Embed Size (px)
Citation preview

Prof. Tom Ziegler - Department of ChemistryUniversity of Calgary-Calgary,Alberta,Canada T2N 1N4
Density Functional Theory. Approaching Chemistry from First Principle
Thursday Februar 22, 2007 19.00 - 21.00 pm.

400 b.c.
John Dalton (1766 - 1844 )
Early Atomic Theory

"We are perhaps not far removed from the time when we shall be able tosubmit the bulk of chemical phenomena to calculation."-
Joseph Louis Gay-Lussac, Memoires de la Societe d'Arcueil 2:207 (1808)

"Every attempt to employ mathematical methods in the study of chemical questions must be considered profoundly irrational and contrary to the spirit of chemistry.
- A. Comte, Philosophie Positive, 1830.

Atomic Theory
Nield Bohr (1885-1962)
Ernest Rutherford (1871-1937)

H(ri)ψ(ri)= Eψ(ri)
i=1,3N
Quantum Wave-Mechanics
Erwin Rudolf Josef Alexander Schrödinger
1887-1961
Werner Karl Heisenberg
1901-1976
1926

"The underlying physical laws necessary for the mathematical theory of..the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much to complicated to solve."
`P.A.M. Dirac
(1929)
H(ri)ψ(ri)= Eψ(ri)
=1,3i N
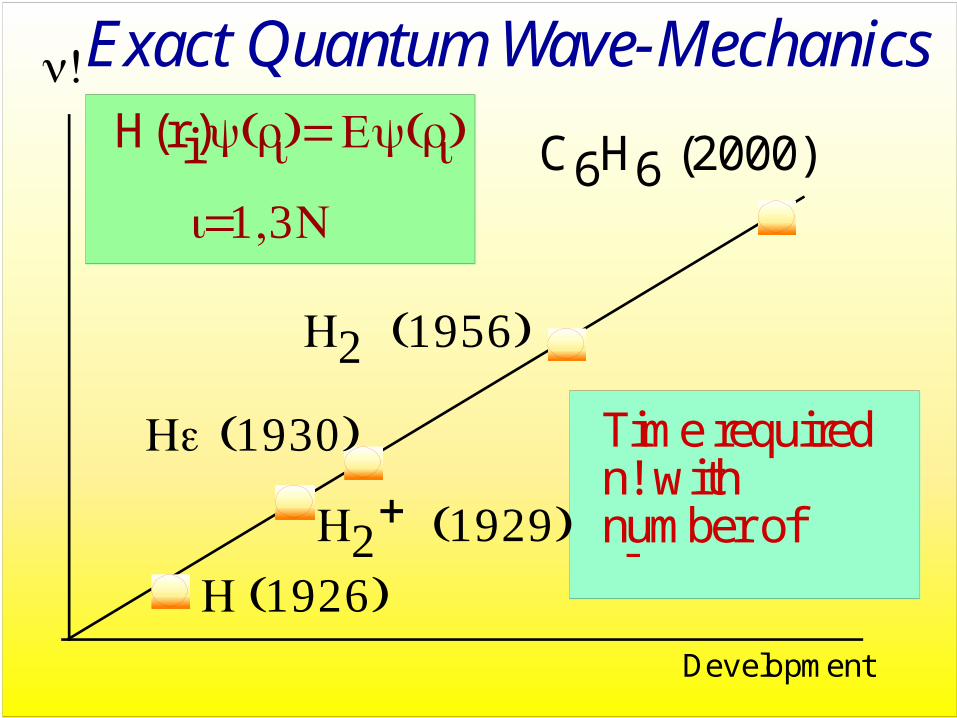
Exact Quantum Wave-MechanicsH(ri)ψ(ri)= Eψ(ri)
i=1,3N
H (1926) H2
+ (1929)
n!
He (1930)
H2 (1956)
Time requiredn! with number of electrons n
C6H6 (2000)
Development
Electron Correlation ?
? ?
Exchange-correlation hole ?

Approximate Quantum Wave-Mechanics
HartreeAtomic Theory
R.K.MullikanMolecular Orbital theory
Some too good to be true
Some theories are too true to be good
L.Pauling J. PopleSystematic ImprovementsValence Bond theory

Approximate Wave mechanics
Hartree Fock (2000 atoms)
log(t)
2
Møller-Plesset (100 atoms)4
Quadratic CI ( 10 atoms)
Dead End

Density Functional theory
Thomas-Fermi-Dirac (1929)Model expression of total energy in terms of electron density
E(ρ)
Fermi
W. Kohn
Kohn-Hohenberg-Sham(1964)
Exact relationship between electrondensity and molecular energy ..
..but, form of relationship not known
E(ρ)
? ?
Exchange-correlation hole ?

Approximate density functional theoriesfor exchange and correlation
HFSLocal exchange
LSDLocal exchange +Local correlation
LSD/NLLocal exchange +
Local correlation+ Nonlocal corrections
Exchange energyfrom electron gas
Exchange+correlationfrom electron gas
Nonlocal Exchange:Becke,A.D. Phys.Rev. 1988,A38
Nonlocal CorrelaionPerdew,J. Phys.Rev. 1986,B33

Calgary
Program Implementations for first generation DFT 1973-1983
Montreal A'dam
Florida
Excellent Molecular geometries and electronic properties
Poor bond and atomization energies
DFT Underground

Axel Becke Queens University
John PerdewTulane
Second generation Gradient Corrected Functionals
1983-1992

Calgary
Implementations for second generation DFT 1983-1993
Montreal A'dam
Florida
Excellent Molecular geometries and electronic properties
Good bond and atomization energies
Zurich
San Diego
DFT-underground
Chicago
Many properties

Nobel Prizes in Chemistry1998
Walter Kohn
E(ρ) = <E ψ|Η|ψ> John Pople

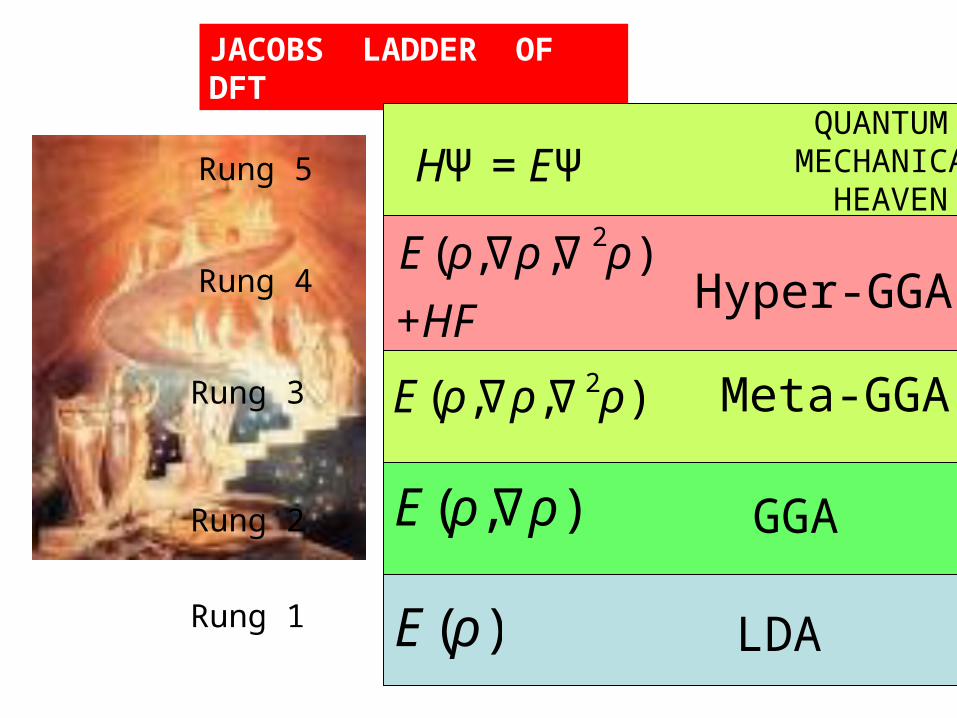
JACOBS LADDER OF DFT
QUANTUM MECHANICAL
HEAVEN
€
E(ρ)
€
E(ρ,∇ρ )
€
E(ρ,∇ρ,∇ 2ρ)
LDA
GGA
Meta-GGA
€
E(ρ,∇ρ,∇ 2ρ)
+HFHyper-GGA
Rung 1
Rung 2
Rung 3
Rung 4
Rung 5
€
HΨ =EΨ
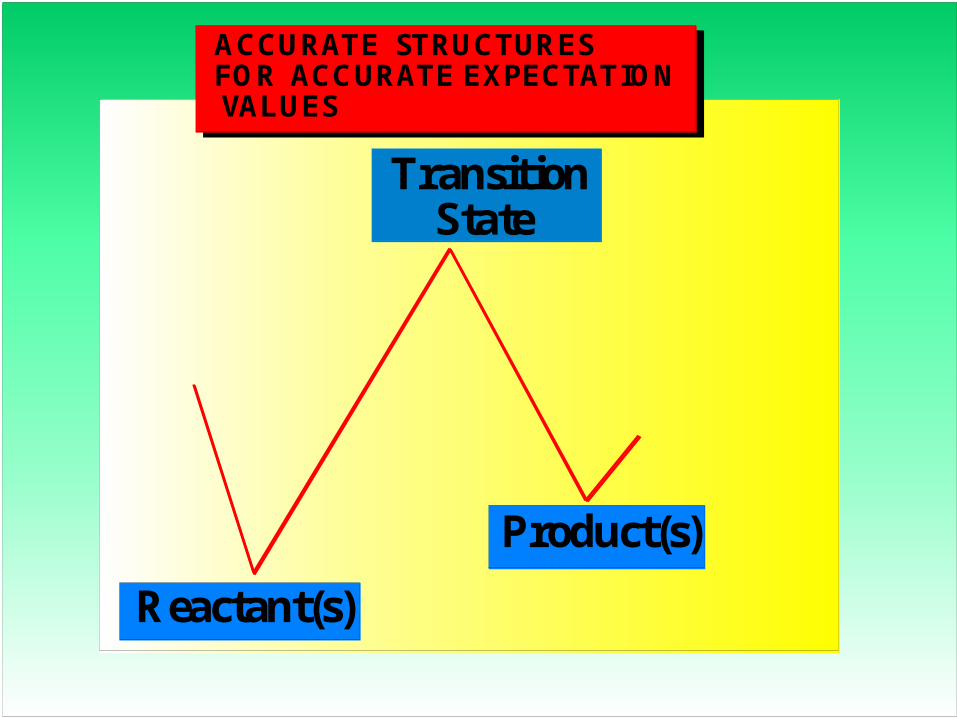
ACCURATE STRUCTURESFOR ACCURATE EXPECTATION VALUES
Reactant(s)
Product(s)
Transition State

