Embed Size (px)
Citation preview

Poxviruses, Biodefense and Bioinformatics
Working towards a better understanding of viral pathogenesis and evolution

PBR Bioinformatics
Managing Complexity– Technology development
Enhancing Understanding– Research

PBR Managing Complexity
Data– Acquisition– Storage– Manipulation– Retrieval

PBR Managing Complexity…
Data Analysis– Development and Utilization of
• Analytical tools
• Visualization tools

PBR Enhancing Understanding
What distinguishes one organism from another?
Sequence Molecular Biology Physiology Pathogenesis Epidemiology Evolution
Will the genomic sequence provide an explanation for the differences?

PBR What is Bioinformatics?
Computer-aided analysis of biological information Discerning the characteristic (repeatable) patterns
in biological information that help to explain the properties and interactions of biological systems.
Caveat:– In the end, bioinformatics (a.k.a. computers) can only
help in making inferences concerning biological processes.
– These inferences (or hypotheses) have to be tested in the laboratory

The Poxvirus Bioinformatic Resource
www.poxvirus.org
PBR

PBR PBR Collaborators
UAB– Elliot Lefkowitz
St. Louis University– Mark Buller
University of Victoria– Chris Upton
ATCC– Charles Buck
Medical College of Wisconsin– Paula Traktman

PBR The UAB MGBF ContingentMolecular and Genetic Bioinformatics Facility
Programmers– Jim Moon– Don Dempsey– Uma Dave– Bei Hu
Students– Chunlin Wang
Fellows– Shankar Changayil– Xiaosi Han

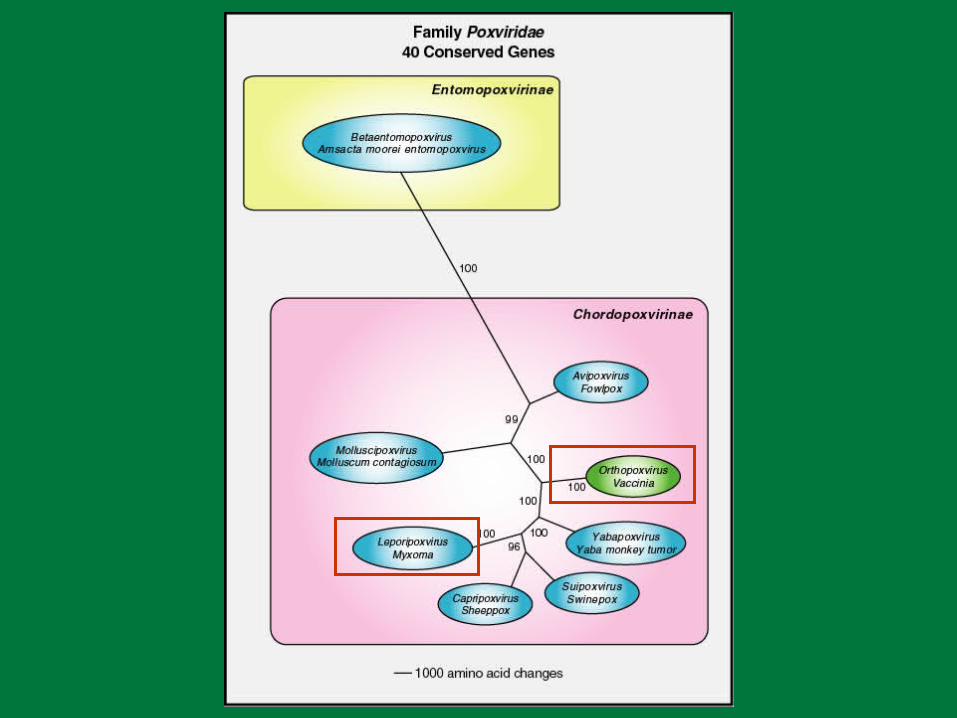
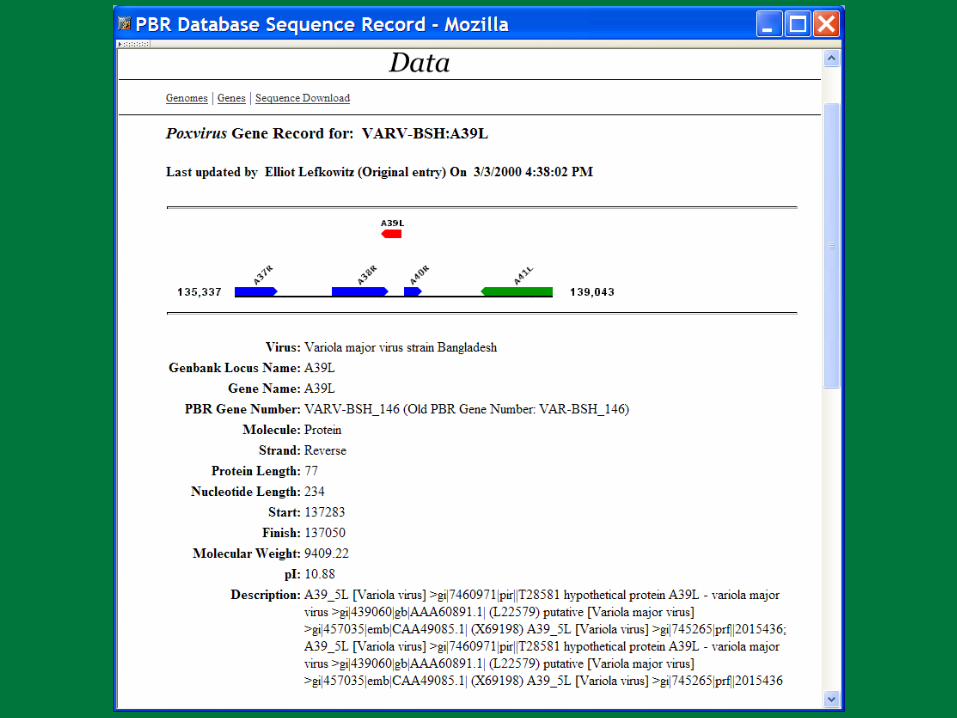
PBR Poxviruses
Large dsDNA genome– 150,000 – 300,000 base pairs– 150 – 260 genes
Complex virion morphology Cytoplasmic replication Array of immunoevasion strategies. Human pathogens
– Molluscum contagiosum– Variola– Monkeypox

PBR The PBR is Designed to Support
Basic and applied research on Poxviruses including the development of new:
Environmental DetectorsDiagnostic ReagentsAnimal ModelsVaccinesAntiviral Compounds

PBR PBR Design Philosophy
Useful and UsedSupporting all poxvirus investigators
– UAB PBR Web-based application requirements• Web Browser
• Java plugin
In-depth analyses– UVic analytical tools
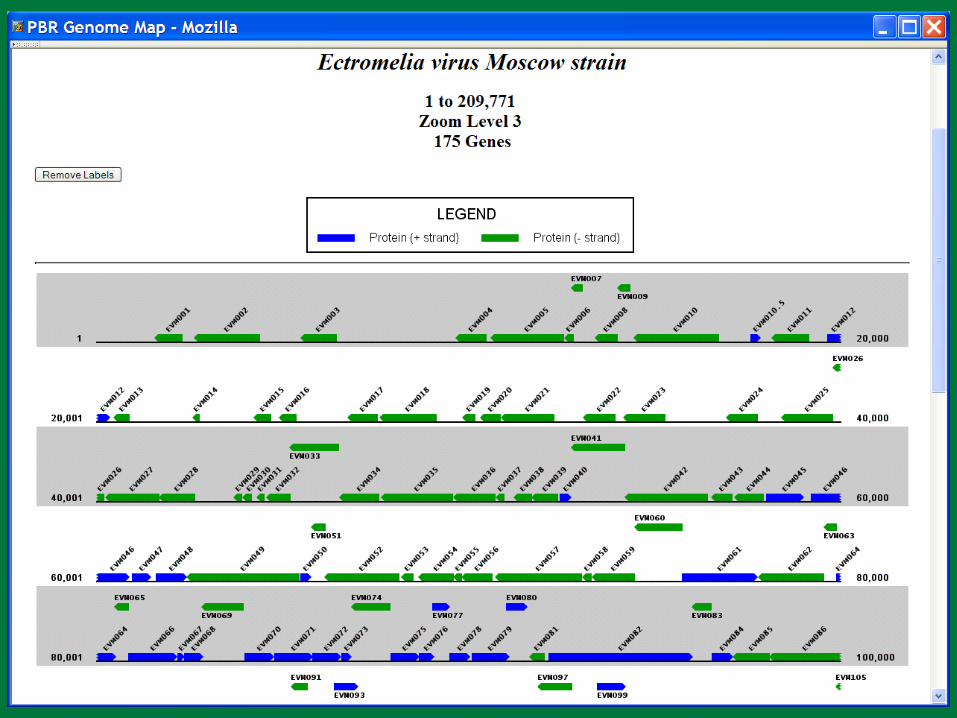
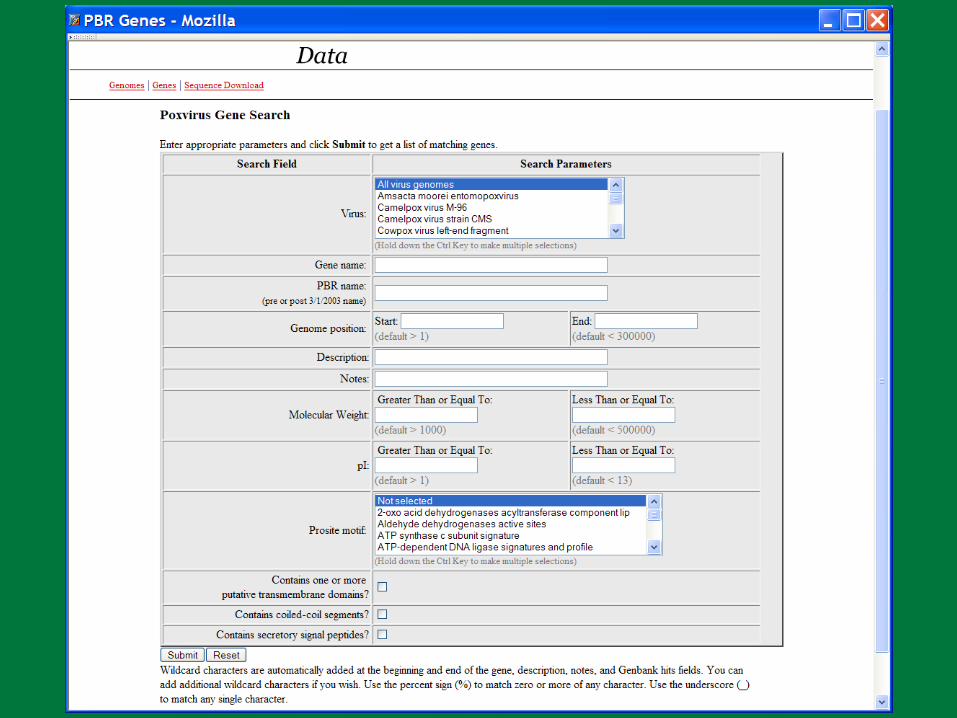
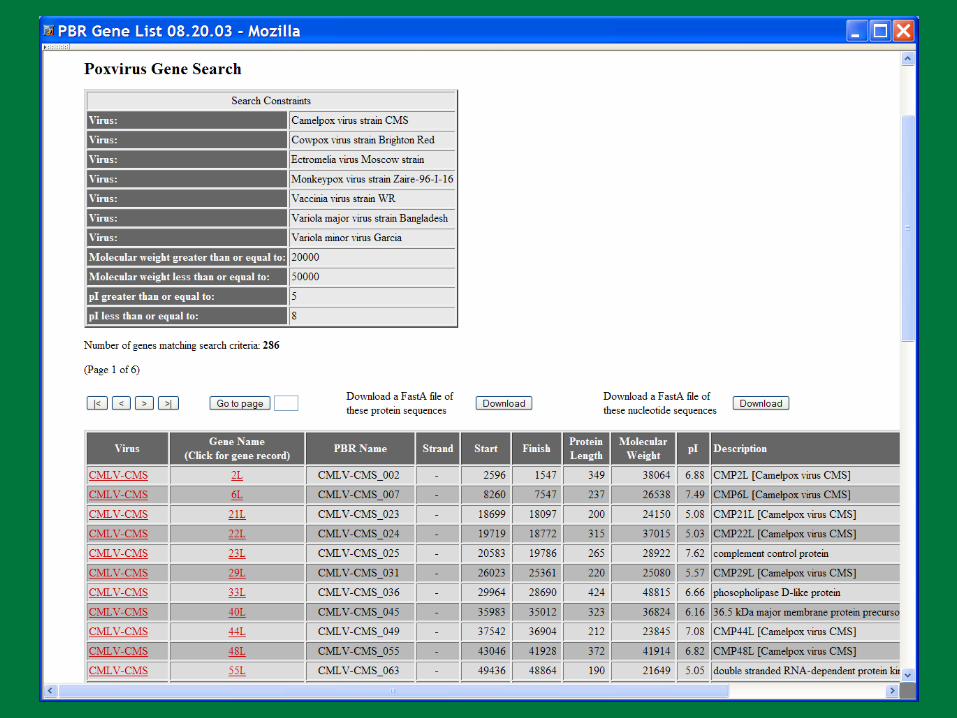
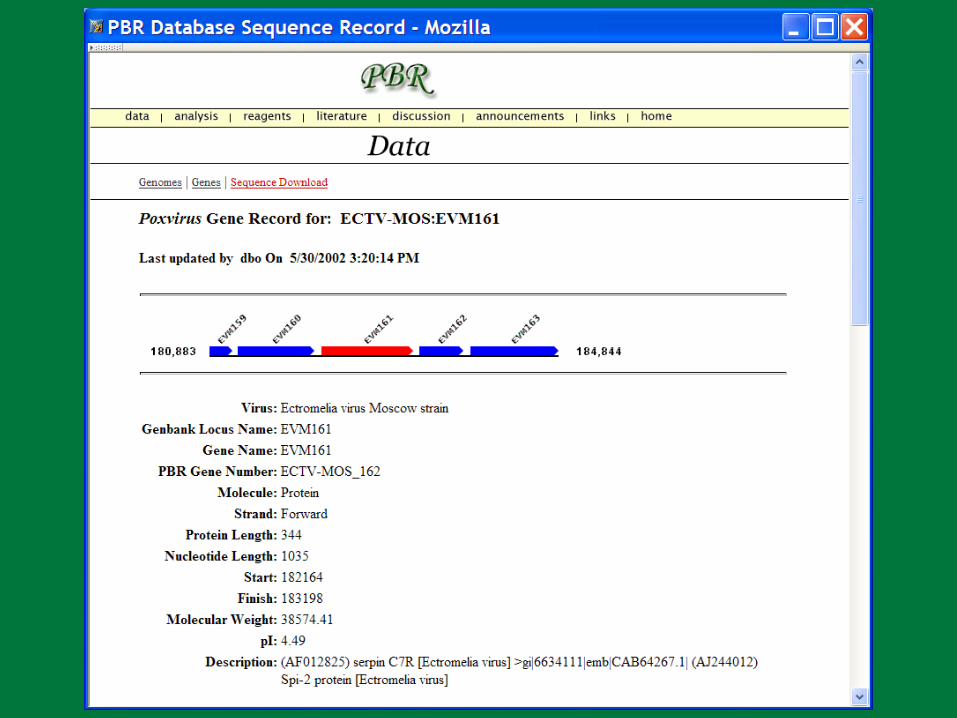

