Embed Size (px)
Citation preview

Clay Minerals (1965) 6, 3.
P R E L I M I N A R Y I N V E S T I G A T I O N S I N T O T H E
S O R P T I O N OF B O R O N BY C L A Y M I N E R A L S
M. E. L. F L E E T
Department of Geology, University of Western Ontario
(Received 22 September 1964)
ABSTRACT: The sorption of boron by clay minerals from natural waters has been studied experimentally. The quantity of boron sorbed per unit weight of clay mineral is dependent on both the salinity and the boron content of the solution. Previous work has shown that illite is the best clay mineral sorbent, though kaolinite and montmorillonite do sorb some boron and this is confirmed by the present work. The new experimental results demonstrate that the amount of boron sorbed by illite is not affected by the original boron content of the mineral and suggest that the process of incorporation of boron into the lattice proceeds in two stages. The bearing of these experimental results on the use of boron as a palaeosalinity indicator in sedimentary rocks is discussed and this leads to the conclusion that the rate of sedi- mentation may also influence the boron content of such rocks.
Boron in the earth's crust is concentrated in the sedimentary rocks (Urey, 1953), but it can be shown that present-day oceans are capable of dissolving an amount of boron equivalent to that present in the sedimentary rocks of the crust. Thus, boron, originally present in the oceans, must have been removed by some other process than precipita- tion of borates, e.g. biogenetically or by adsorption onto the mineral components of sediments. Such evidence as exists suggests that biogenetic incorporation of boron is inadequate as an explanation of the boron contents of sedimentary rocks, and it is generally considered that sorption is the principal mechanism by which boron is removed from natural water into sedimentary materials.
Harder (1959) showed that much of the boron in the finer fractions of argillaceous sediments is located in the illite fraction, and this finding was confirmed by Frederick- son & Reynolds (1960) in their studies on Cretaceous shales. Later these authors suggested that the other clay minerals (kaolinite, chlorite, etc.) contain appreciable amounts of boron through illite is the main host. F rom the results of chemical attack on modern marine sediments, Goldberg & Arrhenius (1958) suggested that boron proxies for silicon in the lattice of illite. Harder (1961) suggested that it proxies for aluminium in tetrahedral lattice sites and these earlier suggestions were subsequently substantiated by infrared studies on synthetic boron-containing micas by Stubican & Roy (1962).
Walker (1962) developed a suggestion by Harder (1959) that boron is installed in the moscovite-type layers of illite in preference to the more degraded layers. He intro-

4 M . E . L . Fleet
duced the concept of 'adjusted boron', which he suggested is related to the salinity of the waters in which deposition of the sediment occurred. Adjusted boron was equated to the observed boron content time 8.5 ] % K~O in the illite or illite-rich rock. In a further development of this approach Walker & Price (1963) converted adjusted boron contents into 'equivalent boron' contents, equivalent boron being the adjusted boron that would exist at equilibrium in an illite containing 5 % K~O from the same salinity medium. By plotting adjusted boron contents of illites from four known environments against the corresponding % K20 they showed that there appears to be a systematic relationship between the two parameters and that three fields can be recognized, which represent non-marine, marine, and evaporite environments. Correlation between the boron content of argilliaceous rocks and the salinity of the waters in which they accumulated has been attempted by various investigators since Landergren's (1945) suggestion that marine and non-marine sediments might be distinguished by their boron content (e.g. Ernst, Krejci-Graf & Wemer, 1958; Frederickson & Reynolds, 1960). However, apparently anomalous cases are not rare and the boron-palaeosalinity relationship is not universally regarded as valid (Hirst, 1962). Eager (1962) showed that the boron content tends to decrease in the marine parts of the Worsborough and Bersham successions of Carboniferous sediments, and drew attention to a possible antipathetic relationship between the contents of boron and organic matter. He concluded that organic matter may have played some part in controlling the distribution of boron in these two cyclothems.
Any hypothesis relating boron contents of sediments or clay minerals to palaeo- salinities assumes some relationship between boron content and overall salinity of the waters from which boron sorption occurs. There is evidence that this is so in modern natural waters. The data quoted by Harvey (1955) suggest a linear relation- ship between boron content and overall salinity for modem sea water. Frederickson & Reynolds (1960) have shown, by analyses of natural waters from the Gulf Coast of U.S.A., that a similar relationship exists over a salinity range from 2%o to 67~ How far the boron] total salinity ratio has remained constant during geological time is a completely open question.
Comparatively little experimental work has been done relevant to this problem, with the exception of the studies conducted by Harder (1961). He demonstrated that clays can take up boron from solution and that boron sorption by iUite depends on the duration of the experiment, the temperature, the boron concentration, and the pH of the solution. He found iUite to be a better sorbent than either kaolinite or montmorillonite.
It is clearly desirable that the boron-palaeosalinity relationship, which could be put to important use, should be supported by thorough experimental investigation. This has hitherto been lacking and this work is an attempt to remedy the deficiency.
E X P E R I M E N T A L P R O C E D U R E
The clay minerals used as sorbents in this work were A.P.I. standards, illite 35, kaolinite 9a and montmoriUonite 11 from the type localities as reported by Kerr et al.
Sorption of boron by clay minerals 5 (1951). The original boron content of these minerals was illite 200 + 10 ppm, kaolinite 7 ppm, and montmorillonite 2"3 ppm.