Metal-ligand bond distances from HF and Xa calculations
Mol Bond HF Xa EXP
Fe(Cp)2 Fe-Cp 1.88 1.60 1.65
Fe(CO)5 Fe-COax 2.04 1.774 1.80 Fe-COeq 1.874 1.798 1.827
Ni(CO)4 Ni-CO 1.921 1.794 1.838
Cr(CO)6 Cr-C 2.00 1.868 1.914
Fan and Ziegler ,J.Chem.Phys. 1991,95,7401
Fe Cr
CO
OC COOC
CO
CO
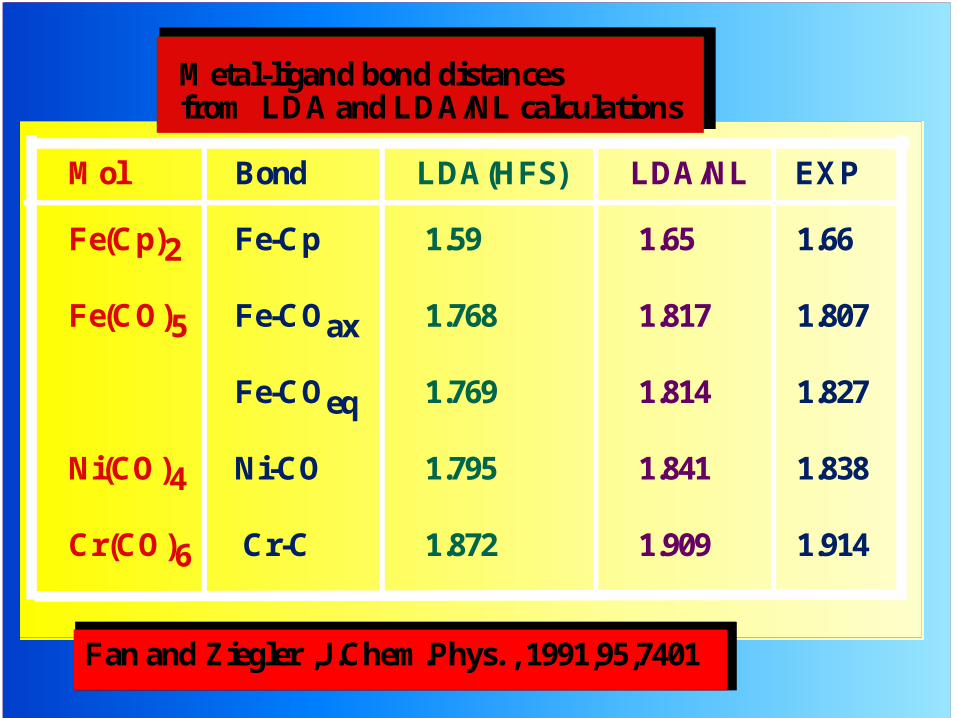
Metal-ligand bond distances from LDA and LDA/NL calculations
Mol Bond LDA(HFS) LDA/NL EXP
Fe(Cp)2 Fe-Cp 1.59 1.65 1.66
Fe(CO)5 Fe-COax 1.768 1.817 1.807
Fe-COeq 1.769 1.814 1.827
Ni(CO)4 Ni-CO 1.795 1.841 1.838
Cr(CO)6 Cr-C 1.872 1.909 1.914
Fan and Ziegler ,J.Chem.Phys. , 1991,95,7401

Method Cr(CO)6 Mo(CO)6 W(CO)6
M-C C-O M-C C-O M-C C-O
LDA 1.866 1.145 2.035 1.144 2.060 1.144
NL-SCF 1.910 1.153 2.077 1.152 2.116 1.154
NL-
SCF+QR 1.910 1.153 2.076 1.153 2.049 1.155
MP2 1.883 1.168 2.066 1.164 2.054 1.166CCSD(T) 1.939 1.178
Exp 1.918 1.141 2.063 1.145 2.058 1.148
Relativity and Structure

that relativistic effects are “of no importance in the consideration of atomic and molecular structure and ordinary chemical reactions.” [P.A.M. Dirac, Proc. R. Soc., London Ser. A 1929, 123, 714.]
"..that relativistic effects are “of no
importance in the consideration of atomic and
molecular structure and ordinary chemical
reactions.” "
`P.A.M. Dirac
(1929)
H(ri)ψ(ri)= Eψ(ri)
=1,3i N
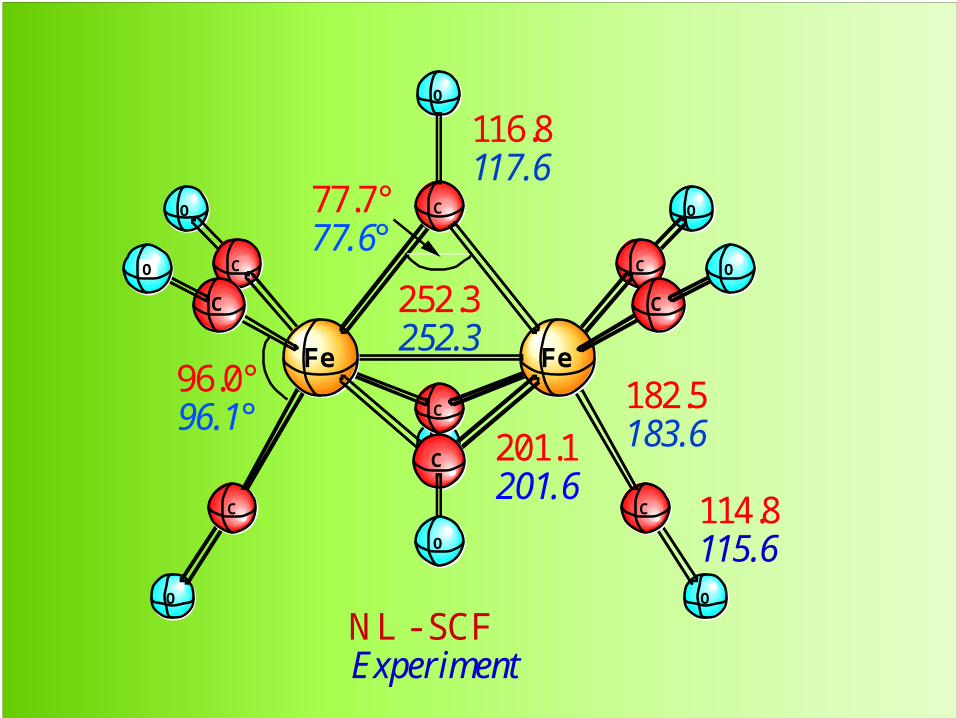
O
O
O O
C
C C
O O
C C
FeFe
C
C
C C
O
O O
252.3252.3
182.5183.6
114.8115.6
201.1201.6
116.8117.6
77.7°77.6°
96.0°96.1°
NL - SCFExperiment

