Embed Size (px)
Citation preview

Title:ProfilingadaptiveimmunerepertoiresacrossmultiplehumantissuesbyRNASequencing
Authors:SergheiMangul*#1,2,IgorMandric*3,HarryTaegyunYang1,NicolasStrauli4,DennisMontoya5,JeremyRotman1,WillVanDerWey1,JiemR.Ronas6,BenjaminStatz1,DouglasYao5,AlexZelikovsky3,RobertoSpreafico2,SagivShifman**7,NoahZaitlen**8,MauraRossetti**9,K.MarkAnsel**10,EleazarEskin**#1,11
Affiliations:
1DepartmentofComputerScience,UniversityofCaliforniaLosAngeles,LosAngeles,CA,USA
2InstituteforQuantitativeandComputationalBiosciences,UniversityofCaliforniaLosAngeles,LosAngeles,CA,USA
3DepartmentofComputerScience,GeorgiaStateUniversity,Atlanta,USA
4BiomedicalSciencesGraduateProgram,UniversityofCalifornia,SanFrancisco,CA,USA
5DepartmentofMolecular,Cell,andDevelopmentalBiology,UniversityofCalifornia,LosAngeles,LosAngeles,CA,
USA
6Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, Los
Angeles,CA90095,USA
7DepartmentofGenetics,TheInstituteofLifeSciences,TheHebrewUniversityofJerusalem,Jerusalem,Israel
8DepartmentofMedicine,UniversityofCalifornia,SanFrancisco,CA,USA
9Immunogenetics Center,Department of Pathology and LaboratoryMedicine,DavidGeffen School ofMedicine,
UniversityofCaliforniaLosAngeles,LosAngeles,CA,USA
10DepartmentofMicrobiologyandImmunology,SandlerAsthmaBasicResearchCenter,UniversityofCalifornia,San
Francisco,SanFrancisco,CA,USA
11DepartmentofHumanGenetics,UniversityofCaliforniaLosAngeles,LosAngeles,USA
#Correspondenceto:[email protected]@cs.ucla.edu
*Equalcontribution
**Equalcontribution

Abstract
Assay-basedapproachesprovideadetailedviewoftheadaptiveimmunesystembyprofilingT
andBcellreceptorrepertoires.However,thesemethodscarryahighcostandlackthescaleof
standard RNA sequencing (RNA-Seq). Here we report the development of ImReP, a novel
computationalmethodforrapidandaccurateprofilingoftheadaptiveimmunerepertoirefrom
regularRNA-Seqdata.Weappliedournovelmethod to8,555samplesacross544 individuals
from 53 tissues from the Genotype-Tissue Expression (GTEx v6) project. ImReP is able to
efficientlyextractTCR-andBCR-derived reads fromRNA-Seqdata. ImRePcanalsoaccurately
assemblethecomplementarydeterminingregions3(CDR3s),themostvariableregionsofBand
T cell receptors, and determine their antigen specificity. Using ImReP, we have created a
systematicatlasofimmunologicalsequencesforBandTcellrepertoiresacrossabroadrangeof
tissuetypes,mostofwhichhavenotbeenstudiedforBandTcellreceptorrepertoires.Wealso
compared the GTEx tissues to track the flow of T- and B-clonotypes across immune-related
tissues,includingsecondarylymphoidorgansandorgansencompassingmucosal,exocrine,and
endocrinesites,andweexaminedthecompositionalsimilaritiesofclonalpopulationsbetween
these tissues. The atlas of T and B cell receptors, freely available at
https://sergheimangul.wordpress.com/atlas-immune-repertoires/, is the largest collection of
CDR3sequencesandtissuetypes.Weanticipatethisrecoursewillenhancefutureimmunology
studiesandadvancedevelopmentoftherapiesforhumandiseases.ImRePisfreelyavailableat
https://sergheimangul.wordpress.com/imrep/.

Introduction
Akeyfunctionoftheadaptiveimmunesystem,whichiscomposedofB-cellsandT-cells, isto
mountprotectivememoryresponses toagivenantigen.BandTcells recognizetheirspecific
antigens through their surface antigen receptors (B and T cell receptors, BCR and TCR,
respectively),whichareuniquetoeachcellanditsprogeny.BCRandTCRarediversifiedthrough
somaticrecombination,aprocessthatrandomlycombinesvariable(V),diversity(D),andjoining
(J)genesegments,andinsertsordeletesnon-templatedbasesattherecombinationjunctions1
(Figure1a).TheresultingDNAsequencesarethentranslatedintotheantigenreceptorproteins.
Thisprocessallowsforanastonishingdiversityofthelymphocyterepertoire(i.e.,thecollection
of antigen receptors of a given individual), with >1013 theoretically possible distinct
immunological receptors1. This diversity is key for the immune system to confer protection
againstawidevarietyofpotentialpathogens2.Inaddition,uponactivationofaB-cell,somatic
hypermutationfurtherdiversifiesBCRsintheirvariableregion.Thesechangesaremostlysingle-
basesubstitutionsoccurringatextremelyhighrates (10–5 to10–3mutationsperbasepairper
generation)3. Isotype switching is another mechanism that contributes to B-cell functional
diversity.Here,antigenspecificityremainsunchangedwhiletheheavychainVDJregionsjoinwith
different constant (C) regions, such as IgG, IgA, or IgE isotypes, and alter the immunological
propertiesofaBCR.
High-throughputtechnologiesenableunprecedentedaccuracywhenprofilingtheBCRandTCR
repertoires.Commonlyusedassay-basedapproachesprovideadetailedviewof theadaptive

immunesystemwithdeepsequencingofamplifiedDNAorRNAfromthevariableregionofBCR
orTCRloci(Rep-Seq)4.Thosetechnologiesareusuallyrestrictedtoonechain,withthemajority
of studies focusing on the beta chain of TCRs and the heavy chain of BCRs. Recent studies2
successfully applied assay-based approaches to characterize the immune repertoire of the
peripheralblood.However,littleisknownabouttheimmunologicalrepertoiresofotherhuman
tissues,includingbarriertissueslikeskinandmucosae.Studiesinvolvingassay-basedprotocols
usually have small sample sizes, thus limiting analysis of intra-individual variation of
immunologicalreceptorsacrossdiversehumantissues.
RNASequencing(RNA-Seq)traditionallyusesthereadsmappedontohumangenomereferences
to study the transcriptional landscape of both single cells and entire cellular populations. In
contrasttoassay-basedprotocolsthatproducereadsfromtheamplifiedvariableregionofBCR
orTCRloci,RNA-Seqisabletocapturetheentirecellularpopulationofthesample,includingB
andTcells.However,duetotherepetitivenatureoflociencodingforBCRsandTCRs,aswellas
theextremelevelofdiversityinBCRandTCRtranscripts,mostmappingtoolsareillequippedto
handle immune repertoire sequences. Despite this, BCR and TCR transcripts often occur in
sufficient numberswithin the transcriptome ofmany tissues to characterize their respective
immunologicalrepertoires5.
In this study, we developed ImReP, a novel computational method for rapid and accurate
profilingoftheadaptiveimmunerepertoirefromregularRNA-Seqdata.Weapplieditto8,555
samplesacross544individualsfrom53tissuesobtainedfromGenotype-TissueExpressionstudy

