Embed Size (px)
Citation preview

6 (2006) 294–312www.elsevier.com/locate/jmicmeth
Journal of Microbiological Methods 6
Production, characterisation and potential application of anovel monoclonal antibody for rapid identification of
virulent Listeria monocytogenes
Stephen Hearty a,b, Paul Leonard a,b, John Quinn a,1, Richard O'Kennedy a,b,⁎
a School of Biotechnology, Dublin City University, Dublin 9, Irelandb National Centre for Sensor Research (NCSR), Dublin City University, Dublin 9, Ireland
Received 29 July 2005; received in revised form 14 December 2005; accepted 19 December 2005Available online 2 February 2006
Abstract
A panel of hybridomas was produced using intact Listeria monocytogenes serotype 1/2a cells as the immunogen. An IgG2amonoclonal antibody (mAb) ‘mAb2B3’ was isolated that reacted with L. monocytogenes but not with a representative panel ofrelated Listeria spp. and non-Listeria spp. Binding activity was greatest against L. monocytogenes serotype 1/2a and wassignificantly enhanced when cells were prepared in Listeria enrichment broth (LEB). The reactive epitope was deduced, byimmunoblot analysis, to be a surface localised protein of approximately 80 kilodaltons (kDa), putatively assumed to be internalin A(InlA). Recombinant InlA protein was subsequently expressed in Escherischia coli. When crude E. coli cell lysates were subjectedto immunoblot analysis, it was demonstrated that the mAb bound specifically to the heterologously expressed recombinant InlAprotein, thus confirming the specificity of the mAb. The mAb was further evaluated in a series of enzyme-linked-immunosorbentassay (ELISA)-based formats and in a surface plasmon resonance (SPR)-based biosensor platform. Both configurations werecapable of differential identification of virulent L. monocytogenes at concentrations greater than or equal to 1×107 cells/ml.Notwithstanding the apparent insensitivity, the results indicate that InlA could be exploited as a marker for highly specificconfirmatory identification of pathogenic L. monocytogenes following primary enrichment of suspect food samples, using the anti-InlA antibody ‘mAb2B3’, described herein.© 2005 Elsevier B.V. All rights reserved.
Keywords: Listeria monocytogenes; Monoclonal antibody; Surface plasmon resonance (SPR)
1. Introduction
Listeria monocytogenes is ubiquitous in the naturalenvironment and is frequently isolated from both
⁎ Corresponding author. School of Biotechnology, Dublin CityUniversity, Dublin 9, Ireland. Tel.: +353 1 7005319; fax: +353 17005412.
E-mail address: [email protected] (R. O'Kennedy).1 Present address: Texas Instruments Inc., 13536 North Central
Expressway, MS 945, Dallas, TX 75243, USA.
0167-7012/$ - see front matter © 2005 Elsevier B.V. All rights reserved.doi:10.1016/j.mimet.2005.12.009
domestic (Duggan and Philips, 1998), and foodprocessing environments (Blackman and Frank, 1996;Gravani, 1999; Aguado et al., 2004). The associatedpotential for significant contamination is exacerbated byvirtue of the organism's ability to tolerate high saltconcentrations, acidic media, modified atmosphericpackaging and the capacity to multiply at refrigerationtemperatures. In 2000, the Centres for Disease Controland Prevention (CDC) reported that of all the foodbornepathogens tracked by the CDC, L. monocytogenes had

295S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
the second highest case fatality rate (21%) and thehighest hospitalisation rate (90.5%) (Anonymous,2003). Thus, L. monocytogenes is of paramount concernwithin the food industry, and improved methods formore reliable, specific and rapid detection and identi-fication of this insidious pathogen are constantly beingsought.
The European Commission has followed the US FDAin adopting a ‘zero tolerance’ attitude towards L.monocytogenes in ‘ready-to-eat’ (RTE) foods in itsproposed draft guidelines on the application of generalprinciples of food hygiene to the management of L.monocytogenes in foods (Anonymous, 2001, 2004).Manufacturers have incorporated the general principlesof food hygiene and, in particular, the Hazard AnalysisCritical Control Point (HACCP)-concept as described inthe Food Hygiene Directive 93/43/EEC. However,HACCP is not a ‘final solution’ to the problem ofListeria in food. It is particularly difficult to generalisecontrol points, with regard to L. monocytogenescontamination in food, due to the bacterium's ubiquitouspresence, robustness and ability to multiply at refriger-ation temperatures. To this effect, in a 1999 FederalRegister notice, the USDA advised manufacturers ofready-to-eat meat and poultry products of the need toreassess HACCP plans to ensure that they adequatelyaddress L. monocytogenes. This is in addition to,suggestions from the USDA for the incorporation offinished product testing as a verification tool (Donnelly,2001). Failure to detect L. monocytogenes in food priorto product release can result in product recalls. Thecost incurred with such recalls is frequently, signifi-cant, in both financial terms, and in relation toconsumer confidence and allegiance. In 2002, 27.4million pounds of turkey produce were recalled underthe Wampler Foods brand of Pilgrims Pride corp.,making it the largest meat recall in the history of theUnited States. This resulted in several fatalities and asubsequent nationwide class-action lawsuit filed inNovember 2002.
In Europe, the Rapid Alert System for Food and Feed(RASFF) was established under Article 8 of Directive92/59/EEC, to provide a procedure to inform thememberstates of when a product represents a serious risk for thehealth and safety of consumers (http://europa.eu.int/comm/food/food/rapidalert/index_en.htm). On 21stFebruary 2002, RASFF received a new legal base(Regulation (EC) No. 178/, 2002) to extend the system toinclude all food and feed products finally destined forhuman consumption. Data for the year ending 2004(http://europa.eu.int/comm/food/food/rapidalert/repor-t2004_en.pdf) indicated that L. monocytogenes was
responsible for 22% of total notifications of microbio-logical contamination. Thus, it is clearly apparent thatboth the food industry and the consumer would benefitfrom an efficient means of rapidly screening end producefor the presence of L. monocytogenes.
Contemporary and traditional methods of detection,specifically pertaining to food samples, rely exclusivelyon the use of enrichment and selective growth media toisolate and enumerate viable bacterial cells from foodsamples, followed by serological and biochemicalconfirmatory tests (e.g., ISO 11 290-1:1996a,b; ISO10 560-1:1999), as validated by Scotter et al. (2001a,b).Beumer and Hazeleger (2003), assessed the diagnosticproblems associated with such ‘horizontal’ methods ofanalysis. Ostensibly, the overwhelming limitation sur-rounds the labour-intensive and time-consuming natureof culture-based methods that can require 5–10 days tocomplete (Klein and Juneja, 1997; Gasanov et al.,2005). As a result, much emphasis is currently beingplaced on expediting the detection of pathogens, such asL. monocytogenes using biosensor-based approaches(Ivnitski et al., 1999; Leonard et al., 2003, 2004, 2005;Deisingh and Thompson, 2004).
The Biacore™ 3000 instrument is an opticalbiosensor that exploits the phenomenon of surfaceplasmon resonance (SPR) (Kretschmann and Raether,1968; Kretschmann, 1971) to monitor molecular bindingevents, in an approach commonly referred to as‘biomolecular interaction analysis’ (Löfås et al., 1991;Liedberg et al., 1995; Malmqvist, 1999; Nagata andHanda, 2000). The main advantage of the SPR-format isthat it allows automated, ‘real-time’ and ‘label-free’analysis of antibody–antigen interactions. It is antic-ipated that such technologies could readily expeditethe process of confirming the presence of pathogenicL. monocytogenes in food samples following a periodof primary enrichment, thereby obviating the need forselective plating and subsequent biochemical orgenetic confirmatory identification. If the develop-ments in sensor technology are to be fully exploitedfor this purpose, the availability of highly specificanti-L. monocytogenes antibodies is an absoluteprerequisite.
Many attempts have been made to produce Listeria-specific antibodies (Bhunia, 1997). However, most are oflimited potential for the specific immunodetection of L.monocytogenes. Several reportedly react with numerousListeria genus members (Butman et al., 1988; Mattinglyet al., 1988; Siragusa and Johnson, 1990; Torensma etal., 1993; Loiseau et al., 1995), while others react with L.monocytogenes, in addition to non-pathogenic L.innocua (Bhunia et al., 1991; Bubert et al., 1994;

