Embed Size (px)
Citation preview

The Pennsylvania State University
The Graduate School
College of Agricultural Sciences
PREPARATION AND CHARACTERIZATION OF LIGNIN-
PROTEIN COVALENT LINKAGES
A Dissertation in
Biorenewable Systems
by
Brett Galen Diehl
©2014 Brett Galen Diehl
Submitted in Partial Fulfillment
of the Requirements
for the Degree of
Doctor of Philosophy
May 2014

ii
The dissertation of Brett Galen Diehl was reviewed and approved* by the following:
Nicole R. Brown
Associate Professor of Wood Chemistry
Dissertation Adviser
Chair of Committee
John E. Carlson
Professor of Molecular Genetics
Jeffrey M. Catchmark
Associate Professor of Agricultural and Biological Engineering
Emmanuel Hatzakis
Director of NMR facility
John Ralph
Special Member
Professor of Biochemistry
University of Wisconsin at Madison
Paul Smith
Head of Biorenewable Systems department
*Signatures are on file in the Graduate School.

iii
Abstract
Lignin is a natural aromatic polymer that is bio-synthesized in the cell walls of almost all
land plants. Great strides have been made in understanding lignin’s biological origins and
chemical and physical properties. However, many unanswered questions remain. For example,
the extent to which lignin interacts with other cell wall components, such as proteins, is largely
unknown. In order to help address this question, the preparation and characterization of lignin-
protein covalent linkages is reported here for the first time. Chapter 1 provides a more detailed
introduction, justification, and literature review.
Chapter 2 focuses on the preparation of low molecular weight lignin-protein model
compounds. The compounds were not prepared under biomimetic conditions. Instead, the
primary focus of this study was on the characterization of the model compounds, leading to the
identification of diagnostic lignin-protein NMR chemical shifts.
Chapter 3 describes the characterization of lignin-protein linkages prepared under
biomimetic conditions of lignin DHP formation. NMR showed that cysteine and tyrosine
containing peptides covalently crosslink with lignin, while other amino acids do not. IR and EDS
were useful for showing the general incorporation of protein into the lignin, but were incapable
of distinguishing covalent and non-covalent interactions.
Chapter 4 describes the interaction between lignin and gelatin protein. It was found, using
EDS and IR, that gelatin was incorporated into lignin DHP. However, a lack of diagnostic NMR
signatures revealed that the crosslinking was likely dominated by non-covalent interactions such
as physical entanglement. This seems likely, as gelatin is lacking in both cysteine and tyrosine
residues, which were shown to be the only reactive amino acids towards lignin.
Chapter 5 details attempts at identifying lignin-protein linkages in wild type Arabidopsis.
Arabidopsis was grown to maturity, then lignin was extracted from cell wall material using
acidified dioxane. Elemental analysis was used to show that the lignin was contaminated with
about 3.75% protein; however, NMR was not able to identify lignin-protein covalent linkages.
Chapter 6 details some future experiments that could be used to explore lignin-protein
linkages, and it is hoped that this work will pave the way for such studies.

iv
TABLE OF CONTENTS
List of Figures…………………………………………………………………………………....vii
List of Tables……………………………………………………………………………………viii
Abbreviations……………………………………………………………………………………..ix
Acknowledgements…………………………………………………………………………..........x
Chapter 1. Introduction ...………………………………………………………………………... 1
1.1. Problem statement ...………………………………………………………………… 1
1.2. Literature review ……………………………………………………………………. 1
1.2.1. Lignin biosynthesis ……………………………………………………….. 1
1.2.2. Plant cell wall structural proteins …………………………………………. 6
1.2.3. Evidence for lignin-protein linkages …………………………………….. 10
1.3. Methods for investigating lignin-protein linkages ………………………………… 12
1.3.1. Preparation of lignin-protein compounds ……………………………….. 12
1.3.1. Characterization of lignin-protein compounds ………………………….. 16
1.4. References …………………………………………………………………………. 21
Chapter 2. Towards lignin-protein crosslinking: Amino acid adducts of a lignin model quinone
methide …………………………………………………………………………………………. 25
2.1. Abstract ……………………………………………………………………………. 25
2.2. Introduction ………………………………………………………………………... 25
2.3. Experimental ………………………………………………………………………. 28
2.3.1. Materials ………………………………………………………………… 28
2.3.2. Model compound preparations ………………………………………….. 28
2.3.3. Model compound properties …………………………………………….. 29
2.3.4. Nuclear magnetic resonance spectroscopy ……………………………… 42
2.3.5. Mass spectrometry ………………………………………………………. 42
2.3.6. Computational methods …………………………………………………. 43
2.4. Results and discussion …………………………………………………………….. 44
2.4.1. Preparation of quinone methide-amino acid adducts ……………………. 44
2.4.2. Solution-state NMR of compounds 3-9 and density functional theory
calculations for compounds 10 and 11 …………………………………. 46
2.4.3. Adduct isomer determination ……………………………………………. 50
2.5. Conclusions ………………………………………………………………………... 50
2.6. Acknowledgements ………………………………………………………………... 51
2.7. References …………………………………………………………………………. 51
Chapter 3. Lignin crosslinks with peptides under biomimetic conditions ……………………... 55
3.1. Abstract ……………………………………………………………………………. 55
3.2. Introduction ………………………………………………………………………... 55
3.3. Experimental ………………………………………………………………………. 57

v
3.3.1. Materials ………………………………………………………………… 57
3.3.2. Synthesis of lignin DHP and lignin-peptide adducts ……………………. 57
3.3.3. Scanning electron microscopy and energy dispersive X-ray spectroscopy 57
3.3.4. Nuclear magnetic resonance spectroscopy ……………………………… 58
3.3.5. Fourier-transform infrared spectroscopy ………………………………... 58
3.4. Results and discussion …………………………………………………………….. 58
3.4.1. Preparation and yields of the lignin-peptide adducts ……………………. 58
3.4.2. Lignin-peptide morphology ……………………………………………... 59
3.4.3. Lignin-peptide linkage identification ……………………………………. 60
3.4.4. Supporting techniques for lignin-peptide characterization ……………… 64
3.5. Conclusions ………………………………………………………………………... 66
3.6. Acknowledgments …………………………………………………………………. 67
3.7. References …………………………………………………………………………. 67
Chapter 4. Preparation and characterization of lignin-gelatin complexes ……………………... 71
4.1. Abstract ……………………………………………………………………………. 71
4.2. Introduction ………………………………………………………………………... 71
4.3. Experimental ………………………………………………………………………. 73
4.3.1. Materials ………………………………………………………………… 73
4.3.2. DHP and DHP-Gel syntheses …………………………………………… 74
4.3.3. Fourier-transform infrared spectroscopy ………………………………... 74
4.3.4. X-ray photoelectron spectroscopy ………………………………………. 74
4.3.5. Scanning electron microscopy and energy dispersive X-ray spectroscopy 75
4.3.6. Nuclear magnetic resonance spectroscopy ……………………………… 75
4.4. Results and discussion …………………………………………………………….. 75
4.4.1. Preparation of DHP-Gel adducts ………………………………………... 75
4.4.2. Fourier-transform infrared spectroscopy of DHP-Gel adducts ………….. 76
4.4.3. Morphology and nitrogen content of DHP-Gel adducts ………………… 77
4.4.4. Nuclear magnetic resonance spectroscopy of DHP-Gel adducts ………... 80
4.5. Conclusions ………………………………………………………………………... 82
4.6. Acknowledgments …………………………………………………………………. 82
4.7. References …………………………………………………………………………. 83
Chapter 5. Searching for lignin-protein linkages in Arabidopsis ……………………………… 86
5.1. Abstract ……………………………………………………………………………. 86
5.2. Introduction ………………………………………………………………………... 86
5.3. Experimental ………………………………………………………………………. 87
5.3.1. Growth and lignin extraction from Arabidopsis ………………………… 87
5.3.2. Elemental analysis of Arabidopsis lignin ……………………………….. 88
5.3.3. Nuclear magnetic resonance spectroscopy of Arabidopsis lignin ………. 88
5.4. Results and discussion …………………………………………………………….. 88

vi
5.4.1. Lignin extractions from Arabidopsis ……………………………………. 88
5.4.2. Protein content of Arabidopsis extracts …………………………………. 89
5.4.3. Nuclear magnetic resonance spectroscopy of Arabidopsis lignin ………. 90
5.5. Conclusions ………………………………………………………………………... 91
5.6. Acknowledgments …………………………………………………………………. 92
5.7. References …………………………………………………………………………. 92
Chapter 6. Conclusions ………………………………………………………………………… 93
6.1. Research summary ………………………………………………………………… 93
6.2. Future endeavors …………………………………………………………………... 94
6.3. References …………………………………………………………………………. 97

vii
List of Figures
1.1. Three ‘common’ and three ‘uncommon’ monolignols …………………………………...….3
1.2. Resonance forms of monolignol radicals ………………………………………………….....3
1.3. Typical lignin inter-unit linkages …………………………………………………….……....4
1.4. Formation via radical coupling of β-ether QMs during lignin polymerization ……………...5
1.5. Nucleophilic amino acids that could potentially react with lignin QMs …………………….5
1.6. Tyrosine radicals and cross-coupled products ……………………………………………….7
1.7. Lignin-protein complex formed via lignin-carbohydrate linkage ....…………………………9
1.8. Preparation of a lignin β-ether model compound and its corresponding QM analog ...…….13
1.9. Preparation of lignin DHP ...………………………………………………………………..14
1.10. General structure of peptides added to lignin DHP preparations ...……………………….14
1.11. 1H NMR spectrum of lignin DHP ...……………………………………………………….17
1.12. HSQC spectrum of lignin DHP ...………………………………………………………….18
1.13. FT-IR ATR spectrum of lignin DHP ...……………………………………………………19
1.14. SEM image of lignin DHP ...………………………………………………………………20
2.1. Formation of β-ether QMs via radical coupling, and their rearomatization ...……………...26
2.2. Guaiacylglycerol-β-guaiacyl ether 1 and its derived quinone methide (QM) 2 ...………….27
2.3. QM-AA model compounds …………………………………………………………………45
2.4. Overlaid HMQC side chain regions of compounds 3 and 5 ...……………………………...48
2.5. HSQC NMR spectrum of lignin DHP with overlaid α- and β-correlation data of 3-11 ...….49
3.1. Lignin-peptide crosslinking mechanism ……………………………………………………56
3.2. SEM images of DHP and lignin-peptide adducts ...………………………………………...60
3.3. HSQC NMR of lignin-CGG adduct ...………………………………………………………61
3.4. HSQC NMR of lignin-YGG adduct ...……………………………………………………...62
3.5. HSQC NMR of lignin-HGG adduct ..………………………………………………………63
3.6. FT-IR spectra of DHP and lignin-peptide adducts …………………………………………65
4.1. FT-IR of neat DHP and DHP-Gel adducts …………………………………………………77
4.2. SEM of neat DHP and DHP-Gel adducts …………………………………………………..78
4.3. Morphology and nitrogen atomic percentages for DHP-Gel adducts ……………………...79
4.4. HSQC NMR spectrum of DHP-Gel1 ……………………………………………………….81
5.1. Optical microscopy of solvents extracted and ball milled Arabidopsis cell wall material …89
5.2. HSQC NMR spectrum of Arabidopsis lignin ………………………………………………81
6.1. Cell wall models ……………………………………………………………………………95
6.2. Synthetic route to α-13C coniferyl alcohol ………………………………………………….96
6.3. Standard lignin α-shifts and α-shifts of lignin-protein linkages ……………………………97

viii
List of Tables
2.1. 1H and 13C NMR chemical shifts for lignin-amino acid adducts …………………………...47
2.2. Observed and DFT calculated α-13C NMR chemical shifts for compound 3 ………………50
3.1. Yield data for the DHP and lignin-peptide adducts ………………………………………...59
3.2. Inter-unit linkage ratios of the DHP and lignin-peptide adducts …………………………...64
3.3. EDS elemental analysis data for DHP and the lignin-peptide adducts ……………………..66
4.1. Nucleophilic amino acid abundance (g/100 g dry, ash-free protein) in gelatin …………….72
4.2. Preparation and yields of DHP and DHP-Gel adducts ……………………………………..76
4.3. Inter-unit linkage ratios of DHP and DHP-Gel adducts ……………………………………81
5.1. Estimated protein content of Arabidopsis and extracted lignin …………………………….89
5.2. Inter-unit linkage ratios of Arabidopsis acidic dioxane lignin ……………………………...90

ix
Abbreviations
AA – amino acid
AGP – arabinogalactan protein
ATR – attenuated total reflectance
DFT – density functional theory
DHP – dehydrogenation polymer
EDS – energy dispersive X-ray spectroscopy
FT-IR – Fourier-transform infrared spectroscopy
GRP – glycine-rich protein
HRGP – hydroxyproline-rich glycoprotein
NMR – nuclear magnetic resonance
PRP – proline-rich protein
QM – quinone methide
SEM – scanning electron microscopy
XPS – X-ray photoelectron spectroscopy

x
Acknowledgements
Here, at the end of my doctoral dissertation, I would like to take the opportunity to thank the
people, without whose assistance this work would not have been possible. This list is not
exhaustive, and I apologize greatly for any unintentional omissions.
First, I would like to thank my advisor, Dr. Nicole Brown, and my committee members, Drs.
John Ralph, Jeff Catchmark, Emmanuel Hatzakis, and John Carlson. I would also like to thank
Professor Emeritus Alan Benesi, who graciously served on my committee until his happy and
healthy retirement. Without guidance and patience from these individuals, this work would not
have been possible.
I would like to thank Dr. John Ralph’s entire research group, especially Matt Regner and Yuki
Tobimatsu, who not only helped with my research but also made life immensely enjoyable when
I visited John’s lab in May of 2012. I plan to revisit Madison as often as possible.
I would like to thank the seemingly countless number of individuals who have helped me with
myriad technical matters throughout the course of my PhD. To the folks in the Materials
Characterization Lab, Josh Stapleton, Trevor Clark, Vince Bojan, Tim Tighe, Julie Anderson,
Melisa Yashinski, and Joe Stitt, for assistance in collecting and interpreting an endless tide of
spectral data, my deepest thanks. To Wenbin Luo and the members of the Scott Showalter group,
sincere thanks for assistance with all manner of NMR technical support.
My funding sources and the programs and research they fostered were instrumental toward the
completion of this dissertation. I would like to acknowledge the USDA National Needs
Fellowship, which provided tuition and research funding support for several years. A very
special thanks is warranted to the DOE sponsored Center for Lignocellulose Structure and
Formation (CLSF) and all of the members therein. The center provided not only funding and
facilities to support this research, but also the breadth and depth of intellectual power necessary
to inspire all its members to perform to their highest capabilities. A very special thanks is also
warranted to the NSF CarbonEARTH fellowship program. This program provided me with
financial support, but more importantly, opportunities and memories that will last a lifetime.
Many thanks to all of my fellow CarbonEARTH’ers for advice, support, and fun times.
I would like to thank my lab mates, Paul Munson and Curtis Frantz, for companionship
throughout the seemingly endless process of graduate school, and for research related insights.
Finally, I would like to thank my parents for their love and patience, and for instilling in me the
skills I need to make it through life’s challenges. I am indebted to them in ways that can never be
repaid.

1
Chapter 1
Introduction
1.1. Problem statement
The purpose of this research was to investigate the reactivities of amino acids and
possibly proteins toward lignin, ultimately resulting in the formation of lignin-protein crosslinks.
It has been suggested that proteins located in plant cell walls may interact with lignin in many
ways. For example, enzymatic proteins such as peroxidases and laccases are necessary for lignin
polymerization. Furthermore, structural proteins (non-enzymatic proteins that assist in cell wall
scaffolding) may assist in the initial stages of lignin deposition, which occurs in the cell wall
corner region. Several mechanisms could be envisioned, with the structural proteins playing a
relatively passive role, or an active one in which they template lignin polymerization, perhaps
influencing the inter-unit linkage sequence of the final lignin polymer. Lignin-protein linkages
may also play a role in genetically engineered plant lines. Research has shown that plants with
up-regulated cell wall protein expression sometimes exhibit altered physical and chemical
properties, including enhanced lignin extractability, which may be due to increased levels of
lignin-protein linkages.
In spite of the potential implications of lignin-protein crosslinks in both wild type and
mutant plant lines, there have been few studies addressing the fundamental aspects of lignin-
protein linkages and their formation. For example, prior to this work, it was largely unknown
which amino acids (if any) were reactive toward lignin, how stable lignin-protein linkages were,
and how the linkages could be identified using standard analytical tools. The goal of this work
was to address and answer some of those questions, mostly through in vitro studies, while trying
to keep in mind the future necessity of lignin-protein identification in vivo.
The remainder of this chapter provides a literature review, which focuses on lignin
biosynthesis, plant cell wall structural proteins, evidence for lignin-protein linkages, and a
section detailing the methods used in this study for lignin-protein linkage preparation and
characterization. The second, third, and fourth chapters are presented in manuscript form and
should be considered stand-alone publications. The final chapter presents a summary of pertinent
findings, discusses limitations, and provides suggestions for future work.
1.2. Literature review
1.2.1. Lignin biosynthesis
Plant cell walls are composed of a network of interacting polymers, namely cellulose,
hemicelluloses, pectins, lignin, and structural proteins (Cosgrove, 2005; McQueen-Mason and
Cosgrove, 1994). Of these, lignin is the most abundant aromatic biopolymer, and the second
most abundant biopolymer overall (Boerjan et al., 2003). The lignin polymer is unique among

2
the plant cell wall polymers in that it is composed of phenylpropanoid monomers known as the
so-called monolignols, which undergo radical polymerization via a mechanism that is apparently
not under biological control beyond the generation of the lignin radicals themselves. Lignin
polymerization exhibits incredible plasticity among and within species. It is thought that lignin
benefits the plant by providing strength and rigidity to the cell wall, enhanced water
conductivity, and pathogen resistance. Pathogen resistance in particular is provided due to the
recalcitrance of lignin towards degradation. Unfortunately, this recalcitrance negatively impacts
human efforts to effectively use plant cell wall materials as biorenewable resources. Specifically,
lignin recalcitrance affects the pulp and paper industry, the developing biofuels industry, the
agricultural industries, and the chemical industries, all of whom seek higher value products from
lignin (Chapple et al., 2007; Chen and Dixon, 2008; Jung and Allen, 1995; Jung, 1989; Li et al.,
2008; Stewart et al., 2006). Thus, a greater understanding of lignin chemistry and biochemistry is
desirable towards controlling and minimizing its recalcitrance, as well as engineering lignin-
based products.
The process of lignification begins with the biosynthesis of the monolignols. Typically,
monolignols are biosynthesized from phenylalanine via a series of enzymatic steps (Boerjan et
al., 2003). There is evidence that the monolignols may be stored and/or transported to the cell
wall as monolignol glucosides (i.e., the phenolic hydroxyl of the monolignol is blocked by 4-O-
glycosylation); however, this may not always be the case. In addition, the mode of monoglignol
transport into the cell wall is unknown (i.e., golgi-derived vesicles versus plasma membrane
pumps) (Boerjan et al., 2003). As noted above, the process of lignification exhibits plasticity, and
this is evidenced by the variability of monolignol biosynthesis and expression. The three most
common lignin monomers are shown in Fig 1.1. The expression of these monolignols varies
among plant taxa. For example, gymnosperm lignin is almost entirely composed of G-units (i.e.,
coniferyl alcohol based) with some traces of H-units (p-coumaryl alcohol based), dicotyledonous
angiosperm lignin is mainly composed of G- and S-units (sinapyl alcohol based), and
monocotyledonous angiosperm lignin is composed of all three units, as well as ferulates,
sinapates, and p-coumarates. Other monolignols are biosynthesized and incorporated into the
lignins of both wildtype and mutant plant lines. For example, lignin found in the seed coats of
some wildtype vanilla orchids and cacti is almost completely composed of caffeyl alcohol (Chen
et al., 2012). In caffeic acid/5-hydroxyconiferaldehyde O-methyltransferase (COMT) deficient
mutants, 5-hydroxyconiferyl alcohol is incorporated into the lignin polymer (Li et al., 2000;
Ralph et al., 2001). And in cinnamyl alcohol dehydrogenase (CAD) deficient mutants,
coniferaldehyde and other aldehydes are incorporated into the lignin polymer (Ralph et al.,
2001).

3
Fig 1.1. Three ‘common’ and three ‘uncommon’ monolignols. From left to right: p-coumaryl
alcohol, coniferyl alcohol, sinapyl alcohol, caffeyl alcohol, 5-hydroxyconiferyl alcohol, and
coniferaldehyde. The side chain carbons of the monolignols are often referred to as α, β, and γ-
positions (see leftmost structure). This nomenclature will be used throughout the document.
Once the monolignols are shuttled to the cell wall, polymerization occurs via enzymatic
dehydrogenation followed by radical recombination. Glycosyl hydrolases are implicated in the
removal of the glucose residue from monolignol glucosides (Boerjan et al., 2003).
Dehydrogenation is then catalyzed by peroxidases and/or laccases. The exact peroxidase and/or
laccase isozymes responsible for monolignol oxidation have yet to be elucidated and may vary
among species (Boerjan et al., 2003). Hydrogen peroxide is necessary for peroxidase catalyzed
monolignol oxidation, and the source of this peroxide is uncertain, though NADPH oxidases may
play a role. Again, further research in this area is necessary. The monolignol radical is stabilized
by resonance (Fig 1.2), a direct consequence of which is the multiple lignin inter-unit linkage
types that are observed (Fig 1.3).
Fig 1.2. Resonance forms of monolignol radicals. R typically represents H or OCH3.
There is currently no evidence for enzymatic control over the process of monolignol
radical recombination (Ralph et al., 2008). Instead, the relative ratios of lignin inter-unit linkages
can vary substantially and can be influenced by many factors, including but not limited to,
monolignol composition (i.e., which monolignols are present), monolignol concentration,
oxidant concentration (e.g., H2O2), catalyst/enzyme concentration, pH, the polymerization matrix
(i.e., is the lignin polymerizing in a hemicellulose-rich, pectin-rich, or protein-rich environment,
or bulk water, etc.), and other physical and chemical concerns (Boerjan et al., 2003; Cathala et
al., 1998). In general though, the predominant inter-unit linkage type in native lignins is the so-
called β-ether (β-O-4) linkage, with varying quantities of other linkages, including
phenylcoumaran (β-5), resinol (β-β), dibenzodioxocin (5-5/β-O-4/α-O-4), spirodienone (β-1),

4
biphenyl ether (4-O-5), biphenyl (5-5), and β-ether/α-aryl ether (β-O-4/α-O-4) (Boerjan et al.,
2003; Capanema et al., 2005; Vanholme et al., 2010).
Fig 1.3. Typical lignin inter-unit linkages. Linkage ratios vary and are influenced by many
factors. Linkage ratios depicted here are not indicative of ratios observed in native lignins.
In the case of the predominant β-ether linkage, radical recombination results in the
formation of an unstable quinone methide (QM) intermediate (Fig 1.4) that cannot be trapped
intramolecularly, but instead must be trapped by an external nucleophile (in contrast to β-5 and
β-β QMs, which can be trapped intramolecularly). The nucleophile is most often water, yielding
the β-ether/α-OH structure. However, other cell wall nucleophiles are known to quench the QM.
For example, lignin has long been understood to covalently crosslink with plant cell wall
components such as hemicelluloses through nucleophilic reactions (via hydroxyl or carboxylic
acid groups) with the α-carbon of lignin QMs (Balakshin et al., 2011; Leary, 1980; Miyagawa et
al., 2012; Ralph et al., 2009; Toikka et al., 1998; Yuan et al., 2011).

5
Fig 1.4. Formation via radical coupling of β-ether QMs during lignin polymerization. L = lignin
polymer, Nuc = nucleophile, R = H or OCH3.
The crosslinking of lignin with cell wall constituents other than hemicelluloses has not
been well investigated. Cell wall structural proteins contain amino acid residues with
nucleophilic side chains that could react with lignin QMs (Harrak et al., 1991; Jose and
Puigdomenech, 1993; Ryser et al., 1997; Kieliszewski et al., 2011). Specifically, the amino acids
cysteine (Cys), lysine (Lys), histidine (His), aspartic acid (Asp), glutamic acid (Glu), serine
(Ser), threonine (Thr), tyrosine (Tyr) and hydroxyproline (Hyp) (Fig 1.5) all contain nucleophilic
side chain groups. Cell wall proteins containing these amino acids vary in quantity among
species and cell types, ranging from as low as 1-2% to 20% dry weight basis (Albersheim et al.,
2010; Cassab et al., 1988). They have previously been postulated to crosslink with lignin, and it
has been suggested that they may serve as nucleation sites or templates during lignification, but
this has not been adequately tested (Boerjan et al., 2003; Cassab et al., 1988; Harrak et al., 1991;
Albersheim et al., 2010; Beat et al., 1989). If true, this mechanism could provide spatial and
temporal control over lignin deposition and architecture (Beat et al., 1989). The following
section will discuss the various classes of cell wall structural proteins that contain nucleophilic
amino acids and are likely to be in close spatial proximity to lignin within the cell wall.
Fig 1.5. Nucleophilic amino acids (nuc side chain groups are highlighted in green) that could
potentially react with lignin QMs. In their free amino acid forms (shown here), the α-amine and
α-acid groups could also be nucleophilic; therefore, these groups were blocked to prevent
competing reactions in the studies described below. From left to right, starting at the top:

6
cysteine (Cys), lysine (Lys), histidine (His), aspartic acid (Asp), glutamic acid (Glu), serine
(Ser), threonine (Thr), tyrosine (Tyr), hydroxyproline (Hyp). Amino acid stereochemistry is not
shown; L-isomers dominate in nature.
1.2.2. Plant cell wall structural proteins
Plant cell wall structural proteins account for a relatively small percentage (dry weight
basis) of the total cell wall material in mature tissues. Early studies showed that primary cell
walls of dicots typically contain 5-10% protein and 2% hydroxyproline (Hyp), which originates
primarily from extensins (Lamport, 1974; Talmadge et al., 1973). Once secondary walls are
deposited, the relative protein content drops. The following section describes the classes of cell
wall proteins that may potentially interact with lignin, as well as proposed interaction
mechanisms. Proteomics of specific plant species of interest are discussed in a later section.
There are two broad classes of cell wall structural proteins that seem most likely to
interact with lignin: glycine-rich proteins (GRPs), and the proline/hydroxyproline-rich
glycoproteins, which are often further subdivided into the proline-rich proteins (PRPs),
hydroxyproline-rich glycoproteins (HRGPs), and arabinogalactan-proteins (AGPs). These
protein classes are evolutionarily related, resulting in structural and functional similarities. Some
evidence has indicated that these proteins may interact with lignin, or even serve as nucleation
sites for lignification in the cell corners and/or the general compound middle lamella. However,
conclusive evidence for lignin-protein linkages has yet to be described.
Glycine-rich proteins (GRPs) are a diverse group of proteins that are often expressed in
plant cell walls. As their moniker implies, they are glycine-rich and typically contain between
60% and 70% glycine, which is much higher than most other enzymatic or structural proteins
found in plants or animals. They most commonly occur in tracheary elements of protoxylem and
metaxylem tissues, and are involved in diverse cellular processes during plant development and
adaptation to environmental change (Chen et al., 2007; Ringli et al., 2001). Their function varies
among cell types, as does their structure, which is the basis for the most current GRP
classification system. Class I GRPs may contain a signal peptide followed by a highly conserved
(GGX)n region, where X is often Ala, Ser, Val, His, Phe, Tyr or Glu. Class II GRPs may also
contain a characteristic cysteine-rich C-terminal. Class III GRPs typically contain fewer glycine-
rich regions compared to other GRPs. Class IV GRPs are RNA-binding and contain either an
RNA-recognition motif or a cold-shock domain. And class V GRPs are glycine-rich with mixed
glycine repeat patterns that are not typically observed in the other classes (Mangeon et al., 2010).
For in-depth information regarding GRP tissue expression pattern, subcellular localization,
structure, and function, three excellent reviews are Sachetto-Martins et al. (2000), Ringli et al.
(2001), and Mangeon et al. (2010).
Based on the amino acid composition of GRPs, two modes of lignin-GRP crosslinking
may be envisioned. The first mode of crosslinking is through QM-nucleophile reactions, the

7
chemistry of which was discussed in a preceding section. In GRPs, the amino acids most likely
to react with lignin in this manner are His, Glu, Ser, and Tyr. Another potential lignin-GRP
crosslinking mechanism is through oxidative coupling of lignin with amino acid moieties,
specifically tyrosine. It has been shown that GRPs are often tyrosine-rich (up to 10% Tyr), and
they crosslink in an intra- and inter-peptide manner via peroxidase mediated reactions (Ringli et
al., 2001; Ryser et al., 2004). The tyrosine radical and experimentally observed tyrosine cross-
coupled products are shown in Fig 1.6. Such intra- and inter-peptide linkages are also observed
with PRPs and HRGPs, as discussed below. When lignin is in close proximity to GRPs, lignin-
tyrosine crosslinking via this oxidative mechanism may result. Alternatively, lignin-tyrosine
radical coupling may be discouraged if the oxidation potentials of the monolignols and tyrosine
are quite different. This seems likely, given that monolignols exhibit radical delocalization over
five resonance forms (Fig 1.2), while tyrosine only exhibits four (Fig 1.6) (Cong et al., 2013).
The work described here mainly focuses on the preparation and characterization of lignin-peptide
linkages formed through QM-nucleophile chemistry, but some attempts were made to identify
putative lignin-tyrosine radical mediated linkages, as well. More work in this area is warranted.
Fig 1.6. Tyrosine radicals and cross-coupled products. Top row: tyrosine radical resonance
forms. Middle row: isodityrosine, dityrosine, and pulcherosine. Bottom row: di-isodityrosine.