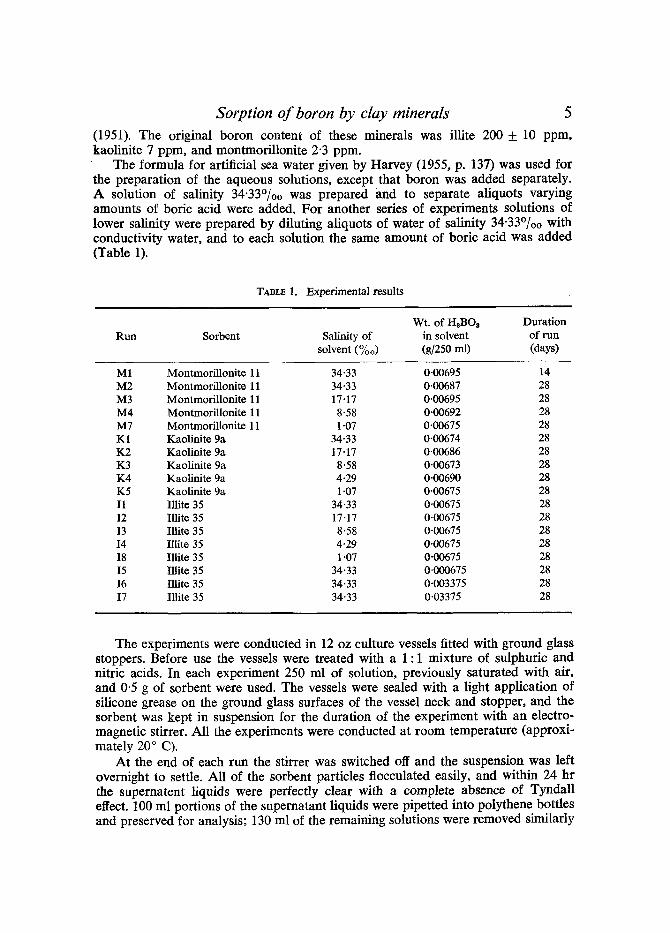
The formula for artificial sea water given by Harvey (1955, p. 137) was used for the preparation of the aqueous solutions, except that boron was added separately. A solution of salinity 34.33%0 was prepared and to separate aliquots varying amounts of boric acid were added. For another series of experiments solutions of lower salinity were prepared by diluting aliquots of water of salinity 34"33%0 with conductivity water, and to each solution the same amount of boric acid was added (Table 1).
TABLE 1. Experimental results
Wt. of 1-IsBOa Duration Run Sorbent Salinity of in solvent of run
solvent (%0) (g/250 ml) (days)
M1 Montmorillonite 11 34"33 0-00695 14 M2 Montmorillonite 11 34"33 0.00687 28 M3 Montmorillonite 11 17.17 0.00695 28 M4 Montmorillonite 11 8-58 0.00692 28 M7 Montmorillonite 11 1.07 0.00675 28 K1 Kaolinite 9a 34.33 0.00674 28 K2 Kaolinite 9a 17.17 0.00686 28 K3 Kaolinite 9a 8'58 0-00673 28 K4 Kaolinite 9a 4-29 0.00690 28 K5 Kaolinite 9a 1.07 0.00675 28 I1 Illite 35 34.33 0.00675 28 12 Illite 35 17.17 0.00675 28 I3 Illite 35 8.58 0.00675 28 14 Illite 35 4-29 0.00675 28 I8 Illite 35 1.07 0.00675 28 15 Illite 35 34.33 0.000675 28 I6 Illite 35 34.33 0.003375 28 17 Illite 35 34.33 0.03375 28
The experiments were conducted in 12 oz culture vessels fitted with ground glass stoppers. Before use the vessels were treated with a 1:1 mixture of sulphuric and nitric acids. In each experiment 250 ml of solution, previously saturated with air, and 0.5 g of sorbent were used. The vessels were sealed with a light application of silicone grease on the ground glass surfaces of the vessel neck and stopper, and the sorbent was kept in suspension for the duration of the experiment with an electro- magnetic stirrer. All the experiments were conducted at room temperature (approxi- mately 20~ C).
At the end of each run the stirrer was switched off and the suspension was left overnight to settle. All of the sorbent particles flocculated easily, and within 24 hr the supernatent liquids were perfectly clear with a complete absence of Tyndall effect. 100 ml portions of the supernatant liquids were pipetted into polythene bottles and preserved for analysis; 130 ml of the remaining solutions were removed similarly

6 M . E . L . Fleet
and the vessels were then left overnight to allow the disturbed sediment to settle, after which most of the remaining supernatant liquids were removed with a pipette. The final fraction of solution was removed from each vessel with tissue papers. The samples of sediment were dried out over a radiator, removed from the vessels, and then lightly ground and mixed in an agate mortar.
It was originally hoped that the amount of sorbed boron would be deter- mined by directly comparing the concentration of H3BOa in the solutions after the experiments with the original concentrations. Preliminary investigations re- vealed that this was impractical. In the early runs H3BO3, after drying to constant weight over phosphorus pentoxide, was added to the saline solutions as solid. The concentrations of HzBOa in the recovered solutions were determined by the carmine- red method of Hatcher & Wilcox (1950). In these runs recovered boron was generally higher than original boron by 1-3%. This is probably to be attributed to slight breakdown of HsBO~ to H20 and B2Oz during the drying process. In the later runs the H3BO8 was added in the form of a solution of known concentration (determined by the carmine-red method). Even the differences here between original and final boron concentrations (Tables 1 and 2) were so small that the analytical error of the colorimetric method was unacceptably large by comparison. For this reason the amount of sorbed boron was determined from the boron content of the sorbents.
The boron content of the sorbent powders after the experiments is derived from three sources: (1) boron originally present in the sorbents, (2) boron acquired by sorption from the solutions, and (3) boron originally present in the small amount of interstitial solution remaining in the sorbent after removal of the supernatant liquid and precipitated out during subsequent drying.