Biological moleculesMetal SurfacesSolids
Biological MoleculesMetal SurfacesSolids
ACCURATE ENERGETICS
Reactant(s)
Product(s)
Transition State

Unsaturated SpeciesGenerating Coordinatively
Unsaturated Species
COLnM LnM
Saturated18-electronsystem
Unsaturated16-electroncatalyst
+ CO
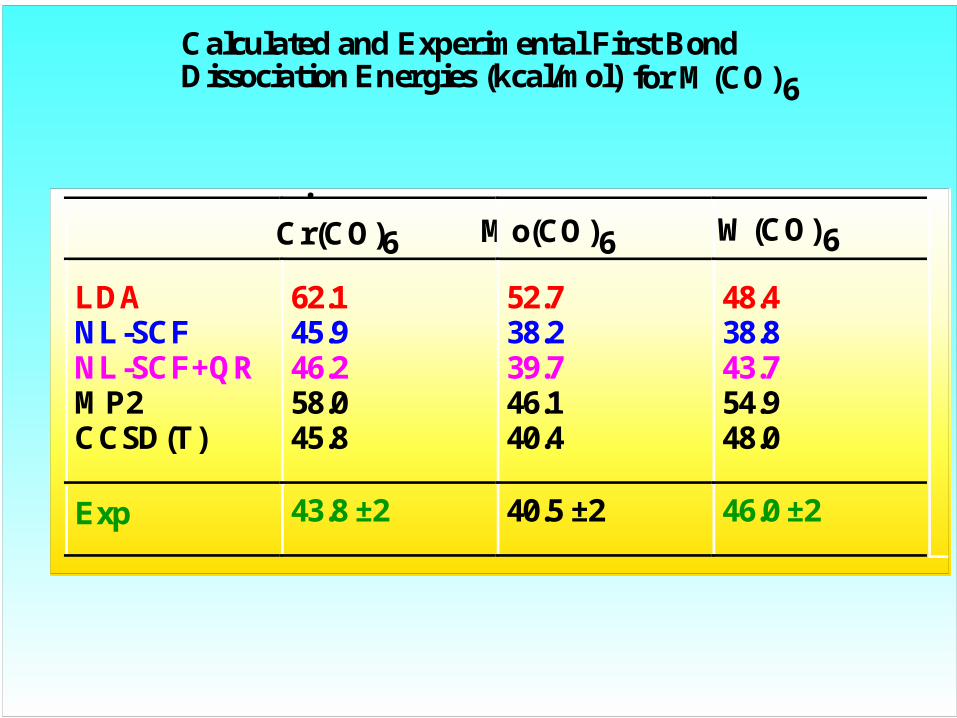
Calculated and Experimental First Bond Dissociation Energies (kcal/mol) for M(CO)6
.
Cr(CO)6 Mo(CO)6 W(CO)6
LDA 62.1 52.7 48.4NL-SCF 45.9 38.2 38.8NL-SCF+QR 46.2 39.7 43.7MP2 58.0 46.1 54.9CCSD(T) 45.8 40.4 48.0
Exp 43.8 ±2 40.5 ±2 46.0 ±2
10
20
30
40a
OO
ΔE (kcal/mol)
10
20
30
40
Ni Pd Pt
Ni Pd Pt
c
ΔE (kcal/mol)
10
20
30
40
Ni Pd Pt
Fe Ru Os
d
10
20
30
40b
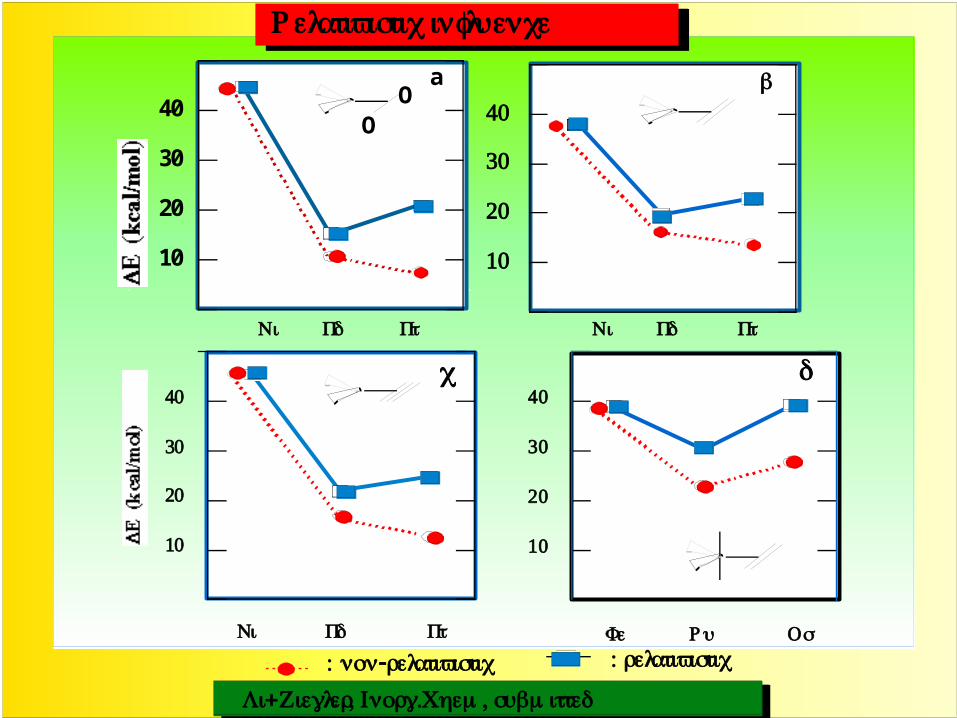
Relativistic influence
: non-relativistic : relativistic
Li+Ziegler, Inorg.Chem, submitted

that relativistic effects are “of no importance in the consideration of atomic and molecular structure and ordinary chemical reactions.” [P.A.M. Dirac, Proc. R. Soc., London Ser. A 1929, 123, 714.]
"..that relativistic effects are “of no
importance in the consideration of atomic and
molecular structure and ordinary chemical
reactions.” "
`P.A.M. Dirac
(1929)
H(ri)ψ(ri)= Eψ(ri)
=1,3i N

Biological molecules ( Hydrogen Bond Strength)
Metal Surfaces (Adsorption Energies)
Solids (Cohesive Energy)
Energetics ???
Vibrations
BondBreaking
Conformations
Excitations IonizationsandAffinities
Spin-flip
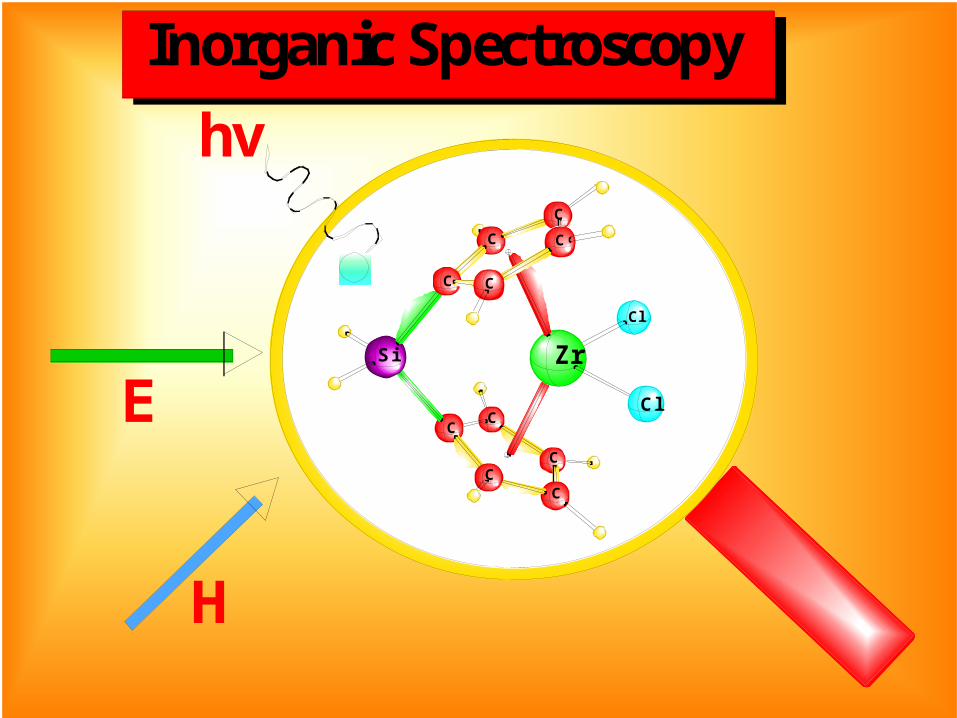
H
E
hv
Cl
C
C
C
C
C
Si Zr
C
C
C
C
Cl
C
Inorganic Spectroscopy