296 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
Kathariou et al., 1994; Sølve et al., 2000). Inability todifferentiate between live and dead cells is also apervading limitation (Butman et al., 1988; Siragusa andJohnson, 1990). Kathariou and co-workers successfullygenerated antibodies capable of reliably discriminatinglive from dead L. monocytogenes serotype 4b cells, yetthese antibodies failed to recognise other L. monocyto-genes serovars, including the 1/2 (a, b and c) varieties,that consistently predominate in food isolates (Boerlinand Piffaretti, 1991; Rocourt, 1994; Delgado da Silva etal., 2001; Pak et al., 2002; Okutani et al., 2004).
Thus, there is still a major requirement for antibodiescapable of reliably and specifically confirming thepresence of viable and virulent L. monocytogenes cells,for the development of immunoassays/immunosensorssuitable for routine analysis.
2. Materials and methods
2.1. Bacterial strains
The following Listeria strains were provided by GaryWyatt (Institute for Food Research (IFR), Norwich, UK)and were originally sourced from the either the NationalCollection of Type Cultures (NCTC), Central PublicHealth Laboratory, 61 Colindale Avenue, London NW95HT, UK or the American Type Culture Collection(ATCC), Manassas, Virginia, USA: L. monocytogenes1/2a (NCTC 4886), L. monocytogenes 1/2c (NCTC5348), L. monocytogenes 4a (NCTC 5214), L. mono-cytogenes 4b (NCTC 4885), L. innocua 6a (NCTC11288), L. innocua 6b (NCTC 11289), L. welshimeri(NCTC 11857) and L. seeligeri (ATCC 35967). Thestrain designated L. ivanovii PHLS was also obtainedfrom Gary Wyatt at IFR. It was originally sourced as a‘serotype unconfirmed’ L. ivanovii isolate, by the UKPublic Health Laboratory Service (UK PHLS). L.ivanovii PA (NCTC 11846) was obtained from thePublic Analyst (PA) laboratory, Dublin, Ireland. The‘non-Listeria’ bacterial strains; Escherischia coli(NCIMB 9485), Enterobacter aerogenes (NCIMB10102), Enterococcus faecilis (NCIMB 775), Bacillussubtilis (NCIMB 1650), Bacillus cereus (NCIMB 3329)were supplied by the Technical Support Laboratory,School of Biotechnology, DCU and were originallysourced from the National Collection of Food, Industrialand Marine Bacteria (NCIMB) Ltd., 23 St. MacharDrive, Aberdeen AB24 3RY Scotland, UK.
The commercial strain ‘E. coli XL10-Gold®’,(genotype: TETR Δ(mcrA) 183 Δ(mcrCdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte[F' proAB lacIqZΔM15 Tn10 (TetR) Amy CamR]) was
obtained from Stratagene, North Torrey Pines Rd., LaJolla, Ca. USA.
2.2. Production of monoclonal antibody
Six week old female BALB/c mice (Harlan UK Ltd.,Bicester, Oxon, OX6 0TP) were immunised via theperitoneal cavity with 1×107 formalin-inactivated L.monocytogenes serotype 1/2a cells, prepared 1 :1 withsterile-filtered PBS and Freund's Complete adjuvant(FCA). Subsequent boosts were prepared in sterile-filtered PBS and administered by intraperitoneal (i.p.)injection over a period of 5 months. The final boost wasadministered in sterile-filtered PBS on day 1 beforefusion, again via the i.p. route. Splenocytes from the im-munised mouse were harvested, resuspended in DMEM(Sigma, D5671) lacking fetal calf serum (FCS), countedand fused with Sp2/0 myeloma cells at a ratio of 5–10 :1in the presence of 50% (w/v) PEG (Sigma, P7181).Hybridomas were selectively cultured in DMEM con-taining FCS (5% (v/v)), Briclone (5% (v/v)) (Archport,Dublin, Ireland) and HAT supplement.
Monoclonal antibody was purified from hybridomasupernatant by affinity chromatography using a ProteinA-sepharose 4B column and desalted using a PD10column (Amersham Pharmacia Biotech). The rabbitpolyclonal antibodies designated ‘anti-Lm1’ and ‘anti-Lm2’ were produced ‘in-house’ and have been previ-ously described (Leonard et al., 2004, 2005).
2.3. Estimation of antibody activity towards cells (directcell-binding ELISA)
All bacterial cells were grown to exponential phase inBHI (Oxoid, CM225) or Listeria enrichment broth(LEB) base (Oxoid, CM0863), as specified in therespective results section, and then harvested andwashed three times in 0.5% of the original culturevolume of PBS. The wells of a Nunc Maxisorp™ platewere coated with 100 μl aliquots of respective test cellsat a concentration of 1×109 cells/ml, or where a mixedcell suspension was used, i.e., L. welshimeri, L. seeligeriand L. innocua (‘L. wsi’), a cumulative cell concentra-tion of 3×109 cells/ml in PBS. The plates wereincubated at 37 °C for 1 h and then blocked with a5% (w/v) solution of dry skimmed milk (PremierInternational Foods Ltd., UK) in PBS. A 100 μl sampleof test hybridoma supernatant or purified monoclonalantibody, diluted in PBS/T (PBS containing 0.05% (v/v)Tween 20) was added to the wells of the plate. Sampleswere incubated at 37 °C, for 1 h, on a rocking platform.The wells were washed and 100 μl of secondary

297S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
antibody (alkaline phosphatase-labeled goat anti-mouseIgG2a antibody), diluted in PBS/T containing 1% (w/v)milk powder (PBS/TM), was added. After a 1 hincubation at 37 °C the wells were washed and 100 μlof SigmaFast™ para-nitrophenyl phosphate (pNPP)substrate (Sigma, N2770) was added per well. Thesubstrate was left to develop in the dark at 37 °C for 30min. Absorbance readings were read at 405 nm using amicroplate reader (Titertek Twinreader® PLUS,EFLAB, Finland). The binding response levels againsteach respective cell type were normalised by dividingthe absorbance level obtained in test wells by thatobtained in parallel control wells (i.e., wells that weretreated with diluent buffer containing zero monoclonalprimary antibody).
2.4. Extraction of Listeria surface proteins
Representative members of the Listeria genus weregrown to exponential phase in BHI or LEB and thenharvested and washed three times in 0.5% of the originalculture volume of PBS. Surface proteins were solubi-lised by resuspending the washed cell pellet in 2% (w/v)SDS at 37 °C for 45 min, while shaking at 125 rpm.
2.5. SDS polyacrylamide gel electrophoresis andWestern blotting
Polyacrylamide gel electrophoresis (PAGE) wascarried out using the discontinuous system, in thepresence of sodium dodecyl sulphate (SDS), as de-scribed by Laemmli (1970). A 12.5% (w/v) resolvingand 5% (w/v) stacking acrylamide gels were usedthroughout. Proteins were transferred from acrylamidegels to nitrocellulose membranes using a BioRad wetblotter, operating at 72 V for 90 min, in electrophoresisbuffer containing 20% (v/v) methanol. The membranewas blocked with PBS containing 5% (w/v) skimmedmilk powder overnight at 4 °C. Purified monoclonalantibody at a 1 /2000 dilution in PBS/TM was thenadded to the membrane and incubated for 1.5 h at roomtemperature on a rocking platform. The membrane waswashed four times in PBS/Tand then incubated with a 1 /2500 dilution of alkaline phosphatase-labelled, goat anti-mouse IgG2a detection antibody, in PBS/TM for 1 h atroom temperature, on a rocking platform. The blot wasdeveloped using 5-bromo-4-chloro-3′-indolyphosphate/nitro blue tetrazolium chloride (BCIP/NBT) reagent(Sigma, B3804). Colour development was stopped bywashing extensively with distilled water. The molecularweight markers used throughout were SigmaMarker™(wide range, unstained) (Sigma, M4038) and Blue-
Ranger® (prestained) (Pierce, 26681) and these arereferred to as M1 and M2, respectively.
2.6. Cloning of inlA gene sequence
Genomic DNA (gDNA), was extracted from a freshculture of L. monocytogenes strain EGD, serovar 1/2a(accession number AJ012346) using guanidium thiocy-anate, in a modification of the method recommended byPitcher et al. (1989). The inlA gene-specific primerswere designed for compatible ligation into the QIAex-press® Type ATG construct, pQE-60 (Qiagen). Theprimer sequences TAFII (5′ CATG-CCATGG-GA-AAG-ACG-GTC-TTA-GGA-AAA-AC 3′) and TABI(5′ CGC-GGATCC-TGA-TTC-TTT-TGA-ATT-ATA-AGG 3′) were chosen to amplify the region encodingthe major extracellular domain of the InlA protein. Aselected pQE-60 plasmid, containing the cloned inlAgene sequence was transformed into ultracompetent(Inoue et al., 1990) E. coli XL-10 Gold® cells. Thetransformed colonies were grown to exponential phaseand then induced with 1 mM IPTG for a further 5 h at 30°C. The cells were lysed by sonication in PBS and theHis-tagged recombinant InlA was purified from thecytoplasmic extract by IMAC, in a modification of themethod described by Leonard et al. (2005).
2.7. Inhibition ELISA
Nunc Maxisorp™ plates were coated and blocked aspreviously described. Serial dilutions of the appropriatetest cells were prepared in PBS/T and 100 μl aliquots ofeach were then placed in individual 500 μl microcen-trifuge tubes. A similar volume of purified monoclonalantibody, diluted 1 /2000 in PBS/T was then added toeach tube. The sample tubes were incubated at 37 °Cfor 1 h on a rocking platform. The tubes were inverted at10-min intervals to ensure homogenous mixing. Sam-ples were then transferred to the coated and blockedmicrowells in 100 μl aliquots and incubated for a further1 h at 37 °C, on a rocking platform. Wells were thenwashed as before and 100 μl aliquots of a 1 /2000dilution of alkaline phosphatase-labeled goat anti-mouse IgG2a secondary antibody in PBS/TM wasadded to the each well and the plate incubated as before.Following washing, pNPP substrate was added to thewells. After a 30-min incubation at 37 °C, theabsorbance values were measured at 405 nm. Absor-bance values at each antigen concentration (A) werethen divided by the absorbance measured in thepresence of zero antigen (A0) to give normalisedabsorbance readings. A plot of the normalised

