Embed Size (px)
Citation preview

1310 Vol. 36, No. 8Biol. Pharm. Bull. 36(8) 1310–1316 (2013)
© 2013 The Pharmaceutical Society of Japan
Regular Article
Promoter Methylation Profiles between Human Lung Adenocarcinoma Multidrug Resistant A549/Cisplatin (A549/DDP) Cells and Its Progenitor A549 CellsRuiling Guo,*,a Guoming Wu,b,# Haidong Li,a Pin Qian,c Juan Han,d Feng Pan,a Wenbi Li,a Jin Li,b and Fuyun Ji*,b
a Department of Respiratory Diseases, 324th Hospital of the People’s Liberation Army; d Department of Emergency Medicine, 324th Hospital of People’s Liberation Army; Chongqing 400020, China: b Institute of Human Respiratory Diseases, Xinqiao Hospital, Third Military Medical University; and c Institute of Field Internal Medicine, Xinqiao Hospital, Third Military Medical University; Chongqing 400037, China. Received February 19, 2013; accepted May 27, 2013
Although aberrant DNA methylation has been implicated in the pathophysiology of lung cancer, the role of methylation in multidrug resistance (MDR) of lung cancer has remained unclear. To investigate whether certain distinct DNA methylation pattern is associated with acquired MDR of lung adenocarcinoma, meth-ylated-DNA immunoprecipitation-chromatin immunoprecipitation (MeDIP-ChIP) was utilised to compare the genome-wide promoter methylation of the human lung adenocarcinoma MDR A549/cisplatin (A549/DDP) cells with its progenitor A549 cells. The comparison identified 3617 genes with differentially methylated promoter, of which 1581 were hypermethylated and 2036 were hypomethylated. Then, bisulphite sequenc-ing polymerase chain reaction (PCR) (BSP) and quantitative reverse transcription (RT)-PCR (Q-PCR) were used to validate the promoter methylation of five candidate genes and to determine whether the expression of genes was associated with the promoter methylation. BSP confirmed that the promoter methylation incidence of the hypermethylated genes, G protein-coupled receptor 56 isoform 3 (GPR56), metallothionein 1G (MT1G), and RAS association domain family gene 1 (RASSF1), was significantly higher in A549/DDP cells compared with A549 cells (p<0.001, p=0.0099, and p=0.0165), whereas no significant difference was found in that of the other two genes, CCNL2 and BAD (p=0.0594 and p=0.5546). Additionally, Q-PCR showed that the mRNA expression of the three hypermethylated genes was significantly lower in A549/DDP cells compared with A549 cells (all p<0.001). In conclusion, this study reported for the first time that a distinct promoter methylation pattern is associated with MDR of lung adenocarcinoma A549/DDP cells and suggested that GPR56, MT1G, and RASSF1 might be the potential methylation markers associated with acquired MDR of lung adenocarcinoma.
Key words lung adenocarcinoma; multidrug resistance; DNA methylation; promoter
Lung cancer has replaced liver cancer to become the lead-ing cause of cancer-related deaths in China, accounting for 22.7% of all cancer deaths.1) The rates of morbidity and mor-tality continue to rise rapidly and the lung cancer patients will reach one million in 2025 if no effective control measures were taken.2) Currently, lung adenocarcinoma has become the major pathologic type of lung cancer, the incidence of which accounts for 30 to 40% of all lung cancer cases in China and exceeds 50% in many areas of Europe.1–3) Chemotherapy is still an indispensable key factor in the standardised treatment of lung adenocarcinoma presently, even though rise of target therapy has been greatly improved the treatment of certain group of lung adenocarcinoma patients such as people with epidermal growth factor receptor (EGFR) mutation. However, regretfully, acquired multidrug resistance (MDR) induced by chemotherapy is the major barrier to the successful chemo-therapeutic treatment.4) Hence, understanding of the potential MDR mechanisms of lung adenocarcinoma is essential to dis-cover novel chemotherapy drugs and improve the efficacy of chemotherapy treatment.
So far, most of MDR studies have been focused on the single genes to investigate the MDR mechanism of lung ad-enocarcinoma and obtained many important results.5–8) How-
ever, these chemotherapy resistance models are demonstrated to be relatively simplistic with the generally poor clinical outcomes.9,10) Interestingly, the growing evidences of epigen-etic alterations in gene expression have provided an indication that DNA methylation status of a series of genes changed either simultaneously or sequentially might be involved in the chemotherapy-resistant phenotype of lung adenocarcinoma. First, many studies showed that DNA methylation is far more vulnerable than the DNA sequence to external factors. These alterations might be the earliest event in tumourigenesis, which could lead to a growth advantage for tumour cells and influence the direction of transformation.11) Second, DNA methylation changes can occur rapidly, which results in resis-tance arising quickly following chemotherapy treatment.12,13) Furthermore, the expression of multiple genes could be simul-taneously affected by DNA methylation.14) Therefore, DNA methylation could be the driving force in acquired MDR,15,16) which have been confirmed in resistant tumour cell line of breast adenocarcinoma,17) murine neuroblastoma cells,18) and drug-resistant ovarian and colon tumour xenografts.19) These findings gave us clue that the DNA methylation might also play important roles in the development of the MDR polygenic phenotype of lung adenocarcinoma cells.