EXCITED STATES
Photo Chemistry
hv
Electron Transfer
Ziegler,Rauk,BaerendsTheor.Chim.Acta1977,43,261
Excitations
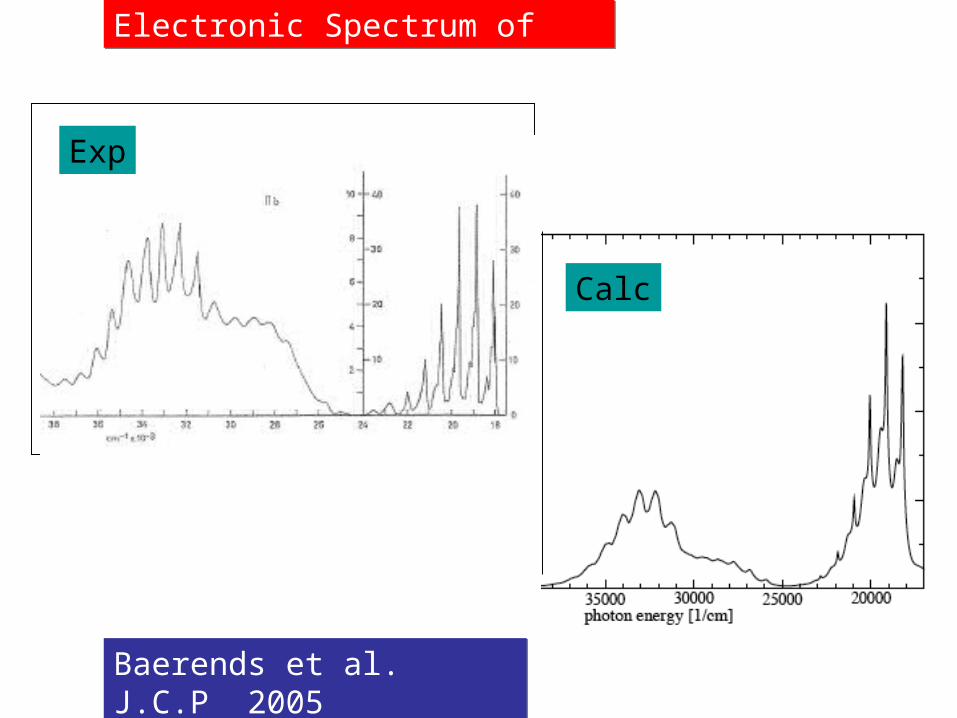
Baerends et al. J.C.P 2005 Baerends et al. J.C.P 2005
Exp
Calc
Electronic Spectrum of Permanganate