The samples were leached with hot dilute hydrochloric acid, which liberated boron from sources 2 and 3 and probably from part of 1. To correct for leached original boron, blanks on clay mineral samples that had not been subjected to sorption were run with every batch of determinations. The leaching was carried out in soft glass sample tubes (dimensions 3 • �88 in) the top halves of which had been drawn out to form a central constricted region and a funnel-shaped mouth. A quantity of dried sample from each sorption experiment (0-025-0.050 g for the iUite samples; 0.1000 g for the kaolinite and montmoriUonite) was accurately weighed into the tube and 5 ml of 5 % hydrochloric acid added from a pipette before the tube was sealed at the constriction. Each tube was shaken and then placed in an oven at 105 ~ C for a period of 2-5-3 hr, after which it was cooled, shaken again and left overnight at room temperature. After opening the tube its contents were transferred to a conical centrifuge tube and centrifuged. Two ml of the supernatant liquid were transferred to a conical flask with a pipette and the HsBO3 content determined colorimetrically by the carmine-red method.
Correction for the boron acquired from the interstitial solution requires a knowledge of the amount of interstitial liquid remaining in the samples before drying. This was indirectly calculated by determining the amount of another element (e.g. Na, C1) acquired from the solvent and using the known ratio of that element to boron in the solution to calculate the corresponding amount of boron and the amount of liquid. Both Na and C1 were investigated as possible monitoring elements. Na con- tent in the solutions was determined by flame photometry. The results (Table 2) indicated that Na + exchange had affected the Na content of the sorbents, esoecially with montmorillonite and C1 was chosen as the preferred monitor. Total C1 deter-
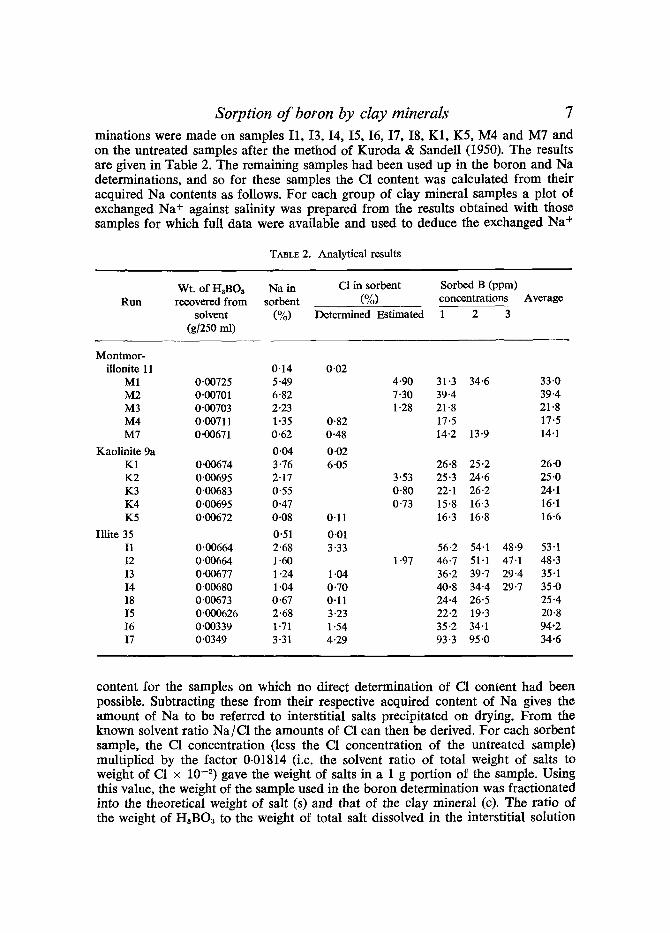
Sorption of boron by clay minerals 7 minations were made on samples I1, I3, I4, I5, I6, I7, I8, K1, K5, M4 and M7 and on the untreated samples after the method of Kuroda & SandelI (1950). The results are given in Table 2. The remaining samples had been used up in the boron and Na determinations, and so for these samples the C1 content was calculated from their acquired Na contents as follows. For each group of clay mineral samples a plot of exchanged Na + against salinity was prepared from the results obtained with those samples for which full data were available and used to deduce the exchanged Na +
TABLE 2. Analytical results
Wt. of H3BOa Na in C1 in sorbent Sorbed B (ppm) Run recovered from sorbent (%) concentrat ions Average
solvent (%) Determined Estimated 1 2 3 (g/250 ml)
Montmor- illonite 11 O. 14 0.02
M1 0"00725 5"49 4"90 31 "3 34'6 M2 0"00701 6"82 7"30 39"4 M3 0'00703 2"23 1 "28 21 "8 M4 0"00711 1 "35 0'82 17"5 M7 0"00671 0"62 0"48 14"213'9
Kaolinite 9a 0.04 0-02 K1 0.00674 3"76 6.05 26.8 25.2 K2 0.00695 2.17 3-53 25.3 24.6 K3 0.00683 0.55 0.80 22.1 26-2 K4 0.00695 0.47 0.73 15.8 16.3 K5 0.00672 0.08 0.11 16-3 16.8
Illite 35 0.51 0.01 I1 0.00664 2.68 3.33 56-2 54.1 I2 0.00664 1"60 1-97 46.7 51.1 13 0.00677 1.24 1.04 36.2 39.7 I4 0.00680 1"04 0.70 40.8 34.4 I8 0-00673 0.67 0.11 24.4 26.5 I5 0.000626 2.68 3-23 22-2 19.3 I6 0.00339 1"71 1.54 35.2 34.1 I7 0.0349 3.31 4.29 93.3 95.0
48.9 47.1 29.4 29.7
33.0 39.4 21.8 17.5 14.1
26.0 25.0 24.1 16.1 16-6
53-1 48.3 35.1 35.0 25 -4 20.8 94.2 34.6
content for the samples on which no direct determination of CI content had been possible. Subtracting these from their respective acquired content of Na gives the amount of Na to be referred to interstitial salts precipitated on drying. From the known solvent ratio Na/C1 the amounts of C1 can then be derived. For each sorbent sample, the C1 concentration (less the C1 concentration of the untreated sample) multiplied by the factor 0.01814 (i.e. the solvent ratio of total weight of salts to weight of CI • 10 -2) gave the weight of salts in a 1 g portion of the sample. Using this value, the weight of the sample used in the boron determination was fractionated into the theoretical weight of salt (s) and that of the clay mineral (c). The ratio of the weight of H3BO3 to the weight of total salt dissolved in the interstitial solution

8 M . E . L . Fleet
was calculated from the recovered H3BO3 content and the weight of salts in the original solution. The ratio • 's' • 1,000,000 gave the correction for the boron acquired from the interstitial solution in ppm H3BO3.