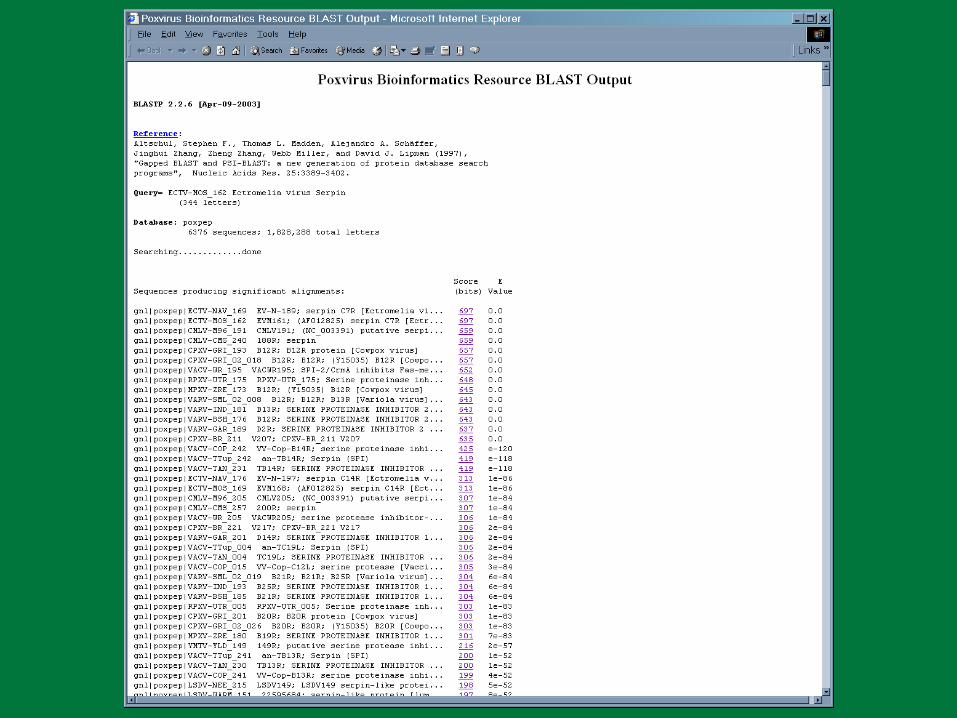
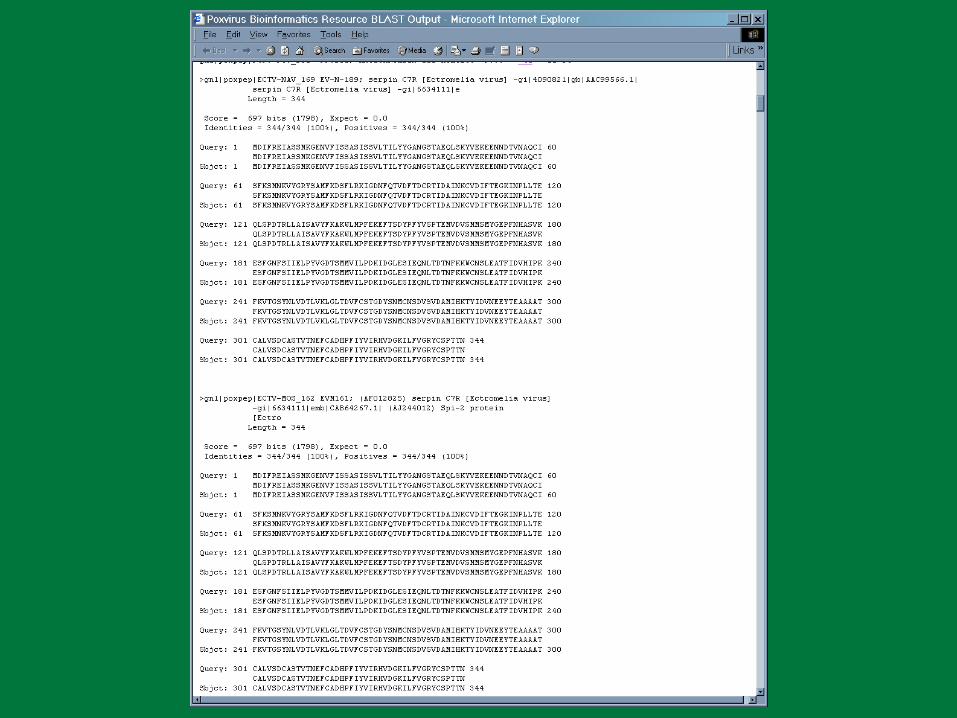
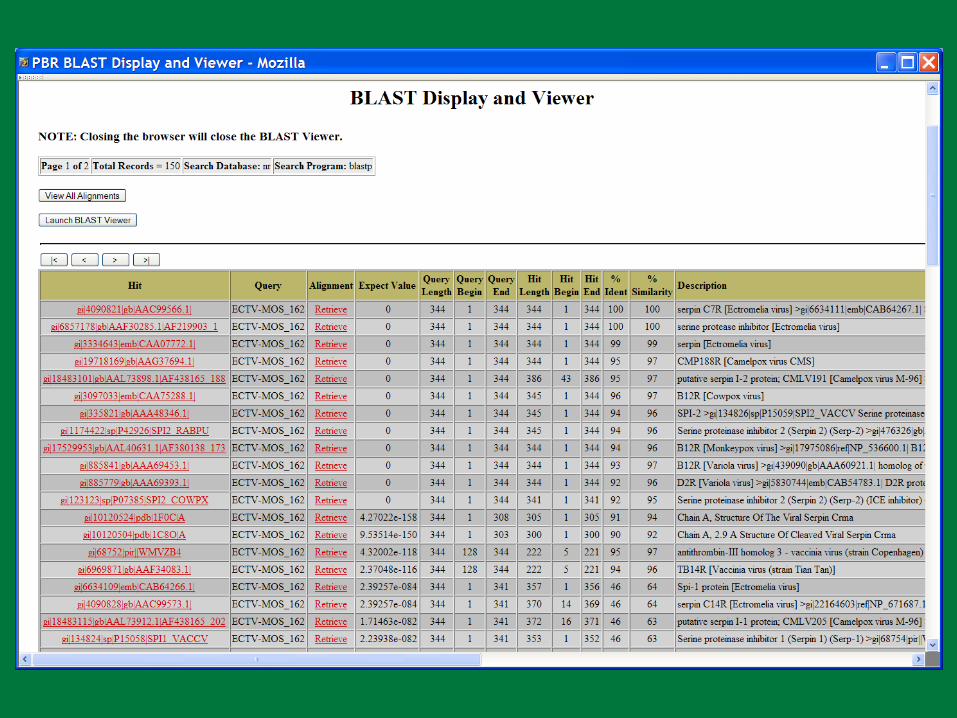
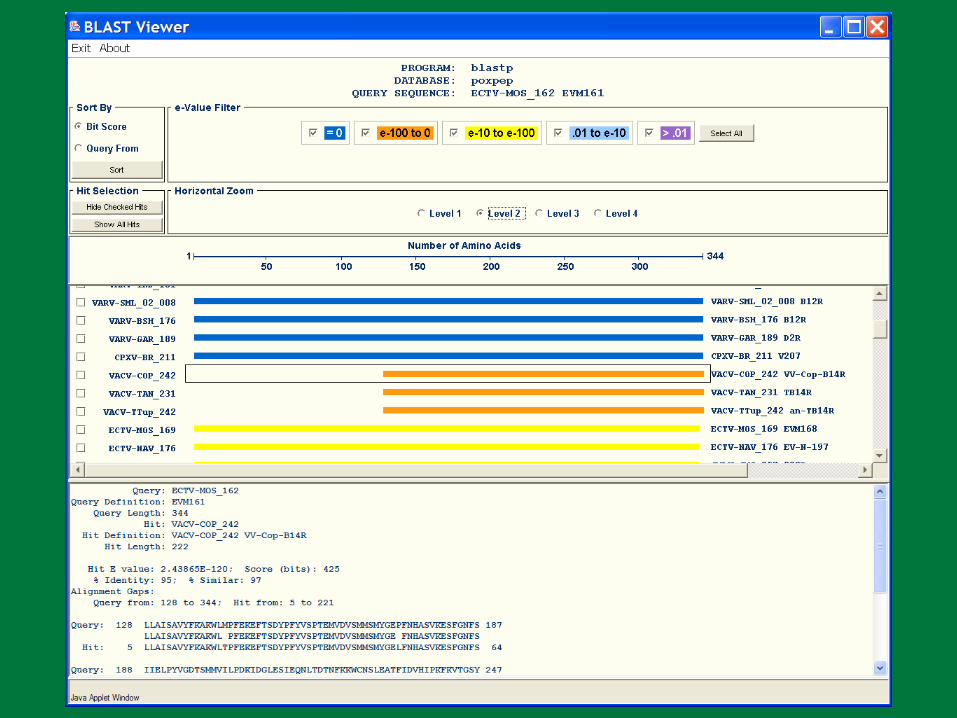
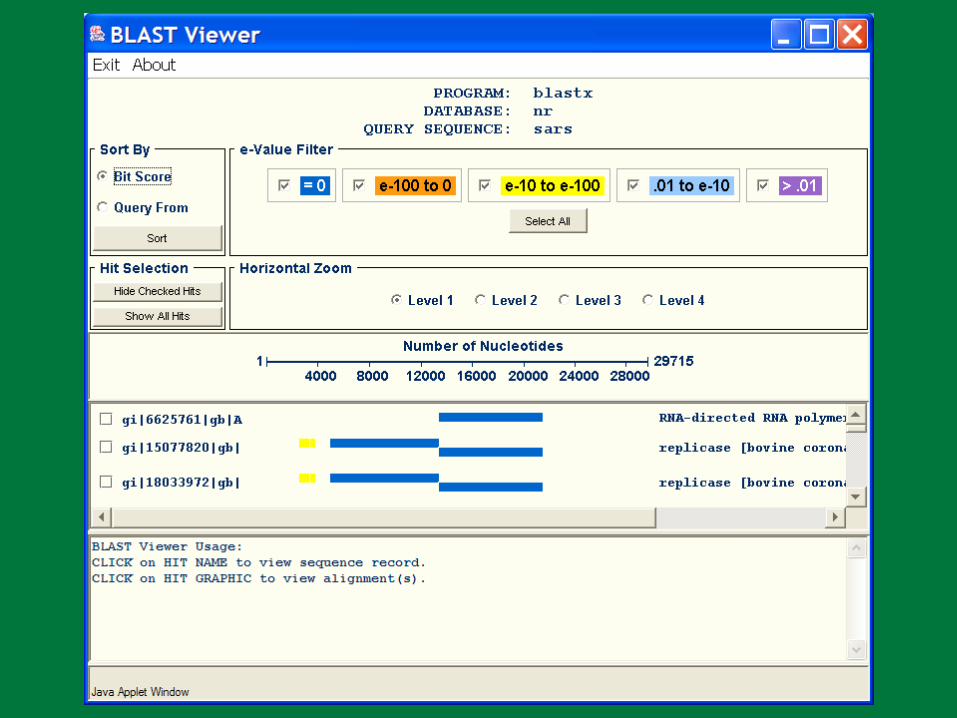
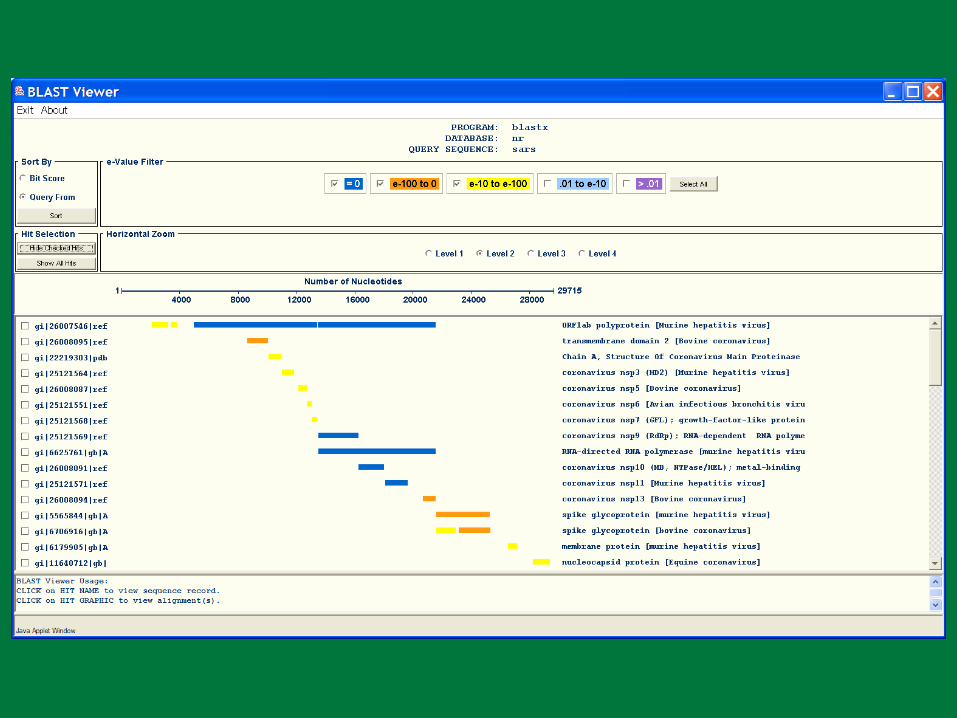
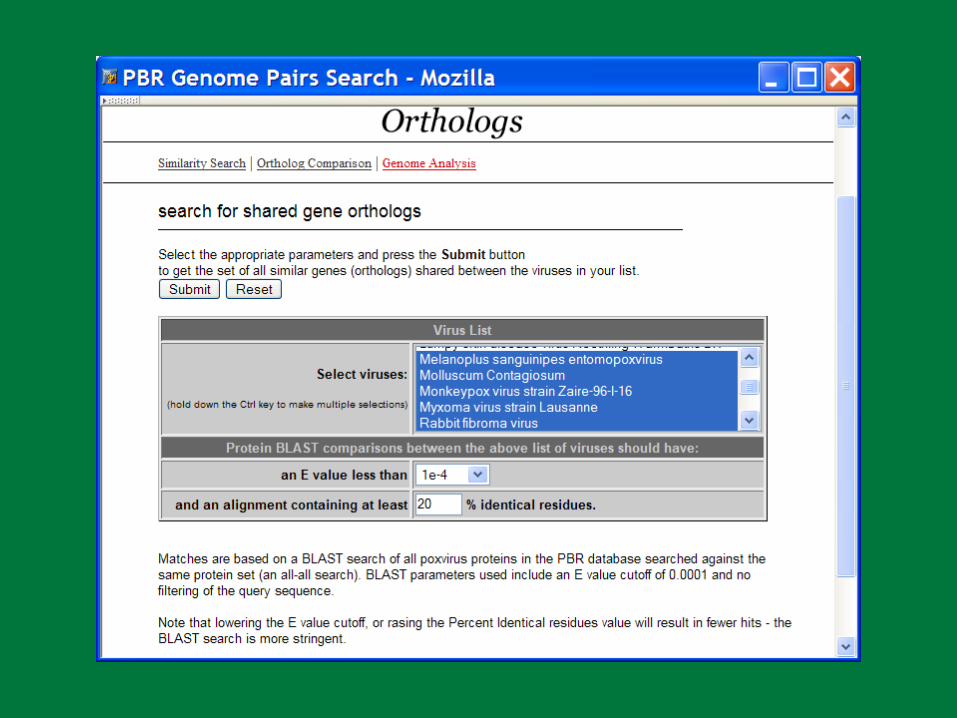
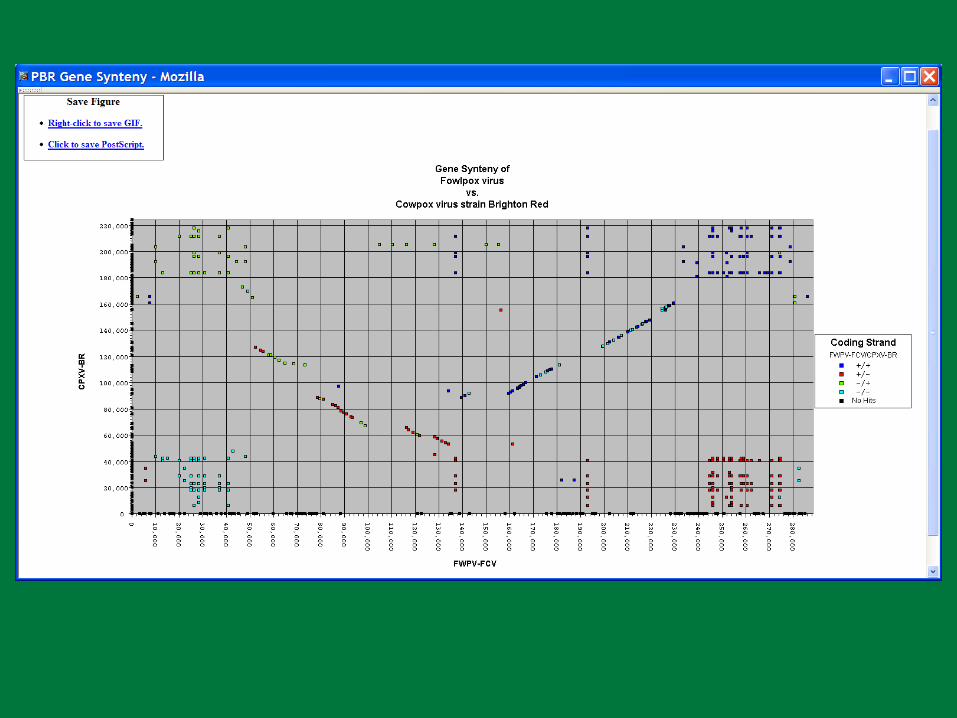
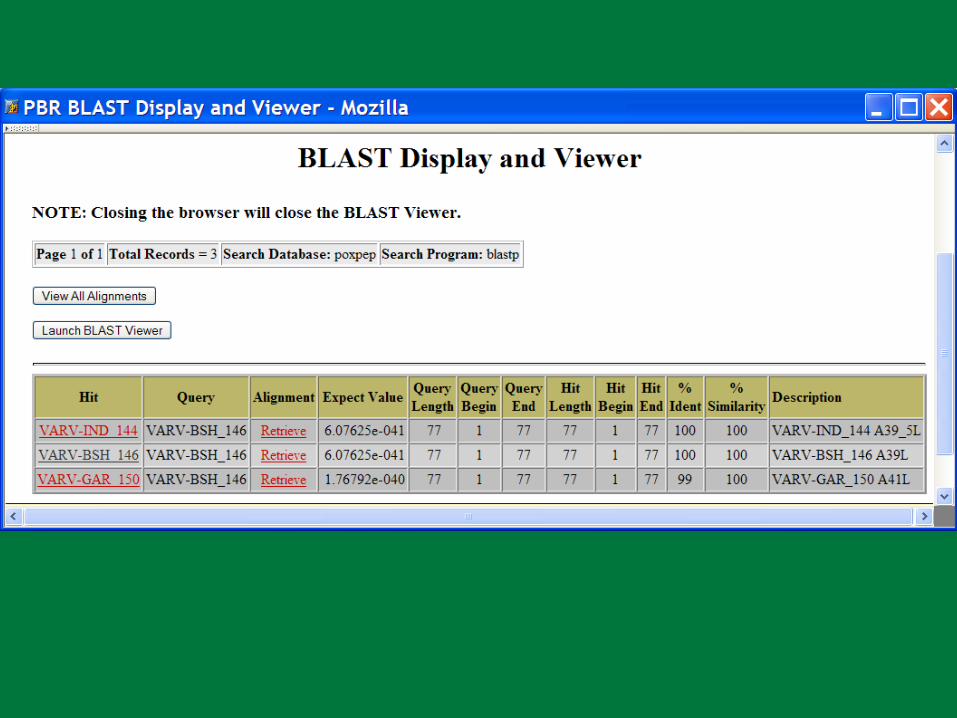
PBR BLAST
Search a sequence database for primary sequence similarities to some query sequence
Provides a measure of the significance of the similarity
Does not necessarily imply common evolutionary origin
Developed at NCBI– Altschul, S.F., Gish, W., Miller, W., Myers, E.W. &
Lipman, D.J. (1990) "Basic local alignment search tool." J. Mol. Biol. 215:403-410.
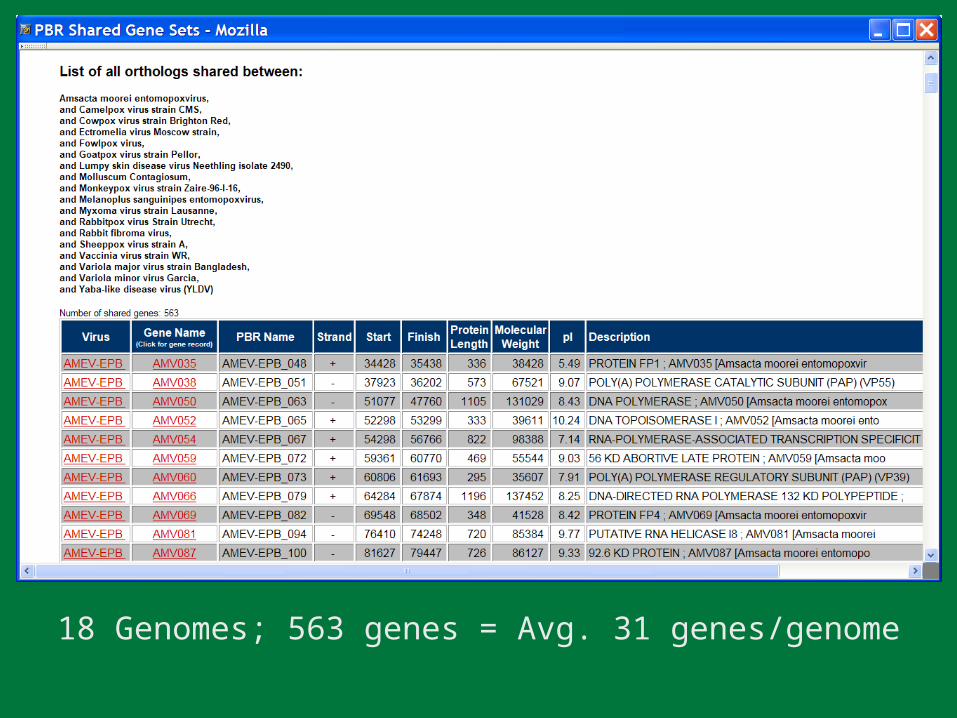
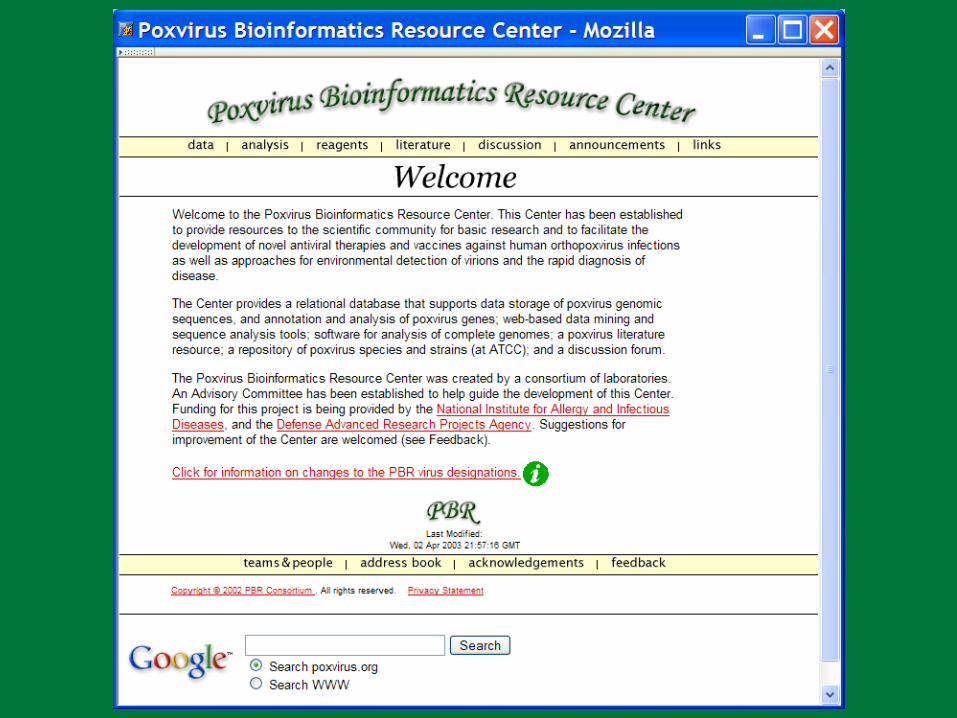
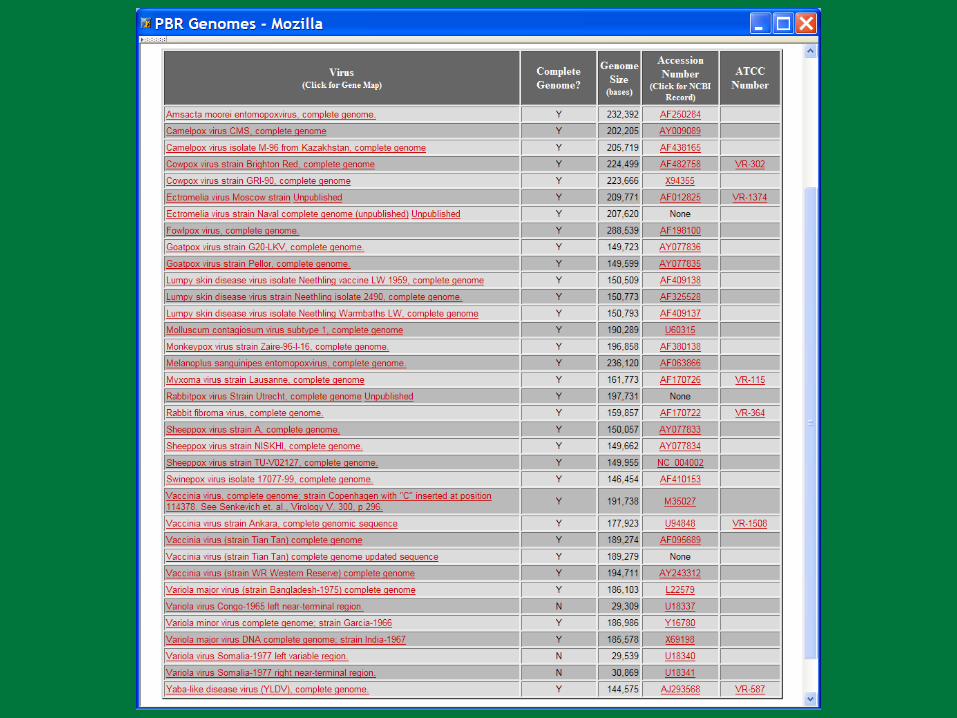
18 Genomes; 563 genes = Avg. 31 genes/genome