cm-1
}Oxygent14t2a1
Metal-d5t2
e
Orbital Diagram
0
1
2
3
4
5
6a1-5t23t2-e
4t2-5t2
t1-5t2
4t2-e
t1-e
AOC
Determinantsonly
1T21T2
1T2
1T2
1T2
1T21T1
1T1
1T1
1T1
1T1
50 000
40 000
30 000
20 000
10 000
eV
0
sharp
broad
sharp
broad
broad
weak
Experiment
Determinantsand integrals
Ground state
3T13T2
cm -1
Charge Transfer Spectrumof Permanganate
R.Dickson + T.Ziegler, Int. J. Quantum Chem., 1995
Mn
O O
O O
ACCURATE FORCE FIELDS
Entropy of activationSpectroscopic finger printing
Molecular Mechanics Force Fields
A.Berces and T.ZieglerTopics in Current ChemistrySpringer 1996
Vibrations
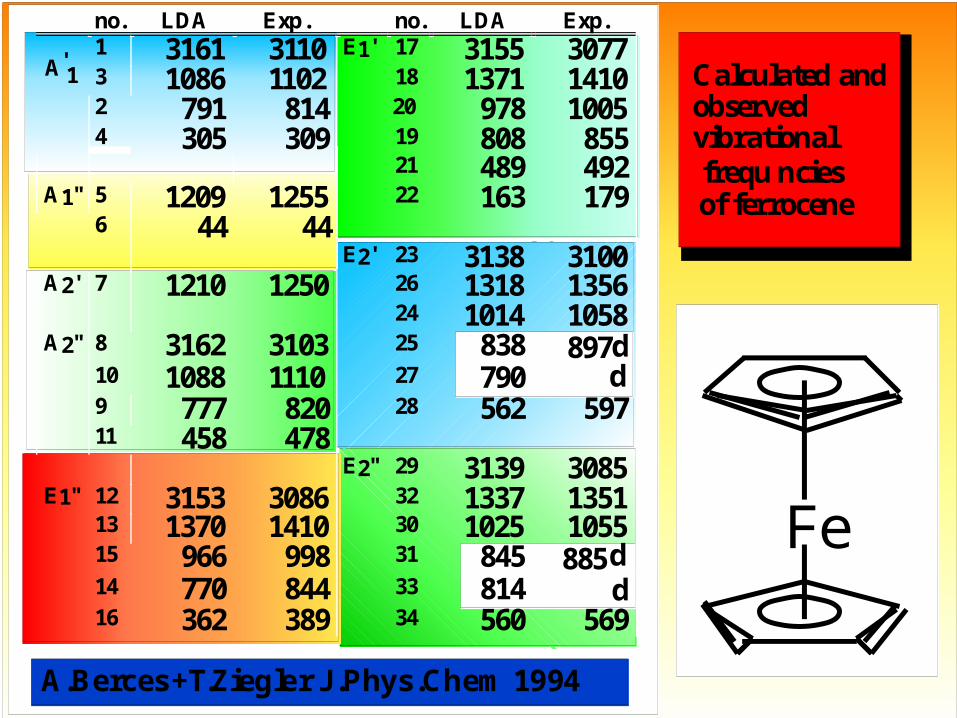
Calculated and observed vibrationalfrequ ncies
of ferrocene
no. LDA Exp. no. LDA Exp.
A 1'
1 3161 3110 E1' 17 3155 30773 1086 1102 18 1371 14102 791 814 20 978 10054 305 309 19 808 855
21 489 492A1" 5 1209 1255 22 163 179
6 44 44E2' 23 3138 3100
A2' 7 1210 1250 26 1318 135624 1014 1058
A2" 8 3162 3103 25 838 897d10 1088 1110 27 790 d9 777 820 28 562 59711 458 478
E2" 29 3139 3085E1" 12 3153 3086 32 1337 1351
13 1370 1410 30 1025 105515 966 998 31 845 885d14 770 844 33 814 d16 362 389 34 560 569
A.Berces+T.Ziegler J.Phys.Chem 1994
Fe
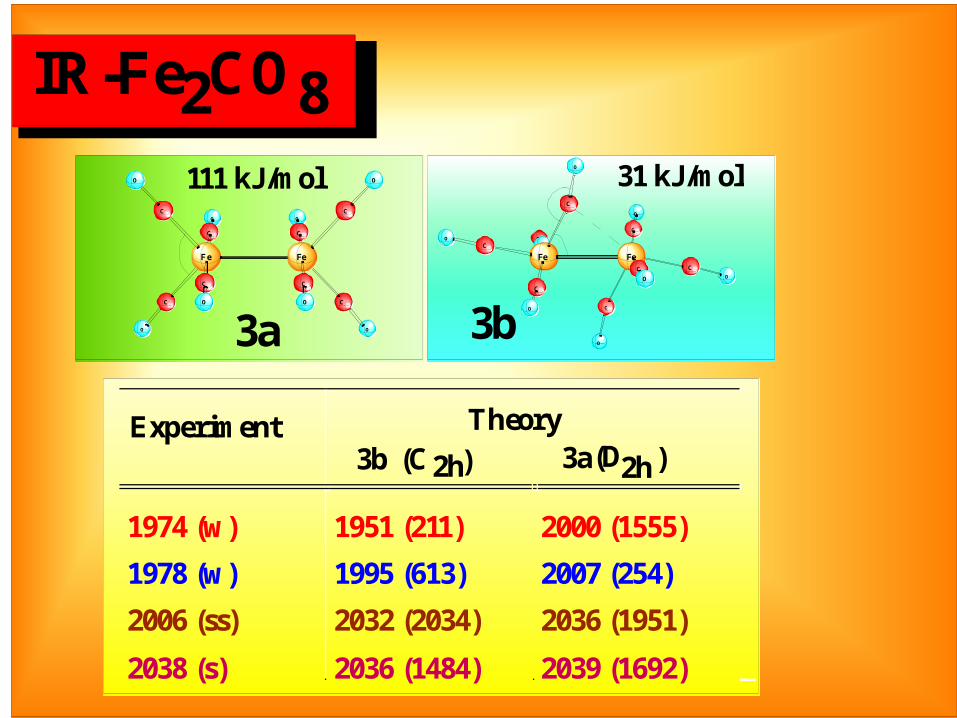
IR-Fe2CO8
O O
C C
O O
C C
Fe Fe
C C
O O
C C
O O
3a
O
O
C
C
O
C
O
C
O
Fe Fe
C
C
O
C
C
O
O
3b
Experiment Theory3b (C2h) 3a (D2h)
1974 (w) 1951 (211) 2000 (1555)
1978 (w) 1995 (613) 2007 (254)
2006 (ss) 2032 (2034) 2036 (1951)
2038 (s) 2036 (1484) 2039 (1692)
111 kJ/mol 31 kJ/mol
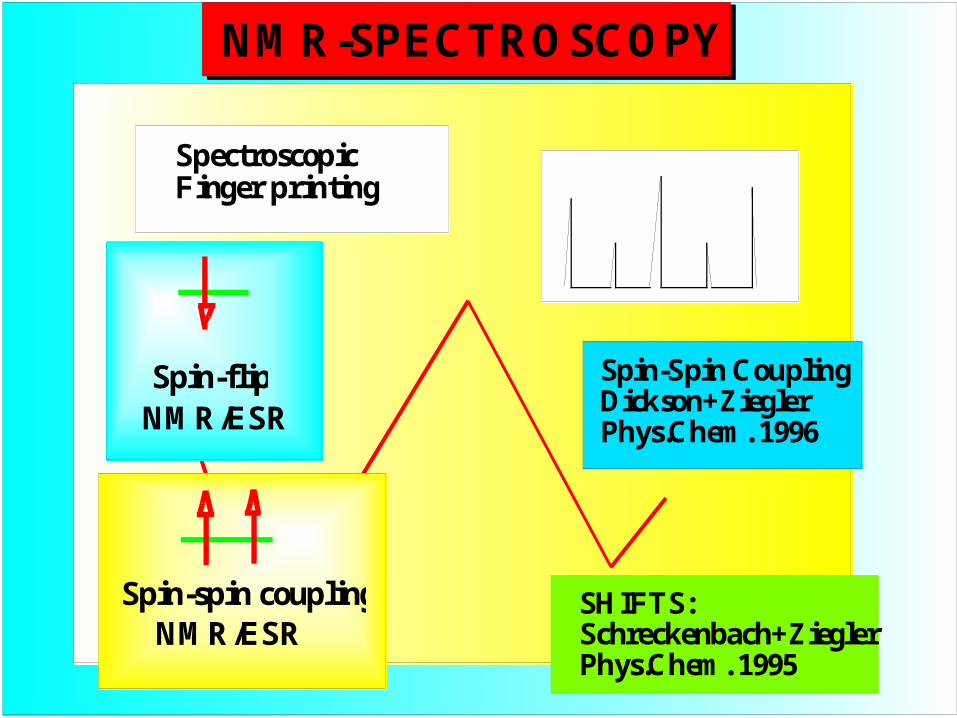
NMR-SPECTROSCOPY
Spectroscopic Finger printing
SHIFTS:Schreckenbach+ZieglerPhys.Chem. 1995
Spin-Spin CouplingDickson+ZieglerPhys.Chem. 1996
Spin-flipNMR/ESR
NMR/ESRSpin-spin coupling
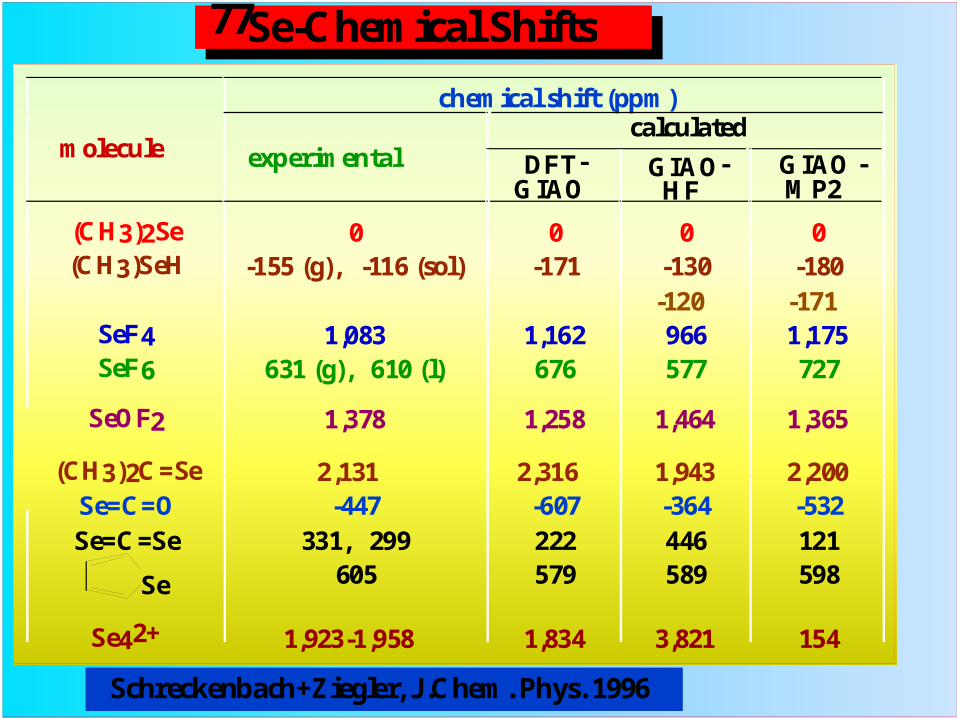
molecule
chemical shift (ppm)
experimentalcalculated
DFT-GIAO
GIAO-HF
GIAO -MP2
(CH3)2Se 0 0 0 0(CH3)SeH -155 (g), -116 (sol) -171 -130
-120-180
-171SeF4 1,083 1,162 966 1,175SeF6 631 (g), 610 (l) 676 577 727
SeOF2 1,378 1,258 1,464 1,365
(CH3)2C=Se 2,131 2,316 1,943 2,200Se=C=O -447 -607 -364 -532Se=C=Se 331, 299 222 446 121
605 579 589 598
Se42+ 1,923-1,958 1,834 3,821 154
Se
Se-Chemical Shifts77
Schreckenbach+Ziegler, J.Chem. Phys. 1996
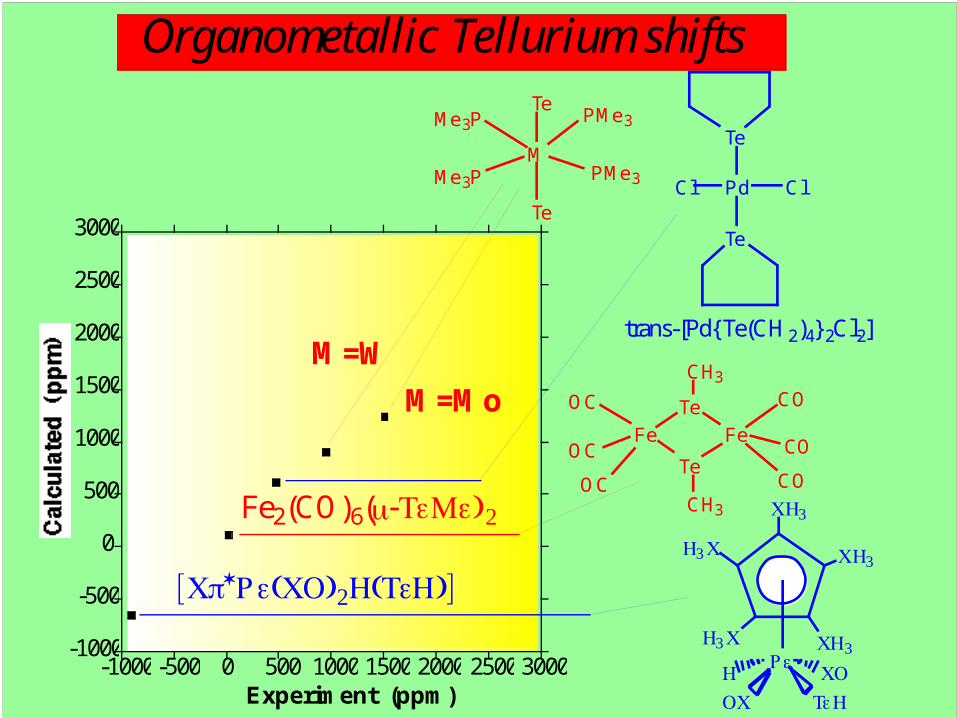
Organometallic Tellurium shifts
-1000
-500
0
500
1000
1500
2000
2500
3000
-1000-500 0 500 1000 1500 2000 2500 3000
Calculated (ppm)
Experiment (ppm)
M
Te
Te
PMe3
PMe3
Me3P
Me3P
M=W
M=Mo
Cl Pd Cl
Te
Te
trans-[Pd{ Te(CH2)4} 2Cl2]
Te
Fe Fe
Te
CO
CO
CO
OC
OC
OC
CH3
CH3Fe2(CO)6(μ-TeMe)2 CH3
CH3
CH3H3C
H3C
Re
TeHOC
COH
[Cp*Re(CO)2H(TeH)]

