Embed Size (px)
Citation preview

Biol. Chem., Vol. 392, pp. 405–413, May 2011 • Copyright � by Walter de Gruyter • Berlin • New York. DOI 10.1515/BC.2011.039
2010/233
Article in press - uncorrected proof
Post-transcriptional regulation of human cathepsin L
expression
Shivani Mittal, Riyaz A. Mir and Shyam S.Chauhan*
Department of Biochemistry, All India Institute of MedicalSciences, Ansari Nagar, New Delhi 110029, India
* Corresponding authore-mail: [email protected]
Abstract
The expression of cathepsin L, a lysosomal protease, isknown to be elevated in cancer and other pathologies. Mul-tiple splice variants of human cathepsin L with variable59UTRs exist, which encode for the same protein. Previouslywe have observed that variant hCATL A (bearing the longest59UTR) was translated in vitro with significantly lower effi-ciency than variant hCATL AIII (bearing the shortest59UTR). Contrary to these findings, results of the presentstudy reveal that in cancer cells, hCATL A mRNA exhibitshigher translatability in spite of having lower stability thanAIII. This is the first report demonstrating a highly contrast-ing trend in translation efficiencies of hCATL variants inrabbit reticulocytes and live cells. Expression from chimericmRNAs containing 59UTRs of A or AIII upstream to lucif-erase reporter cDNA established the A UTR to be the soledeterminant for this effect. Transient transfections of bicis-tronic plasmids and mRNAs confirmed the presence of afunctional Internal Ribosome Entry Site in this UTR. Ourdata suggest that differential stability and translation initia-tion modes mediated by the 59UTRs of human cathepsin Lvariants are involved in regulating its expression.
Keywords: bicistronic mRNA; IRES; translation; 59UTR.
Introduction
Cathepsin L (CATL) is elevated in almost all types of humancancers (Chauhan et al., 1991). Upon over expression, themajority of this protease is secreted by transformed cells, forwhich its carboxyl terminus is essential (Chauhan et al.,1998). It promotes tumour invasion and metastasis bydegrading extracellular matrix proteins (Frade et al., 1998;Kos and Lah, 1998; Rousselet et al., 2004), participating inproteolytic cascades and inactivating cell adhesion proteins(Gocheva and Joyce, 2007). In ras transformed cells, it islocalised in the nucleus and is involved in augmenting cellcycle progression (Goulet et al., 2007). VEGF has beenshown to induce its expression in glioblastoma cells (Keer-thivasan et al., 2007). Its involvement has also been shownin endothelial progenitor cell-mediated neovascularisation
(Urbich et al., 2005). Also, blockage of its secretion led todecreased tumour growth and vascularisation in nude mice(Rousselet et al., 2004; Frade et al., 2008). Thus CATL hasemerged as a key player in tumour progression and studiesfocussing on its regulation are of major significance.
Human CATL is encoded by at least five mRNA variants,which are transcribed off the same gene located on chro-mosome 9q21-22, by two different promoters (Chauhan etal., 1993; Bakhshi et al., 2001; Seth et al., 2003). The pri-mary transcript originating from the proximal promoter isalternatively spliced into four mRNA species, namelyhCATL A, AI, AII and AIII, containing variable lengths ofexon I, which forms major portions of their 59 untranslatedregions (59UTRs). The hCATL A variant contains full lengthexon I (280 nucleotides), whereas AI, AII and AIII lack 27,90 and 145 nucleotides, respectively, from the 39 end of thisexon (Gal and Gottesman, 1988; Rescheleit et al., 1996).
We previously observed differential in vitro translationalefficiencies of the above mentioned hCATL splice variants(Arora and Chauhan, 2002). The shortest variant, hCATLAIII, was translated with the maximum efficiency whereasthe longest variant, hCATL A, exhibited minimum transla-tional efficiency. A similar variation in their relative abun-dance was also observed in five cancer lines derived fromdifferent human organs. The present study was thereforedesigned to gain further insights into the post transcriptionalregulatory mechanisms in cancer cells resulting in differen-tial transcript levels and variable translational outputs.