Correction for the effect of the matrix was made by determining the ratio of the concentration of H3BO3 (in ppm) recorded for the blank to the weight of clay minerals used in the blank and then multiplying this ratio by 'c'.
The corrected weights of H3BO3 were sorbed by 'c' g of sorbent and from these figures the amount of boron (as ppm B) sorbed by a clay mineral in one of the experimental runs can be readily calculated. The results of the various experimental runs are given in Table 2.
Duplicate and triplicate determinations give some idea of the reproducibility of the method (Table 2). The possibility of interference by Si ~+, Fe 3+ and AP + in the colorimetric determination was considered. Leachates subjected to an R20~ precipitation before the carmine-red treatment had optical densities only very slightly lower than the same leachates determined directly. Artificial standards prepared to the composition of montmorillonite 11 but with boron contents of 49-3 and 29-5 ppm gave boron contents of 48"5 and 31.5 ppm respectively when analysed by this method. Two samples of montmoriUonite 11 were leached with dilute hydrochloric acid with added HaBO3 equivalent to an overall sample concentration of 37"4 ppm B. The analysed boron content in both cases was 37"6 ppm.
DISCUSSION AND I N T E R P R E T A T I O N OF E X P E R I M E N T A L RESULTS
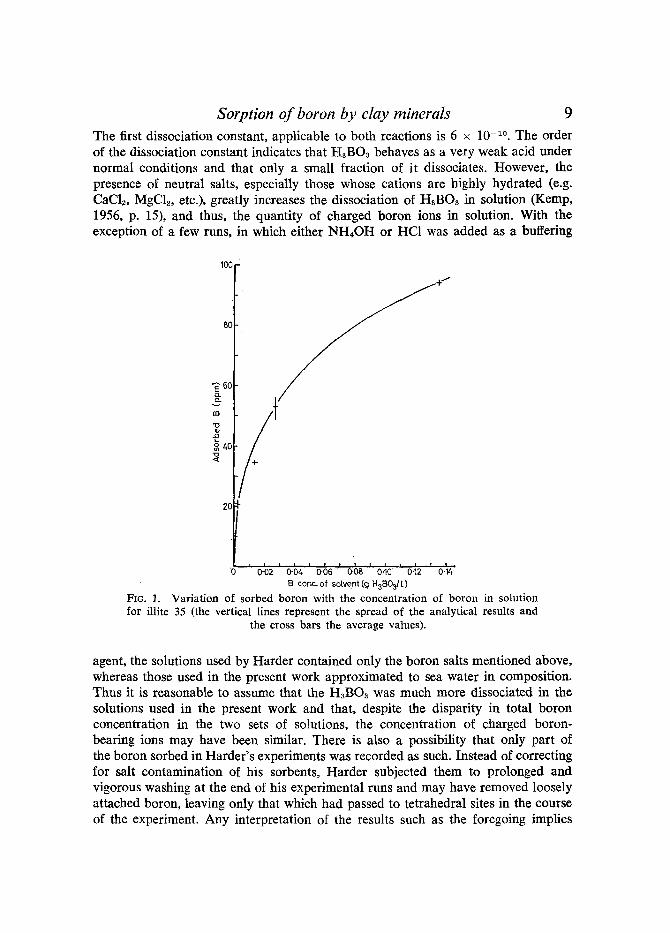
The results of the experiments conducted to determine the effects of different boron concentrations in solutions of constant salinity (34-3%0), runs I1, I5, I6, I7, are shown graphically in Fig. 1. As expected, the amounts of boron sorbed by illite increased with increasing boron concentration in the solution. A 50-fold increase in boron concentration in solution produced nearly a 5-fold increase in the quantity of boron sorbed. It is instructive to compare these results with those of Harder (1961). He found, for runs lasting 30-35 days, the amounts of boron sorbed on illite, kaolinite, and montmorillonite to be 20-40, 4.5, and 3.3 ppm respectively. These results are of the same order of magnitude as those found in the present work for runs of comparable duration and solutions of boron content of approximately 0.027 g H3BO3/1 (see Table 2). However, Harder used solutions that had boron concentrations of 10 g/1 H3BO~ and Na2B407 (approximately 400 times more con- centrated than the solutions used in the present work). This effect cannot be explained by assuming that the sorption process is independent of the boron concentration of the solution, because this is disproved both by Harder's work and the relevant results of the present work (Fig. 1). A much more likely explanation is that the chemical state of boron in solution was different.
In dilute solutions H3BO3 dissociates according to the reaction: HaBO~ .. ~ H2BO3- + H +
or, more likely: H~BO3 + 2H20 , "~ H30 + + B(OH)4- (Kemp, 1956, p. 19).
Sorption of boron by clay minerals 9 The first dissociation constant, applicable to both reactions is 6 • 10 -1~ The order of the dissociation constant indicates that H~BO~ behaves as a very weak acid under normal conditions and that only a small fraction of it dissociates. However, the presence of neutral salts, especially those whose cations are highly hydrated (e.g. CaCI~, MgCI~, etc.), greatly increases the dissociation of HaBO8 in solution (Kemp, 1956, p. 15), and thus, the quantity of charged boron ions in solution. With the exception of a few runs, in which either NH4OH or HC1 was added as a buffering
too
+I"
8o
~6o r n
~ 40 ~ .