PBR PBR Knowledge Database
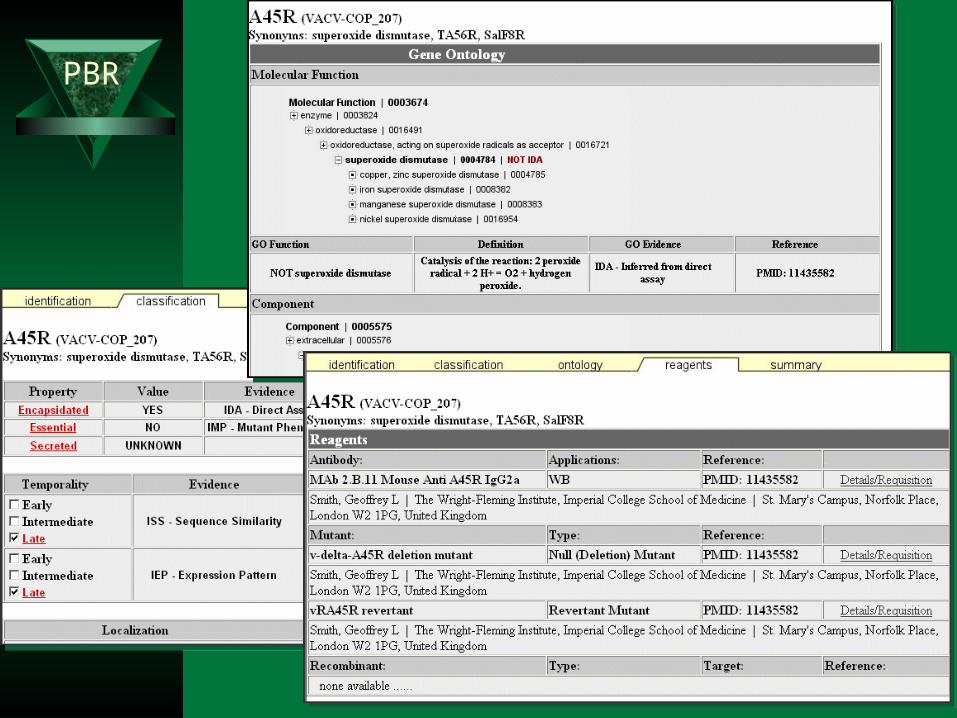
Mini review of available structure-function information – Human-curated database based on the literature
Bibliographic information Available scientific resources
• clones, mutants, and antibodies Empirically-derived properties
– MW, pI . . .– Post-translational modifications– Expression
Functional Assignments– Gene Ontology controlled vocabulary
• Molecular function• Biological Process• Cellular component
– Virulence Ontology
PBR