Until now, except a limited number of genomic regions or genes found to be methylated in MDR of lung cancer,20,21) few studies have reported a genome-wide promoter methylation
* To whom correspondence should be addressed. e-mail: [email protected]; [email protected]
The authors declare no conflict of interest.# Equal contribution with first author.

August 2013 1311
analysis to investigate whether certain distinct promoter meth-ylation pattern is associated with MDR of lung adenocarcino-ma. Thus, in the present study, to investigate the relationship between DNA methylation and MDR of lung adenocarcinoma, methylated-DNA immunoprecipitation-chromatin immunopre-cipitation (MeDIP-ChIP) was utilised to compare the promoter methylation profiles of the human lung adenocarcinoma MDR A549/DDP cells with its progenitor A549 cells. Then, bisul-phite sequencing polymerase chain reaction (PCR) (BSP) was performed to validate the results obtained from MeDIP-ChIP and quantitative reverse transcription (RT)-PCR (Q-PCR) was carried out to investigate whether the expression of the hy-permethylated/hypomethylated genes was associated with the promoter methylation. The study preliminarily established a distinct DNA methylation pattern of the lung adenocarcinoma MDR cells and revealed several epigenetically inactivated genes, which might be the potential candidate genes or meth-ylation markers involved in the MDR of lung adenocarcinoma.
MATERIALS AND METHoDS
Cell Lines and Cell Culture The MDR cell line A549/DDP was established as described as Guo.22) Briefly, the pro-genitor A549 cells were first treated with a high-dose shock of cisplatin (DDP) (1.0 µg/mL) and then stepwise selected for more than 6 months with increasing concentrations of cisplat-in at a range of 0.05 to 1.0 µg/mL in RPMI-1640 medium (Hy-clone, Logan, UT, U.S.A.) with 10% foetal calf serum (Gibco, NY, U.S.A.) in a 37°C humidified incubator supplied with 5% Co2. Then, the selected cells that demonstrated cross-resis-tance to hydroxycamptothecin, vincristine, and 5-fluorouracil (MDR A549/DDP) and A549 cells were regularly maintained in RPMI-1640 medium supplemented with 10% foetal calf serum in a 37°C humidified incubator supplied with 5% CO2.
MeDIP-ChIP MeDIP was performed as previously described.23) Briefly, genomic DNA from A549/DDP cells and A549 cells were extracted using QIAamp DNA Blood Mini Kit according to the manufacturer’s recommendation (QIAGEN, Maryland, U.S.A.), respectively, and fragmented by Bioruptor (Diagenode, Belgium). Immunoprecipitation of methylated DNA was performed using anti-5-methyl cytidine (mouse) and Biomag™ magnetic beads coupled anti-mouse immunoglobulin G (IgG). After the immunoprecipitated DNA was eluted and purified by phenol chloroform extraction, Input and IP DNA were labelled with Cy5- and Cy3-labeled random 9-mers, respectively, and hybridised to NimbleGen human genome annotations (HG18) RefSeq promoter arrays (Roche NimbleGen, Madison, WI, U.S.A.), which is single array design containing all known well-characterized 18028 RefSeq promoter regions (from about −2200 to +500 bp of the transcription start sites (TSSs)) totally covered by ca. 385000 probes. Triplicate sets of hybridisation were performed from three independent MeDIP experiments for each cell line. Finally, scanning was performed using a GenePix 4000B Mi-croarray Scanner (Axon Instruments, Union City, CA, U.S.A.).
Tiling Array Data Analysis The raw data were extracted as pair files by NimbleScan software.24) Then, a modified algorithm for capturing microarray enrichment (ACME) algorithm25) is employed where a fixed-length window (750 bp) is slid along the length of each chromosome, testing at each probe using a one-sided Kolmogorov–Smirnov (KS) test
whether the surrounding window is enriched for high-intensity probes relative to the rest of the array. Each probe has a corre-sponding p-value score (−log 10) and a threshold with positive signal difference is set to select regions that are enriched (i.e., methylated) in the test sample. Thus, transfrags or differen-tially methylated regions (DMRs) were generated by interval analysis with a p-value minimum threshold of 2, maximum spacing between nearby probes within a peak of 500 bp. When we get the DMRs data, we map them with genomic transcripts and make comparison analysis between 2 samples using NimbleGen SignalMap. Annotations of RefSeq were retrieved from the NCBI website. The correlation of log 2 MeDIP/Input DNA ratios between replicates were computed using values from MaxTen calculations as described previously.26)
Promoter Methylation Analysis by BSP Genomic bisul-phite sequencing was performed to confirm the sensitivity of the observed DMRs. The genomic DNA was extracted from the A549 and A549/DDP cells using QIAamp DNA Blood Mini Kit according to the manufacturer’s recommendation (QIAGEN, Maryland, U.S.A.), respectively, and 400 ng ge-nomic DNA was treated with sodium bisulphite using EZ DNA Methylation-Gold Kit (Zymo Research, orange, CA, U.S.A.). Eighty to one hundred nanograms bisulphite-treated DNA was used for PCR amplification. The target regions of the relevant gene promoters and the primers, which were designed using the MethPrimer programme (http://www.urogene.org/methprimer/index1.html), were shown in Table 1. Then, the BSP products were cloned into a pMD®18-T vector according to the manufacturer’s instructions (TaKaRa, Dalian, China). For each cell line and each gene, five positive clones were randomly selected for subsequent sequencing. After this, the amplicon sequence data were aligned to the human refer-ence genome, and the extent of methylation (DNA methylation levels) was determined by comparing the total number of Cs (methylated) to Ts (unmethylated) for each CpG site.
