Embed Size (px)
Citation preview

Parkinson’s disease (PD) is a neurodegenerative dis-order that is characterized, in part, by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta. It affects 1.5% of the global population over 65 years of age. The lack of dopamine causes the classical motor symptoms of bradykinesia, rigidity and resting tremors. These symptoms are improved by cur-rent dopamine replacement strategies, which include levodopa (l-DOPA, the precursor of dopamine) and dopamine receptor agonists, as well as monoam-ine oxidase B (MAOB) inhibitors and catechol O-methyltransferase inhibitors. After several years of disease progression, treatment is complicated by the onset of motor fluctuations and dyskinesia, which leads to alternating periods of reduced mobility and abnor-mal involuntary movements1. Continuous delivery of the dopamine agonist apomorphine (subcutaneously) or l-DOPA (intraduodenally), and surgical strategies such as deep brain stimulation (DBS), provide relief in patients with severe motor fluctuations2–5. Some patients receiving dopaminergic drugs develop abnor-mal behaviours, including impulse control disorders or dopamine dysregulation syndrome6.
Along with dopamine, the neurodegenerative process involves other neurotransmitters such as noradrenergic, serotonergic and cholinergic systems. Non-motor symp-toms such as depression, dementia, sleep abnormalities
and autonomic failure — which often dominate the clin-ical, caregiver and financial burden of the later stages of PD — are probably the consequence of degen-eration of both dopaminergic and non-dopaminergic systems7.
Almost 50 years after its introduction, l-DOPA remains the most effective form of oral symptomatic treatment for motor symptoms. However, l-DOPA has less impact on non-motor symptoms7, and so current therapeutic challenges involve the management of these symptoms. Beyond symptomatic relief, treatments that have neuroprotective or disease-modifying properties remain a critical unmet clinical need. Slowing the rate of loss of nigral dopaminergic neurons would sim-plify the management of motor features, and probably improve the quality of life of patients over the dura-tion of their illness. Similarly, delaying the develop-ment of non-motor features would transform PD into a much more benign clinical condition. Overcoming the limitations of current treatments and advancing the field of drug discovery and development will depend on multiple convergent advancements; these include transformative discoveries on disease pathophysiology and animal models of a higher quality, which would enable a better translation of preclinical research from the laboratory to the clinic. In addition, improvements in the methodologies used in the design of clinical
*Service de Neurologie et Centre de référence atrophie multisystématisée, CHU de Bordeaux, Avenue Magellan F‑33604 Pessac France.Correspondence to W.G.M. e‑mail: wassilios.meissner@chu‑bordeaux.frdoi:10.1038/nrd3430
BradykinesiaAbnormally slow voluntary movements.
RigidityIncreased muscle tone during passive limb movements, which is frequently associated with a ‘cogwheel‘ phenomenon.
DyskinesiaLevodopa-induced involuntary movements that affect the head, trunk and limbs.
Priorities in Parkinson’s disease researchWassilios G. Meissner*‡§, Mark Frasier||, Thomas Gasser¶, Christopher G. Goetz#, Andres Lozano**, Paola Piccini‡‡, José A. Obeso§§, Olivier Rascol||||, Anthony Schapira¶¶, Valerie Voon##, David M. Weiner***, François Tison*‡§ and Erwan Bezard‡§
Abstract | The loss of dopaminergic neurons in the substantia nigra pars compacta leads to the characteristic motor symptoms of Parkinson’s disease: bradykinesia, rigidity and resting tremors. Although these symptoms can be improved using currently available dopamine replacement strategies, there is still a need to improve current strategies of treating these symptoms, together with a need to alleviate non-motor symptoms of the disease. Moreover, treatments that provide neuroprotection and/or disease-modifying effects remain an urgent unmet clinical need. This Review describes the most promising biological targets and therapeutic agents that are currently being assessed to address these treatment goals. Progress will rely on understanding genetic mutations or susceptibility factors that lead to Parkinson’s disease, better translation between preclinical animal models and clinical research, and improving the design of future clinical trials.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 377
© 2011 Macmillan Publishers Limited. All rights reserved

Paradoxic kinesisA sudden and brief relief of motor signs due to a sudden stress — for example, a fire alarm.
Off-period dystoniaSustained muscle contractions and postures during off-periods. Off-period dystonia relates to subtherapeutic levels of levodopa and is both caused and relieved by levodopa. It frequently manifests as early-morning foot dystonia or diphasic painful dystonia of the lower limbs.
Subthalamic nucleusA glutamatergic relay within the basal ganglia that becomes overactive in Parkinson’s disease. Overexpression of glutamic acid decarboxylase — the key enzyme of GABA (γ-aminobutyric acid) synthesis — may dampen the overactivity of subthalamic neurons.
studies are needed, especially with regard to the iden-tification and validation of outcome measures that are more objective.
In this Review, we discuss the key challenges of treating PD, novel emerging agents and targets as potential treatment strategies, and how to better design clinical trials — including the development of objective end points for trials, which will assess the efficacy of neuroprotective agents. We also discuss why progress in understanding genetic mutations and susceptibility factors that lead to or increase the risk of PD and a better translation between preclinical animal models and clinical research are priorities for overcoming the limitations of current therapies.
The industrial landscapeCurrent therapeutic development in PD includes approaches such as re-formulations (for example, extended release formulation) of existing drugs that are approved for PD, re-positioning of compounds that are approved for other indications (such as the antihypertensive drug isradipine, the antiepileptic topiramate or methylpheni-date) and development of novel small-molecule and gene therapy-based approaches. The therapeutic development pipelines appear to be vigorous on the surface. However, once dopaminergic compounds are removed from the pipe-line (FIG. 1), the current landscape is far less encouraging. Such dopaminergic therapies include new formulations of existing drugs, which are more likely to provide incre-mental rather than profound improvements over existing therapies. Many of the therapies that are currently under development — including both dopaminergic and non-dopaminergic compounds — are focused on improve-ment of motor control, fluctuations and dyskinesias. Far fewer approaches address the other two key unmet clinical needs, specifically: alleviating non-motor symptoms; and disease modification and/or neuroprotection (see the sec-tion entitled ‘Future treatments’).
Motor symptoms of PDThe main motor symptoms of PD were first described in the essay on shaking palsy by James Parkinson in 1817 (REF.
8). Among these symptoms, bradykinesia, resting tremors and rigidity are the core features of PD9, and these symp-toms can be controlled for many years using l-DOPA and other dopaminergic agents. However, motor fluctuations and dyskinesias appear in most patients, either as part of disease progression or as a result of chronic dopaminergic therapy10 (or as a result of both), and so alleviating dyski-nesia remain unmet clinical needs.
Motor fluctuations and dyskinesias. Motor fluctuations include the ‘wearing-off ’ phenomenon (a gradual waning of the effect of dopaminergic treatment on motor symp-toms before the next dose), ‘on/off ’ fluctuations (whereby symptoms can reappear and disappear quickly and unexpectedly, freezing of gait, paradoxic kinesis and early-morning dystonia11. As the disease progresses, fluctuations between ‘on’ and ‘off ’ periods (that is, when symptoms increase (off-period) or decrease (on-period)) become more apparent in patients. Although most patients with PD fear dyskinesias at the onset of disease, motor fluctua-tions also make a substantial contribution to the reduced quality of life of patients with advanced-stage PD. Several compounds that delay the metabolism of dopaminergic drugs or act on non-dopaminergic targets are currently under development to target the ‘wearing-off ’ phenom-enon (TABLES 1,2).
There are three forms of dyskinesias that commonly occur with l-DOPA use — peak-dose dyskinesia, dipha-sic dyskinesia and off-period dystonia — which negatively affect the quality of life of patients in advanced stages of the disease12. Peak-dose dyskinesia occurs when plasma l-DOPA levels are highest, diphasic dyskinesia refers to the abnormal involuntary movements that occur tran-siently at the onset and the end of l-DOPA efficacy, and off-period dystonia occurs when a patient receives subther-apeutic levels of l-DOPA. Recent advances in treatment have lessened but not entirely eliminated severe disabling dyskinesias. Specific examples of such advances include DBS of the subthalamic nucleus, continuous subcutane-ous infusion of apomorphine and continuous duodenal infusion of l-DOPA. The identification of agents that can acutely suppress existing disabling dyskinesias and of agents that do not induce dyskinesias is currently a major focus of drug development in PD (see the section entitled ‘Future treatments’).
Aside from DBS and continuous infusion of apomor-phine or l-DOPA, current treatment options for dyski-nesias are limited: only one drug — amantadine — has achieved the designation of ‘efficacious’ as determined by the Movement Disorder Society Evidence-Based Medical Review13. This designation, however, is based on the results of several small, underpowered clinical studies. Several other agents have been studied, but each agent has failed when it has been tested in larger, double-blind placebo-controlled trials (for example, sarizotan14). Three major limitations have hindered the development of new treatments. First, the pharmacology of dyskinesias is not completely understood. Second, there is a lack of
Author addresses‡Université de Bordeaux, Institut des Maladies Neurodégénératives, UMR 5293, F‑33000 Bordeaux, France.§CNRS, Institut des Maladies Neurodégénératives, UMR 5293, F‑33000 Bordeaux, France.||Michael J. Fox Foundation for Parkinson’s Research, New York, New York 10004, USA.¶Department of Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen and DZNE — German Center for Neurodegenerative Diseases, D‑72076 Tübingen, Germany. #Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois 60612, USA.**Department of Neurosurgery, Toronto Western Hospital, University of Toronto, Toronto, Ontario M5T 2S8, Canada. ‡‡Centre for Neuroscience, Imperial College London, London, SW7 2AZ, UK.§§Movement Disorders Group, Neurosciences Division, CIMA, and Department of Neurology and Neurosurgery, Clínica Universitaria and CIBERNED, University of Navarra, 31008 Pamplona, Spain. ||||Departments of Clinical Pharmacology and Neurosciences, INSERM CIC9302, University Hospital of Toulouse, 31000 Toulouse, France.¶¶Department of Clinical Neurosciences, Institute of Neurology, University College London, London NW3 2PF, UK.¶#Behavioral and Clinical Neurosciences Institute, University of Cambridge, Cambridge CB2 0SZ, UK.***EMD Serono, Rockland, Massachusetts 02370, USA.
R E V I E W S
378 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

validated clinical outcome measures that are responsive to treatment, despite the availability of multiple dyski-nesia rating scales15. Third, the development of a unitary, sensitive and robust rating scale is challenging because of the different types of dyskinesias, their temporal patterns, anatomical distributions and associated disabilities.