8
Of the GRPs, those filling cell wall structural functions may be in closest spatial
proximity to lignin. Previous research has shown that GRPs may interact with lignin, though
covalent linkage formation has not been clearly demonstrated. In 1989, Beat et al. noted that
GRPs and lignin were localized to the same cell types within Phaseolus vulgaris (common
bean), and it was hypothesized that the GRPs might provide nucleation sites for lignification via
tyrosine residues. The benefits to the plant would include spatial and temporal control of various
lignin properties including density and three-dimensional pattern (Beat et al., 1989). Similar
results were obtained by Ye and Varner in 1991, this time with regards to soybean (Ye and
Varner, 1991b). In 2004, Ryser et al. demonstrated that GRPs act as linkages between secondary
cell wall thickenings, mainly composed of lignin, in protoxylem elements of seed plants as the
cells passively expand following apoptosis (Ryser et al., 2004). Yet no attempt was made to
determine how the GRPs anchor to the lignin-rich thickenings. Interestingly, in 2007, Chen et al.
showed that an Arabidopsis GRP (AtGRP9) exhibits subcellular localization comparable with
that of AtCAD5, a major Arabidopsis cinnamyl alcohol dehydrogenase localized to the cell wall.
Yeast two-hybrid analysis also revealed that the two proteins interacted strongly, suggesting that
GRPs may play a role in lignin monolignol synthesis, which occurs prior to lignin
polymerization.
Proline-rich proteins (PRPs) display great heterogeneity in their amino acid sequences,
but they all contain amino acids with nucleophilic side chains such as Lys, His, Glu, Ser, and Tyr
(Jose and Puigdomenech, 1993), potentially allowing for QM-nucleophile crosslinking or lignin-
tyrosine oxidative crosslinking. Ryser et al. (1997) stated, "localization of PRPs in lignified
secondary walls and the secretion of the protein during lignification support the hypothesis of Ye
et al. (1991a) that PRP localization is related to the pattern of lignification." They also made the
bold claim that, "it may be speculated that PRPs function as a scaffold for lignin deposition via
their tyrosine groups followed by oxidative cross-linking of lignin monomers" (Ryser et al.,
1997). A similar conclusion was reached with regards to primary cell walls by Harrak et al.
(1999), as it was found that a certain PRP located in wild tomato is down-regulated in response
to drought, as is lignin production. The authors concluded that lignin and protein potentially
interact with one another on the basis that they are up-regulated and down-regulated together and
are located within the same cellular compartment (Harrak et al., 1999).
Hydroxyproline-rich glycoproteins (HRGPs) contribute to tissue integrity and tensile
strength. The most abundant and well-studied HRGPs are the extensins, which are defined by
Ser-Hyp4 glycomodules. The proline hydroxyl groups and glycomodules (typically consisting of
one through four arabinose residues) are post-translationally added and their placement and
abundance is determined by the sequence of the peptide chain (Cannon et al., 2008; Kieliszewski
et al., 2011). Extensins are generally tyrosine-rich, enabling them to crosslink via extensin
peroxidase. These networks involve short motifs, where isodityrosine forms very short
intramolecular crosslinks. This isodityrosine moiety may then react with a tyrosine residue to
form pulcherosine, or react with another isodityrosine residue to form the tyrosine tetramer, di-

9
isodityrosine (Fig 1.6) (Kieliszewski et al., 2011). It is conceivable that lignin could crosslink
with these tyrosine residues via the radical mechanism described previously. In addition,
nucleophilic amino acids are abundant in HRGPs, and include Cys, Lys, His, Tyr, Thr, Asp, and
Ser and Hyp residues that remain un-glycosylated, which may allow for QM-nucleophile
crosslinking. It is also possible that the hydroxyproline-bound arabinose groups may crosslink
with lignin, as the primary hydroxyl of arabinose has been shown to react with lignin QMs in
vitro (Toikka et al., 1998). If this occurs in vivo, then lignin might be indirectly coupled to
HRGPs via lignin-carbohydrate linkages (Fig 1.7). Observing this scenario may be difficult using
standard lignin characterization techniques such as HSQC NMR, and warrants further
investigation.
Fig 1.7. Hypothetical lignin-protein complex formed via lignin-carbohydrate linkage. Protein
fragment sequence is Ser-Hyp-Hyp-Hyp, with varying degrees of arabinose glycosylation.
Lignin-carbohydrate linkage forms through reaction of the arabinose primary hydroxyl (C6-OH)
with the electrophilic α-carbon of the lignin QM.
The arabinogalactan-proteins (AGPs) are much more highly glycosylated than the PRPs
and HRGPs, with type II arabino-3,6-galactans (5 – 25 kDa) accounting for 90% to 98% (w/w)
of the AGP (Ellis et al., 2010). The miniscule protein component is often rich in Hyp, Pro, Ala,
Ser, and Thr. Of these, Hyp, Ser, and Thr could potentially be reactive toward lignin QMs.
However, it seems unlikely that lignin-AGP crosslinking would occur via addition of
nucleophilic amino acids to the QM, both because the protein component is so insignificant and
because the oligosaccharides likely encase the protein, shielding it from inter-polymer
interactions. Crosslinking between lignin and AGPs would likely occur through the mechanism
shown in Fig 1.7.
GRPs, PRPs, HRGPs, and AGPs are abundant plant cell wall structural proteins. Liyama
et al. (1994) stated, "there is evidence that both HRGPs and Gly-rich proteins are associated with
lignin and possibly act as foci for lignin polymerization. However, no information as to the

10
nature of possible covalent linkages or their biosynthetic route is available". There may be at
least two mechanisms for lignin-protein crosslinking in plant cell walls. One mechanism
involves radical crosslinking, perhaps via lignin and tyrosine moieties, while the second
mechanism involves reactions of nucleophilic amino acid side chains with lignin QM
intermediates. The latter mechanism is the primary focus of the research described here, but
lignin-protein oxidative coupling will also be studied where possible.
1.2.3. Evidence for lignin-protein linkages
In the previous section it was shown that lignin and cell wall structural proteins are often
co-localized within the plant cell wall, leading some researchers to speculate on the formation of
lignin-protein complexes. These lignin-protein linkages have proven difficult to detect
conclusively, especially in vivo. Nevertheless, evidence (which is largely anecdotal) suggests that
lignin-protein linkages may occur. The prevailing theory of lignin biosynthesis supports this
hypothesis. Under the prevailing theory, monolignol radicals couple to form lignin inter-unit
linkages under conditions that are free of enzymatic control. This results in the formation of the
predominant β-ether linkage and the subsequent QM intermediate (Fig 1.4), which reacts quickly
with the most abundant and/or most chemically compatible nucleophile. Because the quenching
of the QM is under chemical control, the QM could be expected to react with any nearby
nucleophile, including nucleophiles located on proteins. Indeed, the quenching of QMs by
nucleophiles that are often present on amino acids has been studied in non-lignin systems. For
example, the thiol group of glutathione reacts with an o-QM generated from the flavonoid
quercetin (Awad et al., 2000), the thiol group of cysteine reacts with the relatively unreactive p-
QM, 2,6-di-tert-butyl-4-methylene-2,5-cyclohexadienone (Bolton et al., 1997), and thiols and
thiolates react with QMs derived from anthracyclines (Ramakrishnan and Fisher, 1983).
Similarly, amines (but not amino acids) have been shown to trap lignin QMs (Ralph and Young,
1983). A wide array of acid and hydroxyl-containing reagents react with p-QMs (Leary et al.,
1977). And primary (and to a much lesser extent, secondary) hydroxyl groups of carbohydrates
react with lignin QMs (Toikka et al., 1998). Thus, given the general ability of soft (and even
relatively hard) nucleophiles to quench QMs, and given that lignin QM quenching is under
simple chemical control, it seems plausible that similar reactions could occur in vivo between
lignin QMs and nucleophilic amino acids.
In vitro experiments have provided some evidence for lignin-protein coupling. In 1978
and 1982, F. W. Whitmore published three articles regarding lignin-protein interactions
(Whitmore, 1978a, 1978b, and 1982). Whitmore isolated cell walls of Pinus elliottii (slash pine)
in such a way that native peroxidase enzymes were left intact and active. Lignin dehydrogenation
polymer was then added to one group of cell walls (control), and coniferyl alcohol was added to
another (experimental). Upon extraction, the experimental lignin contained significantly more
protein than the control, providing evidence that proteins were incorporated into lignin during
polymerization and not merely physically entangled in lignin following polymerization.
Whitmore then determined that hydroxyproline interacted more strongly with the lignin than

11
other amino acids, perhaps by forming ether linkages. He hypothesized that extensin was most
responsible for lignin-protein crosslinking (Whitmore, 1978, 1982). However, failure to directly
observe the proposed lignin-protein linkage rendered the results inconclusive. With quantitative
1D and 2D NMR experiments now commonplace, it is perhaps time to revisit these experiments
in order to more accurately ascertain the exact nature of the lignin-protein interactions.
More recently, evidence for lignin-protein interactions has been obtained through the use
of dynamic mechanical analysis (DMA) and Fourier-transform infrared spectroscopy (FT-IR).
Salmen and Petterson (1995) found that only one glass transition was observed for protein and
lignin within the primary cell wall, indicating an association that is roughly homogenous in
nature. Upon treatment with a protease, the glass transition temperature increased due to removal
of protein and subsequent increase in the relative concentration of the thermally stable lignin
polymer. Between 2006 and 2008, Stevanic and Salmen used DMA and FT-IR to study the
primary cell walls of Norway spruce, resulting in three publications. The first article found that,
"strong interactions were evident between lignin and protein, between cellulose and xyloglucan,
and between cellulose and pectin" (Stevanic and Salmen, 2006). A similar conclusion was
reached in the second publication, with the authors stating, "to a certain extent, all the polymers
in the surface material...took part in the stress transfer...indicating an intimately linked network
structure" (Stevanic and Salmen, 2008a). Finally, the third publication reported similar findings,
namely that there appear to be lignin-protein and lignin-pectin interactions within the primary
cell wall (Stevanic and Salmen, 2008b). DMA and dynamic FT-IR can indicate that polymer-
polymer interactions exist, but the exact nature of these interactions cannot be determined using
these methods, so further studies are warranted. It has been shown that horseradish peroxidase
enzymes can crosslink, or at least strongly interact, with a growing lignin polymer. This may be
why active peroxidases persist in lignified plant cells even after apoptosis (Evans and
Himmelsbach, 1991). Kaewtip et al. (2010) showed an interaction between wheat gluten and
lignin, and postulated that thiol groups on cysteine residues reacted with the double bonds of
lignin to form lignin-protein linkages. Unfortunately, they were unable to conclusively confirm
such linkages. It is interesting to note that, using FT-IR, blood plasma protein was observed to
hydrogen bond to lignin (Polus-Ratajczak et al., 2003). It is important to keep in mind that in
addition to covalent crosslinking, non-covalent interactions between lignin and protein could
play an important role in the structure and function of plant cell walls.
In summary, previous work has shown that a variety of nucleophiles react with non-lignin
QMs, indicating the possibility for lignin-protein linkages to form via QM-nucleophile
chemistry. Furthermore, evidence has shown that lignin interacts with proteins under in vitro
conditions as well as in native plant cell walls. Yet there has been no attempt to directly observe
in vitro or in vivo lignin-protein linkages using modern techniques such as multidimensional
NMR. Given the economic importance of lignin and its ubiquitous nature within the biosphere,
increased knowledge of its structure and function should be a priority. The work described here

12
extends our fundamental understanding of lignin chemistry by characterizing lignin-protein
covalent linkages as well as lignin-protein non-covalent interactions.
1.3. Methods for investigating lignin-protein linkages
1.3.1. Preparation of lignin-protein compounds
Lignin-protein model compounds were first prepared and characterized under relatively
simple, in vitro conditions. The simplest lignin-protein model compounds (in terms of chemical
structure and molecular weight) were prepared by reacting single nucleophilic amino acids with
a lignin model quinone methide (QM). A nucleophile (meaning, “nucleus loving”) is broadly
defined as a chemical group containing a partial negative charge that is relatively free to react
with a complementary group of opposite charge called an electrophile (meaning, “electron
loving”). As described above, some amino acids contain nucleophilic side chains (Fig 1.5), as
well as nucleophilic α-amine and α-acid groups. In order to prevent side reactions, these α-amine
and α-acid groups were chemically blocked, resulting in the side chain groups becoming the sole
nucleophilic species in the amino acids. It was hypothesized that the amino acids would react
with a lignin QM, which is an unstable electrophile that forms during lignin polymerization
according to the mechanism shown in Fig 1.4. The model lignin QM used here (Fig 1.8) was
chosen because it can be prepared cleanly, it is relatively small and simple (chemically
speaking), and it is structurally representative of QMs that form in native guaiacyl-based lignins
(Kawai et al., 1999; Landucci et al., 1981; Ralph and Young, 1983). Cross-coupling reactions
were carried out in dichloromethane to obtain the desired lignin-protein model compounds and to
prevent addition of nucleophilic solvent to the QM. Chapter 2 provides detailed descriptions of
the preparation and characterization of these lignin-amino acid compounds.

13
Fig 1.8. Preparation of a lignin β-ether model compound and its corresponding QM analog. The
amino acids shown in Fig 1.5 were then reacted with the QM to form lignin-protein model
compounds via reaction of the amino acid nucleophilic side chain with the electrophilic α-carbon
of the lignin QM.
In order to explore lignin-protein coupling under more biomimetic conditions, tripeptides
were added to lignin dehydrogenation polymer (DHP) during the lignin polymerization process.
Lignin DHP has been used for decades to approximate the natural lignification process. It is
usually prepared by slowly combining lignin monomer, peroxidase enzymes, and hydrogen
peroxide over the course of hours or days (Fig 1.9). This results in a synthetic lignin that is
chemically similar to native lignin, though DHP typically exhibits increased resinol and
phenylcoumaran structures and a corresponding reduction in β-ether structures compared to
native lignins (Freudenberg, 1968; Terashima et al., 1995). For this study, DHP was prepared
according to previously published methods using coniferyl alcohol as the sole lignin monomer,
dilute hydrogen peroxide as initiator, and horseradish peroxidase as enzymatic catalyst
(Terashima et al., 1995). The pH of the DHP solution was 6.5, which is standard for DHP
preparations and only slightly higher than biologically relevant pH (4.5 - 6.0) (Cosgrove, 2005).

14
Fig 1.9. Preparation of lignin DHP. Over the course of several days, a peristaltic pump combines
coniferyl alcohol and horseradish peroxidase (and in this case, peptides) with dilute hydrogen
peroxide, forming lignin DHP (cream-colored solution in flask on left).
Peptides were added to the lignin polymerization reaction with the general formula of
XGG (Fig 1.10), where X was any of the amino acids shown in Fig 1.5. The C-termini and N-
termini of the peptides were blocked via amidation and esterification, respectively, to ensure that
the amino acid of interest (i.e., residue X) was the only nucleophilic moiety. Glycine was chosen
as the "place holder" residue due to its expected inertness towards lignin. Peptide length was
limited to three residues in order to inhibit the formation of large lignin-peptide complexes that
may have been insoluble and thus difficult to characterize (e.g., liquid state NMR may have
become impractical). Peptides were added in 25% mol/mol ratio to the lignin monomer
(coniferyl alcohol) because it was previously reported that lignin DHPs contain between 20 and
30% β-ether linkages (Tobimatsu, 2012). Thus, the ratio of nucleophilic residues to lignin β-
ether QMs was expected to be approximately 1:1 over the course of the polymerization reaction.
In summary, this experiment was designed to explore the ability of amino acids to outcompete
water and other nucleophiles for addition to the QM under aqueous conditions. Chapter 3
provides detailed descriptions of the preparation and characterization of these lignin-peptide
compounds.
Fig 1.10. General structure of peptides added to lignin DHP preparations. X represents the amino
acid nucleophilic side chain.

15
Lignin DHP was prepared in the presence of gelatin protein under conditions similar to
those described above. Though gelatin is of animal origin, the lignin-gelatin complex was
expected to be informative for several reasons. First, gelatin is both glycine and hydroxyproline-
rich, as are many plant cell wall structural proteins. Second, gelatin has a rather high molecular
weight (20 kDa – 100 kDa depending on gelatin type), and is thus more similar in size to cell
wall structural proteins compared to tripeptides. And third, gelatin was previously shown to
interact with lignin, though the presence or absence of covalent linkages was not definitively
determined (Whitmore, 1978b). Gelatin contains amino acids that could potentially be
nucleophilic towards lignin (see Chapter 4); however, two key amino acids, namely cysteine and
tyrosine, are almost entirely lacking. Chapter 4 provides a detailed description of the preparation
and characterization of these lignin-gelatin complexes.
Finally, in an attempt to identify lignin-protein linkages formed under natural conditions
of lignin biosynthesis, Arabidopsis (wild-type Columbia-0) plants were grown to maturity (8
weeks), then lignin was extracted from the inflorescence stems and characterized. The cell wall
proteome of Arabidopsis has been studied more extensively than most other plant species, with
20 published papers and 500 proteins with predicted signal peptide identified (Albenne et al.,
2013). Inconsistencies surrounding the Arabidopsis cell wall proteome remain, and much more
work is needed. For example, the size of the cell wall proteome for five-day-old cell suspension
cultures has been estimated at anywhere between 33 and 96 proteins (Chivasa et al., 2002; Feiz
et al., 2006; Kwon et al., 2005; Robertson et al., 1997), while one study, which characterized
three-day-old cell suspension cultures, estimated the proteome at 792 (Bayer et al., 2006)! It has
been estimated that structural proteins account for 1.6% of the Arabidopsis cell wall proteome
(Albenne et al., 2013). The quantity of structural protein in mature Arabidopsis cell wall, in
terms of dry weight percentage, is unclear. In order to estimate the protein content of
Arabidopsis and extracted Arabidopsis lignin, nitrogen analysis was performed on various
Arabidopsis extracts. This allowed for protein estimation by multiplying the nitrogen percentage
by a factor of 6.25, assuming that all nitrogen in the sample was due to protein (Chang et al.,
2008; Fukushima and Hatfield, 2001).
Lignin was extracted from Arabidopsis following a previously described acidic dioxane
method (Fukushima and Hatfield, 2004). In short, Arabidopsis inflorescence stem material was
pre-ground in a Wiley mill, extracted (water, ethanol, chloroform, and acetone) in a Soxhlet
apparatus, then ball milled in a cryomill. This cell wall material was then extracted by refluxing
with 90:10 dioxane/2 M HCl, to obtain a crude lignin extract. The crude lignin extract was
“purified” by precipitation in water followed by multiple washings with diethyl ether to yield
~30-35 mg lignin per g of Arabidopsis cell wall material. It has been postulated that this
extraction method selectively cleaves α-ether linkages, which should raise concerns regarding
the cleavage of putative lignin-protein linkages, as well. However, this method was deemed
useful for several reasons. First, it was not possible to extract lignin using the typical milled
wood lignin procedure of refluxing the sample in 96:4 dioxane/water. This method has been

16
employed for decades; however, during preliminary investigations with Arabidopsis, only ~2 mg
of lignin was extracted per 1 g of Arabidopsis cell wall material, which is extremely inefficient
and yields far too little lignin for effective characterization. Furthermore, lignin-protein linkages
are expected to be low in quantity in wild type plants, so observing the putative linkages in
cellulolytic enzyme lignins or whole cell walls seems unlikely due to very low signal to noise.
Chapter 5 provides a detailed description of the extraction and characterization of the
Arabidopsis lignin.
1.3.2. Characterization of lignin-protein compounds
The lignin-protein model compounds and Arabidopsis lignin extracts were characterized
using a variety of complementary methods. Perhaps the single most useful of these, at least in
terms of ability to directly detect lignin-protein covalent linkages, is nuclear magnetic resonance
(NMR) spectroscopy. NMR relies on exploiting the quantum mechanical property of spin. When
atomic nuclei with an odd number of protons and/or neutrons are placed in a magnetic field the
magnetic nuclear spins align with the field. A radio frequency (RF) pulse is then applied to the
sample and the nuclear spins align perpendicular to the magnetic field. The nuclear spins
spontaneously relax, realigning with the magnetic field in a finite amount of time through a
series of complex relaxation phenomena based on their local environment. In doing so, they re-
emit radio frequencies at slightly different wavelengths than the original RF pulse, determined by
the local chemical environment of each nucleus. This leads (following Fourier-transform) to the
generation of the NMR spectrum, expressed in ppm.
As noted above, any atomic nucleus with an odd number of protons and/or neutrons is, in
principle, NMR active, though in reality active isotopes exhibit varying degrees of sensitivity to
the NMR technique, and the natural abundance of the varying isotopes is also of critical
importance. For the study of lignin-protein linkages, the most useful atomic isotopes are proton
(1H), carbon-13 (13C, because the most abundant isotope of carbon, 12C, is not NMR active), and
potentially nitrogen-15 (15N, because 14N gives broad NMR peaks). Lignin and proteins also
contain oxygen; however, the NMR active isotope of oxygen (17O) is extremely low in natural
abundance and is quite insensitive to the NMR technique. Thus, 17O NMR is almost never
employed.
There are many NMR techniques, based on the various active nuclei as well as various
pulse programs. Furthermore, NMR data can be acquired as 1-dimensional (1D), 2-dimensional
(2D), or higher dimensional spectra, and in either the solid or liquid state. For the study of lignin-
protein linkages, 1D and 2D liquid state spectra are likely the most useful, but require solubility,
which is sometimes limited. The simplest NMR experiments (in terms of pulse programs, not
necessarily in terms of spectral interpretation) for the study of lignin are the 1D 1H and 13C
experiments. 1H spectra can be collected within minutes, and provide information on functional
groups within a range of ~0-12 ppm. This technique is very useful for the study of small, simple
molecules. However, for complex molecules such as lignin, the relatively narrow ppm range

17
results in significant chemical shift degeneracy (Fig 1.11). The 13C NMR experiment exhibits a
broad chemical shift range of ~0-200 ppm and is therefore more diagnostic for determining
lignin chemical structure compared to the 1H experiment. However, the low sensitivity of the 13C
experiment due to the low natural abundance and the low magnetogyric ratio of the 13C nucleus,
means that relatively large sample quantities (tens of milligrams) are required. This, coupled
with extremely long 13C T1 relaxation times, result in experimental times of many hours or even
days to collect high resolution, quantitative spectra. Despite these disadvantages, the usefulness
of quantitative 13C NMR in determining lignin structure has been well documented (Capanema et
al., 2004; Capanema et al., 2005; Holtman and Kadla, 2004; Holtman et al., 2006).
Fig 1.11. 500 MHz 1H NMR spectrum of lignin DHP collected in DMSO-d6/pyridine-d5.
In addition to 1D NMR, 2D NMR has proven quite useful for elucidating lignin chemical
structure. For example, the heteronuclear single quantum coherence (HSQC) technique (Fig
1.12) shows peaks that correspond to direct 1H-13C coupling, and it has the advantage of
relatively high sensitivity while at the same time largely eliminating the chemical shift
degeneracy that arises in 1D spectra. The HSQC technique has been used, sometimes in
conjunction with quantitative 13C NMR, to identify novel lignin structures and/or interpolymer
crosslinking, for example in the case of the so-called lignin-carbohydrate complexes that arise
from lignin-polysaccharide coupling (Balakshin et al., 2007; Balakshin et al., 2011; Chen et al.,
2012; Kim and Ralph, 2010; Mansfield et al., 2012). The following chapters show that this
technique is also quite useful for the investigation of lignin-protein coupling. Other 2D NMR
techniques useful for investigating lignin-protein linkages include heteronuclear multiple

18
quantum coherence (HMQC), which shows direct 1H-13C coupling but uses a different pulse
program than HSQC, heteronuclear multiple bond correlation (HMBC), which shows long-range
(typically 2 and 3-bond) through-bond 1H-13C coupling, correlation spectroscopy (COSY) or
total correlation spectroscopy (TOCSY), which show long-range through-bond 1H-1H coupling,
and nuclear Overhauser effect spectroscopy (NOESY), which shows 1H-1H through-space
interactions.
Fig 1.12. 500 MHz 1H-13C HSQC spectrum of lignin DHP collected in DMSO-d6/pyridine-d5.
Each shift is indicative of a unique inter-unit linkage type or other functional group, which
collectively represent the lignin polymer.
Fourier-transform infrared (FT-IR) spectroscopy is another useful lignin characterization
technique. It has the advantages of quick spectral acquisition (seconds to minutes) with just a few
milligrams of sample, especially if an attenuated total reflectance (ATR) accessory is used. In
attenuated total reflectance, the IR beam passes through a crystal (typically germanium, zinc
selenide, silicon, or diamond) and total internal reflection occurs. An evanescent wave, which
penetrates several microns into the sample, is established at the boundary of the crystal. The
sample absorbs some wavelengths of IR radiation stronger than others, resulting in the IR
spectrum. An FT-IR ATR spectrum of lignin DHP is shown in Fig 1.13, and spectral

19
assignments are shown where possible (Faix and Beinhoff, 1988). IR is useful for showing
protein incorporation into lignin because proteins exhibit unique IR signatures. The most
diagnostic of these occur near 1540 and 1658 cm-1, which are attributed to N-H deformation with
C-N stretching, and C=O stretching, respectively (Socrates, 2001). In addition, the overall shape
of the OH/NH region is altered upon protein incorporation, generally becoming sharper, and
sometimes exhibiting an enhanced shoulder at 3200 cm-1, attributed to N-H stretching in amide
functional groups (Socrates, 2001). Unfortunately, unlike NMR, direct detection of lignin-protein
linkages may not be possible with IR. This is because IR shifts diagnostic of lignin-protein
linkages are likely to be of very low intensity and located within the crowded fingerprint region.
Thus, IR may be useful for showing protein incorporation into lignin, but not necessarily capable
of elucidating the mechanism of lignin-protein interaction (i.e., covalent vs. non-covalent).
Fig 1.13. FT-IR ATR spectrum of lignin DHP.
Scanning electron microscopy (SEM) can be used to determine how protein incorporation
affects the physical morphology of lignin. Transmission electron microscopy (TEM) can also be
used, but SEM has the advantage of negligible sample preparation. Furthermore, advantages of
TEM, such as the ability to collect diffraction spectra, are nullified by the amorphous nature of
lignin. An SEM image of lignin DHP is shown in Fig 1.14. SEM has been used in the past to
show that lignin morphology is altered by the presence of cellulose (Micic et al., 2003), and to
investigate native lignin morphology within the cell wall (Terashima et al., 2004; Terashima and
Yoshida, 2006).

20
Fig 1.14. SEM image of lignin DHP. Scale = 1 µm.
Elemental analysis, in various forms, is an important analytical tool for characterizing
lignin. Due to the chemical structures of the monolignol constituents, neat lignin contains only
the elements carbon, oxygen, and hydrogen. These three elements also compose the lignin-
carbohydrate complexes, which often form in planta. However, in addition to these three
elements, proteins also contain nitrogen. Thus, if a lignin contains nitrogen, then protein
incorporation/contamination should be suspected. It is common to perform bulk elemental
analyses on extracted lignins to determine protein content (N% is multiplied by a factor of 6.25
to obtain protein percentage, assuming all nitrogen in the sample is from protein) (Chang et al.,
2008; Fukushima and Hatfield, 2001). In addition to purely bulk elemental analyses, energy
dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) can be used
to obtain elemental data. In EDS, elemental composition is determined by bombarding the
sample with electrons, then analyzing characteristic X-rays emitted from the sample. One of the
advantages of EDS is that it can be collected in the SEM instrument while imaging the sample.
This allows for comparison of sample morphology and elemental composition across the sample
on the micron scale (the EDS spot size can be ~1 mm in diameter with an information depth of
~1-2 µm depending upon e- accelerating voltage). XPS is essentially the reverse process of EDS,
as it determines elemental composition by bombarding the sample with X-rays then observing
ejected electrons with characteristic energy levels. Because electrons have a far shorter mean free
path than X-rays, the information depth of XPS is only about 10 nm. This allows for elemental
analysis of the surface region. Comparison of the EDS and XPS data can then be used to show
variations in elemental composition throughout the samples.
The following chapters will show that the experiments and characterization techniques
described above are useful for investigating lignin-protein linkages, specifically under in vitro
conditions. Future research should address the possibility of lignin-protein linkage formation in
native plant systems.

21
1.4. References
Albenne, C.; Canut, H.; Jamet, E. Frontiers in Plant Science 2013, 4, article 111.
Albersheim, P.; Darvill, A.; Roberts, K.; Sederoff, R.; Staehelin, A. Principles of Cell Wall
Architecture and Assembly, in: Plant Cell Walls. 2010. Garland Science, New York, New York,
pp. 227-272.
Awad, H.M.; Boersma, M.G.; Vervoort, J.; Rietjens, I.M.C.M. Arch. Biochem. Biophys. 2000,
378, 224.
Balakshin, M.; Capanema, E.; Chang, H. Holzforschung 2007, 61, 1.
Balakshin, M.; Capanema, E.; Gracz, H.; Chang, H.; Jameel, H. Planta 2011, 233, 1097.
Bayer, E.M.; Bottrill, A.R.; Walshaw, J.; Vigouroux, M.; Naldrett, M.J.; Thomas, C.L.; et al.
Proteomics 2006, 6, 301-311.
Beat, K.; Templeton, M.D.; Lamb, C.J. Proc. Natl. Acad. Sci. USA 1989, 86, 1529.
Boerjan, W.; Ralph, J.; Baucher, M. Annu. Rev. Plant Biol. 2003, 54, 519.
Bolton, J.L.; Turnipseed, S.B.; Thompson, J.A. Chem.-Biol. Interact. 1997, 107, 185.
Cannon, M. C.; Terneus, K.; Hall, Q.; Tan, L.; Wang, Y.; Wegenhart, B.L.; Chen, L.; Lamport,
D.T.A.; Chen, Y.; Kieliszewski, M.J. PNAS 2008, 105(6), 2226.
Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. J. Agric. Food Chem. 2004, 52, 1850.
Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. J. Agric. Food Chem. 2005, 53, 9639.
Cassab, I.G.; Varner, J.E. Ann. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 321.
Cathala, B.; Saake, B.; Faix, O.; Monties, B. Poly. Deg. and Stab. 1998, 59, 65.
Chang, X.F.; Chandra, R.; Berleth, T.; Beatson, R.P. J. Agric. Food Chem. 2008, 56, 6825.
Chapple, C.; Ladisch, M.; Meilan, R. Nat. Biotechnol. 2007, 25, 746.
Chen, F.; Dixon, R.A. In Vitro Cell. Dev. Biol.: Anim. 2008, 44, S28.
Chen, A.; Zhong, N.; Qu, Z.; Wang, F.; Liu, N.; Xia, G. J. Plant Res. 2007, 120, 337.
Chen, F.; Tobimatsu, Y.; Havkin-Frenkel, D.; Dixon, R.A.; Ralph, J. PNAS USA 2012, 109(5),
1772.
Chivasa, S.; Ndimba, B.K.; Simon, W.J.; Robertson, D.; Yu, X.L.; Knox, J.P., et al.
Electrophoresis 2002, 23, 1754-1765.