(GTExv6)6.Thedatawasderivedfrom38solidorgantissues,11brainsubregions,wholeblood,
andthreecell lines. ImReP isabletoefficientlyextractTCR-andBCR-derivedreadsfromthe
RNA-Seq data and accurately assemble the complementarity determining regions 3 (CDR3s).
CDR3 are the most variable regions of B and T cell receptors and determine the antigen
specificity.UsingImReP,wecreatedasystematicatlasofimmunologicalsequencesforB-andT-
cellrepertoriesacrossabroadrangeoftissuetypes,mostofwhichwerenotpreviouslystudied
for B- and T-cell repertoires. We also examined the compositional similarities of clonal
populationsbetweenthetissuestotracktheflowofTandBclonotypesacrossimmune-related
tissues, including secondary lymphoid and organs that encompass mucosal, exocrine, and
endocrinesites.OurproposedapproachisnotsuperiorincomparisontotargetedTCRorBCR;
rather, itprovidesauseful tool formining large-scaleRNA-Seqdatasets forstudyofadaptive
immunerepertoires.

Results
ImReP:atwostageapproachforadaptiveimmunerepertoiresreconstruction
WeappliedImRePto0.6trillionRNA-Seqreads(92Tbp)from8,555samplestoassembleCDR3
sequencesofBandTcellreceptors(TableS1).TheRNA-SeqdatawasgeneratedbytheGenotype-
Tissue Expression Consortium (GTEx v6). First, we mapped RNA-Seq reads to the human
referencegenomeusingashort-readaligner(performedbyGTExconsortium6)(Figure1).Next,
weidentifyreadsspanningtheV(D)JjunctionofBandTcellreceptorsandassembleclonotypes
(agroupofcloneswithidenticalCDR3aminoacidsequences).HereImRePused0.02trillionhigh
qualityreadsthatsuccessfullymappedtoBCRgenes,successfullymappedtoTCRgenes,orwere
unmappedreadsthatfailedtomaptothehumanreferencegenome(Figures1aandS1).
ImReP is a two-stageapproach toassembleCDR3 sequencesanddetect correspondingV(D)J
recombinations(Figure1b).Inthefirststage,ImRePutilizesreadsthatsimultaneouslyoverlapV
andJgenesegmentstoinfertheCDR3sequences.WedefinetheCDR3asthesequenceofamino
acidsbetweenthecysteineontherightofthejunctionandphenylalanine(forallTCRchainsand
immunoglobulinlightchains)ortryptophan(forIGH)ontheleftofthejunction.Inthesecond
stage, ImReP utilizes reads that overlap a single gene segment containing a partial CDR3
sequence.ImRePthenusesasuffixtreetoperformpairwisecomparisonofthereadsandjoin
thereadsbasedonoverlapintheCDR3region.Further,ImRePusesaCASTclusteringtechnique7
toaccuratelyassembleclonotypes forPCRandsequencingerrors.WemapDgenes (for IGH,
TCRB,andTCRG)ontoassembledCDR3sequencesandinfercorrespondingV(D)Jrecombination.
AdetaileddescriptionofthemethodologyimplementedwithImRePisprovidedintheExtended

Experimental Procedures Section. ImReP is freely available at
https://sergheimangul.wordpress.com/imrep/.
To validate the feasibility of using RNA-Seq to study the adaptive immune repertoire, we
simulatedRNA-SeqdataasamixtureoftranscriptomicreadsandreadsderivedfromBCRand
TCR transcripts (Figure S3). BCR and TCR transcripts are simulated based on random
recombinationofVand Jgenesegments (obtained from IMGTdatabase8)withnon-template
insertionattherecombinationjunction(FigureS2).WeassessedtheabilityofImRePtoextract
CDR3-derived reads from the RNA-Seq mixture by applying ImReP to a simulated RNA-Seq
mixture.Whileoursimulationapproachmaynotcompletelysummarizethevariousnuancesand
eccentricitiesofactualimmunerepertoires,itallowsustoassesstheaccuracyofourtool.ImReP
is able to identify 99% of CDR3-derived reads from the RNA-Seq mixture, suggesting it is a
powerful tool for profiling RNA-Seq samples of immune-related tissues. Details about the
simulationdataareprovidedintheExtendedExperimentalProceduressection.
Next,wecomparedImRePwithothermethodsdesignedtoassembleimmunerepertoires.We
alsoinvestigatedthesequencingdepthandreadlengthrequiredtoreliablyassembleTCRand
BCR sequences from RNA-Seq data. Our simulations suggest that both read length and
sequencingdepthhaveamajor impactonprecision-recall ratesofCDR3 sequenceassembly.
ImRePisabletomaintainan80%precisionrateforthemajorityofsimulatedscenarios.Average
CDR3coveragethatishigherthan8allowsImRePtoarchivearecallratecloseto90%foraread
length above 75bp (Figure 2a). Increasing coverage has a positive effect on the number of

assembledclonotypesachievedbyImRePforbothBandTcellreceptors.Ingeneral,weobserve
higherprecision-recallratesofCDR3sequenceassemblyforTCRsincomparisontoBCRs(Figure
2a-b).
We compared the performance of ImReP to MiXCR (RNA-Seq mode)9, TRUST10, TraCeR11,
V’DJer12, IgBlast-basedpipeline13, and iSSAKE14. These toolsweredeveloped to assemble the
hypervariablesequencesintheTandBcellreceptorsdirectlyfromRNA-Seqdata.Wesupplied
eachofthosetoolswiththeoriginalRNA-Seqreadsasrawormappedreads,dependingonthe
softwaredevelopers’recommendations.).TRUSTandTraCeRdonotsupporttheanalysisofBCR
sequencesandwereexcludedfromthecomparisonbasedfortheIGHdata.iSSAKEisnolonger
supportedandwasnotrecommendedforuse.Unfortunately,weobtainedemptyoutputafter
running V’DJer, and increasing coverage in the simulated data did not solve the problem.
Alternativeapproaches,suchasIMSEQ15,cannotbeapplieddirectlytoRNA-Seqreadsbecause
theywere originally designed for targeted sequencing of B or T cell receptor loci. Thus, to
independentlyassessandcompareaccuracywithImReP,weonlyranIMSEQwiththesimulated
readsderivedfromBCRorTCRtranscripts(FigureS1).Scriptsandcommandstorunalltoolsused
inthisstudyareprovidedintheExtendedExperimentalProceduresandareavailableonlineat
https://github.com/smangul1/Profiling-adaptive-immune-repertoires-across-multiple-human-
tissues-by-RNA-Sequencing.ImRePconsistentlyoutperformedexistingmethodsonIGHdatain
bothrecallandprecisionratesforthemajorityofsimulatedparameters.ImRePandMiXCRshow
similarperformanceonTCRAdataandoutperformothermethods.Notably,ImRePwastheonly