VibrationsExcitations
IonizationsandAffinities
Spin-flipNMR/ESR
Spin-spin coupling
NMR/ESR
Spectroscopic Energetics

Tungsten compounds
W(CO)6
W(CO)5PF3
W(CO)5PCl3
W(CO)5WI3
cp-W(CO)3HWF6
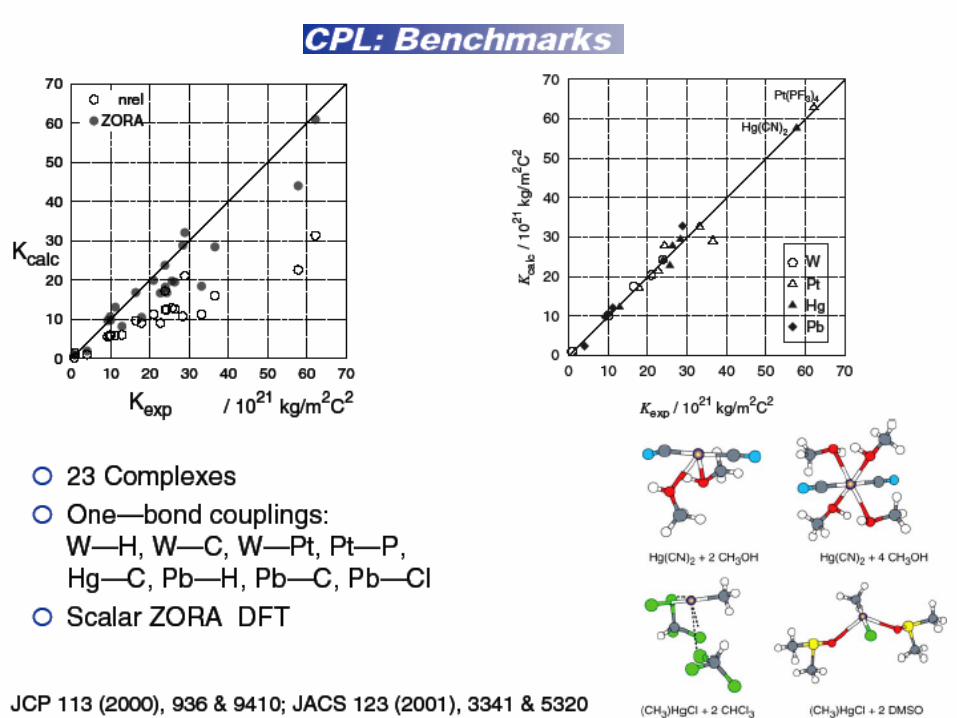
J.Autschbach , T. Ziegler, JCP (2000), 113.9410
Results I : scalar ZORA
Compound Coupling K(NR,GGA) K(Zora,LDA) K(Zora,GGA) K(Exp)
W(CO)6 W-C 603 985 1001 997
W(CO)5PF3 W-P 1726 2383 2435 2380
W(CO)5PCl3 W-P 1126 1997 2041 2090
W(CO)5PCI3 W-P 972 1688 1745 1639
CpW(CO)3H W-H 26 103 96 73
WF6 W-F 151 -96 -94 87
Biological molecules ( ESR of metallo-proteins)
Metal Surfaces (Ir of absorbed molecules)
Solids (NMR of Solids)
Astronomy (Molecules in out-space)
Investigation ofCatalysis
Catalytic Research in Calgary
SpectroscopicFingerprinting
NMR,ESR,IR,UV
Direct Static CalculationsPaths, Barriers,Structures
DynamicsTemperature,Entropy
EnviromentalEffectsSolvents,Counter-ions

Static DFT: Walking on the potential energy surface Static DFT: Walking on the potential energy surface
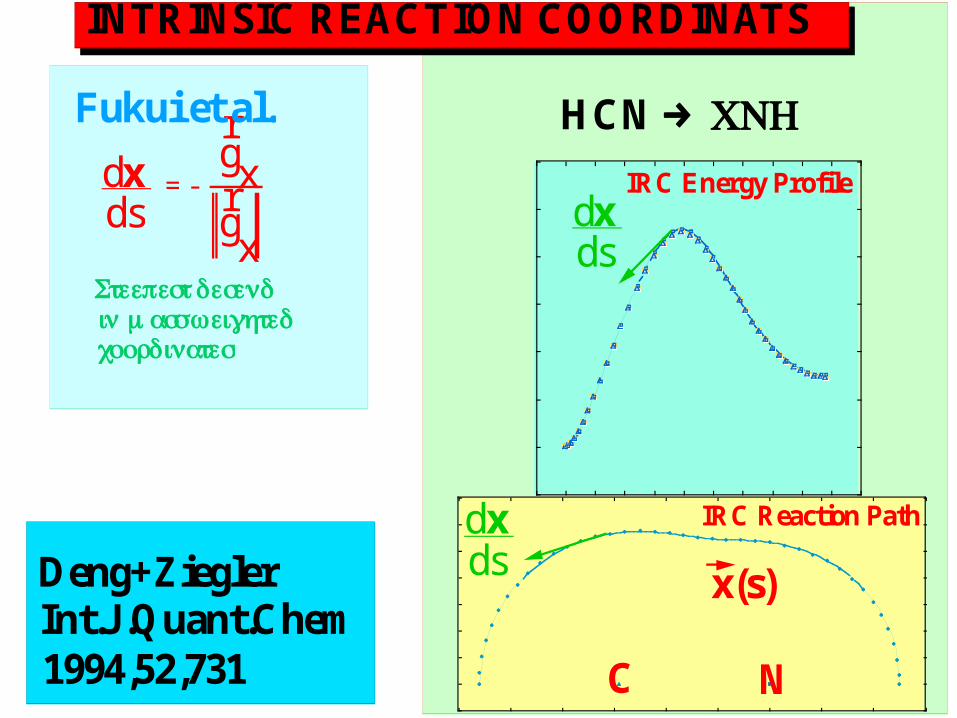
INTRINSIC REACTION COORDINATS
= -
dxds
r g xr g x
dxds
Deng+ZieglerInt.J .Quant.Chem1994,52,731
IRC Energy Profile
x(s)
C N
dxds
IRC Reaction Path
Fukui et al. HCN → CNH
Steepest desendin mass weightedcoordinates