Molecular Evolution and GenomicAnalyses of Poxviruses

PBR Objectives
To better understand the role individual genes and groups of genes (or other genetic elements) play in poxvirus (especial smallpox ) host range and virulence
Try to describe and understand poxvirus diversity via reconstruction of the families evolutionary history
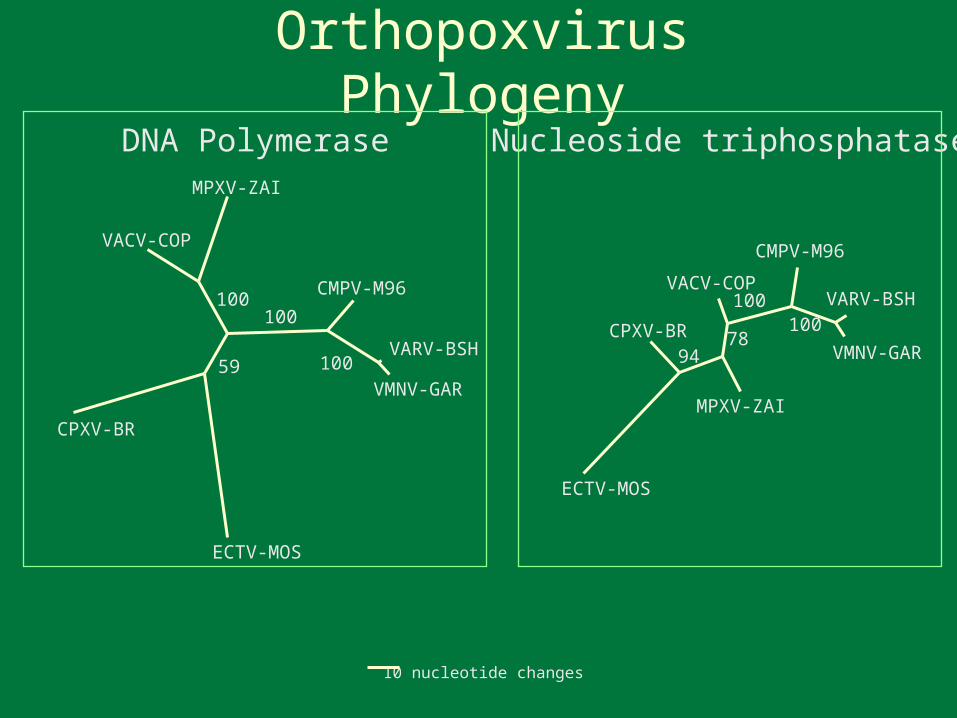
10 nucleotide changes
Orthopoxvirus PhylogenyDNA Polymerase
CMPV-M96
VARV-BSH
VMNV-GAR
ECTV-MOS
CPXV-BR
VACV-COP
MPXV-ZAI
100
100100
59
Nucleoside triphosphatase
MPXV-ZAI
CMPV-M96
VARV-BSH
VMNV-GAR
ECTV-MOS
CPXV-BR
VACV-COP
100100
7894

Orthopoxvirus Phylogeny
132 gene tree possible
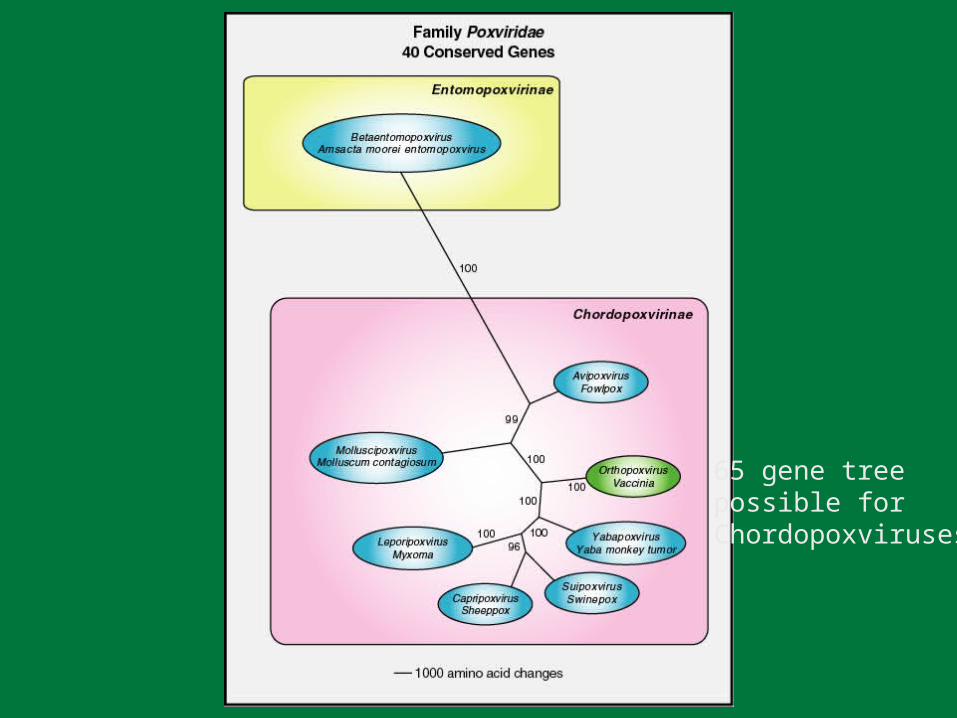
65 gene treepossible forChordopoxviruses