methodwithacceptableperformanceon IGHdataat50bpread length,reconstructingwitha
higherprecisionratesignificantlymoreCDR3clonotypesthanothermethods.
Todemonstrate the feasibilityof applyingnon-specificRNASequencing toassemble immune
repertoiresequences,weusedtheTCRB-Seqdatapreparedfromthreesamplesofkidneyrenal
clearcellcarcinoma(KIRC)byLi,Bo,etal.10.WedownloadedmatchingRNA-Seqsamplesfrom
theTCGAportal.Intotal,weobtained301million2x50bpreadsfromthreeRNA-Seqsamples.
First,wepreparedtheCDR3sequencesobtainedfromTCRB-Seqandconsideredonlycomplete
CDR3s,whichwedefinedasasequenceofaminoacidsstartingwithcysteine(C)andendingwith
phenylalanine (F).We considered theprepared, completeCDR3sobtained fromTCRB-Seq as
totalimmunerepertoire.
WeusedImReP,MIXCR,TRUST,andIMSEQtoassembleCDR3softheTCRBchain.Weexcluded
V’DJer,becauseitonlysupportsimmunoglobulinchainsandisnotsuitabletoassembleCDR3s
fromTcellreceptors.ImReP,MIXCR,andTRUSTassembledcomparablenumbersofcomplete
CDR3sthatfullymatchCDR3sfromthetotalimmunerepertoireobtainedbyTCRB-Seq;IMSEQ
assemblednoneoftheCDR3sequences(Figure2candFigureS4).Onaverage,54%ofCDR3s
assembledby ImRePfullymatchCDR3sfromTCRB-Seq.Ourmethodwasabletorecover0.1-
0.9%ofthetotalimmunerepertoireobtainedbyTCRB-Seqfromkidneytissue.Othertissues,
including spleen andwhole blood, contain a higher fraction of T cells and allow RNA-Seq to
capture a higher fraction of the total immune repertoire. One should note, the number of
completeCDR3sfullymatchingCDR3sobtainedbyTCRB-Seqinourstudy(reportedinFigure2c

andFigureS4)arenotfullycomparablewiththeresultsreportedinLi,Bo,etal.10,whereCDR3
sequencesareconsideredtomatchCDR3sfromTCRB-Seqifatleast6aminoacidsarematched.
Scripts and commands utilized to process the data and run repertoire assembly tools are
provided in Extended Experimental Procedures and are available online at
https://github.com/smangul1/Profiling-adaptive-immune-repertoires-across-multiple-human-
tissues-by-RNA-Sequencing.
WefurthervalidatedtheabilityofImRePtoaccuratelyinfertheproportionofimmunecellsin
thesampled tissue.Wehypothesized that the fractionofB-andT-cells in thesamplewillbe
proportional with the fraction of receptor-derived reads in our RNA-Seq data. We used a
transcriptome-basedcomputationalmethod,SaVant16,whichusescell-specificgenesignatures
(independentofBCRorTCRtranscripts)toinfertherelativeabundanceofBorTcellswithineach
tissue sample. We found that B and T cell signatures inferred by SaVant showed positive
correlationwiththeamountofBCR(r=0.77,P<0.001)orTCR(r=0.86,P<0.001)transcripts,
respectively (Figure 2c,d). An exception to this correlation was for tissues that contain the
highestdensityofBorTcells:spleen,wholeblood,smallintestine(terminalileum),lung,and
EBV-transformedlymphocytes(LCLs).

chr2 chr7 chr14 chr22
IGL
TCRATCRD
IGH
TCRG
TCRB
IGKV N D N J C
CDR3CDR1 CDR2
MappedreadsUnmappedreads
mRNAencodingforTandBcellreceptorchains
VYFICASSEASSLGGSGGYTACCSYEQYFGPG
Vgene J gene
…SQTSVYFCASSE SYEQYFGPGTRLT…
CDR3
SQTSVYFICASSEASSLGGSGGY
Vgene…SQTSVYFCASSE
read
SGGYTACCSYEQYFGPG
J gene
SYEQYFGPGTRLT…
read1read2
TACCSYEQYFGPGYG
M
GM
LQ
! read2
Suffixtree
ImReP
a
b
c
d
readsmatchingonlyVgeneareencodedassuffixtree