CrCl2O2 + CH3OH → CrCl2(OH)2+ CH2O
+
Ha
a
b
cd
e
f
g
h
i
TS1
Alcohol OxidationO-H Addition
#
Reactants
Addition Product
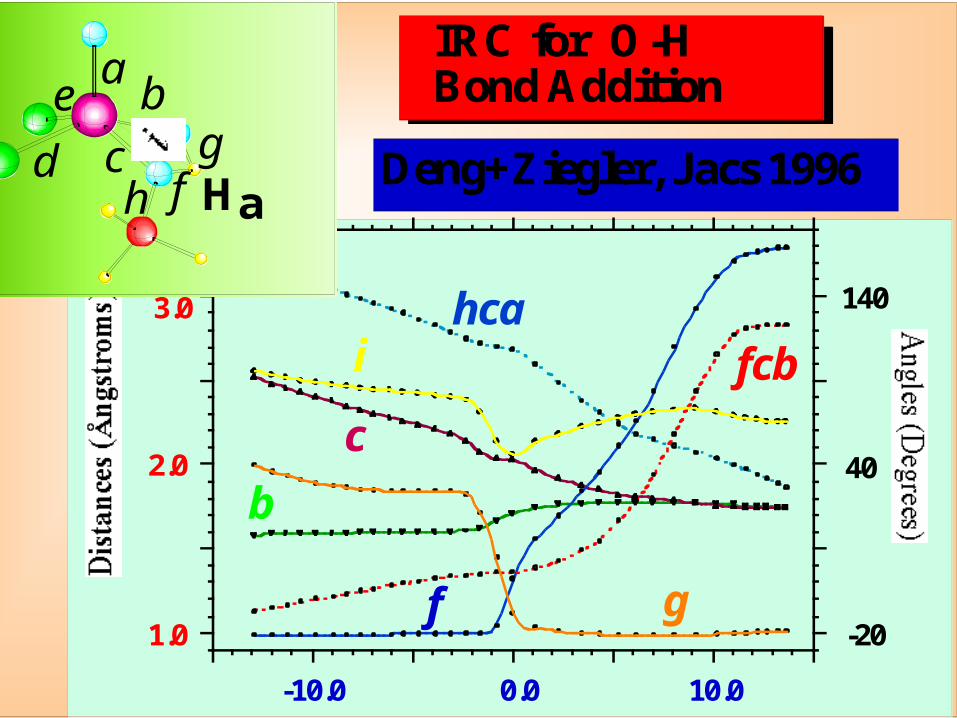
IRC for O-H Bond Addition
Deng+Ziegler, Jacs 1996
1.0
2.0
3.0
-20
40
140
Distances (Ångstroms) Angles (Degrees)
0.0-10.0 10.0
Ha
ab
cd
e
fg
h
i
f g
b
ihca
c
fcb

QuickTime™ and aH.264 decompressor
are needed to see this picture.
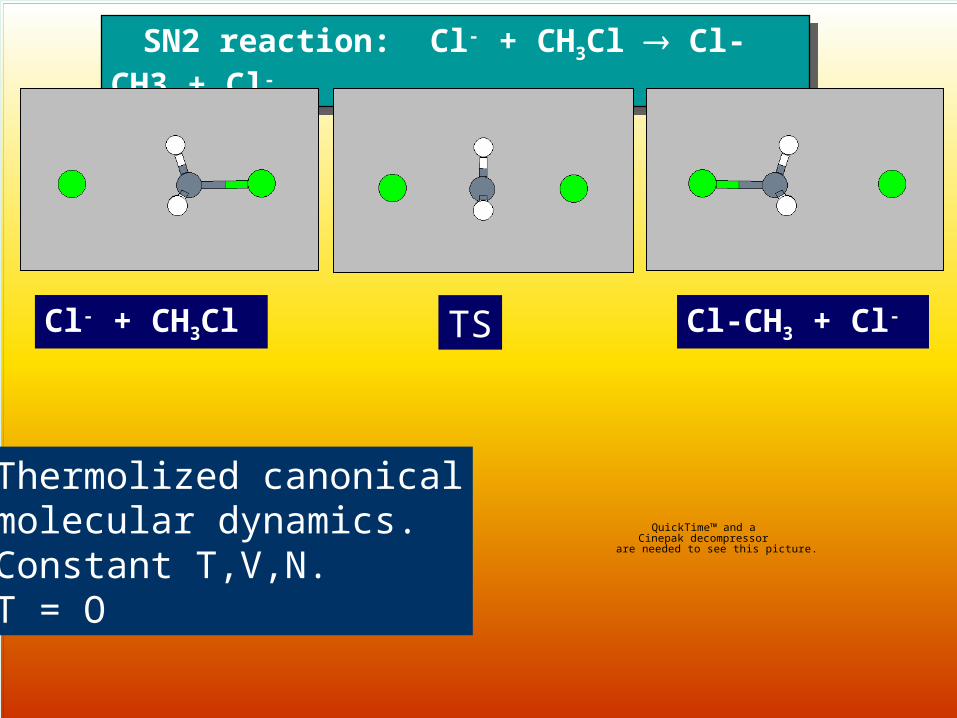
SN2 reaction: Cl- + CH3Cl Cl-CH3 + Cl- SN2 reaction: Cl- + CH3Cl Cl-CH3 + Cl-
Cl- + CH3Cl TS Cl-CH3 + Cl-
Thermolized canonicalmolecular dynamics. Constant T,V,N.T = O
QuickTime™ and aCinepak decompressor
are needed to see this picture.

SN2 reaction: Cl- + CH3Cl Cl-CH3 + Cl- SN2 reaction: Cl- + CH3Cl Cl-CH3 + Cl-
Cl- + CH3Cl TS Cl-CH3 + Cl-
IRC-MD (P TS P):
Thermolized canonicalmolecular dynamics. Constant T,V,N.T > O
QuickTime™ and aCinepak decompressor
are needed to see this picture.

Thermodynamic Integrationn A constraint is applied to the reaction
coordinate during the dynamicsn During the simulation, the value of the
constraint is changed, leading the systemover the barrier
n Free energy differences along the RC arecalculated by
λ
=-Force δ /E δ λ
Δ F =
∂ E
∂ λ
λ
∫
λ , T
d λ
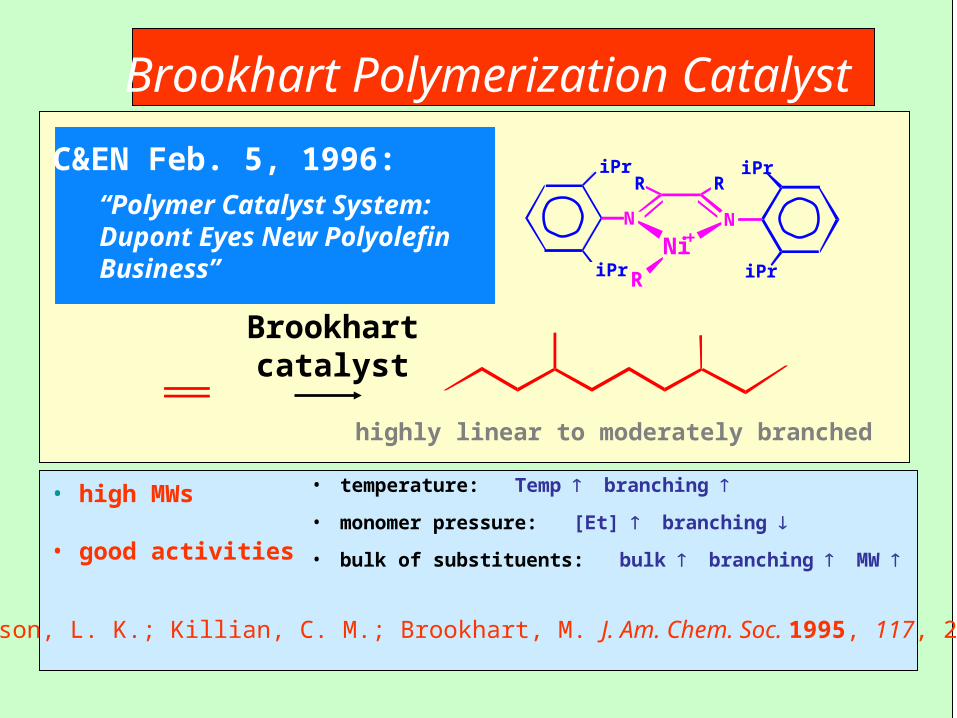
Brookhart Polymerization Catalyst
C&EN Feb. 5, 1996:“Polymer Catalyst System:Dupont Eyes New Polyolefin Business”
Brookhartcatalyst
highly linear to moderately branched
Johnson, L. K.; Killian, C. M.; Brookhart, M. J. Am. Chem. Soc. 1995, 117, 2343.
• high MWs
• good activities
N
Ni+N
RRiPr
iPriPr
iPr
R
• temperature: Temp branching
• monomer pressure: [Et] branching
• bulk of substituents: bulk branching MW