Gene Expression by Q-PCR Total RNA was isolated using TRIzol according to the manufacturer’s recommenda-tion (Invitrogen, Carlsbad, CA, U.S.A.). Two milligrams aliquots were reverse transcribed using an AMV First Strand cDNA Synthesis Kit according to the manufacturer’s instruc-tions (Roche Applied Science, Mannheim, Germany). The SYBR green-based Q-PCR was then performed in triplicate using an ABI SteponePlus Real-Time PCR Systems (Applied Biosystems, Foster, CA, U.S.A.) and the the level of gene expression was normalised by glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primers for Q-PCR were listed in Table 2. PCR cycling conditions consisted of 5 min at 95°C followed by 40 cycles of 15 s of denaturation at 95°C, 30 s of annealing at 55°C and 30 s of extension at 72°C. The relative expression values were computed by the ΔΔCt method.27)
Statistical Analysis Analysis for MeDIP-ChIP was per-formed using the ACME algorithm as previously described.25) Analysis for BSP was performed using chi-square test four-fold table. The expression of the five candidate genes and GADPH in A549/DDP and A549 cells determined by Q-PCR was analysed by two-tailed Student’s t-test. p<0.05 was con-sidered statistically significant.
RESULTS
Identification of Differentially Methylated Genes in
1312 Vol. 36, No. 8
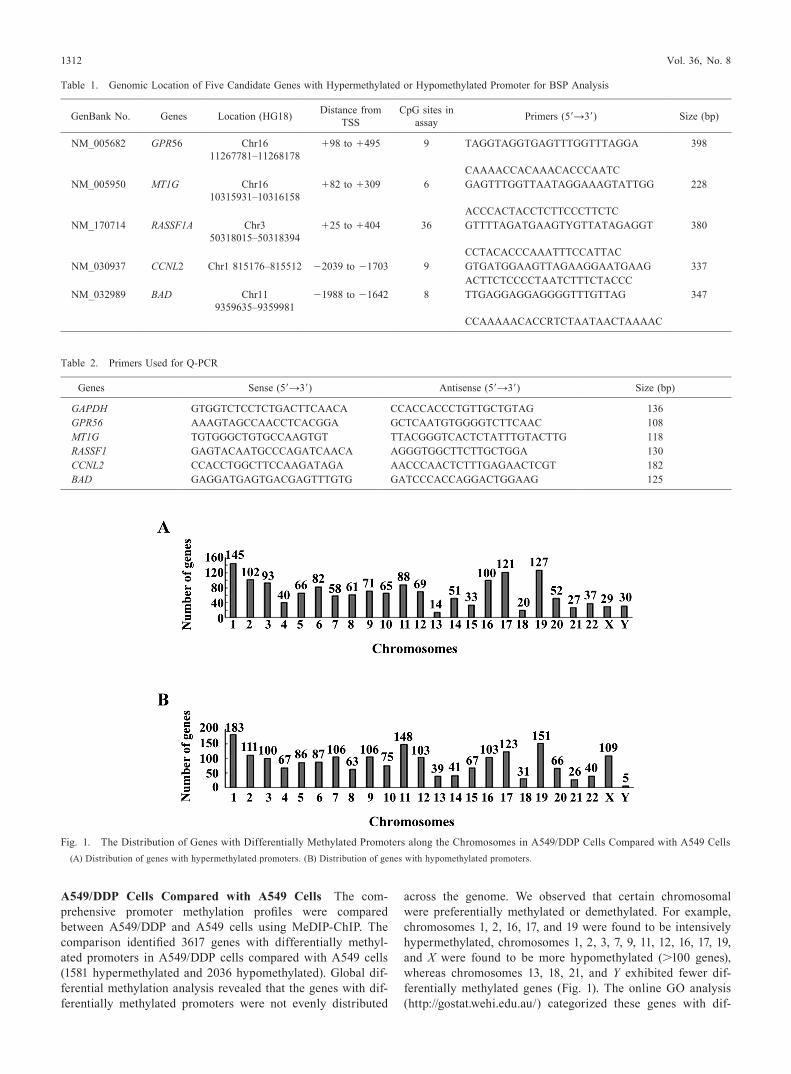
A549/DDP Cells Compared with A549 Cells The com-prehensive promoter methylation profiles were compared between A549/DDP and A549 cells using MeDIP-ChIP. The comparison identified 3617 genes with differentially methyl-ated promoters in A549/DDP cells compared with A549 cells (1581 hypermethylated and 2036 hypomethylated). Global dif-ferential methylation analysis revealed that the genes with dif-ferentially methylated promoters were not evenly distributed
across the genome. We observed that certain chromosomal were preferentially methylated or demethylated. For example, chromosomes 1, 2, 16, 17, and 19 were found to be intensively hypermethylated, chromosomes 1, 2, 3, 7, 9, 11, 12, 16, 17, 19, and X were found to be more hypomethylated (>100 genes), whereas chromosomes 13, 18, 21, and Y exhibited fewer dif-ferentially methylated genes (Fig. 1). The online Go analysis (http://gostat.wehi.edu.au/) categorized these genes with dif-
Table 1. Genomic Location of Five Candidate Genes with Hypermethylated or Hypomethylated Promoter for BSP Analysis
GenBank No. Genes Location (HG18) Distance from TSS
CpG sites in assay Primers (5′→3′) Size (bp)
NM_005682 GPR56 Chr16 11267781–11268178
+98 to +495 9 TAGGTAGGTGAGTTTGGTTTAGGA 398
CAAAACCACAAACACCCAATCNM_005950 MT1G Chr16
10315931–10316158+82 to +309 6 GAGTTTGGTTAATAGGAAAGTATTGG 228
ACCCACTACCTCTTCCCTTCTCNM_170714 RASSF1A Chr3
50318015–50318394+25 to +404 36 GTTTTAGATGAAGTYGTTATAGAGGT 380