Figure 1. Overview of ImReP. (a) Schematic representation of human adaptive immune repertoire. Adaptive
immune repertoire consists of four T-cell receptor loci (blue color, T cell receptor alpha locus (TCRA); T-
cell receptorbeta locus (TCRB);T-cell receptordelta locus (TCRD);andT-cell receptorgamma locus (TCRG))and
three immunoglobulin loci (red color, Immunoglobulin heavy locus (IGH); Immunoglobulin kappa locus (IGK);
Immunoglobulinlambdalocus(IGL).Alternativename–BCR,Bcellreceptor).B-andT-cellreceptorscontainmultiple
variable (V,greencolor),diversity (D,presentonly in IGH,TCRB,TCRG,violetcolor), joining(J,yellowcolor)and
constant(C,bluecolor)genesegments.V(D)Jgenesegmentsarerandomly jointedandnon-templatedbases(N,
darkredcolor)areinsertedattherecombinationjunctions.TheresultingsplicedT-orB-cellrepertoiretranscript
incorporatestheCsegmentandistranslatedintotheantigenreceptorproteins.RNA-Seqreadsarederivedfrom
the rearranged immunoglobulin IG and TCR loci. Reads entirely aligned to genes of B- and T-cell receptors are
inferredfrommappedreads(blackcolor).Readswithextensivesomatichypermutationsandreadsspanningthe
V(D)J recombinationare inferred fromtheunmappedreads (greycolor).Complementaritydetermining region3
(CDR3)isthemostvariableregionofthethreeCDRregionsandisusedtoidentifyT/B-cellreceptorclonotypes—a
group of clones with identical CDR3 amino acid sequences. (b) Receptor derived reads spanning V(D)J
recombinationsare identified fromunmappedreadsandassembled into theCDR3sequences.Wefirst scanthe
aminoacidsequencesofthereadanddeterminetheputativeCDR3boundariesdefinedbylastconservedcysteine
encodedby theVgeneand theconservedphenylalanine (forTCR)or tryptophan (forBCR)of Jgene.Given the
putativeCDR3boundaries,wechecktheprefixandsuffixofthereadtomatchthesuffixofVandprefixofJgenes,
respectively.(c-d)IncaseareadoverlapswithonlytheVorJgene,weperformthesecondstageofImRePtomatch
suchreadsbasedontheoverlapofCDR3sequenceusingsuffixtree.
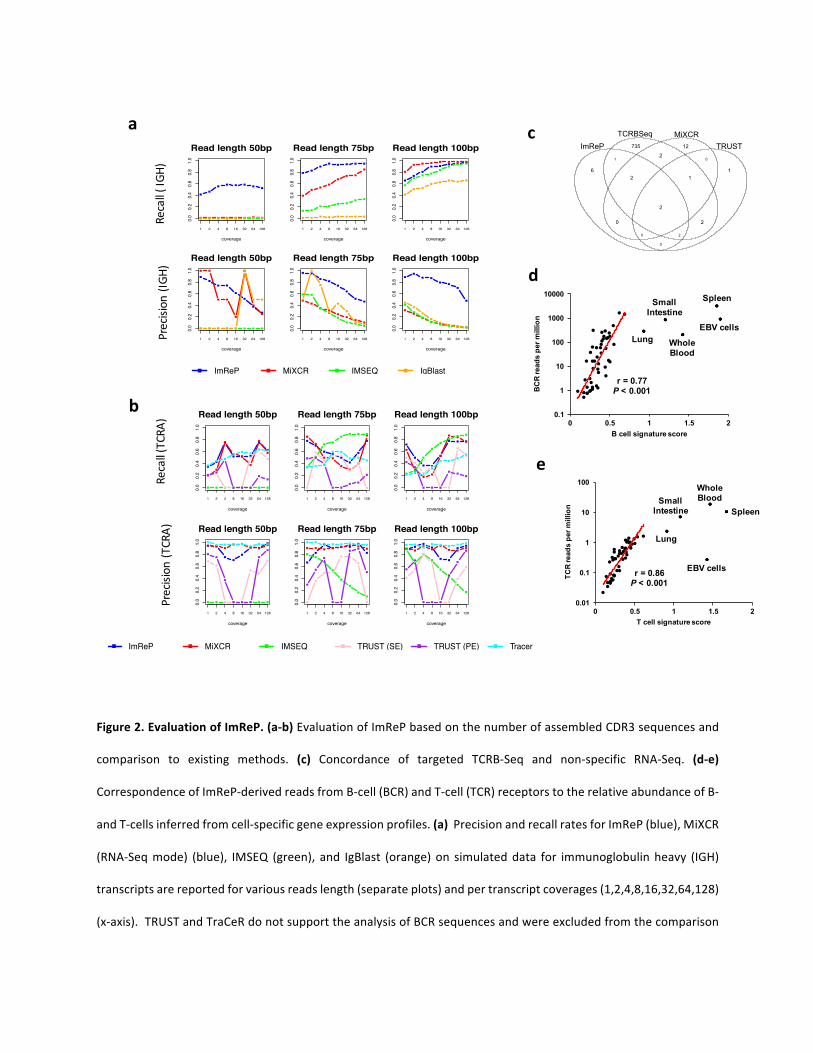
Figure2.EvaluationofImReP.(a-b)EvaluationofImRePbasedonthenumberofassembledCDR3sequencesand
comparison to existing methods. (c) Concordance of targeted TCRB-Seq and non-specific RNA-Seq. (d-e)
CorrespondenceofImReP-derivedreadsfromB-cell(BCR)andT-cell(TCR)receptorstotherelativeabundanceofB-
andT-cellsinferredfromcell-specificgeneexpressionprofiles.(a)PrecisionandrecallratesforImReP(blue),MiXCR
(RNA-Seqmode) (blue), IMSEQ (green), and IgBlast (orange)on simulateddata for immunoglobulinheavy (IGH)
transcriptsarereportedforvariousreadslength(separateplots)andpertranscriptcoverages(1,2,4,8,16,32,64,128)
(x-axis).TRUSTandTraCeRdonotsupporttheanalysisofBCRsequencesandwereexcludedfromthecomparison
Final figures 2c,d
0.1
1
10
100
1000
10000
0 0.5 1 1.5 2
BC
R re
ads
per m
illio
n
B cell signature score
0.01
0.1
1
10
100
0 0.5 1 1.5 2
TCR
read
s pe
r mill
ion
T cell signature score
Spleen
EBV cellsWhole Blood
Small Intestine
Lung
Spleen
EBV cells
Whole BloodSmall
Intestine
Lung
r = 0.77P < 0.001
r = 0.86P < 0.001
d
a
Recall(IG
H)Precision(IGH
)
e
Final figures 2c,d
0.1
1
10
100
1000
10000
0 0.5 1 1.5 2
BC
R re
ads
per m
illio
n
B cell signature score
0.01
0.1
1
10
100
0 0.5 1 1.5 2
TCR
read
s pe
r mill
ion
T cell signature score
Spleen
EBV cellsWhole Blood
Small Intestine
Lung
Spleen
EBV cells
Whole BloodSmall
Intestine
Lung
r = 0.77P < 0.001
r = 0.86P < 0.001
b
Recall(TCRA
)Precision(T
CRA)
ImRePTCRBSeq MiXCR
TRUST
6
735 12
1
1
0
0
2
2
0
2
20
1
2
c
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
ImReP MiXCR IMSEQ IgBlast
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
ImReP MiXCR IMSEQ IgBlast
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
ImReP MiXCR IMSEQ TRUST (SE) TRUST (PE) Tracer
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 50bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 75bp
coverage
1 2 4 8 16 32 64 128
0.0
0.2
0.4
0.6
0.8
1.0
Read length 100bp
coverage
1 2 4 8 16 32 64 128
ImReP MiXCR IMSEQ TRUST (SE) TRUST (PE) Tracer