2(
o 0.'02 o-;4 0.;6' o.;B 'o.~o 'o-~2 o.~' B conc, of sotvent(g H3B0311. )
FIG. l. Variat ion of sorbed boron with the concentration of boron in solution for illite 35 (the vertical lines represent the spread of the analytical results and
the cross bars the average values).
agent, the solutions used by Harder contained only the boron salts mentioned above, whereas those used in the present work approximated to sea water in composition. Thus it is reasonable to assume that the H~BOa was much more dissociated in the solutions used in the present work and that, despite the disparity in total boron concentration in the two sets of solutions, the concentration of charged boron- bearing ions may have been similar. There is also a possibility that only part of the boron sorbed in Harder's experiments was recorded as such. Instead of correcting for salt contamination of his sorbents, Harder subjected them to prolonged and vigorous washing at the end of his experimental runs and may have removed loosely attached boron, leaving only that which had passed to tetrahedral sites in the course of the experiment. Any interpretation of the results such as the foregoing implies
10 M. E. L. Fleet
that, initially, the sorption process is inherently chemical in nature--an adsorption of ions onto surfaces of opposite charge. It will be shown later that there are other reasons for believing that this must he so.
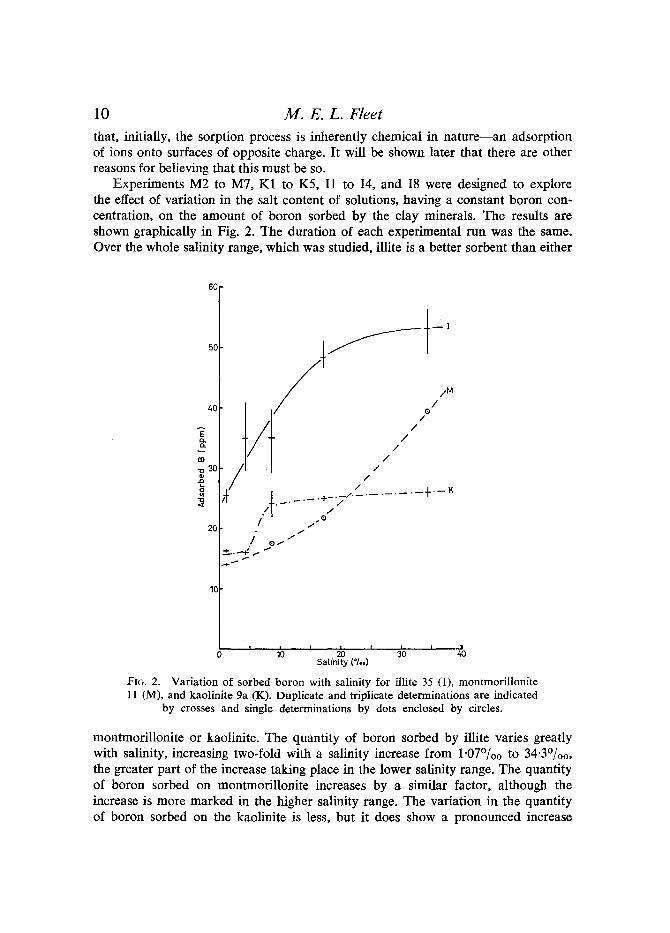
Experiments M2 to M7, K1 to K5, I1 to I4, and I8 were designed to explore the effect of variation in the salt content of solutions, having a constant boron con- centration, on the amount of boron sorbed by the clay minerals. The results are shown graphically in Fig. 2. The duration of each experimental run was the same. Over the whole salinity range, which was studied, illite is a better sorbent than either
6(J
"E~40 / / //////o//M m
-a 30 g ~ .,~ . . . . . . . +S. . . . . . . . . . . + - ~
/ / " ~ 20
1C
I I I 0 lb ~'0 3'0 ' Satinity (*/,,)
Fta. 2. Variation of sorbed boron with salinity for illite 35 (1), montmorillonite 11 (M), and kaolinite 9a (K). Duplicate and triplicate determinations are indicated
by crosses and single determinations by dots enclosed by circles.
montmorillonite or kaolinite. The quantity of boron sorbed by illite varies greatly with salinity, increasing two-fold with a salinity increase from 1-07%o to 34"3%0, the greater part of the increase taking place in the lower salinity range. The quantity of boron sorbed on montmoriUonite increases by a similar factor, although the increase is more marked in the higher salinity range. The variation in the quantity of boron sorbed on the kaolinite is less, but it does show a pronounced increase
Sorption of boron by clay minerals 11 between salinities 4"3 and 8"6%0. These results are in harmony with the suggestion that the dissociation of H3BOa in solution is increased by dissolved neutral salts, but this effect cannot be the sole one governing this adsorption of boron in these experiments because the major increase in sorbed boron takes place over different salinity ranges in the different clay minerals. In chemical adsorption the surface state (i.e. charge density) of the adsorbent will also be important. The boron ions in solution are likely to be negatively charged and, therefore, the clay minerals must have areas of effective positive charge on their surfaces for adsorption to occur. The ease with which the clays flocculated in all the solutions suggests that only residual charges were present on their surfaces. However, the variations in the salt concentration would be expected to affect the surface charges of the clay
1000
o
200
100 �9 0"001
Fro. 3.
J
t r t t t I t t f I I I I I t , I I 0'005 0"010 0'050 0"100
Log c
Variat ion of log a (expressed as ppm H3BO~) with log c for illite 35.
minerals to some extent and slight changes of this nature, specific to each clay mineral, could possibly produce the observed variations.