298 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
absorbance reading versus cell concentration (cell/ml)was used to construct the calibration curve.
2.8. Biacore studies-(instrumentation)
Analysis was carried out on a Biacore 3000™instrument using a CM5 sensor chip (Biacore AB, 2Meadway Court, Meadway Technology Park, Steve-nage, Herts, SG1 2EF, England). The running buffer forall Biacore experiments (unless otherwise specified) wasHBS buffer, pH 7.4, containing 10 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], 150 mMNaCl, 3.4 mM EDTA, and 0.05% (v/v) Tween 20. Therunning buffer was freshly prepared, filtered (0.22 μmcut-off) and degassed using a vacuum filtration appa-ratus (Millipore sintered glass filtration unit), beforeuse. Preconcentration experiments were performedprior to immobilisation, according to the manufacturers'instructions.
2.9. Anti-InlA monoclonal antibody immobilisationconditions
The carboxymethylated dextran matrix on thesensor chip was activated by mixing equal volumesof 100 mM NHS (N-hydroxysuccinimide) and 400mM EDC (N-ethyl-N-(dimethyl-aminopropyl) carbo-diimide hydrochloride), and injecting the mixture overthe sensor chip surface for 10 min at a flowrate of 5μl/min. The affinity-purified monoclonal antibody wasdiluted in 10 mM sodium acetate, pH 4.0, at aconcentration of 100 μg/ml. This solution was theninjected over the activated chip surface for 24 min at 5μl/min. Unreacted NHS groups were capped byinjection of 1 M ethanolamine hydrochloride, pH8.5, for 11 min. Loosely bound material was removedusing a 30 s pulse of 5 mM HCl and 5 mM NaOH,sequentially.
A standard, purified murine IgG2a antibody (Sigma,M9144) was immobilised on the flow-cell immediatelydownstream of the test flow-cell at a similar level. Thisfacilitated reliable on-line referencing.
2.10. Direct binding analysis
All bacterial cells tested were prepared from culturesthat had been grown in LEB broth at 37 °C. Cells wereharvested, washed twice in PBS buffer, and adjusted tothe desired cell concentration in PBS. Cells were passedover the sensor surface at 1 μl/min and surface-regeneration was mediated by a 45 s pulse of 5 mMNaOH.
2.11. AFM image analysis
Bacterial cells that bound monoclonal antibodyimmobilized on the dextran matrix of a CM5 chipwere fixed in situ as follows. Briefly, the CM5 chip wasundocked from the Biacore instrument, after havingbeen exposed to a completed cycle of bacterial sampleanalysis and a buffer wash flow. The chip was thenimmediately placed in a glass petri dish (sample surfacefacing upwards) containing 25 ml 2.5% (v/v) glutaral-dehyde in 100 mM cacodylate buffer (pH 7.4) and leftovernight at 4 °C. The chip was removed from theglutaraldehyde solution and washed twice, in 40 mlfresh cacodylate buffer. The chip was then incubated infreshly prepared 1% (w/v) osmium tetroxide incacodylate buffer for 90 min at room temperature.Following post-fixing, the chip was washed briefly infresh cacodylate buffer, before being exposed to agraded series of ethanol in ultra-pure water (25%, 50%,75%, 90% and 100% (v/v)) for 10 min periods, at roomtemperature. After the final incubation in 100% ethanol,the dehydrated chip was removed from the PVCmounting, prior to imaging. AFM images were obtainedusing a Nano-R™ AFM instrument (Pacific Nanotech-nology, CA. USA).
3. Results and discussion
3.1. Production of monoclonal antibodies to L.monocytogenes
Given the ‘zero tolerance’ criteria with respect toacceptable levels of L. monocytogenes in food, detectionmethods should be able to reliably detect one cell in a25 g sample of food. Thus, an enrichment cultivationstep will almost always be required to increase cellnumbers to detectable levels. The overriding objectiveof this work was to isolate an antibody capable ofspecifically binding to live L. monocytogenes cells. Inorder to maximise the chance of isolating such anantibody, formalin-inactivated L. monocytogenes sero-type 1/2a cells were used to immunise test mice.Recombinant antibodies such as scFvs have beensuccessfully isolated from naïve, synthetic phagemidlibraries that react with Listeria spp. by Benhar et al.(2001). Furthermore, an scFv that reacts specificallywith L. monocytogenes has also been described (Paoli etal., 2004). However, although the latter represents atremendous feat, both scFvs were characterised asphage-displayed chimeric molecules. It is our experi-ence that scFv's isolated from such libraries against L.monocytogenes whole cells have been difficult to stably

299S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
express as functional soluble entities (Leonard, 2003),an observation also reflected in the literature (Petrenkoand Sorokulova, 2004). It would appear that the keystrength of phagemid libraries (i.e., extreme diversity)renders them ‘non-ideal’ reservoirs from which toisolate scFvs against unique surface-displayed epitopes.It is difficult to conceptualise the selective enrichment ofone particular scFv-phage clone (typically from a poolof 1011 clones) against a species-specific bacterialsurface epitope among a panoply of proteins andcarbohydrates, many of which are common across thegenus, without extensive multiple panning. This fre-quently leads to gene loss or partial deletions within thephagemid (de Bruin et al., 1999) resulting in unstable,non-functional scFv molecules.
The initial 600 hybridoma-containing wells werescreened using a comparative, negative-screeningELISA format, where one set of wells was coated withL. monocytogenes serotype 1/2a cells (1×108 cells/well)and a duplicate set of wells was coated with an equalmixture of L. welshimeri, L. seeligeri and L. innocuacells (3×108 cumulative cells/well), prepared under thesame conditions. From this preliminary screening, 39clones were identified that demonstrated preferentialbinding to L. monocytogenes. Following scale-up, thehybridoma populations were re-screened. A proportionof the 39 clones tested exhibited substantial cross-reactivity (Fig. 1). However, a significant percentagealso, still demonstrated an enhanced response to L.monocytogenes cells. In particular, clone no. 39 wasfound to simultaneously generate the greatest responseagainst L. monocytogenes, and the lowest response tothe heterogenous population of non-pathogenic strains.This clone was further isolated and stabilised by limitingdilution, and was designated ‘mAb2B3’.
0.112
0.612
1.112
1.612
2.112
2.612
A40
5 n
m
1 3 5 7 9 11 13 15 17 19
Clone
L. wsi L.
Fig. 1. Screening of supernatants from preliminary hybridoma panel. The lighthe heterogeneous population of non-pathogenic L. welshimeri, L. seeligeri arepresent the supernatant activity towards L. monocytogenes serotype 1/2a c
3.2. Characterisation of monoclonal antibody
The cell binding activity of mAb2B3 was testedagainst a representative selection of pathogenic Listeriaspp. that had been grown in BHI and LEB broth media.The absorbance values recorded (A), were normalisedagainst the background response (A0), according to A/A0, and plotted against the corresponding cell speciestested (Fig. 2). It was apparent that the activity ofmAb2B3 was most potent against L. monocytogenesserotype 1/2a cells and strongest when cells wereprepared in LEB, rather than BHI medium (Fig. 2, top).No activity towards L. ivanovii was detected in eithermedia. Subsequently, a panel of Listeria spp., inaddition to representative gram-positive and gram-negative species, were employed in a direct cell-bindingELISA (Fig. 3). Binding was only apparent in wellscoated with L. monocytogenes strains. The strongresponse to L. monocytogenes serotype 1/2a wasundoubtedly due to the fact that this was also theserotype used for immunisation and hybridoma screen-ing. However, the cells used for immunisation andscreening had also been cultivated in BHI broth. Thehigher response to LEB-cultured serotype 1/2a cellssuggested some degree of up-regulation of themAb2B3-specific antigen. The response to the L.monocytogenes strains 1/2c, 4a and 4b was weaker,yet still significantly higher than the background levelobserved in wells coated with non-L. monocytogenesstrains. Interestingly, the serotype 4a strain was the onlyspecies that demonstrated a weaker response whencultured in LEB medium, rather than BHI. Moresignificantly, the response against the serotype 4b straintested was substantially enhanced by growth in LEBmedium. This is important, given the frequent
21 23 25 27 29 31 33 35 37 39
number
monocytogenes
t bars in the chart foreground represent the supernatant activity towardsnd L. innocua (‘L. wsi’), while the shaded bars in the chart backgroundells.

1
3
5
7
9
11
13
15
L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii
L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii
L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii
L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii
L. monocytogenes1/2a
L. monocytogenes1/2c
L. monocytogenes4a
L. monocytogenes4b
L. ivanovii
A/A
o
1
3
5
7
9
11
13
15
A/A
o
1 1
3
5
7
9
11
13
15
A/A
o
1
3
5
7
9
11
13
15
A/A
o
1
3
5
7
9
11
13
15
A/A
o
1
3
5
7
9
11
13
15
A/A
o
A B
Fig. 2. Reactivity-profiles of purified mAb2B3. The antibody was tested against representative pathogenic Listeria spp. grown in either BHI (panel A)or LEB (panel B) broth media. The top profiles represent ELISAwells coated with 1×108 freshly prepared cells, washed and resuspended in PBS.The centre profile represents the same cell samples following SDS-mediated surface protein extraction, washed and resuspended in PBS, while thebottom profiles refer to the freshly prepared samples following heat treatment (80 °C) for 15 min.
300 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
association of L. monocytogenes 4b strains withsymptomatic listeriosis in humans.
When the cell wall proteinaceous content waspartially solubilised by treatment with detergent (Fig.2, centre) the activity was universally reduced, thus,reaffirming the cell-wall localisation of the reactiveprotein. Rather than a specific up-regulation of theantigenic protein, it was postulated that the overallcellular topography is influenced by the type of growthmedium used, with more accessible reactive epitopesites dominating in LEB-cultured cells. Subjecting thecells to a temperature of 80 °C for 15 min, substantiallyreduced the reactivity (Fig. 2, bottom). This could not beentirely attributed to protein denaturation, since reactiv-
ity was observed following denaturing SDS-PAGE(Fig. 4). This substantiated the postulation that success-ful binding of mAb2B3 antibody to L. monocytogeneswas dependent not only on expression of the specificantigen, but also on the surface topography andaccessibility of the epitope site. This was viewed as asignificant advantage for the development of a L.monocytogenes-specific immunoassay, since it couldpotentially reduce false positive results arising fromnon-viable, heat inactivated L. monocytogenes cells.
Western blot analysis of SDS-extracted surfaceproteins from the same panel of Listeria spp. revealedthat the antibody reacted specifically with an ∼80 kDa-surface-localised protein (Fig. 4), only present in the L.