basedfortheIGHdata.(b)PrecisionandrecallratesforImReP(blue),MiXCR(RNA-Seqmode)(red),TRUST(default,
paired-endmode)(pink),TRUST(single-endmode)(violet),IMSEQ(green),andTraCeR(aqua)onsimulateddatafor
Tcellreceptoralpha(TCRA)transcripts arereportedforvariousreads length(separateplots)andpertranscript
coverages (1,2,4,8,16,32,64,128) (x-axis). (c) Concordance of targeted TCRB-Seq and non-specific RNA-Seq
performedonthreeTCGAsamples(onlyoneisshown)fromkidneyrenalclearcellcarcinoma(KIRC).Venndiagram
on TCGA-CZ-5463 sample presents number of matching CDR3s reported by immunoSEQ Analyzer
(http://www.adaptivebiotech.com/)andCDR3sequencesassembledfromnon-specificRNA-SeqdatabyImReP,
MiXCR(RNA-Seqmode),TRUST(default, paired-end mode). Results on other samples are presented in Figure
S4. (d)ScatterplotofthenumberofallBCRreadsper1millionRNA-Seqreads(y-axis)andB-cellsignaturescore
inferredbySaVant(x-axis).(e)ScatterplotofthenumberofallTCRreadsper1millionRNA-Seqreads(y-axis)andB-
cellsignaturescoreinferredbySaVant(x-axis).Pearsoncorrelationcoefficient(r)andP-valuearereported.
Characterizingtheadaptiveimmunerepertoireacross53GTExtissues
ImReP identified over 26million reads overlapping 3.8million distinct CDR3 sequences that
originatefromdiversehumantissues.ThemajorityofassembledCDR3sequencesderivedfrom
BCRs, with 1.7 million from the immunoglobulin heavy chain (IGH), 0.9 million from the
immunoglobulinkappachain(IGK),and1.0millionfromtheimmunoglobulinlambdachain(IGL).
AsmallerfractionofCDR3sequencesderivedfromTCRs,with0.2millionsequencesfromalpha
andbetaTCRs(TCRAandTCRB).ThevastmajorityofallassembledCDR3shadalowfrequency
inthedata.98%ofCDR3sequenceshadacountof lessthan10reads,andthemedianCDR3
sequencecountwas1.4.CDR3sequencesderivedfromIGKwerethemostabundantacrossall
tissues,accountingonaveragefor54%oftheentireB-cellpopulation(FigureS5).IntheT-cell

population,alphaandbetajointlyaccountedfor83%ofthepopulation.DeltaT-cellpopulation
wastherarest,accountingforlessthanonepercentoftheentireTcellpopulation(FigureS6).
We compared the length and amino acid composition of the assembled CDR3 sequences of
immunoglobulinandT-cellreceptorchains(Figure3a-g).Consistentwithpreviousstudies,we
observedthatimmunoglobulinlightchainshavenotablyshorterandlessvariableCDR3lengths
comparedtoheavychains17(Fig.3h).Thetissuetypeappearstohavenoeffectonthelength
distributionofCDR3sequencesofBCRs(FigureS7).DifferencesintheCDR3lengthdistributions
for TCRs cannot be estimated due to the small number of available TCRs. Sequencing
composition18ofCDR3regionsofbetaT-cellsassembledbyImRePrecapitulatesonedetectedby
TCRsequencing2fromthewholebloodwithtwodominantCASSandCSARmotifsinthebeginning
of the sequence (Fig. 3f). In line with other studies19, both light chains exhibited a reduced
amountofsequencingdiversity(Fig.3b-c).

Figure3.Lengthandaminoacidcompositionof theassembledCDR3sequencesof immunoglobulinandT-cell
receptorchains.Thesequencelogo(usingWebLogo)ofaminoacidcompositionrepresentationforCDR3sequences
withmean length.Theheightoftheaminoacidwithinthestack indicatestherelativefrequency.Distributionof
CDR3sequencelengthisestimatedusingskerneldensity.(a)Sequencelogoof15-amino-acidCDR3sequenceof
IGH.(b)Sequencelogoof11-amino-acidCDR3ofIGK.(c)Sequencelogoof12-amino-acidCDR3sequenceofIGL.(d)
Sequence logoof14-amino-acidCDR3sequenceofTCRA.(e)Sequence logoof14-amino-acidCDR3sequenceof
TCRB. (f) Sequence logo of 17-amino-acid CDR3 sequence of TCRD. (g) Sequence logo of 13-amino-acid CDR3
IGH
IGK
IGL
TCRD
TCRB
TCRA
a
h TCRG
b
c
d
e
f
g
Length(amino acids)

sequenceofTCRG.(h)DistributionofCDR3sequencelengthisestimatedusingskerneldensityseparatelyforeach
chainofT-andB-cellreceptors.
We observed per sample an average of 1331 distinct clonotypes for BCRs and 20 distinct
clonotypessequencesforTCRs.Wenormalizedthenumberofdistinctclonotypesbythetotal
numberofRNA-Seqreads,whichwecallnumberofclonotypesperonemillionreads(CPM).As
thenumberofdistinctclonotypesdoesnot increase linearlywiththesequencingdepth,CPM
metricshouldnotbeusedinstudiescomparingclonotypediversityacrossvariousphenotypes.
Instead,CPMisintendedtobeaninformativemeasureofclonaldiversityadjustedforsequencing
depth.
We used per sample alpha diversity (Shannon entropy) to incorporate the total number of
distinctclonotypesandtheirrelativefrequenciesintoasinglediversitymetric.Amongalltissues,
spleenhasthelargestB-cellpopulation,withamedianof1301BCR-derivedreadsperonemillion
RNA-Seqreads.ItalsohasthemostdiversepopulationofBcellswithmedianpersamplealpha
diversityof7.6correspondingto1025CPM(Figure4andTableS1).Wholebloodhasboththe
largestandmostdiverseT-cellpopulation(Figure4andTableS1).Organsthatpossessmucosal,
exocrine,andendocrinesites(n=24)harborarichclonotypepopulationwithamedianof87CPM
persample.Minorsalivaryglandshavethehighest immunediversity inthegroup(alpha=7.1)
andsurpassthediversityoftheterminalIleumcontainingPeyer’sPatches,whicharesecondary
lymphoidorgans(TableS1).

Tissuesnotrelatedtotheimmunesystem,includingadipose,muscle,andtheorgansfromthe
centralnervoussystem,containedamedianof6CPMpersample,whicharemostlikelydueto
thebloodcontentofthetissues20.ThehighestnumberofdistinctCDR3sequencesamongnon-
lymphoidorganswaspresent in theomentum,amembranousdouble layerofadiposetissue
containingfat-associatedlymphoidclusters.Asexpected21,Epsteinbarvirus(EBV)-transformed
lymphocytes(LCL)harboredalargehomogeneouspopulationofBcellclonotypes(TableS1and
FigureS7).

