Embed Size (px)
Citation preview

Anesthesiology, V 119 • No 6 1370 December 2013
ABSTRACT
Background: Microglial activation is implicated in delayed tissue damage after traumatic brain injury (TBI). Activation of microglia causes up-regulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, with the release of reactive oxygen species and cytotoxicity. Propofol appears to have antiinflammatory actions. The authors evaluated the neuroprotective effects of propofol after TBI and examined in vivo and in vitro whether such actions reflected modula-tion of NADPH oxidase.Methods: Adult male rats were subjected to moderate lateral fluid percussion TBI. Effect of propofol on brain microg-lial activation and functional recovery was assessed up to 28 days postinjury. By using primary microglial and BV2 cell cultures, the authors examined propofol modulation of lipo-polysaccharide and interferon-γ–induced microglial reactiv-ity and neurotoxicity.
Results: Propofol improved cognitive recovery after TBI in novel object recognition test (48 ± 6% for propofol [n = 15] vs. 30 ± 4% for isoflurane [n = 14]; P = 0.005). The functional improvement with propofol was associated with limited microglial activation and decreased cortical lesion volume and neuronal loss. Propofol also attenuated lipopolysaccha-ride- and interferon-γ–induced microglial activation in vitro, with reduced expression of inducible nitric oxide synthase, nitric oxide, tumor necrosis factor-α, interlukin-1β, reactive oxygen species, and NADPH oxidase. Microglial-induced neurotoxicity in vitro was also markedly reduced by propo-fol. The protective effect of propofol was attenuated when the NADPH oxidase subunit p22phox was knocked down by small interfering RNA. Moreover, propofol reduced the expression of p22phox and gp91phox, two key components of NADPH oxidase, after TBI.Conclusion: The neuroprotective effects of propofol after TBI appear to be mediated, in part, through the inhibition of NADPH oxidase.
What We Already Know about This Topic
• Partofthedeleteriouseffectoftraumaticbraininjuryismedi-ated bymicroglial activation and release of reactive oxygenspecies
What This Article Tells Us That Is New
• Propofolreducesfunctionaldeficitaftertraumaticbraininjuryinananimalmodel
• These neuroprotective effects aremediated in part by inhi-bition ofmicroglial nicotinamide adenine dinucleotide phos-phateoxidase
◆ ThisarticleisaccompaniedbyanEditorialView.Pleasesee:SandersRD,CoburnM,PandharipandePP:Neuraland im-muneconsequencesoftraumaticbraininjury:Doespropofolreducetheimpact?ANESThESiology2013;119:1241–3.
Copyright © 2013, the American Society of Anesthesiologists, Inc. Lippincott Williams & Wilkins. Anesthesiology 2013; 119:1370-88
* Postdoctoral Research Fellow, † Assistant Professor, ‡ Research Associate, § Postdoctoral Research Fellow, ║ Research Assistant, # David S. Brown Professor and Director of the Center for Shock Trauma and Anesthesiology Research (STAR), Department of Anes-thesiology and Center for STAR, University of Maryland School of Medicine, Baltimore, Maryland.
Received from the Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, Maryland. Submitted for publication March 15, 2013. Accepted for publication August 8, 2013. This work was supported by grants R01 NS061839 and NS037313 from National Institutes of Health, Bethesda, Maryland (to Dr. Faden). Dr. Luo received departmental salary support. The authors declare no competing interests. The first two authors con-tributed equally to this article.
Address correspondence to Dr. Faden: Department of Anesthe-siology and Center for Shock Trauma and Anesthesiology Research (STAR), University of Maryland School of Medicine, HSF II, Room S247, 20 Penn Street, Baltimore, Maryland 21201. [email protected]. Information on purchasing reprints may be found at www.anes-thesiology.org or on the masthead page at the beginning of this issue. ANeSTHeSIoLogy’s articles are made freely accessible to all readers, for personal use only, 6 months from the cover date of the issue.
Propofol Limits Microglial Activation after Experimental Brain Trauma through Inhibition of Nicotinamide Adenine Dinucleotide Phosphate Oxidase
Taoluo,M.D.,Ph.D.,*JunfangWu,B.M.,Ph.D.,†ShrutiV.Kabadi,Ph.D.,‡BorisSabirzhanov,Ph.D.,§Kelseyguanciale,B.S.,║Mariehanscom,B.S.,║JulianeFaden,B.A.,║KatherineCardiff,B.S.,║CharlesJeremyBengson,B.S.,║Alani.Faden,M.D.#
CRITICAL CARE MEDICINE
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1371 Luo et al.
CRITICAL CARE MEDICINE
M ICROGLIA are the resident macrophage-like cells in the central nervous system and play an important role
in the brain’s innate immunity and inflammatory responses.1 Although microglia have essential protective roles, activated microglia can contribute to neuronal cell death through the production of cytotoxic factors, such as nitric oxide, tumor necrosis factor-α (TNF-α), interlukin-1β, and reactive oxy-gen species (ROS).2,3
Microglial and astroglial activation is prominent after traumatic brain injury (TBI) and contribute to secondary pathophysiological changes. Both clinical and animal studies indicate that TBI causes chronic microglial activation begin-ning days after injury and continuing for months to years.4,5 These activated microglia contribute to chronic neurodegen-eration and related behavioral abnormalities after injury.4,6
Recent studies suggested that the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays a critical role in the modulation of microglial phenotype and sub-sequent inflammatory responses.7,8 The NADPH oxidase complex is composed of two membrane-bound subunits (p22phox and gp91phox), as well as four cytoplasmic sub-units (p40phox, p47phox, p67phox, and the small G-protein Rac).9 Classically activated microglia generate ROS through NADPH oxidase, which likely contributes to delayed tissue damage after TBI.10,11 Genetic or pharmacological modula-tion of NADPH oxidase can attenuate the detrimental con-sequences of microglial activation.8,12
The intravenous general anesthetic propofol is frequently used in the management of clinical head injury, including for surgical intervention, mechanical ventilation, or diag-nostic imaging. Propofol has been shown to modulate vari-ous aspects of the host’s inflammatory response.13 In animal models of endotoxemia, propofol reduced mortality rate and organ injury; promoted expression of Annexin A1, a gluco-corticoid-dependent antiinflammatory protein; and inhib-ited activation of p38 mitogen–activated protein kinases and release of inflammatory factors (interlukin-1β, interlukin-6, and TNF-α).14–16 These protective effects may therefore reflect its antiinflammatory capacity and antioxidant activ-ity.14,17 In vitro, propofol can protect BV2 microglia cells against lipopolysaccharide-induced inflammation through down-regulation of toll-like receptor 4 expression and inac-tivation of glycogen synthase kinase-3β.18 Propofol has also been shown to reduce endotoxic inflammation in vivo and in vitro by inhibiting the interconnected ROS/Akt/IκB kinase β/nuclear factor-κB signaling pathways.15 More recently, Ye et al.19 reported that propofol strongly reduced the responses of BV2 microglia cells to lipopolysaccharide.
In the current study, we examined the antiinflamma-tory effects of propofol after lateral fluid percussion TBI in rats. In parallel in vitro studies, lipopolysaccharide and interferon-γ models of microglial activation were used in primary microglial cultures and the BV2 murine microglial cell line to investigate the impact of propofol on microg-lial reactivity and neurotoxicity. We show that propofol
administration improves long-term cognitive outcomes after TBI and attenuates microglial-associated inflammation both in vitro and in vivo, at least in part, through inhibition of NADPH oxide.
Materials and Methods
Anesthesia Procedures and Surgical PreparationsAll procedures were performed under protocols approved by the University of Maryland School of Medicine Animal Care and Use Committee. Adult male Sprague–Dawley rats weighing 300–340 g (Harlan Laboratories, Indianapolis, IN) were randomly assigned to four groups: isoflurane-TBI, isoflurane-sham, propofol-TBI, or propofol-sham. The ani-mals were anesthetized in an induction chamber saturated with 4% isoflurane (Forene; Abbott Laboratories, Abbott Park, IL) in a supply gas mixture of 70% compressed air and 30% oxygen, intubated, and mechanically ventilated (Harvard Apparatus, Holliston, MA) in oxygen and air (fraction of inspired oxygen = 0.33). A temperature probe was placed into the rectum for monitoring and maintenance of temperature at 37°C on a heating pad. Catheters were inserted into the dorsal tail vein for drug administration and the ventral tail artery for arterial blood pressure monitoring, respectively. Upon completion of the endotracheal intuba-tion, propofol (PropoFlo; Abbott Laboratories) was con-tinuously infused via the tail vein (1.3 mg kg−1 min−1 after a bolus injection of 2 mg/kg) in the propofol group, whereas the rats in the isoflurane group were exposed to 2.5% (2 minimum alveolar concentration in rats). Animals with iso-flurane received saline 0.9% (0.13 ml kg−1 min−1) to match the volume of infusion in the propofol group. Propofol infu-sion at 600–650 μg kg−1 min−1 is approximately 1 minimum alveolar concentration equivalent to isoflurane.20 The time-line of the in vivo experimental design is shown in figure 1.
Rat Lateral Fluid Percussion Trauma ModelOur custom-designed refined lateral fluid percussion trauma model has been previously described in detail.21 In brief, a 5-mm craniotomy was created between the lambda and bregma sutures over the left parietal cortex where a female leur-loc was cemented in place. An isotonic saline-filled fluid
Fig.1. A timeline of the in vivo experimental design. IHC = immunohistochemistry; MWM = Morris water maze; NOR = novel object recognition; TBI = traumatic brain injury.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1372 Luo et al.
Propofol Reduces Microglial Activation after TBI
percussion device with a 5-mm tube was attached by means of a male leur-loc fitting. A brief 2.0–2.2-atm pressure pulse was given when a pendulum struck a piston at the oppo-site end of the device. This procedure results in consistent, moderate brain injury of parietal cortex as previously delin-eated.22 Sham animals received a similarly located craniot-omy but no percussion trauma.
Morris Water Maze TestsSpatial learning and memory was assessed using the acquisi-tion paradigm of the standard Morris water maze (MWM) test on postinjury days (PIDs) 14, 15, 16, and 17 as previ-ously detailed by us.23,24 The MWM protocol included hid-den platform training (acquisition) and standard probe test. A white circular pool was divided into four quadrants using the computer-based AnyMaze video tracking system (Stoelt-ing Co., Wood Dale, IL). The maze was surrounded by vari-ous distinct extra-maze cues on the walls of the room. A transparent platform was submerged 5 cm below the surface of the opaque water. Spatial learning and memory perfor-mance was assessed by determining the latency (in seconds) to locate the submerged hidden platform with a 90-s limit per trial for 4 consecutive days (PID, 14–17). Reference memory was assessed by a probe trial carried out on PID 18, as the time spent (in seconds) with a 60 s limit in the quadrant where the platform had been hidden during the acquisition phase.
Novel Object Recognition TestNovel object recognition was conducted on PID 21 to evalu-ate retention or intact memory and exploratory behavior as previously detailed by us.24–27 The apparatus consists of an open field (40 × 80 cm2) with two adjacently located imagi-nary circular zones. The zones are equally spaced from the sides in the center of the square and designated as “old object” and “novel object” zones, using the AnyMaze video tracking system (Stoelting Co., Wood Dale, IL). Two 5-min trials were performed. The first (training) trial was performed with two old objects in both zones and the second (testing) trial with one old object and one novel object present in the respective zones of the open field. There was an intertrial interval of 1 h, during which the animals were returned to their home cages. The time spent with each object was recorded manually, and the cognitive outcomes were determined as the “discrimina-tion index” for the second trial, which was calculated using the following formula: % discrimination index = Time spent in novel object zone × 100/(time spent in old object zone + time spent in novel object zone).25
Tissue Processing and ImmunohistochemistryAnimals were anesthetized with sodium pentobarbital (100 mg/kg, intraperitoneal injection) on PID 28 and trans-cardially perfused with 200 ml of 0.9% saline followed by 300 ml of 4% paraformaldehyde. The brain was removed and postfixed in 4% paraformaldehyde overnight and
cryoprotected in 30% sucrose. Coronal sections were cut and serially collected throughout the injured brain. Standard fluorescent immunohistochemistry was performed on 20 μm sections. The following primary antibodies were used: rabbit anti-Iba-1 (1:1,000; Wako Chemicals, Richmond, VA), and mouse anti-gp91phox (1:200; BD Transduction Laboratories, Franklin Lakes, NJ). Counterstaining was performed with 4’, 6-diamidino-2-phenylindole (1 μg/ml; Sigma-Aldrich, St. Louis, MO). Fluorescence microscopy was performed using a Leica (TCS SP5 II) confocal micro-scope system (Leica Microsystems Inc., Buffalo Grove, IL).