1
3
5
7
9
11
13
15
L. m
onoc
ytoge
nes 1
/2a
L. m
onoc
ytoge
nes 1
/2c
L. m
onoc
ytoge
nes 4
a
L. m
onoc
ytoge
nes 4
b
L. iv
anov
ii
L. in
nocu
a 112
88
L. in
nocu
a 86
054
L. w
elshim
eri
L. se
eligir
i
B. sub
tilis
E. faec
ilis
E. aer
ogen
es
A/A
o
Fig. 3. Cross-reactivity profiles of purified mAb2B3. The antibody was tested against representative Listeria spp. and non-Listeria spp. that had beencultured in LEB broth media. The concentration of each cell sample was adjusted in PBS to give a final coating concentration of 1×108 cells/well. Allresponse values were normalised against the mean background absorbance values obtained in parallel control wells. Binding of mAb2B3 was onlyobserved in the wells coated with L. monocytogenes cells.
301S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
monocytogenes strains. This was putatively assumed tobe Internalin A (InlA). Although InlA is exclusive toL. monocytogenes, a significant number of internalinhomologues have been identified within the L. ivanoviispecies (Engelbrecht et al., 1998a,b) comprising thecharacteristic LRR domain (Kajava, 1998), which,itself is typical, only of virulence-associated appen-dages in prokaryotes. Cell wall-anchored proteins arewidespread in gram-positive bacteria (Navarre andSchneewind, 1999) and many would share thecommon LPXTG motif and similarly hydrophobic C-terminal domain found in InlA. However, since this
1 2 3 4 5
1 2 3 4 5 6 7 8 9
84 kDa
M2
M2
Gel A
Gel B
84 kDa
Fig. 4. Determination of the specific immunoreactivity of mAb2B3. SDS-extrmembers grown under identical conditions in either BHI (A) or LEB (B) brot(3), L. monocytogenes 4a; (4), L. monocytogenes 4b; (5), L. ivanovii PA (6), Lleft peripheral lane (M2) contains a series of protein standards of known mo
region is buried within the cell wall and thusinaccessible, it was considered highly improbable thatthis region housed the epitope recognised by mAb2B3and, correspondingly, was predicted to be unlikely tomediate cross-reactivity.
3.3. Production of recombinant InlA protein
In order to conclusively confirm that InlAwas indeedthe protein being recognised by mAb2B3, the geneencoding the major surface exposed region of themature protein was amplified from L. monocytogenes
1 2 3 4 5M2Blot A
Blot B1 2 3 4 5 6 7 8 9M2
acted protein fractions (∼100–150 μg/ml) from various Listeria genush media. Lane (1), L. monocytogenes 1/2a; (2), L. monocytogenes 1/2c;. innocua; (7), L. innocua; (8), L. welshimeri and (9), L. seeligeri. Thelecular weight.

302 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
serotype 1/2a genomic DNA, cloned into the pQE60expression vector and the construct then transformedinto E. coli XL-10 Gold® cells. Following induction,transformants harbouring this construct clearly demon-strated up-regulated heterologous expression of an∼80 kDa protein in their respective cell lysate fractionswhen compared to lysate from pQE60 transformantslacking the inlA insert, (Fig. 5A). Western blot analysisof these same lysates confirmed that mAb2B3 onlyreacted with the heterologously expressed recombinantInlA (Fig. 5B) thus, confirming that mAb2B3 wasspecific for InlA.
The suitability of InlA as a virulence marker for L.monocytogenes is substantiated by several associatedcharacteristics. Fundamentally, the inlA gene itself isconstantly and specifically present in L. monocytogenes,regardless of the origin, serovar and virulence of theisolate (Poyart et al., 2003) and the InlA protein isexpressed constitutively at a basal level. There is also adirect correlation between InlA expression and inva-siveness (Dramsi et al., 1993). Primary intestinalinvasion is dependent on InlA-mediated tropism towardhuman host intestinal epithelia, by virtue of its specificinteraction with human epithelial cadherin (hE-CAD)about the Pro16 residue (Lecuit et al., 1999, 2001) and itis widely distributed on the cell surface during all stagesof growth in broth culture (Lebrun et al., 1996).Transcription of the inlA gene operon can proceed
121.5 k
84 kDa66 kDa
47.9 k
A1 2 3 4 M1
Fig. 5. Detection of rInlA in cell lysate using mAb2B3. Panel A: lanes 1, 3 aharbouring pQE60 plasmid with the rInlA-encoding gene sequence inserted;pQE60 plasmid minus the rInlA encoding gene insert. All samples were growthe gel loading volume was 15 μl. There appeared to be distinct up-regulationcontrol sample, lane 2 (indicated by arrows). Panel B represents a Western blothe results from the gel in panel A and confirmed that mAb2B3 was specificweight markers.
independently of the PrfA regulator protein (Leimeister-Wächter et al., 1992; Lingnau et al., 1995; Sheehan etal., 1995; Lalic-Mülthaler et al., 2001; Johansson et al.,2002) and thus it is not subject to the same strictthermoregulation that is intrinsically linked to PrfAexpression (Leimeister-Wächter et al., 1992; Johanssonet al., 2002). The other main virulence genes thatinclude; plcA, hly, mpl, actA and plcB, are arranged on a9-kb chromosomal pathogenicity island referred to asListeria pathogenicity island 1 or LIPI-1 (Vázquez-Boland et al., 2001a,b). Expression of these genesclosely correlates with that of prfA, and appreciableamounts of the nascent proteins are only achieved whengrowth conditions mimic those of the intracellularenvironment, such as was experimentally achieved byaddition activated charcoal to growth media (Ripio etal., 1996; Ermolaeva et al., 2004). InlA expression is notsubject to this ‘charcoal-effect’ and, thus, sampleenrichment can proceed in rich media and at 37 °C,resulting in more rapid attainment of a detectable levelof L. monocytogenes cells without fear of compromisingthe key antigen expression level.
Like InlA, the p60 (Kuhn and Goebel, 1989; Köhleret al., 1991) and p66 (Bhunia et al., 1991) proteins areexpressed at significant levels during enriched growth at37 °C, independent of PfrA levels (Bubert et al., 1997a,b; Wiêckowska-Szakiel et al., 2002; Geng et al., 2003).While p60 homologous proteins are present throughout
Da
Da
BM2 1 2 3 4
nd 4 consist of cell extracts derived from E. coli XL-10 Gold® cloneslane 2 contains extract from an E. coli XL-10 Gold® clone harbouringn under identical conditions and all were induced with 1 mM IPTG andof an 80–84 kDa protein in lanes 1, 3 and 4 that was not apparent in thet of the same samples, probed with mAb2B3 and it clearly corroboratesfor InlA. The inner peripheral lanes (M1 and M2) consist of molecular

303S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
the genus (Bubert et al., 1992a,b) an antibody has beensuccessfully produced against a L. monocytogenes-specific p60 peptide sequence (Bubert et al., 1994).However, it has been indicated that as little as 25% ofexpressed p60 protein is cell-associated (Ruhland et al.,1993) and the antibody was subsequently shown to onlypoorly react with this form of the protein (Bubert et al.,1994). Similarly, although p66 homologous proteinsequences have been demonstrated in L. monocytogenesand L. innocua (Bhunia et al., 1991), an antibody hasbeen generated that preferentially reacts with a p66epitope that is confined to L. monocytogenes (Bhuniaand Johnson, 1992). It has been suggested that thisantibody fails to react with certain L. monocytogenesserotypes (Bhunia, 1997).
Although InlB expression is intrinsically linked toInlA expression, PfrA-independent transcription of theinlB gene is more variable during growth at 37 °C(Lingnau et al., 1995) and more importantly, it is onlyvery loosely associated with the cell wall and poorlyaccessible to antibodies (Jonquières et al., 1999). Thisrenders InlB-targeted immunodetection inherently lessreliable.
3.4. ELISA-based assay for L. monocytogenes cells
The potential for use of mAb2B3 in the developmentof an immunoassay for the specific detection of L.monocytogenes cells was demonstrated using aninhibition-ELISA format, similar to that described by
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1e7 1eConcentratio
A/A
o
Fig. 6. Calibration curve for inhibition-ELISA-based detection of live L. mparameter equation, using BIAevaluation™ software.
Eriksson et al. (1995) and Leonard et al. (2005), for thedetection of Pseudomonas fluorescens and L. mono-cytogenes, respectively.
Upon inspection of the linear portion of thecalibration curve, the dynamic range appeared quitenarrow and did not extend below 4×107 cells/ml. Whenfitted with a four-parameter equation, the lower limit ofdetection was extended to 1.85×107 cells/ml (Fig. 6).The sensitivity and working range could perhaps beimproved by adopting an alternative format. A possiblecandidate may be a sandwich immunoassay thatincorporates a sequential capture and signal-generatinglabelled-antibody binding event. A polyclonal genus-specific antibody could potentially fill the role of eitherthe signal-generating antibody component, as haspreviously been demonstrated by Gavalchin et al.(1991), or the capture-antibody component, withsubsequent binding of species-specific, fluorescently-labelled monoclonal antibody (Sewell et al., 2003).
However, the incorporation of such broadly reactiveantibodies could conceivably impinge on the specificityof the assay. Considering that the initial cell contami-nation level is going to be substantially augmented by anincorporated enrichment incubation step, and the factthat unlike other L. monocytogenes virulence proteins,InlA expression is not substantially down-regulated bygrowth in nutrient-rich media at 37 °C (Bohne et al.,1996; Bubert et al., 1999), a slight improvement insensitivity is not essential and definitely not warranted atthe expense of assay specificity. Alternatively, it has also
8 1e9n (cells/ml)
onocytogenes cells in PBS (n=6). The data was fitted with a four-