CCTACACCCAAATTTCCATTACNM_030937 CCNL2 Chr1 815176–815512 −2039 to −1703 9 GTGATGGAAGTTAGAAGGAATGAAG 337
ACTTCTCCCCTAATCTTTCTACCCNM_032989 BAD Chr11
9359635–9359981−1988 to −1642 8 TTGAGGAGGAGGGGTTTGTTAG 347
CCAAAAACACCRTCTAATAACTAAAAC
Table 2. Primers Used for Q-PCR
Genes Sense (5′→3′) Antisense (5′→3′) Size (bp)
GAPDH GTGGTCTCCTCTGACTTCAACA CCACCACCCTGTTGCTGTAG 136GPR56 AAAGTAGCCAACCTCACGGA GCTCAATGTGGGGTCTTCAAC 108MT1G TGTGGGCTGTGCCAAGTGT TTACGGGTCACTCTATTTGTACTTG 118RASSF1 GAGTACAATGCCCAGATCAACA AGGGTGGCTTCTTGCTGGA 130CCNL2 CCACCTGGCTTCCAAGATAGA AACCCAACTCTTTGAGAACTCGT 182BAD GAGGATGAGTGACGAGTTTGTG GATCCCACCAGGACTGGAAG 125
Fig. 1. The Distribution of Genes with Differentially Methylated Promoters along the Chromosomes in A549/DDP Cells Compared with A549 Cells(A) Distribution of genes with hypermethylated promoters. (B) Distribution of genes with hypomethylated promoters.

August 2013 1313
ferentially methylated promoters into different biological func-tions, including transcription factor activity, plasma membrane part, sequence-specific DNA binding, transcription regulator activity, intrinsic to plasma membrane, ion transport, integral to plasma membrane, embryonic morphogenesis, gastrulation (Bonferroni p<0.05), and a series of signalling pathways with p<0.05, including Wnt signalling pathway, tight junction, ad-herens junction, the TGF-beta signalling pathway, and others (Bonferroni p<0.05) (Fig. 2, Tables 3–4, supplementary Table 1–2). However, no significantly methylated promoters of some previously reported MDR genes such as MDR-1, LPR, MRP of
lung cancer were detected in the present study.Validation of the Promoter Methylation in Five Can-
didate Genes To validate the results obtained from the MeDIP-ChIP analysis, five differentially methylated promot-er-associated genes, three of which were hypermethylated (GPR56, MT1G, and RASSF1) and two of which were hypo-methylated (CCNL2 and BAD), were selected for BSP due to their special roles in tumorigenesis, tumour growth, or stress reaction, BSP results confirmed significantly increased pro-moter methylation of GPR56, MT1G, and RASSF1 in A549/DDP cells compared with A549 cells (p<0.0001, p=0.0099,
Fig. 2. Signal Pathways Involved in Genes with Differentially Methyl-ated Promoters in A549/DDP Cells Compared with A549 Cells
(A) Number of genes with hypermethylated promoters. (B) Number of genes with hypomethylated promoters. 1. Wnt signalling pathway; 2. Tight junction; 3. Adherens junction; 4. TGF-beta signalling pathway; 5. Small cell lung cancer; 6. Neuroactive ligand–receptor interaction; 7. Glycerolipid metabolism; 8. Regulation of actin cytoskeleton; 9. Heparan sulphate biosynthesis; 10. Fc gamma R-mediated phagocytosis; 11. Glycerophospholipid metabolism; 12. Basal cell carcinoma; 13. Vibrio cholerae infection; 14. Neurotrophin signalling pathway; 15. Calcium sig-nalling pathway; 16. Axon guidance.