In addition, dyskinesias are highly susceptible to the effects of placebo, and therefore calculations of sample
size and power estimates for a clinical trial must include an expectation of improvement with placebo treatment14. Interestingly, the placebo effect may vary considerably between different regional sites in multinational clinical trials14, and the likelihood of a patient being assigned to receive placebo may also influence the clinical out-come through stimulation of physiological mechanisms — for example, the release of dopamine and other
Figure 1 | Drugs in development for the treatment of Parkinson’s disease. Compounds are illustrated according to their mode of action (dopaminergic, non-dopaminergic or gene therapy) and phase of development (Phase I through to marketed). Non-dopaminergic drugs include agents that act on serotonin, glutamate, adenosine and adrenergic receptors, as well as those that act on ion channels. It has recently been shown that rasagiline may delay disease progression; however, the clinical significance of these results is a matter of ongoing debate96 (see section entitled ‘Design of future clinical trials’). Data were obtained from public sources, namely press releases and company websites. *This drug was removed from the US market in 2007. ER, extended release; G-CSF, granulocyte colony-stimulationg factor; IR, immediate release; PDGF, platelet-derived growth factor; SAM, S-actenosyl-l-methionine.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 379
© 2011 Macmillan Publishers Limited. All rights reserved
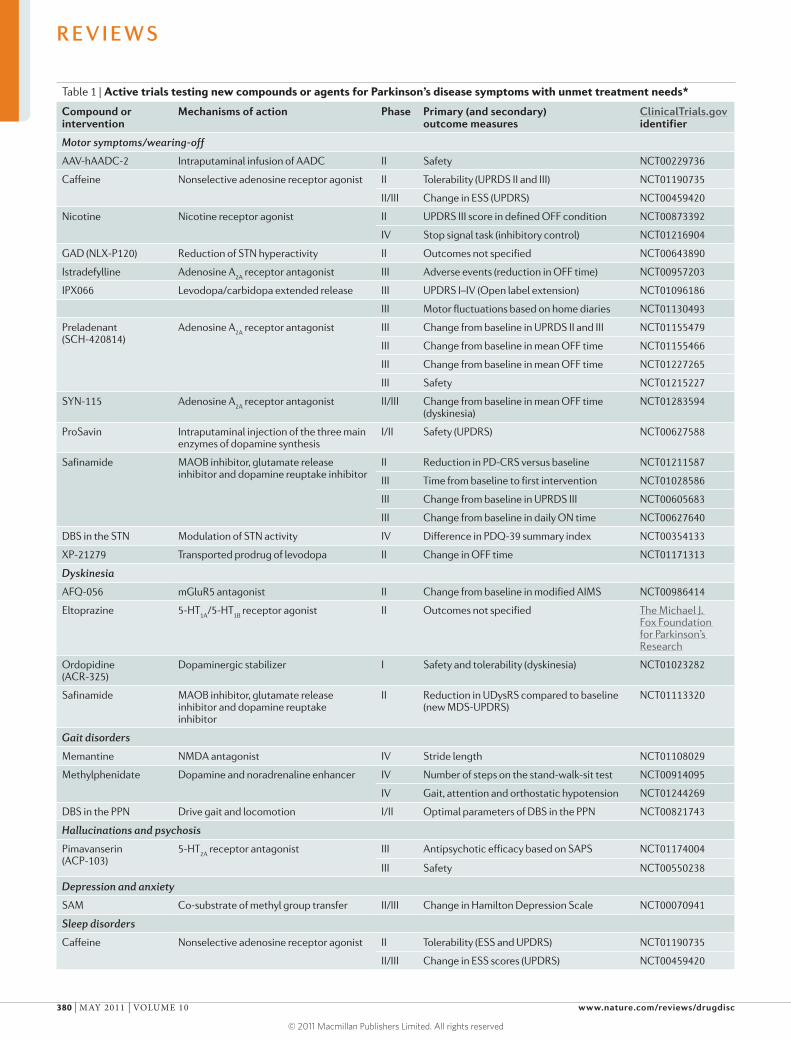
Table 1 | Active trials testing new compounds or agents for Parkinson’s disease symptoms with unmet treatment needs*
Compound or intervention
Mechanisms of action Phase Primary (and secondary) outcome measures
ClinicalTrials.gov identifier
Motor symptoms/wearing-off
AAV-hAADC-2 Intraputaminal infusion of AADC II Safety NCT00229736
Caffeine Nonselective adenosine receptor agonist II Tolerability (UPRDS II and III) NCT01190735
II/III Change in ESS (UPDRS) NCT00459420
Nicotine Nicotine receptor agonist II UPDRS III score in defined OFF condition NCT00873392
IV Stop signal task (inhibitory control) NCT01216904
GAD (NLX-P120) Reduction of STN hyperactivity II Outcomes not specified NCT00643890
Istradefylline Adenosine A2A
receptor antagonist III Adverse events (reduction in OFF time) NCT00957203
IPX066 Levodopa/carbidopa extended release III UPDRS I–IV (Open label extension) NCT01096186
III Motor fluctuations based on home diaries NCT01130493
Preladenant (SCH-420814)
Adenosine A2A
receptor antagonist III Change from baseline in UPRDS II and III NCT01155479
III Change from baseline in mean OFF time NCT01155466
III Change from baseline in mean OFF time NCT01227265
III Safety NCT01215227
SYN-115 Adenosine A2A
receptor antagonist II/III Change from baseline in mean OFF time (dyskinesia)
NCT01283594
ProSavin Intraputaminal injection of the three main enzymes of dopamine synthesis
I/II Safety (UPDRS) NCT00627588
Safinamide MAOB inhibitor, glutamate release inhibitor and dopamine reuptake inhibitor
II Reduction in PD-CRS versus baseline NCT01211587
III Time from baseline to first intervention NCT01028586
III Change from baseline in UPRDS III NCT00605683
III Change from baseline in daily ON time NCT00627640
DBS in the STN Modulation of STN activity IV Difference in PDQ-39 summary index NCT00354133
XP-21279 Transported prodrug of levodopa II Change in OFF time NCT01171313
Dyskinesia
AFQ-056 mGluR5 antagonist II Change from baseline in modified AIMS NCT00986414
Eltoprazine 5-HT1A
/5-HT1B
receptor agonist II Outcomes not specified The Michael J. Fox Foundation for Parkinson’s Research
Ordopidine (ACR-325)
Dopaminergic stabilizer I Safety and tolerability (dyskinesia) NCT01023282
Safinamide MAOB inhibitor, glutamate release inhibitor and dopamine reuptake inhibitor
II Reduction in UDysRS compared to baseline (new MDS-UPDRS)
NCT01113320
Gait disorders
Memantine NMDA antagonist IV Stride length NCT01108029
Methylphenidate Dopamine and noradrenaline enhancer IV Number of steps on the stand-walk-sit test NCT00914095
IV Gait, attention and orthostatic hypotension NCT01244269
DBS in the PPN Drive gait and locomotion I/II Optimal parameters of DBS in the PPN NCT00821743
Hallucinations and psychosis
Pimavanserin (ACP-103)
5-HT2A
receptor antagonist III Antipsychotic efficacy based on SAPS NCT01174004
III Safety NCT00550238
Depression and anxiety
SAM Co-substrate of methyl group transfer II/III Change in Hamilton Depression Scale NCT00070941
Sleep disorders
Caffeine Nonselective adenosine receptor agonist II Tolerability (ESS and UPDRS) NCT01190735
II/III Change in ESS scores (UPDRS) NCT00459420
R E V I E W S
380 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved
neurotransmitters16. The development of study designs and dyskinesia rating scales that are resistant to placebo influences would be important advances in this field. To address this issue, a clinical study has been designed to clearly define the therapeutic impact of amantadine on dyskinesia (ClinicalTrials.gov identifier: NCT01071395). This trial aims to precisely evaluate the effect of receiving placebo treatment in a clinical trial on patient outcome and to determine a hierarchy of numerous rating scales based on their absolute and relative capability to detect changes in dyskinesias during treatment. This trial will thus identify the best rating scale (or scales) for evaluat-ing dyskinesia (BOX 1) while benchmarking the effect of amantadine in the treatment of dyskinesia.
Other motor issues. Freezing of gait, postural instabil-ity, abnormal postures of the trunk and neck, as well as dysarthria and dysphagia are motor manifestations that typically arise 10 or more years after the onset of PD17,18. They cause substantial disability in daily life and no effective treatments are yet available. There is a noticeable paucity of drug development efforts for these important symptoms, which is due in large part to their
poorly understood pathophysiology. Small clinical trials with methylphenidate and DBS of the pedunculopontine nucleus have suggested some efficacy for fall frequency and freezing of gait19–21.
Non-motor symptomsSome non-motor symptoms were already described by James Parkinson in 1817 in his essay on the shak-ing palsy8, and they are increasingly being recognized as features of the clinical picture and PD pathology. These include neuropsychiatric symptoms, autonomic failure and sleep abnormalities, and they dominate the clinical, caregiver and financial burden in advanced stages of PD. Efficacious treatments remain an unmet clinical need; the development of therapies to manage the non-motor symptoms of PD is therefore a priority.
Impulse control disorders and the dopamine dysregu-lation syndrome. Behaviours that include pathological gambling, compulsive shopping, hypersexuality and binge eating — which are collectively termed impulse control disorders — are present in ~15% of patients with PD and are associated with the use of dopamine
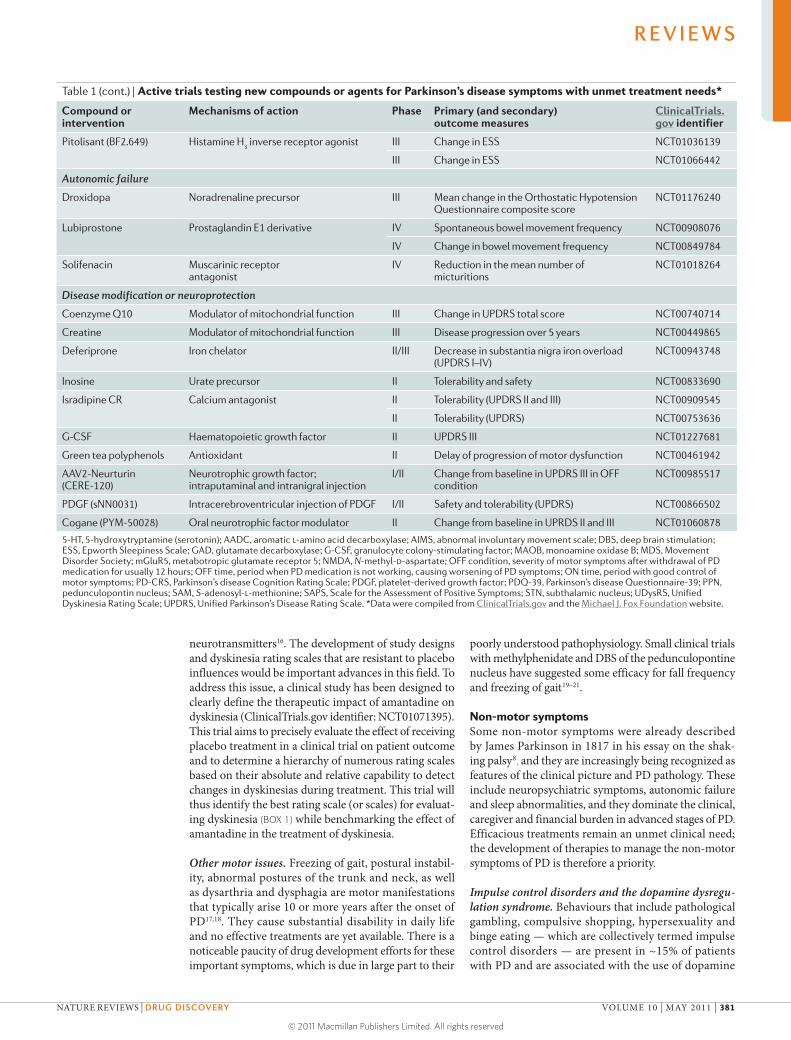
Table 1 (cont.) | Active trials testing new compounds or agents for Parkinson’s disease symptoms with unmet treatment needs*
Compound or intervention
Mechanisms of action Phase Primary (and secondary) outcome measures
ClinicalTrials.gov identifier
Pitolisant (BF2.649) Histamine H3 inverse receptor agonist III Change in ESS NCT01036139
III Change in ESS NCT01066442
Autonomic failure
Droxidopa Noradrenaline precursor III Mean change in the Orthostatic Hypotension Questionnaire composite score
NCT01176240
Lubiprostone Prostaglandin E1 derivative IV Spontaneous bowel movement frequency NCT00908076
IV Change in bowel movement frequency NCT00849784
Solifenacin Muscarinic receptor antagonist
IV Reduction in the mean number of micturitions
NCT01018264
Disease modification or neuroprotection
Coenzyme Q10 Modulator of mitochondrial function III Change in UPDRS total score NCT00740714
Creatine Modulator of mitochondrial function III Disease progression over 5 years NCT00449865
Deferiprone Iron chelator II/III Decrease in substantia nigra iron overload (UPDRS I–IV)
NCT00943748
Inosine Urate precursor II Tolerability and safety NCT00833690
Isradipine CR Calcium antagonist II Tolerability (UPDRS II and III) NCT00909545
II Tolerability (UPDRS) NCT00753636
G-CSF Haematopoietic growth factor II UPDRS III NCT01227681
Green tea polyphenols Antioxidant II Delay of progression of motor dysfunction NCT00461942
AAV2-Neurturin (CERE-120)
Neurotrophic growth factor; intraputaminal and intranigral injection
I/II Change from baseline in UPDRS III in OFF condition
NCT00985517
PDGF (sNN0031) Intracerebroventricular injection of PDGF I/II Safety and tolerability (UPDRS) NCT00866502
Cogane (PYM-50028) Oral neurotrophic factor modulator II Change from baseline in UPRDS II and III NCT01060878
5-HT, 5-hydroxytryptamine (serotonin); AADC, aromatic l-amino acid decarboxylase; AIMS, abnormal involuntary movement scale; DBS, deep brain stimulation; ESS, Epworth Sleepiness Scale; GAD, glutamate decarboxylase; G-CSF, granulocyte colony-stimulating factor; MAOB, monoamine oxidase B; MDS, Movement Disorder Society; mGluR5, metabotropic glutamate receptor 5; NMDA, N-methyl-d-aspartate; OFF condition, severity of motor symptoms after withdrawal of PD medication for usually 12 hours; OFF time, period when PD medication is not working, causing worsening of PD symptoms; ON time, period with good control of motor symptoms; PD-CRS, Parkinson’s disease Cognition Rating Scale; PDGF, platelet-derived growth factor; PDQ-39, Parkinson’s disease Questionnaire-39; PPN, pedunculopontin nucleus; SAM, S-adenosyl-l-methionine; SAPS, Scale for the Assessment of Positive Symptoms; STN, subthalamic nucleus; UDysRS, Unified Dyskinesia Rating Scale; UPDRS, Unified Parkinson’s Disease Rating Scale. *Data were compiled from ClinicalTrials.gov and the Michael J. Fox Foundation website.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 381
© 2011 Macmillan Publishers Limited. All rights reserved

agonists, l-DOPA and higher doses of l-DOPA22. With regard to these behaviours, many questions remain to be investigated, including the role of l-DOPA versus dopamine agonists, tonic versus phasic dopaminergic activity, and the involvement of specific dopamine receptors and neuroplastic adaptations. Studies to assess the cognitive processes underlying these behav-iours are small and have not yet addressed the role of incentive motivation, compulsivity or hedonic tone. A recent small and blinded crossover trial showed that amantadine may be effective in the treatment of patho-logical gambling in PD, thus suggesting a potential role for glutamatergic mechanisms in impulse control disorders23. Future clinical studies, together with pre-clinical research, will allow us to identify risk factors, underlying mechanisms and potential treatments for these disorders.