0 200 400 600 800 1000
CLONOTYPIC RICHNESSOFBCELLS, CPM
IGH IGK IGL
02468101214
SpleenSmallIntestine- TerminalIleumWholeBloodArtery- CoronaryArtery- AortaArtery- TibialMinorSalivaryGlandColon- TransverseStomachColon- SigmoidLungBreast- MammaryTissueEsophagus- MucosaThyroidKidney- CortexBladderFallopianTubeVaginaCervix- EndocervixLiverCervix- EctocervixProstateUterusOvaryPancreasPituitarySkin- NotSunExposed…Skin- SunExposed(Lowerleg)AdrenalGlandTestisCells- EBV-transformed…Cells- TransformedfibroblastsAdipose- Visceral(Omentum)Esophagus- Gastroesophageal…Adipose- SubcutaneousHeart- AtrialAppendageEsophagus- MuscularisHeart- LeftVentricleMuscle- SkeletalNerve- TibialBrain- Spinalcord(cervicalc-1)Brain- AmygdalaBrain- CortexBrain- HypothalamusBrain- Caudate(basalganglia)Brain- SubstantianigraBrain- HippocampusBrain- Putamen(basalganglia)Brain- AnteriorcingulatecortexBrain- Nucleusaccumbens(basal…Brain- CerebellumBrain- FrontalCortex(BA9)Brain- CerebellarHemisphere
TCRA TCRB
TCRD TCRG
a b
Secondarylymphoidorgans
Bloodassociatedsites
Mucosal,exocrineandendocrineorgans
Celllines
Adiposeandmuscleorgans
Organsofcentralnervoussystem
Tissuestypes
CLONOTYPIC RICHNESSOFTCELLS,CPM

Figure 4.Adaptive immune repertoires acrossmultiple human tissues. Adaptive immune repertoires of 8,555
samplesacross544individualsfrom53bodysitesobtainedfromGenotype-TissueExpressionstudy(GTExv6).We
groupthetissuesbytheirrelationshiptotheimmunesystem.Thefirstgroupincludesthelymphoidtissues(n=2,red
colors).Thesecondgroupincludesbloodassociatedsitesincludingwholebloodandbloodvessel(n=4,redcolor).
Thethirdgrouparetheorgansthatencompassesmucosal,exocrineandendocrineorgans(n=21,lavendercolor).
The fourth group are cell lines (n=3, grey color). The filth group are adipose or muscle tissues and the
gastroesophagealjunction(n=7,bluecolor).Thesixthgroupareorgansfromcentralnervoussystem(n=14,green
color).HistogramreportsclonotypicrichnessofTandBcells,calculatedasnumberofdistinctaminoacidsequences
ofCDR3peronemillionRNA-Seqreads(CPM).(a)MedianCPMsarepresentedindividuallyforTcellreceptoralpha
chain(TCRA),T-cellreceptorbetachain(TCRB),T-cellreceptordeltachain(TCRD),andT-cellreceptorgammachain
(TCRG).(b)MediannumberofdistinctaminoacidsequencesofCDR3arepresentedindividuallyforimmunoglobulin
heavychain(IGH),immunoglobulinkappachain(IGK),immunoglobulinlambdachain(IGL).
Individual-andtissue-specificT-andB-cellclonotypes
Aminoacidsequencesofclonotypesexhibitedextremeinter-individualdissimilarity,with88%of
clonotypesuniquetoasingleindividual(private)(Figure5a).Theremaining~400,000clonotypes
weresharedbyatleasttwoindividuals(public).Thenumberofindividualssharingclonotypes
variedacrossTandBcellreceptors,withimmunoglobulinlightchainshavingthehighestnumber
ofpublicclonotypes.Twenty-fivepercentofallIGKclonotypeswerepublic,andthenumberof
individuals sharing the IGK clonotype sequences can be as high as 471 (Figure 5b). T cell
clonotypeshadonaverage7%publicclonotypes,withthehighestnumberofpublicclonotypes
amongthedeltachainofgamma-deltaTCRs(12%).ThelimitedcapacityofRNA-Seqtocoverlow

abundant clonotypes may misclassify public clonotypes as private. Consistent with previous
studies10,22,weobservepublicclonotypestobesignificantlyshorterinlengththantheprivate
ones(p-value<2x10-16).Forexample, IGHchainpublicclonotypeshadanaveragelengthof13
aminoacids,andprivateclonotypeshadanaveragelengthof16.Wealsoexaminedwhetherthe
publicclonotypesweremoreoftensharedacrosstissueswithinanindividual.Only14%ofthe
~240,000 clonotypes shared across tissues were public. The majority of clonotypes were
individual-andtissue-specific(Figure5c).Thefulllistofpublicclonotypesisdistributedwiththe
‘AtlasofT-andB-cellrepertoires’thataccompaniesthismanuscript.

Figure5.PrivateandpublicTandBcellclonotypes.(a)Distributionoffrequenciesofprivate(n=1)andpublic(n>1)
clonotypesacross544 individuals.Wecollectclonotypes fromall tissuesof thesame individual intoa singleset
correspondingtothatindividual.(b)Themostpublicclonotypes(sharedacrossmaximumnumberof individuals)
and corresponding VJ recombination are presented for IGH, IGK, IGL, TCRA, TCRB, and TCRG. (c) Clonotypes
sequences are classified into public clonotypes (shared across individuals), private (individual specific), tissue-
specific,andclonotypessharedacrossmultipletissues.Thenumberofclonotypesfallingintoeachpairofcategories
isreportedacrossallTandBcellreceptorchains.
CQQYGSSPRTFIGKV3-20 IGKJ2471
CARGHYGMDVWIGHV3-53 IGHJ658
CQAWDSSTVVFIGLV3-1 IGLJ2443
CAGADRLQTGMRGAFTRAV8-7 TRAJ19
90
CVIKYFTRBV10-3 TRBJ2-367
CAAWDYYKKLFTRGV10 TRGJ1(2)44
a b
cTissuespecific Sharedacross
tissues
Individualspecific(private) 3.1x106 0.2x106
Sharedacrossindividuals(public)
0.4x106 0.03x106
-1 1 3 5 7 9 11 13 15 17Numbe
rofclono
type
sequ
ences
Numberofindividualssharing theclonotypesequence
IGH IGK IGL TCRA TCRB TCRD TCRG
106
105
104
103
102
10
1
BCRs
TCRs

FlowofTandBcellclonotypesacrosshumanGTExtissues
The large number of individuals available through the study allow us to establish a pairwise
relationshipbetweenthetissuesandtracktheflowofTandBclonotypesacrosshumantissues.
WobservedasignificantincreaseoftheCDR3sequencessharedacrosspairsoftissuesfromthe
same individuals. Further,weobserved thispattern consistently forall chainsofBandT cell
receptors(p-value<2x10-16)(Figure6aandTableS2).Weobserveadifferentamountofshared
CDR3sequencesacrossdifferent typesofBCRsandTCRswithan increase in immunoglobulin
light chains. Decreased number of TCR reads compared to the BCRs makes it unfeasible to
comparethenumberofsharedCDR3acrossTandBcells.Onaverage,weobserve21.0CDR3
sequencestobesharedacrossapairoftissuesfromthesameindividuals.Pairsoftissuesfrom
differentindividualsshareonaverage10.6CDR3sequences(Figure6aandTableS2).
ToestablishtheflowofB-andT-cellclonotypesacrossvarioustissues,wecomparedclonotype
populationsbetweenandwithinthesameindividuals.Welimitedthisanalysistopairsoftissues
forwhichwehadatleast10individuals(870pairsoftissuesoutof1378possiblepairs).Weused
betadiversity (Sørensen–Dicesimilarity index) tomeasurecompositional similaritiesbetween
thetissuesintermsofgainorlossofCDR3sequences(Figure6b-c).Forthemajorityofthe870
availabletissuepairs,weobservenoBCRorTCRsequencesincommon,whichcorrespondsto
betadiversityof0.0.