Table 3. Signal Pathways Involved in Genes with Hypermethylated Pro-moter Identified by MeDIP-ChIP Analysis in Lung Adenocarcinoma MDR A549/DDP Cells Compared with Its Progenitor A549 Cells
Signal pathways Genes
Wnt signaling path-way
WNT5B, PPP2R5B, WNT3A, CAMK2G, CSNK1A1L, SMAD3, SMAD2, FZD5, DAAM2, RBX1, CSNK2A2, CCND1, CCND3, PPP2CA, RAC1, LRP6, CAMK2D, NFATC4, PPP3CA, WNT9A, WNT7A, APC, TBL1Y
Tight junction PARD6A, CLDN7, RAB3B, CLDN18, MAGI2, CLDN5, MPP5, MYH6, ACTN3, PTEN, CLDN23, CSNK2A2, PPP2CA, RRAS2, ASH1L, CLDN1, EXoC3, MYH14, PPP2R2C, AKT2, SPTAN1
Adherens junction EGFR, CSNK2A2, PTPRF, PVRL3, TGFBR1, PVRL2, RAC1, SMAD3, SMAD2, ACTN3, WASL, WAS
TGF-beta signaling pathway
E2F4, E2F5, TGFBR1, GDF5, TGFB3, SMAD3, SMAD2, RBX1, INHBB, ID2, ZFYVE16, PPP2CA, BMP8B
Small cell lung cancer
CoL4A4, TRAF2, CCND1, CoL4A2, CoL4A1, NFKB1, APAF1, TRAF5, PTEN, CHUK, TRAF3, AKT2
Table 4. Signal Pathways Involved in Genes with Hypomethylated Promoter Identified by MeDIP-ChIP Analysis in Lung Adenocarcinoma MDR A549/DDP Cells Compared with Its Progenitor A549 Cells
Signal pathway Genes
Neuroactive ligand–receptor interaction CALCR, DRD1, TSPo, THRB, GABRB2, GRIK2, NPY2R, DRD5, oPRK1, GABBR1, GRIN3B, GABBR2, VIPR1, P2RY8, S1PR3, AGTR1, P2RY6, KISS1R, HRH3, P2RY1, S1PR5, GABRQ, HTR1E, GHR, GABRA2, PTGER3, CCKBR, GRIA4, GRM1, NPY5R, CRHR1, SSTR4, CRHR2, SSTR2, PRLR, P2RX1, AVPR1B, HTR6, GPR50, UTS2R, HTR2C, ADRA1D, F2R
Glycerolipid metabolism DGKA, CEL, AGPAT6, DGAT1, PPAP2C, DGKG, LIPG, DGKZ, AGPAT4, AGK, AGPAT3, AGPAT2
Regulation of actin cytoskeleton FGF19, FGFR2, FGD1, FGF18, ENAH, FGFR3, MYL5, FGF11, INSRR, ACTG1, PAK7, PAK3, GSN, CSK, TMSL3, GIT1, VAV3, RoCK1, LIMK1, MAP2K2, MYLK3, MYL12B, MYH9, FGF20, VAV1, PPP1CB, ARPC5L, CFL1, SCIN, PDGFRA, PDGFRB, TMSB4X, PIP4K2A, F2R
Heparan sulphate biosynthesis B3GAT3, HS3ST3A1, B3GALT6, XYLT1, HS3ST2, HS3ST1, HS3ST3B1, GLCEFc gamma R-mediated phagocytosis PRKCA, PLD2, VAV3, LYN, LIMK1, PPAP2C, NCF1C, VAV1, AMPH, JMJD7-PLA2G4B, DoCK2,
PLCG1, GAB2, GSN, ARPC5L, SCIN, CFL1, INPP5DGlycerophospholipid metabolism PLD2, PPAP2C, LYPLA1, PISD, DGKA, GPD1L, JMJD7-PLA2G4B, AGPAT6, DGKG, DGKZ,
PHoSPHo1, AGPAT4, AGPAT3, AGPAT2Basal cell carcinoma FZD8, SMo, STK36, WNT11, PTCH2, HHIP, FZD2, AXIN2, WNT6, SHH, TCF7L1, DVL1Vibrio cholerae infection PRKCA, ATP6V0C, TCIRG1, ACTG1, PRKACG, MUC2, ATP6V1C2, PLCG1, PDIA4, KCNQ1,
PRKX, ATP6V1FNeurotrophin signalling pathway IRAK1, MAP2K2, MAPK11, FoXo3, BAD, PTPN11, MAGED1, NTRK3, BDNF, MAP3K5,
CAMK4, PLCG1, RPS6KA2, NTRK1, NTRK2, YWHAQ, CALM3, SH2B3, SHC3, CSK, NGFCalcium signalling pathway GNA15, DRD1, GNA11, DRD5, PRKX, PRKACG, AGTR1, PDE1B, PRKCA, PTGER3, CCKBR,
BST1, MYLK3, GRM1, CD38, PLCG1, P2RX1, CAMK4, HTR6, AVPR1B, PDGFRA, CALM3, PDGFRB, HTR2C, ADRA1D, F2R, CACNA1B
Axon guidance RoCK1, EFNB3, LIMK1, GNAI1, PLXNB3, DPYSL2, CXCL12, EPHB1, PAK7, SEMA6A, SEMA5B, RGS3, EPHA8, UNC5A, PAK3, FYN, CFL1, SEMA4D, RHoD, UNC5C, NFATC2

1314 Vol. 36, No. 8
and p=0.0165, respectively) (Figs. 3A–C), while no difference of the promoter methylation level of CCNL2 and BAD was found between A549/DDP cells and A549 cells (p=0.0594 and p=0.5546, respectively) (Figs. 3D, E).