Lesion Volume and Neuronal Number AssessmentSections were stained with cresyl violet (FD NeuroTechnolo-gies, Baltimore, MD), dehydrated, and mounted for analysis (n = 6–8 per group). Lesion volume was quantified based on the Cavalieri method of unbiased stereology using Stereolo-ger 2000 program software (Systems Planning and Analysis, Alexandria, VA). The lesion volume was quantified by out-lining the missing tissue on the injured hemisphere using the Cavalieri estimator with a grid spacing of 0.1 mm. From 96 total 60-μm sections, every fourth section was analyzed beginning from a random start point.
The total number of surviving neurons was quantified in the cortex, thalamus, CornuAmmonis 1–3, and dentate gyrus subregions of the hippocampus using the optical frac-tionator method of unbiased stereology. The optical dissec-tor had a size of 50 μm by 50 μm in the x- and y-axes, respectively, with a height of 10 μm and guard zone of 4 μm from the top of the section. A grid spacing of 400 μm in the x-axis and 400 μm in the y-axis was used, resulting in an area fraction of one-sixty-fourth. The estimated number of surviving neurons in each field was divided by the volume of the region of interest to obtain the neuronal cellular density, expressed as counts/mm3.
Stereological Assessment of Cortical MicrogliaBrain sections were obtained as described above and immu-nostained for microglia marker ionized calcium-binding adapter molecule 1 (Iba-1). Stereoinvestigator software (MBF Biosciences, Williston, VT) was used to count the number of cortical microglia in each of the three microg-lial morphologic phenotypes (namely ramified, hypertro-phic, and bushy) using the optical fractionator method of unbiased stereology.4,28 The sampled region was the ipsi-lateral cortex between −1.22 mm and −2.54 mm from the bregma and dorsal to a depth of 2.0 mm from the surface. Every fourth 60-mm section was analyzed beginning from a random start point. Sections were analyzed using a Leica DM4000B microscope (Leica; Leica Microsystems Inc.). The optical dissector had a size of 50 × 50 mm2 in the x- and y-axes with a height of 10 mm and guard zone of 4 mm from the top of the section. Dissectors were positioned every 150 mm in the x- and y-axes. Microglial phenotypic classification was based on the length and thickness of the
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1373 Luo et al.
CRITICAL CARE MEDICINE
projections, the number of branches, and the size of the cell body as described previously.4,29 The volume of the region of interest was measured using the Cavalieri estimator method with a grid spacing of 150 µm for the cortex. The estimated number of microglia in each phenotypic class was divided by the volume of the region of interest to obtain cellular density expressed in counts/mm3.
Western BlotAt 7 days after injury, a 5-mm area surrounding the lesion epicenter on the ipsilateral cortex was rapidly dissected and stored at −80°C until processing. The samples were lysed in Radio-Immunoprecipitation Assay buffer and centrifuged at 15,000 rpm for 10 min at 4°C. The supernatant was removed, and protein concentration was determined using the Pierce bicinchoninic acid Protein Assay kit (Thermo Sci-entific, West Palm Beach, FL) with a bovine serum albu-min standard. Each sample contains proteins from one animal. Equal amounts of protein were electrophoretically separated on 4–12% NuPAGE Novex Bis-Tris gradient gels (Invitrogen, Grand Island, NY) and transferred to nitrocel-lulose membranes (Invitrogen). After blocking in 5% nonfat milk for 1 h at room temperature, membranes were incu-bated with respective antibodies overnight at 4°C followed by horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Pittsburgh, PA) for 1.5 h at room tempera-ture. The immunoreactivity was detected using SuperSignal West Dura Extended Duration Substrate (Thermo Scien-tific) and quantified by band densitometry of scanned films using the Gel-Pro Analyzer program (Media Cybernetics, Inc., Rockville, MD). Some blots were further stripped in a stripping buffer (Thermo Scientific) for 45 min at 55°C. The loading and blotting of equal amounts of protein were verified by reprobing the membrane with antiglyceraldehyde 3-phosphate dehydrogenase (Chemicon, Billerica, MA).
Microglial Cell CulturesPrimary microglia were cultured from the cerebral cortices of 1- to 3-day-old rat pups as described.30 In brief, the cere-bra were dissected, chopped, triturated, and plated on tissue culture flasks that had been coated with poly-D-lysine (50 ng/ml; Sigma-Aldrich). The cells were grown in Dulbecco’s Modified Eagle’s Medium/F12 (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin/streptomycin at 37°C with 5% CO2. When the cells had grown to confluence, the flasks were shaken at 100 rpm for 1 h at 37°C to isolate microglia. The immortalized murine BV2 microglial cells were grown and maintained in Dulbec-co’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 incubator.
Drug TreatmentsPropofol (12.5–200 μM; Sigma-Aldrich) reconstituted in 0.02% dimethylsulfoxide (final concentration) was applied to microglia for 1 h before lipopolysaccharide (10–50 ng/ml;
Sigma-Aldrich) or recombinant mouse interferon-γ (0.5 ng/ml; R&D Systems, Minneapolis, MN) stimulation. The γ-aminobutyric acid (GABA) A receptor antagonist picrotoxin or bicuculline (500 μM; Sigma-Aldrich) was administered 30 min before propofol administration. All drugs were pre-pared and stored according to the manufacturer’s instructions.
Nitric Oxide AssayThe nitrite in the culture supernatant was measured as an indicator of nitric oxide production using the Griess reagent assay (Invitrogen) according to the manufacturer’s instructions.
TNF-α and Interlukin-1β AssayA sandwich enzyme-linked immunosorbent assay was used for detecting the TNF-α (BD OptEIA Set; BD Biosciences, San Diego, CA) and interlukin-1β levels (Duoset kit; R&D Systems) in culture supernatant according to the manufac-turer’s instructions. Cytokine concentrations were calcu-lated using standard curves generated from recombinant TNF-α and interlukin-1β, respectively, and the results were expressed in picogram per milliliter.
Cell Viability AssayCell viability was assessed using a microculture 3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich)–based colorimetric assay. After 24 h incubation with propofol and lipopolysaccharide, 3-(4,5-dimethylthi-azol-2-yl)-2,5-diphenyltetrazolium bromide was added to cell cultures to give a final concentration of 357 μg/ml, and the samples were incubated for 2 h at 37°C in 5% CO2. The supernatant was then removed, and the formazan crystals pro-duced in viable cells were solubilized with 150 μl of dimethyl-sulfoxide (Sigma-Aldrich). Finally, the absorbance of each well was read at 570 nm using the Synergy HT multi-mode micro-plate reader (Biotek, Winooski, VT). The relative cell viability (%) was expressed as a percentage of the untreated control.
Measurement of Intracellular ROSIntracellular ROS levels were measured by 5-(and-6)-chlo-romethyl-2’,7’- dichlorodihydrofluoresceindiacetate (Invi-trogen). In brief, after 1 h of pretreatment with propofol, primary microglia were stimulated with lipopolysaccha-ride (50 ng/ml) for 24 h, and BV2 cells were stimulated with lipopolysaccharide (10 ng/ml) for 6 h. The cells were then incubated with 10 μM 5-(and-6)-chloromethyl-2’,7’- dichlorodihydrofluoresceindiacetate for 45 min at 37°C in 5% CO2. Fluorescence was measured using excitation and emission wavelengths of 485 and 528 nm, respectively. Data are presented as raw data in arbitrary units.
NADPH Oxidase Activity AssayNADPH oxidase activities were measured by a spectropho-tometric assay of cytochrome creduction.31 In brief, BV2 cells were pretreated with propofol followed by coincubation
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1374 Luo et al.
Propofol Reduces Microglial Activation after TBI
with lipopolysaccharide for 4 h. The membrane proteins were extracted from treated cells and the membrane fractions (50 μg) diluted in Dulbecco’s Modified Eagle’s Medium without phenol red was distributed in 96-well culture plates. Cyto-chrome c (100 mΜ; Sigma-Aldrich) and NADPH (100 μm; Sigma-Aldrich) were added in the presence or absence of superoxide dismutase (200 units/ml; Sigma-Aldrich) and incubated at 37°C for 30 min. Cytochrome c reduction was measured by reading absorbance at 550 nm on the micro-plate reader. The relative NADPH oxidase activity was expressed as a percentage of the untreated control.
Neurotoxicity AssayRat primary cortical neuronal cultures were derived from E18 rat cortices as previously described.32 At 24 h after stim-ulation, the medium from the primary cultured microglia was removed and the cells were grown in fresh media for 48 h. The conditioned media harvested from the stimulated microglia was added to primary cortical neurons for an additional 24 h. Cell death was measured by lactate dehy-drogenase release assay (CytoTox96TM nonradioactive cytotoxicity assay; Promega, Madison, WI) according to the manufacturer’s instructions. Data are presented as a percent-age of control-treated values.
Transient Transfection with Small Interfering RNATransfection of cells with small interfering RNA (siRNA) against p22-phox, gp91-phox, and control-siRNA (Santa Cruz Biotechnology, Inc., Dallas, TX) was performed using the Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. In brief, subconfluent BV2 cells grown in antibiotic-free medium were transfected with 1.2 pmol of siRNA per well for 96-well plate and with 50 pmol for 60-mm dish, respectively. After 6 h of transfection, cells were washed and pretreated with propofol for 1 h, stimu-lated with lipopolysaccharide, and cultured for an additional 24 h for nitric oxide and TNF-α assay. Gene knockdown was verified by Western blotting.
Statistical AnalysisData obtained from independent measurements are pre-sented as the mean ± SEM. For the acquisition trials of the
MWM test, repeated-measures one-way ANOVAs between-subjects were conducted, followed by multiple pairwise comparisons between groups using the Student–Newman–Keuls post hoc test. One-way ANOVA analysis followed by Student–Newman–Keuls post hoc test or two-tailed unpaired Student t test was performed for the other behavioral tests, Western blotting, microglial phenotype, and in vitro assays. Statistical analysis was performed using SigmaPlot Program, Version 12 (Systat Software, San Jose, CA) or GraphPad Prism software, version 4.00 for windows (GraphPad Soft-ware, Inc., San Diego, CA). Differences were considered sig-nificant at a P value of less than 0.05.
ResultsAnimal PhysiologyThere were no significant differences in body weight and duration of surgery among the four groups. The hemody-namic parameters from all groups of animals including intraoperative heart rate and mean arterial blood pressure remained ±20% of the baseline values in each study group, respectively; there were no significant differences between groups (table 1). The average values of rectal temperature and SpO2 were normal in all groups for the duration of the surgery.
Propofol Anesthesia Improves Cognitive Recovery and Reduces Lesion Size after TBITo evaluate the neuroprotective potential of propofol anes-thesia against TBI-induced cognitive functional impair-ments, rats were administrated propofol by intravenous injection, and 2.5% isoflurane anesthesia served as a con-trol. As there were no significant differences across all tests between propofol- and isoflurane-anesthetized sham ani-mals, the data shown for the sham group are combined in order to minimize the use of animals. Spatial learning and memory was tested using the acquisition phase of MWM test. Fluid percussion resulted in learning impair-ments on days 15, 16, and 17 postinjury (fig. 2A). The factors of “PIDs (F(3,152) = 33.6; P < 0.001) and groups (F(2,152) = 7.943; P < 0.001) were found to be statisti-cally significant after performing repeated-measures one-way ANOVA between-subjects. However, the interaction
Table 1. Intraoperative Physiological Parameters in Animals Anesthetized with Isoflurane or Propofol
Start Surgery Craniotomy Before LFP After LFP End of Surgery
MAP Isoflurane 88 ± 2 91 ± 1 93 ± 2 93 ± 1 91 ± 1 Propofol 86 ± 2 94 ± 2 95 ± 2 95 ± 1 92 ± 1HR Isoflurane 312 ± 8 308 ± 6 306 ± 6 301 ± 7 300 ± 8 Propofol 306 ± 8 315 ± 7 308 ± 9 302 ± 8 294 ± 10
Shown as mean ± SEM (N = 13 for isoflurane and n = 14 for propofol). Analysis was performed by repeated-measures two-way ANOVA between-subjects, followed by Student–Newman–Keuls post hoc test, P > 0.05.HR = heart rate; LFP = lateral fluid percussion; MAP = mean arterial pressure.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1375 Luo et al.