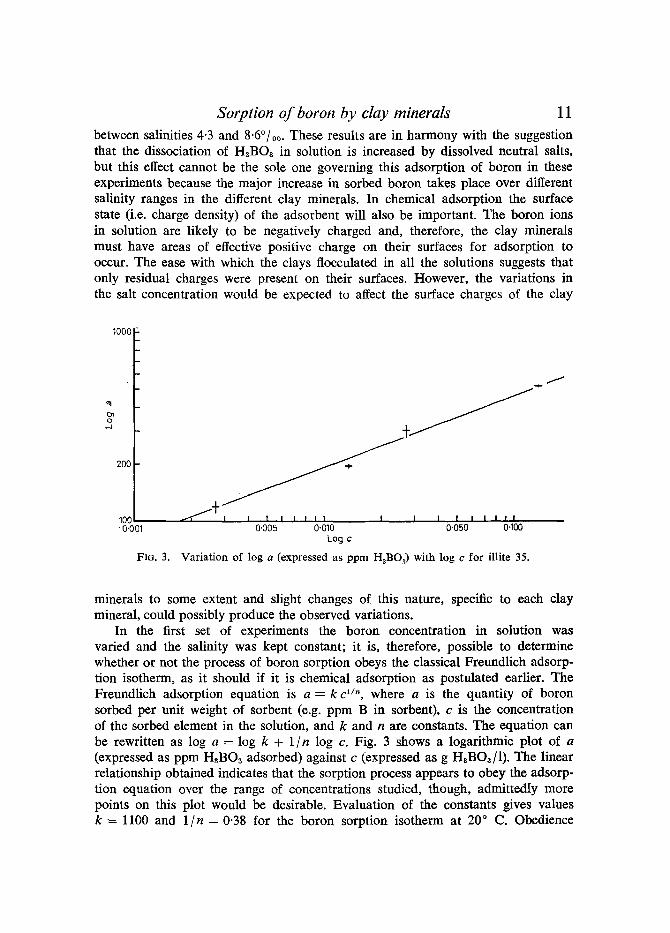
In the first set of experiments the boron concentration in solution was varied and the salinity was kept constant; it is, therefore, possible to determine whether or not the process of boron sorption obeys the classical Freundlich adsorp- tion isotherm, as it should if it is chemical adsorption as postulated earlier. The Freundlich adsorption equation is a = k c 1/'~, where a is the quantity of boron sorbed per unit weight of sorbent (e.g. ppm B in sorbent), c is the concentration of the sorbed element in the solution, and k and n are constants. The equation can be rewritten as log a = log k + 1 I n log c. Fig. 3 shows a logarithmic plot of a (expressed as ppm H,BOa adsorbed) against c (expressed as g HaBO~/I). The linear relationship obtained indicates that the sorption process appears to obey the adsorp- tion equation over the range of concentrations studied, though, admittedly more points on this plot would be desirable. Evaluation of the constants gives values k = 1100 and 1 I n = 0-38 for the boron sorption isotherm at 20 ~ C. Obedience
12 M. E. L. Fleet
to the Freunlich adsorption equation make it very likely that the sorption process studied is chemical adsorption.
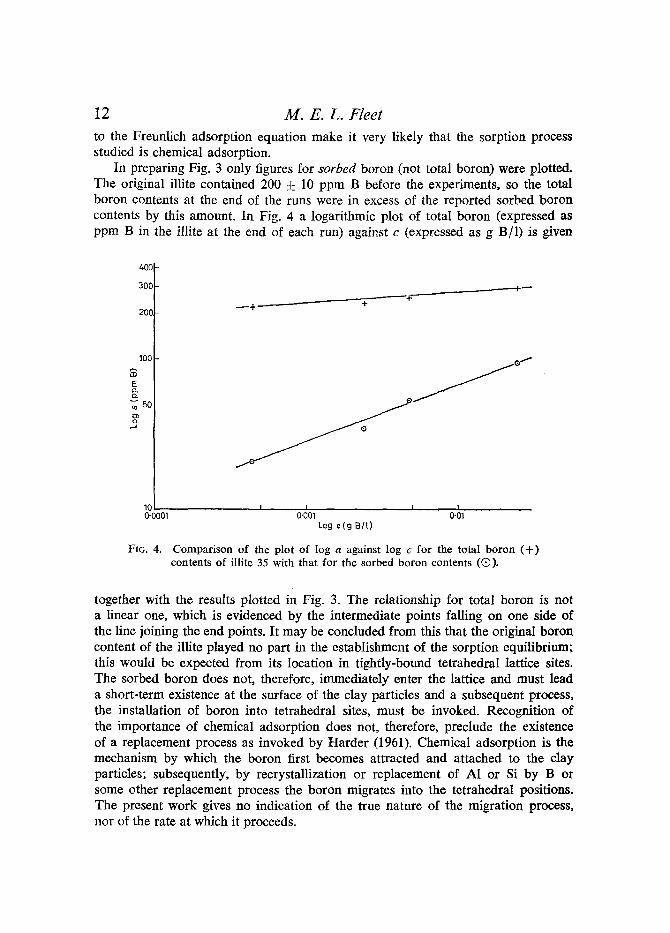
In preparing Fig. 3 only figures for sorbed boron (not total boron) were plotted. The original illite contained 200 + 10 ppm B before the experiments, so the total boron contents at the end of the runs were in excess of the reported sorbed boron contents by this amount. In Fig. 4 a logarithmic plot of total boron (expressed as ppm B in the illite at the end of each run) against c (expressed as g B/ l ) is given
400
300
200
100
$ E g_
---.--,-~--t- ~
10 I I I I 0"0001 0"001 0'01
Log c (g B/t.)