22
Cong, F.; Diehl, B.G; Hill, J.L.; Brown, N.R.; Tien, M. Phytochem. 2013, 96, 449-456.
Cosgrove, D.J. Nat. Rev. Mol. Cell Biol. 2005, 6, 850.
Ellis, M.; Egelund, J.; Schultz, C.J.; Bacic, A. Plant Phys. 2010, 153, 403.
Evans, J.J.; Himmelsbach, D.S. J. Agrc. Food Chem. 1991, 39, 830.
Faix, O. J. of Wood Chem. and Tech. 1988, 8(4), 505.
Feiz, L.; Irshad, M.; Pont-Lezica, R.F.; Canut, H.; Jamet, E. Plant Methods 2006, 2, 10.
Freudenberg, K. The constitution and biosynthesis of lignin, in: Freudenberg, K., Neish, A.C.
(Eds) Constitution and Biosynthesis of Lignin. 1968. Springer-Verlag, Berlin, Germany.
Fukushima, R.S.; Hatfield, R.D. J. of Ag. and Food Chem. 2001, 49(7), 3133.
Harrak, H.; Chamberland, H.; Plante, M.; Bellemare, G.; Lafontaine, J.G.; Tabaeizadeh, Z. Plant
Phys. 1999, 121, 557.
Holtman, K.M.; Chang, H.; Jameel, H.; Kadla, J.F. J. of Wood Chem. and Tech. 2006, 26, 21.
Holtman, K.M.; Kadla, J.F. J. Agric. Food Chem. 2004, 52(4), 720.
Jose, M.; Puigdomenech, P. New Phytol. 1993, 125, 259.
Jung, H.G. Agron. J. 1989, 81, 33.
Jung, H.G.; Allen, M.S. J. Anim. Sci. 1995, 73, 2774.
Kaewtatip, K.; Menut, P.; Auvergne, R.; Tanrattanakul, V.; Morel, M.; Guilbert, S. J. of Ag. and
Food Chem. 2010, 58, 4185.
Kawai, S.; Okita, K.; Sugishita, K.; Tanaka, A.; Ohashi, H. J. Wood Sci. 1999, 45, 440.
Kieliszewski, M.; Lamport, D.T.A.; Tan, L.; Cannon, M.C. Annu. Plant Rev. 2011, 41, 321.
Kim, H.; Ralph, J. Org. Biomol. Chem. 2010, 8, 576.
Kwon, H.K.; Yokoyama, R.; Nishitani, K. Plant Cell Physiol. 2005, 46, 843-857.
Lamport, D.T.A. 1974. 30th Symp. Soc. Dev. Biol., pp. 113-130.
Landucci, L.L.; Geddes, S.A.; Kirk, T.K. Holzforschung 1988, 35, 66.
Leary, G.J. Wood Sci. Technol. 1980, 14, 21.
Leary, G.; Miller, I.J.; Thomas, W.; Woolhouse, A.D. J. Chem. Soc., Perkin Trans. 2 1977, 13,
1737.
Li, L.; Popko, J.L.; Umezawa, T.; Chiang, V.L. J. Biol. Chem. 2000, 275, 6537.

23
Li, X.; Weng, J.K.; Chapple, C. Plant J. 2008, 54, 569.
Liyama, K.; Lam, T.B.; and Stone, B.A. Plant Phys. 1994, 104, 315.
Mangeon, A.; Junqueira, R.M.; Sachetto-Martins, G. Plant Signaling and Behavior 2010, 5(2),
99.
Mansfield, S.D.; Kim, H.; Lu, F.; Ralph, J. Nat. Prot. 2012, 7(9), 1579.
McQueen-Mason, S.; Cosgrove, D.J. PNAS USA 1994, 91, 6574.
Micic, M.; Radotic, K.; Jeremic, M.; Leblanc, R.M. Macromol. Biosci. 2003, 3, 100.
Miyagawa, Y.; Takemoto, O.; Takano, T.; Kamitakahara, H.; Nakatsubo, F. Holzforschung 2012,
66, 459.
Polus-Ratajczak, I.; Mazela, B.; and Golinkski, P. Annals of Warsaw Agricultural University,
Forestry and Wood Technology 2003, 53, 296.
Ralph, J.; Brunow, G.; Harris, P.J.; Dixon, R.A.; Schatz, P.F.; Boerjan, W. Chapter 2.
Lignification: are lignins biosynthesized via simple combinatorial chemistry or via proteinaceous
control and template replication. In Recent Advances in Polyphenol Research. 2008. Blackwell
Publishing.
Ralph, J.; Lapierre, C.; Marita, J.M.; Kim, H.; Lu, F.; Hatfield, R.D.; Ralph, S.; Chapple, C.;
Franke, R.; Hemm, M.R.; Van Doorsselaere, J.; Sederoff, R.R.; O’Malley, D.M.; Scott, J.T.;
MacKay, J.J.; Yahiaoui, N.; Boudet, A.; Pean, M.; Pilate, G.; Jouanin, L.; Boerjan, W.
Phytochem. 2001, 57(6), 993.
Ralph, J.; Schatz, P.F.; Lu, F.; Kim, H.; Akiyama, T.; Nelsen, S.F. Quinone Methides in
Lignification, in: Rokita, S.E. (Ed.), Quinone Methides. 2009. John Wiley & Sons, Hoboken,
New Jersey, pp. 385-420.
Ralph, J.; Young, R.A. J. Wood Chem. Technol. 1983, 3(2), 161.
Ralph, S.A.; Ralph, J.; Landucci, L.L. NMR Database of Lignin and Cell Wall Model
Compounds. Available at URL http://ars.usda.gov/Services/docs.htm?docid=10491 (November
2004).
Ramakrishnan, K.; Fisher, J. J. Am. Chem. Soc. 1983, 105, 7187.
Ringli, C.; Keller, B.; Ryser, U. Cell. and Mol. Life Sci. 2001, 58, 1430.
Ryser, U.; Schorderet, M.; Guyot, R.; Keller, B. J. of Cell Sci. 2004, 117, 1179.
Ryser, U.; Schorderet, M.; Zhao, G.; Studer, D.; Ruel, K.; Hauf, G.; Keller, B. The Plant J. 1997,
12(1), 97.

24
Sachetto-Martins, G.; Franco, L.O.; Oliveira, D.E. Biochimica et Biophysica Acta 2000, 1492,
10.
Salmen, L.; Petterson, B. Cellulose Chem. and Tech. 1995, 29, 331.
Socrates, G. Infrared and Raman characteristic group frequencies, third edition. 2001. George
Wiley and Sons, LTD. West Sussex, England.
Stevanic, J.S.; Salmen, L. Cellulose Chem. and Tech. 2006, 40(9-10), 761.
Stevanic, J.S.; Salmen, L. Cellulose 2008, 15, 285.
Stevanic, J.S.; Salmen, L. J. of Pulp and Paper Sci. 2008, 34(2), 107.
Stewart, J.J.; Kadla, J.F.; Mansfield, S.D. Holzforschung 2006, 60, 111.
Talmadge, K.W.; Keegstra, K.; Bauer, W.D.; Albersheim, P. Plant Physiol. 1973, 51, 158-173.
Terashima, N.; Atalla, R.H.; Ralph, S.A.; Landucci, L.L.; Lapierre, C.; Monties, B.
Holzforschung 1995, 49, 521.
Terashima, N.; Awano, T.; Takabe, K.; Yoshida, M. C. R. Biologies 2004, 327, 903.
Terashiam, N.; Yoshida, M. Cell. Chem. and Tech. 2006, 40(9-10), 727.
Tobimatsu, Y.; Elumalai, S.; Grabber, J.H.; Davidson, C.L.; Pan, X.; Ralph, J. ChemSusChem.
2012, 5(4), 676.
Toikka, M.; Jussi, S.; Teleman, A.; Brunow, G. J. Chem. Soc., Perkin Trans. 1 1998, 1, 3813.
Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Plant Phys. 2010, 153, 895.
Whitmore, F.W. Phytochemistry 1978a, 17, 421.
Whitmore, F.W. Plant Science Letters 1978b, 13, 241.
Whitmore, F.W. Phytochemistry 1982, 21(2), 315.
Ye, Z.; Song, Y.; Marcus, A.; Varner, J. E. The Plant J. 1991, 1(2), 175.
Ye, Z.; Varner, J.E. The Plant Cell 1991, 3, 23.
Yuan, T.; Sun, S.; Xu, F.; Sun, R. J. Agric. Food Chem. 2011, 59, 10604.

25
Chapter 2
Towards lignin-protein crosslinking: Amino acid adducts of a lignin model quinone
methide
(Published in Cellulose, available here)
2.1. Abstract
The polyaromatic structure of lignin has long been recognized as a key contributor to the rigidity
of plant vascular tissues. Although lignin structure was once conceptualized as a highly
networked, heterogeneous, high molecular weight polymer, recent studies have suggested a very
different configuration may exist in planta. These findings, coupled with the increasing attention
and interest in efficiently utilizing lignocellulosic materials for green materials and energy
applications, have renewed interest in lignin chemistry. Here we focus on quinone methides—
key intermediates in lignin polymerization—that are quenched via reaction with cell-wall-
available nucleophiles. Reactions with alcohol and uronic acid groups of hemicelluloses, for
example, can lead to lignin-carbohydrate crosslinks. Our work is a first step toward exploring
potential quinone methide (QM) reactions with nucleophilic groups in cell wall proteins. We
conducted a model compound study wherein the lignin model compound guaiacylglycerol-β-
guaiacyl ether 1, was converted to its QM 2, then reacted with amino acids bearing nucleophilic
side-groups. Yields for the QM-amino acid adducts ranged from quantitative in the case of QM-
lysine 3, to zero (no reaction) in the cases of QM-threonine 10 and QM-hydroxyproline 11. The
structures of the QM-amino acid adducts were confirmed via 1D and 2D nuclear magnetic
resonance (NMR) spectroscopy and density functional theory calculations, thereby extending the
lignin NMR database to include amino acid crosslinks. Some of the QM-amino acid adducts
formed both syn- and anti-isomers, whereas others favored only one isomer. Because the QM-
threonine 10 and QM-hydroxyproline 11 compounds could not be experimentally prepared under
conditions described here but could potentially form in vivo, we used density functional theory to
calculate their NMR shifts. Characterization of these model adducts extends the lignin NMR
database to aid in the identification of lignin-protein linkages in more complex in vitro and in
vivo systems, and may allow for the identification of such linkages in planta.
2.2. Introduction
Plant cell walls are composed of a network of interacting polymers, namely cellulose,
hemicelluloses, pectins, lignin, and structural proteins (Cosgrove 2005; McQueen-Mason and
Cosgrove 1994). Of these, lignin is the major aromatic component, derived from monolignols—
phenylpropanoid units whose biosynthesis exhibits incredible plasticity (Boerjan et al. 2003;
Ralph et al. 2004; Vanholme et al. 2010). Lignin’s mode of polymerization is unique among the
cell wall polymers. Resonance stabilized radicals are enzymatically generated from the
monolignols, and as the radical-bearing structures couple combinatorially, a heterogeneous
polymer containing many types of inter-unit linkages forms. The variety of the inter-unit

26
linkages contributes notable recalcitrance to the plant cell wall, stymying not only natural
degradation, but also affecting the economics of many industrial sectors, including the pulp and
paper industry, the developing biofuels industry, agricultural industries, and chemical industries,
which all seek higher value products from lignin (Chapple et al. 2007; Chen and Dixon 2008;
Jung 1989; Jung and Allen 1995; Li et al. 2008; Stewart et al. 2006).
Inter-unit linkages are not, however, the sole factor influencing lignin’s recalcitrance in
planta. Lignin may be crosslinked with other polymers in the plant wall. Hydroxyl and uronic
acid groups of polysaccharides bear mildly nucleophilic groups that can react with a key lignin
intermediate—the α-carbon of quinone methides (QMs) (Balakshin et al. 2011; Leary 1980;
Miyagawa et al. 2012; Ralph et al. 2009; Toikka et al. 1998; Yuan et al. 2011). These QMs form
each time a monolignol radical couples at its β-position and, because β-coupling is prevalent, the
importance of QMs in lignin structure cannot be understated. In certain cases, particularly β-5-
and β-β-coupling, QM intermediates are quickly trapped intramolecularly, producing
phenylcoumaran and resinol units (Leary 1980; Ralph et al. 2009). However, in the case of the
predominant β-O-4-coupling, which produces β-ether linkages, the QM’s α-carbon becomes
susceptible to external nucleophilic attack (Fig 2.1) (Leary 1980; Ralph et al. 2009). This
reactivity of the QM is the focus of the current study.
Fig 2.1. Formation of β-ether QMs via radical coupling, and their rearomatization during lignin
polymerization. L = lignin polymer, Nuc = nucleophile (e.g., H2O, and also here Cys, Lys, His,
Asp, Glu, Tyr or Ser), R = H or OCH3
The crosslinking of lignin with cell wall constituents other than hemicelluloses has not been
well investigated. Cell wall structural proteins, including glycine-rich proteins (GRPs), proline-
rich proteins (PRPs), and hydroxyproline-rich glycoproteins (HRGPs), all contain amino acid
residues with nucleophilic side-chains that could react with lignin QMs (Harrak et al. 1991; Jose
and Puigdomenech 1993; Kieliszewski et al. 2011; Ryser et al. 1997). Cell wall proteins vary in
quantity among species and cell types, ranging from as low as 1-2% to 20% on a dry weight
basis in wild type plants (Albersheim et al. 2010; Cassab and Varner 1988). In 1978 and 1982,
Whitmore showed evidence for the formation of lignin-protein linkages in isolated cell walls of
slash pine. Further literature sources suggest that structural proteins may crosslink with lignin, or
possibly even nucleate, or provide a template for, lignin structure, but these ideas have not been
adequately tested (Albersheim et al. 2010; Beat et al. 1989; Boerjan et al. 2003; Cassab and

27
Varner 1988; Harrak et al. 1991). If true, this mechanism could provide spatial and temporal
control over lignin deposition and architecture (Beat et al. 1989). Furthermore, it has recently
been suggested that over-expression of cell wall proteins could result in increased lignin-protein
linkage formation, which may affect cell wall physical and chemical properties, for example
increased sugar extractability (Liang et al. 2008; Xu et al. 2013). However, identifying such
linkages in planta would be difficult without first determining diagnostic lignin-protein
spectroscopic signatures under simpler, more controlled conditions.
As a first step toward investigating potential lignin-protein linkages in planta, we conducted
a model compound study to characterize products formed when the lignin model compound
guaiacylglycerol-β-guaiacyl ether 1 was converted to its QM 2 (Fig 2.2), then reacted with amino
acids bearing nucleophilic side-groups. Thiols, amines, acids and alcohols have been shown to
quench QMs in a diverse array of systems. The thiol group of glutathione reacts with an o-QM
generated from the flavonoid, quercetin (Awad et al. 2000); the thiol group of cysteine reacts
with the relatively unreactive p-QM, 2,6-di-tert-butyl-4-methylene-2,5-cyclohexadienone
(Bolton et al. 1997); and thiols and thiolates react with QMs derived from anthracyclines
(Ramakrishnan and Fisher 1983). Similarly, amines have been shown to trap lignin QMs (Ralph
and Young 1983). A wide array of acid- and hydroxyl-containing compounds react with p-QMs
(Leary et al. 1977), and primary (and to a much lesser extent, secondary) hydroxyl groups of
carbohydrates may react with QM 2 (Toikka et al. 1988). However, similar nucleophile-QM
adducts have not been characterized in lignin-protein systems.
Fig 2.2. Guaiacylglycerol-β-guaiacyl ether 1 and its derived quinone methide (QM) 2
The nucleophilic amino acids investigated here—cysteine (Cys), lysine (Lys), histidine (His),
aspartic acid (Asp), glutamic acid (Glu), tyrosine (Tyr), serine (Ser), threonine (Thr) and
hydroxyproline (Hyp)—occur in plant cell wall structural proteins and may react to form lignin-
protein crosslinks in vivo (Jose and Puigdomenech 1993; Kieliszewski et al. 2011). Because cell
wall proteins are thought to exist in the wall prior to lignification, the α-amine and α-acid groups
of the amino acids were protected to mimic their inclusion within a peptide. This allowed
reactions of the nucleophilic side-chains to be determined without the complication of competing
reactions from the terminal α-amine and α-acid groups. The QM-amino acid adducts (Fig 2.3)
were characterized by nuclear magnetic resonance (NMR) spectroscopy, density functional

28
theory (DFT), mass spectrometry, and UV/Visible (UV/Vis) spectrophotometry. The
characterization of these model adducts extends the lignin NMR database to aid in the
identification of lignin-protein linkages in more complex in vitro and in vivo systems (Ralph et
al. 2004).
2.3. Experimental
2.3.1 Materials
All chemicals used in the preparation of compounds 1 and 2, and lignin dehydrogenation
polymer (DHP), were purchased from Sigma. All amino acids used in the preparation of
compounds 3-9 were purchased from Sigma with the exception of Boc-L-histidine methyl ester,
which was purchased from Indofine Chemical Company.
2.3.2. Model compound preparations
Compound 1 was prepared according to previous methods, as was its QM analog (2)
(Kawai et al. 1999; Landucci et al. 1981; Ralph and Young 1983). Protected amino acids (1.05
eq) were added directly to the anhydrous solution of 2 in dichloromethane at room temperature.
In the case of Lys, which was obtained as Nα-acetyl-L-lysine methyl ester hydrochloride,
triethylamine (~5 eq) was added in order to deprotonate the terminal amine and facilitate
dissolution. NMR was used to show that triethylamine was not reactive towards the QM. A stir
bar was added, the flask was stoppered, and the atmosphere was rendered inert by alternating
between vacuum and dry nitrogen several times. The reaction was monitored visually;
dissipation of the yellow hue indicated consumption of the QM. Intermittently, the reaction was
also monitored by TLC (1:1 ethyl acetate/hexanes). Lys and His reacted with the QM within
minutes. Other amino acids reacted more slowly with the QM and were allowed to stir overnight
(Cys, Asp, Glu, Tyr) or for several days (Ser, Thr, Hyp), again, with intermittent monitoring by
TLC. When TLC revealed that the reaction had reached equilibrium the mixture was evaporated
to dryness. In the case of Lys, the reaction went to completion (complete consumption of the
QM) and the crude products were evaporated to dryness and submitted to NMR without further
purification. QM reactions with other amino acids did not go to completion. In the case of QM-
Thr and QM-Hyp, TLC and NMR showed no reaction even over the course of several weeks.
The products were purified via flash chromatography using silica gel and 1:1 ethyl
acetate/hexanes as eluent. The purified products were then characterized using nuclear magnetic
resonance (NMR) spectroscopy, mass spectrometry, and density functional theory (DFT). In the
case of QM-His (5), the product could not be chromatographically separated (a range of eluent
solvent systems were attempted) from α-O-aryl products formed presumably due to self-
dimerization of the QM (2); however, mass spec and 2D NMR techniques were still able to
confirm the identity of the QM-His product. In the case of QM-Ser (8), the product could not be
fully separated from unreacted serine. The neat serine shifts as well as the shifts of compound 8
are labeled in the NMR spectra (see below).

29
Lignin guaiacyl-based dehydrogenation polymer (DHP) was synthesized according to a
previously published method (Terashima et al. 1995). The DHP was characterized via HSQC
NMR as described below and was found to contain shifts typical of native lignin and DHP
(Capanema et al. 2004; Kim and Ralph 2010).
2.3.3. Model compound properties
QM-Cys, 3 (2-tert-Butoxycarbonylamino-3-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-
methoxy-phenoxy)-propylsulfanyl]-propionic acid methyl ester). Pale white oil (yield: 73% after
purification). Theoretical mass: 537.20 g/mol (+ H+: 538.21 g/mol). Observed m/z + H+: 538.21.
Major isomer (86%): 1H NMR (400 MHz, acetone-d6): δ = 1.39 (9H, s, H7), 2.82 (2H, m,
H1), 3.53 (1H, m, Hγ), 3.67 (3H, s, H4), 3.74 (1H, m, Hγ), 3.80 (3H, s, OMeB), 3.87 (3H, s,
OMeA), 4.34 (1H, t, J = 5.78, Hα), 4.42 (1H, m, H2), 4.65 (1H, m, Hβ), 6.77 (1H, m, HA5), 6.85
(1H, m, HB6), 6.90 (1H, m, HB4), 6.94 (1H, m, HA6), 6.97 (1H, m, HB5), 7.05 (1H, m, HB3),
7.34 (1H, m, HA2). Minor isomer (14%): 1H NMR (400 MHz, acetone-d6): δ = 1.42 (9H, s,
H7), 2.74 (2H, m, H1), 3.58 (1H, m, Hγ), 3.65 (1H, s, H4), 3.72 (1H, m, Hγ), 3.79 (3H, s,
OMeB), 3.86 (3H, s, OMeA), 4.55 (1H, m, Hβ). Major isomer (86%): 13C NMR (75.5 MHz,
acetone-d6): δ = 28.50 (C7), 33.70 (C1), 51.41 (Cα), 52.40 (C4), 54.13 (C2), 56.11 (OMeA),
56.25 (OMeB), 62.01 (Cγ), 79.54 (C6), 83.87 (Cβ), 113.51 (CA2), 113.78 (CB5), 114.80 (CA5),
117.64 (CB3), 121.61 (CB6), 122.79 (CB4), 123.37 (CA6), 130.87 (CA1), 146.78 (CA4), 148.03
(CA3), 149.07 (CB1), 151.50 (CB2), 155.95 (C5), 172.20 (C3). Minor isomer (14%): 13C
NMR (75.5 MHz, acetone-d6): δ = 113.63 (CA2), 115.41 (CA5), 117.88 (CB3), 121.79 (CB6),
131.00 (CA1), 156.33 (C5). Major isomer (86%): 1H NMR (400 MHz, DMSO-d6/pyridine-
d5): δ = 1.34 (9H, s, H7), 2.66-2.86 (2H, m, H1), 3.43 (1H, m, Hγ), 3.56 (3H, s, H4), 3.58 (1H,
m, Hγ), 3.70 (3H, s, OMeA), 3.78 (3H, s, OMeB), 4.18-4.27 (1H, m, H2), 4.33 (1H, m, Hα), 4.70
(1H, m, Hβ), 5.08 (1H, s, γ-OH), 6.76 (1H, m, HA5), 6.85 (1H, m, HB5), 6.88 (1H, m, HB4),
6.91 (1H, m, HA6), 6.92 (1H, m, HB6), 7.10 (1H, m, HB3), 7.34 (1H, m, HA1), 7.39 (1H, d, J =
8.07, NH), 9.19 (1H, s, A4-OH). 13C NMR (75.5 MHz, DMSO-d6/pyridine-d5): δ = 28.09
(C7), 32.23 (C1), 50.27 (Cα), 51.93 (C4), 53.47 (C2), 55.35 (OMeB), 55.74 (OMeA), 60.68 (Cγ),
81.78 (Cβ), 112.74 (CB6), 113.53 (CA2), 114.60 (CA5), 115.34 (CB3), 120.78 (CB5), 121.44
(CB4), 122.48 (CA6), 129.22 (CA1), 130.90 (C6), 146.05 (CA4), 147.43 (CA3), 148.01 (CB1),
149.87 (CB2), 155.37 (C5), 171.78 (C3). Minor isomer (14%): 1H NMR (400 MHz, DMSO-
d6/pyridine-d5): δ = 4.35 (1H, m, Hα), 4.61 (1H, m, Hβ). 13C NMR (75.5 MHz, DMSO-
d6/pyridine-d5): δ = 28.12 (C7), 32.42 (C1), 50.60 (Cα), 51.97 (C4), 53.69 (C2), 60.96 (Cγ),
81.65 (Cβ), 155.63 (C5), 171.74 (C3).

30

31
QM-Lys, 4 (2-Acetylamino-6-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-methoxy-
phenoxy)-propylamino]hexanoic acid methyl ester). Pale white oil (yield: quantitative, no
purification necessary). Theoretical mass: 504.25 g/mol (+ H+: 505.25 g/mol). Observed m/z +
H+: 505.25. 1H NMR (300 MHz, acetone-d6): δ = 1.4 (4H, m, H2, H3), 1.52 (2H, m, H4), 1.90
(3H, s, H7), 2.41 (2H, m, H1), 3.50 (1H, m, Hγ), 3.63 (3H, s, H9), 3.69 (1H, m, Hγ), 3.79 (3H, s,
OMeA), 3.86 (3H, s, OMeB), 3.97 (1H, d, J = 6.82 Hz, Hα), 4.18 (1H, m, Hβ), 4.38 (1H, m, H5),
6.77 (1H, m, HB6), 6.85 (1H, m, HA6), 6.93 (1H, m, HA5), 6.96 (1H, m, HB5), 6.96 (1H, m,
HB4), 7.09 (1H, m, HA2), 7.14 (1H, m, HB3), 7.39 (1H, d, J = 7.47, acetyl-NH). 13C NMR
(75.5 MHz, acetone-d6): δ = 22.55 (C7), 24.09 (C3), 30.24 (C2), 32.34 (C4), 47.34 (C1), 52.03
(C9), 52.97 (C5), 56.09 (OMeA), 56.09 (OMeb), 62.19 (Cγ), 64.62 (Cα), 87.12 (Cβ), 111.96
(CA2), 113.16 (CB5), 115.35 (CB6), 118.98 (CB3), 121.75 (CA6), 121.75 (CB4), 122.99 (CA5),
133.12 (CA1), 146.70 (CA4), 148.33 (CA3), 149.45 (CB1), 151.63 (CB2), 170.11 (C6), 173.60
(C8). 1H NMR (300 MHz, DMSO-d6/pyridine-d5): δ = 1.21 (4H, m, H2, H3), 1.58 (2H, m,
H4), 1.88 (3H, s, H7), 2.31 (2H, m, H1), 3.45 (1H, m, Hγ), 3.59 (3H, s, H9), 3.67 (1H, m, Hγ),
3.71 (3H, s, OMeA), 3.79 (3H, s, OMeB), 3.90 (1H, d, J = 6.82 Hz, Hα), 4.21 (1H, m, Hβ), 4.27
(1H, m, H5), 5.00 (1H, s, γ-OH), 6.76 (2H, m, HA5, HA6), 6.84 (1H, m, HB5), 6.90 (1H, m,
HB4), 6.95 (1H, m, HB6), 6.98 (1H, m, HA2), 7.15 (1H, m, HB3), 8.30 (1H, d, J = 7.47, acetyl-
NH), 9.12 (1H, s, A4-OH). 13C NMR (75.5 MHz, DMSO-d6/pyridine-d5): δ = 23.24 (C7),
23.26 (C3), 29.21 (C2), 30.94 (C4), 46.61 (C1), 51.62 (C9), 52.04 (C5), 55.41 (OMeA), 55.46
(OMeb), 60.82 (Cγ), 63.00 (Cα), 85.89 (Cβ), 111.66 (CA2), 112.29 (CB6), 115.15 (CA5), 116.99
(CB3), 120.71 (CA6), 120.78 (CB5), 121.57 (CB4), 131.4 (CA1), 145.83 (CA4), 147.58 (CA3),
148.60 (CB1), 149.93 (CB2), 169.56 (C6), 172.96 (C8).

