the metal ion charge—the so-called charge solvation structure. In such a structure, the protein interacts with the charge without becoming charged itself.
In the other structure, the protein can form a salt bridge, an electrostatic interaction with alternating positive and negative charges. Salt bridges are among the strongest protein interactions in solution. They stabilize a zwitterion structure, one in which a species contains charges but has zero net charge. "If s difficult to say whether this interaction is going to stabilize or destabilize a protein," Berkeley's Williams said. "One of the advantages of gas-phase measurements is that we can actually understand the intrinsic stability of these salt bridges. Then, by adding wa-
NOVEMBER 27,2000 C&EN 23

science/technology
By using mass spectrometry to determine solvent accessibility, UC San Diego's Komives and coworkers identified the "hot spot" on thrombin that is critical for binding to thrombomodulin. This backbone ribbon diagram of thrombin shows the active site in green, the hot spot (completely solvent-excluded residues) in red, partially solvent-excluded areas in pink, and unchanged residues in dark blue. Information could not be obtained for the gray regions. A buried strand that undergoes an uninterpretable change is in light blue.
ter, we can predict whafs going to happen to the salt bridge when ifs in a protein structure."
Bowers has found that in the absence of solvent in the gas phase, zwitterion and salt bridge stability decrease with increasing peptide chain length. "Basically it means that, all things being equal, as the size of the peptide increases the more likely charge solvation is going to occur and the less likely a salt bridge is going to occur," he said. "That doesn't mean ifs always going to happen that way, because the system can be teetering on the edge of wanting to form a salt bridge."
Most of Bowers' work to date has been with alkali metals. The next step is to move to transition-metal ions, which can be important for the function of some proteins. "That's a tougher problem because transition metals don't just sit there as a charge center. They also like to react and dig in with their d orbit-als," Bowers said.
Williams' group investigated the effects of adding water on the metal-protein interaction. With one or two water molecules, valine forms a charge solvation structure. By the time six water molecules have been added, the amino acid forms a salt bridge structure, just as in bulk solution.
Using MS to probe protein structure in the solution phase is more analogous to what is done with NMR than probing the gas-phase behavior. One example of mass spectrometry being used in this way is to measure hydrogen-deuterium (H/D) exchange at amide peptide linkages. Each exchange of a hydrogen atom with a deuterium atom increases the mass by one.
David L. Smith, a chemistry professor at University of Nebraska, Lincoln, explained that H/D exchange in proteins falls into two regimes, depending on both the exchange kinetics and the protein unfolding/refolding kinetics.
If the protein refolding is much faster than the H/D exchange, the protein must unfold and refold many times before the amide linkages are exchanged. The mass spectrum would consist of a random distribution of deuterium atoms and a single envelope of isotope peaks. If the reverse were true, the protein would unfold only once, and the mass spectrum would consist of a bimodal distribution that reveals the population of folded and unfolded protein molecules.
H/D exchange is used in both MS and NMR. Smith said that mass spectrometry has several advantages for this application. It can distinguish between the two different types of exchange kinetics. In addition, it is more sensitive than NMR and can be used to analyze very large proteins. And it can be used to detect exchange at the most rapidly exchanging peptide amide hydrogens.
In contrast, the advantage of NMR for H/D exchange is that it can determine the exchange rates at individual peptide amide linkages.
"Both MS and NMR are high-tech methods," Smith told C&EN. "As a result, the choice in many cases will depend on one's area of expertise. For the analysis of relatively small, highly soluble proteins, the two methods give complementary information."
Andrew D. Robertson, a biochemist at the University of Iowa College of Medicine, said mass spectrometry coupled with H/D exchange is the only method that can identify
some aspects of protein dynamics, such as correlated motions. He explained that if amino acids participate in the same conformational fluctuation, only two masses will be observed in the mass spectrum—the unchanged mass and the mass that is shifted by one mass unit for each amino acid involved in the motion. If the motions are not correlated, then the complete set of possible masses will be observed.
At the conference, Smith described the use of H/D exchange and mass spectrometry to monitor GroELmediated folding of malate dehydrogenase (MDH). GroEL, a large assembly of 14 identical subunits that form a barrel shape, is a chaperone that helps proteins fold and unfold. The mass spectrum contains two envelopes of isotope peaks, representing unfolded and folded MDH. Smith used the populations of these two states to determine the rate constant for the folding of MDH inside GroEL. According to Smith, the presence of only two envelopes reveals that MDH folding is primarily a two-state process.