N
NiN C
iPr
iPriPr
iPr
C
ClH
Cl H
HCl
HCl
Cl
H
ClH
Cl
Cl
HH
H
HClCl
HH
Cl Cl
H
H
ClClCl
Cl
HH
Cl
HHCl
+ B-
Cl
H
ClH
Cl
H
ClH
Cl
Cl
HH
Cl
Cl
HH
Cl
H
Cl H
H
HClCl
HH
Cl Cl
Including Steric Bulk and Solvation :Typical Polymerization System

Polar copolymerization – diimine catalysts
QuickTime™ and aCinepak decompressor
are needed to see this picture.

N
NiNH H+
no substituents
0 K simulation(static)
no solvent
no counter-ion
Including Steric Bulk and SolvationTraditional Computational Models
gas phasemodel system
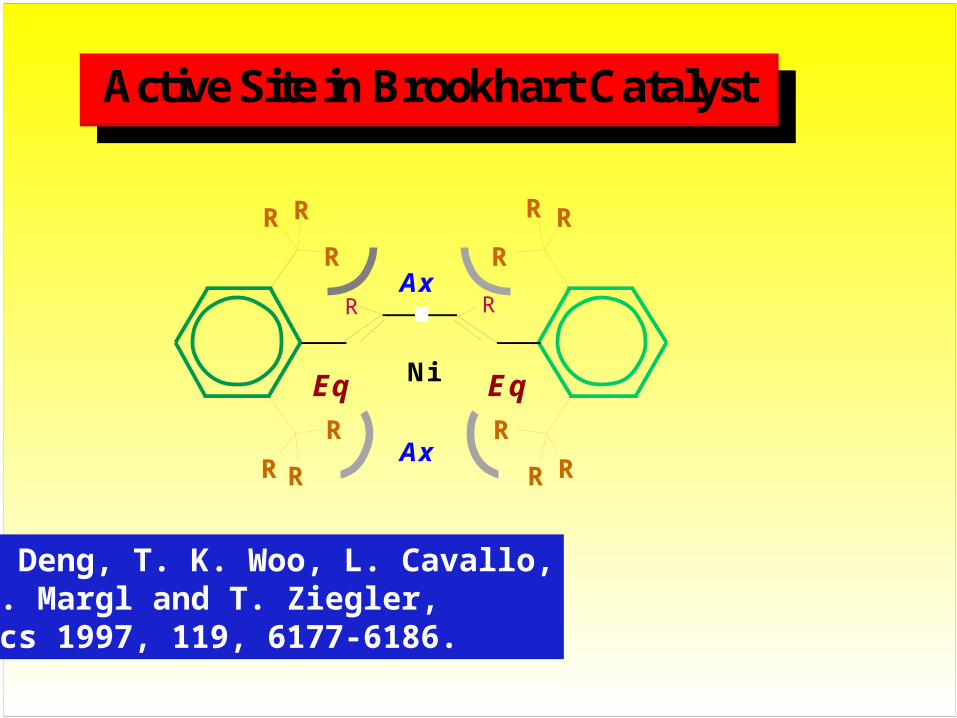
Ax
NiEq Eq
Ax
R
RR
R
RR
R
R R
R
R R
Active Site in Brookhart Catalyst
R R
L. Deng, T. K. Woo, L. Cavallo, P. Margl and T. Ziegler, Jacs 1997, 119, 6177-6186.

Including Steric Bulk and SolvationContinuum Model
explicit solvent
QM solute
_
+
+
_ ++
_+
+
_
++
_
+
+
_+
+ _ +
+
_
+
+
_++
_+
+
_+
+_ +
++
+
+
++
+
continuum
QM solute
__
_
__
__
a b
1. COSMO : Klamt, A.; Schuurmann, G. J. Chem. Soc. Perkin Trans. 1993, 2, 799.
2. PCM : Tomasi, J. Chem. Rev. 1994, 94, 2027.

energy relative to free species(kcal/mol)
2.0 3.0 4.0 5.0 6.0 7.0
olefin midpoint-Ni distance (Å)
5 : Ar = 2,6-C6H3(i-Pr)2, R'=H
6 : Ar = 2,6-C6H3(i-Pr)2, R'=Me
4 : Ar = H, R'=H (pure QM)
0
-5
-10
-15
-20
Enthalpy of Capture

0.0
2.0
4.0
6.0
8.0
10.0
12.0
∆F (kcal/mol)
2.50 3.00 3.50 4.00 4.50 5.00 5.50
Ni - Olefin midpoint distance (Å)
654
Free Energy of Capture

Ni
R=MeR=H
Ni
A QM/MM study of Monomer Capture in Brookhart’s Catalyst
R’ substituent effect found to be both electronic and steric effect
indirect steric effect
Woo,Ziegler J,Phys.Chem. A,1997


PRF
Mitsui

Moving to Calgary Canada

Calgary Skyline

University of Calgary

Banff Rocky Mountain National Park

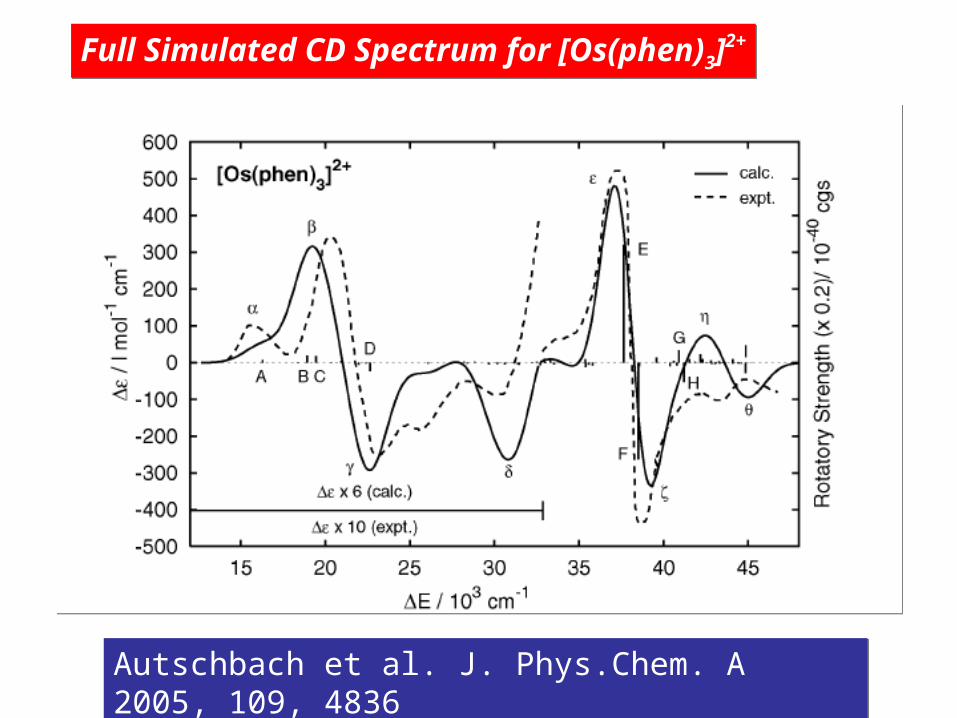
Autschbach et al. J. Phys.Chem. A 2005, 109, 4836 Autschbach et al. J. Phys.Chem. A 2005, 109, 4836
Full Simulated CD Spectrum for [Os(phen)3]2+Full Simulated CD Spectrum for [Os(phen)3]2+





