32
QM-His, 5a (2-tert-Butoxycarbonylamino-3-{3-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-
(2-methoxy-phenoxy)-propyl]-3H-imidazol-4-yl}-propionic acid methyl ester) and QM-His, 5b
(2-tert-Butoxycarbonylamino-3-{1-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-methoxy-

33
phenoxy)-propyl]-1H-imidazol-4-yl}-propionic acid methyl ester) (Note: Some of the NMR shift
assignments for this compound were based on interpretation of the 2D HMQC and HMBC NMR
spectra because, as noted above, the QM-His products could not be chromatographically
separated from a lignin QM dimer, leading to some shift degeneracy in the 1H and 13C 1D
spectra.) Pale white oil (yield: 45% by NMR). Theoretical mass: 571.25 g/mol (+ H+: 572.26
g/mol). Observed m/z + H+: 572.26. 1H NMR (400 MHz, acetone-d6): 1.35 (9H, s, H12), 2.97
(2H, m, H6), 3.44 (1H, m, Hγ), 3.54 (3H, s, H9), 3.56 (1H, m, Hγ), 3.63 (6H, s, OMe), 4.37 (1H,
m, H7), 4.96 (1H, m, Hβ), 5.66 (1H, m, Hα), 6.74 (1H, m, HA5), 6.77 (1H, m, HB3), 6.86 (1H,
m, HB4), 6.88 (1H, m, HA6), 6.89 (1H, m, HB5), 6.90 (1H, m, H5), 7.06 (1H, m, HB6), 7.11
(1H, m, HA2), 7.55 (1H, s, H5), 7.81 (1H, s, H2). 13C NMR (75.5 MHz, acetone-d6): 27.80
(C12), 29.40 (C6), 51.00 (C9), 54.06 (C7), 55.60 (OMe), 59.90 (Cγ), 60.97 (Cα), 78.40 (C11),
81.45 (Cβ), 111.40 (CB6), 111.60 (CA2), 112.60 (CB5), 115.00 (CA5), 116.56 (C5), 117.30
(C4), 120.20 (CA6), 120.80 (CB3), 122.40 (CB4), 129.80 (CA1), 135.20 (C5), 138.00 (C2),
147.50 (CA3), 147.60 (CB1), 147.80 (CB4), 150.80 (CB2), 155.40 (C10), 172.60 (C8). 1H NMR
(400 MHz, DMSO-d6/pyridine-d5): 1.33 (9H, m, H12), 2.89 (2H, m, H6), 3.46 (3H, s, OMeA),
3.47 (2H, m, Hγ), 3.47 (3H, s, H9), 3.67 (3H, s, OMeB), 4.37 (1H, m, H7), 5.02 (1H, m, Hβ),
5.70 (1H, m, Hα), 6.76 (1H, m, HA5), 6.76 (1H, m, H5), 6.82 (1H, m, HA6), 6.89 (1H, m, HB5),
7.05 (1H, m, HB3), 7.08 (1H, m, HB6), 7.13 (1H, m, HA2), 7.13 (1H, m, HB4), 7.62 (1H, s,
H2), 7.81 (1H, d, J = 8.42 Hz, acetyl-0NH), 9.23 (1H, s, A4-OH). 13C NMR (75.5 MHz,
DMSO-d6/pyridine-d5): 28.04 (C12), 29.77 (C6), 51.20 (C9), 53.98 (C7), 55.45 (OMeA), 55.63
(OMeβ), 59.31 (Cγ), 60.20 (Cα), 78.29 (C11), 80.20 (Cβ), 111.62 (CB6), 111.81 (CA2), 112.47
(CB5), 114.80 (C5), 114.83 (CA5), 116.60 (CB3), 120.69 (CA6), 130.00 (CA1), 134.98 (C2),
136.44 (C4), 147.20 (CA4), 147.88 (CB1), 148.05 (CA3), 150.66 (CB2), 155.41 (C10), 172.63
(C8).
QM-Asp, 6 (2-tert-Butoxycaronylamino-succinic acid 1-benzyl ester 4-[3-hydroxy-1-(4-
hydroxy-3-methoxy-phenyl)-2-(2-methoxy-phenoxy)-propyl] ester). Pale white oil (yield: 58%
after purification). Theoretical mass: 625.25 g/mol (+ Na+: 648.24 g/mol). Observed m/z + Na+:
648.24. Major isomer (74%): 1H NMR (400 MHz, acetone-d6): δ = 1.37 (9H, s, H6), 2.92

34
(2H, m, H2), 3.6 (1H, m, H3), 3.65 (1H, m, Hγ), 3.75 (1H, m, Hγ), 3.80 (3H, s, OMeB), 3.84
(3H, s, OMeA), 4.61 (1H, m, Hβ), 5.10 (2H, s, H8), 6.06 (1H, d, J = 4.42, Hα), 6.82 (1H, m,
HA5), 7.03 (1H, m, HB3), 6.84 (1H, m, HB5), 6.94 (1H, m, HA6), 6.94 (1H, m, HB4), 6.96 (1H,
m, HB6), 7.17 (1H, m, HA2), 7.34 (5H, m, H10, H11, H12, H13, H14), 7.65 (1H, s, A4-OH). 13C NMR (75.5 MHz, acetone-d6): δ = 28.42 (C6), 37.13 (C2), 51.23 (C3), 56.20 (OMeA),
56.16 (OMeB), 61.18 (Cγ), 67.37 (C8), 75.82 (Cα), 79.57 (C5), 83.60 (Cβ), 112.22 (CA2),
113.48 (CB6), 115.14 (CA5), 119.10 (CB3), 121.65 (CA6), 121.65 (CB5), 128.69 (CB4), 128.72
(CA1), 129.18 (C10-C14), 136.81 (C9), 147.32 (CA3), 147.97 (CA4), 151.75 (CB1), 151.80
(CB2), 156.11 (C4), 169.98 (C1), 171.71 (C7). Minor isomer (26%): 1H NMR (400 MHz,
acetone-d6): δ = 4.51 (1H, m, Hβ), 6.14 (1H, m, Hα). 13C NMR (75.5 MHz, acetone-d6): δ =
76.41 (Cα), 84.62 (Cβ), 119.27 (CB3), 169.81 (C1), 171.67 (C7). Major isomer (74%): 1H
NMR (400 MHz, DMSO-d6/pyridine-d5): δ = 1.33 (9H, s, H6), 2.82 (2H, m, H2), 3.57 (1H, m,
Hγ), 3.66 (1H, m, Hγ), 3.70 (3H, s, OMeB), 3.76 (3H, s, OMeA), 4.55 (1H, m, H3), 4.67 (1H, m,
Hβ), 5.09 (2H, s, H8), 6.04 (1H, m, Hα), 6.81 (1H, m, HA5), 6.83 (1H, m, HB3), 6.86 (1H, m,
HB5), 6.89 (1H, m, HA6), 6.91 (1H, m, HB6), 7.09 (1H, m, HA2), 7.11 (1H, m, HB4), 7.37 (5H,
m, H10, H11, H12, H13, H14), 7.48 (1H, d, J = 3.42, NH), 9.36 (1H, s, A4-OH). 13C NMR (75.5
MHz, DMSO-d6/pyridine-d5): δ = 27.98 (C6), 35.60 (C2), 50.24 (C3), 55.44 (OMeA), 55.54
(OMeB), 59.63 (Cγ), 65.98 (C8), 74.75 (Cα), 78.45 (C5), 81.39 (Cβ), 111.96 (CA2), 112.76
(CB6), 114.84 (CA5), 116.77 (CB4), 120.69 (CA6), 120.69 (CB3), 120.69 (CB5), 127.19 (CA1),
127.53 (C12), 127.66 (C13), 127.78 (C11), 127.95 (C14), 128.29 (C10), 135.98 (C9), 144.32
(C4), 146.65 (CA4), 147.30 (CA3), 147.67 (CB1), 150.12 (CB2), 169.12 (C1), 171.79 (C7).
Minor isomer (26%): 1H NMR (400 MHz, DMSO-d6/pyridine-d5): δ = 4.55 (1H, m, Hβ),
6.11 (1H, m, Hα). 13C NMR (75.5 MHz, DMSO-d6/pyridine-d5): δ = 75.08 (Cα), 82.70 (Cβ),
112.58 (CB6), 116.87 (CB4), 147.27 (CA3), 150.16 (CB2), 168.92 (C1).

35
QM-Glu, 7 (2-tert-Butoxycaronlyamino-pentanedioic acid 1-tert-butyl ester 5-[3-hydroxy-1-(4-
hyroxy-3-methoxy-phenyl)-2-(2-methoxy-phenoxy)-propyl] ester). Pale white oil (yield: 47%
after purification). Theoretical mass: 605.28 g/mol (+ Na+: 628.27 g/mol). Observed m/z + Na+:
628.25. Major isomer (74%): 1H NMR (400 MHz, acetone-d6): δ = 1.42 (9H, s, H10), 1.46
(9H, s, H7), 1.92, 2.08 (2H, m, H3), 2.47 (2H, t, J = 8.02, H2), 3.69, 3.79 (2H, m, Hγ), 3.84 (3H,
s, OMeB), 3.88 (3H, s, OMeA), 4.08 (1H, m, H4), 4.62 (1H, m, Hβ), 5.78 (1H, s, γ-OH), 6.06
(1H, d, J = 5.11, Hα), 6.79 (1H, m, HA5), 6.85 (1H, m, HB5), 6.94 (1H, m, HA6), 6.94 (1H, m,
HB4), 6.96 (1H, m, HB6), 7.02 (1H, m, HB3), 7.17 (1H, m, HA2). 13C NMR (75.5 MHz,
acetone-d6): δ = 27.69 (C3), 28.07 (C10), 28.49 (C7), 31.19 (C2), 54.57 (C4), 56.21 (OMeA),
56.21 (OMeB), 61.27 (Cγ), 75.23 (Cα), 79.17 (C9), 81.46 (C6), 83.89 (Cβ), 112.29 (CA2),
113.55 (CB6), 115.15 (CA5), 119.02 (CB3), 121.68 (CB5), 121.68 (CA6), 123.31 (CB4), 129.32
(CA1), 147.32 (CA4), 147.96 (CA3), 148.97 (CB1), 151.78 (CB2), 156.40 (C5), 171.93 (C1),
174.01 (C8). Minor isomer (26%): 1H NMR (400 MHz, acetone-d6): δ = 4.53 (1H, m, Hβ),
6.13 (1H, m, Hα). 13C NMR (75.5 MHz, acetone-d6): δ = 75.93 (Cα), 84.85 (Cβ), 115.53

36
(CA5), 119.26 (CB3), 129.72 (CA1), 147.53 (CA4), 148.20 (CA3), 172.10 (C1). Major isomer
(74%): 1H NMR (400 MHz, DMSO-d6/pyridine-d5): δ = 1.34 (9H, s, H10), 1.36 (9H, s, H7),
1.83 (2H, m, H3), 2.39 (2H, m, H2), 3.58, 3.66 (2H, m, Hγ), 3.71 (3H, s, OMeB), 3.76 (3H, s,
OMeA), 3.93 (1H, m, H4), 4.67 (1H, m, Hβ), 5.10 (1H, s, γ-OH), 6.02 (1H, m, Hα), 6.81 (1H, m,
HA5), 6.82 (1H, m, HB3), 6.85 (1H, m, HB5), 6.89 (1H, m, HA6), 6.92 (1H, m, HB6), 7.08 (1H,
m, HA1), 7.10 (1H, m, HB4), 7.28 (1H, d, J = 7.86, NH), 9.34 (1H, s, A4-OH). 13C NMR (75.5
MHz, DMSO-d6/pyridine-d5): δ = 26.04 (C3), 27.48 (C10), 28.05 (C7), 30.36 (C2), 53.60
(C4), 55.45 (OMeA), 55.57 (OMeB), 59.71 (Cγ), 74.23 (Cα), 78.09 (C6), 80.37 (C9), 81.58 (Cβ),
111.91 (CA2), 112.77 (CB6), 114.90 (CA5), 116.73 (CB4), 120.02 (CB3), 120.56 (CB5), 120.70
(CA6), 127.53 (CA1), 146.61 (CA4), 147.30 (CA3), 147.72 (CB1), 150.13 (CB2), 155.65 (C5),
171.20 (C8), 171.38 (C1). Minor isomer (26%): 1H NMR (400 MHz, DMSO-d6/pyridine-d5):
δ = 1.97 (2H, m, H3), 4.56 (1H, m, Hβ), 6.08 (1H, m, Hα). 13C NMR (75.5 MHz, DMSO-
d6/pyridine-d5): δ = 26.31 (C3), 30.13 (C2), 53.86 (C4), 74.68 (Cα), 78.00 (C6), 80.24 (C9),
82.64 (Cβ), 111.35 (CA2), 115.21 (CA5), 128.32 (CA1), 146.72 (CA4), 147.47 (CA3), 148.42
(CB1), 149.98 (CB2), 154.74 (C5), 171.65 (C1).

37
QM-Ser, 8 (2-Benzyloxycarbonylamino-3-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-
methoxy-phenoxy)-propoxy]-propionic acid methyl ester). Pale white oil (39% after
purification). Theoretical mass: 555.21 g/mol (+ Na+: 578.20 g/mol). Observed m/z + Na+:
578.20. Major isomer (68%): 1H NMR (400 MHz, acetone-d6): δ = 3.65 (2H, m, H1), 3.70
(3H, s, H4), 3.76 (3H, s, OMeB), 3.81 (3H, s, OMeA), 3.81 (2H, m, Hγ), 4.32 (1H, m, Hβ), 4.43
(1H, m, H2), 4.60 (1H, m, Hα), 5.09 (2H, m, H6), 6.80 (1H, m, HB6), 6.82 (1H, m, HB3), 6.88
(1H, m, HA5), 6.89 (1H, m, HA6), 6.89 (1H, m, HB4), 6.92 (1H, m, HB5), 7.00 (1H, m, HA2),
7.15, (1H, m, H10), 7.38 (5H, m, H8, H9, H11, H12). 13C NMR (75.5 MHz, acetone-d6): δ =
52.35 (C4), 55.59 (C2), 56.20 (OMe), 61.74 (Cγ), 66.87 (C6), 69.92 (C1), 82.25 (Cα), 85.28
(Cβ), 111.70 (C10), 112.00 (CA2), 113.49 (CB5), 115.23 (CB6), 119.27 (CA5), 121.73 (CA6),
122.03 (CB3), 123.09 (CB4), 128.67 (C8, C12), 129.22 (C9, C11), 130.62 (CA1), 138.05 (C7),
147.23 (CA4), 148.17 (CA3), 149.20 (CB1), 151.76 (CB2), 157.06 (C5), 171.87 (C3). Minor
isomer (32%): 1H NMR (400 MHz, acetone-d6): δ = 3.72 (2H, m, Hγ), 3.79 (3H, s, OMeB),
3.83 (3H, s, OMeA), 4.55 (1H, m, Hα). 13C NMR (75.5 MHz, acetone-d6): δ = 55.42 (C2),
61.33 (Cγ) 69.47 (C1), 82.40 (Cα), 85.68 (Cβ), 111.94 (CA2), 119.15 (CA5), 130.19 (CA1),
147.29 (CA4), 148.95 (CB1), 156.95 (C5). Major isomer (68%): 1H NMR (400 MHz, DMSO-
d6/pyridine-d5): δ = 3.39 (1H, m, Hγ), 3.56-3.65 (2H, m, H1), 3.65 (3H, s, H4), 3.67 (1H, m,
Hγ), 3.65 (3H, s, OMeB), 3.72 (3H, s, OMeA), 4.41 (1H, m, H2), 4.46 (1H, m, Hβ), 4.59 (1H, m,
Hα), 5.08 (2H, s, H6), 6.73-6.83 (1H, m, HA5), 6.74-6.80 (1H, m, HA6), 6.80-6.87 (1H, m
HB4), 6.81 (1H, m, HB6), 6.82-6.93 (1H, m, HB5), 6.94-7.07 (1H, m, HA2), 6.97-7.06 (1H, m,
HB3), 7.25-7.38 (5H, m, H7-H12), 7.81 (1H, d, J = 8.42, NH), 9.23 (1H, s, A4-OH). 13C NMR
(75.5 MHz, DMSO-d6/pyridine-d5): δ = 54.20 (C1), 55.29 (OMeA), 55.38 (OMeB), 51.81 (C4)
60.08 (Cγ), 65.80 (C6), 67.90 (C1), 80.91 (Cα), 82.50 (Cβ), 111.48 (CA2), 112.75 (CB5), 114.82
(CA5), 115.06 (CB6), 116.38 (CB4), 120.57 (CA6), 120.59 (CB3), 127.80-128.38 (C7-C12),
128.72 (CA1), 137.05 (C7), 146.36 (CA4), 147.84 (CB1), 149.55 (CB2), 149.93 (CA3), 156.30
(C5), 170.81 (C3). Minor isomer (32%): 1H NMR (400 MHz, DMSO-d6/pyridine-d5): δ =

38
4.42 (1H, m, Hβ), 4.55 (1H, m, Hα). 13C NMR (75.5 MHz, DMSO-d6/pyridine-d5): δ = 80.69
(Cα), 82.90 (Cβ), 111.84 (CA2), 115.90 (CB4), 128.54 (CA1).

39
QM-Tyr, 9 (2-tert-Butoxycarbonlyamino-3-{4-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-
(2-methoxy-phenoxy)-propoxy]-phenyl}-propionic acid methyl ester). Pale white oil (yield: 45%
after purification). Theoretical mass: 597.26 g/mol (+ H+: 598.27 g/mol). Observed m/z + H+:
598.29. 1H NMR (300 MHz, acetone-d6): δ = 1.33 (9H, d, J = 3.28 Hz, H13), 2.88 (1H, m, H7),
3.00 (1H, m, H7), 3.62 (3H, d, J = 3.74, H10), 3.78 (3H, s, OMeB), 3.81 (3H, s, OMeA), 3.81
(1H, m, Hγ), 3.91 (1H, m, Hγ), 4.31 (1H, m, H8), 4.55 (1H, m, Hβ), 5.13 (1H, d, J = 5.13, Hα),
6.07 (1H, d, J = 7.66, γ-OH), 6.78 (1H, m, HA5), 6.81 (1H, m, HB5), 6.87 (2H, m, H3, H5), 6.93
(1H, m, HB4), 6.95 (2H, m, HB3, HB6), 6.97 (1H, m, HA6), 7.06 (2H, m, H2, H6), 7.17 (1H, m,
HA2), 7.55 (1H, s, Ph-OH). 13C NMR (100 MHz, acetone-d6): δ = 28.42 (C13), 37.29 (C7),
52.07 (C10), 56.02 (C8), 56.17 (OMe), 61.19 (Cγ), 79.22 (C12), 79.42 (Cα), 85.40 (Cβ), 112.09
(CA2), 113.50 (CB6), 115.29 (CA5), 116.81 (C3, C5), 119.21 (CB5), 121.43 (CA6), 121.70
(CB3), 123.22 (CB4), 130.32 (CA1), 130.82 (C2, C4, C6), 147.12 (CA4), 148.13 (CA3), 149.10
(CB1), 151.82 (CB2), 156.10 (C11), 157.77 (C1), 173.22 (C9). 1H NMR (400 MHz, DMSO-
d6/pyridine-d5): δ = 1.27 (9H, s, H13), 2.71-2.95 (2H, m, H7), 3.62 (3H, s, H4), 3.67 (3H, s,
OMeB), 3.71 (3H, s, OMeA), 3.73 (1H, m, Hγ), 3.55 (3H, s, H10), 3.79 (1H, m, Hγ), 4.18 (1H, m,
H8), 4.66 (1H, m, Hβ), 5.08 (1H, s, γ-OH), 5.49 (1H, d, J = 4.20, Hα), 6.76 (1H, m, HA5), 6.81
(3H, m, HB3, H3, H5), 6.84 (1H, m, HB5), 6.89 (2H, m, HA6, HB6), 7.05 (2H, m, H2, H6), 7.06
(1H, m, HB4), 7.11 (1H, m, HA2), 9.21 (1H, s, A4-OH). 13C NMR (75.5 MHz, DMSO-
d6/pyridine-d5): δ = 28.10 (C13), 35.50 (C7), 51.60 (C10), 55.47 (OMeB), 55.50 (C8), 55.60
(OMeA), 59.78 (Cγ), 78.26 (C12), 78.30 (Cα), 82.69 (Cβ), 112.02 (CA2), 112.81 (CB6) 115.01
(CA5), 115.79 (C3, C5), 116.43 (CB4), 120.73 (CA6), 120.75 (CB3), 121.57 (CB5), 128.42
(CA1), 129.65 (C2, C6), 129.96 (C4), 146.35 (CA4), 147.37 (CA3), 148.03 (CB1), 150.06
(CB2), 156.30 (C1), 172.56 (C9).

40

41
QM-Thr, 10 (2-tert-Butoxycarbonylamino-3-[3-hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-
methoxy-phenoxy)-propoxy]-butyric acid methyl ester). Not experimentally observed. DFT-
calculated 1H NMR (syn-isomer, DMSO force field): 0.8 (H1), 3.2 (Hβ), 3.3 (Hγ), 3.3 (H3),
3.5 (OMeB), 3.6 (Hγ), 3.6 (H2), 3.6 (OMeA), 4.4 (Hα), 6.1 (HB6), 6.7 (HA5), 6.7 (HB3), 6.7
(HB5), 6.9 (HA2), 6.9 (HA6), 7.0 (HB4). DFT-calculated 13C NMR (syn-isomer, DMSO force
field): 15.2 (C1), 52.1 (OMeA), 52.1 (OMeB), 55.9 (Cγ), 60.7 (C3), 69.2 (C2), 70.7 (Cα), 88.5
(Cβ), 109.0 (CA2), 112.4 (CB3), 113.4 (CA5), 121.5 (CB5), 123.6 (CA6), 124.5 (CB6), 125.7
(CB4), 131.4 (CA1), 144.5 (CA4), 145.8 (CA3), 145.9 (CB1), 151.9 (CB2). DFT-calculated 1H
NMR (anti-isomer, DMSO force field): 0.9 (H1), 3.1 (Hγ), 3.2 (H3), 3.3 (H2), 3.3 (OMeA), 3.4
(Hβ), 3.5 (OMeB), 3.9 (Hγ), 4.3 (Hα), 4.8 (HB6), 6.4 (HB5), 6.6 (HB3), 6.8 (HA5), 6.8 (HA6),
6.8 (HB4), 7.2 (HA2). DFT-calculated 13C NMR (anti-isomer, DMSO force field): 51.9
(OMeB), 52.6 (OMeA), 73.7 (Cα), 60.9 (Cγ), 90.4 (Cβ), 112.1 (CB3), 113.9 (CA5), 114.3 (CA2),
121.2 (CA6), 121.5 (CB5), 122.4 (CB6), 123.8 (CB4), 130.1 (CA1), 144.8 (CA4), 146.1 (CA3),
149.3 (CB2), 149.3 (CB1).
QM-Hyp, 11 (4-[3-Hydroxy-1-(4-hydroxy-3-methoxy-phenyl)-2-(2-methoxy-phenoxy)-
propoxy]-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester 2-methyl ester). Not experimentally
observed. DFT-calculated 1H NMR (syn-isomer, DMSO force field): 3.2 (Hβ), 3.5 (OMeB),
3.6 (OMeA), 4.7 (Hα), 3.4 (Hγ), 3.5 (Hγ), 6.0 (HB6), 6.1 (H2), 6.6 (HA5), 6.7 (HB3), 6.7 (HB5),
6.8 (HA6), 7.0 (HB4), 7.2 (HA2), 7.2 (H4). DFT-calculated 13C NMR (syn-isomer, DMSO
force field): 52.0 (OMeB), 52.2 (OMeA), 56.3 (Cγ), 80.5 (Cα), 87.7 (Cβ), 105.1 (C3), 109.8
(CA2), 112.4 (CB3), 112.5 (C1), 113.0 (CA5), 119.1 (C4), 121.6 (CB5), 122.3 (CA6), 124.5
(CB6), 125.5 (CB4), 132.7 (CA1), 144.1 (CA4), 145.3 (CA3), 147.1 (CB1), 147.6 (C2), 151.2
(CB2). DFT-calculated 1H NMR (anti-isomer, DMSO force field): 3.4 (OMeA), 3.4 (Hγ), 3.5
(OMeB), 3.6 (Hβ), 3.7 (Hγ), 4.0 (H2), 4.6 (Hα), 4.8 (HB6), 6.1 (H4), 6.5 (HB5), 6.6 (HB3), 6.8
(HA5), 6.8 (HA6), 6.9 (HB4), 7.2 (HA2). DFT-calculated 13C NMR (anti-isomer, DMSO

42
force field): 51.9 (OMeB), 52.4 (OMeA), 60.9 (Cγ), 78.7 (Cα), 90.9 (Cβ), 95.7 (C3), 99.8 (C1),
112.2 (CB3), 112.3 (C4), 113.0 (CA2), 113.9 (CA5), 119.5 (CA6), 121.8 (CB5), 122.4 (CB6),
124.1 (CB4), 131.8 (CA1), 144.5 (CA4), 145.9 (CA3), 139.0 (C2), 149.1 (CB1), 149.2 (CB2).
2.3.4. Nuclear magnetic resonance spectroscopy
NMR spectra were collected in both acetone-d6 (spectra shown above) and DMSO-
d6/pyridine-d5 (4:1 v/v, 500 ul). DMSO-d6/pyridine-d5 was chosen because it is a preferred
solvent for NMR of lignin DHP, milled wood lignin (MWL), and whole cell walls; using the
same solvent system allows for accurate shift comparisons (Kim and Ralph 2010). In general,
negligible shift migration was observed between the two solvent systems. NMR spectra were
acquired on Bruker DPX-300 (300 MHz 1H resonance freq.), DRX-400 (400 MHz 1H resonance
freq.), AV-III-500 (500 MHz 1H resonance freq.) with a cryogenically-cooled probe and inverse
probe geometry (i.e. proton coils closest to sample), AV-III-600 (500 MHz 1H resonance freq.)
with a cryogenically-cooled probe, and AV-III-850 (850 MHz 1H resonance freq.) with a
cryogenically-cooled probe. Spectral processing was performed in Bruker's Topspin 3.1
software. Standard Bruker pulse programs were employed: 1H (8-16 scans), 13C (5k-10k scans),
HMQC (Bruker pulse program 'inv4gptp’, 64 scans), and HMBC (Bruker pulse program
'inv4gslplrnd’, 64 scans). Spectra were calibrated to the central solvent peaks (acetone: 2.05/29.8
ppm; dimethyl sulfoxide: 2.50/39.5 ppm). In the case of lignin DHP, NMR spectra were acquired
on a Bruker Biospin (Billerica, MA, USA) AVANCE 500 (500 MHz 1H resonance freq.)
spectrometer fitted with a cryogenically-cooled probe having inverse geometry, i.e., with the
proton coils closest to the sample. Spectra were processed with Bruker’s Topspin 3.1 software,
using the central solvent peak as internal reference (δH/δC: dimethyl sulfoxide (DMSO),
2.50/39.5 ppm). The synthetic lignin DHP (~50 mg) was placed in an NMR tube (ID: 4.1 mm),
swollen homogeneously in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul) with the aid of
ultrasonication (~3h), and then subjected to adiabatic 2D-HSQC (‘hsqcetgpsisp2.2’) experiments
using the parameters described by Mansfield et al. (2012). Processing used typical matched
Gaussian apodization in F2 (LB = -0.3, GB = 0.001), and squared cosine-bell and one level of
linear prediction (32 coefficients) in F1 (Mansfield et al. 2012).
2.3.5. Mass spectrometry
Exact masses for compounds 3-6 (see online resource) were calculated using
ChemBioDraw Ultra 13.0. Mass spectrometric analysis was performed on a Waters LCT Premier
time-of-flight (TOF) mass spectrometer (Waters Corporation (Micromass Ltd.), Manchester,
UK), using MassLynx™ software Version 4.0. Samples were introduced using a Waters 2695
high performance liquid chromatograph. Sample analysis utilized flow injection analysis (FIA).
The mobile phase used was 90% acetonitrile (LC-MS grade) and 10% aqueous ammonium
acetate (10mM). The flow rate was 0.25 mL/min. The nitrogen drying gas temperature was set to
300 °C at a flow of 7 L/min. The capillary voltage was 2200 V. The mass spectrometer was set
to scan from 100-1000 m/z in positive ion mode, using electrospray ionization (ESI).

43
2.3.6. Computational methods
Eight conformational isomers of QM-Cys, QM-Thr, and QM-Hyp, and sixteen
conformational isomers of QM-His were built using Materials Studio 6.0 (Accelrys Inc., San
Diego, CA). Eight of the QM-His models exhibited a CαQM-N1His bond and eight models
exhibited CαQM-N3His bond; these models allowed us to determine which CαQM-NHis bond was
occurring and to determine if an observed chemical shift (α13C) at 78.8 ppm was due to a C-N
bond. Each set of eight models (i.e., compound 3 (QM-Cys), compound 5a (QM-His(N3)),
compound 5b (QM-His(N1)), compound 10 (QM-Thr), or compound 11 (QM-Hyp)) contained
two of each of the stereoisomers (R,R), (SS), (R,S), and (S,R), where the former two
stereoisomers are syn and the latter two stereoisomers are anti. These models were built to
determine if the calculated NMR chemical shifts could differentiate the observed shifts for the
syn and anti stereoisomers of QM-His and QM-Cys. Experimental NMR shifts for QM-Thr and
QM-Hyp were not obtained because Thr and Hyp did not react with the QM; however, we
reported the calculated shifts for these compounds (below) as potential references for other
researchers to use.
Each model was energy minimized without symmetry or atomic constraints using the
density functional theory (DFT) method M05-2X, coupled with the 6-311++G(2df,2p) basis set
using the program Gaussian 09 (Curtiss et al. 2001; Frisch et al. 2009; Hohenberg and Kohn
1964; Kohn and Sham 1965; Zhao et al. 2006). Following the geometry optimization
calculations, frequency calculations assured that each model attained a potential energy surface
(PES) minimum, where no imaginary frequencies were present (Frisch et al. 2009).
Subsequent gauge-independent atomic orbital (GIAO) calculations using Gaussian 09 at
the mPW1PW91/6-31G(d) theory level provided the NMR magnetic shielding tensors (α13C and
α1H) for the energy-minimized structures (Adamo and Barone 1998; Buhl et al. 1999;
Cheeseman et al. 1996; Karadakov 2008; Lodewyk et al. 2012; Schreckenbach and Ziegler 1995;
Wolinski et al. 1990). Because our experiments were conducted in dimethylsulfoxide (DMSO),
the GIAO calculations were also performed in a dielectric continuum of DMSO using a self-
consistent reaction field (SCRF) and the integral equation formalism variant of the polarized
continuum model (IEFPCM) (Cances et al. 1997; Gogonea 1998). Note that the structures were
not energy minimized within the polarized continuum because prior work showed that doing so
did not improve the precision of the calculations (Watts et al. 2011). A multi-standard NMR
method using benzene for sp2-hybridized C- and H-atoms, and methanol for sp3-hybridized C-
and H-atoms led to the α13C and α1H results (Sarotti and Pellegrinet 2009; Sarotti and Pellegrinet
2012; Watts et al. 2011). Benzene and methanol were energy minimized using M05-2X/6-
311++G(2df,2p) and underwent subsequent GIAO calculations using mPW1PW91/6-31G(d).
The precision of the multi-standard method versus the single-standard method (e.g.,
tetramethylsilane as the standard) is illustrated when comparing single-standard results recently
reported by Mostaghni et al. (2013) with the multi-standard results of Watts et al. (2011). Both

44
groups reported the δ13C for β-O-4 linkages in lignin model compounds; however, the mean
unsigned errors, root-mean-squared errors, and maximum errors reported by Mostaghni et al.
(2013) were approximately 10, 12, and 23 ppm, while those reported by Watts et al. (2011) were
approximately 2, 3, and 8 ppm. Therefore, the multi-standard method produced results that were
more precise than those produced by the single-standard method for lignin model compounds
with β-O-4 linkages.
For each C- or H-nucleus, we used δxcalc= σref - σcalc + δref to calculate the chemical shift
of each H- and C-nucleus of interest (δxcalc) in the GG-amino acid models (Sarotti and Pellegrinet
2009; Sarotti and Pellegrinet 2012). Here, σref is the calculated tensor of the C- or H- nucleus of
the standard (i.e., methanol or benzene), σcalc is the calculated tensor of the nucleus of interest
from the GG-amino acid model, and δref is the experimental chemical shift of the C- and H-
nuclei in benzene or methanol dissolved in DMSO (Gottlieb et al. 1997). The chemical shifts for
each C- and H-nucleus was thermodynamically weighted using the relative, calculated Gibbs
free energy of each model to account for the thermodynamic abundance of each model (Barone
et al. 2002). The calculated δ13C and δ1H results were then correlated with their respective NMR
data.
2.4. Results and discussion
2.4.1. Preparation of quinone methide-amino acid adducts
A lignin β-ether QM 2 was prepared cleanly from guaiacylglycerol-β-guaiacyl ether 1, as
previously described (Kawai et al. 1999; Landucci et al. 1981; Ralph and Young 1983). One of
nine amino acids bearing a nucleophilic side-group was then added to the QM, with each
reaction monitored by thin layer chromatography. It was observed that amino acids with amine-
containing side-chains (Lys and His) reacted with the QM quickly (within minutes), whereas
thiol-, acid-, and hydroxyl-containing amino acids reacted slowly (over hours or days). In the
case of the secondary hydroxyl-containing amino acids (Thr and Hyp) no cross-coupling was
observed (i.e., compounds 10 and 11 did not form), despite attempts to catalyze the cross-
coupling reaction (refer to the electronic supplement for detailed reaction protocols). Products
were purified via column chromatography and yields ranged from quantitative in the case of
compound 3 (QM-Lys) to zero (no reaction) in the cases of compounds 10 and 11 (QM-Thr and
QM-Hyp). Cross-coupling reactions were carried out in dichloromethane to produce the desired
lignin-protein adducts.