In separate work, Smith was able to monitor the folding status of GroEL, which is too large to be followed by NMR H/D exchange MS showed that the apical and intermediate domains of GroEL unfolded before the equatorial domain [Biochemistry, 39,4250 (2000)].
Elizabeth A. Komives, a biochemist
1200 1600 2000 2400 2800 3200 3600 4000 4400
The mass spectrum recorded for the collision-induced dissociation products of the intact 50S subunit of Escherichia coli ribosomes shows the charge states of both individual proteins (L) and pentameric (P) and hexameric (H) complexes of proteins from the stalk region (modified from Proc. Natl. Acad. Sci. USA [97, 5185 (2000)]). The inset structure is the cryogenic electron microscopy reconstruction of the intact E. coli ribosome oriented from the back of the 50S subunit The ribosome is shown as blue mesh with a subset of the 55 proteins shown in red and RNA in yellow.
2 4 NOVEMBER 27,2000 C&EN

at the University of California, San Diego, monitors protein-protein interactions with H/D exchange and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. She maps the protein interaction surface by finding which amide hydrogens exchange on rapidly exchanging surfaces.
Her group also has been able to determine which parts of the surface are partially blocked from the solvent and which are completely blocked. "The ones that are completely solvent excluded exchange very slowly when the protein is complexed with its partner," she said. 'The ones that are partially solvent accessible appear to continue exchanging even when the binding partner is there." The researchers verified this by raising the pH, which increases the rate of H/D exchange. The protons that were solvent inaccessible stfll had the same slow rate of exchange, but the accessible protons exchanged more rapidly.
Komives likes MALDI because it is simpler than electrospray (another ionization technique) in both experimental design (no chromatography is re
quired) and in data interpretation (peptides are only singly charged so there is only one peak per peptide). One of the disadvantages of using MALDI, Komives said, is that it yields only about 70% of the available information because it doesn't capture the less abundant peptides. "In every experimental technique you miss some of the information," Komives told C&EN. "The question is, what are you willing to tolerate in terms of missing information? Usually, we can piece together what the interface is, even when we have one or two pieces of information that are missing."
Komives used MS to study the phos-phorylated form of the protein methyl-esterase CheB. The phosphorylated state is the active form of the protein, but it can't be crystallized. This protein has a regulatory and a catalytic domain. She wanted to know how the interface between the two domains opened up. To do this, she compared the changes that happened upon phosphorylation with what happened if the regulatory domain was simply chopped off.
When the regulatory domain was re
moved, the domain interface on the catalytic domain, which had not been solvent accessible, became highly solvent accessible. When the protein was phosphorylated, two of the five peptides at the interface became more solvent accessible. Those two peptides were right at the edge of the catalytic domain.
According to Komives, a popular model for the action of this protein has been that the phosphorylation disrupts the interaction between the two domains and effectively "pops the lid." Komives* results appear to indicate that instead of popping open, the regulatory domain simply slips a little to provide access to the catalytic domain. "What we're saying is that you don't really just open it up," Komives told C&EN. "If you chop the regulatory domain off, you don't get maximal activity. It makes sense that the regulatory domain is there partially to position the substrate, but it's out of the way enough that the substrate can get in."
Carol V. Robinson, a structural biologist at the University of Oxford, uses MS to analyze megadalton complexes,
BcMa&u* .otfsW-
COME GET IT!
That's right! You can now view the most current two months
of Analytical Chemistry's "News & Features" section - FREE! Go to http://pubs.acs.org/ac, click on the "News & Features" but
ton, and you'll get instant access to....full-text feature articles,
interviews with leaders in the field, in-depth product reviews,
descriptions of new technologies, detailed listing of analytical conferences and meetings,
plus book and software reviews. Come and get the latest news
and information happening in analytical chemistry -
it's FREE!
http://pubs.acs.org/ac
NOVEMBER 27,2000 C&EN 2 5
COME SEE IT...

science/ technology
such as ribosomes and viral capsids. One of the challenges there is making sure that such huge particles survive the mass spectrometry process.
"If we weren't maintaining the complex initially, then the fact that proteins could be seen to be interacting with each other would be meaningless," she told C&EN. Speaking about the viral capsid, she said, "If we're going to make meaningful deductions about changes in the conformation, we need to prove initially that the particle remains intact and the integrity is maintained during transmission in the mass spectrometer." The particles can be maintained through careful control of the pressure inside the mass spectrometer.