Expression of Five Candidate Genes Aberrant promoter methylation is usually linked to an altered chromosomal state and, thus, to transcriptional gene silencing.28) Therefore, Q-PCR was carried out to investigate whether the expression of the five candidate genes was regulated by the promoter methylation. Q-PCR analysis demonstrated that except BAD (p=0.426), the expression of the other four genes were all significantly downregulated in A549/DDP cells compared with A549 cells (all p<0.001) (Fig. 4).
DISCUSSIoN
MDR is one of the major clinical obstacles to the suc-cessful treatment of lung adenocarcinoma. It appears to be a polygenic phenomenon in which epigenetic-mediated changes might be the driving force leading to this phenotype.29) To in-vestigate whether certain distinct DNA methylation patterns is associated with the MDR phenotype of lung adenocarcinoma, MeDIP-ChIP was utilised to compare the promoter methyla-tion profiles of the human lung adenocarcinoma MDR A549/DDP cells with its parental A549 cells. Totally, the promoters of 3617 genes were found to be differentially methylated, of which 1581 were hypermethylated and 2036 were hypomethyl-ated in A549/DDP cells compared with A549 cells. To verify the MeDIP-ChIP results and investigate the association the expression of genes with its promoter methylation status, five
Fig. 3. Analog Representation of the Promoter Methylation Level of Five Candidate Genes in A549/DDP Cells Compared with A549 Cells by BSP Analysis
Each circle represents one CpG; the shaded circles indicate the presence of methyl-cytosine, and the empty circles indicate the absence of methylation. The numbers indicating the locations of the CpG sites in each gene. The methylation index (MI) is calculated by dividing all of the CpGs analysed by the total number of methylated CpGs detected. The incidence of promoter methylation of the three genes (GPR56, MT1G, and RASSF1) increased significantly in A549/DDP cells compared with A549 cells (p<0.0001, p=0.0099, and p=0.0165, respectively) (A–C), while the promoter methylation of the other two genes (CCNL2 and BAD) in A549/DDP cells was not dif-ferent from that in A549 cells (p=0.0594 and p=0.5546, respectively) (D, E).

August 2013 1315
potential methylation markers of lung adencarcinoma MDR, three genes with hypermethylated promoter (GPR56, MT1G, and RASSF1) and two genes with hypomethylated promoter (CCNL2 and BAD) were selected due to their special role in tumorigenesis, tumour growth, or stress reaction, using BSP and Q-PCR, respectively. The results indicated that the pro-moter methylation of GPR56, MT1G, and RASSF1 by BSP was consistent with that of MeDIP-ChIP, accompanied with tran-scriptional downregulation. However, the promoter methyla-tion level of CCNL2 and BAD decreased but did not reach sta-tistical significance in A549/DDP cells compared with A549 cells. Then, anticipated transcriptional upregulation of the two genes was not observed. Instead, the expression of CCNL2 was lower in A549/DDP cells compared with A549 cells, and which of BAD was unchanged.
RASSF1 and MT1G, two of the hypermethylated genes, were previously found to be involved in lung cancer prog-ress and prognostic, especially for non-small-cell lung cancer (NSCLC), and regulated by promoter methylation.30,31) RAS association domain family gene 1 (RASSF1) is a recently discovered RAS family-related gene that modulates multiple apoptotic and cell-cycle checkpoint pathways and promotes microtubule stability. RASSF1A, one isoform of RASSF1, has been identified as a tumour suppressor gene whose inactiva-tion has been implicated in the development of many cancers, especially lung cancer. Metallothionein 1G (MT1G) belongs to a class of metal-binding proteins (MTs) and participates in several cellular processes, such as metal ion stability, detoxi-fication, oxidation, and damage resistance. G protein-coupled receptor 56 isoform 3 (GPR56) is an orphan G protein-cou-pled receptor that has been shown to be implicated in brain development and play various roles in endogenous cancer progression,32) and suppress tumour growth and metastasis in human melanoma cell line xenograft models.33) But their roles in MDR of lung cancer is still unclear.34,35) Hence, this is the first study to report that the promoter methylation of these three genes was significantly higher in MDR cells of lung adenocarcinoma compared with its progenitor cells and the expression of these three genes was downregulated by the promoter hypermethylation. Therefore, we deduced that the three genes might be involved in the development of acquired MDR of lung adenocarcinoma. The mechanisms need be fur-ther investigated.
Interestingly, in the present study, Cyclin L2 (CCNL2), of which no promoter methylation was found between MDR cells with its progenitor cells by BSP, was found to be down-regulated. The distance between the mCpGs and the TSS might be one of the most important reasons to explain this phenominon36): the distance between the mCpGs and the TSS of CCNL2 is relatively longer (−1000 to −2200 bp), whereas that of the three hypermethylated genes (RASSF1, MT1G, and GPR56)) is relatively shorter (−200 to +500 bp). The phenom-enon also provides evidence that the expression of gene is not correlated with its promoter methylation.37–40)
There are several limitations in the present study. First, several genes (e.g., MDR-1) that had previously been reported to be methylated in MDR cells were not identified by the MeDIP-ChIP. one of the possible reasons is that the promoter regions of these genes might not be covered by the HG18 methylation array. Additionally, heterogeneity might generate certain deviations.41,42) Second, though the promoters of 3617
genes were found to be differentially methylated and three candidate genes (GPR56, MT1G, and RASSF1) with differen-tially methylated promoter might be the potential methylation markers associated with acquired MDR of lung adenocarci-noma, whether other genes or signalling pathways were in-volved in the acquired MDR of A549/DDP cells need further investigation.