304 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
been shown that a monoclonal antibody can be used asboth the capture and signal generating components of asandwich immunoassay for detection of L. monocyto-genes and L. innocua (Sølve et al., 2000). This is a facetof sandwich immunoassays that is restricted to theanalysis of large, multi-epitope bearing analytes, such ascells. The high copy number and wide distribution ofaccessible InlA on the surface of L. monocytogenesmake this protein a potentially suitable target for such anassay. Alternatively, the use of a SPR-based biosensorplatform may overcome many of the inherent problemsassociated with ELISA (e.g., multiple reagents, labellingof selected reagents, lengthy washing and incubationsteps).
3.5. Optimisation of biosensor platform
The test and control antibody surfaces were immo-bilised with the respective antibodies by amine couplingmediated by EDC/NHS activation (Johnsson et al.,1991; O'Shannessey et al., 1992). Approximately29,000 RUs of mAb2B3 antibody was efficientlycoupled. It was felt that due to the large size of thecells (∼2×5 μm) and the net negative charge associatedwith the bacterial cell wall, the highly negativelycharged and relatively viscous (at least in relation tosuch bulky cellular material) carboxymethylated dextranhydrogel would compromise efficient binding ofbacterial cells to the immobilised mAb2B3. Preliminarystudies were conducted using a C1 sensor chip thatcomprised a planar carboxymethylated surface with nodextran hydrogel. However, the significantly reducedimmobilisation capacity, with respect to mAb2B3 (datanot shown), afforded by the planar surface prompted thedecision to conduct the analysis using a CM5 sensorchip. A further pertinent point of initial concern was thatthe mAb2B3 antibody would be quite heterogeneouslyoriented both on and within the hydrogel as a result ofthe random covalent cross-linking afforded by the aminecoupling method. The controlled orientation of anti-bodies on sensor surfaces using protein A/G and Fc-specific antibodies has previously been investigated (Luet al., 1996; Quinn et al., 1999). However, given thedimensions of the bacterial cell in relation to the surface-localised evanescent field it was felt that such anorientation would potentially render the cell attachmentto the distal antigen-binding (Fab) domain of theimmobilised antibody, beyond the effective sensingrange. Recent reports on similar applications haveclearly demonstrated that a covalently immobilisedantibody surface facilitated vastly superior cell-bindingcapacity than when the same antibody was immobilised
using a protein A-capture strategy (Fratamico et al.,1998; Lathrop et al., 2003). A further concern whenusing such a configuration was the associated require-ment to constantly replenish the detector antibodybetween analyses, leading to high consumption ofvaluable, purified monoclonal antibody. In view of theabove considerations it was decided to proceed withmAb2B3 amine coupled at a high level to a CM5surface. Recent reports of successful binding of liveHelicobacter pylori cells to a receptor ligand, covalentlyattached to the generic CM5 surface, have validated theapplicability of the direct cell-capture format (Clyne etal., 2004).
3.6. Direct cell binding analysis
A flow rate of 1 μl/min was employed for all sampleanalysis in order to maximise the mAb2B3-L. mono-cytogenes interaction exposure and an interaction timeof 15 min was chosen so as to allow the cell-antibodybinding interaction to approach equilibrium. Followinga series of exploratory experiments, the concentrationsof L. monocytogenes cells selected for testing rangedfrom 1.07×109 to 1.23×107 cells/ml and a representa-tive, overlaid binding profile is demonstrated in Fig. 7.The upper cut-off concentration was defined to reducethe risk of the instruments intricate micro-fluidic valveassembly becoming clogged with cellular material. Eachsample was mixed manually by pipette aspirationimmediately prior to injection and a portion of eachsample examined using an optical microscope to checkfor cell clumping.
It was felt that the distribution and accessibility of theInlA protein on the L. monocytogenes cell surface wasof paramount importance in mediating whole cellbinding at the sensor biointerface. Clear advantages oftargeting somatic (O) antigens and the influence of theassociated cell-surface topographical microenviron-ment, with specific reference to biosensor-based cellbinding, have also been cited by Olsen et al. (2003).This importance derives from the fact that a minimum,threshold avidity is required to enable cell binding andretention of cells at the biointerface. This minimumthreshold is in turn, dictated by the accumulativeresistive forces that comprise shear, ionic, gravitationaland lateral elevation forces (Quinn, 1999).
The cell-binding : response signal ratios and detectionrange were consistent with the findings of severalindependent researchers, who have investigated similar,electrochemical-based biosensor approaches for detec-tion of bacterial cells (O'Sullivan et al., 1999; Park et al.,2000; Kim and Park, 2003). Using this particular

-200
0
200
400
600
800
1000
1200
1400
1600
-200 0 200 400 600 800 1000 1200
1.07 x 109 cells/ml
4.74 x 108 cells/ml
3.16 x 108 cells/ml
2.11 x 108 cells/ml
1.41 x 108 cells/ml9.36 x 107 cells/ml6.24 x 107 cells/ml4.16 x 107 cells/ml2.77 x 107 cells/ml1.85 x 107 cells/ml1.23 x 107 cells/ml
Res
po
nse
(R
U)
Time (s)
Fig. 7. Overlayed binding profile for increasing concentrations of L. monocytogenes cells. L. monocytogenes cells were passed over the mAb2B3-CM5 sensor chip surface at a flow rate of 1 μl/min for 15 min. The ‘bound’ response levels were taken 60 s after the end of the sample injection.
305S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
calibration curve, cell concentrations could reliably bedetermined down to ‘at least’ 1.23×107 cells/ml.However, reliable quantification may be an overlyrestrictive criterion on which to assess the assayperformance. Furthermore, quantification may not bean absolute prerequisite, since current legislationessentially dictates that a specific, ‘presence-or-absence’test is required. Following optimised enrichment ofsuspected contaminated samples, it would be reasonablyexpected that the enriched test sample would containcell concentrations in excess of 1×107 cells/ml.
3.7. Biosensor specificity
It is acknowledged that certain L. monocytogenesstrains produce soluble, truncated InlA (Jonquières etal., 1998). However, such strains are normally associ-ated with asymptomatic carriage (Olier et al., 2003).Expression of fully functional, cell wall-anchored InlAprotein is a dominant trait of clinically significant strains(Jacquet et al., 2004). Thus, by effectively targetingcellular InlA, a higher echelon of discrimination, withrespect to clinical significance, may be afforded.
The extensive heterogeneity between ELISA andSPR-based analysis formats, dictated that the biosensorconfiguration be challenged with a panel of relatedListeria spp. and representative non-Listeria spp., of
both the gram-negative and gram-positive varieties. Thismethod, when challenged individually with the selectedpanel of potential cross-reacting species, at a similarconcentration to that of the highest L. monocytogenesconcentration tested (∼1.1×109 cells/ml), generatednegligible responses (Fig. 8). In fact none of the teststrains generated a response above 10 RUs, which canbe considered background. A further key advantageassociated with this method is the apparent ability topreferentially bind live cells. Heat treatment at 80 °Cresulted in a rapid decrease in the apparent bindinglevels (Fig. 9). It might, thus, be assumed that the InlAprotein is being partially or completely solubilised and/or denatured, or that the surface topography of thebacterial cell is altered, concomitantly rendering theInlA protein less accessible.
Listeria genus-specific polyclonal antibodies havebeen successfully used to demonstrate the genericpotential of amended SPR-based detection of Listeria(Koubová et al., 2001; Leonard et al., 2003). Suchantibodies would, however, limit identification to thegenus level at best. In an effort to elicit signalenhancement following direct binding of bacteria tothe immobilised capture-antibody, several authors havesuggested merit in adopting a sandwich approachwhereby, a secondary, genus-specific polyclonal anti-body is applied (Fratamico et al., 1998; Bokken et al.,

-100 100 300 500 700 900-10
-8
-6
-4
-2
0
2
4
6
8
10
Time (s)
Res
po
nse
(R
U)
A -1
0
1
2
3
4
5
6
7
8
9
900 920 940 960 980 1000 1020 1040 1060
Res
po
nse
(R
U)
Time (s)
B. subtilis
L. innocua 6a
E. faecilis
E. coli
L. ivanovii PA
L. welshimeriL. seeligeriL. ivanovii PHLS
B. cereusE. entereogenesL. innocua 6b
B
Fig. 8. Binding response of a representative sample of non-L. monocytogenes cells. Cell concentrations were adjusted to ∼1×109 cells/ml in PBSbuffer prior to analysis. The overlaid, complete binding profile for each test strain is demonstrated (A), with the region corresponding to the finalresponse values magnified and labelled (B). None of the strains tested generated a response value above 10 RUs.
306 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
2003). Polyclonal antibody concentrations of at least50 μg/ml were required to achieve significant enhance-ment. With such a large consumption of polyclonal
-100
0
100
200
300
400
-200 0 200 400
Tim
Res
po
nse
(R
U)
No heat treatment
80 °C, 10 mins
80 °C, 15 mins
Fig. 9. Binding response profile of L. monocytogenes cells following heat inacsurface, generating a response of 317.1 RUs. Following treatment at 80 °C for 1respectively. This indicated a clear, inverse proportional relationship between
reagent, the cost factor and batch-to-batch heterogeneitymay hinder routine testing potential. We investigated theeffect the of sandwich enhancement using two anti-
600 800 1000 1200
e (s)
317.1 RU
52.9 RU
4.9 RU
tivation. Approximately 2.11×108 cells/ml were injected across the test0 and 15min, respectively, the response decreased to 52.9 and 4.9 RUs,degree of heat inactivation and absolute binding response levels.

-50
0
50
100
150
200
250
300
350
-100 200 500 800 1100 1400
Res
po
nse
(R
U)
Time (s)
Anti-Lm1polyclonal Ab
Anti-Lm2polyclonal Ab
Cells
Fig. 10. Sandwich approach to signal enhancement using polyclonal antibodies. Approximately 270 RUs of fresh L. monocytogenes 1/2a cells werecaptured on the mAb2B3 at a flow rate of 1 μl/min. A 50 μl aliquot of each of the anti-L. monocytogenes (Anti-Lm1 and Anti-Lm2) polyclonalantibodies was then sequentially passed over the captured cells at 100 μg/ml and 10 μl/min. No enhancement of the binding response was observed.
307S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
Listeria polyclonal antibodies. However, as Fig. 10clearly shows, no enhancement was achieved. This wasnot an entirely surprising result given the similarfindings of Lathrop et al. (2003).
3.8. Comparative assessment of biosensor-based cellbinding technique
The possibility of using SPR and evanescent wavetechnology to monitor whole cell–ligand biding inter-actions is a relatively new concept. While someinteresting applications have been cited in the literature(Holmes et al., 1997; Quinn et al., 1997, 2000;
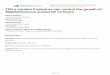
Fig. 11. Image analysis of the sensor chip surface with mAb2B3-captured L. mThe injected cell concentration corresponded to 4.74×108 cells/ml.
Kawashima et al., 2003; Clyne et al., 2004; Pourshafieet al., 2004) most research has undoubtedly concentrat-ed on the refinement of the technique for rapid andreliable detection of pathogenic cells. Crucial to thesuccess of this approach, and indeed any immunodetec-tion method, is the specificity of the antibody beingemployed. The monoclonal antibody used here(mAb2B3) performed admirably, with no cross-reactiv-ity apparent.
The AFM image depicted in Fig. 11 clearly show L.monocytogenes cells captured on the CM5 surface. Themoderate affinity (KD=1.93×10
−7 M, data not shown)for the mAb2B3 antibody with recombinant InlA
onocytogenes cells. The surface topography was observed using AFM.

