Embed Size (px)
Citation preview

Protein purification: the basics
Arvind Varsani

Reasons for protein purification
• To identify the FUNCTION of a protein
• To identify the STRUCTURE of a protein
• To use the use the purified product –INTERMIDIATE- in downstream reactions / processing
• To produce a COMMERCIAL product

Selection of protein source
• Starting material can be from
– Animal tissue– Plant material– Biological fluids (e.g. blood, milk, sera)
RECOMBINANT expression
– Fermentation cultures (yeast, fungi, bacteria)– Cell cultures (animal cells, plant cells, insect cells)

Important
• Protein in low concentration in natural sources– Need to induce expression
Or express recombinantly in various expression systems
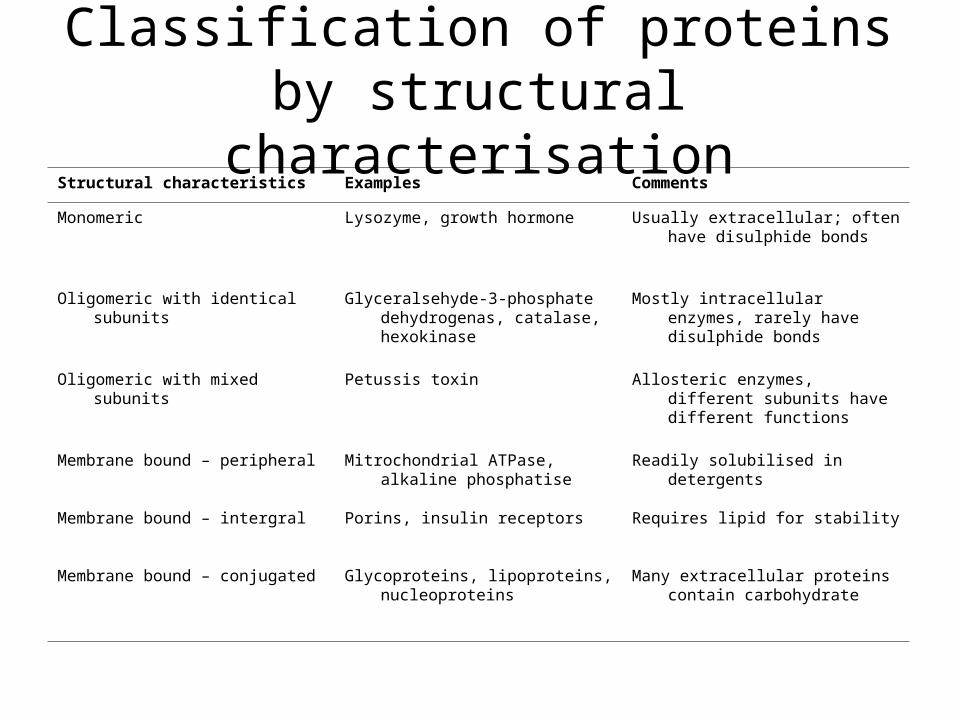
Classification of proteins by structural characterisation
Structural characteristics Examples Comments
Monomeric Lysozyme, growth hormone Usually extracellular; often have disulphide bonds
Oligomeric with identical subunits Glyceralsehyde-3-phosphate dehydrogenas, catalase, hexokinase
Mostly intracellular enzymes, rarely have disulphide bonds
Oligomeric with mixed subunits Petussis toxin Allosteric enzymes, different subunits have different functions
Membrane bound – peripheral Mitrochondrial ATPase, alkaline phosphatise
Readily solubilised in detergents
Membrane bound – intergral Porins, insulin receptors Requires lipid for stability
Membrane bound – conjugated Glycoproteins, lipoproteins, nucleoproteins
Many extracellular proteins contain carbohydrate

Classification of proteins by function
Function Examples
Amino acid storage Seed proteins (e.g. gluten), milk proteins (e.g. caesin)
Structural – inert Collagen, keratin
Structural – with activity Actin, myosin, tubulin
Binding – soluble Albumin, hemoglobin, hormones
Binding – insoluble Surface receptors (e.g. insulin receptor), antigens (e.g. viral coat proteins)
Binding – with activity Enzymes, membrane transporters (e.g. ion pumps
Rel
ativ
e ab
unda
nce