PD dementia. Dementia occurs in more than 80% of patients with PD after 20 years of disease duration18. Once dementia occurs, it indicates a short time to death, irrespective of age and disease duration24. From an anato-mopathological standpoint, PD dementia is believed to be due to a variable combination of the extension of Lewy pathology to limbic and cortical structures with concomitant Alzheimer’s disease (AD)-related neurofi-brillary tangles and amyloid-β plaque pathology24,25. The recent observation that lower levels of amyloid-β1–42 in the cerebral spinal fluid may predict a more rapid cog-nitive decline supports the contribution of AD-related pathologies to the cognitive impairment that is seen in patients with PD26.
Clinical criteria for diagnosing possible and prob-able PD dementia have recently been proposed25 (BOX 2). These definitions are likely to facilitate the inclusion in future clinical trials of patient populations that are more homogeneous, and they are important prerequisites for prospective studies that aim to assess the natural history of PD dementia. We believe that these criteria for diag-nosing PD dementia will enable specific points in disease progression to be defined, particularly in the elderly, and they could be used as a measure of clinically meaningful outcomes in clinical trials of disease-modifying strate-gies in patients with advanced-stage PD. Mild cognitive impairment is the most important risk co-factor, along with age, for predicting PD dementia, and the signs that constitute mild cognitive impairment have recently been better characterized in a large multicentre cohort study27. In addition, improved criteria for diagnosing mild cogni-tive impairment in PD are urgently needed to stimulate efforts of finding drugs that will act at earlier pathologi-cal phases before overt dementia becomes apparent, to identify the most appropriate cohort of patients for con-ducting clinical trials of these drugs, and to allow regula-tory approval of such drugs.
The goals of current treatments in PD dementia are to provide relief from cognitive, behavioural and other neuropsychiatric symptoms without worsening motor impairment or altering the relief of symptoms that is provided by l-DOPA. Strategies to achieve this rely on fine-tuning the balance between dopaminergic and non-
dopaminergic (prominently cholinergic) neurotransmis-sion. Anticholinesterases, particularly rivastigmine, have been shown to have efficacy on cognitive impairment when they have been assessed by clinical global impres-sion and global cognitive scales28. N-methyl-d-aspartate (NMDA) receptor antagonists such as memantine have shown some efficacy in a pilot study29. However, in a larger, placebo-controlled trial, positive effects of this drug were only observed in patients with Lewy body dementia (BOX 2), not in those with PD dementia30. As memantine is also effective in AD31, and patients with Lewy body dementia display more AD pathology than those with PD dementia32, memantine might exert stronger effects in patients with a higher amyloid-β burden. Another explanation for these results may be the lower baseline neuropsychiatric inventory scores in patients with PD receiving memantine compared to patients with PD receiving placebo. We notice that ongoing clinical trials for PD dementia that are regis-tered in the ClinicalTrials.gov database are sparse and not particularly innovative, given the public health bur-den it represents.
Other neuropsychiatric features. Beyond demen-tia, major non-motor neuropsychiatric symptoms include depression, anxiety, apathy and psychosis33,34. Management of hallucinations and psychosis in patients with PD is a challenge. Although the atypical neurolep-tic clozapine — a 5-hydroxytryptamine (serotonin) 2A (5-HT2A) receptor and 5-HT2C receptor antagonist — remains the most effective treatment, its haematological side effects require careful monitoring35. With regard to depression, double-blind placebo-controlled trials have highlighted that dopaminergic and non-dopaminergic transmitter systems contribute to depression in PD36–38, and that tricyclic antidepressants — which are dual uptake inhibitors of noradrenaline and serotonin — may be more effective at treating PD-induced depres-sion than selective serotonin reuptake inhibitors37. This observation is not surprising in view of the higher loss of dopaminergic and noradrenergic neurons in the locus coeruleus, ventral striatum, thalamus, anterior cingulated cortex and amygdala in individuals with PD who suffer from depression, compared with individuals with PD who do not suffer from depression39. Symptoms of depression in antidepressant-naive patients with PD further correlate with higher levels of serotonin trans-porter binding sites in raphe nuclei and limbic struc-tures, possibly reflecting lower extracellular serotonin levels40. Taken together, these findings point to a specific pathophysiological pattern in PD-associated depression, and suggest that drugs that combine dopaminergic, noradrenergic and possibly serotonergic actions may be the most promising for the treatment of PD-associated depression.
Autonomic failure and sleep disorders. Autonomic failure — which manifests as symptomatic orthostatic hypotension, urinary incontinence and constipation — and sleep disorders are prevalent in patients with PD. Several treatments such as sildenafil, macrogol,
R E V I E W S
382 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved
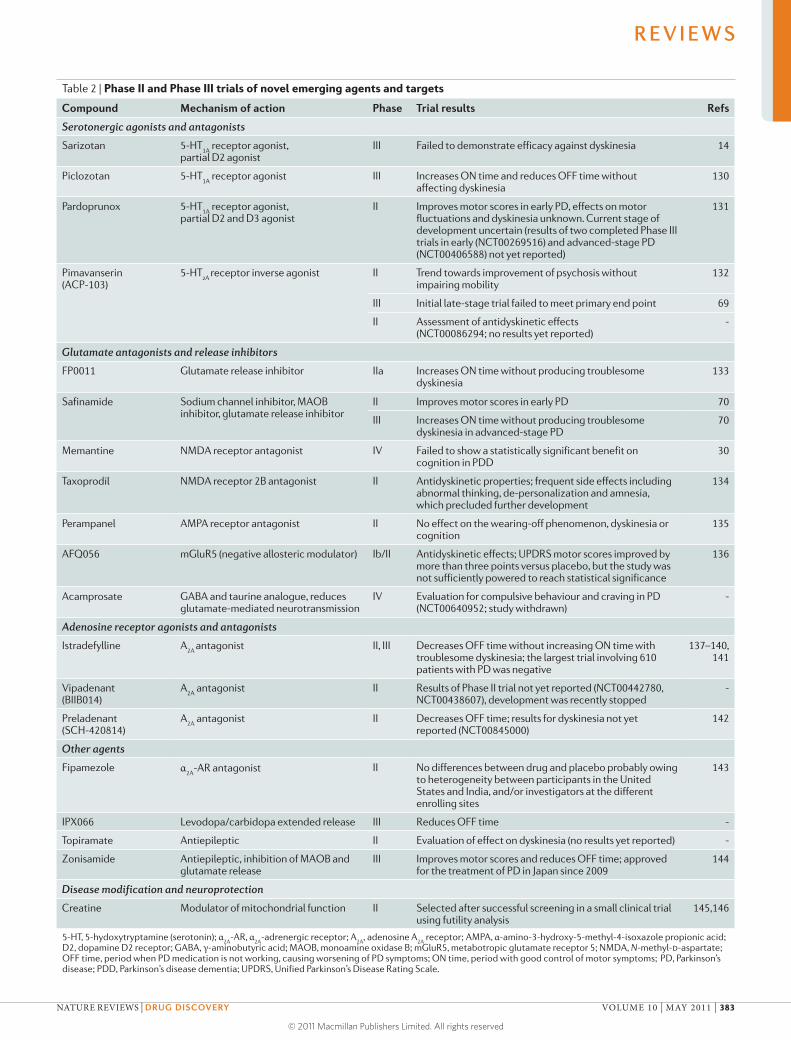
Table 2 | Phase II and Phase III trials of novel emerging agents and targets
Compound Mechanism of action Phase Trial results Refs
Serotonergic agonists and antagonists
Sarizotan 5-HT1A
receptor agonist, partial D2 agonist
III Failed to demonstrate efficacy against dyskinesia 14
Piclozotan 5-HT1A
receptor agonist III Increases ON time and reduces OFF time without affecting dyskinesia
130
Pardoprunox 5-HT1A
receptor agonist, partial D2 and D3 agonist
II Improves motor scores in early PD, effects on motor fluctuations and dyskinesia unknown. Current stage of development uncertain (results of two completed Phase III trials in early (NCT00269516) and advanced-stage PD (NCT00406588) not yet reported)
131
Pimavanserin (ACP-103)
5-HT2A
receptor inverse agonist II Trend towards improvement of psychosis without impairing mobility
132
III Initial late-stage trial failed to meet primary end point 69
II Assessment of antidyskinetic effects (NCT00086294; no results yet reported)
-
Glutamate antagonists and release inhibitors
FP0011 Glutamate release inhibitor IIa Increases ON time without producing troublesome dyskinesia
133
Safinamide Sodium channel inhibitor, MAOB inhibitor, glutamate release inhibitor
II Improves motor scores in early PD 70
III Increases ON time without producing troublesome dyskinesia in advanced-stage PD
70
Memantine NMDA receptor antagonist IV Failed to show a statistically significant benefit on cognition in PDD
30
Taxoprodil NMDA receptor 2B antagonist II Antidyskinetic properties; frequent side effects including abnormal thinking, de-personalization and amnesia, which precluded further development
134
Perampanel AMPA receptor antagonist II No effect on the wearing-off phenomenon, dyskinesia or cognition
135
AFQ056 mGluR5 (negative allosteric modulator) Ib/II Antidyskinetic effects; UPDRS motor scores improved by more than three points versus placebo, but the study was not sufficiently powered to reach statistical significance
136
Acamprosate GABA and taurine analogue, reduces glutamate-mediated neurotransmission
IV Evaluation for compulsive behaviour and craving in PD (NCT00640952; study withdrawn)
-
Adenosine receptor agonists and antagonists
Istradefylline A2A
antagonist II, III Decreases OFF time without increasing ON time with troublesome dyskinesia; the largest trial involving 610 patients with PD was negative
137–140, 141
Vipadenant (BIIB014)
A2A
antagonist II Results of Phase II trial not yet reported (NCT00442780, NCT00438607), development was recently stopped
-
Preladenant (SCH-420814)
A2A
antagonist II Decreases OFF time; results for dyskinesia not yet reported (NCT00845000)
142
Other agents
Fipamezole α2A
-AR antagonist II No differences between drug and placebo probably owing to heterogeneity between participants in the United States and India, and/or investigators at the different enrolling sites
143
IPX066 Levodopa/carbidopa extended release III Reduces OFF time -
Topiramate Antiepileptic II Evaluation of effect on dyskinesia (no results yet reported) -
Zonisamide Antiepileptic, inhibition of MAOB and glutamate release
III Improves motor scores and reduces OFF time; approved for the treatment of PD in Japan since 2009
144
Disease modification and neuroprotection
Creatine Modulator of mitochondrial function II Selected after successful screening in a small clinical trial using futility analysis
145,146
5-HT, 5-hydoxytryptamine (serotonin); α2A
-AR, α2A
-adrenergic receptor; A2A
, adenosine A2A
receptor; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; D2, dopamine D2 receptor; GABA, γ-aminobutyric acid; MAOB, monoamine oxidase B; mGluR5, metabotropic glutamate receptor 5; NMDA, N-methyl-d-aspartate; OFF time, period when PD medication is not working, causing worsening of PD symptoms; ON time, period with good control of motor symptoms; PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; UPDRS, Unified Parkinson’s Disease Rating Scale.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 383
© 2011 Macmillan Publishers Limited. All rights reserved

α-synucleinA protein with unknown physiological function that forms abnormal intraneuronal aggregates in Parkinson’s disease.