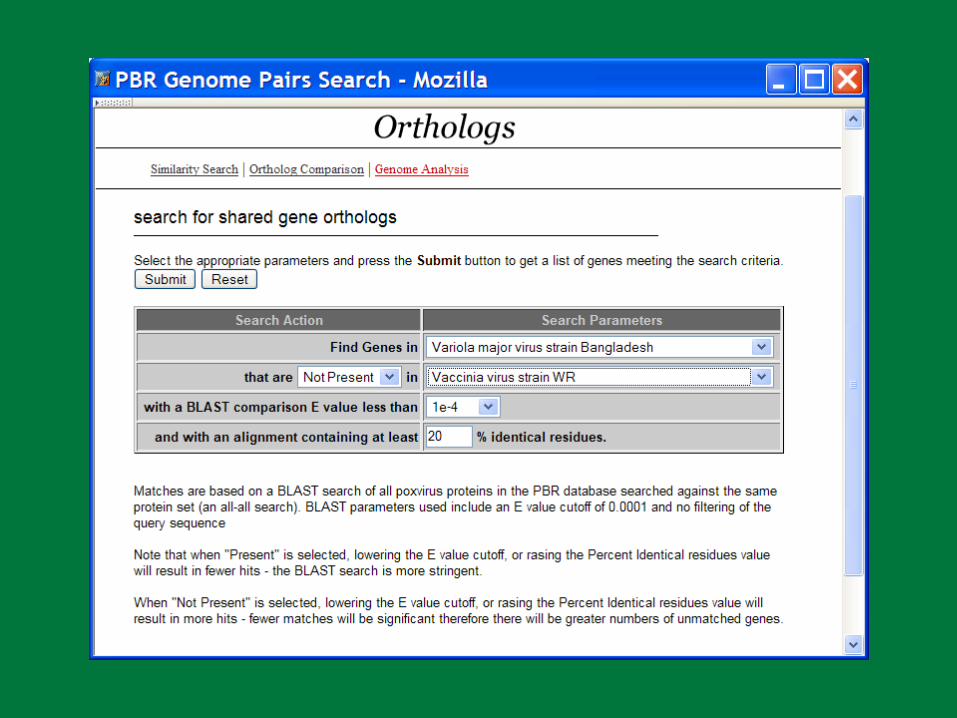
PBR Horizontal Gene Transfer
The acquisition of genetic material from another organism that becomes a “permanent” addition to the recipient’s genome
Many poxvirus genes involved in immune evasion may have been acquired thorough HGT
Detection of HGT– Alternative base composition– Alternative codon usage pattern– Alternative evolutionary inheritance pattern
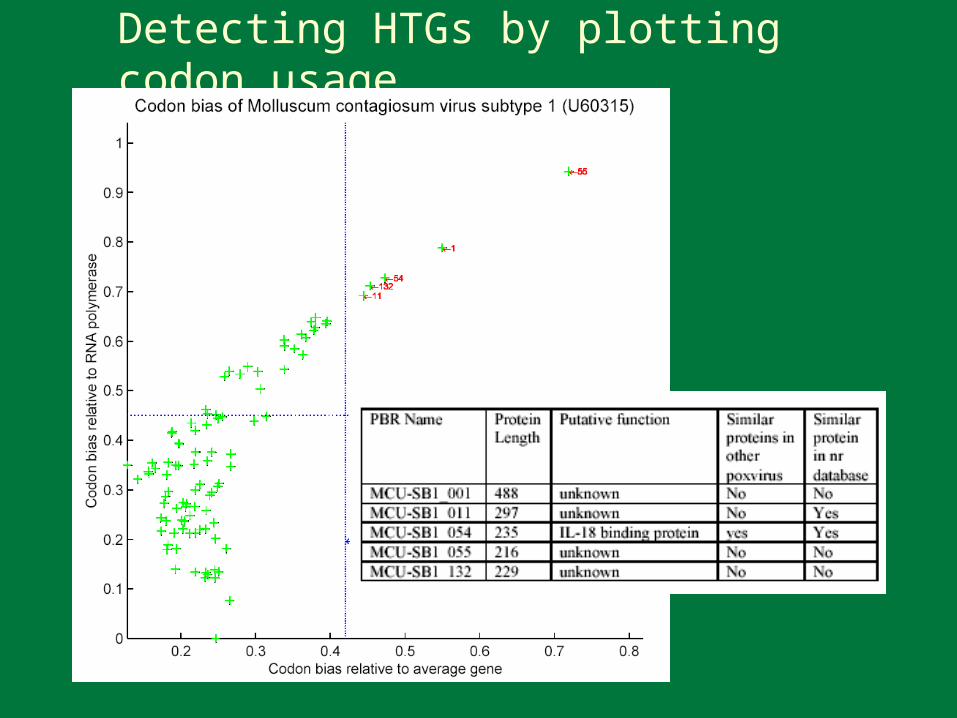
Detecting HTGs by plotting codon usage
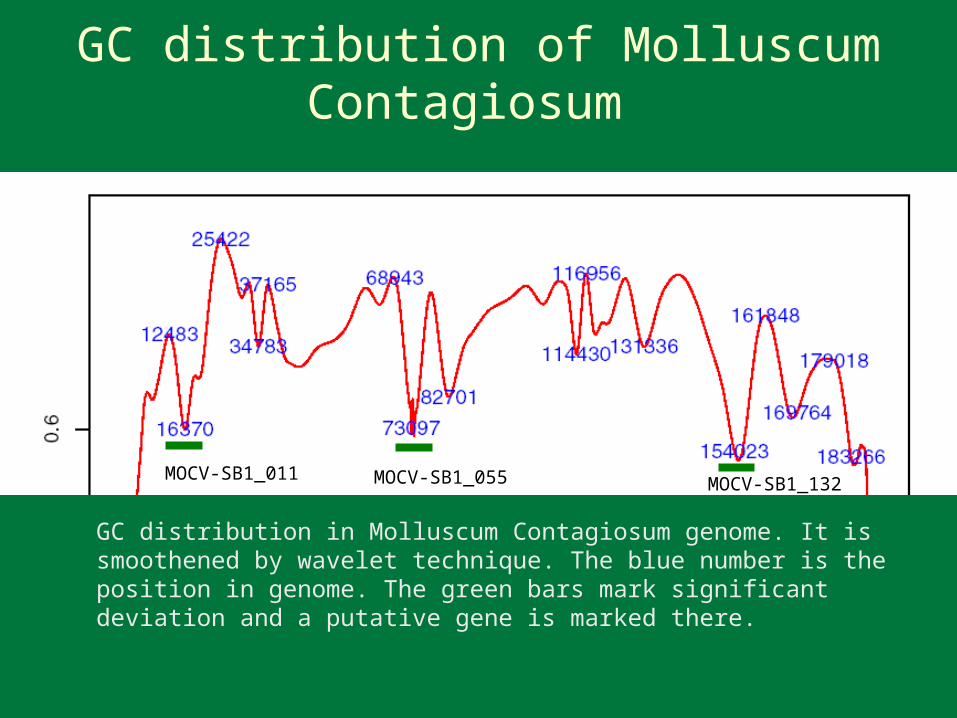
MOCV-SB1_011 MOCV-SB1_055 MOCV-SB1_132
GC distribution in Molluscum Contagiosum genome. It is smoothened by wavelet technique. The blue number is the position in genome. The green bars mark significant deviation and a putative gene is marked there.
GC distribution of Molluscum Contagiosum

VARV Proteins with Similarity to Human Proteins 3-beta-hydroxysteroid dehydrogenase Ankyrin CD47 antigen Carbonic Anhydrase Casein kinase 1 Complement control protein DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide DNA ligase Glutaredoxin Hypothetical protein JNK-stimulating phosphatase Kelch-like protein Lymphocyte activation-associated protein Makorin zinc-finger protein Myosin heavy chain Plasminogen activator inhibitor Profilin RNA polymerase Ribonucleotide reductase M2 SNF2 transcription activator Serine proteinase inhibitor Squamous cell carcinoma antigen Superoxide dismutase Thymidine kinase Tumor necrosis factor receptor
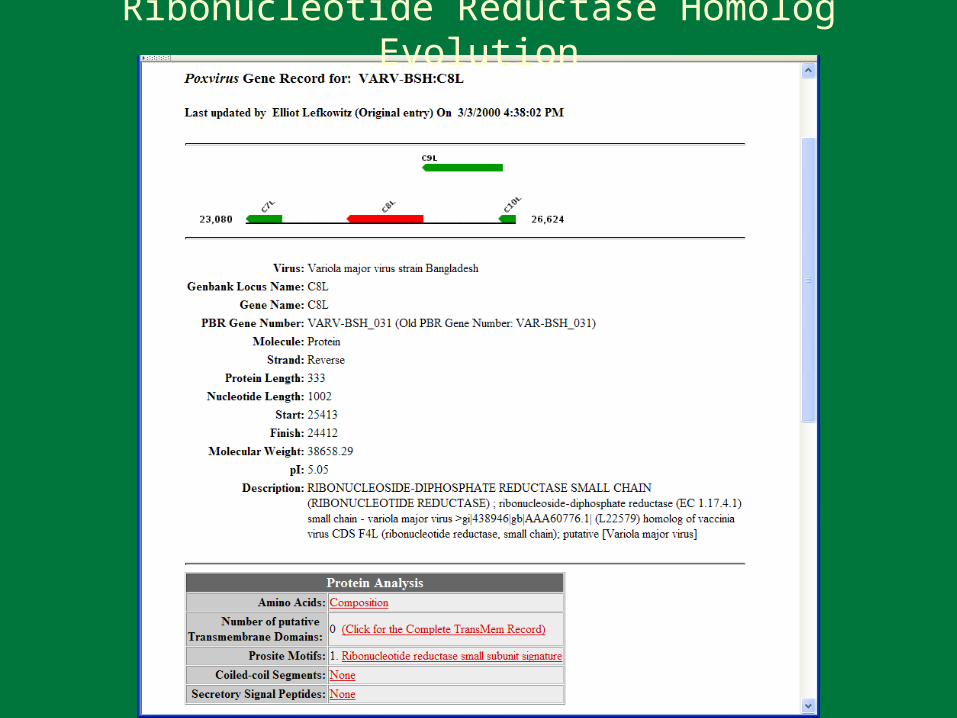
Ribonucleotide Reductase Homolog Evolution
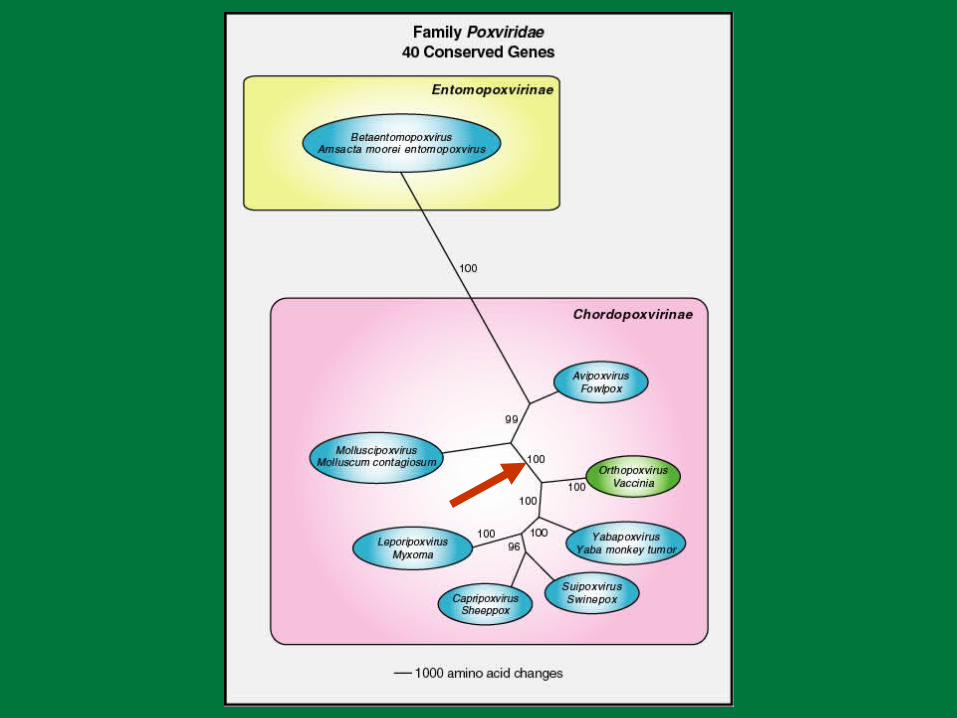
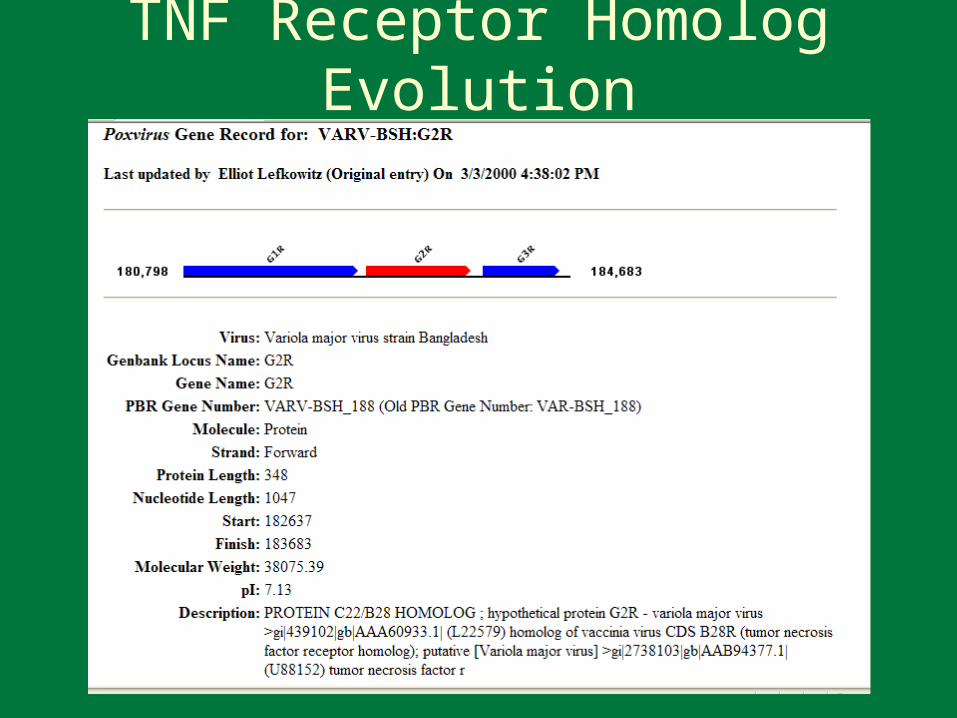
TNF Receptor Homolog Evolution