45

46
Fig 2.3. QM-AA model compounds. Lignin-cysteine (QM-Cys) 3, lignin-lysine (QM-Lys) 4,
lignin-histidine (QM-His) 5, lignin-aspartic acid (QM-Asp) 6, lignin-glutamic acid (QM-Glu) 7,
lignin-serine (QM-Ser) 8, lignin-Tyrosine (QM-Tyr) 9, lignin-threonine (QM-Thr) 10, and
lignin-hydroxyproline (QM-Hyp) 11 adducts derived from QM 2
2.4.2. Solution-state NMR of compounds 3-9 and density functional theory calculations for
compounds 10 and 11
Reaction products were characterized using solution-state 1D 1H and 13C NMR, as well as
2D heteronuclear multiple quantum coherence (HMQC) and heteronuclear multiple-bond
correlation (HMBC) experiments. Full spectral assignments for compounds 3-9 are given in the
methods sections (sections 2.3.3 and 2.3.4). Interpretation of these results is consistent with
structures 3-9 (Fig 2.3), indicating that Cys, Lys, His, Asp, Glu, Ser and Tyr all add to QM 2 in
vitro. Density functional theory (DFT) was used to predict NMR shifts for compounds 10 (QM-
Thr) and 11 (QM-Hyp), which did not form under the synthetic conditions employed here.
Table 2.1 shows the lignin α and β 1H and 13C shifts for compounds 3-11. The γ-shifts of
these compounds are almost entirely degenerate and are therefore considered non-diagnostic.
Because threonine and hydroxyproline are abundant in cell wall structural proteins (especially
hyp, which can account for up to 33% of the amino acid profiles of some structural proteins), the
authors perceived that estimations of the QM-Thr and QM-Hyp NMR chemical shifts could still
be useful. Thus, NMR shifts for compounds 10 and 11 were calculated using DFT. As a control,
DFT was also used to calculate NMR shifts for compounds 3 and 5 (Fig 2.4), showing
comparison to experimental results. Calculated 13C shifts were generally in agreement with
experimentally observed shifts. For example, calculated 13C α-shifts overestimated the observed
shifts by only 0.8-3.1 ppm. Calculated 13C β-shifts overestimated the observed shifts by 5.4-9.8
ppm. Similar discrepancies in DFT calculated β-shifts of β-ether compounds have been
previously reported, and further work is necessary to refine these calculations (Watts et al. 2011).
Calculated 1H shifts consistently underestimated the experimentally observed shifts by about 0.5-
1 ppm. Thus, the calculated 1H shifts for compounds 10 and 11 are not reproducing the observed 1H shifts; however it could be possible with future work to develop a method to correlate the
calculated and experimental 1H shifts, because of the consistent underestimation of the
experimental 1H shifts by the calculated shifts. Lodewyk et al. (2012) described a method for
using empirical scaling factors to obtain improved correlation between experimental and
calculated 1H and 13C shifts; however, doing so is beyond the scope of the present work. In
addition to the use of scaling factors, further research to develop multi-standard methods that are
based on DFT results is necessary. This work could require the development and assessment of
DFT methods, as well as basis sets to obtain methods to calculate 1H shifts more precisely.

47
Table 2.1. 1H and 13C NMR chemical shifts for lignin-amino acid adducts.
α-shifts β-shifts
Experimental Calculated Experimental Calculated
Compound 1H/13C 1H/13C 1H/13C 1H/13C
3 (QM-Cys)a 4.3/50.3
4.4/50.6
3.8/53.4
3.5/52.9
4.7/81.8
4.6/81.7
3.7/89.6
4.0/87.1
4 (QM-Lys) 3.9/63.0 4.2/85.9
5 (QM-His)b 5.7/60.2 5.0/61.0 5.0/80.2 3.9/90.0
6 (QM-Asp)a 6.0/74.8
6.1/75.1
4.7/81.4
4.6/82.7
7 (QM-Glu)a 6.0/74.2
6.1/74.7
4.7/81.6
4.6/82.6
8 (QM-Ser)a 4.6/80.9
4.6/80.7
4.4/82.5
4.4/82.9
9 (QM-Tyr) 5.5/78.3 4.7/82.7
10 (QM-Thr)c n/a 4.4/70.7
4.3/73.7 n/a
3.2/88.5
3.4/90.4
11 (QM-Hyp)c n/a 4.7/80.5
4.6/78.7 n/a
3.2/87.7
3.6/90.9
Key: a, products exhibited two stereoisomers, shifts for the major isomer are shown first; b, only
the calculated shifts of anti-5b are shown, see the electronic supplement for calculated shifts of
additional isomers of 5; c, syn-isomer shifts are shown first.

48
Fig 2.4. Overlaid HMQC side chain regions of compounds 3 and 5. The α- and β-shifts are
labeled; methoxyl and γ-shifts are not labeled due to substantial shift degeneracy. Grey shifts are
non-diagnostic. DFT calculated α-shifts (red squares) and β-shifts (blue squares) are shown for
compounds 3, 5a and 5b (both threo and erythro stereoisomers are shown). Calculated 13C shifts
correlate relatively well with experimentally observed 13C shifts, though not well enough to
allow for assignment of stereochemistry in the experimentally observed product shifts.
Calculated 1H shifts are underestimated by about 0.5-1.0 ppm. Further research is necessary to
refine the predicative abilities of DFT for 1H shifts of lignin compounds.
Fig 2.5 highlights the location of diagnostic HMQC NMR peak contours of the lignin-amino
acid adducts overlaid on the spectrum of a synthetic lignin (a so-called dehydrogenation
polymer, or DHP). Differences in chemical shifts among the lignin-amino acid adducts are most
salient for the α-positions and, as expected, less for those from the β-positions. Most of the
lignin-amino acid shifts are readily distinguishable from correlations of native structures in
lignin; however, the α-shifts of compound 4 (QM-Lys) are degenerate with phenylcoumaran γ-
shifts. In this case, identifying a lignin-lysine crosslink may be possible by observing the lignin-
lysine β-shifts. The α- and β-shifts of compound 9 (QM-Tyr) are degenerate with benzyl aryl
ether linkages (so called α-O-aryl linkages) sometimes observed in synthetic and native lignin
polymers. These lignin-lignin linkages form when QMs are quenched by phenolic moieties, and
degeneracy is not surprising given the structural similarities among tyrosine and the lignin
monomers, p-coumaryl, coniferyl, and sinapyl alcohols. This may make it difficult to distinguish
lignin-tyrosine crosslinking from lignin-lignin α-O-aryl linkages in native lignins.
Though not depicted graphically, the lignin-peptide linkages described herein are largely free
from overlap with previously described polysaccharide shifts in both angiosperms and

49
gymnosperms. However, a few of the lignin-amino acid shifts may overlap with signatures
attributed to lignin-carbohydrate linkages. For example, the α-shifts of compounds 6 and 7
exhibit degeneracy with lignin-carbohydrate benzyl esters (α-shifts at 6.1/75.0 ppm) due to
structural similarity (Balakshin et al. 2011; Toikka et al. 1998). Likewise, the α-shifts of 8 and 11
exhibit degeneracy with lignin-carbohydrate benzyl ethers (α-shifts located at 4.6/80.5 ppm)
(Balakshin et al. 2011; Toikka et al. 1998). Thus, caution should be exercised when attempting to
discern certain lignin-protein and lignin-carbohydrate linkages using 1D and 2D NMR
techniques. The results of the current study indicate that NMR identification of lignin-protein
linkages, especially linkages of the benzyl thioether and benzyl amine types, should be possible
in whole cell walls or lignin extracts provided the linkages are adequately abundant (Kim and
Ralph 2010; Mansfield et al. 2012).
Fig 2.5. HSQC NMR spectrum of a lignin DHP with overlaid α- and β-correlation data from
compounds 3-11 represented by red (α) and blue squares (β)

50
2.4.3. Adduct isomer determination
Of purely fundamental interest, we attempted to resolve the stereochemistry of the products
by the use of DFT, but these efforts were largely unsuccessful. For example, in the case of QM-
Cys, 3, the root mean-squared error (RMSE) between experimental and calculated shifts was too
large to reliably assign the isomers (Table 2.2). Although it may have been possible to improve
the DFT results through the addition of conformational isomers, the added computational cost
may not have reduced the calculated RMSE to experimental uncertainty levels. Hence additional
attempts to resolve stereoisomers (compounds 6, 7, 8) were abandoned; likewise, DFT was not
used to identify which stereoisomer was produced in 4 and 9 (only one product was observed in
each case). Previously, addition of primary amines were shown (via diagnostic NMR of
tetrahydro-1,3-oxazine derivatives) to strongly (>90%) favor formation of the syn-isomer (Ralph
and Young 1983), so product 4 is likely syn.
Table 2.2. Observed and DFT calculated α-13C NMR chemical shifts for compound 3.
α-13C chemical shifts (ppm)
Observed Calculated RMSE
50.27 52.90 - (3, syn) 2.7
50.60 53.40 - (3, anti) 2.8
In the case of 5 (QM-His), one α-shift was observed, occurring at 5.7/60.2 ppm. The His
system is an interesting one to consider given the tautomerization in the His imidazole group and
the potential for various regio-isomeric products (Nagy et al. 2005). In the HMQC and HMBC
spectra (see methods section) the α-1H shows correlations to positions 2 and 5 of the imidazole
ring, though α-1H correlations to position 5 are weak and partially degenerate with correlations to
carbon A6. The NMR results suggest the formation of both compounds 5a and 5b, resulting from
either N1 or N3 addition, but quantification of these compounds via NMR was rendered
impossible due to the aforementioned shift degeneracy. The Gibbs free energy-based Boltzmann
factors in the gas-phase suggested that compound 5b is thermodynamically prevalent relative to
compound 5a (93.5% to 6.5%, respectively), and prior work by Watts et al. (2011) suggested that
models with greater thermodynamic abundance generally provided α-13C results that were better
correlated with experimental NMR data.
2.5. Conclusions
This study is the first to report on the synthesis of lignin-protein model compounds and
contributes to the growing lignin NMR database. QM-amino acid adducts were synthesized and
characterized. Namely, Cys, Lys, His, Asp, Glu, Ser, Tyr, Thr, and Hyp were reacted with a
lignin model quinone methide—an important intermediate in lignification. The selected quinone
methide 2 represents the structure and reactivity of QMs native to lignin. The amino acids were

51
selected because of their nucleophilic side-groups; furthermore, these amino acids are common
in plant cell wall structural proteins and represent functional groups (amines, thiols, acids, and
alcohols) that are known to react with quinone methides (Awad et al. 2000; Bolton et al. 1997;
Ramakrishnan and Fisher 1983). The selected amino acids quenched the QM with varying
efficiencies (in general, amine > thiol > acid > hydroxyl) under neutral organic solvent
conditions. The secondary alcohols (Thr, Hyp) did not react under the selected conditions.
Using the results from these model compounds to identify any lignin-protein crosslinks in
planta is our goal. Based on the results herein, lignin-protein NMR shifts should be well
dispersed and, in most cases, distinct even within the complex NMR spectra of polymerized
lignin (Fig 2.5). This suggests that the linkages may be detectable in planta if they exist in
significant quantities.
Although density functional theory was used to predict NMR chemical shifts of lignin-
protein crosslinks, the calculated chemical shifts did not display the level of accuracy required to
distinguish stereoisomers. Future studies are needed to improve the correlation between these
DFT calculations and experimentally observed shifts.
2.6. Acknowledgements
This research was supported as part of The Center for Lignocellulose Structure and
Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy,
Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001090, and
the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-
07ER64494). The authors would like to thank and acknowledge the Center for Lignocellulose
Structure and Formation (CLSF) and the members thereof. Student fellowships were provided by
the USDA National Needs Program and the National Science Foundation. The authors would
like to thank Dr. Alan Benesi and Dr. Wenbin Luo for assistance in acquiring NMR spectra of
the lignin model compounds, Dr. James Miller for acquiring mass spec data, and Dr. Josh
Stapleton for providing assistance with UV/Vis. The primary author would also like to
acknowledge Paul Munson and Curtis Frantz for valuable discussion, and valuable interactions
with Dan Gall and other members of the Wisconsin lab.
2.7. References
Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675.
Albersheim, P.; Darvill, A.; Roberts, K.; Sederoff, R.; Staehelin, A. 2010. Principles of Cell Wall
Architecture and Assembly, in: Plant Cell Walls. Garland Science, New York, New York, pp.
227-272.
Awad, H. M.; Boersma, M. G.; Vervoort, J.; Rietjens, I. M. C. M. Arch. Biochem. Biophys. 2000,
378, 224-233.

52
Barone, G.; Duca, D.; Silvestri, A.; Gomez-Paloma, L.; Riccio, R.; Bifulco, G. Chem.--Eur. J.
2002, 8(14), 3240–3245.
Beat, K.; Templeton, M. D.; Lamb, C. J. Proc. Natl. Acad. Sci. USA 1989, 86, 1529-1533.
Balakshin, M.; Capanema, E.; Gracz, H.; Chang, H.; Jameel, H. Planta 2011, 233, 1097-1110.
Boerjan, W.; Ralph, J.; Baucher, M. Annu. Rev. Plant Biol. 2003, 54, 519-546.
Bolton, J. L.; Turnipseed, S. B.; Thompson, J. A. Chem.-Biol. Interact. 1997, 107, 185-200.
Buhl, M.; Kaupp, M.; Malkina, O.L.; Malkin, V.G. J. Comput. Chem. 1999, 20, 91–105.
Cances, E.; Mennucci, B.; Tomasi, J. J. Chem. Phys. 1997, 107(8), 3032–3041.
Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. J. Agric. Food Chem. 2004, 52, 1850-1860.
Cassab, I. G.; Varner, J. E. Ann. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 321-353.
Chapple, C.; Ladisch, M.; Meilan, R. Nat. Biotechnol. 2007, 25, 746-748.
Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. J. Chem. Phys. 1996, 104, 5497–5509.
Chen, F.; Dixon, R. A. In Vitro Cell. Dev. Biol.: Anim. 2008, 44, S28-S29.
Cosgrove, D. J. Nat. Rev. Mol. Cell Biol. 2005, 6, 850-861.
Curtiss, L.A.; Redfern, P.C.; Raghavachari, K.; Pople, J.A. J. Chem. Phys. 2001, 114(1), 108–
117.
Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.;
Montgomery, Jr., J.A. 2009. Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford,
Connecticut.
Gogonea, V. 1998. Self-Consistent Reaction Field Methods: Cavities, in: Schleyer, P.v.R.,
Schreiner, P.R., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P., Schaefer III, H.F. (Eds.),
Encyclopedia of Computational Chemistry. Wiley, New York, New York, pp. 2560–2574.
Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62(21), 7512–7515.
Harrak, H.; Chamberland, H.; Plante, M.; Bellemare, G.; Lafontaine, J. G.; Tabaeizadeh, Z. Plant
Phys. 1991, 121, 557-564.
Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136(3B), B864-B871.
Jose, M.; Puigdomenech, P. New Phytol. 1993, 125, 259-282.

53
Jung, H. G.; Allen, M. S. J. Anim. Sci. 1995, 73, 2774-2790.
Jung, H. G. Agron. J. 1989, 81, 33-38.
Karadakov, P.B. 2008. Ab Initio Calculation of NMR Shielding Constants, in: Webb, G.A. (Ed.),
Modern Magnetic Resonance. Springer, New York, New York, pp. 63–70.
Kawai, S.; Okita, K.; Sugishita, K.; Tanaka, A.; Ohashi, H. J. Wood Sci. 1999, 45, 440-443.
Kieliszewski, M.; Lamport, D. T. A.; Tan, L.; Cannon, M. C. Annu. Plant Rev. 2011, 41, 321-
342.
Kim, H.; Ralph, J. Org. Biomol. Chem. 2010, 8, 576-591.
Kohn, W.; Sham, L.J. Phys. Rev. 1965, 140(4A), A1133–A1138.
Landucci, L. L.; Geddes, S. A.; Kirk, T. K. Holzforschung 1981, 35, 66-69.
Leary, G. J. Wood Sci. Technol. 1980, 14, 21-34.
Leary, G.; Miller, I. J.; Thomas, W.; Woolhouse, A. D. J. Chem. Soc., Perkin Trans. 2 1977, 13,
1737-1739.
Liang, H.; Frost, C.J.; Wei, X.; Brown, N.R.; Carlson, J.E.; Tien, M. Clean 2008, 36(8), 662-
668.
Li, X.; Weng, J. K.; Chapple, C. Plant J. 2008, 54, 569-581.
Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Chem. Rev. 2012, 112(3), 1839–62.
Mansfield, S. D.; Kim, H.; Lu, F.; Ralph, J. Nat. Protoc. 2012, 7(9), 1579-1589.
McQueen-Mason, S.; Cosgrove, D. J. Proc. Natl. Acad. Sci. USA 1994, 91, 6574-6578.
Miyagawa, Y.; Takemoto, O.; Takano, T.; Kamitakahara, H.; Nakatsubo, F. Holzforschung 2012,
66, 459-465.
Mostaghni, F.; Abbas T.; Seyed, A.M. Spectrochimica Acta Part A: Molecular and Biomolecular
Spectroscopy. 2013, 110, 430-436.
Nagy, P. I.; Tejada, F. R.; Messer, W. S. Jr. J. Phys. Chem. 2005, 109, 22588-22602.
Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P. F.; Marita, J. M.; Hatfield, R.
D.; Ralph, S. A.; Christensen, J. H. Phytochem. Rev. 2004, 3, 29-60.
Ralph, J.; Schatz, P. F.; Lu, F.; Kim, H.; Akiyama, T.; Nelsen, S. F. 2009. Quinone Methides in
Lignification, in: Rokita, S. E. (Ed.), Quinone Methides. John Wiley & Sons, Hoboken, New
Jersey, pp. 385-420.

54
Ralph, J.; Young, R. A. J. Wood Chem. Technol. 1983, 3(2), 161-181.
Ralph, S.A., Ralph, J., Landucci, L.L. NMR Database of Lignin and Cell Wall Model
Compounds. Available at URL http://ars.usda.gov/Services/docs.htm?docid=10491 (November
2004).
Ramakrishnan, K.; Fisher, J. J. Am. Chem. Soc. 1983, 105, 7187-7188.
Ryser, U.; Schorderet, M.; Zhao, G.; Studer, D.; Ruel, K.; Hauf, G.; Keller, B. The Plant J. 1997,
12(1), 97-111.
Sarotti, A.M.; Pellegrinet, S.C. J. Org. Chem. 2009, 74(19), 7254–7260.
Sarotti, A.M.; Pellegrinet, S.C. J. Org. Chem. 2012, 77(14), 6059–65.
Schreckenbach, G.; Ziegler, T. J. Chem. Phys. 1995, 99(2), 606–611.
Stewart, J. J.; Kadla, J. F.; Mansfield, S. D. Holzforschung 2006, 60, 111-122.
Terashima, N.; Atalla, R.H.; Ralph, S.A.; Landucci, L.L.; Lapierre, C.; Monties, B.
Holzforschung 1995, 49, 521-527.
Toikka, M.; Jussi, S.; Teleman, A.; Brunow, G. J. Chem. Soc., Perkin Trans. 1. 1998, 1, 3813-
3818.
Vanholme, R.; Morreel, K.; Ralph, J.; Boerjan, W. Plant Phys. 2010, 153, 895-905.
Watts, H. D.; Mohamed, M. N. A.; Kubicki, J. D. J. Phys. Chem. B. 2011, 115(9), 1958–1970.
Wolinski, K.; Hinton, J.F.; Pulay, P. J. Am. Chem. Soc. 1990, 112(23), 8251–8260.
Xu, Y.; Chen, C.; Thomas, T.P.; Azadi, P.; Diehl, B.; Tsai, C.; Brown, N.; Carlson, J.E.; Tien,
M.; Liang, H. Plant Cell Rep. 2013, 32, 1827-1841.
Yuan, T.; Sun, S.; Xu, F.; Sun, R. J. Agric. Food Chem. 2011, 59, 10604-10614.
Zhao, Y.; Schultz, N.E.; Truhlar, D.G. J. Chem. Theory Comput. 2006, 2(2), 364–382.

55
Chapter 3
Lignin crosslinks with peptides under biomimetic conditions
(Target journal for publication is Biomacromolecules)
3.1. Abstract
The work presented here investigates the crosslinking of various nucleophilic amino acids with
lignin under aqueous conditions, thus providing insight as to which amino acids might crosslink
with lignin in planta. Lignin dehydrogenation polymer (DHP) was prepared in aqueous solutions
that contained peptides with the general structure XGG, where X represents an amino acid with a
nucleophilic side chain. Fourier-transform infrared spectroscopy and energy dispersive X-ray
spectroscopy showed that peptides containing cysteine and tyrosine were incorporated into the
DHP, while peptides containing other nucleophilic amino acids were not. Scanning electron
microscopy showed that the physical morphology of the DHP was altered by the presence of
peptides, regardless of peptide incorporation. Nuclear magnetic resonance (NMR) spectroscopy
showed that cysteine-containing peptide crosslinked with lignin at the lignin α-position, whereas
in the case of the lignin-tyrosine adduct the exact crosslinking mechanism could not be
determined. This is the first study to use NMR to confirm crosslinking between lignin and
peptides under biomimetic conditions. The results of this study may indicate the potential for
lignin-protein linkage formation in planta, particularly between lignin and cysteine and/or
tyrosine-rich proteins.
3.2. Introduction
Lignin is an abundant, aromatic biopolymer that forms in the lignocellulosic matrices of
plant cell walls. Its free radical polymerization mechanism and heterogeneous nature make it
unique within the plant kingdom. Lignin is economically important to the pulp and paper
industries, the agricultural industries, and the biofuels and biorenewables industries, all of whom
are hampered by its recalcitrance against extraction and/or degradation (Boerjan, 2003; Stewart,
2006; Chen, 2008; Li, 2008; Chapple, 2007; Jung, 1989; Jung, 1995). Many aspects of
lignification are still poorly understood, in spite of its abundance and economic relevance. For
example, the extent to which lignin interacts with surrounding cell wall polymers, particularly
proteins, is largely unknown.
It is understood that lignin forms covalent crosslinks with plant cell wall components,
particularly hemicelluloses (Balakshin, 2011; Miyagawa, 2012; Toikka, 1998; Yuan, 2011). One
prevalent mechanism for lignin-carbohydrate linkage formation is through the reaction of a
nucleophilic moiety (e.g., an hydroxyl or carboxylic acid group) with the electrophilic α-carbon
of the lignin quinone methide (QM) intermediate (Leary, 1980; Ralph, 2009). The crosslinking
of lignin with other cell wall components, such as proteins, has not been well investigated,
despite the fact that lignin-protein linkages may play important roles in wild type and transgenic
plant lines. In most wild type plant lines the pattern of lignin deposition indicates the presence of
so-called nucleation sites within specific regions of the plant cell wall (e.g., the cell corners), but
the nature of these nucleation sites remains unknown (Boerjan et al., 2003). It has been suggested
that nucleation sites may be rich in structural proteins, perhaps leading to lignin-protein

56
crosslinking, but this hypothesis has not been adequately tested. Furthermore, lignin-protein
linkages may affect the physical and chemical properties of transgenic plant lines. For example, a
recently engineered line of Populus secretes a tyrosine-rich peptide into the cell wall. Increased
sugar extractability was observed in these Populus lines upon protease digestion of the walls, and
it was hypothesized that this was due to lignin-protein linkage formation. However, the putative
lignin-protein linkages have yet to be identified (Liang et al., 2008; Xu et al., 2013). Diehl et al.
(2014) recently showed that amino acids bearing nucleophilic side chains, namely Cys, Lys, His,
Asp, Glu, Ser, and Tyr all react with a lignin model QM in dichloromethane. The study identified
diagnostic NMR shifts of lignin-peptide compounds, but did not investigate the propensity for
such linkages to form under biomimetic conditions (i.e., conditions of higher molecular weight
lignin formation with peptides in aqueous media). In order to expand upon these results, the
work described here investigates the propensities for various amino acids (in peptide chains) to
crosslink with lignin dehydrogenation polymer, which is a biomimetic lignin model compound
(Terashima et al., 1995). It is anticipated that this will assist in future studies to help elucidate the
interactions between lignin and proteins in planta.
In order to investigate the propensity for lignin-peptide crosslinking under biomimetic
conditions, lignin dehydrogenation polymer (DHP) was prepared in aqueous solutions containing
peptides. Each peptide had the general structure X-glycine-glycine (XGG), with X being cysteine
(C), lysine (K), histidine (H), aspartic acid (D), glutamic acid (E), serine (S), tyrosine (Y),
threonine (T), or hydroxyproline (Hyp). These amino acids were previously identified as being
reactive (or potentially reactive in the case of T and Hyp) toward lignin QMs (Diehl et al., 2014).
The general peptide structure and predicted mode of lignin-peptide crosslinking is shown in Fig
3.1. The C-termini and N-termini of the peptides were blocked via amidation and esterification,
respectively, to ensure that the amino acid of interest (i.e., residue X) contained the only
nucleophilic moiety. Glycine was chosen as the "place holder" residue due to its expected lack of
reactivity toward lignin. The lengths of the peptides were limited to three residues because
reaction of larger peptides with DHPs results in the formation of lignin-peptide complexes that
are insoluble and thus difficult to characterize (e.g., liquid state NMR becomes impractical)
(results not yet published). Peptides were added in 25% mol/mol ratio to the lignin monomer
(coniferyl alcohol) because it was previously reported that lignin DHPs contain between 20 and
30% β-ether linkages (Tobimatsu, 2012). Thus, the ratio of nucleophilic amino acids to lignin β-
ether QMs was expected to be approximately 1:1 over the course of the polymerization reaction.

57
Fig 3.1. Proposed lignin-peptide crosslinking mechanism.
Lignin-peptide crosslinks form when nucleophilic side chains of amino acids react with quinone
methides formed during lignin β-ether coupling. R = H or OMe, L = lignin.
Fourier-transform infrared spectroscopy (FT-IR), scanning electron microscopy (SEM),
energy dispersive X-ray spectroscopy (EDS), and nuclear magnetic resonance spectroscopy
(NMR) were used to characterize the lignin-peptide adducts. FT-IR and, more recently, NMR,
have become staples of lignin characterization (Capanema et al., 2004; Faix, 1988; Kim and
Ralph, 2010). Multidimensional NMR techniques (e.g., heteronuclear single quantum coherence
(HSQC)) are particularly useful because the shift degeneracies observed in 1D spectra are largely
eliminated. Furthermore, diagnostic NMR shifts of lignin-peptide model compounds have
previously been assigned (Diehl et al., 2014). SEM imaging of synthetic and native lignins has
not garnered much research attention, but the technique was employed here in order to monitor
morphological differences between neat DHP and the lignin-peptide adducts (Micic et al., 2003;
Terashima et al., 2004). It was convenient to also collect EDS elemental analysis data while the
lignin-peptide samples were in the SEM instrument, with the presence of nitrogen suggesting
peptide incorporation because neat lignin contains only carbon, hydrogen, and oxygen. Through
the use of these techniques, this study provides new insights into the propensities and
mechanisms of lignin-peptide linkage formation. It is expected that this will be useful toward the
continued study of lignin formation in both native and mutant plant lines.
3.3. Experimental
3.3.1. Materials
All chemicals necessary for DHP preparation were purchased from Sigma with the
exception of the peptides (>95% purity), which were purchased from Peptide 2.0
(www.peptide2.com).
3.3.2. Synthesis of lignin DHP and lignin-peptide adducts
Guaiacyl-based DHP was synthesized according to a previously published method in
sodium phosphate buffer (pH 6.5) using coniferyl alcohol as the sole lignin monomer
(Terashima, 1995). The DHP crude product was centrifuged (10k g, 20 min, 4 °C) and the pellet
washed four times with distilled water. The DHP product was then lyophilized to yield dry DHP
(typical yields 60-70%), which was characterized via NMR as described below and was found to
contain shifts typical of G-DHP (Capanema, 2004; Kim, 2010).
Lignin-peptide adducts were prepared as above, with the exception that 25% peptide to
coniferyl alcohol (mol/mol basis) was added to the flask containing coniferyl alcohol prior to the
start of the reaction. The crude reaction products were centrifuged and lyophilized as described
above to yield tan powders. These adducts were characterized using IR, SEM, EDS and NMR.
3.3.3. Scanning electron microscopy and energy dispersive X-ray spectroscopy
Scanning electron microscopy images were collected on a field emission SEM (FESEM -
FEI NanoSEM 630) at 2 or 3 kV under high vacuum (1.7 x 10-6 Torr). Samples were not sputter
coated prior to imaging. Characteristic X-rays were collected with an X-Max silicon drift
detector (Oxford Instruments) inside the FESEM at 10 kV under low vacuum conditions (0.6

58
Torr) in order to prevent sample charging. Elements were selected and quantified using Aztec
Energy Analyser Software (Oxford Instruments).
3.3.4. Nuclear magnetic resonance spectroscopy
The neat peptides (25 mg) were dissolved in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul), and
proton (16 scans), carbon (4k scans), HMQC (64 scans) and HMBC (32 scans) spectra were
collected using standard Bruker pulse programs on a Bruker DRX-400 (400 MHz 1H resonance
freq.) using the central solvent peak [δH/δC: dimethyl sulfoxide (DMSO), 2.50/39.50 ppm] as
internal standard. In the case of DHP and the lignin-peptide adducts, NMR spectra were acquired
on a Bruker Biospin (Billerica, MA, USA) AVANCE 500 (500 MHz 1H resonance freq.)
spectrometer fitted with cryogenically-cooled gradient probes having inverse geometry, i.e., with
the proton coils closest to the sample. Spectra were processed with Bruker’s Topspin 3.1
software, using the central solvent peak as internal reference [δH/δC: dimethyl sulfoxide
(DMSO), 2.50/39.5 ppm]. The DHP or lignin-peptide adducts (~45 mg) were placed in an NMR
tube (ID: 4.1 mm), dissolved in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul), and subjected to
adiabatic HSQC (‘hsqcetgpsisp2.2’) experiments, and, in the case of DHP-YGG, also subjected
to HMBC (‘hmbcgpndqf’), COSY (‘cosygpqf’), and NOESY (‘noesyesgpph’) experiments in an
attempt to determine the lignin-tyrosine crosslinking mechanism. Processing used typical
matched Gaussian apodization in F2 (LB = -0.3, GB = 0.001), and squared cosine-bell and one
level of linear prediction (32 coefficients) in F1 (Mansfield, 2012). For an estimation of the
various inter-unit linkage types in DHP and DHP-peptide adducts (Table 2; β-ether/α-OH, β-
ether/α-O-aryl, β-ether/α-peptide, phenylcoumaran, pinoresinol, and dibenzodioxocin), the well
resolved Cα-Hα contours were integrated; no correction factors were used.
3.3.5. Fourier-transform infrared spectroscopy
Lignin DHP, neat peptides, and lignin-peptide adducts were analyzed using a Bruker
Vertex V70 Spectrometer (Bruker Optics Billerica MA) equipped with an MVP-Pro diamond
single reflection ATR accessory (Harrick Scientific Pleasantville NY), and 100 scans at 6 cm-1
resolution were averaged for each sample using a DTGS detector and scan frequency of 5 kHz.
In all cases, the spectrum of the clean diamond crystal was used as the reference spectrum. All
spectral manipulations were performed using OPUS 6.0 (Bruker Optics, Billerica MA).
3.4. Results and discussion
3.4.1. Preparation and yields of the lignin-peptide adducts
Lignin DHP was prepared in aqueous solutions containing tripeptides (25%
peptide:coniferyl alcohol mol/mol basis). Each tripeptide contained one nucleophilic amino acid
and blocked N- and C-termini in order to mimic inclusion within a larger protein and to prohibit
potential side reactions. The lignin-peptide adducts were collected via centrifugation, washed,
and characterized via SEM, NMR, EDS, and FT-IR. The results, detailed below, indicate
covalent incorporation of CGG and YGG peptides into the lignin polymer, while other peptides
did not show significant reactivity.
Yields for the DHP and lignin-peptide adducts are shown in Table 3.1. Yield A was
determined by dividing the mass of recovered solids by the total starting mass (i.e., combined
mass of lignin monomer and peptide), while yield B was determined by dividing the mass of