Sporadic PDA form of Parkinson’s disease (PD) that is unrelated to genetic mutations or other known aetiologies.
midodrine, fludrocortisone, droxidopa, modafinil and methylphenidate have been shown to provide some relief for constipation, orthostatic hypotension, sexual dysfunction, urinary urgency and sleep disorders in placebo-controlled trials in PD. However, most of these benefits are only classified as unproven41, thus demon-strating the need for an improvement in development efforts and resources.
Future treatmentsThe assessment of new targets for the symptomatic relief of motor and non-motor signs is in progress; however, the main challenge remains the development of treatments that are disease-modifying or neuroprotective. Therefore, the discovery of treatments that reduce the progression of cell death in both dopaminergic and non-dopaminergic neurons is an important focus of research. In this respect, strategies targeting α-synuclein using drugs or passive immunization are promising approaches in PD, but the translational value of current preclinical models remains unsatisfactory. Similar approaches that target amyloid-β biology in AD have taken a considerable period of time to reach the clinical trials stage42, so the development of α-synuclein-targeting strategies may take some time. Investigating how genetic mutations lead to PD is another promising approach. Both strategies may help to over-come the numerous hurdles in developing effective neu-roprotective treatments for PD, including the need for an improved understanding of the aetiology and patho-genesis of PD.
Genetics as a basis for novel treatments. The discovery of hereditary forms of PD has generated tremendous excite-ment and renewed energy in PD research. Although cur-rently less than 15% of all PD cases can be ascribed to monogenic causes, it is hoped that novel drug targets will be discovered as a result of pathophysiological insights from the identification of the role of genetic mutations that are responsible for hereditary forms of PD. It is also hoped that future therapies targeted towards these muta-tions will have broader efficacy against sporadic PD than currently available symptomatic treatments.
Most patients have sporadic PD, which is characterized by mitochondrial dysfunction as well as oxidative and nit-rosative stress43. Complex I of the mitochondrial electron
transport chain is selectively dysfunctional in dopaminer-gic neurons in sporadic PD44,45. Several other mechanisms, such as neuroinflammation, glutamatergic excitotoxicity, decreased levels of neurotrophic factors and environmen-tal factors are also thought to contribute to PD.
Discoveries related to the identification or role of genes that are implicated in the pathogenesis of PD include point mutations46–48 (at the PARK1 locus), as well as duplications49 and triplications50 (at the PARK4 locus) in the gene that encodes for α-synuclein (SNCA), which has been implicated as a rare cause of PD in families with a history of PD. Mutations in the gene that encodes leucine-rich repeat serine/threonine protein kinase 2 (LRRK2; PARK8 locus) are a much more common cause of autosomal dominant PD51. The gene encodes a mem-ber of the mitogen-activated protein kinase (MAPK) protein family, which suggests involvement of intracel-lular phosphorylation cascades in the pathogenesis of the disease52. Mutations in several genes are linked to recessive early-onset variants of PD — these include par-kin (PARK2 locus)53, the oncogene DJ1 (PARK7 locus)54, and PTEN-induced putative kinase protein 1 (PINK1; PARK6 locus)55. These genes implicate various cellular subsystems in the pathogenesis of PD, such as mitochon-drial function and oxidative stress, and the proteasomal protein degradation pathways.
There is emerging evidence that common vari-ants in some of these genes may have a direct role in disease aetiology. That is, they act as risk factors for common sporadic forms of PD; this has been shown most convincingly for the gene encoding α-synuclein56. Mutations in other genes such as those that encode Gaucher’s disease-associated glucocerebrosidase A57 and the mitochondrial protease HTRA2 (also known as OMI)58 may also substantially influence disease risk. Moreover, the identification of the Tau locus in a recent genome-wide association study in PD56 provides a tantalizing clue as to possible shared mechanisms of disease susceptibility and disease expression with AD, and suggests that Tau-based therapeutics that are being developed for AD could potentially also be tested in individuals with PD. Thus, an increasingly complex network of genes that contribute to disease risk and progression is emerging. These findings provide the ‘genetic entry points’ for identifying molecular targets
Box 1 | Dyskinesia rating scales
Dyskinesia rating scales can rely on observations by a physician, the use of home diaries in which the patient records the time spent with dyskinesias over several days, or they can combine both of these methods. A systematic review by a task force of the Movement Disorders Society concluded that the Abnormal Involuntary Movement Scale (AIMS) and the Rush Dyskinesia Rating Scale (RDRS) formally fulfil the criteria known as ‘recommended’,15 but both scales still have considerable limitations. The AIMS, which was initially developed for the evaluation of tardive dyskinesia in psychiatric patients, has been modified by several authors for its use in Parkinson’s disease (PD); however, but such modifications raise issues with the scale’s overall clinimetric properties. The RDRS focuses on disability and the impact of dyskinesia on specific activities of daily living.
The newly developed Unified Dyskinesia Rating Scale (UDysRS) combines elements of the AIMS and RDRS into a single measure to assess both impairment and disability126; it contains a self‑assessment by the patient as well as an examination by the physician. Another scale of high potential value once further testing is performed is the PD Dyskinesia Scale‑26, which is a patient‑based measure for quantifying the impact of dyskinesias on specific activities of daily living and quality of life of patients with PD127. Successful registration of new dyskinesia therapies will not only require demonstration of safety and efficacy but also assessments of the impact of the treatment on these outcomes.
R E V I E W S
384 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

and potential readouts that may serve as objective biological markers, which are necessary for designing disease-modifying treatments.
Studying genetically defined clinical cohorts — PD-affected and non-affected carriers of mutations in the LRRK2 gene51 in particular — may add to our current understanding of the time course of striatal dopamine loss59 and PD progression. As disease pen-etrance is approximately 80% at age 70 (REF. 60), studying non-affected carriers provides a unique opportunity to study presymptomatic disease markers in longitudinal cohorts, and to assess the potential efficacy of future disease-modifying or neuroprotective strategies.
A need for clinically relevant animal models. Validation of disease-modifying treatments before clinical testing implies that they have to convincingly interfere with cell death mechanisms that are relevant to PD in an in vivo experimental model. In this regard, current neurotoxin-based and genetically modified animal models have con-siderable limitations.
Although models that use neurotoxins such as 1-methly-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine are the models of choice for assessing therapies that provide symptom control, they have considerable limitations for assessing disease-
modifying and neuroprotective treatments. Some of these drawbacks are linked to the experimental design of the models rather than to the neurotoxin them-selves61. It was hoped that the introduction of PD-related mammalian genetic models — which were supported by successful dopaminergic neuron degeneration in invertebrate and nematode models — would provide a disease-relevant context for testing potential neuro-protective therapies. However, a common feature of the genetically modified rodent models is the paucity or absence of cell death in the substantia nigra pars com-pacta and the absence of Lewy pathology. Nonetheless, the mutations do replicate some features of the auto-somal dominant forms of PD (for example, mutations in the genes encoding α-synuclein62 and LRRK2 (REF. 63)) or the autosomal recessive forms of PD (for example, mutations in parkin and DJ1 (REF. 64)). In contrast to neurotoxin-based models, which show acute neuronal death, genetically modified rodent models were thought to allow progressive degeneration, in the same way as in human PD, hence it was disappointing to discover that this was not the case.
Although genetically modified rodents still need to demonstrate a significant degeneration and a related parkinsonian phenotype, the viral vector-driven expres-sion of α-synuclein induces death of dopaminergic cells in rodents65 and primates66, which suggests that a cer-tain titre for successfully transducing α-synuclein is needed and/or that dopaminergic cell death involves cell-autonomous and non-cell-autonomous processes, as viral vectors may also drive expression of α-synuclein in glia. Such information should be taken into account when developing new animal models, as viral studies have shown that the mouse substantia nigra is actually prone to degeneration. We believe that future strategies should include obtaining combinations of mutations by breeding mice with single or dual mutations, and chal-lenging existing models with environmental stressors such as neurotoxins or neuroinflammation inducers in animals with an established and stable genetic back-ground (a minimum of eight to ten backcrossing in the desired strain).
A complementary proposal for explaining the absence of dopaminergic neuron loss in genetically modified mice is that there are compensatory mechanisms that prevent the loss of dopaminergic neurons during the lifespan of a mouse. Such absence of overt cell loss has also been observed in the early days of neurotoxin-based mouse models. Bypassing such compensatory mecha-nisms could be achieved by conditional overexpression or conditional knockout of PARK-related genes; that is, the mutation becomes active only during adulthood, which is analogous to the way in which neurotoxins are injected into adult models, rather than in utero or into neonates, to prevent adaptations from occurring during development. This strategy might create genetic mod-els of PD in which degeneration of dopaminergic neu-rons occurs during adulthood.
Interestingly, however, most autosomal dominant and autosomal recessive models display a disruption of stri-atal dopaminergic neurotransmission that is associated
Box 2 | Diagnostic criteria for PD dementia and Lewy body dementia
Definition of dementia associated with PDThe diagnosis of Parkinson’s disease (PD) dementia25 relies on the occurrence of a dementia syndrome (known as a core feature) with insidious onset and slow progression that develops within the context of established PD.
The diagnosis of probable PD dementia requires the presence of both core features along with associated features of a typical profile of cognitive deficit (two out of four cognitive domain: deficits in attention, executive and visuospatial functions, and impaired free recall memory that is usually improved with cueing) as well as one characteristic behavioural symptom (apathy, depressed or anxious mood, hallucinations, delusions or excessive daytime sleepiness). For a confirmed diagnosis of probable PD dementia, none of the features should make the diagnosis uncertain (for example, a short time interval between motor and cognitive symptoms) and none of the features should suggest that other conditions or diseases may be the cause of mental impairment.
Possible PD dementia is diagnosed when associated clinical features are lacking or atypical, or if any of the features that are present make the diagnosis uncertain. The sensitivity and specificity of these distinctions remain to be ascertained.
Definition of dementia with Lewy bodiesThe diagnosis of ‘dementia with Lewy bodies’ (DLB)128 requires the presence of a progressive cognitive decline that is of sufficient magnitude to interfere with normal social and occupational function. Memory impairment does not necessarily occur in early stages, whereas deficits in attention, frontal–subcortical skills and visuospatial ability may be prominent. The three core features of DLB are: fluctuating cognition with pronounced variations in attention and alertness; recurrent visual hallucinations that are typically well‑formed and detailed; and spontaneous features of parkinsonism. Suggestive features (the presence of rapid eye movement sleep behaviour disorder, severe neuroleptic sensitivity and low dopamine transporter uptake) have been included in the third revision of the clinical consensus criteria. The arbitrary ‘one year rule’ — which qualifies the onset of dementia within one year of parkinsonism as DLB and beyond one year as PD dementia — is only recommended for clinical studies in which a clear distinction is needed between DLB and PD dementia.
The diagnosis of probable DLB requires the presence of the central feature plus two core features, whereas the diagnosis of possible DLB requires the presence of the central features plus one core feature. If one or more suggestive features are observed in the presence of one or more core features, a diagnosis of probable DLB can be made.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 385
© 2011 Macmillan Publishers Limited. All rights reserved

Prodromal phaseThe early phase of the disease before the appearance of full-blown symptoms. In Parkinson’s disease, it means the time before the onset of motor signs.
with subtle motor and non-motor deficits. They might therefore constitute models of the prodromal phase of PD and, as such, they may be useful for target valida-tion. Consequently, we find it surprising that very little attempt has been made to test neuroprotective strategies in these models by taking the alterations already defined in each of them as end points — for example, altered striatal dopaminergic transmission or the subtle motor and non-motor deficits.
Novel emerging agents and targets. Although a major aim of PD research and development is to find agents that are disease-modifying or neuroprotective, in the shorter term it is also necessary to find agents that improve symptoms, as until a disease-modifying therapy is found, patients will still need control of their disease symptoms67,68.