TNF Receptor GenBank nr Hits
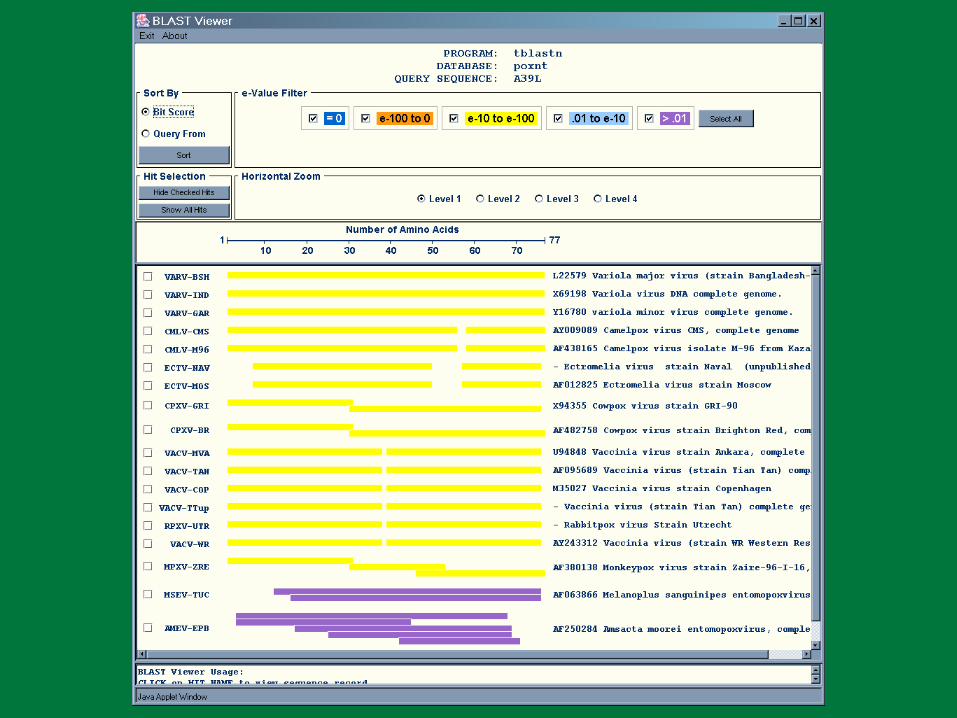
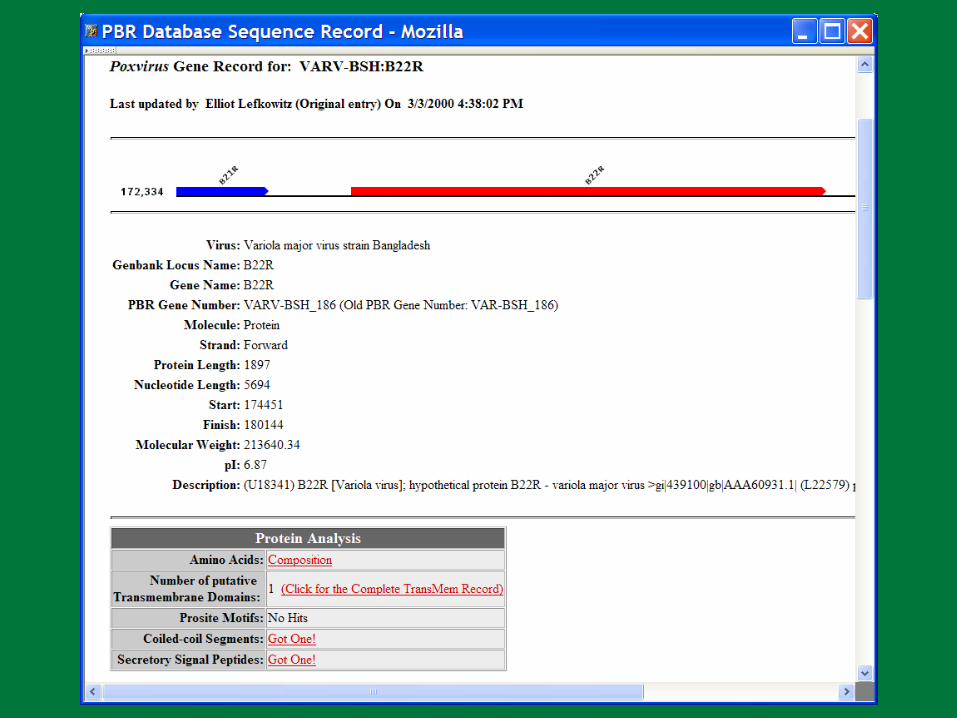

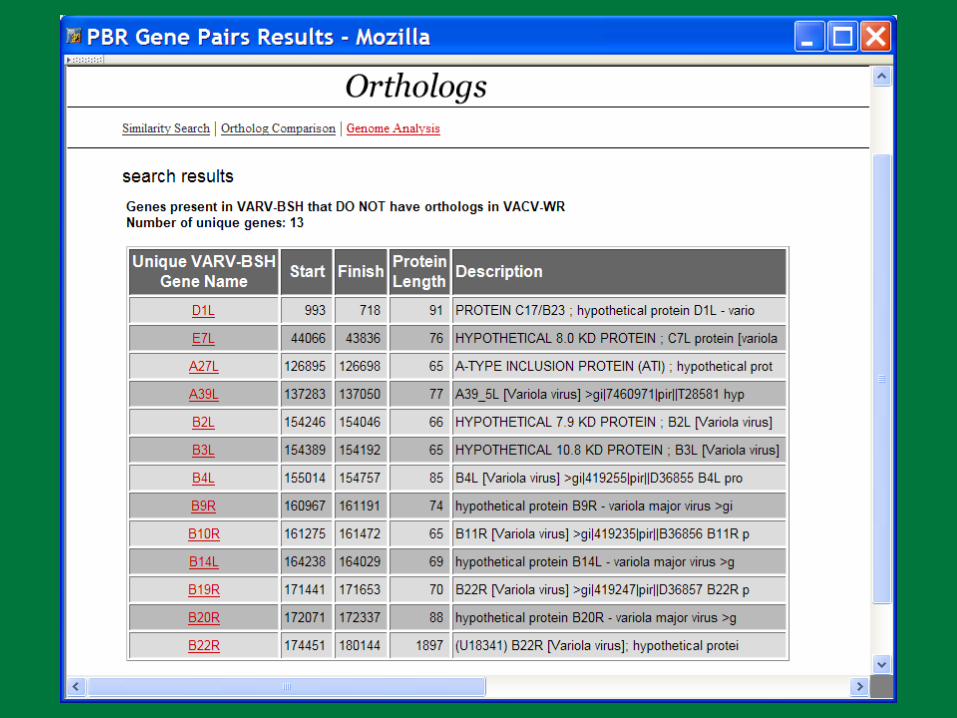
VARV B22R BLASTN Results
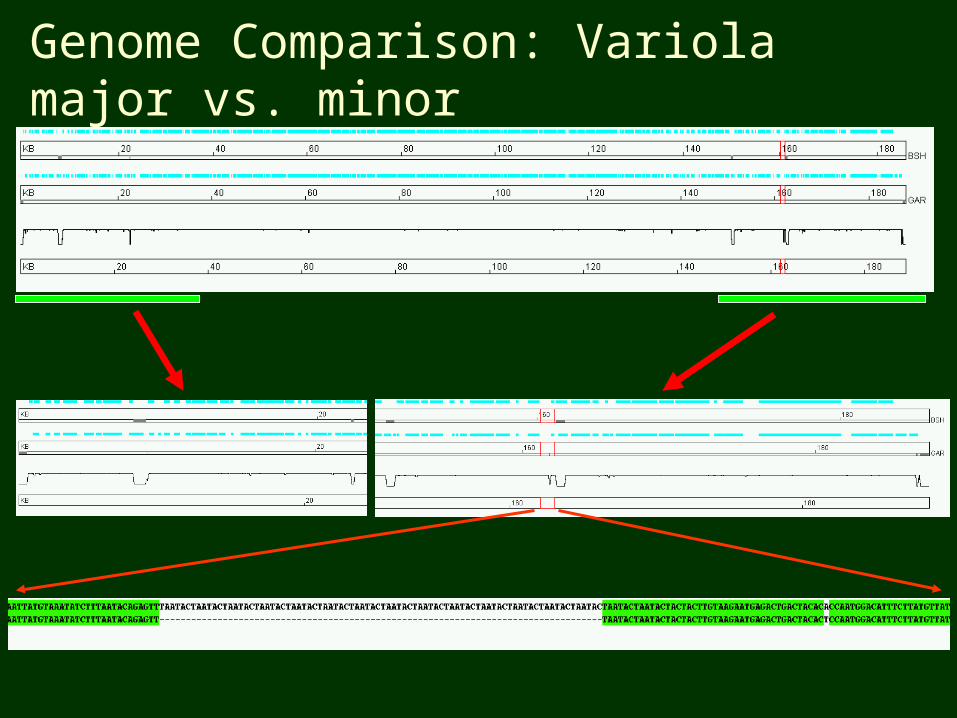
Genome Comparison: Variola major vs. minor
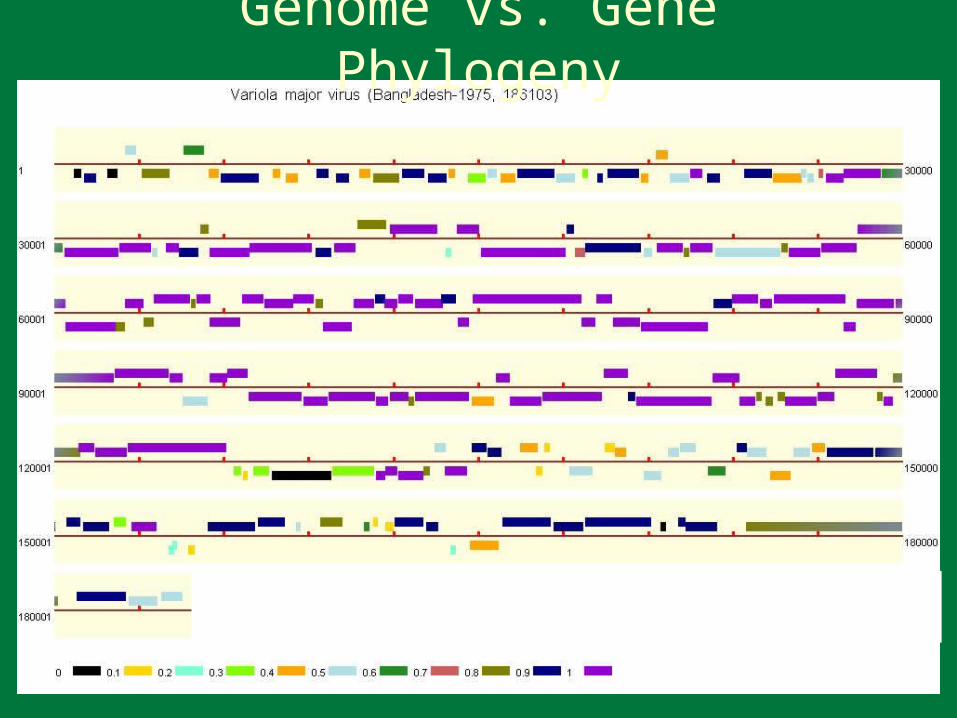
Genome vs. Gene Phylogeny