CRITICAL CARE MEDICINE
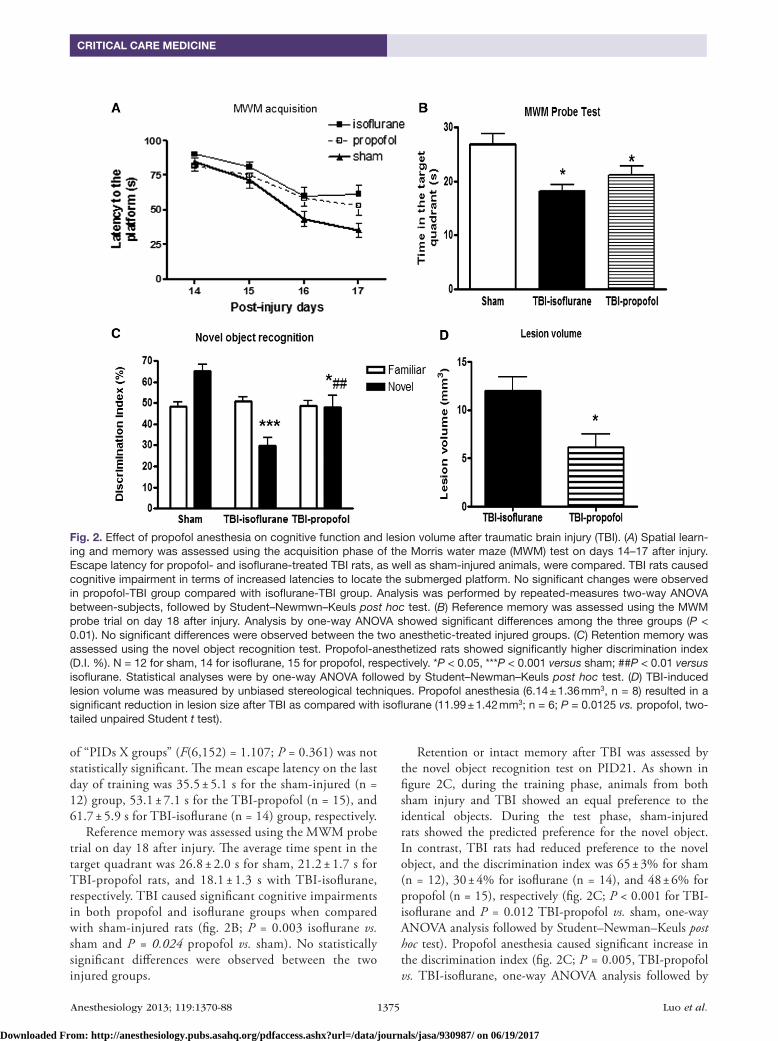
of “PIDs X groups” (F(6,152) = 1.107; P = 0.361) was not statistically significant. The mean escape latency on the last day of training was 35.5 ± 5.1 s for the sham-injured (n = 12) group, 53.1 ± 7.1 s for the TBI-propofol (n = 15), and 61.7 ± 5.9 s for TBI-isoflurane (n = 14) group, respectively.
Reference memory was assessed using the MWM probe trial on day 18 after injury. The average time spent in the target quadrant was 26.8 ± 2.0 s for sham, 21.2 ± 1.7 s for TBI-propofol rats, and 18.1 ± 1.3 s with TBI-isoflurane, respectively. TBI caused significant cognitive impairments in both propofol and isoflurane groups when compared with sham-injured rats (fig. 2B; P = 0.003 isoflurane vs. sham and P = 0.024 propofol vs. sham). No statistically significant differences were observed between the two injured groups.
Retention or intact memory after TBI was assessed by the novel object recognition test on PID21. As shown in figure 2C, during the training phase, animals from both sham injury and TBI showed an equal preference to the identical objects. During the test phase, sham-injured rats showed the predicted preference for the novel object. In contrast, TBI rats had reduced preference to the novel object, and the discrimination index was 65 ± 3% for sham (n = 12), 30 ± 4% for isoflurane (n = 14), and 48 ± 6% for propofol (n = 15), respectively (fig. 2C; P < 0.001 for TBI-isoflurane and P = 0.012 TBI-propofol vs. sham, one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test). Propofol anesthesia caused significant increase in the discrimination index (fig. 2C; P = 0.005, TBI-propofol vs. TBI-isoflurane, one-way ANOVA analysis followed by
Fig. 2. Effect of propofol anesthesia on cognitive function and lesion volume after traumatic brain injury (TBI). (A) Spatial learn-ing and memory was assessed using the acquisition phase of the Morris water maze (MWM) test on days 14–17 after injury. Escape latency for propofol- and isoflurane-treated TBI rats, as well as sham-injured animals, were compared. TBI rats caused cognitive impairment in terms of increased latencies to locate the submerged platform. No significant changes were observed in propofol-TBI group compared with isoflurane-TBI group. Analysis was performed by repeated-measures two-way ANOVA between-subjects, followed by Student–Newmwn–Keuls post hoc test. (B) Reference memory was assessed using the MWM probe trial on day 18 after injury. Analysis by one-way ANOVA showed significant differences among the three groups (P < 0.01). No significant differences were observed between the two anesthetic-treated injured groups. (C) Retention memory was assessed using the novel object recognition test. Propofol-anesthetized rats showed significantly higher discrimination index (D.I. %). N = 12 for sham, 14 for isoflurane, 15 for propofol, respectively. *P < 0.05, ***P < 0.001 versus sham; ##P < 0.01 versus isoflurane. Statistical analyses were by one-way ANOVA followed by Student–Newman–Keuls post hoc test. (D) TBI-induced lesion volume was measured by unbiased stereological techniques. Propofol anesthesia (6.14 ± 1.36 mm3, n = 8) resulted in a significant reduction in lesion size after TBI as compared with isoflurane (11.99 ± 1.42 mm3; n = 6; P = 0.0125 vs. propofol, two-tailed unpaired Student t test).
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1376 Luo et al.
Propofol Reduces Microglial Activation after TBI
Student–Newman–Keuls post hoc test), indicating improve-ment in retention memory performance.
TBI-induced lesion volume was measured by unbiased stereological techniques (fig. 2D). Histological assessment showed that propofol anesthesia (6.14 ± 1.36 mm3, n = 8) resulted in a significant reduction in lesion size after TBI as compared with isoflurane (11.99 ± 1.42 mm3; n = 6; P = 0.0125 vs. propofol, two-tailed unpaired Student t test).
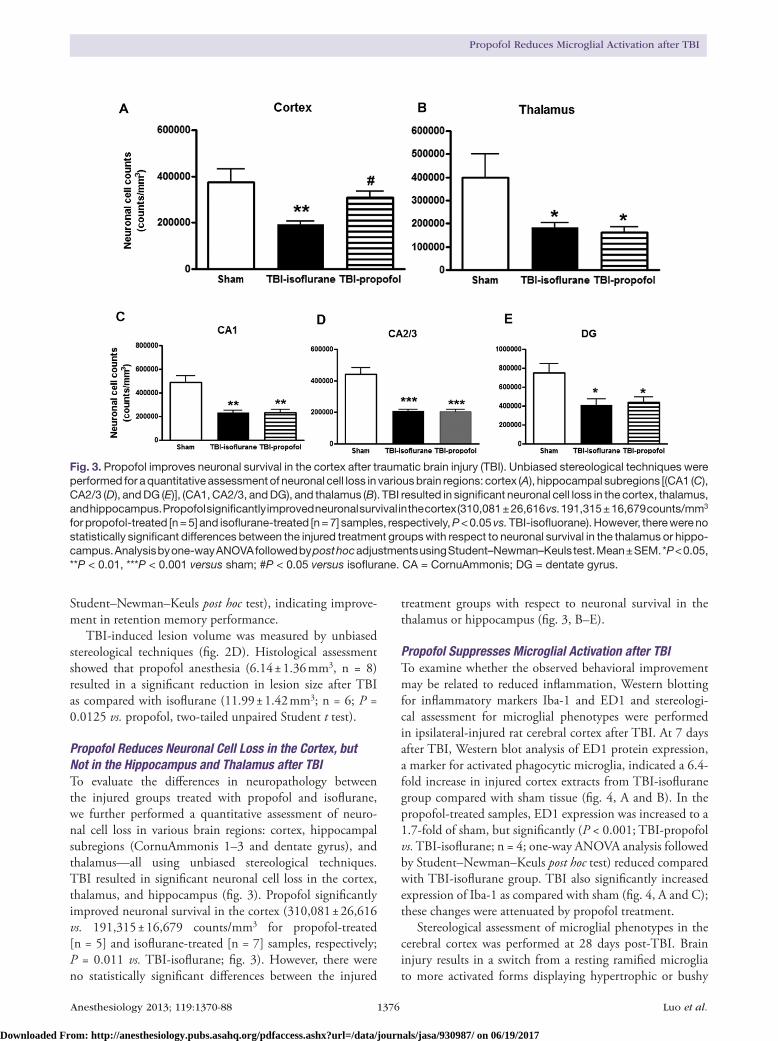
Propofol Reduces Neuronal Cell Loss in the Cortex, but Not in the Hippocampus and Thalamus after TBITo evaluate the differences in neuropathology between the injured groups treated with propofol and isoflurane, we further performed a quantitative assessment of neuro-nal cell loss in various brain regions: cortex, hippocampal subregions (CornuAmmonis 1–3 and dentate gyrus), and thalamus—all using unbiased stereological techniques. TBI resulted in significant neuronal cell loss in the cortex, thalamus, and hippocampus (fig. 3). Propofol significantly improved neuronal survival in the cortex (310,081 ± 26,616 vs. 191,315 ± 16,679 counts/mm3 for propofol-treated [n = 5] and isoflurane-treated [n = 7] samples, respectively; P = 0.011 vs. TBI-isoflurane; fig. 3). However, there were no statistically significant differences between the injured
treatment groups with respect to neuronal survival in the thalamus or hippocampus (fig. 3, B–E).