Protein properties and their effect on development of purification strategiesSample and target protein properties Influence on purification strategy
Temperature stability Need to work rapidly at lower temperatures
pH stability Selection of buffers for extraction and purification (conditions for ion exchange, affinity or reverse phase chromatography)
Organic solvents Selection of condition for reverse phase chromatography
Detergent requirements Consider effects on chromatographic steps and the need for detergent removal. consider choice of detergent
Salt (ionic strength) Selection of conditions for precipitation techniques and hydrophobic interaction chromatography
Co-factors for stability or activity Selection of additives, pH, salts and buffers
Protease sensitivity Need for fast removal of proteases or addition of inhibitors
Sensitivity to metal ions Need to add EDTA or EGTA in buffers
Redox sensitivity Need to add reducing agents
Molecular weight Selection of gel filtration media
Charge properties Selection of ion exchange conditions
Biospecific affinity Selection of ligand for affinity medium
Post translational modifications Selection of group specific affinity medium
Hydrophobicity Selection of medium for hydrophobic interaction chromatography
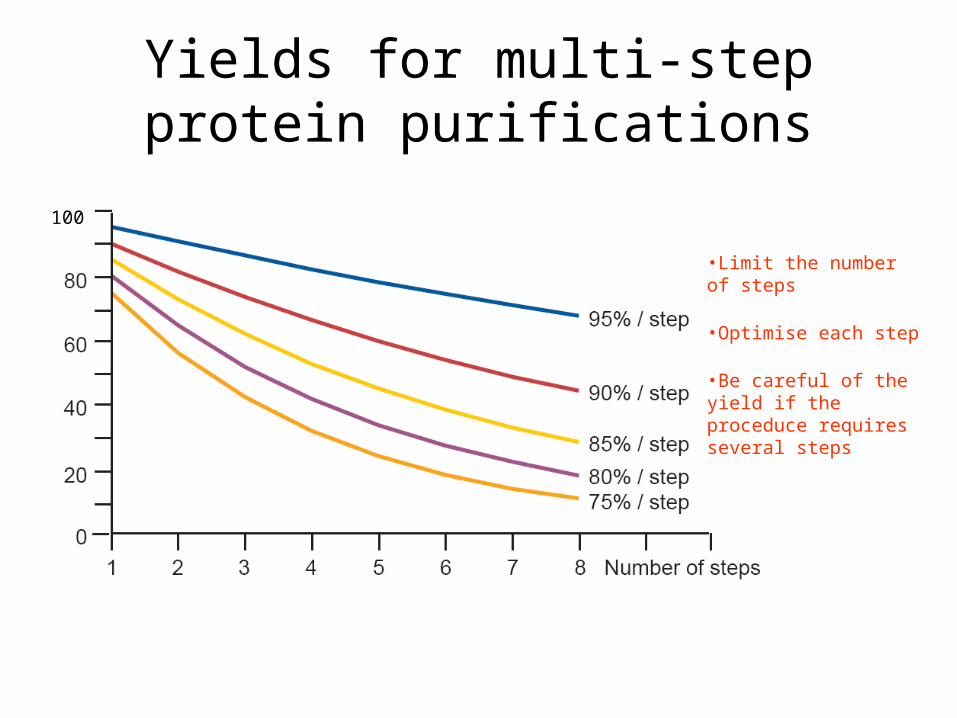
Yields for multi-step protein purifications
100
•Limit the number of steps
•Optimise each step
•Be careful of the yield if the proceduce requires several steps

Key steps in purification
• Release of target protein from starting material
• Removal of solids to leave the protein in the supernatant
• Concentration of the protein
• Removal of contaminants to achieve the desired purity
• Stabilization of the target protein

Three phase purification strategy
The final purification process should ideally consist of sample preparation, including extraction and clarification when required, followed by 3 major purifications step. The number of steps will depend on the purification strategy, purity requirements and intended use of the protein

Protein analysis
• Tracking protein of interest and determining the yield during purification– Intended use of protein / source of starting
material• Physical studies e.g. x-ray, NMR, EM• End product – pharmaceuticals

Analysis of protein purity
• Total protein
• Specific quantification– Activity assays– Binding assays
• Detection of impurities– HPLC– Gel electrophoresis
• Protein mass spectrometry