Agonists of 5-HT1A and 5-HT1B receptors, as well as antagonists of 5-HT2A and 5-HT2C receptors, are in development for the treatment of motor fluctua-tions and dyskinesia (TABLES 1,2). Out of these agents, the compound that reached the highest clinical phase is the 5-HT1A receptor agonist sarizotan. However, it failed to demonstrate efficacy against dyskinesia14 in two late-stage trials, and development has been discon-tinued (BOX 3). Reasons for the lack of efficacy may have included prominent placebo effects14 and dose limita-tions owing to the low potency of sarizotan and partial antagonism at dopamine D2 receptors. Pimavanserin (ACP-103), a 5-HT2A receptor inverse agonist, is cur-rently being assessed for the treatment of hallucinations and psychosis in PD (TABLES 1,2). A placebo effect that occurred in patients who were enrolled at study cen-tres in specific geographical regions was considered to contribute to the failure of the Phase III clinical trial69, thus suggesting that such regional placebo effects occur with both motor (for example, dyskinesias) and neu-ropsychiatric outcomes in PD trials. Another seroton-ergic compound, flibanserin, which is a 5-HT2A receptor antagonist and a 5-HT1A receptor agonist, will soon be tested for antidyskinetic activity in an early-stage proof-of-concept trial (BOX 3).
Glutamate release inhibitors and antagonists of NMDA, AMPA (α-amino-5-hydroxy-3-methyl-4-isoxaz- ole propionic acid) and metabotropic glutamate receptors (mGluRs) have been tested or are currently being evaluated in patients with PD for the treatment of dementia, motor signs (including dyskinesia) and gait disorders (TABLES 1,2). The most advanced compound is safinamide, which is currently in late-stage clinical trials for motor control, and is also being explored for potential efficacy against dyski-nesias and cognitive impairment in PD70. Other promising agents are the negative allosteric modulators of mGluR5: AFQ-056 and ADX48621. The latter will soon enter early-stage assessments for dyskinesias (BOX 3).
The investigation of drugs that act on adenosine A2A receptors has received great interest owing to their rela-tively selective localization on striatal medium spiny neu-rons of the indirect striatopallidal pathway71. The most advanced compound in this category is preladenant (SCH-420814) (TABLES 1,2). Other A2A antagonists have
been tested or are currently in development (TABLES 1,2). Epidemiological data link the consumption of caffeine, which is a nonspecific adenosine receptor antagonist (it acts on A1 and A2 receptors) to a reduced risk of develop-ing PD72. Caffeine prevents neurodegeneration in several preclinical models of parkinsonism (reviewed in REF. 73), but clinical studies have failed to show any effect of caf-feine intake on markers of disease progression74. A1 recep-tors are involved in sleep promotion (reviewed in REF. 75). Accordingly, A1 receptor antagonists might be useful for the treatment of excessive daytime sleepiness, whereas A1 receptor agonists could be a target for insomnia.
Other symptomatic treatments for motor or non-motor signs include antiepileptics, α2-adrenergic recep-tor antagonists (BOX 3) and histamine H3 receptor inverse agonists (TABLES 1,2).
The blood–brain barrier remains an obstacle for drug development in PD. Accordingly, agents that do not cross the blood–brain barrier have to be delivered stereotacti-cally. This approach has been used in PD for the delivery of glutamic acid decarboxylase (GAD) to the subthalamic nucleus via gene transfer76. Indeed, patients with PD who received unilateral GAD delivery showed sustained improvements in motor symptoms76, and confirmatory data in a larger, recently completed study (ClinicalTrials.gov identifier: NCT00643890) using bilateral injection of GAD are pending. Intraputaminal delivery of the three main enzymes that are responsible for dopamine synthesis (tyrosine hydroxylase, aromatic l-amino acid decarboxylase and GTP-cyclohydrolase 1) restored extra-cellular dopamine levels and improved motor symptoms in non-human primates77. An open-label Phase I study of this treatment in patients with PD is ongoing78.
Disease modification and neuroprotection. Disease-modifying and neuroprotective treatments remain unmet clinical needs. In this section, we discuss ongoing efforts and future strategies aimed at targeting key events that are involved in neuronal cell death in PD pathology (reviewed in REF. 79). Accordingly, ongoing studies are investigating the effect of the mitochondrial modulators creatine (BOX 4) and coenzyme Q10, as well as the L-type calcium antagonist isradipine. In addition, two agents that are both believed to decrease oxidative stress are under investigation: green tea polyphenols (reviewed in REF. 80) and deferiprone — an iron chelator (TABLE 1).
In recent years, glial cell line-derived neurotrophic fac-tor (GDNF) and neurturin, a GDNF-related growth factor, have been assessed in patients with PD. Their delivery to the brain has been a key issue during their development, and multiple approaches are being explored, including intrac-erebral and intraputaminal infusions of GDNF, as well as stereotactically injected viral vectors that drive the expres-sion of neurturin in transfected neurons81–85. Although an open-label pilot study using intraputaminal GDNF infusion reported a statistically significant improvement in motor scores81, a large placebo-controlled trial failed to reproduce these results82. Some explanations for this could be the methodological differences between the trials and a placebo bias in the open-label pilot study86. Viral delivery of neurturin failed to reach its primary objective (Unified
R E V I E W S
386 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

PD Rating Scale (UPDRS) motor scores in the OFF condi-tion at 12 months of follow-up) in a robust Phase II trial that compared putaminal delivery with sham surgery83. This was due, in part, to greater than expected placebo effects, bearing in mind that placebo effects are highest in surgical intervention trials in PD (42% versus 14% in medication studies)86. Further analysis of a subset of 30 patients who were monitored for up to 18 months showed clinically significant differences for the primary outcome measures84. Post-mortem studies of two patients who died within 90 days after operation owing to unrelated events revealed evidence of putaminal (nerve terminal) neurturin expression. However, there was no evidence of increased neurturin protein or tyrosine hydroxylase expression in the cell bodies of the substantia nigra85.
By contrast, non-human primates (MPTP-treated and young and aged controls) revealed clear expres-sion of neurturin in the substantia nigra pars compacta with robust induction of tyrosine hydroxylase following putaminal delivery85. This distinction implies a profound difference in the status and function of nigrostriatal neu-rons — namely, a disruption in the retrograde transport pathway from the striatum to nigral cell bodies — in patients with advanced-stage PD compared to the gold
Box 3 | Further information on the development status of compounds
ADX‑48621Addex Pharmaceuticals was recently awarded a US$900,000 grant from the Michael J. Fox Foundation to help fund a Phase II study of dipraglurant-IR (ADX-48621) for the treatment of dyskinesia; this study is expected to start soon (see the Addex Pharmaceuticals website for further information).
FipamezoleSanthera Pharmaceuticals have regained full development rights for fipamezole in the United States from Biovail Laboratories International. In the press release on 25 October 2010, Santhera Pharmaceuticals stated that they were prepared for Phase III development of fipamezole for the treatment of dyskinesia in Parkinson’s disease (see the Santhera Pharmaceuticals website for further information). Santhera and Ipsen entered into a licensing agreement for fipamezole in 3 September 2010 (see the Santhera Pharmaceuticals website for further information).
FlibanserinThis drug was initially developed for women with hypoactive sexual desire disorder. The development of this drug for that indication was stopped in October 2010 after a non‑approval by the US Food and Drug Administration (FDA) (see the Boehringer Ingelheim website for further information).
Monitoring Force, a contract research organization in Germany, filed an orphan drug status application to the European Medicines Agency in November 2009 for the indication treatment‑relevant for dyskinesia in Parkinson’s disease (PD). The company is preparing a similar application for the FDA and planning its first clinical trial in patients with PD (see the CheckOrphan web site for further information).
IPX066Two Phase III trials (APEX-PD and ADVANCE-PD) have been completed. The results of both studies were issued in press releases on 18 November 2010 and on 14 March 2011 (see the IMPAX Laboratories website for further information). According to these reports, IPX066 substantially improves activities of daily living and motor signs in early disease and reduces OFF time in advanced‑stage PD compared to the immediate‑release drug levodopa/carbidopa (Sinemet; Bristol‑Myers Squibb). IMPAX Pharmaceuticals intends to file a new drug application to the FDA before the end of 2011.
SarizotanFurther development of this drug for the treatment of PD was stopped in 2006 (see the The Merck Group website for further information).
standard animal models used for translational research. Taken together, these observations suggest that longer patient follow-ups, and concomitant administration to the putamen and substantia nigra, should be considered when designing future clinical trials to assess the efficacy of neurotrophic factors in PD. One such trial with neu-rturin is currently underway (ClinicalTrials.gov identi-fier: NCT00985517).
In contrast to the stereotactical delivery of GDNF and neurturin, cogane (PYM-50028), which is believed to be an inducer of neurotrophic factor, is currently being assessed (ClinicalTrials.gov identifier: NCT01060878).
We believe that therapies based on single drugs are not likely to provide measurable disease modification or neuroprotection in PD. Rather, a combination of com-pounds that interfere simultaneously with different key events of PD pathology seems to be the most promising strategy. This approach would, however, require collabo-ration between the pharmaceutical companies that hold the patent rights of the different compounds.
DBS. Stereotactical approaches have been used for many decades to treat motor symptoms in PD87. In recent years, DBS has become an established treatment in advanced stages of PD2,3. New anatomical targets for DBS, such as the pedunculopontine nucleus, are cur-rently being explored in patients with PD who have gait disorders20,21. Beyond the search for new targets, smart DBS techniques such as coordinated reset stimulation are under development88. These techniques will, in their final form, allow individualized on-demand modulation of abnormal activity in the basal ganglia–cortical net-works of patients with PD.
Design of future clinical trialsPharmacological and non-pharmacological interven-tions for PD are often initially tested in open-label pilot studies. Although the predictability and reliability of open-label early translational studies have been chal-lenged because of the important placebo bias of such designs86,89, they remain useful for the early characteriza-tion of safety and tolerability, and for potentially refining the appropriate range of doses before embarking on large trials. Following this, randomized placebo-controlled clinical trials and randomized active comparator clini-cal trials are undertaken, which have been used for many years in the assessment of the symptomatic effects of PD treatments in classical Phase II and Phase III develop-ment programmes.
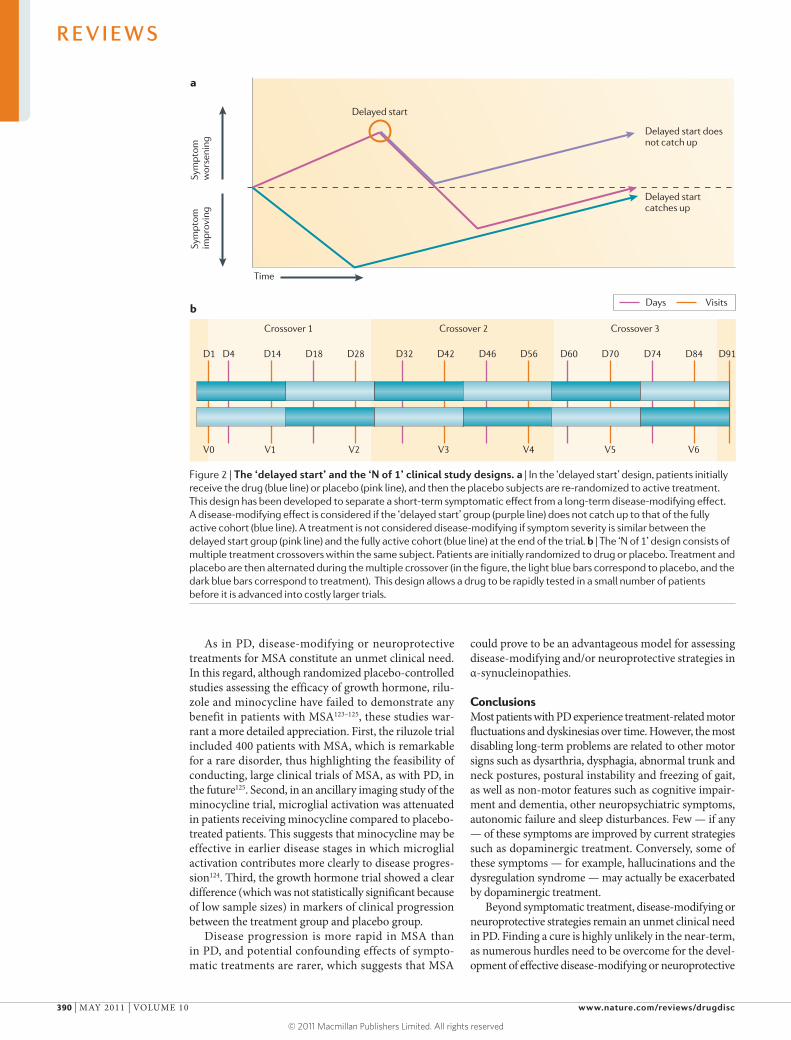
Novel designs for early translational studies are cur-rently being explored in PD. For example, the ‘N of 1’ design (FIG. 2a) is an innovative concept that is useful for the early-stage assessment of symptomatic drugs before embarking into costly larger trials. The study design uses repeated and alternating administration of drug and pla-cebo to the same subject. However, there are several con-straints of this trial design; these include the necessity of a temporally stable clinical condition, the requirement of drugs to have a rapid onset and short duration of action, and the logistics of repeated visits to the study centre by the patient and investigator over a short period of time.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 387
© 2011 Macmillan Publishers Limited. All rights reserved

The need for large, pragmatic ‘real-life’ trials to assess the long-term impact of a form of treatment is increas-ingly being recognized. These trials could become mandatory for maintaining regulatory approval based on the results of Phase III trials. Such requirements will need innovative models for sharing the financial bur-den among the pharmaceutical industry, government agencies, academic centres and PD research founda-tions. Given the important implications of such studies, patient support groups and private donors may also need to invest in these clinical research efforts.