In summary, to our knowledge, the present study reported for the first time the genome-wide analysis of promoter meth-ylation in the human lung adenocarcinoma MDR A549/DDP cells. Additionally, GPR56, MT1G, and RASSF1, three genes with differentially methylated promoter, might be the potential methylation markers associated with acquired MDR of lung adenocarcinoma.
Acknowledgments The research was funded by Grants from the Medical Science Foundation of Chengdu Military Area of China (No. MB09010), and grants from the Chong-qing Natural Science Foundation (No. CSTC2012JJA1595) awarded to Ruiling Guo; Grants from the National Natural Science Foundation of China (No. 81170044), and Grants from the Natural Science Foundation of Third Military Medical University (No. 2010XLC29) awarded to Fuyun Ji. The fund-ing agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manu-script.
REFERENCES
1) Zhi XY. Advances in surgical treatment of non-small cell lung can-cer. Oncology Progress, 7, 379–386 (2009a).
2) Zheng S, Qian P, Li F, Qian G, Wang C, Wu G, Li Q, Chen Y, Li J, Li H, He B, Ji F. Association of mitochondrial DNA variations with lung cancer risk in a Han Chinese population from southwestern China. PLoS ONE, 7, e31322 (2012).
3) Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male : female differences diminish-ing and adenocarcinoma rates rising. Int. J. Cancer, 117, 294–299 (2005).
4) Solyanik GI. Multifactorial nature of tumor drug resistance. Exp. Oncol., 32, 181–185 (2010).
5) Zeng-Rong N, Paterson J, Alpert L, Tsao MS, Viallet J, Alaoui-Jamali MA. Elevated DNA repair capacity is associated with in-
Fig. 4. Expression of the Five Candidate Genes in A549/DDP Cells Compared with A549 Cells Using Q-PCR
Q-PCR analysis demonstrated that except BAD, the expression of the other four genes were all significantly downregulated in A549/DDP cells compared with A549 cells (all p<0.001). The light blue and navy blue indicate the hypermethyl-ated genes (GPR56, MT1G, and RASSF1) and the hypomethylated genes (CCNL2, and BAD) identified in A549/DDP by MeDIP-ChIP, respectively. The Q-PCR ex-periments were performed in triplicate for each sample.

1316 Vol. 36, No. 8
trinsic resistance of lung cancer to chemotherapy. Cancer Res., 55, 4760–4764 (1995).
6) Pommier Y, Sordet o, Antony S, Hayward RL, Kohn KW. Apopto-sis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene, 23, 2934–2949 (2004).
7) Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene, 22, 7369–7375 (2003).
8) Mashima T, Tsuruo T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist. Updat., 8, 339–343 (2005).
9) oza AM. Clinical development of P glycoprotein modulators in on-cology. Novartis Found. Symp., 243, 103–115, discussion, 115–118, 180–185 (2002).
10) Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int., 5, 30 (2005).
11) Baylin SB, ohm JE. Epigenetic gene silencing in cancer—A mech-anism for early oncogenic pathway addiction? Nat. Rev. Cancer, 6, 107–116 (2006).
12) Nyce J. Drug-induced DNA hypermethylation and drug resistance in human tumors. Cancer Res., 49, 5829–5836 (1989).
13) Baker EK, El-osta A. The rise of DNA methylation and the im-portance of chromatin on multidrug resistance in cancer. Exp. Cell Res., 290, 177–194 (2003).
14) Roberti A, La Sala D, Cinti C. Multiple genetic and epigenetic interacting mechanisms contribute to clonally selection of drug-resistant tumors: current views and new therapeutic prospective. J. Cell. Physiol., 207, 571–581 (2006).
15) Teodoridis JM, Strathdee G, Brown R. Epigenetic silencing medi-ated by CpG island methylation: potential as a therapeutic target and as a biomarker. Drug Resist. Updat., 7, 267–278 (2004).
16) Brown R, Glasspool R. Epigenetic modulation of resistance to che-motherapy? Ann. Oncol., 18, 1429–1430 (2007).
17) Chekhun VF, Lukyanova NY, Kovalchuk o, Tryndyak VP, Pogrib-ny IP. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethyl-ated targets. Mol. Cancer Ther., 6, 1089–1098 (2007).
18) Qiu YY, Mirkin BL, Dwivedi RS. Inhibition of DNA methyltrans-ferase reverses cisplatin induced drug resistance in murine neuro-blastoma cells. Cancer Detect. Prev., 29, 456–463 (2005).
19) Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacyti-dine-induced demethylation of the hMLH1 gene promoter. Cancer Res., 60, 6039–6044 (2000).
20) Liu R, Hong Z. Relationship between methylation status of multi-drug resistance protein (MRP) and multi-drug resistance in lung cancer cell lines. Chin. J. Cancer Res., 19, 277–282 (2007).
21) Guo R, Wu G, Ji F, Xu S. Methylation of DNA mismatch repair gene MLH1 and MSH2 in acquired multidrug-resistance of human small cell lung cancer cells H446. Acta Academiae Medicinae Mili-taris Tertiae, 29, 2247–2249 (2007).