59
recovered solids by the starting mass of lignin monomer only. Thus, yield B is only valid for
lignin-peptide reactions in which peptide incorporation into the lignin was negligible. Notably,
the yields were very high when DHP was prepared in the presence of non-covalently reactive
peptides (i.e., all peptides other than CGG and YGG). The reason for this was unclear.
In the cases of the considerably reactive peptides (i.e., CGG and YGG) the yields of
recoverable DHP were depressed. For DHP-CGG, a likely explanation is that the thiol group of
the CGG peptide inhibited the catalytic ability of horseradish peroxidase, thus hampering
polymerization (Tobimatsu et al, 2009; Veitch, 2004). It is not known why the yield was
depressed in the case of DHP-YGG. The authors perceived that in the cases of DHP-CGG and
DHP-YGG a portion of the lignin-peptide adducts may have been aqueous soluble and held in
solution during the centrifugation process. However, extraction of the aqueous supernatants with
ethyl acetate and chloroform followed by NMR analyses did not show evidence for lignin-
protein complexes. Drying down the aqueous supernatant, re-suspending the solids in DMSO-
d6/pyridine-d5, and analyzing the products via NMR similarly failed to provide evidence for
lignin-protein crosslinking. This confirmed the depression of DHP yields in the cases of DHP-
CGG and DHP-YGG.
Table 3.1. Yield data for the DHP and lignin-peptide adducts
CA (mg) pep (mg) yield (mg) Yield A (%) Yield B (%)
DHP 200.0 0.0 130.0 65.0 65.0
DHP-CGG 200.0 88.4 100.0 34.7 -
DHP-KGG 200.0 83.6 174.5 61.5 87.3
DHP-HGG 200.0 86.1 176.0 61.5 88.0
DHP-DGG 200.0 80.0 180.0 64.3 90.0
DHP-EGG 200.0 83.9 197.9 69.7 99.0
DHP-SGG 200.0 72.2 172.8 63.5 86.4
DHP-YGG 200.0 93.3 114.2 38.9 -
DHP-TGG 200.0 76.1 179.1 64.9 89.6
DHP-HypGG 200.0 79.4 177.4 63.5 88.7
Yield A was determined by dividing the mass of recovered solids by the total starting mass (i.e.,
combined mass of lignin monomer and peptide). Yield B was determined by dividing the mass of
recovered solids by the starting mass of lignin monomer only. CA = coniferyl alcohol, pep =
peptide.
3.4.2. Lignin-peptide morphology
Scanning electron microscopy was used to compare the morphologies of DHP and the
lignin-peptide adducts (Fig 3.2). The DHP particles clumped together to form nearly perfect
spheres, as reported previously (Micic et al, 2003; Micic et al, 2004). Comparatively, spheres of
lignin-peptide adducts tended to form large, amorphous domains. This alteration of morphology
was observed regardless of whether the peptide in question was incorporated into the lignin. This
change in morphology was unexpected but was most likely due to non-covalent interactions

60
occurring between the peptides and the growing lignin chain. Further research is necessary to
determine the influence of non-covalent inter-polymer interactions during lignin polymerization.
Fig 3.2. SEM images of DHP (top), then, proceeding from left to right and top to bottom, DHP-
CGG, DHP-YGG, DHP-HypGG, DHP-DGG, DHP-EGG, DHP-KGG, DHP-HGG, DHP-SGG,
and DHP-TGG. Scale bar: 2 µm.
3.4.3. Lignin-peptide linkage identification
Fig 3.3 shows the heteronuclear single quantum coherence (HSQC) spectrum of DHP-
CGG. This 2D NMR technique is particularly useful for lignin analysis because the shift
degeneracy observed in 1D NMR spectra is largely avoided. Novel shifts are shown in green,
red, and blue, while standard lignin shifts are shown in black (Capanema, 2004; Kim, 2010).
Reference shifts of neat CGG (purple) were added to the spectrum during processing; these shifts
were not observed in the DHP-CGG spectrum. Some peptide shifts migrated as a result of DHP-
CGG crosslinking. For example, the cys-13C/1Hα shift (originally at 4.5/56.0 ppm in neat CGG)
migrated to 4.6/52.8 ppm, and the cys-13C/1Hβ shift (originally at 2.8/26.6 ppm in neat CGG)
migrated to 2.8/32.9 ppm in the DHP-CGG adduct. These shifts migrated upon lignin-peptide
crosslinking due to their proximity to the thiol group, which is the reactive center of the CGG

61
peptide. Shifts of proton and carbon atoms located far from the reactive thiol were largely
unaffected by crosslinking (e.g., shifts at 3.9/42.8 and 3.8/42.5 ppm).
Two novel lignin shifts, found at 4.4/50.1 ppm (Fig 3.3, red peak) and 4.8/81.3
ppm (Fig 3.3, blue peak), confirmed covalent crosslinking of DHP with CGG. Similar lignin α-
shifts (4.3/50.4 ppm) and β-shifts (4.7/81.7 ppm) were previously reported when a single
cysteine residue was reacted with a lignin model quinone methide to yield a structure similar to
that shown in Fig 3.3 (Diehl et al., 2014). The minute differences in shift locations can be
attributed to changes in chemical environment between a small lignin model compound and a
high molecular weight lignin. Volume integration of the HSQC contours showed that
approximately 33% of the β-ether linkages in DHP-CGG exhibited cysteine functionality at the
α-carbon, while the remaining β-ether linkages exhibited typical α-hydroxyl functionality and a
minor fraction of α-aryl ether (α-O-aryl) moieties. This indicated that cysteine was an efficient
trapper of lignin QMs under biomimetic conditions.
Fig 3.3. Side-chain and aromatic regions (inset) of the HSQC NMR spectrum of DHP-CGG.
Black shifts are typical of G-DHPs, green shifts correspond to peptide α- and β-signals, and red
and blue shifts correspond to lignin α- and β-signals in β-ether/α-cysteine structures (top left).
Purple shifts were added during processing to indicate shifts of neat CGG peptide.
Fig 3.4 shows the HSQC spectrum of DHP-YGG. As with the DHP-CGG adduct,
incorporation of YGG peptide into lignin was evidenced by the appearance of diagnostic
chemical shifts (Fig 3.4, green and orange contours). Reference shifts of neat YGG (purple, solid
yellow, and solid orange contours) were added during processing. The authors perceived that
given the similarity of tyrosine and coniferyl alcohol, crosslinking of YGG with DHP may have
occurred via two mechanisms.

62
The first potential mechanism involves oxidation of the phenolic hydroxyl of tyrosine by
horseradish peroxidase, followed by recombination of the tyrosine radical with a radical on the
lignin polymer. This mechanism may be unfavorable because radicals generated on tyrosine
could be shuttled to coniferyl alcohol, which exhibits an additional resonance structure compared
to tyrosine, presumably making it more stable (Cong et al., 2013). In addition to HSQC NMR,
we submitted the DHP-YGG adduct to heteronuclear multiple bond correlation (HMBC),
correlation spectroscopy (COSY), and nuclear Overhauser effect spectroscopy (NOESY)
techniques (spectra not shown), but were unable to conclusively assign NMR shifts of lignin-
tyrosine linkages formed in this manner. This may indicate that the mechanism is not valid under
our experimental conditions, and/or may illustrate the inadequacy of NMR to resolve shift
degeneracy between lignin-tyrosine linkages and typical lignin shifts.
A second crosslinking mechanism is possible when the phenolic hydroxyl of tyrosine
quenches the lignin quinone methide to form the α-aryl ether structure shown in Fig 3.4. Again,
shift degeneracy may complicate the investigation of this mechanism, as a lignin-tyrosine model
compound exhibited similar NMR shifts (α-1H/13C: 5.5/78.3 ppm in DMSO/pyridine) to α-aryl
ether linkages known to occur in neat DHPs (α-13C: 79.01 ppm in DMSO) (Diehl et al., 2014;
Ralph, Ralph and Landucci, 2004). In an attempt to overcome this issue, the well-resolved
HSQC α-signals of neat DHP and DHP-YGG adduct were integrated. It was observed that α-aryl
ether shifts comprised approximately 4.2% of the total α-signal in DHP-YGG but only 1.9% in
neat DHP synthesized under similar conditions. This increase could be due to imprecision in the
HSQC volume integration or random variation among DHP syntheses (other lignin-peptide
adducts displayed similarly high α-aryl ether signals), making it unclear if the structure shown in
Fig 3.4 formed in the DHP-YGG adduct. In summary, the NMR, EDS, and IR data (shown
below) strongly suggest that the YGG peptide crosslinked with lignin DHP; however, the
mechanism of lignin-tyrosine crosslinking is still uncertain.

63
Fig 3.4. Side-chain and aromatic regions (inset) of the HSQC NMR spectrum of DHP-YGG.
Black shifts are typical of G-DHPs, green shifts correspond to peptide α- and β-signals, and red
and blue shifts correspond to lignin α- and β-signals in β-ether/α-tyrosine structures (top left)
and/or lignin-lignin α-O-aryl structures. Purple shifts were added during processing to indicate
shifts of neat YGG peptide. Within the aromatic region, solid yellow and orange shifts (added
during processing) were assigned to the aromatic ring of tyrosine in neat YGG. It can be seen
that the contours (orange) representative of tyrosine ring positions 3 and 5 shift downfield as a
result of lignin-tyrosine crosslinking. The specific lignin-tyrosine crosslinking mechanism could
not be determined by NMR, and the structure shown is one of several possibilities.
It was notable that in the case of lignin-peptide adducts other than DHP-CGG and DHP-
YGG, peptide peaks could always be observed when viewing the HSQC contours quite low (i.e.,
near the signal to noise limit). Fig 3.5 shows the HSQC spectrum of DHP-HGG. This sample
showed the highest concentration of peptide after DHP-CGG and DHP-YGG. A putative lignin-
α-histidine crosslink was observed at 5.7/60.4 ppm, in good agreement with the α-shift of a
lignin-histidine model compound (5.7/60.2 ppm) (Diehl et al., 2014). Volume integration showed
that the abundance of the lignin-α-histidine shift only accounted for ~0.1% of the total lignin α-
signal. It is noteworthy that this low abundance of peptide was detected by HSQC NMR but not
readily detected by IR or EDS, thus illustrating the sensitivity of multidimensional NMR toward
investigating lignin-protein linkages.
Other lignin-peptide adducts exhibited less abundant NMR peptide shifts than DHP-
HGG. This indicated that negligible lignin-peptide crosslinking had occurred, in concurrence
with IR and EDS data (below).

64
Fig 3.5. Side-chain region of the HSQC NMR spectrum of DHP-HGG. Black shifts are typical of
G-DHPs, green shifts correspond to peptide α- and β-signals, and red and blue shifts correspond
to lignin α- and β-signals in β-ether/α-histidine structures (top left). Purple shifts were added
during processing to indicate shifts of neat HGG peptide.
Volume integration of the HSQC contour signals allowed for comparison of the various
lignin inter-unit linkages among DHP and lignin-peptide adducts (Table 3.2). The DHP
contained linkage ratios typical of DHPs (Terashima et al., 1995 and 2009; Tobimatsu et al.,
2012). Linkage ratios varied among the lignin-peptide adducts, but decreased β-ether content
with increased pinoresinol (β-β) content was generally observed. This occurred regardless of
covalent reactivity towards the lignin DHP, demonstrating the ability of a matrix material (in this
case peptides) to influence lignin structure during polymerization.
Table 3.2. Inter-unit linkage ratios of the DHP and lignin-peptide adducts
HSQC signal ratios
β-ether/α-OH β-ether/α-O-aryl β-ether/α-pep β-5 β-β Dibenz.
DHP 27.3 1.9 - 50.3 19.2 1.2
DHP-CGG 8.6 1.5 5.1 50.8 32.4 1.6
DHP-DGG 10.1 5.5 0.1 54.1 30.2 tr
DHP-EGG 13.1 4.6 0.1 54.1 27.2 0.9
DHP-KGG 4.7 3.1 tr 62.3 29.9 tr
DHP-HGG 20.4 0.9 0.1 57.7 20.3 0.6
DHP-SGG 11.1 2.3 tr 52.5 34.1 tr
DHP-YGG 11.5 4.2 51.7 32.5 tr
DHP-TGG 21.8 0.9 tr 54.1 22.2 1.0
DHP-HypGG 17.7 2.7 tr 53.2 26.2 0.2
Lignin inter-unit linkage ratios (as percentage of total α-signal) for DHP and lignin-peptide
adducts. In the case of DHP-YGG the DHP-α-peptide shift was degenerate with standard lignin
α-O-aryl shifts, thus the β-ether/α-O-aryl and β-ether/α-pep quantities were combined. tr, trace
(<0.1%).
3.4.4. Supporting techniques for characterization of lignin-peptide entanglement
In addition to NMR, which can provide direct evidence of covalent crosslinking, other
techniques can be used to show peptide incorporation into lignin. Fig 3.6 shows FT-IR spectra of
neat DHP and the lignin-peptide adducts. The neat DHP IR spectrum exhibited bands typical of
lignin DHPs (Faix, 1988). The DHP-CGG and DHP-YGG spectra exhibited three peaks
indicative of peptide incorporation into the lignin. The shoulder near 3200 cm-1 was attributed to
N-H stretching in amide functional groups, the peak at 1658 cm-1 increased dramatically and was
attributed to increased C=O stretching due to the incorporation of amide functional groups, and
the shoulder at 1540 cm-1 was attributed to N-H deformation with C-N stretching, again
indicating incorporation of amide functionalities (Socrates, 2001). It is notable that these shifts
displayed greater intensity in the DHP-CGG adduct compared to the DHP-YGG adduct,
suggesting greater incorporation of CGG peptide. These peaks were not observed in the IR
spectra of other lignin-peptide adducts, suggesting a lack of peptide incorporation. In the case of
DHP-CGG and DHP-YGG, incorporation of peptide into the lignin polymer caused an increase

65
in the band at 1505 cm-1, which was previously assigned to aromatic skeletal vibrations (Faix,
1988). This increase in peak height was not observed in other lignin-peptide adducts and the
origin of this increased peak intensity is unclear. Though the IR results suggested incorporation
of CGG and YGG peptides into lignin DHP, peaks directly attributable to lignin-peptide linkages
were not identified. These results demonstrate that IR is a quick and reliable technique for
indicating lignin-peptide interactions in general, but may be insufficient for determining the
presence or absence of covalent crosslinks.
Fig 3.6. FT-IR spectra of DHP and lignin-peptide adducts.
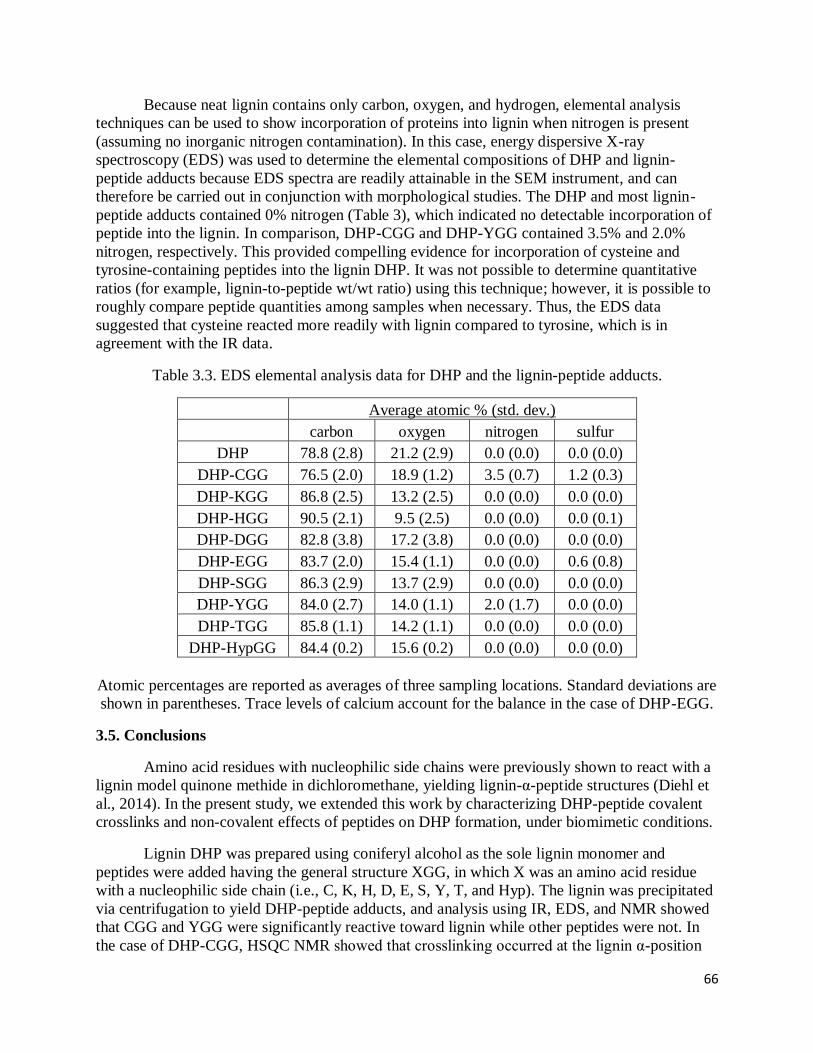
66
Because neat lignin contains only carbon, oxygen, and hydrogen, elemental analysis
techniques can be used to show incorporation of proteins into lignin when nitrogen is present
(assuming no inorganic nitrogen contamination). In this case, energy dispersive X-ray
spectroscopy (EDS) was used to determine the elemental compositions of DHP and lignin-
peptide adducts because EDS spectra are readily attainable in the SEM instrument, and can
therefore be carried out in conjunction with morphological studies. The DHP and most lignin-
peptide adducts contained 0% nitrogen (Table 3), which indicated no detectable incorporation of
peptide into the lignin. In comparison, DHP-CGG and DHP-YGG contained 3.5% and 2.0%
nitrogen, respectively. This provided compelling evidence for incorporation of cysteine and
tyrosine-containing peptides into the lignin DHP. It was not possible to determine quantitative
ratios (for example, lignin-to-peptide wt/wt ratio) using this technique; however, it is possible to
roughly compare peptide quantities among samples when necessary. Thus, the EDS data
suggested that cysteine reacted more readily with lignin compared to tyrosine, which is in
agreement with the IR data.
Table 3.3. EDS elemental analysis data for DHP and the lignin-peptide adducts.
Average atomic % (std. dev.)
carbon oxygen nitrogen sulfur
DHP 78.8 (2.8) 21.2 (2.9) 0.0 (0.0) 0.0 (0.0)
DHP-CGG 76.5 (2.0) 18.9 (1.2) 3.5 (0.7) 1.2 (0.3)
DHP-KGG 86.8 (2.5) 13.2 (2.5) 0.0 (0.0) 0.0 (0.0)
DHP-HGG 90.5 (2.1) 9.5 (2.5) 0.0 (0.0) 0.0 (0.1)
DHP-DGG 82.8 (3.8) 17.2 (3.8) 0.0 (0.0) 0.0 (0.0)
DHP-EGG 83.7 (2.0) 15.4 (1.1) 0.0 (0.0) 0.6 (0.8)
DHP-SGG 86.3 (2.9) 13.7 (2.9) 0.0 (0.0) 0.0 (0.0)
DHP-YGG 84.0 (2.7) 14.0 (1.1) 2.0 (1.7) 0.0 (0.0)
DHP-TGG 85.8 (1.1) 14.2 (1.1) 0.0 (0.0) 0.0 (0.0)
DHP-HypGG 84.4 (0.2) 15.6 (0.2) 0.0 (0.0) 0.0 (0.0)
Atomic percentages are reported as averages of three sampling locations. Standard deviations are
shown in parentheses. Trace levels of calcium account for the balance in the case of DHP-EGG.
3.5. Conclusions
Amino acid residues with nucleophilic side chains were previously shown to react with a
lignin model quinone methide in dichloromethane, yielding lignin-α-peptide structures (Diehl et
al., 2014). In the present study, we extended this work by characterizing DHP-peptide covalent
crosslinks and non-covalent effects of peptides on DHP formation, under biomimetic conditions.
Lignin DHP was prepared using coniferyl alcohol as the sole lignin monomer and
peptides were added having the general structure XGG, in which X was an amino acid residue
with a nucleophilic side chain (i.e., C, K, H, D, E, S, Y, T, and Hyp). The lignin was precipitated
via centrifugation to yield DHP-peptide adducts, and analysis using IR, EDS, and NMR showed
that CGG and YGG were significantly reactive toward lignin while other peptides were not. In
the case of DHP-CGG, HSQC NMR showed that crosslinking occurred at the lignin α-position

67
(Fig 3.4). The crosslinking mechanism of DHP with YGG could not be conclusively elucidated.
SEM imaging showed that DHP-peptide adducts exhibited a unique morphology compared to
neat DHP, regardless of peptide incorporation into the lignin. With regards to lignin inter-unit
lignin ratios, the quantity of β-ether linkages was typically depressed in DHPs synthesized in the
presence of peptides, while the quantity of pinoresinol structures increased. The yields of DHP-
CGG and DHP-YGG were depressed compared to neat DHP yields; however, curiously, the
yields increased when DHP was prepared in the presence of other peptides.
We have shown that cysteine and tyrosine crosslink with lignin under biomimetic
conditions. This suggests that similar crosslinking may occur in the cell walls of both native and
transgenic plant lines. Further research is needed to investigate whether this crosslinking does
occur, and to discover how the plant might control and benefit from such crosslinking (i.e., does
it help stiffen the wall, assist water conduction, provide protection from pathogens, etc). In
addition, a better understanding of lignin-protein linkages could lead to genetic manipulation
(up-regulation or down-regulation of the linkages), as already suggested by Liang et al. (2008)
and Xu et al. (2013). This could reduce lignin recalcitrance, which is currently a barrier to using
lignocellulosic materials in developing industries such as biofuels. It is anticipated that this work
will lead to future studies of lignin-protein linkages in planta, and a more thorough
understanding of how such linkages could be tailored and modified.
3.6. Acknowledgements
This material is based upon work supported as part of The Center for Lignocellulose
Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of
Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-
SC0001090. Student fellowships were provided by the USDA National Needs Program and the
National Science Foundation via the CarbonEARTH program. Many thanks to Julie Anderson
and Melisa Yashinski (PSU MRI) for acquisition of SEM/EDS data and for valuable discussions.
Thanks also to Dr. John Ralph, Yuki Tobimatsu, and Matt Regner for valuable discussions
regarding multidimensional NMR of lignin.
3.7. References
Awad, H. M.; Boersma, M. G.; Vervoort, J.; Rietjens, I. M. C. M. Peroxidase-catalyzed
formation of quercetin quinone methide-glutathione adducts. Arch. Biochem. Biophys. 2000, 378,
224-233.
Balakshin, M.; Capanema, E.; Gracz, H.; Chang, H.; Jameel, H. Quantification of lignin-
carbohydrate linkages with high-resolution NMR spectroscopy. Planta 2011, 233, 1097-1110.
Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Re. Plant Biol. 2003, 54, 519-
546.
Bolton, J. L.; Turnipseed, S. B., Thompson, J. A. Influence of quinone methide reactivity on the
alkylation of thiol and amino groups in proteins: studies utilizing amino acid and peptide models.
Chem. Biol. Interact. 1997, 107, 185-200.

68
Capanema, E. A.; Balakshin, M. Y.; Kadla, J. F. A comprehensive approach for quantitative
lignin characterization by NMR spectroscopy. J. Agric. Food Chem. 2004, 52, 1850-1860.
Chapple, C.; Ladisch, M.; Meilan, R. Loosening lignin's grip on biofuel production. Nat.
Biotechnol. 2007, 25, 746-748.
Chen, F.; Dixon, R. A. Genetic manipulation of lignin biosynthesis to improve biomass
characteristics for agro-industrial processes. In Vitro Cell. Dev. Biol. - Animal. 2008, 44, S28-
S29.
Cong, F.; Diehl, B.G.; Hill, J.L.; Brown, N.R.; Tien, M. Covalent bond formation between amino
acids and lignin: Cross-coupling between proteins and lignin. Phytochem. 2013, 96, 449-456.
Diehl, B.G.; Watts, H.D.; Kubicki, J.D.; Regner, M.R.; Ralph, J.; Brown, N.R. Towards lignin-
protein crosslinking: Amino acid adducts of a lignin model quinone methide. Cellulose 2014,
available online.
Faix O, Beinhoff O. 1988. Journal of Wood Chemistry and Technology, 8 (4): 505-522. "FTIR
spectra of milled wood lignins and lignin polymer models with enhanced resolution obtained by
deconvolution."
Fisher, J.; Abdella, B. R. J.; McLane, K. E. Anthracycline antibiotic reduction by spinach
ferredoxin-NADP+ reductase and ferredoxin. Biochemistry 1985, 24, 3562-3571.
Jung, H. G.; Allen, M. S. Characteristics of plant cell walls affecting intake and digestibility of
forages by ruminants. J. Animal Sci. 1995, 73, 2774-2790.
Jung, H. G. Forage lignins and their effects on fiber digestibility. Agron. J. 1989, 81, 33-38.
Kim, H.; Ralph, J. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-
d6/pyridine-d5. Org. Biomol. Chem. 2010, 8, 576-591.
Leary, G. J. Quinone methides and the structure of lignin. Wood Sci. and Tech. 1980, 14, 21-34.
Liang, H.; Frost, C. J.; Wei, X.; Brown, N. R.; Carlson, J. E.; Tien, M. Improved sugar release
from lignocellulosic material by introducing a tyrosine-rich cell wall peptide gene in poplar.
Clean 2008, 36(8), 662-668.
Li, X.; Weng, J. K.; Chapple, C. Improvement of biomass through lignin modification. Plant J.
2008, 54, 569-581.
Mansfield, S. D.; Kim, H.; Lu, F.; Ralph, J. Whole plant cell wall characterization using
solution-state 2D NMR. Nature Protocols. 2012, 7(9), 1579-1589.
Micic, M.; Radotic, K.; Jeremic, M.; Djikanovic, D.; Kammer, S. B. Study of the lignin model
compound supramolecular structure by combination of near-field scanning optical microscopy
and atomic force microscopy. Colloids and Surfaces B: Biointerfaces 2004, 34, 33-40.
Micic, M.; Radotic, K.; Jeremic, M.; Leblanc, R. M. Study of the self-assembly of the lignin
model compound on cellulose model substrate. Macromol. Biosci. 2003, 3 (2), 100-106.

69
Miyagawa, Y.; Takemoto, O.; Takano, T.; Kamitakahara, H.; Nakatsubo, F. Fractionation and
characterization of lignin carbohydrate complexes (LCCs) of Eucalyptus globulus in residues left
after MWL isolation. Part I: Analyses of hemicellulose-lignin fractionation (HC-L).
Holzforschung 2012, 66, 459-465.
Ralph, S.A., Ralph, J., Landucci, L.L. NMR Database of Lignin and Cell Wall Model
Compounds. Available at URL http://ars.usda.gov/Services/docs.htm?docid=10491 (November
2004).
Ralph, J.; Schatz, P. F.; Lu, F.; Kim, H.; Akiyama, T.; Nelsen, S. F. Quinone methides in
lignification. In Quinone Methides; Rokita, S. E., Ed.; John Wiley & Sons , 2009; pp 385-420.
Ramakrishnan, K.; Fisher, J. Nucleophilic trapping of 7,11-dideoxyanthracyclinone quinone
methides. J. Am. Chem. Soc. 1983, 105, 7187-7188.
Richard, J. P.; Toteva, M. M.; Crugeiras, J. Structure-reactivity relationships and intrinsic
reaction barriers for nucleophile additions to a quinone methide: a strongly resonance-stabilized
carbocation. J. Am. Chem. Soc. 2000, 122, 1664-1674.
Socrates, George. Infrared and Raman characteristic group frequencies, third edition. George
Wiley and Sons, LTD. West Sussex, England. 2001.
Stewart, J. J.; Kadla, J. F.; Mansfield, S. D. The influence of lignin chemistry and ultrastructure
on the pulping efficiency of clonal aspen (Populus termuloides Michx). Holzforschung 2006, 60,
111-122.
Terashima, N.; Akiyama, T.; Ralph, S.; Evtuguin, D.; Neto, C.P.; Parkas, J.; Paulsson, M.;
Westermark, U.; Ralph, J. 2D-NMR (HSQC) difference spectra between specifically 13C-
enriched and unenriched protolignin of Ginkgo biloba obtained in the solution state of whole cell
wall material. Holzforschung 2009, 63, 379-384.
Terashima, N.; Atalla, R. H.; Ralph, S. A.; Landucci, L. L.; Lapierre, C.; Monties, B. New
preparations of lignin polymer models under conditions that approximate cell wall lignification.
Holzforschung 1995, 49, 521-527.
Terashima, N.; Awano, T.; Takabe, K.; Yoshida, M. Formation of macromolecular lignin in
gingko xylem cell walls as observed by FESEM. C.R. Biologies 2004, 327, 903-910.
Tobimatsu, Y.; Elumalai, S.; Grabber, J. H.; Davidson, C. L.; Pan, X.; Ralph, J.
Hydroxycinnamate conjugates as potential monolignol replacements: In vitro lignification and
cell wall studies with rosmarinic acid. ChemSusChem. 2012, 5 (4), 676-686.
Tobimatsu, Y.; Takano, T.; Kamitakahara, H.; Nakatsubo, F. Reactivity of syringyl quinone
methide intermediates in dehydrogenative polymerization I: high-yield production of synthetic
lignins (DHPs) in horseradish peroxidase-catalyzed polymerization of sinapyl alcohol in the
presence of nucleophilic reagents. J. Wood Sci. 2009, 56(3), 233-241.