In the absence of valid objective surrogate markers (see section on ‘Objective end points for neuroprotec-tion trials below), primary outcomes — in trials that have been completed so far — assessing disease modi-fication and neuroprotection were based on clinical measures such as the need for symptomatic treatment in drug-naive patients with PD or progression of dis-ease-related disability as assessed by validated clini-cal rating scales90–92. This explains why the separation of the confounding effects of symptomatic treatment from disease-modifying or neuroprotective actions was a major difficulty in previous trials. In the DATATOP study91 the primary end point was the time taken before the patient needed rescue therapy. Although selegiline, a MAOB inhibitor, delayed the time until l-DOPA rescue therapy was required compared to placebo, the results of this study were inconclusive because of the confounding effects of selegiline on disease symptoms, which were not anticipated in the study design. The ELLDOPA trial92 tried to overcome the methodological limitations of the DATATOP study by including a delayed washout of two weeks at the end of the trial. The analysis was based on a comparison of changes in UPDRS total scores over time, between patients receiving l-DOPA and those receiving placebo. At the end of the washout period, patients who received l-DOPA had lower UPDRS scores compared to patients who received placebo treatment. However, although the plasma half-life of l-DOPA is very short, long-term clinical effects may persist for up to 1 month93. Therefore, the results of the ELLDOPA study were also inconclusive because the washout period may have been too brief; however, longer washout periods are impractical because of ethical concerns. To overcome
this dilemma, other experimental plans, including the delayed-start design, (FIG. 2b) have been utilized. One example is the ADAGIO study, in which patients initially receive drug or placebo, and the placebo subjects are then re-randomized to active treatment94,95. During a second period, all patients receive the study drug. Accordingly, the behaviour of the initial placebo group should never catch up to that of the fully active cohort. Such a design could demonstrate that a given medication may act via two different mechanisms: short-term (symptomatic) and long-term (disease-modifying). Nevertheless, such a design also has its own intrinsic limitations. Wash-in periods (the time necessary to develop the full sympto-matic effect of a tested compound) of the symptomatic effect are based on pre-specified assumptions and the duration of the first placebo phase is limited because drug-naive patients with PD often cannot remain on placebo for more than a few months. Consequently, expected differences between the early- and delayed-start groups are small, owing to the slow rate of progres-sion of motor symptoms in PD, and the clinical relevance of these differences — that is, whether the drug provides disease modification or neuroprotection — is inevita-bly a matter of considerable debate96,97. It has recently been argued that end point-based clinical trial analysis may have been one important reason for the failure of previous studies that sought to assess disease modifica-tion or neuroprotection, and that quantitative modelling over the entire time course of the clinical study, with separation of three concomitant events — that is, disease progression, symptomatic effects and disease modifying properties — may be more appropriate in these trials97.
We propose that the use of large, simply designed and longer term pragmatic trials should be explored and debated, and if they display the appropriate metrics, non-motor outcomes (such as falls, cognitive impairment or quality of life) should be utilized to assess neuroprotec-tion and/or disease modification in PD.
Objective end points for neuroprotection trials. Objective end points based on imaging, cerebrospinal fluid (CSF) and/or blood biomarkers or other techniques are an unmet clinical need in PD (BOX 4). Accordingly, primary end points in past and ongoing trials of potential
Box 4 | Initiatives for developing disease-modifying and/or neuroprotective treatments in PD
There are currently no established, validated biomarkers in Parkinson’s disease (PD), and additional efforts for the identification of such markers are urgently needed. In this regard, the Michael J. Fox Foundation has recently launched the Parkinson’s Progression Markers Initiative (PPMI), which is a large prospective observational cohort study that will follow 400 newly diagnosed patients with sporadic PD and 200 age-matched healthy controls for 3–5 years129. The main objective of this study is to collect clinical, imaging and biological data (from blood, urine and cerebrospinal fluid) and samples of blood, urine and CSF to identify promising biomarkers of PD progression, and potentially validate existing candidate markers. As a precursor initiative of innovative collaboration, the PPMI will create a consortium of academic centres, government agencies, PD foundations, and pharmaceutical and biotechnology companies to collectively design, implement and fund a comprehensive programme to develop markers of PD progression. Furthermore, at the US National Institutes of Health, the National Institute of Neurological Disorders and Stroke (NINDS) has called for projects that coordinate PD‑related activities with handling of data on PD biomarkers.
The NINDS Exploratory Trials in PD (NINDS NET‑PD) initiative approach the issue of neuroprotection by taking large numbers of patients and monitoring them by using simple outcome measures over 5 years. This approach will attempt to determine whether an intervention has a substantive impact on the long‑term progression of PD. The effect of creatine will be assessed in a large study that will include nearly 2,000 subjects.
Delayed washoutA prolonged symptomatic benefit that exceeds the time taken for the elimination of the administered drug from the body.
R E V I E W S
388 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

neuroprotective drugs rely on clinical assessment scales. Here, we discuss current efforts and future directions for the development of biomarkers as objective outcome measures in neuroprotection trials.
Because PD is associated with nigral degeneration and striatal dopamine deficiency, the demonstration of the dysfunction of dopaminergic nerve terminals in the striatum — using positron emission tomography (PET) or single photon emission computed tomography (SPECT) — supports the diagnosis of PD. In addition, it is possible that dopamine imaging could be used as an end point in neuroprotection trials. However, when it was used as a secondary outcome measure to test the effect of symptomatic treatments in several large PD clinical trials92,98,99, there was discordance between clini-cal end points and imaging outcomes. This has raised concerns regarding interactions among the different study drugs, l-DOPA, and the tracers used for imaging. Despite this, dopamine imaging is still being assessed as a potential surrogate marker for PD disease-modifying or neuroprotection trials100,101.
Studies based on18F-fluorodeoxyglucose PET have revealed a specific PD-related pattern of metabolic abnormalities102,103. PET studies that use ligands that are specific for activated microglia can detect brain inflam-mation in PD, which may be useful for assessing the role of anti-inflammatory agents as therapeutic approaches for PD104. In addition, progress has been achieved in visualizing α-synuclein in vivo, which may ultimately allow the progression of Lewy pathology during disease progression to be monitored105. However, no longitudinal data based on repeat imaging during disease progression are available for any of these tracers.
The refinement of magnetic resonance imaging to measure nigral iron content, the use of high-resolution midbrain diffusion tensor imaging, and progress in tran-scranial Doppler ultrasound imaging may be applica-ble in the selection of specific patient subpopulations for proof-of-concept studies. In addition, with further clinical validation, these applications may even provide future innovative secondary clinical end points106–109. As in other neurodegenerative diseases, current PD research also focuses on objective blood and CSF markers110,111. For example, α-synuclein is detectable in the blood and CSF of patients with PD; however, published stud-ies (reviewed in REF. 110) are conflicting, as some have reported decreased levels of α-synuclein, whereas others did not find any change compared to controls. Moreover, α-synuclein levels in the CSF were not related to disease severity in one small study112. The development of multi-plex enzyme-linked immunosorbent assays to detect the different oligomeric forms of α-synuclein, and control-ling major confounders such as blood contamination, may allow researchers to overcome these limitations in the future112–114.
Non-motor signs such as constipation, rapid eye movement sleep behaviour disorder (RBD), olfactory dys-function, anxiety disorders and depression may precede motor signs by more than 20 years43,115. However, most of these symptoms are nonspecific and they also occur under other clinical circumstances; therefore, they cannot
easily be used as biomarkers of PD. The identification of co-factors to these premotor signs that clearly predict con-version to PD with high probability would allow identifi-cation of an at risk population of patients when they are at an earlier disease state before motor symptoms become apparent. The association among RBD, olfactory dysfunc-tion and dopamine transporter imaging is a promising approach116. Research in this area is important because disease-modifying or neuroprotective interventions may have a greater probability of success at earlier stages of PD, before motor symptoms become apparent. Moreover, the study of disease progression and the mechanisms that compensate for the increasing dopamine deficit in these patients could guide future biomarker research. It is unlikely that a single marker will provide sufficient sensi-tivity and specificity for early diagnosis or prognosis. We believe that a combination of different biomarkers will be vital for the development of objective end points for future neuroprotection trials. Given their accessibility, the development of blood biomarkers would be a major advancement. Beyond current techniques, new tools — for example, colonic or skin biopsy — may help with identifying and validating new targets. Moreover, future progress in our pathophysiological understanding of PD will undoubtedly open up new avenues for biomarker research. Given the conflicting results for some promis-ing biomarkers, such as α-synuclein (reviewed in REF. 110), progress would also benefit from a standardization of sampling and imaging protocols. Cost-efficiency should also be considered in the development of new biomark-ers — for example, PET imaging is expensive and only available in some specialized centres.
The clinical model of multiple system atrophy. Multiple system atrophy (MSA) is a rare disorder, with a preva-lence of 2–5 per 100,000 (reviewed in REF.117), and is often initially mistaken for PD. Together with PD and Lewy body dementia, MSA belongs to a group of neu-rodegenerative disorders — the α-synucleinopathies — which are characterized by the abnormal accumulation of α-synuclein. In contrast to PD, in MSA α-synuclein mainly accumulates in glial cells but also in neurons. Although no genetic forms of MSA have been identi-fied so far, variants of the SNCA gene are associated with an increased risk for MSA118, as has been reported for PD56. Individuals with MSA could therefore prove to be an advantageous patient group in which to assess the effect of disease-modifying and/or neuro-protective strategies for treating α-synucleinopathies, including PD.
Crucial milestones have been reached for success-fully conducting clinical intervention trials in patients with MSA. For example, consensus diagnosis criteria are now available119, clinical scales assessing different aspects of motor impairment, activities of daily living and quality of life have been validated120,121, and large international networks have been established in Europe, the United States, Canada and Asia. Moreover, toxin-based and transgenic animal models enable screening for potential drugs and characterization of underlying disease mechanisms122.
Longitudinal dataData that are collected at several time points in order to monitor disease progression over time.
Diffusion tensor imagingA magnetic resonance imaging technique that measures the preferred direction of water diffusion in fibre tracts. It informs about white matter connectivity between different brain areas. Loss of neuronal axons or white matter modifies measures of diffusion tensor imaging.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 389
© 2011 Macmillan Publishers Limited. All rights reserved
As in PD, disease-modifying or neuroprotective treatments for MSA constitute an unmet clinical need. In this regard, although randomized placebo-controlled studies assessing the efficacy of growth hormone, rilu-zole and minocycline have failed to demonstrate any benefit in patients with MSA123–125, these studies war-rant a more detailed appreciation. First, the riluzole trial included 400 patients with MSA, which is remarkable for a rare disorder, thus highlighting the feasibility of conducting, large clinical trials of MSA, as with PD, in the future125. Second, in an ancillary imaging study of the minocycline trial, microglial activation was attenuated in patients receiving minocycline compared to placebo-treated patients. This suggests that minocycline may be effective in earlier disease stages in which microglial activation contributes more clearly to disease progres-sion124. Third, the growth hormone trial showed a clear difference (which was not statistically significant because of low sample sizes) in markers of clinical progression between the treatment group and placebo group.