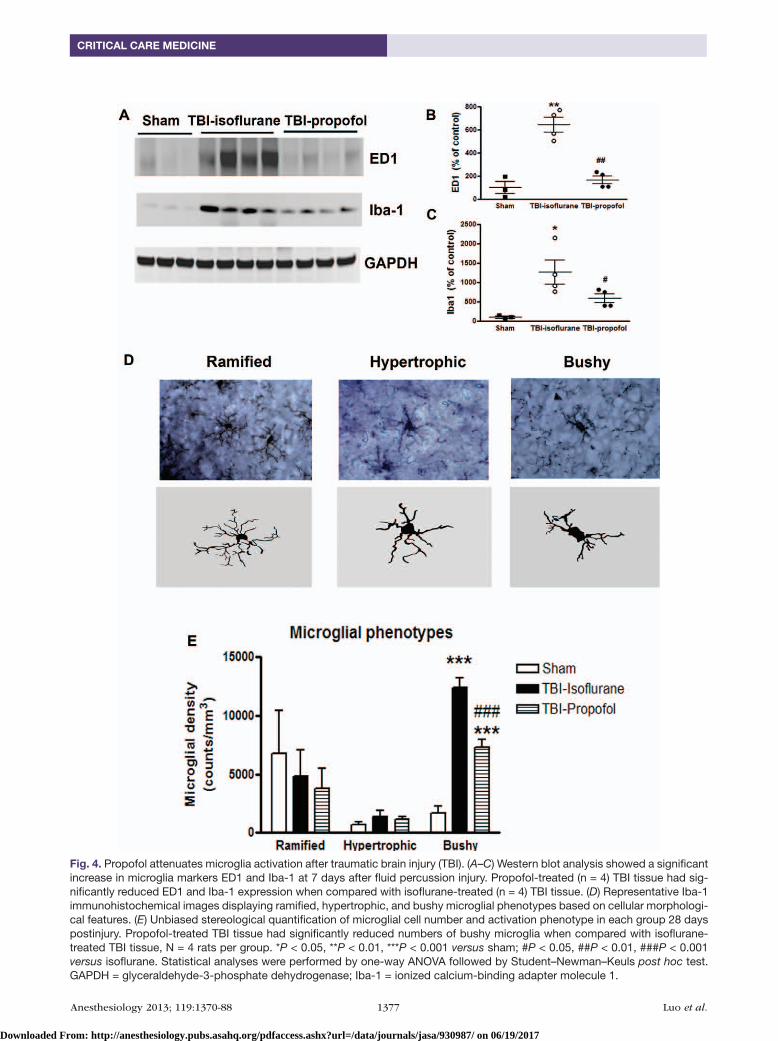
Propofol Suppresses Microglial Activation after TBITo examine whether the observed behavioral improvement may be related to reduced inflammation, Western blotting for inflammatory markers Iba-1 and ED1 and stereologi-cal assessment for microglial phenotypes were performed in ipsilateral-injured rat cerebral cortex after TBI. At 7 days after TBI, Western blot analysis of ED1 protein expression, a marker for activated phagocytic microglia, indicated a 6.4-fold increase in injured cortex extracts from TBI-isoflurane group compared with sham tissue (fig. 4, A and B). In the propofol-treated samples, ED1 expression was increased to a 1.7-fold of sham, but significantly (P < 0.001; TBI-propofol vs. TBI-isoflurane; n = 4; one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test) reduced compared with TBI-isoflurane group. TBI also significantly increased expression of Iba-1 as compared with sham (fig. 4, A and C); these changes were attenuated by propofol treatment.
Stereological assessment of microglial phenotypes in the cerebral cortex was performed at 28 days post-TBI. Brain injury results in a switch from a resting ramified microglia to more activated forms displaying hypertrophic or bushy
Fig. 3. Propofol improves neuronal survival in the cortex after traumatic brain injury (TBI). Unbiased stereological techniques were performed for a quantitative assessment of neuronal cell loss in various brain regions: cortex (A), hippocampal subregions [(CA1 (C), CA2/3 (D), and DG (E)], (CA1, CA2/3, and DG), and thalamus (B). TBI resulted in significant neuronal cell loss in the cortex, thalamus, and hippocampus. Propofol significantly improved neuronal survival in the cortex (310,081 ± 26,616 vs. 191,315 ± 16,679 counts/mm3 for propofol-treated [n = 5] and isoflurane-treated [n = 7] samples, respectively, P < 0.05 vs. TBI-isofluorane). However, there were no statistically significant differences between the injured treatment groups with respect to neuronal survival in the thalamus or hippo-campus. Analysis by one-way ANOVA followed by post hoc adjustments using Student–Newman–Keuls test. Mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; #P < 0.05 versus isoflurane. CA = CornuAmmonis; DG = dentate gyrus.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1377 Luo et al.
CRITICAL CARE MEDICINE
Fig. 4. Propofol attenuates microglia activation after traumatic brain injury (TBI). (A–C) Western blot analysis showed a significant increase in microglia markers ED1 and Iba-1 at 7 days after fluid percussion injury. Propofol-treated (n = 4) TBI tissue had sig-nificantly reduced ED1 and Iba-1 expression when compared with isoflurane-treated (n = 4) TBI tissue. (D) Representative Iba-1 immunohistochemical images displaying ramified, hypertrophic, and bushy microglial phenotypes based on cellular morphologi-cal features. (E) Unbiased stereological quantification of microglial cell number and activation phenotype in each group 28 days postinjury. Propofol-treated TBI tissue had significantly reduced numbers of bushy microglia when compared with isoflurane-treated TBI tissue, N = 4 rats per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; #P < 0.05, ##P < 0.01, ###P < 0.001 versus isoflurane. Statistical analyses were performed by one-way ANOVA followed by Student–Newman–Keuls post hoc test. GAPDH = glyceraldehyde-3-phosphate dehydrogenase; Iba-1 = ionized calcium-binding adapter molecule 1.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1378 Luo et al.
Propofol Reduces Microglial Activation after TBI
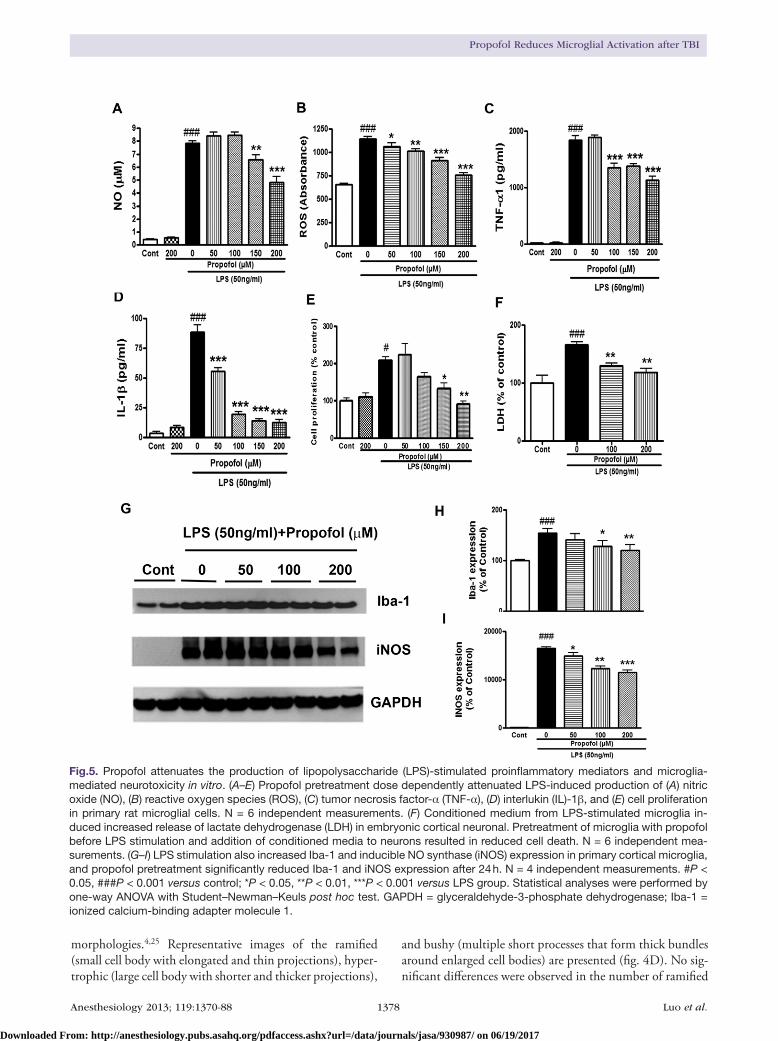
Fig.5. Propofol attenuates the production of lipopolysaccharide (LPS)-stimulated proinflammatory mediators and microglia-mediated neurotoxicity in vitro. (A–E) Propofol pretreatment dose dependently attenuated LPS-induced production of (A) nitric oxide (NO), (B) reactive oxygen species (ROS), (C) tumor necrosis factor-α (TNF-α), (D) interlukin (IL)-1β, and (E) cell proliferation in primary rat microglial cells. N = 6 independent measurements. (F) Conditioned medium from LPS-stimulated microglia in-duced increased release of lactate dehydrogenase (LDH) in embryonic cortical neuronal. Pretreatment of microglia with propofol before LPS stimulation and addition of conditioned media to neurons resulted in reduced cell death. N = 6 independent mea-surements. (G–I) LPS stimulation also increased Iba-1 and inducible NO synthase (iNOS) expression in primary cortical microglia, and propofol pretreatment significantly reduced Iba-1 and iNOS expression after 24 h. N = 4 independent measurements. #P < 0.05, ###P < 0.001 versus control; *P < 0.05, **P < 0.01, ***P < 0.001 versus LPS group. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. GAPDH = glyceraldehyde-3-phosphate dehydrogenase; Iba-1 = ionized calcium-binding adapter molecule 1.
morphologies.4,25 Representative images of the ramified (small cell body with elongated and thin projections), hyper-trophic (large cell body with shorter and thicker projections),
and bushy (multiple short processes that form thick bundles around enlarged cell bodies) are presented (fig. 4D). No sig-nificant differences were observed in the number of ramified
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1379 Luo et al.
CRITICAL CARE MEDICINE
and hypertrophic microglia across the groups. Activated microglial bushy phenotypes were significantly increased in both TBI-propofol and TBI-isoflurane animals compared with sham group. Notably, propofol significantly reduced the number of bushy microglial phenotypes compared with TBI-isoflurane group (fig. 4E; P < 0.001; propofol vs. isoflu-rane; n = 4; one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test).
Propofol Reduces Lipopolysaccharide-stimulated Primary Microglia Activation and NeurotoxicityLipopolysaccharide- or interferon-γ–induced microglia acti-vation in vitro has been extensively used as complementary tools to investigate the mechanisms underlying microglial activation in central nervous system injury. To determine whether propofol exerted a direct effect on microglia-medi-ated neuroinflammation, rat primary microglial cells were cultured in 96-well plates, and propofol was added alone or in combination with lipopolysaccharide. Pretreatment with propofol (50–200 μM) for 1 h before additional 24 h of lipo-polysaccharide (50 ng/ml) stimulation reduced the expres-sion of several independent markers of microglial activation, including ROS, nitric oxide, cell proliferation, TNF-α, and interlukin-1β (fig. 5, A–E). These effects of propofol were dose dependent, with maximal actions at 200 μM. The cell viability remained above 100% of control value with incuba-tion of propofol alone, and propofol by itself had no effect on nitric oxide, ROS, TNF-α, and interlukin-1β release.
Addition of activated microglia to neuronal cultures is known to induce neuronal cell death. In order to determine whether propofol affects microglia-induced neurotoxicity, conditioned medium prepared from lipopolysaccharide-treated microglia with or without propofol was applied to cultured cerebral cortical neurons and the neuronal cell death was assessed by determining lactate dehydrogenase release. As shown in figure 5F, when conditioned medium from lipopolysaccharide-stimulated microglia was added to cul-tured neurons, lactate dehydrogenase release was significantly increased after 24 h. However, pretreatment of microglia with propofol before lipopolysaccharide stimulation and addition of conditioned medium to neurons significantly reduced neu-ronal cell death (fig. 5F; P = 0.002 vs. lipopolysaccharide; n = 6; one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test). Neither lipopolysaccharide nor propofol had any direct effect on neuronal viability (data not shown).
Western blot analysis of Iba-1 and inducible nitric oxide in primary microglia also showed that pretreatment with propo-fol resulted in a dose-dependent reduction of Iba-1 and induc-ible nitric oxide expression (fig. 5, G–I). Thus, propofol limits the effects of lipopolysaccharide on microglia activation.
Propofol Attenuates Lipopolysaccharide– and Interferon-γ–induced Microglial Activation in BV2 CellsBV2 cells are derived from raf/myc-immortalized murine neonatal microglia and are the most frequently used
substitute for primary microglia; their response pattern parallels that of primary microglia. The antiinflammatory effects of propofol were therefore further tested in BV2 cells. Propofol, at clinically relevant concentrations, showed a significant suppression of nitric oxide, ROS, and TNF-α production induced by lipopolysaccharide 10 ng/ml (fig. 6, A–C). Furthermore, when BV2 microglia were stimulated with 0.5 ng/ml interferon-γ, pretreatment with propofol reduced in a dose-dependent manner the expression of sev-eral independent markers of microglial activation—includ-ing nitric oxide, TNF-α, and ROS (fig. 7, A–C).