Molecular Evolution and GenomicAnalyses of Poxviruses
We have a problem…
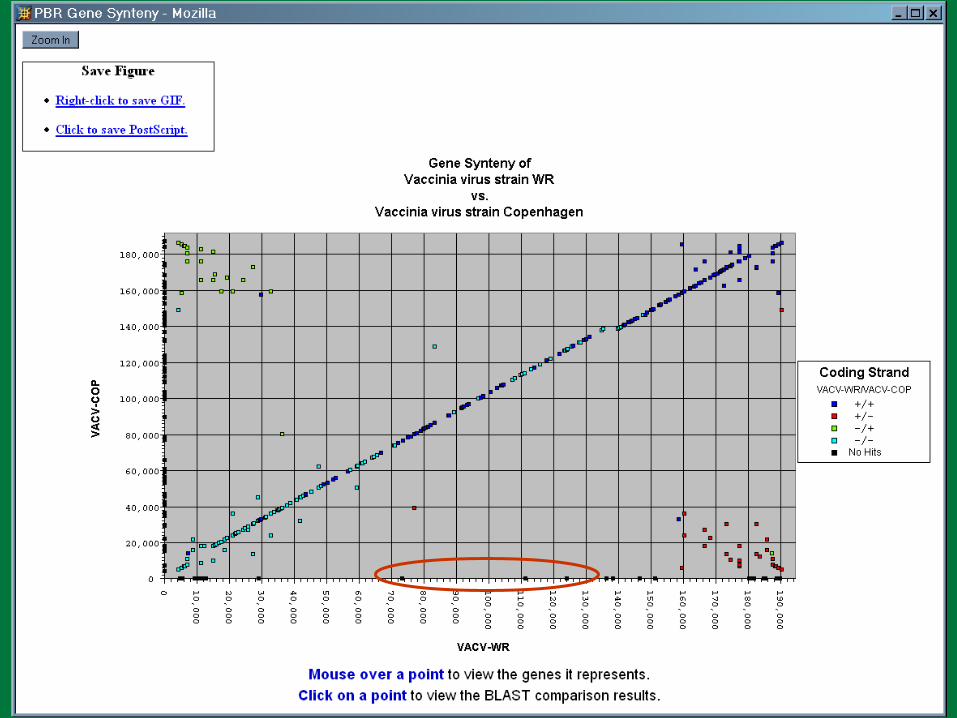
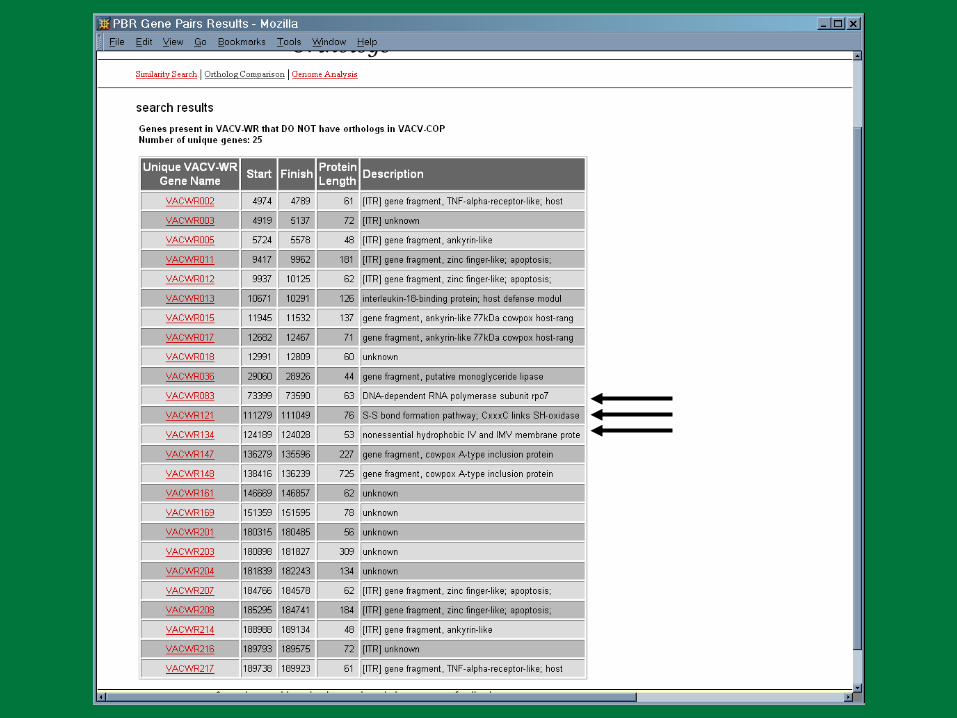
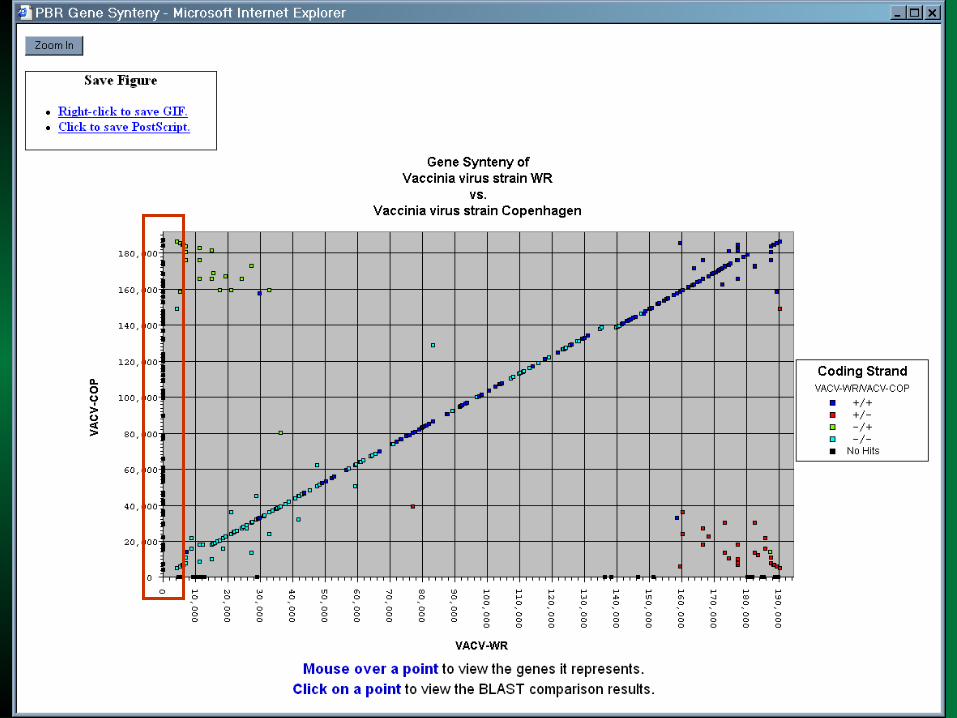
PBR
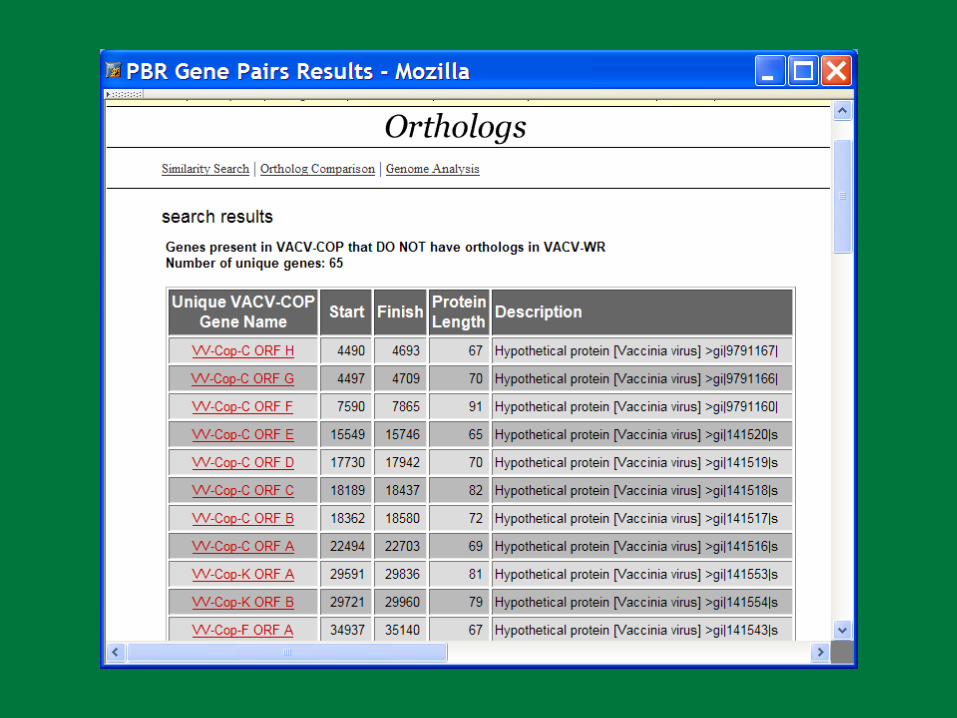

PBR Poxvirus Gene Prediction
Little consistency from one genome to another
Methods employed– Minimum ORF size– Similarity with previously described proteins

PBR Consistently predict and annotate the gene set for all Poxvirus genomes
Development of a comprehensive gene prediction tool– Discovery of new or “missed” genes– Removal of “pseudo” genes
As an added bonus:– Computational annotation of each predicted
gene

PBR What is a gene?
Does it looks like a gene?– Open Reading Frame– Base composition– Codon usage
Is it expressed?– Regulatory signals– Transcription– Translation
Has it been previously recognized?– Similarity searching

PBR Proposal gene finding tool
Combination of a series of complementary gene prediction algorithms DNA Signals
– ORF detection– Base composition– Codon preference– HMM gene models
Similarity searching– BLAST similarity searches– Similarity to identified poxvirus protein domains using an HMM-based domain
database Promoter detection
– Neural Network promoter detection tool Patterns of amino acid sequence conservation
– Biodictionary-based analysis Knowledge-based integration of all predictive methods
– Computational conclusions– Visualization tool for human inspection

Using High Performance Computing to Speedup Bioinformatic
Applications

PBR Features to consider in porting an application to a cluster environment
Balancing the processing workload among nodes is critical to successful implementation
A computational method with a lower percentage load imbalance (PLIB) is more efficient than one with a higher PLIB. The workload is perfectly balanced if PLIB is equal to zero.
Similarity searching workload can be difficult to estimate– Dependent on the nature of both the database and query sequences
• sequence length• number of sequences• complexity of the sequences
100arg
arg
estLoadL
adSmallestLoestLoadLPLIB

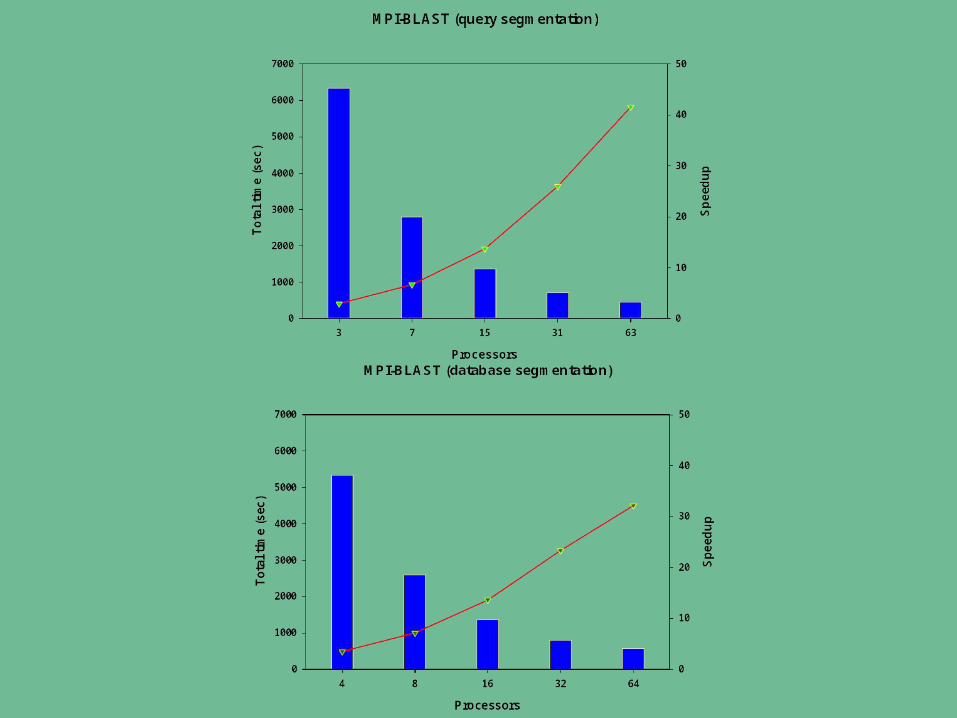
PBR Data Segmentation
Database Sequences– Utilize when the database size is larger than physical memory of
each computational node– Results need to be combined and statistics recalculated– Not possible with some applications (PSI-BLAST)
Query Sequences– Flexible and allows for better balancing of the workload– Statistics remain valid– Database remains intact– Best performance when the database can be fully loaded into
available memory
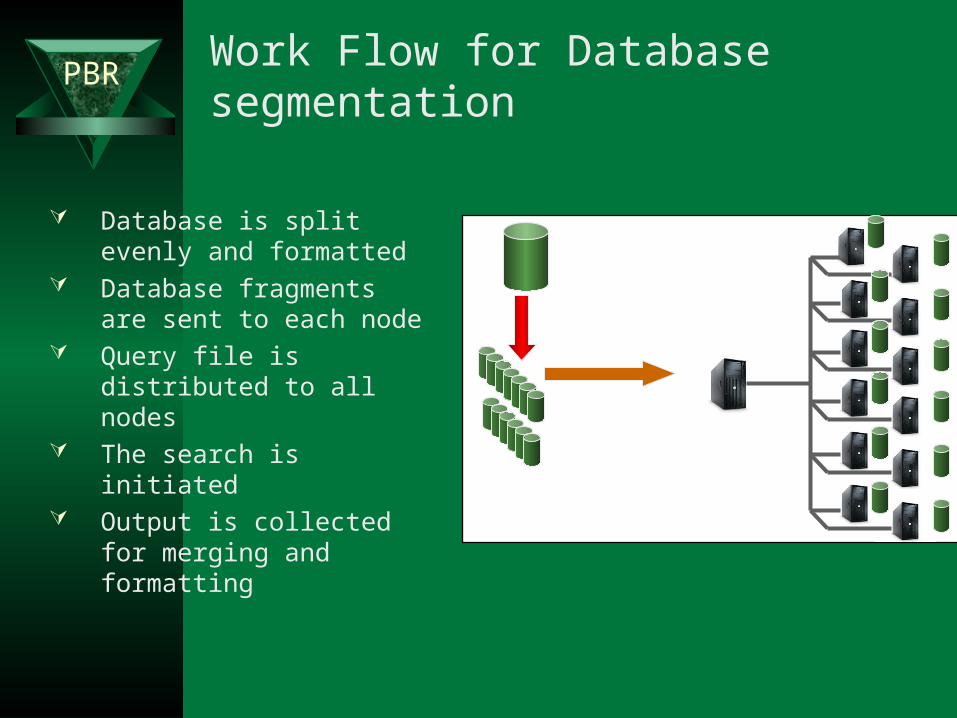
PBR Work Flow for Database segmentation
Database is split evenly and formatted
Database fragments are sent to each node
Query file is distributed to all nodes
The search is initiated Output is collected for
merging and formatting