Disease progression is more rapid in MSA than in PD, and potential confounding effects of sympto-matic treatments are rarer, which suggests that MSA
could prove to be an advantageous model for assessing disease-modifying and/or neuroprotective strategies in α-synucleinopathies.
ConclusionsMost patients with PD experience treatment-related motor fluctuations and dyskinesias over time. However, the most disabling long-term problems are related to other motor signs such as dysarthria, dysphagia, abnormal trunk and neck postures, postural instability and freezing of gait, as well as non-motor features such as cognitive impair-ment and dementia, other neuropsychiatric symptoms, autonomic failure and sleep disturbances. Few — if any — of these symptoms are improved by current strategies such as dopaminergic treatment. Conversely, some of these symptoms — for example, hallucinations and the dysregulation syndrome — may actually be exacerbated by dopaminergic treatment.
Beyond symptomatic treatment, disease-modifying or neuroprotective strategies remain an unmet clinical need in PD. Finding a cure is highly unlikely in the near-term, as numerous hurdles need to be overcome for the devel-opment of effective disease-modifying or neuroprotective
Figure 2 | The ‘delayed start’ and the ‘N of 1’ clinical study designs. a | In the ‘delayed start’ design, patients initially receive the drug (blue line) or placebo (pink line), and then the placebo subjects are re-randomized to active treatment. This design has been developed to separate a short-term symptomatic effect from a long-term disease-modifying effect. A disease-modifying effect is considered if the ‘delayed start’ group (purple line) does not catch up to that of the fully active cohort (blue line). A treatment is not considered disease-modifying if symptom severity is similar between the delayed start group (pink line) and the fully active cohort (blue line) at the end of the trial. b | The ‘N of 1’ design consists of multiple treatment crossovers within the same subject. Patients are initially randomized to drug or placebo. Treatment and placebo are then alternated during the multiple crossover (in the figure, the light blue bars correspond to placebo, and the dark blue bars correspond to treatment). This design allows a drug to be rapidly tested in a small number of patients before it is advanced into costly larger trials.
R E V I E W S
390 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

1. Marsden, C. D., Parkes, J. D., Quinn, N. & Fahn, S. in Neurology 2: Movement Disorders 96–122 (Butterworth Scientific, London, 1982).
2. Deuschl, G. et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N. Engl. J. Med. 355, 896–908 (2006).
3. Krack, P. et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N. Engl. J. Med. 349, 1925–1934 (2003).
4. Garcia Ruiz, P. J. et al. Efficacy of long-term continuous subcutaneous apomorphine infusion in advanced Parkinson’s disease with motor fluctuations: a multicenter study. Mov. Disord. 23, 1130–1136 (2008).
5. Nyholm, D. et al. Duodenal levodopa infusion monotherapy vs oral polypharmacy in advanced Parkinson disease. Neurology 64, 216–223 (2005).
6. Voon, V. et al. Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol. 8, 1140–1149 (2009).This was the first comprehensive review covering clinical features, molecular mechanisms and a specific cognitive mechanism of habit-learning that may underlie conditions that are related to chronic dopaminergic stimulation, such as l-DOPA-induced dyskinesia and impulsive control disorders.
7. Chaudhuri, K. R. & Schapira, A. H. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 8, 464–474 (2009).A comprehensive review of the different non-motor signs in PD and their response to dopaminergic treatment.
8. Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 14, 223–236 (2002).
9. Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatr. 79, 368–376 (2008).
10. Fabbrini, G., Brotchie, J. M., Grandas, F., Nomoto, M. & Goetz, C. G. Levodopa-induced dyskinesias. Mov. Disord. 22, 1379–1389 (2007).
11. Quinn, N. P. Classification of fluctuations in patients with Parkinson’s disease. Neurology 51, S25–S29 (1998).
12. Pechevis, M. et al. Effects of dyskinesias in Parkinson’s disease on quality of life and health-related costs: a prospective European study. Eur. J. Neurol. 12, 956–963 (2005).
13. Goetz, C. G., Poewe, W., Rascol, O. & Sampaio, C. Evidence-based medical review update: pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004. Mov. Disord. 20, 523–539 (2005).
14. Goetz, C. G. et al. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov. Disord. 22, 179–186 (2007).
15. Colosimo, C. et al. Task force report on scales to assess dyskinesia in Parkinson’s disease: critique and recommendations. Mov. Disord. 25, 1131–1142 (2010).
16. Lidstone, S. C. et al. Effects of expectation on placebo-induced dopamine release in Parkinson disease. Arch. Gen. Psychiatry 67, 857–865 (2010).
17. Hely, M. A., Morris, J. G., Reid, W. G. & Trafficante, R. Sydney Multicenter Study of Parkinson’s disease: non-l-dopa-responsive problems dominate at 15 years. Mov. Disord. 20, 190–199 (2005).
18. Hely, M. A., Reid, W. G., Adena, M. A., Halliday, G. M. & Morris, J. G. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov. Disord. 23, 837–844 (2008).
19. Devos, D. et al. Improvement of gait by chronic, high doses of methylphenidate in patients with advanced Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 78, 470–475 (2007).
20. Ferraye, M. U. et al. Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson’s disease. Brain 133, 205–214 (2010).
21. Moro, E. et al. Unilateral pedunculopontine stimulation improves falls in Parkinson’s disease. Brain 133, 215–224 (2010).
22. Weintraub, D. et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3,090 patients. Arch. Neurol. 67, 589–595 (2010).
23. Thomas, A., Bonanni, L., Gambi, F., Di Iorio, A. & Onofrj, M. Pathological gambling in Parkinson disease is reduced by amantadine. Ann. Neurol. 68, 400–404 (2010).
24. Kempster, P. A., O’Sullivan, S. S., Holton, J. L., Revesz, T. & Lees, A. J. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 133, 1755–1762 (2010).This was a clinicopathological study showing that four major disability milestones (frequent falls, hallucinations, dementia and the need for residential care) occur at a similar time from death, irrespective of the age at disease onset and total disease duration, thus making them suitable as clinical end points for treatment trials in late-stage PD.
25. Emre, M. et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 22, 1689–1707 (2007).
26. Siderowf, A. et al. CSF amyloid β1–42 predicts cognitive decline in Parkinson disease. Neurology 75, 1055–1061 (2010).
27. Aarsland, D. et al. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology 75, 1062–1069 (2010).
28. Emre, M. et al. Rivastigmine for dementia associated with Parkinson’s disease. N. Engl. J. Med. 351, 2509–2518 (2004).
29. Aarsland, D. et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 8, 613–618 (2009).
30. Emre, M. et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9, 969–977 (2010).
31. Reisberg, B. et al. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 348, 1333–1341 (2003).
32. Edison, P. et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J. Neurol. Neurosurg. Psychiatr. 79, 1331–1338 (2008).
33. Chaudhuri, K. R., Healy, D. G. & Schapira, A. H. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 5, 235–245 (2006).
34. Aarsland, D., Marsh, L. & Schrag, A. Neuropsychiatric symptoms in Parkinson’s disease. Mov. Disord. 24, 2175–2186 (2009).
35. Pollak, P. et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J. Neurol. Neurosurg. Psychiatr. 75, 689–695 (2004).
36. Barone, P. et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9, 573–580 (2010).
37. Menza, M. et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 72, 886–892 (2009).
38. Devos, D. et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov. Disord. 23, 850–857 (2008).
39. Remy, P., Doder, M., Lees, A., Turjanski, N. & Brooks, D. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 128, 1314–1322 (2005).
40. Politis, M. et al. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology 75, 1920–1927 (2010).
41. Zesiewicz, T. A. et al. Practice Parameter: treatment of nonmotor symptoms of Parkinson disease: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 74, 924–931 (2010).
42. Grill, J. D. & Cummings, J. L. Current therapeutic targets for the treatment of Alzheimer’s disease. Expert Rev. Neurother. 10, 711–728 (2010).
43. Schapira, A. H. & Tolosa, E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nature Rev. Neurol. 6, 309–317 (2010).
44. Schapira, A. H. et al. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827 (1990).
45. Schapira, A. H. et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269 (1989).
46. Polymeropoulos, M. H. et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274, 1197–1199 (1996).This was a milestone paper that linked autosomal dominant PD to chromosome 4q21-q23. The corresponding gene has since then been identified to code for α-synuclein.
47. Kruger, R. et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nature Genet. 18, 106–108 (1998).
48. Zarranz, J. J. et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004).
49. Chartier-Harlin, M. C. et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
50. Singleton, A. B. et al. α-synuclein locus triplication causes Parkinson’s disease. Science 302, 841 (2003).
51. Zimprich, A. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004).
52. Marin, I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol. Biol. Evol. 23, 2423–2433 (2006).
53. Matsumine, H. et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 60, 588–596 (1997).
54. van Duijn, C. M. et al. PARK7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 69, 629–634 (2001).
55. Valente, E. M. et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900 (2001).
56. Simon-Sanchez, J. et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature Genet. 41, 1308–1312 (2009).
57. Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661 (2009).
58. Strauss, K. M. et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 14, 2099–2111 (2005).
59. Nandhagopal, R. et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology 71, 1790–1795 (2008).
60. Hulihan, M. M. et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 7, 2591–2594 (2008).
61. Meissner, W., Hill, M. P., Tison, F., Gross, C. E. & Bezard, E. Neuroprotective strategies for Parkinson’s disease: conceptual limits of animal models and clinical trials. Trends Pharmacol. Sci. 25, 249–253 (2004).
treatments. Novel drug targets are needed, both in terms of molecules and pathways. We believe that the develop-ment of translationally accurate disease models is cru-cial for the development of novel drug targets, as existing models of PD do not adequately capture the progressive nature of the disease or its extension from dopaminergic to non-dopaminergic involvement. Lastly, when a com-pound is considered for clinical testing, several factors
require careful consideration; these include the trial design, patient characteristics and end points. Because MSA has a far more rapid disease progression, fewer potential confounders owing to symptomatic treatment effects and a close resemblance to the neuropathological features of PD, this disease may be a promising first step for testing new agents for disease-modifying properties in related α-synucleinopathies such as PD.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 391
© 2011 Macmillan Publishers Limited. All rights reserved

62. Masliah, E. et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 (2000).
63. Dusonchet, J. et al. A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 2907–2912 (2011).
64. Goldberg, M. S. et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ‑1. Neuron 45, 489–496 (2005).
65. Lo Bianco, C., Ridet, J. L., Schneider, B. L., Deglon, N. & Aebischer, P. α-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc. Natl Acad. Sci. USA 99, 10813–10818 (2002).This was the first convincing non-toxin-based animal model that demonstrated degeneration of nigral dopaminergic neurons by using lentiviral-mediated overexpression of α-synuclein in mice.
66. Kirik, D. et al. Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: a new primate model of Parkinson’s disease. Proc. Natl Acad. Sci. USA 100, 2884–2889 (2003).
67. Schapira, A. H. et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nature Rev. Drug Discov. 5, 845–854 (2006).
68. Fox, S. H., Brotchie, J. M. & Lang, A. E. Non-dopaminergic treatments in development for Parkinson’s disease. Lancet Neurol. 7, 927–938 (2008).
69. Friedman, J. H. et al. A multi-centre, placebo-controlled, double-blind trial to examine the safety and efficacity of pimavanserin in the treatment of psychosis in Parkinson’s disease. Mov. Disord. 25 (Suppl. 2), 292 (2010).
70. Schapira, A. H. Safinamide in the treatment of Parkinson’s disease. Expert Opin. Pharmacother. 11, 2261–2268 (2010).
71. Fink, J. S. et al. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res. Mol. Brain Res. 14, 186–195 (1992).
72. Ascherio, A. et al. Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology 60, 790–795 (2003).
73. Prediger, R. D. Effects of caffeine in Parkinson’s disease: from neuroprotection to the management of motor and non-motor symptoms. J. Alzheimers Dis. 20 (Suppl. 1), 205–220 (2010).
74. Simon, D. K. et al. Caffeine and progression of Parkinson disease. Clin. Neuropharmacol. 31, 189–196 (2008).
75. Bjorness, T. E. & Greene, R. W. Adenosine and sleep. Curr. Neuropharmacol. 7, 238–245 (2009).
76. Kaplitt, M. G. et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369, 2097–2105 (2007).This was an elegant study that used adeno-associated virus-mediated overexpression of GAD — the key enzyme of GABA synthesis — to reduce the abnormal excitatory drive of the subthalamic nucleus in patients with PD.