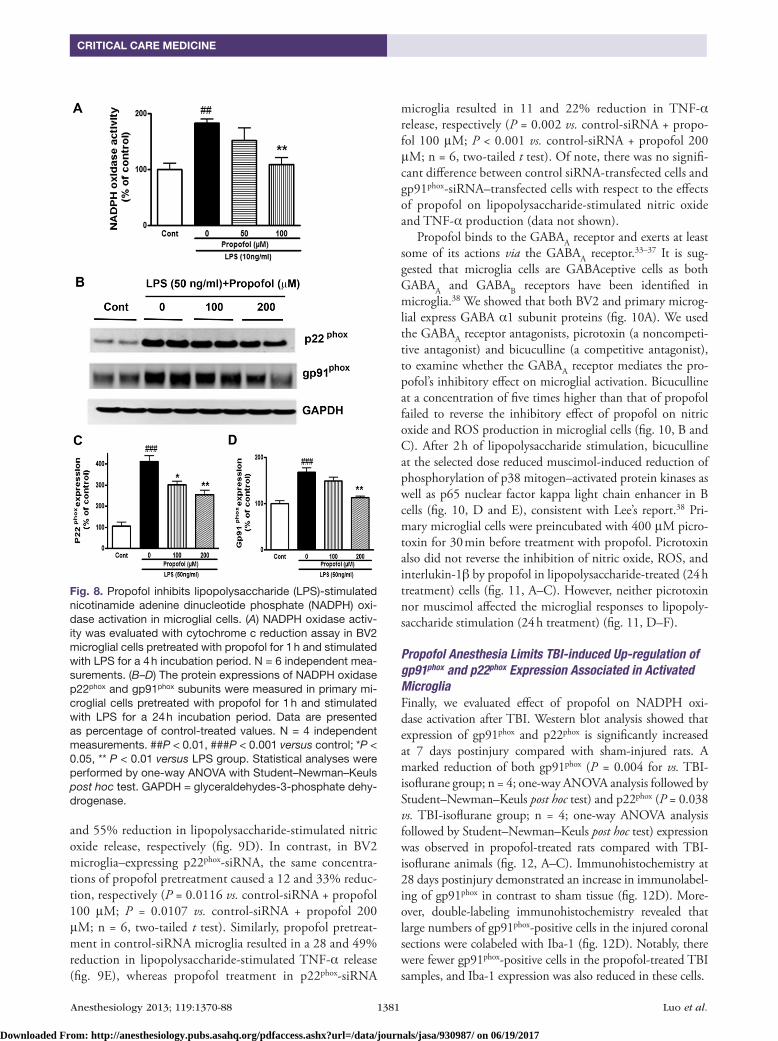
Propofol Reduces Microglial Activation via Inhibition of NADPH Oxidase Activity, but Not the GABAA ReceptorActivation of NADPH oxidase is a major mechanism for intracellular ROS production and subsequent inflamma-tory responses.7,8 To determine whether changes in NADPH oxidase activity might be related to the observed changes in proinflammatory factors by propofol, the enzymatic activ-ity of NADPH oxidase was assessed in lipopolysaccharide-stimulated BV2 microglia, with or without pretreatment with propofol. Lipopolysaccharide caused a 1.8-fold increase in NADPH oxidase activity after 4 h of stimulation com-pared with the control cells (fig. 8A),which was significantly decreased upon propofol treatment (fig. 8A; P = 0.005 vs. lipopolysaccharide; n = 6; one-way ANOVA analysis fol-lowed by Student–Newman–Keuls post hoc test).
Activation of the NADPH oxidase requires membrane translocation of the cytosolic proteins to assemble with the membrane-spanning catalytic subunit flavocytochrome b558, which is composed of gp91phox and p22phox, to form the active system. We next determined the expression of the membrane subunits of the NADPH oxidase complex, p22phox and gp91phox, after lipopolysaccharide stimulation in primary microglia. Western blot analysis showed an increase in p22phox and gp91phox at 24 h after exposure to lipopolysaccharide (fig. 8, B–D). The expression of p22phox and gp91phox was significantly reduced with propofol treatment (fig. 8, B–D), suggesting that propofol may inhibit production of proinflammatory factors in microglia through the inhibition of p22phox and gp91phox. In a parallel experiment, using BV2 cell lines, the levels of p22phox and gp91phox were also dose dependently decreased by propofol in response to lipopolysaccharide treatment (data not shown).
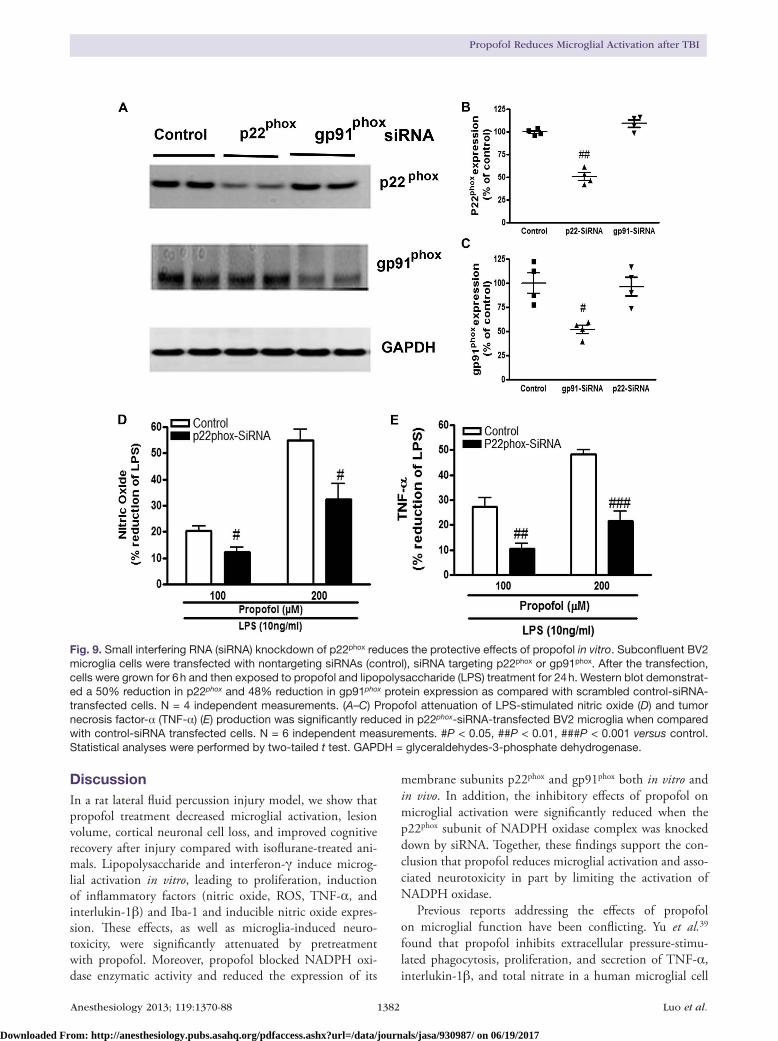
To further identify the role of individual NADPH oxi-dase components involved in propofol antiinflammation, siRNA-mediated silencing of the p22phox messenger RNA and gp91phox messenger RNA was transfected into growing BV2 cells. The expression of p22phox or gp91phox protein was reduced by 50 and 48%, respectively, when compared with control siRNA-transfected cell levels (fig. 9, A–C). Then, effect of propofol on lipopolysaccharide-stimulated nitric oxide and TNF-α production was determined in control-, p22phox-, and gp91phox-siRNA–transfected cells. In BV2 microglia that expressed the scrambled control-siRNA, pro-pofol at concentrations of 100 and 200 μM resulted in a 21
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1380 Luo et al.
Propofol Reduces Microglial Activation after TBI
Fig. 6. Propofol dose dependently attenuates lipopolysac-charide (LPS)-stimulated release of proinflammatory me-diators in BV2 microglial cells. LPS stimulation induced significant increases in the production of (A) nitric oxide (NO), (B) reactive oxygen species (ROS), and (C) tumor necrosis factor-α (TNF-α) in BV2 cells, whereas propofol pretreatment significantly attenuated LPS-stimulated NO, ROS, and TNF-α production. N = 6 independent measure-ments. ### P < 0.001 versus control, ** P < 0.01. ***P < 0.001 versus LPS group. Statistical analyses were per-formed by one-way ANOVA with Student–Newman–Keuls post hoc test.
Fig. 7. Propofol dose dependently attenuates interferon-γ (IFN-γ)-stimulated release of proinflammatory mediators in BV2 microglial cells. IFN-γ stimulation induced significant increase in the production (A) nitric oxide (NO), (B) reactive oxygen species (ROS), and (C) tumor necrosis factor-α (TNF-α) in BV2 cells, whereas propofol pretreatment significantly attenuated IFN-γ-stimulated NO, ROS, and TNF-α produc-tion. N = 6 independent measurements. ###P < 0.001 versus control, *P < 0.05, ***P < 0.001 versus IFN-γ group. Statisti-cal analyses were performed by one-way ANOVA followed by Student–Newman–Keuls post hoc test.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1381 Luo et al.
CRITICAL CARE MEDICINE
and 55% reduction in lipopolysaccharide-stimulated nitric oxide release, respectively (fig. 9D). In contrast, in BV2 microglia–expressing p22phox-siRNA, the same concentra-tions of propofol pretreatment caused a 12 and 33% reduc-tion, respectively (P = 0.0116 vs. control-siRNA + propofol 100 μM; P = 0.0107 vs. control-siRNA + propofol 200 μM; n = 6, two-tailed t test). Similarly, propofol pretreat-ment in control-siRNA microglia resulted in a 28 and 49% reduction in lipopolysaccharide-stimulated TNF-α release (fig. 9E), whereas propofol treatment in p22phox-siRNA
microglia resulted in 11 and 22% reduction in TNF-α release, respectively (P = 0.002 vs. control-siRNA + propo-fol 100 μM; P < 0.001 vs. control-siRNA + propofol 200 μM; n = 6, two-tailed t test). Of note, there was no signifi-cant difference between control siRNA-transfected cells and gp91phox-siRNA–transfected cells with respect to the effects of propofol on lipopolysaccharide-stimulated nitric oxide and TNF-α production (data not shown).