FIG. 4. Comparison of the plot of log a against log c for the total boron (+) contents of illite 35 with that for the sorbed boron contents (q)).
together with the results plotted in Fig. 3. The relationship for total boron is not a linear one, which is evidenced by the intermediate points falling on one side of the line joining the end points. It may be concluded from this that the original boron content of the illite played no part in the establishment of the sorption equilibrium; this would be expected from its location in tightly-bound tetrahedral lattice sites. The sorbed boron does not, therefore, immediately enter the lattice and must lead a short-term existence at the surface of the clay particles and a subsequent process, the installation of boron into tetrahedral sites, must be invoked. Recognition of the importance of chemical adsorption does not, therefore, preclude the existence of a replacement process as invoked by Harder (1961). Chemical adsorption is the mechanism by which the boron first becomes attracted and attached to the clay particles; subsequently, by recrystallization or replacement of A1 or Si by B or some other replacement process the boron migrates into the tetrahedral positions. The present work gives no indication of the true nature of the migration process, nor of the rate at which it proceeds.

Sorption of boron by clay minerals 13
The initial adsorption process is likely to proceed quite rapidly. The results on M1 (14 days) and M2 (28 days) show that during the interval 14-28 days of the second run the amount of sorbed boron only increased from 33"0 to 39"4 ppm, indicating that most of the boron is sorbed quickly. This finding agrees with the results of similar experiments conducted by Harder (1961). Initial sorption by clay minerals in response to the external conditions is, therefore, geologically speaking, immediate, but it does not necessarily follow that the installation of boron into tetrahedral sites in the clay mineral lattices is likewise.
Frederickson & Reynolds (1960) have demonstrated that boron in natural waters increases approximately linearly with salinity. Thus, it appears likely that if the boron concentration experiments had been conducted in solutions of salinities equivalent to their respective boron contents, as in natural waters, the variation in the quantities of sorbed boron would have been much greater.
C O N C L U S I O N
Evidence has been presented by other investigators (Goldberg & Arrhenius, 1958; Harder, 196I) that boron is held very strongly in the iUite lattice. Bonds between ionic boron and the illite lattice directly resulting from chemical adsorption must be very weak, because the sorbed boron can be leached from the fllite quite easily. There- fore, the mechanism by which ilfite acquires boron from natural waters would appear to consist of two processes, an initial chemical adsorption followed by boron in- stallation in the tetrahedral lattice sites.
Deferment of the installation process until post-depositional (diagenetic) re- crystallization of the illite seems unlikely in view of the experimental results. The quantity of boron sorbed by illite in run I1 (53 ppm) is only one-eighth to one-sixth of that of a marine illite (Frederickson & Reynolds, 1960) and the results of runs M1 and M2 discussed above make it unlikely that this discrepancy could be ex- plained by low equilibrium sorption values by a factor of six to eight. It is much more likely that the installation of boron in the tetrahedral sites by migration from the sorption sites begins while the clay minerals are still in contact with the boron- bearing solutions. As boron migrates from the sorption sites, more boron is sorbed from solution onto these sites to maintain sorption equilibrium. The rate of the installation process, although unknown, is clearly important to a full understanding of the mechanism of boron retention by clay minerals. All that can be said at present is that it is slower than the sorption process. This hypothesis implies that the amount of boron taken up from a solution by a clay mineral depends not only on the boron content and salinity of the solution, but also on the period during which the clay mineral is in contact with that solution.
In general terms, the experiments reported in this paper have confirmed the principal findings of Harder (1961).Also, by using simulated natural waters it has been demonstrated that the sorption of boron is increased markedly in saline solutions, regardless of increase of boron in these solutions. Thus these findings would seem to strengthen the hypothesis that clays from a marine environment would have a higher
14 M. E. L. Fleet
boron content than those from a fresh-water one. In addition the rate of sedimentation could also be a factor in determining the amount of boron taken up by the clay mineral in a particular environment, because it has been shown that boron uptake depends on the period during which a clay mineral is in contact with a boron- bearing solution.
When the boron-salinity relationship is tested against palaeontological evidence of palaeosalinity, anomalous cases are occasionally encountered and this is also true when the same relationship is tested in some modern environments where data on the salinity of the waters are available, for example deep sea clays, sediments from the Gulf of Paria (Hirst, 1962). Localized fluctuations in the concentration of boron in natural waters (e.g. due to volcanic activity, etc.) would produce varia- tions in the boron contents of iUite but, at present, there is little evidence in support
10
~ 6
%
§ 0
J
./ !
1C0
x X
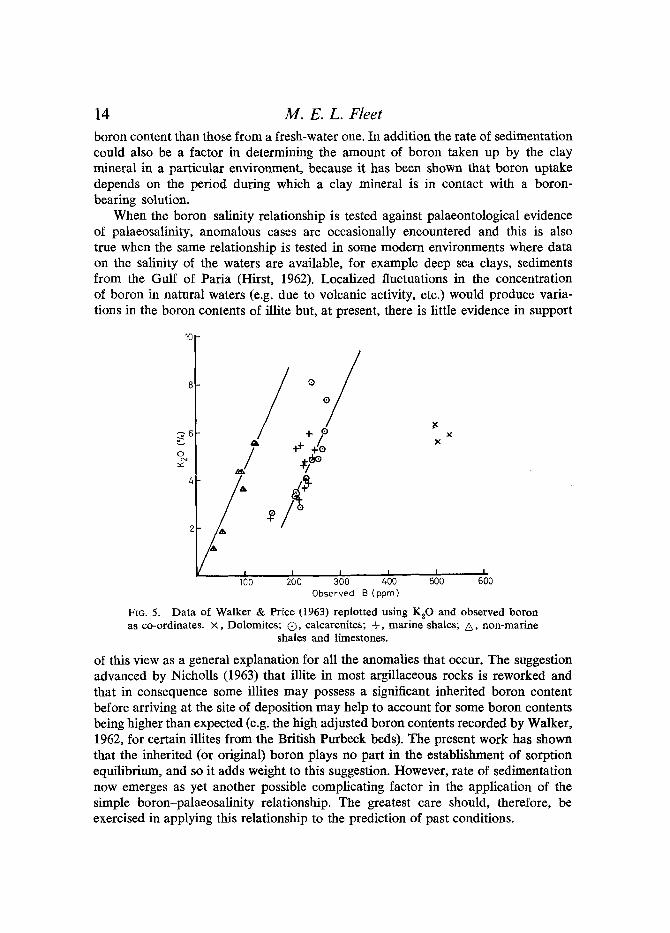
FIG. 5. Data of Walker & Price (1963) replotted using KzO and observed boron as co-ordinates. • Dolomites; Q, ealcarenites; +, marine shales; A, non-marine
shales and limestones.