In this study, we demonstrate for the first time that signi-ficantly higher stability of hCATL AIII compared to hCATLA mRNA is responsible for their differential abundance. Inaddition, we observe higher translational ability of thehCATL A variant in living cells. This is contrary to the invitro situation, where hCATL A exhibits minimum transla-tion potential. Further analysis of the hCATL A mRNArevealed a functional IRES in its 59UTR region, supportinghigher cathepsin L expression.
Results
HCATL A and AIII exhibit differential in vivo
stabilities
Variable decay rates of hCATL mRNA splice variants couldresult in their differential abundance. To ascertain this, therelative half lives of these mRNA variants were determined.For this, stable clones of NIH3T3 cells (mouse fibroblasts),expressing hCATL A or AIII variants, were generated. Thepercentage of the mRNA remaining after different timepoints of actinomycin D treatment was determined by North-
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM
406 S. Mittal et al.
Article in press - uncorrected proof
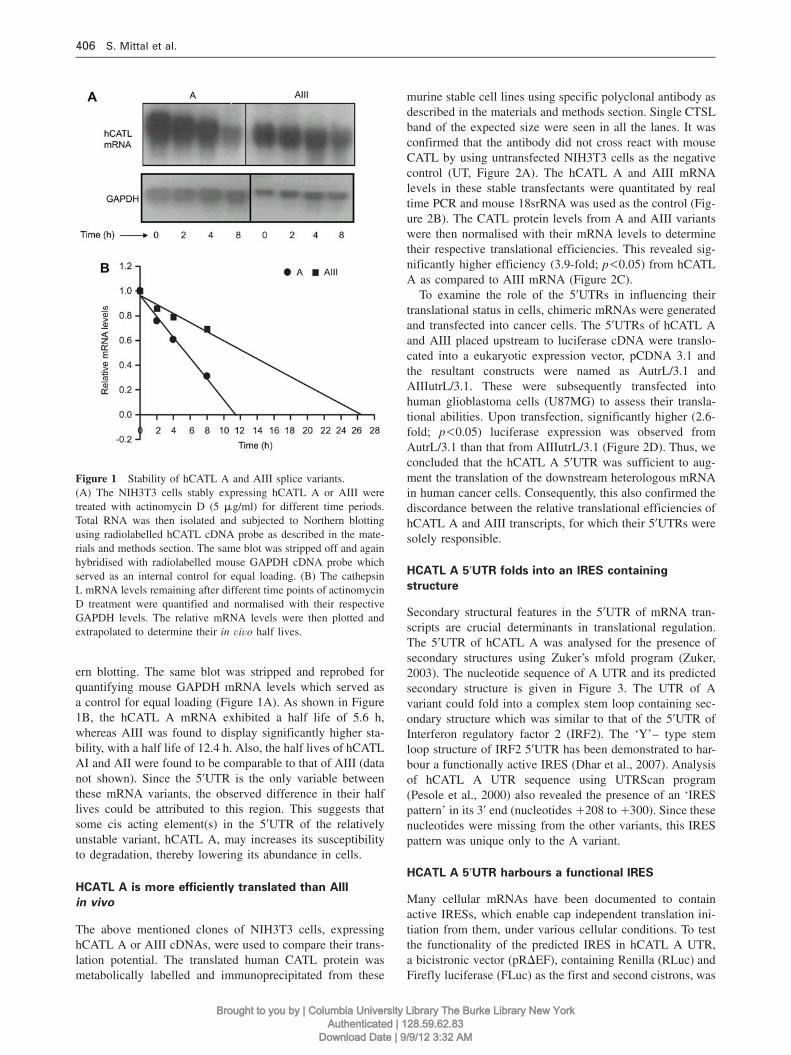
Figure 1 Stability of hCATL A and AIII splice variants.(A) The NIH3T3 cells stably expressing hCATL A or AIII weretreated with actinomycin D (5 mg/ml) for different time periods.Total RNA was then isolated and subjected to Northern blottingusing radiolabelled hCATL cDNA probe as described in the mate-rials and methods section. The same blot was stripped off and againhybridised with radiolabelled mouse GAPDH cDNA probe whichserved as an internal control for equal loading. (B) The cathepsinL mRNA levels remaining after different time points of actinomycinD treatment were quantified and normalised with their respectiveGAPDH levels. The relative mRNA levels were then plotted andextrapolated to determine their in vivo half lives.