22) Guo R, Wu G, Ji F, Huang G, Chen W. Preliminary study of mecha-nisms of docetacel on human lung adenocarcinoma multidrug-resistant cells A549/CDDP and its parental cells. Acta Academiae Medicinae Militaris Tertiae, 27, 610–613 (2005).
23) Weber M, Davies JJ, Wittig D, oakeley EJ, Haase M, Lam WL, Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and trans-formed human cells. Nat. Genet., 37, 853–862 (2005).
24) Johnson DS, Li W, Gordon DB, Bhattacharjee A, Curry B, Ghosh J, Brizuela L, Carroll JS, Brown M, Flicek P, Koch CM, Dunham I, Bieda M, Xu X, Farnham PJ, Kapranov P, Nix DA, Gingeras TR, Zhang X, Holster H, Jiang N, Green RD, Song JS, McCuine SA, Anton E, Nguyen L, Trinklein ND, Ye Z, Ching K, Hawkins D, Ren
B, Scacheri PC, Rozowsky J, Karpikov A, Euskirchen G, Weiss-man S, Gerstein M, Snyder M, Yang A, Moqtaderi Z, Hirsch H, Shulha HP, Fu Y, Weng Z, Struhl K, Myers RM, Lieb JD, Liu XS. Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets. Genome Res., 18, 393–403 (2008).
25) Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol., 411, 270–282 (2006).
26) o’Geen H, Nicolet CM, Blahnik K, Green R, Farnham PJ. Compari-son of sample preparation methods for ChIP-chip assays. Biotech-niques, 41, 577–580 (2006).
27) Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–408 (2001).
28) Li B, Carey M, Workman JL. The role of chromatin during tran-scription. Cell, 128, 707–719 (2007).
29) Glasspool RM, Teodoridis JM, Brown R. Epigenetics as a mecha-nism driving polygenic clinical drug resistance. Br. J. Cancer, 94, 1087–1092 (2006).
30) Wang J, Wang B, Chen X, Bi J. The prognostic value of RASSF1A promoter hypermethylation in non-small cell lung carcinoma: a systematic review and meta-analysis. Carcinogenesis, 32, 411–416 (2011).
31) Chung JH, Lee HJ, Kim BH, Cho NY, Kang GH. DNA methylation profile during multistage progression of pulmonary adenocarcino-mas. Virchows Arch., 459, 201–211 (2011).
32) Xu L, Begum S, Barry M, Crowley D, Yang L, Bronson RT, Hynes Ro. GPR56 plays varying roles in endogenous cancer progression. Clin. Exp. Metastasis, 27, 241–249 (2010).
33) Ke N, Sundaram R, Liu G, Chionis J, Fan W, Rogers C, Awad T, Grifman M, Yu D, Wong-Staal F, Li QX. orphan G protein-coupled receptor GPR56 plays a role in cell transformation and tumorigen-esis involving the cell adhesion pathway. Mol. Cancer Ther., 6, 1840–1850 (2007).
34) Zhao QZ, Dou KF. Methylation of Ras association domain family protein 1, isoform A correlated with proliferation and drug resis-tance in hepatocellular carcinoma cell line SMMC-7721. J. Gastro-enterol. Hepatol., 22, 683–689 (2007).
35) Huang Y, de la Chapelle A, Pellegata NS. Hypermethylation, but not LoH, is associated with the low expression of MT1G and CRABP1 in papillary thyroid carcinoma. Int. J. Cancer, 104, 735–744 (2003).
36) Koga Y, Pelizzola M, Cheng E, Krauthammer M, Sznol M, Ariyan S, Narayan D, Molinaro AM, Halaban R, Weissman SM. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res., 19, 1462–1470 (2009).
37) Boyes J, Bird A. Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. EMBO J., 11, 327–333 (1992).
38) Tycko B. Epigenetic gene silencing in cancer. J. Clin. Invest., 105, 401–407 (2000).
39) Suzuki MM, Bird A. DNA methylation landscapes: provocative in-sights from epigenomics. Nat. Rev. Genet., 9, 465–476 (2008).
40) Cheung HH, Lee TL, Davis AJ, Taft DH, Rennert oM, Chan WY. Genome-wide DNA methylation profiling reveals novel epige-netically regulated genes and non-coding RNAs in human testicular cancer. Br. J. Cancer, 102, 419–427 (2010).
41) Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, olek A, Berlin K, Beck S. DNA methylation profiling of human chromo-somes 6, 20 and 22. Nat. Genet., 38, 1378–1385 (2006).
42) Feinberg AP, ohlsson R, Henikoff S. The epigenetic progenitor ori-gin of human cancer. Nat. Rev. Genet., 7, 21–33 (2006).



























![Synthesis, structures and magnetic properties of cyano ...or.nsfc.gov.cn/bitstream/00001903-5/216669/1/1000014739483.pdfinduced magnetization [26,27]. Furthermore, Tb(III) and Dy(III)](https://img.pdfslide.us/doc/110x75/5cc107ba88c993c70a8b5bea/synthesis-structures-and-magnetic-properties-of-cyano-ornsfcgovcnbitstream00001903-52166691.jpg)