77. Jarraya, B. et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci. Transl. Med. 1, 2ra4 (2009).
78. Jarraya, B. et al. A phase I clinical trial on the safety and efficacy of ProSavin1 a dopamine replacement gene therapy for Parkinson’s disease (PD): an interim report. Mov. Disord. 25 (Suppl. 2), 267 (2010).
79. Gallagher, D. A. & Schapira, A. H. Etiopathogenesis and treatment of Parkinson’s disease. Curr. Top. Med. Chem. 9, 860–868 (2009).
80. Mandel, S. A., Amit, T., Weinreb, O., Reznichenko, L. & Youdim, M. B. Simultaneous manipulation of multiple brain targets by green tea catechins: a potential neuroprotective strategy for Alzheimer and Parkinson diseases. CNS Neurosci. Ther. 14, 352–365 (2008).
81. Gill, S. S. et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nature Med. 9, 589–595 (2003).
82. Lang, A. E. et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 59, 459–466 (2006).
83. Marks, W. J. Jr et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 9, 1164–1172 (2010).
84. Siffert, J. et al. AAV2-neurturin (CERE-120) for advanced Parkinson’s disease (PD): efficacy and safety results from a controlled Phase 2 clinical trial. Soc. Neurosci. Abstr. 326.7 (2009).
85. Bartus, R. T. et al. Bioactivity of AAV2-Neurturin gene therapy (CERE-120): differences between Parkinson’s Disease and nonhuman primate brains. Mov. Disord. 26, 27–36 (2011).
86. Goetz, C. G. et al. Placebo influences on dyskinesia in Parkinson’s disease. Mov. Disord. 23, 700–707 (2008).
87. Gillingham, J. Forty-five years of stereotactic surgery for Parkinson’s disease: a review. Stereotact. Funct. Neurosurg. 74, 95–98 (2000).
88. Hauptmann, C., Popovych, O. & Tass, P. A. Desynchronizing the abnormally synchronized neural activity in the subthalamic nucleus: a modeling study. Expert Rev. Med. Devices 4, 633–650 (2007).
89. Goetz, C. G. et al. Placebo response in Parkinson’s disease: comparisons among 11 trials covering medical and surgical interventions. Mov. Disord. 23, 690–699 (2008).This was an analysis of 11 trials covering medical and surgical interventions for PD that reported the highest likelihood of placebo-associated improvements in patients with motor-fluctuations, in patients receiving surgical interventions and in those with a higher chance of placebo assignment.
90. Rascol, O. “Disease-modification” trials in Parkinson disease: target populations, endpoints and study design. Neurology 72, S51–S58 (2009).
91. The Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 321, 1364–1371 (1989).
92. Fahn, S. et al. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 351, 2498–2508 (2004).
93. Hauser, R. A. & Holford, N. H. Quantitative description of loss of clinical benefit following withdrawal of levodopa-carbidopa and bromocriptine in early Parkinson’s disease. Mov. Disord. 17, 961–968 (2002).
94. Olanow, C. W. et al. A randomized, double-blind, placebo-controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson’s disease (the ADAGIO study): rationale, design, and baseline characteristics. Mov. Disord. 23, 2194–2201 (2008).This was the first clinical study in the field of PD that used the delayed-start design with three primary end points to assess disease-modifying properties of a compound.
95. Olanow, C. W. et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 361, 1268–1278 (2009).
96. Ahlskog, J. E. & Uitti, R. J. Rasagiline, Parkinson neuroprotection, and delayed-start trials: still no satisfaction? Neurology 74, 1143–1148 (2010).
97. Holford, N. H. & Nutt, J. Interpreting the results of Parkinson’s disease clinical trials: time for a change. Mov. Disord. 2 Mar 2011(doi:10.1002/mds.23555).
98. The Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. JAMA 284, 1931–1938 (2000).
99. Whone, A. L. et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann. Neurol. 54, 93–101 (2003).
100. Ravina, B. et al. The role of radiotracer imaging in Parkinson disease. Neurology 64, 208–215 (2005).
101. Morrish, P. K. How valid is dopamine transporter imaging as a surrogate marker in research trials in Parkinson’s disease? Mov. Disord. 18 (Suppl. 7), 63–70 (2003).
102. Eidelberg, D. et al. The metabolic topography of parkinsonism. J. Cereb. Blood Flow Metab. 14, 783–801 (1994).
103. Eckert, T. et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 26, 912–921 (2005).
104. Gerhard, A. et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 21, 404–412 (2006).
105. Kikuchi, A. et al. In vivo visualization of α-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain 133, 1772–1778 (2010).
106. Martin, W. R., Wieler, M. & Gee, M. Midbrain iron content in early Parkinson disease: a potential biomarker of disease status. Neurology 70, 1411–1417 (2008).
107. Vaillancourt, D. E. et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology 72, 1378–1384 (2009).
108. Berg, D., Godau, J. & Walter, U. Transcranial sonography in movement disorders. Lancet Neurol. 7, 1044–1055 (2008).
109. Peran, P. et al. Magnetic resonance imaging markers of Parkinson’s disease nigrostriatal signature. Brain 133, 3423–3433 (2010).
110. van Dijk, K. D. et al. Diagnostic cerebrospinal fluid biomarkers for Parkinson’s disease: A pathogenetically based approach. Neurobiol. Dis. 39, 229–241 (2010).
111. Scherzer, C. R. et al. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc. Natl Acad. Sci. USA 104, 955–960 (2007).
112. Hong, Z. et al. DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133, 713–726 (2010).
113. El-Agnaf, O. M. et al. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 20, 419–425 (2006).
114. Tokuda, T. et al. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75, 1766–1770 (2010).This study showed that oligomeric α-synuclein is a promising biomarker for the diagnosis and early detection of PD.
115. Savica, R., Rocca, W. A. & Ahlskog, J. E. When does Parkinson disease start? Arch. Neurol. 67, 798–801 (2010).This paper provides a concise update of the symptoms of the prodromal phase of PD, which may help to better define a population who are at risk for developing PD and facilitate early diagnosis.
116. Stiasny-Kolster, K. et al. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for α-synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain 128, 126–137 (2005).
117. Stefanova, N., Bucke, P., Duerr, S. & Wenning, G. K. Multiple system atrophy: an update. Lancet Neurol. 8, 1172–1178 (2009).
118. Scholz, S. W. et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol. 65, 610–614 (2009).
119. Gilman, S. et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71, 670–676 (2008).
120. Wenning, G. K. et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov. Disord. 19, 1391–1402 (2004).
121. Schrag, A. et al. Measuring health-related quality of life in MSA: the MSA-QoL. Mov. Disord. 22, 2332–2338 (2007).
122. Stefanova, N., Tison, F., Reindl, M., Poewe, W. & Wenning, G. K. Animal models of multiple system atrophy. Trends Neurosci. 28, 501–506 (2005).
123. Holmberg, B. et al. Safety and tolerability of growth hormone therapy in multiple system atrophy: a double-blind, placebo-controlled study. Mov. Disord. 22, 1138–1144 (2007).
124. Dodel, R. et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [11C] (R)-PK11195 PET (MEMSA-trial). Mov. Disord. 25, 97–107 (2010).
125. Bensimon, G. et al. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 132, 156–171 (2009).
126. Goetz, C. G., Nutt, J. G. & Stebbins, G. T. The Unified Dyskinesia Rating Scale: presentation and clinimetric profile. Mov. Disord. 23, 2398–2403 (2008).
127. Katzenschlager, R. et al. Quantifying the impact of dyskinesias in PD: the PDYS-26: a patient-based outcome measure. Neurology 69, 555–563 (2007).
128. McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65, 1863–1872 (2005).
129. Frasier, M. et al. The Parkinson’s progression markers initiative: a prospective biomarkers study. Mov. Disord. 25 (Suppl. 2), 296 (2010).
130. Sage, J. I. et al. Pilot study of the efficacy and safety of piclozotan in Parkinson’s disease patients with l-DOPA induced motor complications. Mov. Disord. 24 (Suppl. 1), 277 (2009).
R E V I E W S
392 | MAY 2011 | VOLUME 10 www.nature.com/reviews/drugdisc
© 2011 Macmillan Publishers Limited. All rights reserved

131. Bronzova, J. et al. Double-blind study of pardoprunox, a new partial dopamine agonist, in early Parkinson’s disease. Mov. Disord. 25, 730–738 (2010).
132. Meltzer, H. Y. et al. Pimavanserin, a serotonin2A receptor inverse agonist, for the treatment of parkinson’s disease psychosis. Neuropsychopharmacology 35, 881–892 (2010).
133. Rascol, O. et al. A “N=1” randomized placebo-controlled multiple cross-over pilot study of FP0011, a novel antiglutamate agent, in advanced PD. Mov. Disord. 22 (Suppl. 16), 277 (2007).
134. Nutt, J. G. et al. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov. Disord. 23, 1860–1866 (2008).
135. Eggert, K. et al. Safety and efficacy of perampanel in advanced Parkinson’s disease: a randomized, placebo-controlled study. Mov. Disord. 25, 896–905 (2010).
136. Berg, D. et al. AFQ056 treatment of severe levodopa-induced dyskinesias: proof of concept study. Mov. Disord. 25 (Suppl. 2), 290 (2010).
137. Mizuno, Y., Hasegawa, K., Kondo, T., Kuno, S. & Yamamoto, M. Clinical efficacy of istradefylline (KW-6002) in Parkinson’s disease: a randomized, controlled study. Mov. Disord. 25, 1437–1443 (2010).
138. Hauser, R. A. et al. Study of istradefylline in patients with Parkinson’s disease on levodopa with motor fluctuations. Mov. Disord. 23, 2177–2185 (2008).
139. Stacy, M. et al. A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology 70, 2233–2240 (2008).
140. LeWitt, P. A. et al. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 63, 295–302 (2008).
141. Guttman, M. & Group, U.-C. I. Efficacy of istradefylline in Parkinson’s disease patients treated with levodopa with motor response complications: results of the KW-6002-US-018 study. Mov. Disord. 21 (Suppl. 15), 585 (2006).
142. Hauser, R. A. et al. Preladenant in patients with Parkinson’s disease and motor fluctuations: a phase 2, double-blind, randomised trial. Lancet Neurol. 10, 221–229 (2011).
143. LeWitt, P. A. et al. Fipamezole in the treatment of dyskinesia in advanced Parkinson’s disease (FJORD study). Mov. Disord. 25 (Suppl. 2), 300 (2010).
144. Murata, M., Hasegawa, K. & Kanazawa, I. Zonisamide improves motor function in Parkinson disease: a randomized, double-blind study. Neurology 68, 45–50 (2007).
145. NINDS NET-PD Investigators. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin. Neuropharmacol. 31, 141–150 (2008).
146. NINDS NET-PD investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 66, 664–671 (2006).
AcknowledgementsThis Review summarises the main outcomes of an International Symposium that was held in Bordeaux, France, between May 4 and 5 2010. We are indebted to the devotion and efficiency of N. Vignais in helping to organize the Symposium.
Competing interests statementThe authors declare competing financial interests: see Web version for details.
FURTHER INFORMATIONAuthor’s homepage: http://www.imn-bordeaux.orgAddex Pharmaceuticals: http://www.addexpharma.com/pipeline-old/adx48621/Boehringer Ingelheim: http://www.boehringer-ingelheim.com/news/news_releases/press_releases/2010/08_october_2010_fliba.htmlCheckOrphan: http://www.checkorphan.org/grid/news/treatment/monitoring-force-gmbh-requests-acknowledgment-of-flibanserin-as-orphan-drugClinicalTrials.gov: http://www.clinicaltrials.govIMPAX Laboratories: http://www.impaxlabs.comSanthera Pharmaceuticals: http://www.santhera.comThe Merck Group: http://www.merck.de/en/investors/ad_hoc_announcements/ad_hoc_announcements.htmlThe Michael J. Fox Foundation for Parkinson’s Research: http://www.michaeljfox.org
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 10 | MAY 2011 | 393
© 2011 Macmillan Publishers Limited. All rights reserved





