of this view as a general explanation for all the anomalies that occur. The suggestion advanced by Nicholls (1963) that illite in most argillaceous rocks is reworked and that in consequence some illites may possess a significant inherited boron content before arriving at the site of deposition may help to account for some boron contents being higher than expected (e.g. the high adjusted boron contents recorded by Walker, 1962, for certain illites from the British Purbeck beds). The present work has shown that the inherited (or original) boron plays no part in the establishment of sorption equilibrium, and so it adds weight to this suggestion. However, rate of sedimentation now emerges as yet another possible complicating factor in the application of the simple boron-palaeosalinity relationship. The greatest care should, therefore, be exercised in applying this relationship to the prediction of past conditions.
I I I I I 200 300 400 500 600
Observed B (ppm)

Sorption of boron by clay minerals 15 The contention of Walker (1962) and of Walker & Price (1963) that the sorption
of boron by illite is in some way dependent on the potassium content of the illite is open to doubt. A plot of the observed boron (Walker & Price, 1963, Table 1) against the corresponding potassium figures (Fig. 5) shows a pronounced variation of observed boron with potassium content and reveals three salinity fields, representing non-marine, marine, and evaporite environments (equivalent to those reported by Walker & Price). This increase in the observed boron content with potassium may not be associated with the suggested increase in the number of muscovite-type layers in the illites. Two alternative explanations are possible. The potassium content of a rock may reflect the approximate amount of iUite in that rock, or the variation may be due to the simultaneous sorption of boron and potassium. The former is unlikely since Walker & Price assumed that all the rocks considered contain at least 80 % illite, but the latter is worth further consideration. On entering a marine environment a degraded illite may concentrate both boron and potassium by ex- change, the amounts of each installed depending on the rate at which the illite sediments. Thus, potassium and boron contents in illite would be related because enrichment of both constituents is favoured by the same environment. The arrange- ment of the plots into three fields, representing non-marine, marine, and evaporite environments, may then be ascribed to phenomena associated with the relative abundance of potassium and boron and the relative sorption power of illite in the three environments. It is considered that such potassium-observed boron plots may be of real value in interpreting palaeosalinity data, especially to overcome the effects of different sedimentation rates; however, it is not established ff the fields on these plots are broad enough to negate the reworking effect. In the light of this approach, because potassium-boron plots and equivalent boron values serve a similar function, the latter would seem to be an unnecessary complication.
At present the use of boron as a salinity indicator, in the absence of other estab- lished criteria, is open to question. It is hoped that further experimental investigation into the process of boron uptake by clay minerals will reveal, more lucidly, the factors which determine its mechanism and thus permit the correction of boron contents before their use in interpretive sedimentary geochemistry.
A C K N O W L E D G M E N T S
Many thanks are due to Dr G. D. Nicholls for his assistance and advice in the research and in the preparation of this manuscript. The author also acknowledges financial assistance from D.S.I.R.
R E F E R E N C E S
EAGAR R.M.C. (1962) Nature, Lond. 196, 428. ERNST W., KREJCI-GRAF K. & WERNER H. (1958) Geochim. cosmochim, Acta, 14, 211. FREDERICKSON A.F. (1962) Bull. Am. Ass. Petrol. Geol. 46, 518. FREDERtCKSON A.F. & REYNOLDS R.C., JR. (1960) Clays and Clay Minerals. Proc. 8th Conf.,
p. 203. Pergamon Press, Oxford.

16 M. E. L. Fleet GOLDBERG E.D. & ARRHENIUS G.O.S. (1958) Geoehim. eosmochim. Acta 13, 153. HARDER H. (1959) Nachr. Akad. Wiss. G6ttingen, Math.-phys. Kl. 6, 123. HARDER H. (1961) Geochim. eosmochim. Aeta 21, 284. HARVEY H.W. (1955) The Chemistry and Fertility of Sea-Waters. Cambridge University Press. HATCHER J.T. & WILCOX L.V. (1950) Analyt. Chem. 22, 567. HmST D.M. (1962) Geochim. cosmochim. Acta '26, 309. KEMP P.H. (1956) The Chemistry of Borates. Part 1. Borax Consolidated Ltd, London. KERR P.F. et al. (1951) Reference Clay Minerals. Amer. Petrol. Inst. Research Project 49. KURODA P.K. & SANDELL E.B. (1950) Analyt. Chem. 22, 1144. LANOERGREN S. (1945) Ark. Kemi Miner. Geol. 19A, No. 26. NICHOLLS G.D. (1963) Sci. Prog., Lond. 51, 12. STUBICAN V. & ROY R. (1962) Am. Miner. 47, 1166. UREY H.C. (1953) Proc. R. Soe. A 219, 281. WALKER C.T. (1962) Nature, Lond. 194, 1073. WALKER C.T. & PRICE N.B. (1963) Bull. Am. Ass. Petrol. Geol. 47, 833.





