308 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
protein, the extensive distribution of InlA on the cellsurface, and the high level of immobilised captureantibody employed suggest that avidity effects may besignificant in the capture process.
It was postulated that specific dynamic eventswithin the hydrogel that contravene its restrictiveclassification as a classical ‘brush-border’ may aidcell binding. These include, a significant degree ofelasticity within the polymeric dextran (Rief et al.,1997; Piehler et al., 1999) that allows polymer-tetheredantibodies to extend beyond the average, ambientequilibrium configuration (dependent on runningbuffer) and bind to complementary, surface-exposedantigen. Bonds formed by these ‘rare extendedconformations' are further aided by the increasedintrinsic translational and rotational mobility exhibitedby the tethered ligand when compared to the polymeritself (Jeppesen et al., 2001). Thus, notwithstanding therandom nature of the amine-coupling method employedand the associated risk to binding site integrity(Vaughan et al., 2001, 2003), a significant populationof rare extended conformations bearing active antigen-binding domains can mediate attainment of the aviditythreshold required for cell binding.
The apparent poor sensitivity of the SPR formatwith respect to low concentrations of whole micro-organisms (Pourshafie et al., 2004) is acknowledged.Recently, Gray and Bhunia (2005) have directlyaddressed this challenge to biosensor sensitivity byusing an immunobead capture approach to concomi-tantly concentrate low numbers of L. monocytogenescells and isolate the cells from complex samplematrices, thereby enhancing sensitivity and decreasingbackground interference. Nonetheless, it is alsoaccepted that all ‘rapid methods’ will likely requirepre-enrichment of food samples to allow cells reachdetectable concentrations (Karamonová et al., 2003)and it is important to emphasise that the methoddescribed herein, although referred to as a ‘real-time’system, does not purport to obviate the need for sucha period of enrichment incubation.
4. Conclusions
The novel ‘negative-screening’ strategy describedproved eminently efficient at isolating species-specificanti-L. monocytogenes hybridoma clones. This simplemethod could feasibly be employed for the selectiveisolation of monoclonal antibodies against any givenspecies of pathogenic bacteria.
The monoclonal antibody ‘mAb2B3’ reacted highlyspecifically with both native and recombinantly pro-
duced forms of InlA protein. It also recognised both thesoluble and cell wall-bound forms of the protein andretained its capacity to specifically bind L. monocyto-genes whole cells when incorporated into both ELISA-and SPR-based whole cell binding configurations. Thecombined fundamental merits of this novel, highlyspecific antibody and the virulence marker, InlA,compared most impressively with previously reportedanti-Listeria antibodies and putative candidate pathoge-nicity marker proteins.
In summary, the exquisite specificity of the novel L.monocytogenes-specific mAb combined with its equallyrobust performance in ELISA and biosensor-based cellbinding experiments render it a superlative candidate forintegration into future novel and rapid immunosensor-based strategies for improved identification of L.monocytogenes contamination in food.
Acknowledgements
The authors would like to acknowledge support fromINCO COPERNICUS project No. PL979012. Manythanks to Gary Wyatt at the Institute for Food Research,Norwich, UK and both Vincent Young and RachelHewitt at the Public Analysts Laboratory, Dublin,Ireland for providing bacterial strains used in this study.
References
Aguado, V., Vitas, A.I., García-Jalón, A.I., 2004. Characterisation ofListeria monocytogenes and Listeria innocua from a vegetableprocessing plant by RAPD and REA. Int. J. Food Microbiol. 90,341–347.
Anonymous, 2001. Guidelines for the interpretation of results ofmicrobiological analysis of some ready-to-eat foods sampled atpoint of sale. Food Safety Authority of Ireland, Abbey Court,Lower Abbey Street, Dublin 1, Ireland.
Anonymous, 2003. Quantitative assessment of relative risk to publichealth from foodborne Listeria monocytogenes among selectedcategories of ready-to-eat foods. FDA/Centre for Food Safety andApplied Nutrition, USDA/Food Safety and Inspection Service,Centres for Disease Control and Prevention, USA, Executivesummary, viii–xv.
Anonymous, 2004. Draft guidelines on the application of generalprinciples of food hygiene to the management of L. monocytogenesin foods. European Community position paper—preparation forCodex Committee on Food Hygiene. Agenda Item 7: Washington,USA, 29th March–3rd April.
Benhar, I., Eshkenazi, I., Neufeld, T., Opatowsky, J., Shaky, S.,Rishpon, J., 2001. Recombinant single chain antibodies inbioelectrochemical sensors. Talanta 55, 899–907.
Beumer, R.R., Hazeleger, W.C., 2003. Listeria monocytogenes:diagnostic problems. FEMS Immunol. Med. Microbiol. 35,191–197.
Bhunia, A.K., 1997. Antibodies to Listeria monocytogenes. Crit. Rev.Microbiol. 23 (2), 77–107.

309S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
Bhunia, A.K., Johnson, M.G., 1992. Monoclonal antibody specific forListeria monocytogenes associated with a 66-kilodalton cellsurface antigen. Appl. Environ. Microbiol. 58 (6), 1924–1929.
Bhunia, A.K., Ball, P.H., Faud, B.W., Kurz, J.W., Emerson, J.W.,Johnson, M.G., 1991. Development and characterisation of amonoclonal antibody specific for Listeria monocytogenes andListeria innocua. Infect. Immun. 59, 3176–3184.
Blackman, I.C., Frank, J.F., 1996. Growth of Listeria monocytogenesas a biofilm on various food-processing surfaces. J. Food Prot. 59,827–831.
Boerlin, P., Piffaretti, J.-C., 1991. Typing of human, animal, food andenvironmental isolates of Listeria monocytogenes by multilocusenzyme electrophoresis. Appl. Environ.Microbiol. 57, 1624–1629.
Bohne, J., Kestler, H., Uebele, C., Sokolovic, Z., Goebel, W., 1996.Differential regulation of the virulence genes of Listeria mono-cytogenes. Mol. Microbiol. 20, 1189–1198.
Bokken, G.C.A.M., Corbee, R.J., van Knapen, F., Bergwerff, A.A.,2003. Immunochemical detection of Salmonella group B, D and Eusing optical surface plasmon resonance biosensor. FEMSMicrobiol. Lett. 222, 75–82.
Bubert, A., Kuhn, M., Goebel, W., Köhler, S., 1992a. Structural andfunctional properties of the p60 proteins from different Listeriaspecies. J. Bacteriol. 174, 8166–8171.
Bubert, A., Köhler, S., Goebel, W., 1992b. The homologous andheterologous regions within the iap gene allow genus- and species-specific identification of Listeria spp. by polymerase chainreaction. Appl. Environ. Microbiol. 58, 2625–2632.
Bubert, A., Köhler, S., Frank, R., Goebel, W., 1994. Synthetic peptidesderived from the Listeria monocytogenes p60 protein as antigensfor the generation of polyclonal antibodies specific for secretedcell-free L. monocytogenes p60 proteins. Appl. Environ. Micro-biol. 60 (9), 3120–3127.
Bubert, A., Kestler, H., Götz, M., Böckmann, R., Goebel, W., 1997a.The Listeria monocytogenes iap gene as an indicator gene for thestudy of PrfA-dependent regulation. Mol. Gen. Genet. 25,625–654.
Bubert, A., Riebe, J., Schnitzler, N., Schönberg, A., Goebel, W.,Schubert, P., 1997b. Isolation of catalase-negative Listeriamonocytogenes strains from listeriosis patients and their rapididentification by anti-p60 antibodies and/or PCR. J. Clin.Microbiol. 35 (1), 179–183.
Bubert, A., Sokolovic, Z., Chun, S.K., Papatheodoru, L., Simm,Goebel, W., 1999. Differential expression of Listeria monocyto-genes virulence genes in mammalian host cells. Mol. Gen. Genet.261, 232–336.
Butman, B.T., Plank, M.C., Durham, R.J., Mattingly, J.A., 1988.Monoclonal antibodies which identify a genus-specific Listeriaantigen. Appl. Environ. Microbiol. 54, 1564–1569.
Clyne, M., Dillon, P., Daly, S., O'Kennedy, R., May, F.E.B., Westley,B.R., Drumm, B., 2004. Helicobacter pylori interacts with thehuman single-domain trefoil protein TFF1. Proc. Natl. Acad. Sci.U.S.A. 101 (19), 7409–7414.
de Bruin, R., Spelt, K., Mol, J., Koes, R., Quattrocchio, F., 1999.Selection of high-affinity phage antibodies from phage displaylibraries. Nat. Biotechnol. 17, 397–399.
Deisingh, A.K., Thompson, M., 2004. Biosensors for the detection ofbacteria. Can. J. Microbiol. 50 (2), 69–77.
Delgado da Silva, M.C.D., Destro, M.T., Hofer, E., Tibana, A., 2001.Characterisation and evaluation of some virulence markers ofListeria monocytogenes strains isolated from Brazilian cheesesusing molecular, biochemical and serotyping techniques. Int. J.Food Microbiol. 63, 275–280.
Directive 92/59/EEC, 1992. European Parliament and Council OfficialJournal of the European Communities 1992, L 268/1, 14thSeptember, 1992.
Directive 93/43/EEC, 1993. European Parliament and Council OfficialJournal of the European Communities 1993, L 175/1, 19th July,1993.
Donnelly, C.W., 2001. Listeria monocytogenes: a continuing chal-lenge. Nutr. Rev. 59 (6), 183–194.
Dramsi, S., Kocks, C., Forestier, C., Cossart, P., 1993. Internalin-mediated invasion of epithelial cells by Listeria monocytogenes isregulated by the bacterial growth state, temperature, and thepleiotropic activator prfA. Mol. Microbiol. 9 (5), 931–941.
Duggan, J., Philips, C.A., 1998. Listeria in the domestic environment.Nutr. Food Sci. 2, 73–79.
Engelbrecht, F., Dickneite, C., Lampidis, R., Götz, M., DasGupta,U., Goebel, W., 1998a. Sequence comparison of the chromo-somal regions encompassing the internalin C genes (inlC) of L.monocytogenes and L. ivanovii. Mol. Gen. Genet. 257,186–197.
Engelbrecht, F., Dominguez-Bernal, G., Dickneite, C., Hess, J.,Greiffenberg, L., Lampidis, R., Raffelsbauer, D., Kaufmann, J.,Kreft, J., Vásquez-Boland, J.A., Goebel, W., 1998b. A novel PrfA-regulated chromosomal locus of Listeria ivanovii encoding twosmall, secreted internalins is essential for virulence in mice. Mol.Microbiol. 30, 405–417.
Eriksson, P.V., Di Paola, G.N., Pasetti, M.F., Manghi, M.A., 1995.Inhibition enzyme-linked immunoassay for detection of Peudo-monas fluorescens on meat surfaces. Appl. Environ. Microbiol. 61(1), 397–398.
Ermolaeva, S., Novella, S., Vega, Y., Ripio, M.-T., Scortti, M.,Vázquez-Boland, J., 2004. Negative control of Listeria mono-cytogenes virulence genes by a diffusible repressor. Mol.Microbiol. 52 (2), 601–611.
Fratamico, P.M., Strobaugh, T.P., Medina, M.B., Gehring, A.G., 1998.Detection of Escherichia coli O157:H7 using a surface plasmonresonance biosensor. Biotechnol. Tech. 12 (7), 571–576.
Gasanov, U., Hughes, D., Hansbro, P.M., 2005. Methods for isolationand identification of Listeria spp. and Listeria monocytogenes: areview. FEMS Microbiol. Reviews 29 (5), 851–875.
Gavalchin, J., Tortorello, M.L., Malek, R., Landers, M., Batt, C.A.,1991. Isolation of monoclonal antibodies that react preferentiallywith Listeria monocytogenes. Food Microbiol. 8, 325–330.
Geng, T., Kim, K.P., Gomez, R., Sherman, D.M., Bashir, R., Ladisch,M.R., Bhunia, A.K., 2003. Expression of cellular antigens ofListeria monocytogenes that react with monoclonal antibodiesC11E9 and EM-7G1 under acid-, salt- or temperature-inducedstress environments. J. Appl. Microbiol. 95, 762–772.
Gravani, R., 1999. Incidence and control of Listeria in food-processing facilities, In: Ryser, E.T., Marth, E.H. (Eds.), Listeria,Listeriosis, and Food Safety, 2nd ed. Marcel Dekker Inc., New-York, pp. 657–709.
Gray, K.M., Bhunia, A.K., 2005. Specific detection of cytopathogenicListeria monocytogenes using a two-step method of immunose-peration and cytotoxicity analysis. J. Immunol. Methods 60,259–268.
Holmes, S.D., May, K., Johansson, V., Markey, F., Critchley, I.A.,1997. Studies on the interaction of Staphylococcus aureus andStaphylococcus epidermidis with fibronectin using surface plas-mon resonance (BIAcore). J. Microbiol. Methods 28, 77–84.
http://europa.eu.int/comm/food/food/rapidalert/report2004_en.pdf.Inoue, H., Nojima, H., Okayama, H., 1990. High efficiency
transformation of Escherichia coli with plasmids. Gene 96, 23–28.