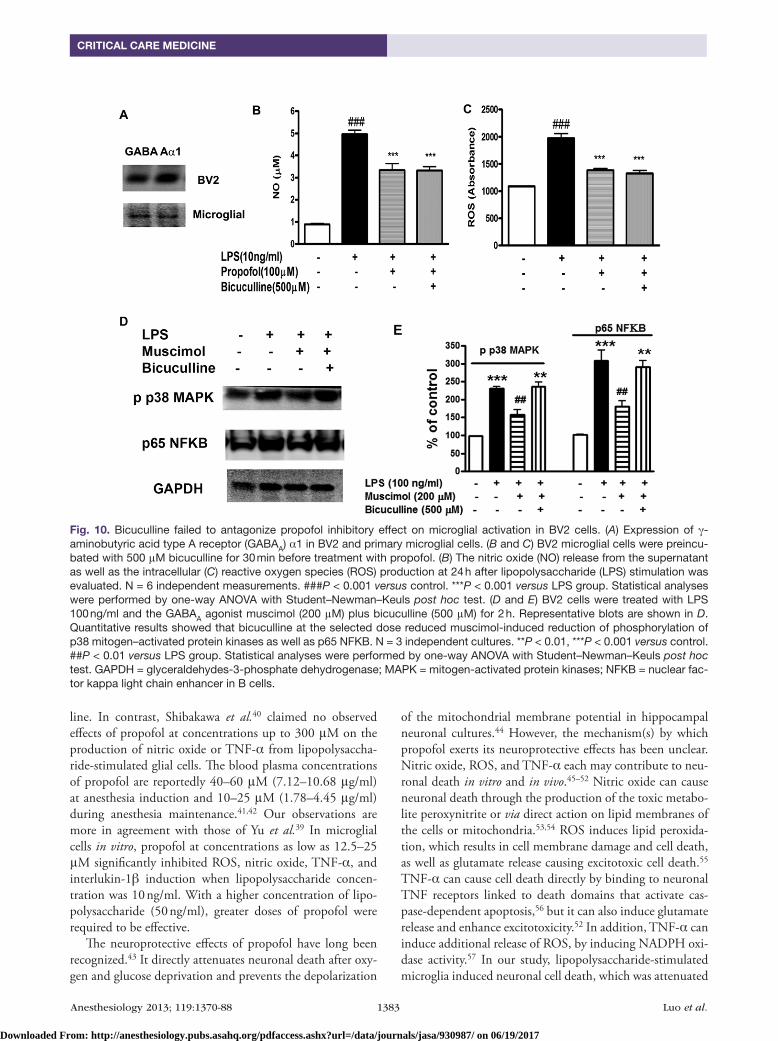
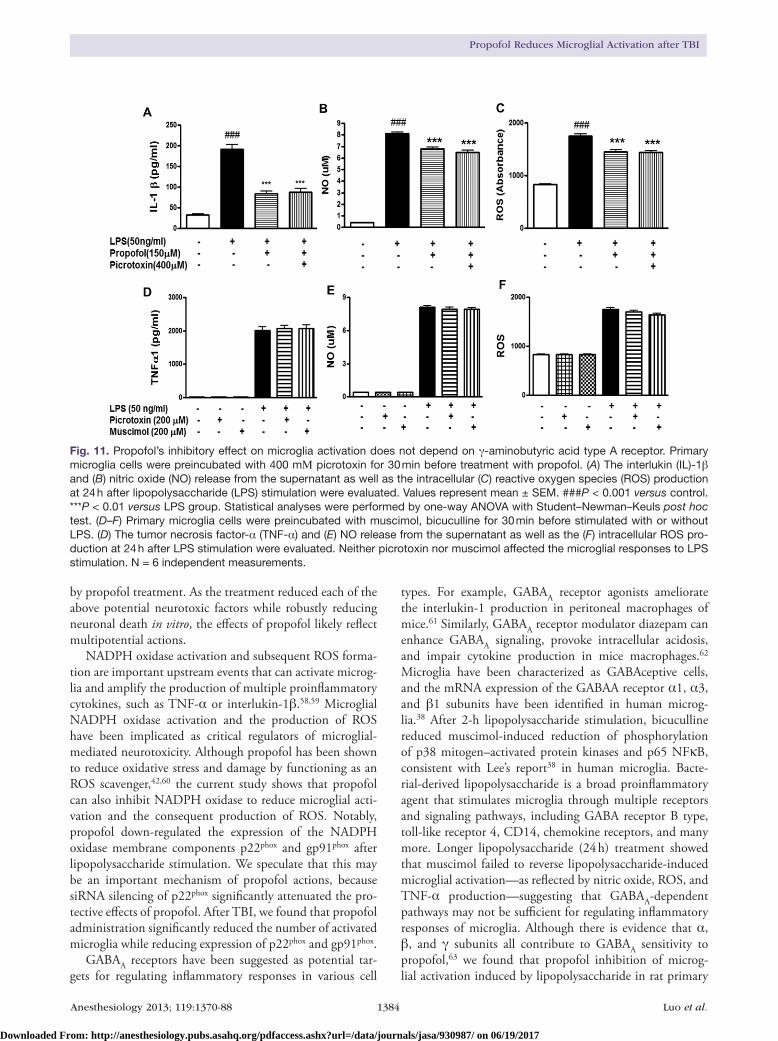
Propofol binds to the GABAA receptor and exerts at least some of its actions via the GABAA receptor.33–37 It is sug-gested that microglia cells are GABAceptive cells as both GABAA and GABAB receptors have been identified in microglia.38 We showed that both BV2 and primary microg-lial express GABA α1 subunit proteins (fig. 10A). We used the GABAA receptor antagonists, picrotoxin (a noncompeti-tive antagonist) and bicuculline (a competitive antagonist), to examine whether the GABAA receptor mediates the pro-pofol’s inhibitory effect on microglial activation. Bicuculline at a concentration of five times higher than that of propofol failed to reverse the inhibitory effect of propofol on nitric oxide and ROS production in microglial cells (fig. 10, B and C). After 2 h of lipopolysaccharide stimulation, bicuculline at the selected dose reduced muscimol-induced reduction of phosphorylation of p38 mitogen–activated protein kinases as well as p65 nuclear factor kappa light chain enhancer in B cells (fig. 10, D and E), consistent with Lee’s report.38 Pri-mary microglial cells were preincubated with 400 μM picro-toxin for 30 min before treatment with propofol. Picrotoxin also did not reverse the inhibition of nitric oxide, ROS, and interlukin-1β by propofol in lipopolysaccharide-treated (24 h treatment) cells (fig. 11, A–C). However, neither picrotoxin nor muscimol affected the microglial responses to lipopoly-saccharide stimulation (24 h treatment) (fig. 11, D–F).
Propofol Anesthesia Limits TBI-induced Up-regulation of gp91phox and p22phox Expression Associated in Activated MicrogliaFinally, we evaluated effect of propofol on NADPH oxi-dase activation after TBI. Western blot analysis showed that expression of gp91phox and p22phox is significantly increased at 7 days postinjury compared with sham-injured rats. A marked reduction of both gp91phox (P = 0.004 for vs. TBI-isoflurane group; n = 4; one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test) and p22phox (P = 0.038 vs. TBI-isoflurane group; n = 4; one-way ANOVA analysis followed by Student–Newman–Keuls post hoc test) expression was observed in propofol-treated rats compared with TBI-isoflurane animals (fig. 12, A–C). Immunohistochemistry at 28 days postinjury demonstrated an increase in immunolabel-ing of gp91phox in contrast to sham tissue (fig. 12D). More-over, double-labeling immunohistochemistry revealed that large numbers of gp91phox-positive cells in the injured coronal sections were colabeled with Iba-1 (fig. 12D). Notably, there were fewer gp91phox-positive cells in the propofol-treated TBI samples, and Iba-1 expression was also reduced in these cells.
Fig. 8. Propofol inhibits lipopolysaccharide (LPS)-stimulated nicotinamide adenine dinucleotide phosphate (NADPH) oxi-dase activation in microglial cells. (A) NADPH oxidase activ-ity was evaluated with cytochrome c reduction assay in BV2 microglial cells pretreated with propofol for 1 h and stimulated with LPS for a 4 h incubation period. N = 6 independent mea-surements. (B–D) The protein expressions of NADPH oxidase p22phox and gp91phox subunits were measured in primary mi-croglial cells pretreated with propofol for 1 h and stimulated with LPS for a 24 h incubation period. Data are presented as percentage of control-treated values. N = 4 independent measurements. ##P < 0.01, ###P < 0.001 versus control; *P < 0.05, ** P < 0.01 versus LPS group. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. GAPDH = glyceraldehydes-3-phosphate dehy-drogenase.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1382 Luo et al.
Propofol Reduces Microglial Activation after TBI
DiscussionIn a rat lateral fluid percussion injury model, we show that propofol treatment decreased microglial activation, lesion volume, cortical neuronal cell loss, and improved cognitive recovery after injury compared with isoflurane-treated ani-mals. Lipopolysaccharide and interferon-γ induce microg-lial activation in vitro, leading to proliferation, induction of inflammatory factors (nitric oxide, ROS, TNF-α, and interlukin-1β) and Iba-1 and inducible nitric oxide expres-sion. These effects, as well as microglia-induced neuro-toxicity, were significantly attenuated by pretreatment with propofol. Moreover, propofol blocked NADPH oxi-dase enzymatic activity and reduced the expression of its
membrane subunits p22phox and gp91phox both in vitro and in vivo. In addition, the inhibitory effects of propofol on microglial activation were significantly reduced when the p22phox subunit of NADPH oxidase complex was knocked down by siRNA. Together, these findings support the con-clusion that propofol reduces microglial activation and asso-ciated neurotoxicity in part by limiting the activation of NADPH oxidase.
Previous reports addressing the effects of propofol on microglial function have been conflicting. Yu et al.39 found that propofol inhibits extracellular pressure-stimu-lated phagocytosis, proliferation, and secretion of TNF-α, interlukin-1β, and total nitrate in a human microglial cell
Fig. 9. Small interfering RNA (siRNA) knockdown of p22phox reduces the protective effects of propofol in vitro. Subconfluent BV2 microglia cells were transfected with nontargeting siRNAs (control), siRNA targeting p22phox or gp91phox. After the transfection, cells were grown for 6 h and then exposed to propofol and lipopolysaccharide (LPS) treatment for 24 h. Western blot demonstrat-ed a 50% reduction in p22phox and 48% reduction in gp91phox protein expression as compared with scrambled control-siRNA-transfected cells. N = 4 independent measurements. (A–C) Propofol attenuation of LPS-stimulated nitric oxide (D) and tumor necrosis factor-α (TNF-α) (E) production was significantly reduced in p22phox-siRNA-transfected BV2 microglia when compared with control-siRNA transfected cells. N = 6 independent measurements. #P < 0.05, ##P < 0.01, ###P < 0.001 versus control. Statistical analyses were performed by two-tailed t test. GAPDH = glyceraldehydes-3-phosphate dehydrogenase.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1383 Luo et al.
CRITICAL CARE MEDICINE
line. In contrast, Shibakawa et al.40 claimed no observed effects of propofol at concentrations up to 300 μM on the production of nitric oxide or TNF-α from lipopolysaccha-ride-stimulated glial cells. The blood plasma concentrations of propofol are reportedly 40–60 μM (7.12–10.68 μg/ml) at anesthesia induction and 10–25 μM (1.78–4.45 μg/ml) during anesthesia maintenance.41,42 Our observations are more in agreement with those of Yu et al.39 In microglial cells in vitro, propofol at concentrations as low as 12.5–25 μM significantly inhibited ROS, nitric oxide, TNF-α, and interlukin-1β induction when lipopolysaccharide concen-tration was 10 ng/ml. With a higher concentration of lipo-polysaccharide (50 ng/ml), greater doses of propofol were required to be effective.
The neuroprotective effects of propofol have long been recognized.43 It directly attenuates neuronal death after oxy-gen and glucose deprivation and prevents the depolarization
of the mitochondrial membrane potential in hippocampal neuronal cultures.44 However, the mechanism(s) by which propofol exerts its neuroprotective effects has been unclear. Nitric oxide, ROS, and TNF-α each may contribute to neu-ronal death in vitro and in vivo.45–52 Nitric oxide can cause neuronal death through the production of the toxic metabo-lite peroxynitrite or via direct action on lipid membranes of the cells or mitochondria.53,54 ROS induces lipid peroxida-tion, which results in cell membrane damage and cell death, as well as glutamate release causing excitotoxic cell death.55 TNF-α can cause cell death directly by binding to neuronal TNF receptors linked to death domains that activate cas-pase-dependent apoptosis,56 but it can also induce glutamate release and enhance excitotoxicity.52 In addition, TNF-α can induce additional release of ROS, by inducing NADPH oxi-dase activity.57 In our study, lipopolysaccharide-stimulated microglia induced neuronal cell death, which was attenuated
Fig. 10. Bicuculline failed to antagonize propofol inhibitory effect on microglial activation in BV2 cells. (A) Expression of γ-aminobutyric acid type A receptor (GABAA) α1 in BV2 and primary microglial cells. (B and C) BV2 microglial cells were preincu-bated with 500 μM bicuculline for 30 min before treatment with propofol. (B) The nitric oxide (NO) release from the supernatant as well as the intracellular (C) reactive oxygen species (ROS) production at 24 h after lipopolysaccharide (LPS) stimulation was evaluated. N = 6 independent measurements. ###P < 0.001 versus control. ***P < 0.001 versus LPS group. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. (D and E) BV2 cells were treated with LPS 100 ng/ml and the GABAA agonist muscimol (200 μM) plus bicuculline (500 μM) for 2 h. Representative blots are shown in D. Quantitative results showed that bicuculline at the selected dose reduced muscimol-induced reduction of phosphorylation of p38 mitogen–activated protein kinases as well as p65 NFKB. N = 3 independent cultures. **P < 0.01, ***P < 0.001 versus control. ##P < 0.01 versus LPS group. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. GAPDH = glyceraldehydes-3-phosphate dehydrogenase; MAPK = mitogen-activated protein kinases; NFKB = nuclear fac-tor kappa light chain enhancer in B cells.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017
Anesthesiology 2013; 119:1370-88 1384 Luo et al.