WeexaminedtheflowofIGHclonotypesacrosstissuesandpresenteditasanetwork(Figure
5b).Among870availabletissuepairs,wehaveidentified56tissuepairswithbetadiversityabove
.001.Spleenwasthemosthighlyconnectedtissue,with17connections,followedbylung,with
16 connections. Clonotypes represents one connected component, meaning that every two
nodesareconnecteddirectlyorviaothernodes.Clonotypepopulationsofspleenandlungare
themostsimilar (0.02betadiversity),otherpairs includeminorsalivaryglandandesophagus
mucosa,terminalileum(smallintestine)andtransversecolon.Weobserveabove200pairsof
tissueswithbetadiversityabove.001forimmunoglobulinlightchains(FigureS9-S10).Themost
similartissuepairsforIGKchainwerespleenandtransversecolon(0.15betadiversity).
WealsoexaminedtheflowofTCRBclonotypesacrosstissues.TCRBclonotypesequenceswere
sharedacross10pairsoftissueswithaveragebetadiversityof0.02.SimilartoBcells,thenetwork
of T cell clonotypes is a single connected component, with spleen being the most highly
connectedtissue(Figure6c).FortheTCRgammachain,weobservebetadiversityof0.0forall
tissuepairs.Atthesametime12%ofTCRgammaclonotypesarepublic,showingthehighestrate
amongallTCRgenes.AtthesequencingdepthprovidedbyRNA-Seq,weareunabletoobserve
TCRdeltaclonotypesharingacrossindividualsandtissues.

Figure6.FlowofTandBcellclonotypesacrossdiversehumantissues.Resultsarebasedonpairsoftissueswithat
least10individuals.(a)Thenumberofclonotypesequencessharedacrosspairsoftissuesfromthesameindividuals
(bluecolor)andfromdifferentindividuals(orangecolor)arepresented.Numberofclonotypessharedacrosstissues
fromthesameindividualsforTCRsis<.1.Numberofclonotypessharedacrosstissuesfromdifferentindividualsfor
TCRsis<.001.(b)-(c)Flowofclonotypesacrossdiversehumantissuesispresentedasanetwork.Eachnodeisatissue
withthesizeproportionaltoamediannumberofclonotypesofthetissue.Thecolorofthenodecorrespondstoa
typeofthetissuetype:lymphoidtissues(yellowcolors),bloodassociatedsites(redcolor),organsthatencompasses
mucosal,exocrineandendocrineorgans(lavendercolor).Compositionalsimilaritiesbetweenthetissuesintermsof
gain or loss of CDR3 sequences aremeasured across valid pairs of tissues using beta diversity (Sørensen–Dice
similarity index). Edges areweighted according to the beta diversity. (b) Flowof IGH clonotypes across diverse
ab
Averagenu
mbero
fclono
types
sharedacrossp
airsoftissues
c
IGH
TCRB
-15
-10
-5
0
5
10
15
IGH
IGK
IGL
TRCA
TRCB
TRCD
TRCG
Pairoftissueswithinthesame individual
Pairoftissues acrossdifferentindividuals

humantissuesispresentedasanetwork.Edgeswithbetadiversity>.001arepresented.(c)FlowofTCRBclonotypes
acrossdiversehumantissuesispresentedasanetwork.Edgeswithbetadiversity>.001arepresented.
ImRePidentifiestissuesampleswithlymphocyteinfiltration
Histologicalimagesoftissuecross-sectionsandpathologists’noteshavebeenusedtovalidate
the ImReP’s ability to detect the samples with a high lymphocyte content, which often
correspondstoadiseasestate.WeexaminedtheIGHclonotypepopulationsfromthyroidtissue
acrossindividuals.ThemediannumberofinferreddistinctCDR3sequencespersamplewas20,
though14.5%ofthesampleshadmorethan500distinctCDR3sequences. Weobservedthe
highestnumberofCDR3sequencesamongallthethyroidsamplesinanindividualwithlatestage
Hashimoto'sthyroiditis,anautoimmunediseasecharacterizedbylymphocyteinfiltrationandT-
cellmediatedcytotoxicity.Accordingtopathologists’notes,Hashimoto'sdiseasewaspresentin
11.2%ofthyroidsamples,withvaryingdegreesofseverity.First,weusedpathologists’notesto
annotate samples as healthy or bearing Hashimoto's disease, and then we compared the
adaptiverepertoirediversitybetweenthesegroups.Weobservedasignificant increaseinthe
numberofdistinct IGHclonotypes insampleswithHashimoto's thyroiditis (p-value=1.5x10-5)
(FigureS11).Thenumberofclonotypesvariedfrom113forfocalHashimoto'sthyroiditisto5621
forlatestageHashimoto'sthyroiditis(Figure7a).Inaddition,highclonotypediversityinkidney
samplesindicatedthepresenceofglomerulosclerosis.Inlungsamples,highclonotypediversity
correspondedtoinflammatorydiseasessuchassarcoidosisandbronchopneumonia.

Weobservednodifferenceinclonaldiversityinmalesandfemalesacrossthetissues,exceptin
breasttissues(p-value<3.2x10-12,BCRs). Increasedclonotypediversityofbreasttissue inmale
individualscorrespondedtogynecomastia,acommondisorderofnon-cancerousenlargementof
malebreasttissue(Figure7b).

Figure7.ImRePisabletoidentifysampleswithhighactivityoflymphocytes.Histologicalimagesoftissuecross-
sectionsandpathologists’noteshavebeenusedtovalidatetheImReP’sabilitytodetectthesampleswithahigh
activityoflymphocytes.(a)SampleswereorderedbyHashimoto'sthyroiditisseverity,asreportedbypathologists’
notes.Histologicalimagesareprovidedtoillustratethediseasestate.Averagenumberofclonotypesisreportedfor
eachdiseasegroup.(b)Boxplotreportingnumberofclonotypesinthebreasttissuesformalesandfemales.Outlier
amongthemalesamplesisillustratedwiththehistologicalimage.
113 4961315
2353
5621
FOCA
LHAS
HIMOTO
S
PATCHY
HA
SHIM
OTO
S
EARLYHA
SHIM
OTO
S
HASH
IMOTO
S
LATESTAGE
HA
SHIM
OTO
S
Num
berofIGH
clo
notypes
DiseaseseverityHistologica
limages
female
male
0
1000
2000
3000
Number of IGH clonotypes
sex_
s
Gynecomastia
b
a