ern blotting. The same blot was stripped and reprobed forquantifying mouse GAPDH mRNA levels which served asa control for equal loading (Figure 1A). As shown in Figure1B, the hCATL A mRNA exhibited a half life of 5.6 h,whereas AIII was found to display significantly higher sta-bility, with a half life of 12.4 h. Also, the half lives of hCATLAI and AII were found to be comparable to that of AIII (datanot shown). Since the 59UTR is the only variable betweenthese mRNA variants, the observed difference in their halflives could be attributed to this region. This suggests thatsome cis acting element(s) in the 59UTR of the relativelyunstable variant, hCATL A, may increases its susceptibilityto degradation, thereby lowering its abundance in cells.
HCATL A is more efficiently translated than AIII
in vivo
The above mentioned clones of NIH3T3 cells, expressinghCATL A or AIII cDNAs, were used to compare their trans-lation potential. The translated human CATL protein wasmetabolically labelled and immunoprecipitated from these
murine stable cell lines using specific polyclonal antibody asdescribed in the materials and methods section. Single CTSLband of the expected size were seen in all the lanes. It wasconfirmed that the antibody did not cross react with mouseCATL by using untransfected NIH3T3 cells as the negativecontrol (UT, Figure 2A). The hCATL A and AIII mRNAlevels in these stable transfectants were quantitated by realtime PCR and mouse 18srRNA was used as the control (Fig-ure 2B). The CATL protein levels from A and AIII variantswere then normalised with their mRNA levels to determinetheir respective translational efficiencies. This revealed sig-nificantly higher efficiency (3.9-fold; p-0.05) from hCATLA as compared to AIII mRNA (Figure 2C).
To examine the role of the 59UTRs in influencing theirtranslational status in cells, chimeric mRNAs were generatedand transfected into cancer cells. The 59UTRs of hCATL Aand AIII placed upstream to luciferase cDNA were translo-cated into a eukaryotic expression vector, pCDNA 3.1 andthe resultant constructs were named as AutrL/3.1 andAIIIutrL/3.1. These were subsequently transfected intohuman glioblastoma cells (U87MG) to assess their transla-tional abilities. Upon transfection, significantly higher (2.6-fold; p-0.05) luciferase expression was observed fromAutrL/3.1 than that from AIIIutrL/3.1 (Figure 2D). Thus, weconcluded that the hCATL A 59UTR was sufficient to aug-ment the translation of the downstream heterologous mRNAin human cancer cells. Consequently, this also confirmed thediscordance between the relative translational efficiencies ofhCATL A and AIII transcripts, for which their 59UTRs weresolely responsible.
HCATL A 59UTR folds into an IRES containing
structure
Secondary structural features in the 59UTR of mRNA tran-scripts are crucial determinants in translational regulation.The 59UTR of hCATL A was analysed for the presence ofsecondary structures using Zuker’s mfold program (Zuker,2003). The nucleotide sequence of A UTR and its predictedsecondary structure is given in Figure 3. The UTR of Avariant could fold into a complex stem loop containing sec-ondary structure which was similar to that of the 59UTR ofInterferon regulatory factor 2 (IRF2). The ‘Y’– type stemloop structure of IRF2 59UTR has been demonstrated to har-bour a functionally active IRES (Dhar et al., 2007). Analysisof hCATL A UTR sequence using UTRScan program(Pesole et al., 2000) also revealed the presence of an ‘IRESpattern’ in its 39 end (nucleotides q208 to q300). Since thesenucleotides were missing from the other variants, this IRESpattern was unique only to the A variant.