Propofol Reduces Microglial Activation after TBI
by propofol treatment. As the treatment reduced each of the above potential neurotoxic factors while robustly reducing neuronal death in vitro, the effects of propofol likely reflect multipotential actions.
NADPH oxidase activation and subsequent ROS forma-tion are important upstream events that can activate microg-lia and amplify the production of multiple proinflammatory cytokines, such as TNF-α or interlukin-1β.58,59 Microglial NADPH oxidase activation and the production of ROS have been implicated as critical regulators of microglial-mediated neurotoxicity. Although propofol has been shown to reduce oxidative stress and damage by functioning as an ROS scavenger,42,60 the current study shows that propofol can also inhibit NADPH oxidase to reduce microglial acti-vation and the consequent production of ROS. Notably, propofol down-regulated the expression of the NADPH oxidase membrane components p22phox and gp91phox after lipopolysaccharide stimulation. We speculate that this may be an important mechanism of propofol actions, because siRNA silencing of p22phox significantly attenuated the pro-tective effects of propofol. After TBI, we found that propofol administration significantly reduced the number of activated microglia while reducing expression of p22phox and gp91phox.
GABAA receptors have been suggested as potential tar-gets for regulating inflammatory responses in various cell
types. For example, GABAA receptor agonists ameliorate the interlukin-1 production in peritoneal macrophages of mice.61 Similarly, GABAA receptor modulator diazepam can enhance GABAA signaling, provoke intracellular acidosis, and impair cytokine production in mice macrophages.62 Microglia have been characterized as GABAceptive cells, and the mRNA expression of the GABAA receptor α1, α3, and β1 subunits have been identified in human microg-lia.38 After 2-h lipopolysaccharide stimulation, bicuculline reduced muscimol-induced reduction of phosphorylation of p38 mitogen–activated protein kinases and p65 NFκB, consistent with Lee’s report38 in human microglia. Bacte-rial-derived lipopolysaccharide is a broad proinflammatory agent that stimulates microglia through multiple receptors and signaling pathways, including GABA receptor B type, toll-like receptor 4, CD14, chemokine receptors, and many more. Longer lipopolysaccharide (24 h) treatment showed that muscimol failed to reverse lipopolysaccharide-induced microglial activation—as reflected by nitric oxide, ROS, and TNF-α production—suggesting that GABAA-dependent pathways may not be sufficient for regulating inflammatory responses of microglia. Although there is evidence that α, β, and γ subunits all contribute to GABAA sensitivity to propofol,63 we found that propofol inhibition of microg-lial activation induced by lipopolysaccharide in rat primary
Fig. 11. Propofol’s inhibitory effect on microglia activation does not depend on γ-aminobutyric acid type A receptor. Primary microglia cells were preincubated with 400 mΜ picrotoxin for 30 min before treatment with propofol. (A) The interlukin (IL)-1β and (B) nitric oxide (NO) release from the supernatant as well as the intracellular (C) reactive oxygen species (ROS) production at 24 h after lipopolysaccharide (LPS) stimulation were evaluated. Values represent mean ± SEM. ###P < 0.001 versus control. ***P < 0.01 versus LPS group. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. (D–F) Primary microglia cells were preincubated with muscimol, bicuculline for 30 min before stimulated with or without LPS. (D) The tumor necrosis factor-α (TNF-α) and (E) NO release from the supernatant as well as the (F) intracellular ROS pro-duction at 24 h after LPS stimulation were evaluated. Neither picrotoxin nor muscimol affected the microglial responses to LPS stimulation. N = 6 independent measurements.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1385 Luo et al.
CRITICAL CARE MEDICINE
microglia, and BV2 cells was not reversed by GABAA recep-tor antagonists bicuculline or picrotoxin, and the antiinflam-matory effect of propofol could not be mimicked by GABAA
receptor agonist muscimol. A recent study from Ye et al.19 reported that propofol abolished the lipopolysaccharide proinflammatory cytokine response in BV2 cells, whereas
Fig. 12. Propofol anesthesia attenuates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase expression after traumatic brain injury (TBI). (A–C) Western blot analysis showed significant increases in membrane-bound components of the NADPH oxidase enzyme, gp91phox and p22phox at 7 days after fluid percussion injury. Propofol anesthesia attenuates NADPH oxidase expression. N = 3 in sham and n = 4 in propofol and isoflurane group, respectively. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; #P < 0.05, ##P < 0.01 versus isoflurane. Statistical analyses were performed by one-way ANOVA with Student–Newman–Keuls post hoc test. (D) Immuno-fluorescence staining showed the membrane-bound component of the NADPH oxidase enzyme, gp91phox (red), colocalized (merged) with Iba-1–positive reactive microglial (green) at 28 days post-TBI that displayed a hypertrophic or bushy cell morphology in the injured cortex of isoflurane-treated TBI samples 28 days post-TBI. Propofol-treated TBI samples have reduced gp91phox-positive reactive microglia at this time point. Bar = 100 μm. DAPI = 4′, 6-diamidino-2-phenylindole; Iba-1 = ionized calcium-binding adapter molecule 1.
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1386 Luo et al.
Propofol Reduces Microglial Activation after TBI
inhalation anesthetics including isoflurane had no effect. As GABAA receptors are known to be a common target for both propofol and isoflurane action,64 our results and those of Ye et al. are consistent and suggest that the GABA system may not be essentially involved in propofol’s antiinflammatory effect. Recent studies have also identified other receptors as potential molecular targets of propofol, including glycine, nicotinic, and M1 muscarinic receptors.65–67 Whether these receptors act as upstream regulators of microglial activation or NADPH oxidase remains to be determined.
Microglial activation and increased inflammatory cyto-kines have been implicated in the cognitive decline asso-ciated with various neurodegenerative diseases.68,69 The activation of microglial cells and neuroinflammation after TBI has been extensively studied by us and many others and has long been linked to changes in cognitive function through both direct and indirect effects on neurons.70 Two recent clinical studies, one using positron emission tomog-raphy scanning to delineate microglial activity and the other pathological, have further underscored the importance of microglial activation in chronic neurodegeneration after TBI.5,71 The effects of propopol on microglial activation were marked, as well as highly significant. The impairment in cognitive performance in the MWM task is an indica-tor of hippocampal damage and the relative contribution of various hippocampal subregions in encoding and retrieval of learning and memory can be quantified by counting neurons in these regions.25,28,72 As there were no significant differences between the two injured treatment groups with respect to TBI-induced neuronal cell loss in various hip-pocampal subregions, this is consistent with the failure of propofol to improve cognitive performance in the MWM test. In contrast, cognitive outcomes in the novel object rec-ognition test reflect both frontal cortical and hippocampal involvement.25,26,73 Propofol treatment significantly reduced the cortical lesion size and improved neuronal survival in the cortex. Therefore, the degree of cognitive improvement observed in novel object recognition task on treatment with propofol after TBI likely reflects its neuroprotective effects in the cortex. In our rat lateral fluid percussion injury model, the primary injury affects the ipsilateral cortex and adjacent penumbra.23,74 As such, the cortex shows the greatest degree of inflammation and cell death. Changes in the hippocam-pus and thalamus are more distant and show less inflamma-tory components. We have identified other approaches that target microglial activation, such as with cell cycle inhibitors that also show protective effects largely restricted to injured cortex.23,74 One major limitation of the study was the appli-cation of the general anesthetics before and during the deliv-ery of TBI, which do not ideally reflect clinical situations. But all TBI studies require anesthetic use.
A potential confounding effect in the current study with regard to group differences is that isoflurane itself may have neuroprotective effects; more specifically beneficial effects have been reported by several groups for isoflurane on
outcome after TBI.75,76 Isoflurane can also reduce lipopoly-saccharide- and interferon-γ–induced microglial activation in vitro, through actions at adenosine triphosphate potassium channels.77,78 However, recent studies have implicated neu-roinflammation and microglial activation in the pathogenesis of postoperative cognitive dysfunction,79–81 with a possible link between exposure to volatile anesthetics and exacerba-tion of neurodegenerative disorders. Interestingly, isoflurane has been associated with cognitive impairment in the absence of any surgical manipulation, whereas propofol has not.82,83 Whether propofol should be preferentially considered for sur-gery in the elderly or for the treatment of TBI is an important question that requires further studies in other model systems.
References 1. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A:
Physiology of microglia. Physiol Rev 2011; 91:461–53
2. Hanisch UK, Kettenmann H: Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007; 10:1387–94
3. Perry VH, Nicoll JA, Holmes C: Microglia in neurodegenera-tive disease. Nat Rev Neurol 2010; 6:193–201
4. Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI: Delayed mGluR5 activation limits neuroinflammation and neurode-generation after traumatic brain injury. J Neuroinflammation 2012; 9:43
5. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ: Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol 2011; 70:374–83
6. Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI: Activation of mGluR5 and inhibition of NADPH oxidase improves func-tional recovery after traumatic brain injury. J Neurotrauma 2013; 30:403–12
7. Cheret C, Gervais A, Lelli A, Colin C, Amar L, Ravassard P, Mallet J, Cumano A, Krause KH, Mallat M: Neurotoxic activa-tion of microglia is promoted by a nox1-dependent NADPH oxidase. J Neurosci 2008; 28:12039–51
8. Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F: Inhibition of NADPH oxidase promotes alternative and anti-inflam-matory microglial activation during neuroinflammation. J Neurochem 2012; 120:292–301
9. Jiang F, Zhang Y, Dusting GJ: NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tol-erance, and tissue repair. Pharmacol Rev 2011; 63:218–42
10. Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW: Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation fol-lowing traumatic brain injury. PLoS One 2012; 7:e34504
11. Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T: Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 2010; 7:41
12. Block ML: NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci 2008; 9(suppl 2):S8
13. Vanlersberghe C, Camu F: Propofol, Handb Exp Pharmacol 2008; 182:227–52
14. Hsu BG, Yang FL, Lee RP, Peng TC, Chen HI: Effects of post-treatment with low-dose propofol on inflammatory responses to lipopolysaccharide-induced shock in conscious rats. Clin Exp Pharmacol Physiol 2005; 32:24–9
15. Hsing CH, Lin MC, Choi PC, Huang WC, Kai JI, Tsai CC, Cheng YL, Hsieh CY, Wang CY, Chang YP, Chen YH, Chen CL, Lin CF: Anesthetic propofol reduces endotoxic inflammation
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1387 Luo et al.