PBR Work Flow for Query Segmentation
Database is distributed to all nodes 90% of the query sequences are split into bins and
distributed among the available nodes– Balanced for sequence length and number
The remaining 10% query of the query sequences are delivered to nodes as they finish the initial search
Individual results are merged and reported

PBR Implementation
Utilizes the LAM/MPI Message Passing Interface package from Indiana University
The application executables are not altered– The implementation wraps the executable and data and sends it to each node – Easily accommodate application updates– Easily extends to similar applications
Currently have implemented two wrappers– BLAST– HMMPFAM
• Sean Eddy, Washington University School of Medicine, St. Louis, Missouri Benchmarks performed on the UAB School of Engineer Linux cluster
– 2 storage servers (IBM x345). – one compile node and 64 compute nodes (IBM x335)
• 2 x 2.4 GHz Xeon processors per node• 2-4 GB of RAM per node• 18 GB SCSI hard drive• connected via Gigabit Ethernet to a Cisco 4006 switch
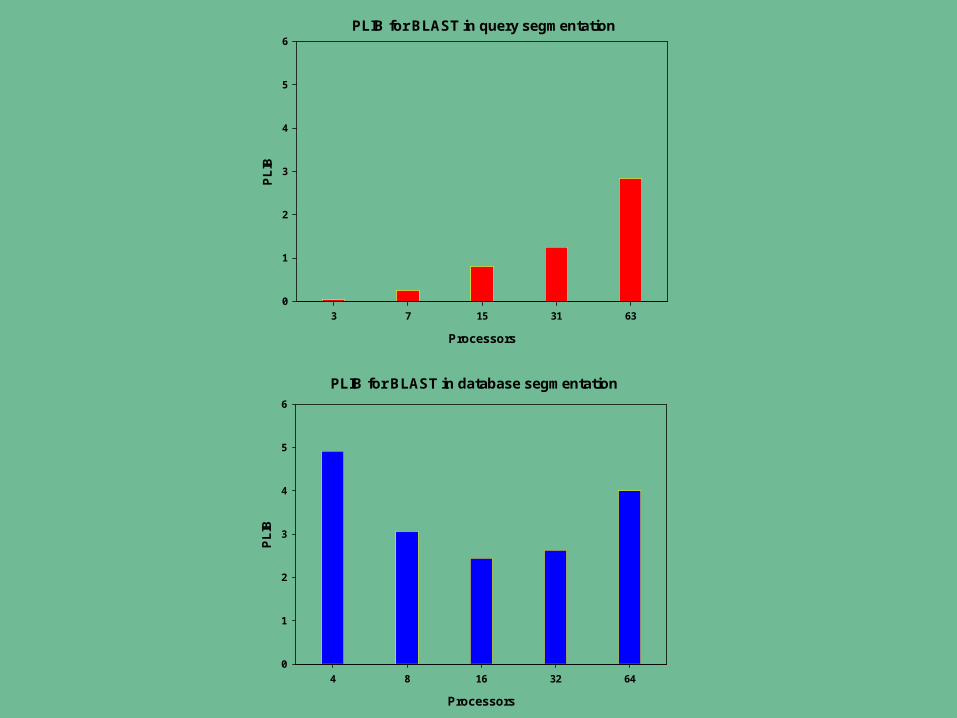
PLIB for BLAST in query segmentation
Processors
3 7 15 31 63
PL
IB
0
1
2
3
4
5
6
PLIB for BLAST in database segmentation
Processors
4 8 16 32 64
PL
IB
0
1
2
3
4
5
6
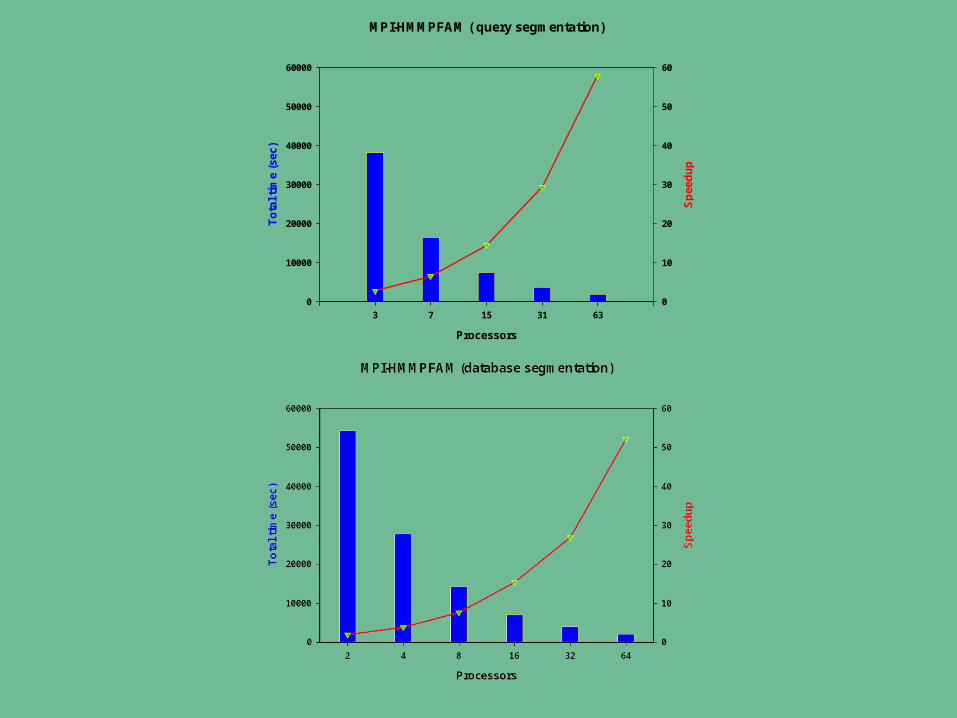
MPI-HMMPFAM ( query segmentation)
Processors
3 7 15 31 63
To
tal t
ime
(sec
)
0
10000
20000
30000
40000
50000
60000
Sp
eed
up
0
10
20
30
40
50
60
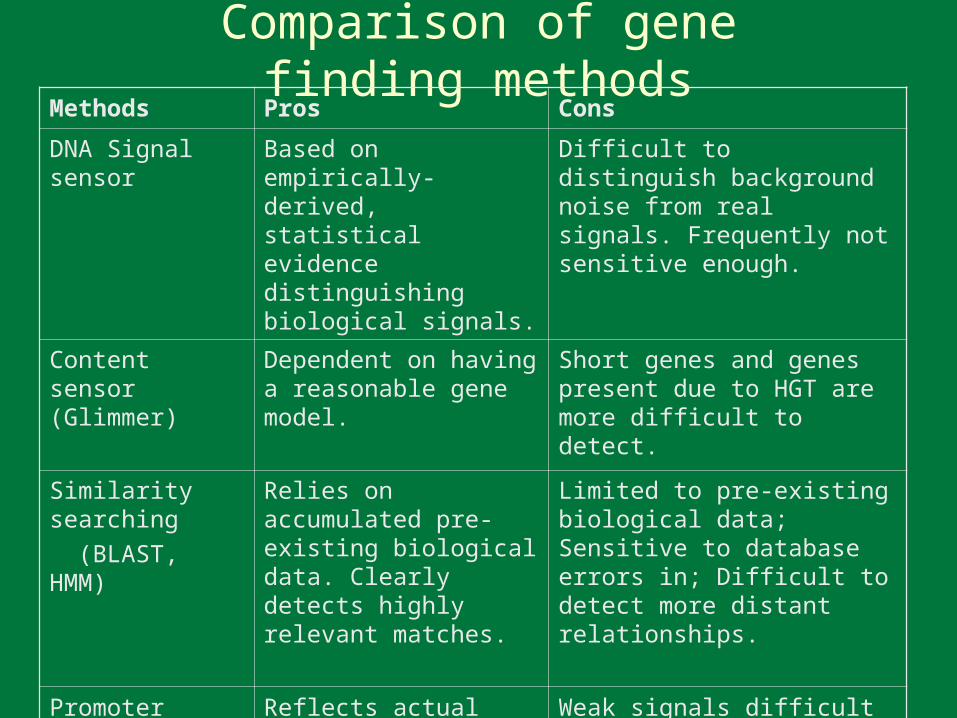
Comparison of gene finding methodsMethods Pros Cons
DNA Signal sensor Based on empirically-derived, statistical evidence distinguishing biological signals.
Difficult to distinguish background noise from real signals. Frequently not sensitive enough.
Content sensor(Glimmer)
Dependent on having a reasonable gene model.
Short genes and genes present due to HGT are more difficult to detect.
Similarity searching
(BLAST, HMM)
Relies on accumulated pre-existing biological data. Clearly detects highly relevant matches.
Limited to pre-existing biological data; Sensitive to database errors in; Difficult to detect more distant relationships.
Promoterdetection
Reflects actual poxvirus biology (gene expression).
Weak signals difficult to detect.
Bio-dictionaries Useful for detecting novel genes.
Difficult to implement; no biological evidence.
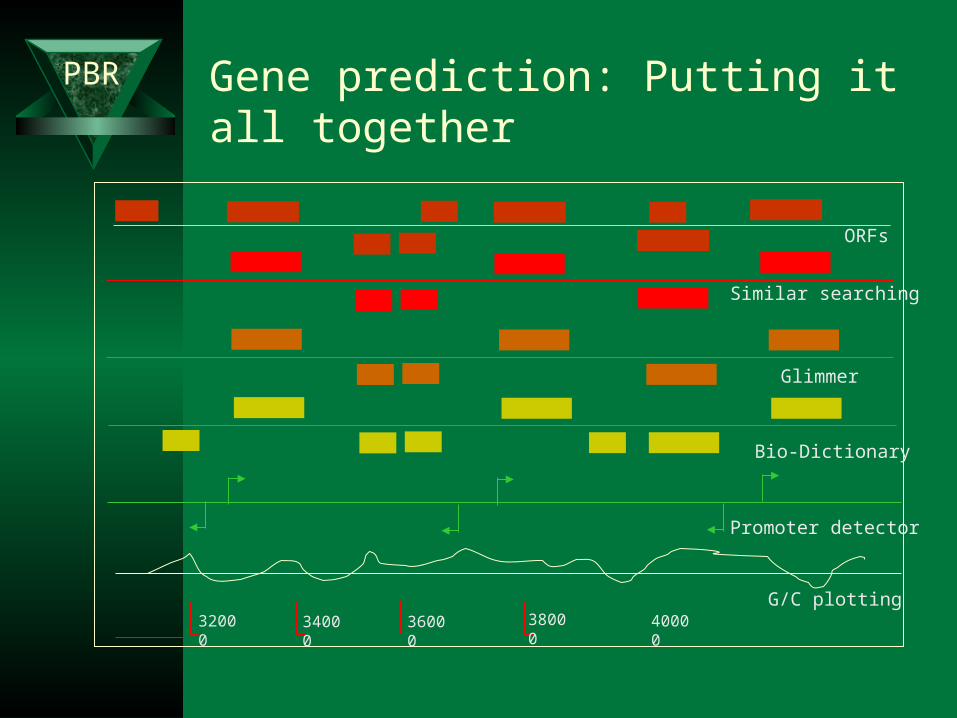
PBR Gene prediction: Putting it all together
32000 3800036000 40000
Similar searching
ORFs
Glimmer
Promoter detector
34000G/C plotting
Bio-Dictionary

PBR Now the real work can begin:
More rigorous comparative analysis– Shared and unique sets of gene composition– SNP analysis of gene differences
Whole genome phylogenetic prediction Individual gene phylogenetic predictionUnique patterns of evolutionary inheritance“Clustering” of evolutionary inheritance
with pathogenesis