Discussion
We have developed a novel computational approach (ImReP) for reconstruction of adaptive
immune repertoires using RNA-Seq data.We demonstrate the ability of ImReP to efficiently
extract TCR- and BCR- derived reads from the RNA-Seq data and accurately assemble
correspondingBCRandTCRclonotypes.TheproposedalgorithmcanaccuratelyassembleCDR3
sequencesofimmunereceptorsdespitethepresenceofsequencingerrorsandshortreadlength.
Simulations generated using various read lengths and coverage depth show that ImReP
consistentlyoutperformsexistingmethodsintermsofprecisionandrecallrates.
We have demonstrated the feasibility of applying RNA-Seq to study the adaptive immune
repertoire.AlthoughRNA-Seqlacksthesequencingdepthoftargetedsequencing(Rep-Seq),it
can compensate by examining a larger sample size. Using ImReP, we have created the first
systematicatlasofimmunologicalsequenceforB-andT-cellreceptorrepertoriesacrossdiverse
humantissues.Thisprovidesarichresourceforcomparativeanalysisofarangeoftissuetypes,
mostofwhichhavenotbeenstudiedbefore.TheatlasofT-andB-cellreceptors,availablewith
thepaper,isthelargestcollectionofCDR3sequencesandtissuetypes.Weanticipatethatthis
database will enhance future studies in areas such as immunology and contribute to the
developmentoftherapiesforhumandiseases.
Using RNA-Seq to study immune repertoires has some advantages, including the ability to
simultaneouslycapturebothTandBcellclonotypepopulationsduringasinglerun.Italsoallows

simultaneousdetectionofoveralltranscriptionalresponsesoftheadaptiveimmunesystem,by
comparingchangesinthenumberofBCRandTCRtranscriptstothemuchlargertranscriptome.
Given large number of large-scale RNA-Seq datasets becoming available,we look forward to
scalinguptheatlasofT-andB-cellreceptorsinordertoprovidevaluableinsightsintoimmune
responsesacrossvariousautoimmunediseases,allergies,andcancers.

References
1. Georgiou,G.etal.Thepromiseandchallengeofhigh-throughputsequencingofthe
antibodyrepertoire.Nat.Biotechnol.32,158–168(2014).
2. Freeman,J.D.,Warren,R.L.,Webb,J.R.,Nelson,B.H.&Holt,R.A.ProfilingtheT-cell
receptorbeta-chainrepertoirebymassivelyparallelsequencing.GenomeRes.19,1817–
1824(2009).
3. Rajewsky,K.,Forster,I.&Cumano,A.Evolutionaryandsomaticselectionoftheantibody
repertoireinthemouse.Science(80-.).238,1088–1094(1987).
4. Benichou,J.,Ben-Hamo,R.,Louzoun,Y.&Efroni,S.Rep-Seq:uncoveringthe
immunologicalrepertoirethroughnext-generationsequencing.Immunology135,183–
191(2012).
5. Mangul,S.etal.DumpsterdivinginRNA-sequencingtofindthesourceofeverylastread.
bioRxiv53041(2016).
6. Ardlie,K.G.etal.TheGenotype-TissueExpression(GTEx)pilotanalysis:Multitissuegene
regulationinhumans.Science(80-.).348,648–660(2015).
7. Ben-Dor,A.,Shamir,R.&Yakhini,Z.Clusteringgeneexpressionpatterns.J.Comput.Biol.
6,281–297(1999).
8. Lefranc,M.-P.etal.IMGT{®},theinternationalImMunoGeneTicsinformationsystem{®}
25yearson.NucleicAcidsRes.gku1056(2014).
9. Bolotin,D.A.etal.MiXCR:softwareforcomprehensiveadaptiveimmunityprofiling.Nat.

Methods12,380–381(2015).
10. Li,B.etal.Landscapeoftumor-infiltratingTcellrepertoireofhumancancers.Nat.
Genet.(2016).
11. Stubbington,M.J.T.etal.Tcellfateandclonalityinferencefromsingle-cell
transcriptomes.Nat.Methods(2016).
12. Mose,L.E.etal.Assembly-basedinferenceofB-cellreceptorrepertoiresfromshortread
RNAsequencingdatawithV’DJer.Bioinformaticsbtw526(2016).
13. Strauli,N.&Hernandez,R.StatisticalInferenceofaConvergentAntibodyRepertoire
ResponsetoInfluenzaVaccine.bioRxiv25098(2015).
14. Warren,R.L.,Nelson,B.H.&Holt,R.A.ProfilingmodelT-cellmetagenomeswithshort
reads.Bioinformatics25,458–464(2009).
15. Kuchenbecker,L.etal.IMSEQ—afastanderrorawareapproachtoimmunogenetic
sequenceanalysis.Bioinformatics31,2963–2971(2015).
16. Lefranc,M.-P.etal.SaVanT:Aweb-basedtoolforthesample-levelvisualizationof
molecularsignaturesingeneexpressionprofiles.NucleicAcidsRes.gku1056(2014).
17. Philibert,P.etal.AfocusedantibodylibraryforselectingscFvsexpressedathighlevelsin
thecytoplasm.BMCBiotechnol.7,1(2007).
18. Crooks,G.E.,Hon,G.,Chandonia,J.-M.&Brenner,S.E.WebLogo:asequencelogo
generator.GenomeRes.14,1188–1190(2004).
19. Hoi,K.H.&Ippolito,G.C.Intrinsicbiasandpublicrearrangementsinthehuman
immunoglobulinVλlightchainrepertoire.GenesImmun.14,271–276(2013).
20. Yu,H.-P.,Chiu,Y.-W.,Lin,H.-H.,Chang,T.-C.&Shen,Y.-Z.Bloodcontentinguinea-pig

tissues:correctionforthestudyofdrugtissuedistribution.Pharmacol.Res.23,337–347
(1991).
21. DeRossi,A.etal.InfectionofEpstein-Barrvirus-transformedlymphoblastoidBcellsby
thehumanimmunodeficiencyvirus:evidenceforapersistentandproductiveinfection
leadingtoBcellphenotypicchanges.Eur.J.Immunol.20,2041–2049(1990).
22. Warren,R.L.etal.ExhaustiveT-cellrepertoiresequencingofhumanperipheralblood
samplesrevealssignaturesofantigenselectionandadirectlymeasuredrepertoiresizeof
atleast1millionclonotypes.GenomeRes.21,790–797(2011).















![Adapters in lymphocyte signaling · immunoreceptor (e.g., the T cell receptor [TCR], the B cell receptor [BCR], or the Fc receptors [FcRs]), and a costim-ulatory signal (see Frauwirth](https://img.pdfslide.us/doc/110x75/5c5fb15d09d3f2a3398b4758/adapters-in-lymphocyte-signaling-immunoreceptor-eg-the-t-cell-receptor-tcr.jpg)