70
Toikka, M.; Jussi, S.; Teleman, A.; Brunow, G. Lignin-carbohydrate model compounds.
Formation of lignin-methyl arabinoside and lignin-methyl galactoside benzyl ethers via quinone
methide intermediates. J. Chem. Soc.; Perkin Trans. 1. 1998, 1, 3813-3818.
Veitch, N. C. Horseradish peroxidase: a modern view of a classic enzyme. Phytochem. 2004, 65,
249-259.
Xu, Y.; Chen, C.; Thomas, T.P.; Azadi, P.; Diehl, B.; Tsai, C.; Brown, N.; Carlson, J.E.; Tien,
M.; Liang, H. Plant Cell Rep. 2013, 32, 1827-1841.
Yuan, T.; Sun, S.; Xu, F.; Sun, R. Charcterization of lignin structures and lignin-carbohydrate
complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy. J. Agric. Food
Chem. 2011, 59, 10604-10614.

71
Chapter 4
Preparation and characterization of lignin-gelatin complexes
(Target journal for publication is Journal of Applied Polymer Science)
4.1. Abstract
Lignin dehydrogenation polymer (DHP) was prepared in the presence of gelatin protein to yield
“DHP-Gel adducts.” The DHP-Gel adducts were characterized using Fourier-transform infrared
spectroscopy (FT-IR), scanning electron microscopy (SEM), energy dispersive X-ray
spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS) and heteronuclear single quantum
coherence nuclear magnetic resonance spectroscopy (HSQC NMR). FT-IR, EDS and XPS
showed that gelatin was incorporated into the lignin even when added in small quantities. In
addition, EDS and XPS showed that gelatin was distributed throughout the DHP when added
during lignin polymerization, but adsorbed to the surface of DHP when added following
polymerization. A lack of diagnostic lignin-protein crosslink signatures in the HSQC NMR
spectra suggested that the lignin-gelatin interaction was largely non-covalent in nature. This may
have implications toward lignification in planta, in which lignin is biosynthesized in a pre-
deposited matrix of polysaccharides and proteins. Cell wall structural proteins, which are
common in the cell corner region of the middle lamella (where lignification begins), may help
“nucleate” lignification without necessarily covalently crosslinking with lignin.
Key words: lignin, gelatin, crosslinking, non-covalent, scanning electron microscopy, energy
dispersive X-ray spectroscopy, X-ray photoelectron spectroscopy, nuclear magnetic resonance
spectroscopy.
4.2. Introduction
Lignin is the most abundant aromatic biopolymer on earth (Boerjan et al., 2003). It is
most commonly biosynthesized in the cell walls of land plants from three monolignols (p-
coumaryl, coniferyl, and sinapyl alcohols) that vary in their degree of aromatic ring
methoxylation. It is commonly believed that lignin imparts three main evolutionary advantages
to the plant: structural rigidity, water conductivity, and pathogen resistance. Lignin is
commercially important to the pulp and paper industry, agricultural industries concerned with
forage digestibility, and the developing biofuels industry, in which it is known to foul cellulose
to ethanol conversion processes (Stewart, 2006; Chen, 2008; Li, 2008; Chapple, 2007; Jung,
1989; Jung, 1995). Lignin is also a potential source of renewable carbon for plastics, carbon
fibers, solvents, and low and high value chemicals, to name a few (Gellerstedt, et al., 2010, Chen
and Sarkanen, 2006, Dorrestijn, et al., 2000, Clark, et al., 2009).
Despite decades of research, some details of lignification are still poorly understood. For
example, it has long been known that within most plants, lignin deposition begins in the cell
corner region of the middle lamella. However, the mechanism by which the plant controls this
pattern of lignin deposition is unknown. Lignification initiation sites (sometimes referred to as
“nucleation sites”) have been postulated, with two commonly hypothesized initiation sites being

72
calcium-pectate complexes (which may bind anionic peroxidases necessary for lignin
polymerization) and cell wall structural proteins (especially extensins, which are abundant in the
cell corners) (Albersheim et al., 2010; Boerjan et al., 2003). Neither of these hypotheses has been
adequately investigated in vitro or in vivo. Several studies have shown that proteins have an
affinity to bind lignin; however, the nature of the lignin-protein binding (i.e., covalent vs. non-
covalent) was not elucidated (Whitmore, 1978a; Whitmore, 1978b; Whitmore, 1982). With
regard to lignin-protein covalent crosslinking, it was recently shown that Cys, Lys, His, Asp,
Glu, Ser, and Tyr crosslink with lignin in non-polar solvents, and that Cys and Tyr crosslink with
lignin even under aqueous, biomimetic conditions (Diehl et al., 2014; Diehl and Brown, in
review). Here, we report the preparation and characterization of lignin DHP in the presence of
gelatin, a glycine and hydroxyproline-rich animal protein.
Though gelatin protein does not originate from plants, the lignin-gelatin complex is
interesting and potentially informative for several reasons. Gelatin is both glycine and
hydroxyproline-rich, as are many plant cell wall structural proteins. Gelatin has previously been
shown to interact with lignin, though the presence or absence of covalent crosslinks was not
definitely determined (Whitmore, 1978b). Several potentially nucleophilic amino acid residues
are found in gelatin; however, cysteine and tyrosine residues are almost entirely lacking (Table
4.1) (Eastoe, 1955). While nucleophilic amino acids other than cysteine and tyrosine have been
shown to react with lignin under ideal conditions in non-polar solvents, only cysteine and
tyrosine have been shown to covalently crosslink with lignin under biomimetic conditions of
DHP preparation (Diehl et al., 2014; Diehl and Brown, in review). The affinity for gelatin to
interact with lignin under aqueous conditions is therefore interesting, as it seems most likely to
arise from physical entanglement and/or non-covalent interactions, or from lignin-protein
crosslinkage types that have not previously been observed under conditions of DHP preparation.
Understanding the interactions occurring within the lignin-gelatin complex may provide insights
into lignin-protein interactions in planta, where lignin and cell wall structural proteins could be
expected to be in close spatial proximity.
Table 4.1. Nucleophilic amino acid abundance (g/100 g dry, ash-free protein) in gelatin.
Residue Porcine Gelatin Bovine Gelatin
Cys 0.00 0.05
Lys 4.14 5.20
His 1.01 0.63
Glu 11.30 12.10
Asp 6.70 6.90
Tyr 0.60 0.14
Ser 4.13 2.90
Thr 2.19 2.20
Hyp 13.50 14.40
Total 43.57 44.52

73
In order to elucidate the lignin-gelatin interaction, five lignin-gelatin complexes (DHP-
Gel adducts) were prepared (see Table 4.2 for DHP-Gel preparations). The DHP was prepared by
slowly combining monolignol (e.g., coniferyl alcohol) and hydrogen peroxide solutions to a
horseradish peroxidase solution, as previously described (Terashima et al., 1995). In addition,
various quantities of porcine (high molecular weight) and bovine (low molecular weight) gelatin
were added to the DHP preparations. DHP-Gel1 and DHP-Gel5 contained equivalent quantities
(wt/wt basis) of monolignol and porcine gelatin, with the only difference being that gelatin was
dissolved in the same flask as the coniferyl alcohol in the case of DHP-Gel1, and was thus added
slowly and continuously to the DHP preparation during polymerization, whereas in the case of
DHP-Gel5 gelatin was added to the DHP preparation following completion of polymerization. It
was perceived that covalent crosslinking, if it occurred, might be more likely in the case of DHP-
Gel1 because the gelatin would potentially be in intimate contact with reactive lignin
intermediates (i.e., quinone methides) over the course of the polymerization reaction. It was also
perceived that addition of gelatin at different times might influence the morphology of the DHP-
Gel adducts. In the cases of DHP-Gel2, DHP-Gel3, and DHP-Gel4, decreasing quantities of
bovine gelatin (2.5:1, 5:1, and 10:1 monolignol to gelatin wt/wt basis) were added to the DHP,
again during the polymerization process. These DHP-Gel adducts allowed for investigation of
lignin interactions with small quantities of low molecular weight gelatin.
The DHP-Gel adducts were characterized using several techniques. Fourier-transform
infrared spectroscopy (FT-IR) was employed because it can show incorporation of protein into
lignin, although it is admittedly deficient at identifying lignin-protein covalent crosslinks (Diehl
et al., in review). Scanning electron microscopy (SEM) was used to determine the physical
morphology of the DHP-Gel adducts. Energy dispersive X-ray spectroscopy (EDS) and X-ray
photoelectron spectroscopy (XPS) were used to both confirm the incorporation of protein into
the lignin (via quantification of nitrogen) as well as to elucidate morphological details. Finally,
heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance (NMR)
spectroscopy was used to investigate potential lignin-gelatin covalent crosslinks. Investigation of
the lignin-gelatin complexes reported here may lead to a better understanding of lignin-protein
interactions in native plant systems.
4.3. Experimental
4.3.1. Materials
Coniferyl alcohol was prepared from coniferaldehyde (Sigma Aldrich) as previously
described (Ludley and Ralph, 1996). Horseradish peroxidase (type I), hydrogen peroxide,
sodium phosphate and porcine and bovine gelatin were purchased from Sigma Aldrich. The
bovine gelatin (Sigma #G6650) had a bloom number of 75, with an estimated molecular weight
of 20 to 25 KDa. The porcine gelatin (Sigma #G2500) had an estimated molecular weight of 100

74
KDa. The peristaltic pump used in the DHP synthesis was a Cole-Parmer Masterflex, model
number 77120-52.
4.3.2. DHP and DHP-Gel preparations
Lignin DHP was synthesized according to a published method with a few modifications
(Terashima et al., 1995). Coniferyl alcohol (200 mg) was added to 200 ml warm sodium
phosphate (0.01 M, pH 6.5) buffer. Horseradish peroxidase (HRP) (4 mg) was added to this flask
after the buffer temperature dropped below 40° C. In a second flask, hydrogen peroxide was
added to 200 ml of buffer to a final concentration of 0.025%. A peristaltic pump was used to
combine the contents of the flasks into a single 500 ml flask that initially contained 2 ml of
buffer and 1 mg of HRP. Addition of reactants was performed at a rate of approximately 6
ml/min and the contents of the collection flask were allowed to stir for an additional 24 hours
upon completion of reactant addition.
DHP-Gel adducts were prepared as above, but porcine or bovine gelatin (quantities
shown in Table 4.2) were added to the flask containing coniferyl alcohol prior to the start of the
reaction. In the case of DHP-Gel5, gelatin was added following DHP polymerization (i.e., gelatin
was added approximately 24 hours after the complete addition of coniferyl alcohol and hydrogen
peroxide).
Neat DHP and DHP-Gel adducts were centrifuged at 10k g for 20 min at 4° C. The
supernatants were discarded and the samples were re-suspended in DI water and centrifuged
again. This was repeated for a total of 5 washings. DHP-Gel adducts were then dried under
vacuum at room temperature to obtain yields shown in Table 2. Solutions of neat gelatin and
gelatin that had been subjected to the oxidative conditions of DHP preparation were centrifuged
as described above and were not found to precipitate.
4.3.3. Fourier-transform infrared spectroscopy
Lignin DHP and DHP-Gel adducts were analyzed using a Bruker Vertex V70
Spectrometer (Bruker Optics Billerica MA) equipped with an MVP-Pro diamond single
reflection attenuated total reflectance (ATR) accessory (Harrick Scientific Pleasantville NY), and
100 scans at 6 cm-1 resolution were averaged for each sample using a DTGS detector and scan
frequency of 5 kHz. In all cases, the spectrum of the clean diamond crystal was used as the
reference spectrum. All spectral manipulations were performed using OPUS 6.0 (Bruker Optics,
Billerica MA).
4.3.4. X-ray photoelectron spectroscopy
The spectra were acquired with a Kratos Axis Ultra, using monochromatic Al-Kα X-rays.
Analysis chamber pressures were in the mid-10-8 torr range during measurements. Samples were
mounted on a 7mm x 7mm piece of Scotch Brand 3M double-sided tape (cat #137). The

75
materials covered the tape well enough to prevent exposure of the glue, and the tape was secured
to a piece of OFHC copper which was slightly larger than the tape. All spectra were acquired
with the analyzer set in hybrid mode, with the charge neutralizer on. The Pass Energy was set at
80 eV for surveys and 20 eV for high-resolution scans. Step sizes were 0.5 eV and 0.1 eV for
survey and high-res scans, respectively. The survey scan dwell time was set at 150 ms, while
values for high-resolutions scans varied from 600-2000 ms depending on the peak intensity.
4.3.5. Scanning electron microscopy and energy dispersive X-ray spectroscopy
Scanning electron microscopy (SEM) images were collected on a field emission SEM
(FESEM - FEI NanoSEM 630) at 2 or 3 kV under high vacuum (1.7 x 10-6 Torr). Samples were
sputter coated with iridium prior to imaging. Characteristic X-rays were collected with an X-Max
silicon drift detector (Oxford Instruments) inside the FESEM at 10 kV under low vacuum
conditions (0.6 Torr) in order to prevent sample charging. Samples were not sputter coated prior
to EDS analysis. Elements were selected and quantified using Aztec Energy Analyser Software
(Oxford Instruments).
4.3.6. Nuclear magnetic resonance spectroscopy
NMR spectra were acquired on a Bruker Biospin (Billerica, MA, USA) AVANCE 500
(500 MHz 1H resonance freq.) spectrometer fitted with a cryogenically-cooled gradient probe
having inverse geometry, i.e., with the proton coils closest to the sample. Spectra were processed
with Bruker’s Topspin 3.1 software, using the central solvent peak as internal reference [δH/δC:
dimethyl sulfoxide (DMSO), 2.50/39.5 ppm]. The DHP or DHP-Gel adducts (~50 mg) were
placed in an NMR tube (ID: 4.1 mm), swelled in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul), and
subjected to adiabatic 2D-HSQC (‘hsqcetgpsisp2.2’) experiments. Processing used typical
matched Gaussian apodization in F2 (LB = -0.3, GB = 0.001), and squared cosine-bell and one
level of linear prediction (32 coefficients) in F1 (Mansfield, 2012). For an estimation of the
various inter-unit linkage types in DHP and DHP-Gel adducts (Table 3; β-ether/α-OH, β-ether/α-
O-aryl, phenylcoumaran, pinoresinol, and dibenzodioxocin), the well resolved Cα-Hα contours
were integrated; no correction factors were used.
4.4. Results
4.4.1. Preparation of DHP-Gel adducts
DHP-Gel adducts were prepared by adding various quantities of porcine or bovine gelatin
to lignin DHP as it polymerized in the cases of DHP-Gel1, DHP-Gel2, DHP-Gel3, and DHP-Gel4,
or following lignin polymerization in the case of DHP-Gel5. Adduct yields following
centrifugation are shown in Table 4.2. The adducts were characterized using IR, EDS, XPS,
SEM, and NMR, as detailed below.

76
Table 4.2. Preparation and yields of DHP and DHP-Gel adducts.
sample CA (mg) PG (mg) BG (mg) yield (mg) yield (%)
DHP 200 0 0 124 62
DHP-Gel1 200 200 0 154 39
DHP-Gel2 200 0 80 139 50
DHP-Gel3 200 0 40 159 66
DHP-Gel4 200 0 20 138 63
DHP-Gel5 200 200 0 112 28
CA: coniferyl alcohol, PG: porcine gelatin, BG: bovine gelatin. Yields were calculated by
dividing the mass of recovered product by the total mass of reactants (i.e., CA + PG or BG). In
the case of DHP-Gel5, porcine gelatin was added following DHP polymerization.
4.4.2. Fourier-transform infrared spectroscopy of DHP-Gel adducts
Fig 4.1 shows FT-IR spectra of neat gelatin, neat DHP, and DHP-Gel adducts. The DHP
FT-IR spectrum exhibited bands typical of lignin DHPs (Faix, 1988). The DHP-Gel adducts
exhibited three peaks indicative of protein incorporation into the lignin. The peaks were located
at approximately 3200 cm-1, 1658 cm-1, and 1540 cm-1, and were previously observed in lignin-
protein adducts prepared by reacting DHPs with tripeptides (Diehl et al., in review). These peaks
were indicative of protein incorporation but were not specifically diagnostic toward covalent
versus non-covalent lignin-protein bonding. Qualitatively, FT-IR spectra showed that DHP-Gel1
apparently contained the most protein, with protein content decreasing through DHP-Gel2, DHP-
Gel3, and DHP-Gel4. Protein content of DHP-Gel5 (gelatin added following lignin
polymerization) appeared to lie between that of DHP-Gel3 and DHP-Gel4.

77
Fig 4.1. FT-IR of neat DHP and DHP-Gel adducts. Y-scaling is arbitrary.
4.4.3. Morphology and nitrogen content of DHP-Gel adducts
Fig 4.2 shows SEM images of neat DHP and DHP-Gel adducts. The neat DHP was
composed of smooth spheres of varying sizes, as previously observed (Micic et al., 2003). In the
cases of DHP-Gel1, DHP-Gel2, DHP-Gel3, and DHP-Gel4, the samples exhibited spherical
morphology, but the sizes and shapes of the spheres varied among samples and within samples.
DHP-Gel4 (lowest concentration of gelatin) exhibited the most variation in particle shape and
size—an affect that was reproducible. The particles within these DHP-Gel adducts exhibited
bumpy surfaces, which can be seen especially well in Fig. 2, DHP-Gel3, inset. Spherical particles
of DHP-Gel5 (gelatin added following polymerization) exhibited smooth surfaces, similar to neat
DHP.

78
Fig 4.2. SEM images of DHP-Gel1 (top left), DHP-Gel2 (top right), DHP-Gel3 (middle left),
DHP-Gel4 (middle right), DHP-Gel5 (bottom left) and neat DHP (bottom right). Bar: 2 µm
(DHP-Gel3 inset bar: 500 nm, DHP-Gel5 inset bar: 1 µm).
It was perceived that gelatin may not have been homogeneously dispersed throughout the
DHP-Gel particles (see Fig 4.3, models A and B). In order to test this hypothesis, elemental
analysis data was obtained using both XPS and EDS. XPS is a surface sensitive technique with
an approximate information depth of only 10 nm. In contrast, EDS has an information depth of
>1 µm when employing an accelerating voltage of 10 kV to a “light” substrate (i.e., an organic
substance such as the lignin-gelatin complex). Because the DHP-Gel particles were generally
several tens or hundreds of nanometers in diameter (Fig 4.2), XPS analyses were expected to
reveal nitrogen content near the surface (i.e., shell) of the particles, while EDS analyses were
expected to reveal nitrogen content throughout multiple particles.
Fig 4.3 shows the atomic nitrogen percentages of the DHP-Gel adducts as determined by
XPS and EDS (averages were determined by sampling three locations per DHP-Gel complex).

79
Two sample t-tests (using unequal variances and α = 0.05) showed that DHP-Gel1, DHP-Gel2,
DHP-Gel3, and DHP-Gel4 contained no significant differences in nitrogen as determined by XPS
and EDS (p-values: DHP-Gel1: 0.215, DHP-Gel2: 0.783, DHP-Gel3: 0.659, DHP-Gel4: 0.212),
suggesting a morphology similar to Fig 4.3, model A. In the case of DHP-Gel5 (gelatin added
following polymerization), the nitrogen content determined by XPS was significantly higher (p =
0.005) than the nitrogen content determined by EDS. This suggested that gelatin preferentially
bound to the lignin surface when added following lignin polymerization, suggesting morphology
similar to Fig 4.3, model B.
Fig 4.3. Morphology and nitrogen atomic percentages for DHP-Gel adducts as determined by
XPS and EDS (averages were determined by sampling three locations per DHP-Gel complex;
error bars are one positive and one negative standard deviation). For DHP-Gel morphological
models (A and B), black circles represent individual lignin modules (thin and thick lines
represent hydrophilic and hydrophobic domains, respectively), which may aggregate to form
macromolecules (Micic et al., 2004). Green lines represent gelatin. XPS and EDS showed that
model A best represented DHP-Gel1, 2, 3, and 4, while model B best represented DHP-Gel5.
Within the plant cell wall, lignification of a given region generally occurs following the
deposition of all other structural wall polymers, including structural proteins (Donaldson, 2001;
Boerjan et al., 2003). In addition, cell wall lignin (especially lignin from the cell corner region of
the middle lamella) adopts a similar morphology to the DHP and DHP-Gel adducts observed
here (Donaldson, 1994; Hafren et al., 2000; Terashima et al., 2004). This suggests that lignin
may surround and/or entangle cell wall proteins during lignification in planta, resulting in

80
morphology similar to Fig 4.3, model A. This physical entanglement, with the potential also for
lignin-protein covalent crosslink formation, may explain why protein is often found as a
contaminant in lignin extractions (Hatfield et al., 1994; Fukushima and Hatfield, 2001).
4.4.3. Nuclear magnetic resonance spectroscopy of DHP-Gel adducts
Fig 4.4 shows the HSQC NMR spectrum of DHP-Gel1, which was representative of the
DHP-Gel adducts. Shifts in orange were added during processing and are representative of
gelatin shifts. Gelatin shifts were visible when viewing the HSQC spectrum at a lower contour
level than shown in Fig 4.4. There are several reasons why the gelatin shifts were less intense
than the lignin shifts. First, the gelatin was the limiting reagent in most cases (except in the cases
of DHP-Gel1 and DHP-Gel5). Second, the gelatin was not completely incorporated into the
lignin. And third, the DHP-Gel adducts were not fully soluble in the NMR solvent system
(DMSO-d6/pyridine-d5). The gelatin shifts could be expected to be depressed in intensity
compared to the lignin shifts if the lignin component is more soluble than the gelatin component.
Diehl et al. (2014) previously identified diagnostic NMR shifts for lignin-protein
crosslinks. None of these shifts were observed in NMR spectra of DHP-Gel adducts, which
suggests that lignin and gelatin did not covalently crosslink. This may not be surprising, as Diehl
et al. (in review) also showed that cysteine and tyrosine were the only amino acid residues to
substantially crosslink with lignin under similar conditions of DHP preparation, and these
residues are almost entirely absent from gelatin (Table 1.1). The apparent lack of DHP-Gel
crosslinking suggested that non-covalent interactions were largely responsible for the observed
DHP-Gel interaction. In the case of DHP-Gel1 and DHP-Gel5, equal quantities of gelatin were
added to the reactions, with the only difference being whether the gelatin was added during
lignin polymerization or after. The DHP-Gel1 (gelatin added during lignin polymerization)
showed greater gelatin incorporation compared to DHP-Gel5 (gelatin added after lignin
polymerization) (Fig 4.1 and Fig 4.3). This increased gelatin incorporation was probably due to
physical entanglement of the gelatin within the lignin, as evidenced by XPS and EDS analyses
(Fig 4.3). Though DHP-Gel covalent crosslinks were not readily identified by NMR it may be
inappropriate to completely rule out the possibility of crosslink formation, albeit in minor
amounts. Isotopically labeled proteins and/or monolignols may be useful toward identifying trace
lignin-protein crosslinkages in further in vitro and in vivo experiments.

81
Fig 4.4. Side chain and aromatic (inset) regions of the HSQC NMR spectrum of DHP-Gel1.
Shifts in orange were added during processing. These shifts are indicative of gelatin and can be
observed when the HSQC spectrum is viewed at a lower contour level than shown here.
Table 4.3 shows estimates of the lignin inter-unit linkage ratios for the DHP and DHP-
Gel adducts as determined by volume integration of the well-resolved HSQC α-shifts. Neat DHP
contained linkage ratios typical of DHPs (Terashima et al., 1995; Terashima et al., 2009;
Tobimatsu et al., 2012). The variation in inter-unit linkage ratios exhibited no clear trend with
regards to the quantity of gelatin added, suggesting that gelatin has no significant effect on the
mechanism of lignin polymerization. The observed variation of inter-unit linkage ratios is most
likely attributable to the fact that DHP syntheses are inherently difficult to reproduce (Cathala et
al., 1998).
Table 4.3. Inter-unit linkage ratios of DHP and DHP-Gel adducts.
HSQC signal ratio (as % of total α-signal)
β-ether/α-
OH
β-ether/α-
aryl β-5 β-β Dibenz.
DHP 27.3 1.9 50.3 19.2 1.3
DHP-Gel1 23.5 0.5 53.4 18.1 4.6
DHP-Gel2 23.2 0.4 55.6 20.3 0.4
DHP-Gel3 13.5 0.7 58.7 25.4 1.7
DHP-Gel4 20.8 4.0 49.2 22.0 4.0
DHP-Gel5 32.8 1.3 45.3 16.4 4.1

82
4.5. Conclusions
DHP-Gel adducts were prepared under biomimetic conditions of lignin polymerization. A
variety of methods was used to characterize the DHP-Gel adducts. FT-IR showed incorporation
of gelatin into lignin DHP, but was unable to definitively show either the presence or absence of
lignin-gelatin covalent linkages. SEM showed that the DHP-Gel adducts generally consisted of
spherical particles ranging from tens to hundreds of nanometers in diameter, with morphological
details varying among samples. XPS and EDS were used in combination to show that gelatin was
relatively evenly dispersed throughout DHP-Gel particles when added during lignin
polymerization (Fig 4.3, model A), but aggregated mostly at the surface of DHP-Gel particles
when added following lignin polymerization (Fig 4.3, model B). It was interesting to note that
covalent crosslinking was not observed by HSQC NMR. This may not be surprising, as Diehl et
al. (in review) previously showed that cysteine and tyrosine were the only amino acid residues to
substantially crosslink with lignin under similar conditions of DHP preparation, and gelat in is
almost entirely lacking in these residues. The observation that gelatin adsorbed to the lignin
surface when added to lignin post-polymerization provided further evidence that covalent
crosslinking was not necessary to account for the lignin-gelatin interaction.
The lignin-gelatin interaction appears to be essentially non-covalent, and gelatin peptide
was dispersed throughout the DHP-Gel particles when it was added over the course of lignin
polymerization. In planta, lignification occurs in a pre-deposited polysaccharide and protein
matrix. Based on the results of this study it seems plausible that cell wall structural proteins may
become surrounded or physically entangled by lignin without necessarily forming covalent
crosslinks. It may be possible for structural proteins to serve as initiation sites of lignification
without lignin-protein covalent bond formation. Based on previous studies, covalent crosslinking
may also occur if cysteine and tyrosine residues are present (Diehl et al., in review).
Alternatively, lignin-protein crosslinking may occur at saccharide residues that are found on
plant cell wall structural proteins (particularly extensins and arabinogalactan proteins) but not on
gelatin (Lamport et al., 2011; Toikka et al., 1998; Wilson and Fry, 1986); the study reported here
did not test such a hypothesis. The intimate physical entanglement of the lignin-protein complex
may make covalent crosslinking favorable by bringing reactive lignin and protein species into
close proximity. The formation of such complexes may not only serve as lignin nucleation sites,
but may also help to rigidify and/or waterproof the cell wall. Further research is needed to
determine the role of covalent and non-covalent lignin-protein complexes in plant cell walls.
4.6. Acknowledgements
This material is based upon work supported as part of The Center for LignoCellulose
Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of
Energy, Office of Science, under Award Number DE-SC0001090. Student fellowships were
provided by the USDA National Needs Program and the National Science Foundation via the
CarbonEARTH program. Many thanks to Julie Anderson, Melisa Yashinski, and Vince Bojan

83
(Penn State Materials Research Institute) for acquisition of SEM, EDS and XPS data and for
valuable discussions, Jenna Ferraraccio for assistance with figure preparation, and John Ralph,
Yuki Tobimatsu, and Matt Regner for acquisition of NMR spectra and valuable discussions
regarding multidimensional NMR of lignin.
4.7. References
Albersheim, P.; Darvill, A.; Roberts, K.; Sederoff, R.; Staehelin, A. Principles of Cell Wall
Architecture and Assembly. In Plant Cell Walls; Garland Science, 2010; pp 227-27
Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Re. Plant Biol. 2003, 54, 519-
546.
Cathala, B.; Saake, B.; Faix, O.; Monties, B. 1998. Evaluation of the reproducibility of the
synthesis of dehydrogenation polymer models of lignin. Polymer Degradation and Stability
59:65-69.
Chapple, C.; Ladisch, M.; Meilan, R. Loosening lignin's grip on biofuel production. Nat.
Biotechnol. 2007, 25, 746-748.
Chen, Y. and S. Sarkanen. 2006. Cellulose Chemistry and Technology, 40(3-4): 149-163. "From
the macromolecular behavior of lignin components to the mechanical properties of lignin-based
plastics."
Chen, F.; Dixon, R. A. Genetic manipulation of lignin biosynthesis to improve biomass
characteristics for agro-industrial processes. In Vitro Cell. Dev. Biol. - Animal. 2008, 44, S28-
S29.
Clark, J. H., Deswarte, F. E. I. and T. J. Farmer. 2009. Biofuels, bioproducts and biorefining, 3:
72-90. "The integration of green chemistry into future biorefineries."
Donaldson, L.A. 1994. Mechanical constraints on lignin deposition during lignification. Wood
Sci. and Technol. 27:111-118.
Donaldson, L.A. 2001. Lignification and lignin topochemistry – an ultrastructural view.
Phytochemistry 57: 859-873.
Dorrestijn, E., Laarhoven, L. J. J., Arends, I. W. C. E. and P. Mulder. 2000. Journal of analytical
and applied pyrolysis, 54: 153-192. "The occurrence and reactivity of phenoxyl linkages in
lignin and low rank coal."
Eastoe, J.E. 1955. The amino acid composition of mammalian collagen and gelatin. Biochemical
Journal 61(4):589-600.
Faix O, Beinhoff O. 1988. Journal of Wood Chemistry and Technology, 8 (4): 505-522. "FTIR
spectra of milled wood lignins and lignin polymer models with enhanced resolution obtained by
deconvolution."