Methods for quantification of proteins in solution
Assay method Useful range Comments
NanoOrange assay 100ng/ml to 10ug/ml Samples can be read up to six hours later without any loss in the
sensitivityLow protein to protein signal variabilityDetection not influenced by reducing agents or nucleic acid
BCA method (Cu reduction)
0.5ug/ml to 1.5mg.ml Samples must be read within 10 mins
Not compatible with reducing agents
Bradford assay (dye binding)
1ug/ml to 1.5 mg/ml Protein precipitates over time
High protein to protein signal variabilityNot compatible with detergents
Lowry assay 1ug/ml to 1.5mg.ml Lengthy, multi-step procedure
Not compatible with detergents, carbohydrates or reducing agents
Absorbance at 280nm 50ng/ml to 2mg/ml High protein to protein signal variability
Detection influenced by nucleic acids and other UV absorbing contaminants

BSA assay (Bicinchoninic acid)• The first step is a Biuret reaction which reduces Cu+2 to Cu+1
• In the second step BCA forms a complex with Cu+1 which it purple colored and is detectable at 562 nm
Bradford assay (coomassie dye binding)
• Absorbance shift in Coomassie Brilliant Blue G-250 (CBBG) when bound to arginine and aromatic residues
• The anionic (bound form) has absorbance maximum at 595 nm whereas the cationic form (unbound form) has and absorbance maximum at 470 nm

Lowry assay (Cu reduction)
Monitors the absorbance of aromatic amino acids, tyrosine and tryptophan or if the wavelength is lowered, the absorbance of the peptide bond. Higher order structure in the proteins will influence the absorption
The first step is a Biuret reaction which reduces Cu+2 to Cu+1The second reaction uses Cu+1 to reduce the Folin-Ciocalteu reagent (phosphomolybdate and phosphotungstate). This is detectable in the range of 500 to 750 nm
Absorbance at 280nm

Enzyme activity assays
• Continuous (kinetic assays)– No separation step

• ELISA
• SDS

Cell disruption / breakage for protein release
• Extraction techniques are selected based on the source of protein (e.g. bacteria, plant, mammalian, intracellular or extra cellular)
• Use procedures that are as gentle as possible. Cell disruption leads to the release of proteolytic enzymes and general acidification
• Selection of an extraction technique often depends on the equipment availability and the scale of operation
• Extractions should be performed quickly, at sub-ambient temperatures in a suitable buffer to maintain pH and ionic strength
• Samples should be clear and free of particles before beginning chromatography

Cell disruption: source variations
• Tissues – variable
• Mammalian cells – easy
• Plant cells – some problems
• Microbial cells – vary, common
• Yeast and fungal cells – more difficult

Cell disruption: methods•Chemical / enzymatic
•Cell lysis (osmotic shock and freeze thaw)
•Enzymatic digestionBlood
cells
Mammalian cells
•Fractional precipitation
•Extra cellular proteins
•Mechanical
•Hand and blade homogenizers
tissue
•Sonicator / disruptors
•Grinding with abrasive
plant/yeast
•Bad beaters / mill
•French press
•micro fluidizer

Lytic enzymes and detergents
• Lysozyme: disrupts bacterial cell walls (hydrolysis of peptidoglycans) leading to cell rupture– Effective with gram positive bacteria, gram negative
generally require pre-treatment with a chelating agent such as EDTA
• Detergents: anionic and non-ionic detergents have been used to permeabilize gram negative cells. Detergents are required for the release of integral membrane proteins.

Simple shear methods
• Glass homogenizer (dounce, ten-broeck)

• sonicator • French pressure cell

Sample clarification
• Centrifugal sedimentation
• Coagulation and flocculation
• Filtration

Sedimentation
• Operates on the basis of density difference between components in a mixture (e.g. solids and liquid)
• Rate of sedimentation is dependent on:– Magnitude of different in component densities– Particle size, shape and concentration– Magnitude of centrifugal force– Flocculating of coagulating cells or organelles

Coagulation and flocculation
• Coagulation– Increase in particle size from the joining of like
particles– Promote by reducing charge repulsion
• Addition of multivalent ions (e.g. Al3+)• Adjust pH to isoelectric point
• Flocculation– increase in particle size by addition of agents acting
as bridges between particles– Generally polyelectrolytes that neutralise surface
charges on particles and then link particles to form aggregates

Concentration of extracts
• Freeze drying
• Dialysis
• PEG precipitation
• Concentration / fractionation by salting out– Ammonium sulphate precipitation
• Ultraflitration– Desalting– Size fractionation

Protein purification
• Affinity chromatography– Binding to immobilised ligands e.g antibodies, co-
factors• Ion exchange chromatography
– Anion (-) and cation (+) exchanger• Hydrophobic interaction chromatography
– Colum coated with hydrophobic fatty acid chains• Size exclusion chromatography
– Gel filtration• Electrophoresis
– SDS





