310 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
ISO 11 290-1, 1996a. Microbiology of Food and Animal FeedingStuffs—Horizontal Method for the Detection and Enumeration ofListeria monocytogenes: Part 1. Detection Method. InternationalOrganisation for Standardisation, Geneva, Switzerland.
ISO 11 290-1, 1996b. Microbiology of Food and Animal FeedingStuffs—Horizontal Method for the Detection and Enumeration ofListeria monocytogenes: Part 2. Enumeration Method. Interna-tional Organisation for Standardisation, Geneva, Switzerland.
ISO 11 560-1, 1999. Milk and Milk Products—Detection of Listeriamonocytogenes. International Organisation for Standardisation,Geneva, Switzerland.
Ivnitski, D., Abel-Hamid, I., Atanasov, P., Wilkins, E., 1999.Biosensors for detection of pathogenic bacteria. Biosens. Bioelec-tron. 14, 599–624.
Jacquet, C., Doumith, M., Gordon, J.I., Martin, P.M.V., Cossart, P.,Lecuit, M., 2004. A molecular marker for evaluating thepathogenic potential of Listeria monocytogenes. J. Infect. Dis.189, 2094–2100.
Jeppesen, C., Wong, J.Y., Kuhl, T.L., Israelachivili, J.N., Mullah, N.,Zalipsky, S., Marques, C.M., 2001. Impact of polymer tetherlength on multiple ligand–receptor bond formation. Science 293,465–468.
Johansson, J., Mandin, P., Renzoni, A., Chiaruttini, C., Sprinter, M.,Cossart, P., 2002. An RNA thermosensor controls expression ofvirulence genes in Listeria monocytogenes. Cell 110, 551–561.
Johnsson, B., Löfås, S., Lindquist, G., 1991. Immobilisation ofproteins to a carboxymethyldextran-modified gold surface forbiospecific interaction analysis in surface plasmon resonancesensors. Anal. Biochem. 198, 268–277.
Jonquières, R., Bierne, H., Cossart, P., 1998. The inlA gene of Listeriamonocytogenes LO28 harbours a nonsense mutation resulting inrelease of internalin. Infect. Immun. 66, 3420–3422.
Jonquières, R., Bierne, H., Fiedler, FR., Gounon, P., Cossart, P., 1999.Interaction between the protein InlB of Listeria monocytogenesand lipoteichoic acid: a novel mechanism of protein association atthe surface of Gram-positive bacteria. Mol. Microbiol. 34 (5),902–914.
Kajava, A.V., 1998. Structural diversity of leucine-rich repeat proteins.J. Mol. Biol. 277, 211–227.
Karamonova, L., Blažka, M., Fukal, L., Rauch, P., Greifova, M.,Horáková, K., Tomáška, M., Roubal, P., Brett, G.M., Wyatt, G.M., 2003. Development of an ELISA specific for Listeriamonocytogenes using a polyclonal antibody raised against a cellextract containing internalin B. Food Agric. Immunol. 15 (3–4),167–182.
Kathariou, S., Mizumoto, C., Allen, R.D., Fok, A.K., Benedict, A.A.,1994. Monoclonal antibodies with a high degree of specificity forListeria monocytogenes serotype 4b. Appl. Environ. Microbiol. 60(10), 3548–3552.
Kawashima, M., Hanada, N., Tagami, J., Senpuku, H., 2003. Real-timeinteraction of oral streptococci with human salivary components.Oral Microbiol. Immunol. 18, 220–225.
Kim, N., Park, I.-S., 2003. Application of a flow type antibody sensorto the detection of Escherichia coli in various foods. Biosen.Bioelectron. 18, 1101–1107.
Klein, P.G., Juneja, V.K., 1997. Sensitive detection of viable Listeriamonocytogenes by reverse transcription-PCR. Appl. Environ.Microbiol. 63 (11), 4441–4448.
Köhler, S., Bubert, A., Vogel, M., Goebel, W., 1991. Expression of theiap gene coding for protein p60 of Listeria monocytogenes iscontrolled on the posttranscriptional level. J. Bacteriol. 173 (15),4668–4674.
Koubová, V., Brynda, E., Karasová, L., Škvor, J., Homola, J.,Dostálek, J., Tobiška, P., Rošický, J., 2001. Detection of foodbornepathogens using surface plasmon resonance biosensors. Sens.Actuators, B, Chem. 74, 100–105.
Kretschmann, E., 1971. The determination of the optical constants ofmetals by excitation of surface plasmons resonance. Z. Phys. 241,313–324.
Kretschmann, E., Raether, H., 1968. Radiative decay of non-radiativesurface plasmons excited by light. Z. Naturforsch. 23, 2135–2136(A).
Kuhn, M., Goebel, W., 1989. Identification of an extracellular proteinof Listeria monocytogenes possibly involved in intracellularuptake by mammalian cells. Infect. Immun. 57, 55–61.
Laemmli, U.K., 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature 227, 680–685.
Lalic-Mülthaler, M., Bohne, J., Goebel, W., 2001. In vitro transcriptionof PrfA-dependent and -independent genes of Listeria mono-cytogenes. Mol. Microbiol. 42 (1), 111–120.
Lathrop, A.A., Jaradat, Z.W., Haley, T., Bhunia, A.K., 2003.Characterisation and application of a Listeria monocytogenesreactive monoclonal antibody C11E9 in a resonant mirrorbiosensor. J. Immunol. Methods 281, 119–128.
Lebrun, M., Mengaud, J., Ohayon, H., Nato, F., Cossart, P., 1996.Internalin must be on the bacterial surface to mediate entry ofListeria monocytogenes into epithelial cells. Mol. Microbiol. 21(3), 579–592.
Lecuit, M., Dramsi, S., Gottardi, C., Fedor-Chaiken, M., Gumbiner,B., Cossart, P., 1999. A single amino acid in the E-cadherinmolecule was responsible for host specificity towards the humanpathogen Listeria monocytogenes. EMBO J. 18 (14),3956–3963.
Lecuit, M., Vandormael-Pournin, S., Lefort, J., Huerre, M., Gounon,P., Dupuy, C., Babinet, C., Cossart, P., 2001. A transgenic modelfor listeriosis: role of internalin in crossing the intestinal barrier.Science 292 (5522), 1722–1725.
Leimeister-Wächter, M., Domann, E., Chakraborty, T., 1992. Theexpression of virulence genes in Listeria monocytogenes isthermoregulated. J. Bacteriol. 174, 947–952.
Leonard, P., 2003. Production of antibodies for use in a biosensor-based assay for Listeria monocytogenes. Ph.D. thesis, Dublin CityUniversity, Ireland.
Leonard, P., Hearty, S., Brennan, J., Dunne, L., Quinn, J., Chakraborty,T., O'Kennedy, R., 2003. Advances in biosensors for detection ofpathogens in food and water. Enzyme Microb. Technol. 32, 3–13.
Leonard, P., Hearty, S., Quinn, J., O'Kennedy, R., 2004. A genericapproach for the detection of whole Listeria monocytogenes cellsin contaminated samples using surface plasmon resonance.Biosens. Bioelectron. 19, 1331–1335.
Leonard, P., Hearty, S., Wyatt, G., Quinn, J., O'Kennedy, R., 2005.Development of a surface plasmon resonance-based immunoassayfor Listeria monocytogenes. J. Food Prot. 68 (4), 728–735.
Lingnau, A., Domann, E., Hudel, M., Bock, M., Nichterlein, T.,Wehland, J., Chakraborty, T., 1995. Expression of the Listeriamonocytogenes EGD inlA and inlB genes, whose products mediatebacterial entry into tissue culture cell lines, by PrfA-dependent and-independent mechanisms. Infect. Immun. 63 (10), 3896–3903.
Liedberg, B., Nylander, C., Lundstrom, I., 1995. Biosensing withsurface plasmon resonance—how it all started. Biosens. Bioelec-tron. 10, 1–9.
Löfås, S., Malmqvist, M., Rönnberg, I., Stenberg, E., Liedberg, B.,Lundström, I., 1991. Bioanalysis with surface plasmon resonance.Sens. Actuators, B, Chem. 5, 79–84.