84
Fukushima, R.S.; Hatfield, R.D. 2001. Extraction and isolation of lignin for utilization as a
standard to determine lignin concentration using the acetyl bromide spectrophotometric method.
Journal of Agricultural and Food Chemistry 49(7):3133-3139.
Gellerstedt, G., Sjoholm, E. and I. Brodin. 2010. The Open Agriculture Journal, 3: 119-124.
"The wood-based biorefinery: A source of carbon fiber?"
Hafren, J., Funino, T., Itoh, T., Westermark, U., Terashima, N. 2000. Ultrastructural changes in
the compound middle lamella of Pinus thunbergii during lignification and lignin removal.
Holzforschung 54:234-240.
Hatfield, R.D.; Jung, H.G.; Ralph, J.; Buxton, D.R.; Weimer, P.J. 1994. A comparison of the
insoluble residues produced by the Klason lignin and acid detergent lignin procedures. J. Sci.
Food Agric. 65:51-58.
Jung, H. G.; Allen, M. S. Characteristics of plant cell walls affecting intake and digestibility of
forages by ruminants. J. Animal Sci. 1995, 73, 2774-2790.
Jung, H.G. Forage lignins and their effects on fiber digestibility. Agron. J. 1989, 81, 33-38.
Lamport, D.T.A.; Kieliszewski, M.J.; Chen, Y.; Cannon, M.C. Plant Phys 2011, 156, 11-19.
Li, X.; Weng, J. K.; Chapple, C. Improvement of biomass through lignin modification. Plant J.
2008, 54, 569-581.
Ludley, F. H. and J. Ralph. 1996. Journal of Agricultural and Food Chemistry, 44: 2942-2943.
"Improved preparation of coniferyl and sinapyl alcohols."
Mansfield, S. D.; Kim, H.; Lu, F.; Ralph, J. Whole plant cell wall characterization using
solution-state 2D NMR. Nature Protocols. 2012, 7(9), 1579-1589.
Micic, M.; Radotic, K.; Jeremic, M.; Leblanc, R. M. Study of the self-assembly of the lignin
model compound on cellulose model substrate. Macromol. Biosci. 2003, 3 (2), 100-106.
Micic, M.; Radotic, K.; Jeremic, M.; Djikanovic, D.; Kammer, S.B. 2004. Study of the lignin
model compound supramolecular structure by combination of near-field scanning optical
microscopy and atomic force microscopy. Colloids and Surfaces B: Biointerfaces 34:33-40.
Stewart, J. J.; Kadla, J. F.; Mansfield, S. D. The influence of lignin chemistry and ultrastructure
on the pulping efficiency of clonal aspen (Populus termuloides Michx). Holzforschung 2006, 60,
111-122.
Terashima, N.; Akiyama, T.; Ralph, S.; Evtuguin, D.; Neto, C.P.; Parkas, J.; Paulsson, M.;
Westermark, U.; Ralph, J. 2D-NMR (HSQC) difference spectra between specifically 13C-
enriched and unenriched protolignin of Ginkgo biloba obtained in the solution state of whole cell
wall material. Holzforschung 2009, 63, 379-384.

85
Terashima, N., Atalla, R.H., Ralph, S.A., Landucci L.L., Lapierre, C., Monties, B. 1995.
Holzforschung, 49: 521-527. "New preparations of lignin polymer models under conditions that
approximate cell wall lignification."
Terashima, N., Awano, T., Takabe, K., Yoshida, M. 2004. Formation of macromolecular lignin
in ginkgo xylem cell walls as observed by field emission scanning electron microscopy. C. R.
Biologies 327: 903-910.
Tobimatsu, Y.; Elumalai, S.; Grabber, J. H.; Davidson, C. L.; Pan, X.; Ralph, J.
Hydroxycinnamate conjugates as potential monolignol replacements: In vitro lignification and
cell wall studies with rosmarinic acid. ChemSusChem. 2012, 5 (4), 676-686.
Toikka, M.; Sipila, J.; Teleman, A.; Brunow, Gosta. 1998. Lignin-carbohydrate model
compounds. Formation of lignin-methyl arabinoside and lignin-methyl galactoside benzyl ethers
via quinone methide intermediates. J. Chem. Soc., Perkin Trans. 1 3813-3818.
Whitmore, F.W. 1978a. Lignin-carbohydrate complex formed in isolated cell walls of callus.
Phytochemistry 17:421-425.
Whitmore, F.W. 1978b. Lignin-protein complex catalyzed by peroxidase. Plant Science Letters
13:241-245.
Whitmore, F.W. 1982. Lignin-protein complex in cell walls of Pinus elliottii: Amino acid
constituents. Phytochemistry 21(2):315-318.
Wilson, L.G.; Fry, J.C. 1986. Extensin—A major cell wall glycloprotein. Plant, Cell and
Environment 9:239-260.

86
Chapter 5
Searching for lignin-protein linkages in Arabidopsis
5.1. Abstract
In order to explore lignin-protein linkages in planta, Arabidopsis thaliana was grown to maturity
and lignin was extracted from the inflorescence stems. Elemental analysis was used to estimate
the protein content of the crude Arabidopsis, the Arabidopsis following solvents extraction, and
the Arabidopsis lignin extracts. Nuclear magnetic resonance techniques were then used to search
for putative lignin-protein covalent linkages. No apparent linkages were identified, but further
work is needed in other wild type and mutant plant species, and isotopically labeled monolignols
may be useful toward investigating lignin-protein linkages in both wild type and mutant plants.
5.2. Introduction
The previous chapters have shown that lignin crosslinks with several amino acids under
ideal conditions (i.e., in neutral, non-polar solvents), and that cysteine and tyrosine crosslink with
lignin under biomimetic conditions of lignin polymerization. Finally, in an attempt to identify
lignin-protein linkages formed under natural conditions of lignin biosynthesis, Arabidopsis
thaliana (wild-type Columbia-0) plants were grown to maturity (8 weeks), then lignin was
extracted from the inflorescence stems and characterized.
Arabidopsis lignin is composed mainly of guaiacyl and syringyl type lignin moieties. The
lignin content of mature Arabidopsis inflorescence stems has been estimated at around 14%
using the Klason method (Chang et al., 2008), and 14-16% using the acetyl bromide method
(Yong Bum Park, unpublished data). The quantity of structural protein in mature Arabidopsis
cell wall, in terms of dry weight percentage, is unclear. However, Chang et al. (2008) previously
showed that extracted Arabidopsis lignin (dioxane and Klason lignin) contained about 3.7%
protein contamination. In order to estimate the quantity of proteins in Arabidopsis, nitrogen
content was determined for mature Arabidopsis inflorescence stems at three levels of sample
preparation: crude ball-milled Arabidopsis, Arabidopsis that was ball-milled and solvents
extracted to yield cell wall material, and extracted Arabidopsis lignin. Nitrogen content can then
be used to determine protein percentage by multiplying by 6.25 (assuming all nitrogen is due to
protein) (Chang et al., 2008; Fukushima and Hatfield, 2001), and in this way the protein content
can be monitored throughout the various steps of Arabidopsis lignin extraction.
Lignin was extracted from Arabidopsis following a previously described acidic dioxane
(ADL) method (Fukushima and Hatfield, 2004). It has been postulated that this extraction
method selectively cleaves α-ether linkages, which should raise concerns regarding the cleavage
of putative lignin-protein linkages, as well. However, this method was deemed useful for several
reasons. First, it was not possible to extract lignin using the typical milled wood lignin procedure
of refluxing the sample in 96:4 dioxane/water. This method has been employed for decades;

87
however, during preliminary investigations with Arabidopsis, only ~2 mg of lignin was extracted
per 1 g of Arabidopsis cell wall material, which is extremely inefficient and yields far too little
lignin for effective characterization. Furthermore, lignin-protein linkages are expected to be low
in quantity in wild type plants, so observing the putative linkages in cellulolytic enzyme lignins
or whole cell walls seems unlikely due to very low signal to noise.
Following lignin extraction and protein content estimation, the Arabidopsis lignin was
characterized using 2D nuclear magnetic resonance (NMR) spectroscopy. The two most
important techniques were heteronuclear single quantum coherence (HSQC) and heteronuclear
multiple bond correlation (HMBC) experiments, which were previously shown to be quite useful
toward elucidating lignin-protein linkages.
5.3. Experimental
5.3.1. Growth and lignin extraction from Arabidopsis
Arabidopsis samples were prepared as follows. After 4 days of cold treatment at 4°C,
Arabidopsis thaliana wild-type (Colombia (Col-0) ecotype) seedlings were grown on 1× MS
medium (Murashige and Skoog, 1962) containing 1% sucrose for 1 week, and 500–600 seedlings
were transferred onto soil and grown for 7–8 more weeks under 70 µmol m-2s-1 light intensity
(day/night: 16/8 h, temperature: 22/16°C). The matured Arabidopsis inflorescence stems were
collected and frozen at -80° C. Prior to lignin extraction, Arabidopsis was prepared by grinding
in a blender, followed by freeze-drying. Samples (typically 2-3 g) were then Soxhlet extracted
with water, ethanol, chloroform, and acetone, for eight hours per solvent. Solvents extracted
Arabidopsis was then Wiley milled to pass a 60 mesh screen, then ball milled in a Retsch
cryomill (1.5 g Wiley milled Arabidopsis for 2 hr at 10 Hz). Lignin was extracted following the
previously described acidic dioxane lignin (ADL) method (Fukushima and Hatfield, 2004).
Briefly, 1 g of solvents extracted Arabidopsis was refluxed for 45 min in 20 ml of 90:10
dioxane/2 M HCl (aq). The solubilized lignin was then filtered through a WhatmanTM glass
microfiber filter, and the cell wall residue was rinsed with 96:4 dioxane/water. The dioxane
filtrates were combined and neutralized with sodium bicarbonate, filtered through a 0.45 µm
nylon membrane filter, then concentrated under reduced pressure. The concentrated lignin
solution (~1-2 ml) was added dropwise to ~40 ml DI water in a centrifuge tube, then centrifuged
at 10k g at 5° C for 20 min. The aqueous supernatant was poured off and saved, then the lignin
pellet was resolubilized using a minimum volume of dioxane. Ether was added to the centrifuge
tube and the sample was centrifuged at 10k g at 5° C for 15 min, and this solubilization followed
by ether washing and centrifugation was repeated for a total of two cycles. The ether
supernatants were then discarded and the lignin was freeze dried and stored at 4° C for future
characterization. Typically, lignin yields were 30-35 mg per 1 g of solvents extracted
Arabidopsis cell wall material.

88
5.3.2. Elemental analysis of Arabidopsis lignin
Nitrogen weight percentages were determined using a CE Instruments EA 1110 CHNS-O
elemental analyzer. Approximately 3 mg of sample were massed to the nearest ten-thousandth of
a milligram and analyzed according to the manufacturer’s instruction. Protein content was
determined by multiplying nitrogen percentage by 6.25, as described previously (Chang et al.,
2008; Fukushima and Hatfield, 2001).
5.3.3. Nuclear magnetic resonance spectroscopy of Arabidopsis lignin
NMR spectra were collected in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul). DMSO-
d6/pyridine-d5 was chosen because it is a preferred solvent for NMR of lignin DHP, milled
wood lignin (MWL), and whole cell walls (Kim and Ralph, 2010). NMR spectra were acquired
on a Bruker Biospin (Billerica, MA, USA) AVANCE 500 (500 MHz 1H resonance freq.)
spectrometer fitted with a cryogenically-cooled gradient probe having inverse geometry, i.e.,
with the proton coils closest to the sample. Spectra were processed with Bruker’s Topspin 3.1
software, using the central solvent peak as internal reference (δH/δC: dimethyl sulfoxide
(DMSO), 2.50/39.5 ppm). Lignin (~20 mg) was placed in an NMR tube (ID: 4.1 mm), swollen
homogeneously in DMSO-d6/pyridine-d5 (4:1 v/v, 500 ul), and then subjected to adiabatic 2D-
HSQC (‘hsqcetgpsisp2.2’) experiments using the parameters described by Mansfield et al.
(2012). Processing used typical matched Gaussian apodization in F2 (LB = -0.3, GB = 0.001),
and squared cosine-bell and one level of linear prediction (32 coefficients) in F1 (Mansfield et
al., 2012).
5.4. Results and discussion
5.4.1. Lignin extractions from Arabidopsis
Arabidopsis was grown to maturity, then inflorescence stems were harvested and
prepared for lignin extraction by solvents extracting and cryomill grinding. The grinding time of
approximately 2 hr per 1.5 g of Arabidopsis resulted in highly variable particle sizes (Fig 5.1).
Longer grinding times may be necessary, although increased sample alteration following
increased grinding times is always cause for concern. The grinding times employed here allowed
for the extraction of ~30-35 mg of acidic dioxane lignin (ADL) per g of Arabidopsis cell wall
material. This was similar to previously reported ADL yields for grassy plants such as alfalfa and
red clover (Fukushima and Hatfield, 2001, 2004). This material was then subjected to protein
content estimation and nuclear magnetic resonance spectroscopy, as described below.

89
Fig 5.1. Optical microscopy of solvents extracted and ball milled Arabidopsis cell wall material.
Particle size distribution is highly variable. Scale bar = 50 µm.
5.4.2. Protein content of Arabidopsis extracts
Protein contents of crude Arabidopsis, solvents extracted Arabidopsis, and Arabidopsis
lignin were estimated by multiplying the measured nitrogen atomic percentages by a factor of
6.25, as previously described (Chang et al., 2008; Fukushima and Hatfield, 2001). It was found
that crude Arabidopsis inflorescence stems contained approximately 5.31% protein, Arabidopsis
that had been solvents extracted contained 4.94% protein, and Arabidopsis ADL contained
approximately 3.75% protein. This was very similar to the protein content determined by Chang
et al. (2008) for dioxane and Klason Arabidopsis lignins, but was considerably higher than that
of Loblolly ADL, which was typically <1%. This may be expected due to the prominent
secondary cell walls in Loblolly and other plants with a strong tree habit, and the general lack of
protein in secondary cell walls. Because of the increased protein content in Arabidopsis (and
presumably grasses and other non-grasses exhibiting a grass-like growth habit), wild type and
mutant Arabidopsis lines may be useful for future investigations of lignin-protein linkages.
Table 5.1. Nitrogen content and estimated protein content of Arabidopsis extracts.
N % Protein %
Crude Arabidopsis 0.85 5.31
Solvents extracted Arabidopsis 0.79 4.94
Arabidopsis acidic dioxane lignin 0.60 3.75

90
5.4.3. Nuclear magnetic resonance spectroscopy of Arabidopsis lignin
Acidic dioxane lignin isolated from Arabidopsis inflorescence stems was analyzed using
HSQC and HMBC NMR techniques in DMSO-d6/pyridine-d5. Table 5.2 shows estimates of the
lignin inter-unit linkage ratios as determined by volume integration of the well-resolved HSQC
α-shifts, and the ratios were typical of dicotyledonous G/S lignins (Capanema et al, 2004). An
HSQC spectrum of Arabidopsis ADL is shown in Fig 5.2; however, the peaks discussed in
further detail below were generally too weak to be observed at the contour levels shown.
Table 5.2. Inter-unit linkage ratios of Arabidopsis ADL.
HSQC signal ratio (as % of total α-signal)
β-ether/α-OH β-ether/α-aryl β-5 β-β Dibenz.
70.6 tr 17.7 9.7 2.1
Estimates of the lignin inter-unit linkage ratios as determined by volume integration of the well-
resolved HSQC α-shifts are shown. tr = trace.
A very weak shift was observed in the HSQC spectrum at 4.4/50.8 ppm. This is close to
the observed α-shift of a lignin-cysteine linkage (4.4/50.6) identified in Chapter 2. However, the
corresponding HMBC spectrum did not identify this shift as an α-shift, and thus the likelihood of
this shift being attributable to a lignin-cysteine linkage seems low. This demonstrates the
importance of using multiple NMR techniques when investigating putative lignin-protein
crosslinks. No other putative lignin-protein shifts were identified in the HSQC and HMBC
spectra. Furthermore, essentially no lignin α-ester and only very minor quantities of α-ether
linkages were observed in the HSQC spectrum. This suggests that even lignin-hemicellulose
linkages were essentially absent from the Arabidopsis ADL. This may have been due to the
harshness of the extraction procedure, and less harsh lignin extraction techniques may be
necessary in order to optimize the chances of lignin-protein linkage identification.

91
Fig 5.2. HSQC NMR of Arabidopsis acidic dioxane lignin. Typical lignin shifts and residual
dioxane and pyridine solvent shifts are labeled. Cinnamyl end groups and dibenzodioxocin
structures were not abundant enough to be seen at the contour levels shown. Lignin-protein
linkages were not apparent even at low contour levels.
5.5. Conclusions
Wild type Arabidopsis was grown to maturity and inflorescence stems were harvested
and lignin extracted using the acidic dioxane (ADL) method. It was found that the ADL
contained a significant protein content (3.75%), so the lignin was characterized using HSQC and
HMBC NMR techniques. No lignin-protein linkages were identified. Furthermore, the lignin
appeared to be essentially free of lignin-carbohydrate linkages. The acidic dioxane extraction
method may have cleaved such linkages if they were in fact present in the wild type Arabidopsis,
and it may be beneficial to use a milder lignin extraction procedure. Alternatively, lignin-protein
linkages may be very low in abundance (or non-existent), and thus below the NMR signal to

92
noise limit. Exploration of lignin-protein linkages in mutant plant lines and/or using isotopically
labeled monolignols may help probe this question.
5.6. Acknowledgements
This material is based upon work supported as part of The Center for LignoCellulose
Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of
Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-
SC0001090. Student fellowships were provided by the USDA National Needs Program and the
National Science Foundation via the CarbonEARTH program. Many thanks to Ephraim Govere
for assistance in acquiring elemental analysis data.
5.7. References
Capanema, E.A.; Balakshin, M. Y.; Kadla, J.F. J. Agric. Food Chem. 2004, 52, 1850-1860.
Chang, X.F.; Chandra, R.; Berleth, T.; Beatson, R.P. J. Agric. Food Chem. 2008, 56, 6825-6834.
Fukushima, R.S.; Hatfield, R.D. J. Agric. Food Chem. 2001, 49, 3133-3139.
Fukushima, R.S.; Hatfield, R.D. J. Agric. Food Chem. 2004, 52, 3713-3720.
Kim, H.; Ralph, J. Org. Biomol. Chem. 2010, 8, 576-591.
Mansfield, S.D.; Kim, H.; Lu, F.; Ralph, J. Nat. Protoc. 2012, 7(9), 1579-1589.
Murashige, T.; Skoog, F. Physiol. Plant 1962, 15, 473-497.

93
Chapter 6
Conclusions
6.1. Research summary
Lignin is a heterogeneous, aromatic polymer that is biosynthesized in the cell walls of
almost all lands plants. It is economically relevant to several industries, especially pulp and
paper, agricultural, and biofuels industries, where lignin’s natural recalcitrance to extraction and
degradation pose problems. Additionally, lignin could potentially be used as a feedstock for
many renewable products, including plastics, activated carbons, carbon fibers, solid and liquid
fuels, and other specialty chemicals, but many of these lignin utilization schemes are beyond the
reach of currently available technology. To this end, it is important that we continue to focus on
both fundamental and applied advances in lignin and its related fields.
The plant cell wall is a complex matrix composed of structural polymers such as
cellulose, hemicelluloses, pectins, lignin, and proteins, as well as lower molecular weight organic
compounds. Lignin polymerization occurs within the pre-deposited polysaccharide and protein
matrix, causing lignin to interact with its local chemical environment. This results not only in
non-covalent interactions, but also in covalent crosslinking with hemicelluloses, and perhaps
other matrix components, such as proteins. It has been hypothesized that lignin-protein linkages
may be important in both wild type and mutant plant lines, yet lignin-protein linkages have not
been previously described. This work helps fill that gap in the literature by describing the
preparation and characterization of lignin-protein linkages.
In the first study, a low molecular weight quinone methide, representative of native lignin
quinone methide structures, was reacted with single amino acids. It was found that under these
ideal conditions, a diverse array of amino acids, including those bearing thiol, amine, carboxylic
acid, and hydroxyl functional groups on their side chains, reacted with lignin quinone methides
to form lignin-protein model compounds. Characterization of these compounds with NMR
resulted in the identification of diagnostic lignin-protein crosslink signatures, which were helpful
in identifying lignin-protein linkages in more complex model systems, and should also be helpful
toward identifying lignin-protein linkages in native lignins.
The second study expanded upon the first by exploring the reactivities of amino acids
toward lignin quinone methide intermediates under biomimetic conditions. Specifically, the
amino acids were incorporated into short peptide chains and exposed to lignin dehydrogenation
polymer as it polymerized in aqueous media. Using NMR, it was possible to show that cysteine
and tyrosine-containing peptides were covalently incorporated into the synthetic lignin polymer,
while other amino acids were almost entirely inert under such biomimetic conditions. This
suggests that cysteine and/or tyrosine-rich proteins may be the most likely to covalently crosslink
with lignin in planta. In this study it was also shown that Fourier-transform infrared

94
spectroscopy and elemental analysis (specifically energy dispersive X-ray spectroscopy in this
case) were useful toward showing lignin-protein interactions.
A third study investigated the interactions between lignin DHP and gelatin protein.
Gelatin was chosen because it shares similar characteristics with plant cell wall structural
proteins, but is much more readily available. In addition, gelatin lacks cysteine and tyrosine,
which were the only two amino acids shown in this work to covalently crosslink with lignin in
aqueous media. Thus, the observed interaction between lignin and gelatin was considered worthy
of investigation. SEM, EDS, and FT-IR showed that gelatin was incorporated into lignin DHP,
but a lack of characteristic lignin-protein NMR signatures suggested that the interaction was
largely (or entirely) non-covalent. This indicates that lignin-protein non-covalent interactions
may be important in planta, and further work is necessary.
Finally, in a fourth study, an attempt was made to identify lignin-protein linkages in the
wild type dicot plant, Arabidopsis. Unfortunately, the cell wall protein content was shown to be
very low in this wild type plant, and NMR was unable to reveal lignin-protein linkages in acidic
dioxane lignin extracts. This does not necessarily rule out the formation of these linkages, but
may merely suggest that linkage abundance is below the detection limit of NMR. Furthermore,
this study did not investigate the abundance or importance of linkages in mutant plant lines, and
additional studies are needed.
Though the work described here has allowed for more efficient and reliable
characterization of lignin-protein linkages in model systems, more work is necessary, especially
regarding native plant systems. The final section of this document will outline some ways in
which future investigations could be carried out.
6.2. Future endeavors
To further investigate lignin-protein linkages in more realistic model systems, it may be
useful to lignify model cell walls, or cell walls that have been isolated from native plants, then
characterize these walls using NMR and other techniques. Cybulska et al. (2010) and
Dammstrom et al. (2005) previously described the preparation of model cell walls using either
pure cellulose, or cellulose with the incorporation of hemicelluloses and/or pectins. In addition,
Uraki et al. (2011) demonstrated the preparation of cellulose-based, honey-comb shaped model
cell walls (Fig 6.1).

95
Fig 6.1. Cell wall models. Left: cellulose, pectin, and xyloglucan model (Dammstrom et al.,
2005). Right: honey-comb cellulose model (Uraki et al., 2011). Incorporation of proteins into
these models, followed by lignin DHP polymerization, may allow for investigations into lignin-
protein crosslinking.
I propose the preparation of similar cell walls, but with the added inclusion of proteins of
interest. Peroxidases could be incorporated into the cell wall models during assembly, or flowed
over the models after assembly. Lignin monomers and hydrogen peroxide would then be flowed
over the cell wall models, causing lignin polymerization to take place. The entire cell wall model
would then be characterized without disturbing the micro and macro structures that formed, as
this might provide the most insight into the lignin polymerization mechanisms. Techniques such
as NMR could be used to explore putative lignin-protein covalent linkages, while other
techniques could be used to explore the topology and distribution of the lignin DHP. The
exploration of potential lignin nucleation sites would be particularly interesting (for example,
lignin may nucleate at sites rich in hemicelluloses, pectins/pectates, peroxidases, structural
proteins, or show no particular nucleation pattern at all). Visualization techniques such as SEM
and AFM could be useful in probing this question. Also, the use of fluorescent monolignols (and
perhaps additional labeling of other components) could be beneficial (Tobimatsu et al., 2011).
Monitoring lignin distribution and topology at various time points of polymerization might also
be helpful towards understanding the lignin polymerization mechanism.
In addition to more advanced model compound studies, studies using mutant plant lines
are also warranted. While lignin-protein linkages may exist in wild type plant lines, and studies
involving wild type plants should not be dismissed, mutant plant lines may show the most
promise toward identifying and characterizing lignin-protein linkages. These mutant plant lines
should be engineered to secrete cysteine and/or tyrosine-rich peptides into the plant cell walls, as
these amino acids have been shown to be most reactive toward lignin in model studies. A mutant
plant line meeting these characteristics has already been prepared, although lignin-protein
covalent linkages have yet to be identified at the time of document preparation (Liang et al.,
2008; Xu et al., 2013). It is hoped that lignin-protein linkages will be further explored in mutant

96
plant lines, as practical implications toward lignin extractability have been demonstrated in pilot
studies.
It seems appropriate to briefly address a method that could be useful toward enhancing
the identification of putative lignin-protein linkages via NMR. It has previously been shown that
NMR is a powerful technique for characterizing plant cell walls and their constituent
components, and this work has shown that the technique can also be applied to the identification
of lignin-protein linkages. However, when exploring lignin-protein linkages in complex chemical
systems, such as those that exist in planta, achieving adequate signal to noise is expected to
become a problem. This is because the ratio of lignin-protein linkages to all other chemical
structures in the sample is expected to be exceedingly low. Isotopic labeling experiments may
help address this problem. Previously, α-13C coniferyl alcohol glucoside (coniferin) was prepared
and fed to live ginkgo (Xie and Terashima, 1991), then the lignin was characterized via NMR
and the α-shifts were shown to exhibit increased signal to noise. A possible route to α-13C
coniferyl alcohol is shown in Fig 6.2 (slightly modified compared to Xie and Terashima’s
original route to α-13C coniferin). This labeled coniferyl alcohol could be useful for identifying
lignin-protein linkages in complex in vitro or in vivo systems, because NMR α-shifts of lignin-
protein linkages (as well as standard lignin α-shifts) would exhibit greater signal to noise
compared to spectra of unlabeled lignins (Fig 6.3).
Fig 6.2. Proposed synthetic route to α-13C coniferyl alcohol.

97
Fig 6.3. Standard lignin α-shifts (red) and α-shifts of lignin-protein linkages (red squares). These
shifts would exhibit greater signal to noise compared to standard lignin shifts (black) in spectra
of α-13C-enriched lignin, perhaps allowing for easier identification of putative lignin-protein
linkages.
Efficient extraction and utilization of lignin is of great economic importance. In spite of
this, there is still much about the lignin polymer that remains unknown. This work has
illuminated fundamental aspects of lignin-protein linkage formation in an attempt to better
understand how lignin interacts with the protein component of plant cell walls. It is hoped that
this research will translate into a practical means of reducing lignin’s recalcitrance toward
degradation and extraction.
6.3. References
Cybulska, J.; Vanstreels, E.; Tri Ho, Q.; Courtin, C.M.; Craeyveld, V.V.; Nicolai, B.; Zdunek,
A.; Konstankiewicz, K. Mechanical characteristics of artificial cell walls. J. of Food Eng. 2010,
96, 287-294.
Dammstrom, S.; Salmen, L.; Gatenholm, P. The effect of moisture on the dynamical mechanical
properties of bacterial cellulose/glucuronoxylan nanocomposites. Polymer 2005, 46, 10364-
10371.
Liang, H.; Frost, C.J.; Wei, X.; Brown, N.R.; Carlson, J.E.; Tien, M. Improved sugar release
from lignocellulosic material by inducing a tyrosine-rich cell wall peptide gene in poplar. Clean
2008, 36(8), 662-668.

98
Tobimatsu, Y.; Davidson, C.L.; Grabber, J.H.; Ralph, J. Fluorescence-tagged monolignols:
Synthesis, and application to studying in vitro lignification. Biomac. 2011, 12, 1752-1761.
Uraki, Y.; Tamai, Y,; Hirai, T.; Koda, K.; Yabu, H.; Shimomura, M. Fabrication of honeycomb-
patterned cellulose material that mimics wood cell wall formation processes. Mat. Sci. and Eng.
C 2011, 31, 1201-1208.
Xie, Y.; Terashima, N. Selective carbon 13-enrichment of side chain carbons of ginkgo lignin
traced by carbon 13 nuclear magnetic resonance. Mokuzai Gakkaishi 1991, 37(10), 935-941.
Xu, Y.; Chen, C.; Thomas, T.P.; Azadi, P.; Diehl, B.; Tsai, C.; Brown, N.; Carlson, J.E.; Tien,
M.; Liang, H. Wood chemistry analysis and expression profiling of a poplar clone expressing a
tyrosine-rich peptide. Plant Cell Rep. 2013, 32, 1827-1841.

Vita
Brett Galen Diehl
Education
May, 2014 Ph.D. Biorenewable Systems
Department of Agricultural and Biological Engineering
The Pennsylvania State University, University Park, PA
May, 2009 B.S. Wood Products, Processing and Manufacturing Option
School of Forest Resources
The Pennsylvania State University, University Park, PA
Research
2009-2014 Graduate Research, Penn State, University Park, PA
• Illuminated fundamental aspects of lignin-protein linkages, which may influence
the physical and chemical properties of plant cell walls impacting industries such
as agriculture, pulp and paper, and biofuels.
2008 NSF Research Experience for Undergraduates, Penn State, University Park, PA
• Researched cellulose-producing Acetobacter xylinum and methods of
generating biofuels from cellulosic materials.
2007 Paid Internship, Armstrong World Industries, Lancaster, PA
• Studied exotic hardwood species using scanning electron microscopy.
Publications
B.G. Diehl, H.D. Watts, J.D. Kubicki, M.R. Regner, J. Ralph, N.R. Brown. Towards lignin-
protein crosslinking: Nucleophilic amino acid adducts of a lignin model quinone methide.
Cellulose, accepted January, 2014, not yet published.
B.G. Diehl, N.R. Brown, C.W. Frantz, M.R. Lumadue, F. Cannon. Effects of pyrolysis
temperature on the chemical composition of refined softwood and hardwood lignins. Carbon
(2013), 60: 531-537.
Y. Xu, C. Chen, T.P. Thomas, P. Azadi, B.G. Diehl, C. Tsai, N. Brown, J.E. Carlson, M. Tien,
H. Liang. Wood chemistry analysis and expression profiling of a poplar clone expressing a
tyrosine-rich peptide. Plant Cell Reports (2013), 32(12): 1827-1841.
F. Cong, B.G. Diehl, J.L. Hill, N.R. Brown, M. Tien. Covalent bond formation between amino
acids and lignin: Cross-coupling between proteins and lignin. Phytochemistry (2013), 96: 449-
456.
All data collected, preparing for publication, target journal is Biomacromolecules: B.G. Diehl,
N.R. Brown. Lignin crosslinks with peptides under biomimetic conditions.
All data collected, preparing for publication, target journal is Journal of Applied Polymer
Science: B.G. Diehl, N.R. Brown. Characterization of lignin-gelatin complexes.









![Extraction and characterization of lignin ... - CARBON LETTcarbonlett.org/Upload/files/CARBONLETT/[081-088]-14.pdf · Extraction and characterization of lignin from black liquor and](https://img.pdfslide.us/doc/110x75/5ae10ead7f8b9af05b8e7480/extraction-and-characterization-of-lignin-carbon-081-088-14pdfextraction.jpg)



