CRITICAL CARE MEDICINE
by inhibiting reactive oxygen species-regulated Akt/IKKβ/NF-κB signaling. PLoS One 2011; 6:e17598
16. Tang J, Chen X, Tu W, Guo Y, Zhao Z, Xue Q, Lin C, Xiao J, Sun X, Tao T, Gu M, Liu Y: Propofol inhibits the activation of p38 through up-regulating the expression of annexin A1 to exert its anti-inflammation effect. PLoS One 2011; 6:e27890
17. Tsao CM, Ho ST, Liaw WJ, Chen A, Wu CC: Combined effects of propofol and dexamethasone on rats with endotoxemia. Crit Care Med 2008; 36:887–94
18. Gui B, Su M, Chen J, Jin L, Wan R, Qian Y: Neuroprotective effects of pretreatment with propofol in LPS-induced BV-2 microglia cells: Role of TLR4 and GSK-3β. Inflammation 2012; 35:1632–40
19. Ye X, Lian Q, Eckenhoff MF, Eckenhoff RG, Pan JZ: Differential general anesthetic effects on microglial cytokine expression. PLoS One 2013; 8:e52887
20. Todd MM, Weeks J: Comparative effects of propofol, pento-barbital, and isoflurane on cerebral blood flow and blood volume. J Neurosurg Anesthesiol 1996; 8:296–303
21. Kabadi SV, Hilton GD, Stoica BA, Zapple DN, Faden AI: Fluid-percussion-induced traumatic brain injury model in rats. Nat Protoc 2010; 5:1552–63
22. Faden AI, Knoblach SM, Cernak I, Fan L, Vink R, Araldi GL, Fricke ST, Roth BL, Kozikowski AP: Novel diketopiperazine enhances motor and cognitive recovery after traumatic brain injury in rats and shows neuroprotection in vitro and in vivo. J Cereb Blood Flow Metab 2003; 23:342–54
23. Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI: Cell cycle inhibition provides neuro-protection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 2005; 102:8333–8
24. Zhao Z, Loane DJ, Murray MG II, Stoica BA, Faden AI: Comparing the predictive value of multiple cognitive, affec-tive, and motor tasks after rodent traumatic brain injury. J Neurotrauma 2012; 29:2475–89
25. Kabadi SV, Stoica BA, Hanscom M, Loane DJ, Kharebava G, Murray Ii MG, Cabatbat RM, Faden AI: CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimen-tal traumatic brain injury. Neurotherapeutics 2012; 9:405–21
26. Broadbent NJ, Gaskin S, Squire LR, Clark RE: Object recogni-tion memory and the rodent hippocampus. Learn Mem 2010; 17:5–11
27. Cross L, Brown MW, Aggleton JP, Warburton EC: The medial dorsal thalamic nucleus and the medial prefrontal cortex of the rat function together to support associative recognition and recency but not item recognition. Learn Mem 2012; 20:41–50
28. Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI: Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab 2012; 32:137–49
29. Soltys Z, Ziaja M, Pawlinski R, Setkowicz Z, Janeczko K: Morphology of reactive microglia in the injured cerebral cor-tex. Fractal analysis and complementary quantitative meth-ods. J Neurosci Res 2001; 63:90–7
30. Wu J, Wrathall JR, Schachner M: Phosphatidylinositol 3-kinase/protein kinase Cdelta activation induces close homolog of adhesion molecule L1 (CHL1) expression in cul-tured astrocytes. Glia 2010; 58:315–28
31. Someya A, Nunoi H, Hasebe T, Nagaoka I: Phosphorylation of p40-phox during activation of neutrophil NADPH oxidase. J Leukoc Biol 1999; 66:851–7
32. Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, Hanscom M, Guanciale K, Faden AI: Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS One 2012; 7:e42129
33. Hara M, Kai Y, Ikemoto Y: Enhancement by propofol of the gamma-aminobutyric acidA response in dissociated
hippocampal pyramidal neurons of the rat. ANESTHESIOLOGY 1994; 81:988–94
34. Richardson JE, Garcia PS, O’Toole KK, Derry JM, Bell SV, Jenkins A: A conserved tyrosine in the beta2 subunit M4 segment is a determinant of gamma-aminobutyric acid type A receptor sensitivity to propofol. ANESTHESIOLOGY 2007; 107:412–8
35. Hollrigel GS, Toth K, Soltesz I: Neuroprotection by propofol in acute mechanical injury: Role of GABAergic inhibition. J Neurophysiol 1996; 76:2412–22
36. Ito H, Watanabe Y, Isshiki A, Uchino H: Neuroprotective prop-erties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiol Scand 1999; 43:153–62
37. Orser BA, Wang LY, Pennefather PS, MacDonald JF: Propofol modulates activation and desensitization of GABAA recep-tors in cultured murine hippocampal neurons. The Journal of neuroscience 1994; 14:7747–60
38. Lee M, Schwab C, McGeer PL: Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011; 59:152–65
39. Yu G, Dymond M, Yuan L, Chaturvedi LS, Shiratsuchi H, Durairaj S, Marsh HM, Basson MD: Propofol’s effects on phagocytosis, proliferation, nitrate production, and cytokine secretion in pressure-stimulated microglial cells. Surgery 2011; 150:887–96
40. Shibakawa YS, Sasaki Y, Goshima Y, Echigo N, Kamiya Y, Kurahashi K, Yamada Y, Andoh T: Effects of ketamine and propofol on inflammatory responses of primary glial cell cul-tures stimulated with lipopolysaccharide. Br J Anaesth 2005; 95:803–10
41. Adachi YU, Satomoto M, Higuchi H, Watanabe K: Rapid fluid infusion therapy decreases the plasma concentration of con-tinuously infused propofol. Acta Anaesthesiol Scand 2005; 49:331–6
42. Murphy PG, Davies MJ, Columb MO, Stratford N: Effect of propofol and thiopentone on free radical mediated oxidative stress of the erythrocyte. Br J Anaesth 1996; 76:536–43
43. Adembri C, Venturi L, Pellegrini-Giampietro DE: Neuroprotective effects of propofol in acute cerebral injury. CNS Drug Rev 2007; 13:333–51
44. Iijima T, Mishima T, Akagawa K, Iwao Y: Neuroprotective effect of propofol on necrosis and apoptosis following oxy-gen-glucose deprivation—Relationship between mitochon-drial membrane potential and mode of death. Brain Res 2006; 1099:25–32
45. He Y, Imam SZ, Dong Z, Jankovic J, Ali SF, Appel SH, Le W: Role of nitric oxide in rotenone-induced nigro-striatal injury. J Neurochem 2003; 86:1338–45
46. Iravani MM, Kashefi K, Mander P, Rose S, Jenner P: Involvement of inducible nitric oxide synthase in inflamma-tion-induced dopaminergic neurodegeneration. Neuroscience 2002; 110:49–58
47. Jeohn GH, Kim WG, Hong JS: Time dependency of the action of nitric oxide in lipopolysaccharide-interferon-gamma-induced neuronal cell death in murine primary neuron-glia co-cultures. Brain Res 2000; 880:173–7
48. Juurlink BH, Paterson PG: Review of oxidative stress in brain and spinal cord injury: Suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med 1998; 21:309–34
49. Lewen A, Matz P, Chan PH: Free radical pathways in CNS injury. J Neurotrauma 2000; 17:871–90
50. Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, Huttunen HJ, Founds H, Sajithlal G: Microglial activation induces cell death, inhibits neurite out-growth and causes neurite retraction of differentiated neuro-blastoma cells. Exp Brain Res 2003; 150:1–8
51. Taylor DL, Jones F, Kubota ES, Pocock JM: Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017

Anesthesiology 2013; 119:1370-88 1388 Luo et al.
Propofol Reduces Microglial Activation after TBI
tumor necrosis factor alpha-induced neurotoxicity in con-cert with microglial-derived Fas ligand. J Neuroscience 2005; 25:2952–64
52. Zou JY, Crews FT: TNF alpha potentiates glutamate neuro-toxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NF kappa B inhibition. Brain Res 2005; 1034:11–24
53. Gibbons HM, Dragunow M: Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res 2006; 1084:1–15
54. Mander P, Brown GC: Activation of microglial NADPH oxi-dase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neu-rodegeneration. J Neuroinflammation 2005; 2:20
55. Barger SW, Goodwin ME, Porter MM, Beggs ML: Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem 2007; 101:1205–13
56. Zhao X, Bausano B, Pike BR, Newcomb-Fernandez JK, Wang KK, Shohami E, Ringger NC, DeFord SM, Anderson DK, Hayes RL: TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res 2001; 64:121–31
57. Li JM, Fan LM, Christie MR, Shah AM: Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascu-lar endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol 2005; 25:2320–30
58. Pawate S, Shen Q, Fan F, Bhat NR: Redox regulation of glial inflammatory response to lipopolysaccharide and interferon-gamma. J Neurosci Res 2004; 77:540–51
59. Mander PK, Jekabsone A, Brown GC: Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol 2006; 176:1046–52
60. Thiry JC, Hans P, Deby-Dupont G, Mouythis-Mickalad A, Bonhomme V, Lamy M: Propofol scavenges reactive oxy-gen species and inhibits the protein nitration induced by activated polymorphonuclear neutrophils. Eur J Pharmacol 2004; 499:29–33
61. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, Steinman L: Inhibitory role for GABA in autoimmune inflam-mation. Proc Natl Acad Sci U S A 2010; 107:2580–5
62. Sanders RD, Godlee A, Fujimori T, Goulding J, Xin G, Salek-Ardakani S, Snelgrove RJ, Ma D, Maze M, Hussell T: Benzodiazepine augmented γ-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med 2013; 41:1627–36
63. Garcia PS, Kolesky SE, Jenkins A: General anesthetic actions on GABA(A) receptors. Curr Neuropharmacol 2010; 8:2–9
64. Franks NP: Molecular targets underlying general anaesthesia. Br J Pharmacol 2006; 147(suppl 1):S72–81
65. Nguyen HT, Li KY, daGraca RL, Delphin E, Xiong M, Ye JH: Behavior and cellular evidence for propofol-induced hypno-sis involving brain glycine receptors. ANESTHESIOLOGY 2009; 110:326–32
66. Violet JM, Downie DL, Nakisa RC, Lieb WR, Franks NP: Differential sensitivities of mammalian neuronal and mus-cle nicotinic acetylcholine receptors to general anesthetics. ANESTHESIOLOGY 1997; 86:866–74
67. Murasaki O, Kaibara M, Nagase Y, Mitarai S, Doi Y, Sumikawa K, Taniyama K: Site of action of the general anesthetic pro-pofol in muscarinic M1 receptor-mediated signal transduc-tion. J Pharmacol Exp Ther 2003; 307:995–1000
68. Biscaro B, Lindvall O, Tesco G, Ekdahl CT, Nitsch RM: Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer’s disease. Neurodegener Dis 2012; 9:187–98
69. Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L: CX3CR1 protein signaling modu-lates microglial activation and protects against plaque-inde-pendent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 2011; 286:32713–22
70. Kumar A, Loane DJ: Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav Immun 2012; 26:1191–201
71. Smith AM, Gibbons HM, Oldfield RL, Bergin PM, Mee EW, Faull RL, Dragunow M: The transcription factor PU.1 is criti-cal for viability and function of human brain microglia. Glia 2013; 61:929–42
72. Redish AD, Touretzky DS: The role of the hippocampus in solving the Morris water maze. Neural Comput 1998; 10:73–111
73. Akirav I, Maroun M: Ventromedial prefrontal cortex is obliga-tory for consolidation and reconsolidation of object recogni-tion memory. Cereb Cortex 2006; 16:1759–65
74. Cernak I, Stoica B, Byrnes KR, Di Giovanni S, Faden AI: Role of the cell cycle in the pathobiology of central nervous sys-tem trauma. Cell Cycle 2005; 4:1286–93
75. Statler KD, Alexander H, Vagni V, Dixon CE, Clark RS, Jenkins L, Kochanek PM: Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult, male rats. J Neurotrauma 2006; 23:97–108
76. Luh C, Gierth K, Timaru-Kast R, Engelhard K, Werner C, Thal SC: Influence of a brief episode of anesthesia during the induction of experimental brain trauma on secondary brain damage and inflammation. PLoS One 2011; 6:e19948
77. Xu X, Kim JA, Zuo Z: Isoflurane preconditioning reduces mouse microglial activation and injury induced by lipo-polysaccharide and interferon-gamma. Neuroscience 2008; 154:1002–8
78. Kim JA, Li L, Zuo Z: Delayed treatment with isoflurane attenu-ates lipopolysaccharide and interferon gamma-induced acti-vation and injury of mouse microglial cells. ANESTHESIOLOGY 2009; 111:566–73
79. Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M: Tumor necrosis factor-alpha triggers a cytokine cas-cade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A 2010; 107:20518–22
80. Terrando N, Eriksson LI, Ryu JK, Yang T, Monaco C, Feldmann M, Jonsson Fagerlund M, Charo IF, Akassoglou K, Maze M: Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol 2011; 70:986–95
81. Terrando N, Rei Fidalgo A, Vizcaychipi M, Cibelli M, Ma D, Monaco C, Feldmann M, Maze M: The impact of IL-1 modula-tion on the development of lipopolysaccharide-induced cog-nitive dysfunction. Crit Care 2010; 14:R88
82. Culley DJ, Baxter MG, Yukhananov R, Crosby G: Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. ANESTHESIOLOGY 2004; 100:309–14
83. Lee IH, Culley DJ, Baxter MG, Xie Z, Tanzi RE, Crosby G: Spatial memory is intact in aged rats after propofol anesthe-sia. Anesth Analg 2008; 107:1211–5
Downloaded From: http://anesthesiology.pubs.asahq.org/pdfaccess.ashx?url=/data/journals/jasa/930987/ on 06/19/2017





