311S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
Loiseau, O., Cottin, J., Robert, R., Tronchin, G., Mahaza, C., Senet, J.M., 1995. Development and characterisation of monoclonalantibodies specific for the genus Listeria. FEMS Immunol. Med.Microbiol. 11, 219–230.
Lu, B., Smyth, M.R., O'Kennedy, R., 1996. Oriented immobilizationof antibodies and its applications in immunoassays and immuno-sensors. Analyst 121, 29R–32R.
Malmqvist, M., 1999. BIACORE: an affinity biosensor system forcharacterisation of biomolecular interactions. Biochem. Soc.Trans. 27, 335–340.
Mattingly, J.A., Butman, B.T., Plank, M.C., Durham, R.J., Robison, B.J., 1988. Rapid monoclonal antibody-based enzyme-linkedimmunosorbent assay for detection of Listeria in food products.J. Assoc. Off. Anal. Chem. 71, 679–681.
Nagata, K., Handa, H. (Eds.), 2000. Real-time Analysis ofBiomolecular Interactions–Applications of BIAcore. Springer-Verlag, Tokyo, Japan.
Navarre, W.W., Schneewind, O., 1999. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wallenvelope. Micrbiol. Mol. Biol. Rev. 63 (1), 174–229.
Okutani, A., Okada, Y., Yamamoto, S., Igimi, S., 2004. Overview ofListeria monocytogenes contamination in Japan. Int. J. FoodMicrobiol. 93 (2), 131–140.
Olier, M., Pierre, F., Rousseaux, S., Lemaître, J.-P., Rousset, A.,Piveteau, P., Guzzo, J., 2003. Expression of truncated internalin Ais involved in impaired internalization of some Listeria mono-cytogenes isolates carried asymptomatically by humans. Infect.Immun. 71 (3), 1217–1224.
Olsen, E.V., Pathirana, S.T., Samoylov, A.M., Barbaree, J.M., Chin, B.A., Neely, W.C., Vodyanoy, V., 2003. Specific and slectivebiosensor for Salmonella and its detection in the environment.J. Immunol. Methods 53, 273–285.
O'Shannessey, D.J., Bringham-Burke, M., Peck, K., 1992. Immobi-lisation chemistries suitable for use in the BIAcore surfaceplasmon resonance detector. Anal. Biochem. 205, 132–136.
O'Sullivan, C.K., Vaughan, R., Guilbault, G.G., 1999. Piezoelectricimmunosensors—theory and applications. Anal. Lett. 32, 2353–2377.
Pak, S.-I., Spahr, U., Jemmi, T., Salman, M.D., 2002. Risk factors forListeria monocytogenes contamination of dairy products inSwitzerland, 1990–1999. Prev. Vet. Med. 53, 55–65.
Paoli, G.C., Chen, C.-Y., Brewster, J.D., 2004. Single-chain Fvantibody with specificity for Listeria monocytogenes. J. Immunol.Methods 289, 147–155.
Park, I.-S., Kim, W.-Y., Kim, N., 2000. Operational characteristics ofan antibody-immobilized QCM system detecting Salmonella spp.Biosens. Bioelectron. 15, 167–172.
Petrenko, V.A., Sorokulova, I.B., 2004. Detection of biological threats.A challenge for directed molecular evolution. J. Microbiol.Methods 58, 147–168.
Piehler, J., Brecht, A., Hehl, K., Gauglitz, G., 1999. Proteininteractions in covalently attached dextran layers. Colloids Surf.,B Biointerfaces 13, 325–336.
Pitcher, D.G., Saunders, N.A., Owen, R.J., 1989. Rapid extraction ofbacterial genomic DNA with guanidium thiocyanate. Letts. Appl.Microbiol. 8, 151–156.
Pourshafie, M.R., Marlund, B.I., Ohlson, S., 2004. Binding interac-tions of Escherichia coli with globtetraosylceramide (globoside)using a surface plasmon resonance biosensor. J. Microbiol.Methods 58, 313–320.
Poyart, C., Trieu-Cuot, P., Berche, P., 2003. The inlA gene required forcell invasion is conserved and specific to Listeria monocytogenes.Microbiology 142, 173–180.
Quinn, J.G., 1999. Real-time biomolecular interaction analysis: novelapplications and developments. Ph.D. thesis, Dublin CityUniversity, Ireland.
Quinn, J.G., O'Kennedy, R., Smyth, M., Moulds, J., Frame, T., 1997.Detection of blood antigens utilising immobilised antibodies andsurface plasmon resonance. J. Immunol. Methods 206, 87–96.
Quinn, J.G., Patel, P., Fitzpatrick, B., Manning, B., Dillon, P., Daly,S., O'Kennedy, R., Alcocer, M., Lee, H., Morgan, M., Lang,K., 1999. The use of regenerable, affinity ligand-based surfacesfor immunosensor applications. Biosens. Bioelectron. 14,587–595.
Quinn, J.G., O'Neil, S., Doyle, S., McAtamney, C., Diamond, D.,McCraith, B.D., O'Kennedy, R., 2000. Development and applica-tion of surface plasmon resonance-based biosensors for thedetection of cell–ligand interactions. Anal. Biochem. 281,135–143.
Regulation (EC) No. 178/2002, 2002. Of The European Parliamentand The Council of 28 January 2002. Official Journal of theEuropean Communities 1992, L 31/1, 1st February, 2002.
Rief, M., Oesterhelt, F., Heymann, B., Gaub, H.E., 1997. Singlemolecule force spectroscopy on polysaccharides by atomic forcemicroscopy. Science 275, 1295–1297.
Ripio, M.-T., Domínguez-Bernal, G., Brehm, K., Berche, P., Suárez,M., Vázquez-Boland, J.A., 1996. Transcriptional activation ofvirulence genes in wild-type strains of Listeria monocytogenes inresponse to a change in the extracellular medium composition.Res. Microbiol. 147, 311–384.
Rocourt, J., 1994. Listeria monocytogenes: the state of the art. DairyFood Environ. Sanit. 14, 70–82.
Ruhland, G.J., Hellwig, M., Wanner, S., Frank, R., Goebel, W., 1993.Cell-surface location of Listeria-specific protein p60-detection ofListeria cells by indirect immunofluorescence. J. Gen. Microbiol.139, 609–616.
Scotter, S.L., Langton, S., Lombard, B., Schulten, S., Nagelkerke, N.,In't Veld, P.H., Rollier, P., Lahellec, C., 2001a. Validation of ISOmethod 11290: Part 1. Detection of Listeria monocytogenes infoods. Int. J. Food Microbiol. 64, 295–306.
Scotter, S.L., Langton, S., Lombard, B., Lahellec, C., Schulten, S.,Nagelkerke, N., In't Veld, P.H., Rollier, P., 2001b. Validation ofISO method 11290: Part 2. Enumeration of Listeria monocyto-genes in foods. Int. J. Food Microbiol. 70, 121–129.
Sewell, A.M., Warburton, D.W., Boville, A., Daley, E.F., Mullen, K.,2003. The development of an efficient and rapid enzyme-linkedfluorescent assay method for the detection of Listeria spp. fromfoods. Int. J. Food Microbiol. 81, 123–129.
Sheehan, B., Klarsfeld, A., Msadek, T., Cossart, P., 1995. Differentialactivation of virulence gene expression by PrfA, the Listeriamonocytogenes virulence regulator. J. Bacteriol. 177 (22),6469–6476.
Siragusa, G.R., Johnson, M.G., 1990. Monoclonal antibody specificfor Listeria monocytogenes, Listeria innocua and Listeriawelshimeri. Appl. Environ. Microbiol. 56, 1897–1904.
Sølve, M., Boel, J., Nørrung, B., 2000. Evaluation of a monoclonalantibody able to detect live Listeria monocytogenes and Listeriainnocua. Int. J. Food Microbiol. 57, 219–224.
Torensma, R., Visser, M.J.C., Aarsman, C.J.M., Poppelier, M.J.J.G., Fluit, A.C., Verhoef, J., 1993. Monoclonal antibodies thatreact with live Listeria spp. Appl. Environ. Microbiol. 59,2713–2716.
Vaughan, R.D., O'Sullivan, C.K., Guilbault, G.G., 2001. Development ofa quartz crystal microbalance (QCM) immunosensor for the detectionof Listeria monocytogenes. Enzyme Microb. Technol. 29, 635–638.

312 S. Hearty et al. / Journal of Microbiological Methods 66 (2006) 294–312
Vaughan, R.D., Carter, R.M., O'Sullivan, C.K., Guilbault, G.G., 2003.A quartz crystal microbalance (QCM) sensor for the detection ofBacillus cereus. Anal. Lett. 36 (4), 731–747.
Vázquez-Boland, J.A., Kuhn, M., Berche, P., Chakraborty, T.,Domínguez-Bernal, G., Goebel, W., González-B., Wehland, J,Kreft, J., 2001a. Listeria pathogenesis and molecular virulencedeterminants. Clin. Microbiol. Rev. 14 (3), 584–640.
Vázquez-Boland, J.A., Domínguez-Bernal, G., González-Zorn, B.,Kreft, J., Goebel, W., 2001b. Pathogenicity islands and virulenceevolution in Listeria. Microbes Infect. 3, 571–584.
Więckowska-Szakiel, M., Bubert, A., Różalski, M., Krajewska, U.,Rudnicka, W., Różalska, B., 2002. Colony-blot assay with anti-p60 antibodies as a method for quick identification of Listeria infood. Int. J. Food Microbiol. 72, 63–71.