Using MS, Robinson and her coworkers have been able to observe more heterogeneous samples than they have been able to crystallize. "We're looking at ribosomes that have the nascent chain attached, but of the population of ribosomes only about 20% would have this property," she said. According to Robinson, they have been able to observe posttranslational modifications of the ribosome and interactions of therapeutic agents with the ribosome.
The major challenge in applying mass spectrometry to such large complexes, Robinson said, is assigning the charge. (MS actually reveals the mass-to-charge ratio, so correctly assigning the mass of a given peak depends on knowing the charge.) She and her coworkers devised a method for iterating the charge across all the peaks in the mass spectrum. For a 2.5 million-dalton particle, the uncertainty in the mass was ±25,200 dalton \J. Am. Chem. Soc, 122, 3550(2000)].
Robinson doesn't believe mass spectrometry will compete with the detailed information available from diffraction methods. However, she thinks the combination will be powerful. "We hope to prepare samples that are more homogeneous using these methods." Her plan is to use MS as a way of selecting particular features for analysis by other methods.
So, where does mass spectrometry fit in with the other techniques used for structural analysis of proteins?
"For the foreseeable future, X-ray crystallography and NMR will continue to be the principal methods for determining high-resolution structures of proteins," Smith told C&EN. "Mass spectrometry coupled with various labeling methods will be important for
structural analyses of proteins that cannot be analyzed by X ray or NMR, and it will be important for analyzing all proteins while performing their natural functions. The latter use is possible because the labeling can be performed very quickly and under a wide range of conditions."^
General Ketone Synthesis Safe For Base-Sensitive Groups Inspired by the way nature uses metal-sulfur interactions in many biochemical processes, chemists at Emory University, Atlanta, have been searching for new synthetic organic methods based on metal-catalyzed transformations of thio-compounds. They've now discovered a unique palladium-catalyzed coupling of thiol esters with boronic acids to give ketones under mild conditions \J. Am. Chem. Soc, 122,11260 (2000)].
"Because our new reaction proceeds even at pH 4.5 (in acetic acid), the conditions are very mild and will tolerate functional groups that would not survive standard cross-coupling systems," notes chemistry professor Lanny S. liebeskind, who is also Emory's senior associate dean for science and research. His coauthor and collaborator of the past five years, senior research fellow Jiri Srogl, initiated their work on activation of sulfur-carbon bonds and metal-sulfur interactions several years ago.
The Emory team's method for carbon-carbon bond formation, says Paul Reider, vice president of process research at Merck, "opens the door to the use of dual metal-mediated reactions analogous to the elegant ways in which nature uses
Chloromethyl group survives coupling reaction
Srogl (left) and Liebeskind
O
R S "
1%Pd(dba)3
3%TFP
1.6 equivalents CuTC
R = p-tolyl dba = dibenzylideneacetone TFP = tris-2-furylphosphine CuTC = copper(l) thiophene-2-carboxylate
metal-containing enzymes. We have been so impressed by the novelty and utility of the Liebeskind chemistry that we have provided research support for its study and development Our belief is that it will provide a tool previously unavailable to the research community."
Stephen L. Buchwald, professor of chemistry at Massachusetts Institute of Technology, agrees there is a good chance chemists will adopt the method. The Emory researchers have demonstrated that it works on difficult examples, he notes, including thioesters that contain chloromethyl groups, trifluo-romethyl groups, or heterocycles with multiple nitrogen atoms.
"The reaction is mechanistically distinct from the Suzuki coupling of boronic acids and organic halides, where base is normally used," liebeskind points out. The researchers are still exploring the mechanism, but they believe the reaction proceeds through a catalytically generated acylpalladium-thiolate, which is
selectively activated for trans-metallation from boron to palladium by copper. Other applications of the protocol are coming soon, Liebeskind says.
"The coupling of boronic acids to thiol esters expands boronic acid coupling into a completely new field," says Gary Alfred, chief technology officer of Frontier Scientific, Logan, Utah. The company, which makes and sells func-tionalized boronic acids, is adding thiol esters for Liebes-kind-Srogl coupling to its product line.
Pamela Zurer
B(OH)2
*CH2CI
2 6 NOVEMBER 27,2000 C&EN