HCATL A 59UTR harbours a functional IRES
Many cellular mRNAs have been documented to containactive IRESs, which enable cap independent translation ini-tiation from them, under various cellular conditions. To testthe functionality of the predicted IRES in hCATL A UTR,a bicistronic vector (pRDEF), containing Renilla (RLuc) andFirefly luciferase (FLuc) as the first and second cistrons, was
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

59UTR mediated regulation of hCATL expression 407
Article in press - uncorrected proof
Figure 2 Translation efficiency of hCATL A and AIII in vivo.(A) Equal number of NIH3T3 cells stably expressing full length hCATL A or hCATL AIII cDNA cloned in pCDNA 3.1 vector were plated.Next day, the cells were metabolically labelled with w35Sx methionine as described in the materials and methods section. Radiolabelledhuman CATL in cell extract was then immunoprecipitated using rabbit antihuman proCATL antibody. Immunoprecipitates were resolvedon 12% SDS PAGE and subjected to autoradiography. Consequently, a single band of human CATL (42 kDa) was seen in each lane exceptfor negative control (UT, untransfected NIH3T3 cells). (B) The expression of hCATL A and AIII mRNAs in the stably transfected NIH3T3cells were quantified by real time RT-PCR using specific hCATL primers. After normalisation with mouse 18s rRNA levels, the hCATLAIII mRNA level is presented as fold increase over hCATLA mRNA. Values are mean"SE from at least three independent experimentsperformed in triplicates. The results were analysed by Student’s t-test and values significantly different (p-0.05) are indicated by ‘*’. (C)The translational efficiencies in NIH3T3 cells were calculated by densitometric quantitation of the immunoprecipitated human CATL bandstranslated from hCATL A and AIII (shown in Figure 1A). The protein values were normalised with the respective mRNA levels of theabove variants determined by Northern blotting to assess the translation efficiency of each variant. Values are mean"SE from at least threeindependent experiments performed in triplicates. Statistical analysis was carried out as described above. (D) U87MG cells were co-transfected with 1 mg of the constructs pAutrL/3.1 or pAIIIutrL/3.1 along with pRL null in a molar ratio of 1:20 respectively. The cellswere lysed 48 h post transfection followed by analysis of lysates for luciferase activities as described in materials and methods. The valuesare presented as mean"SE from at least three independent experiments performed in triplicates. Other details are as given above.
used. The putative IRES containing UTR was cloned inbetween the RLuc and FLuc coding region, downstream toa landscape of secondary structures (represented by DE). Theresultant vector, designated as pRDECF (Figure 4A), wastransiently transfected in two different cancer cell lines. Ele-vated FLuc/RLuc ratios were observed from this vector inboth U87MG (39 fold) and HepG2 (17 fold) cells as com-pared to those from the parent vector (pRDEF), which served
as control (Figure 4B). Thus significantly higher expressionof the downstream FLuc cistron upon insertion of the UTRindicated that the UTR contains a functional IRES.
Bicistronic DNA transfections could give false positiveresults for IRES activity as aberrant splicing or cryptic pro-moter in the UTR region could generate monocistronic FLuctranscripts, resulting in higher FLuc expression. To rule outthis, RT-PCR and Northern blotting were performed using
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

408 S. Mittal et al.
Article in press - uncorrected proof
Figure 3 The A UTR folds into a characteristic IRES containing secondary structure.The hCATL A 59UTR nucleotide sequence shown in the box was analysed using the ‘MFOLD’ web server. The A UTR was found to foldinto a highly complex and stable secondary structure (DGs-115.5 kcal/mol). The Y-shaped secondary structure formed by A UTR sharedsimilarity to that of IRF2 59UTR, which is known to exhibit IRES activity.
RNA extracted from U87MG cells transfected with pRDEFor pRDECF plasmids. Two primer pairs, P1/P2 and P3/P4,were used to look for spurious transcripts from the bicistronicconstructs using RT-PCR. This was also confirmed byNorthern blotting. As shown in Figure 4C, a single band ofexpected size was seen in each lane. This confirmed that thebicistronic transcripts remained largely intact, arguing thatthe translation from the second cistron was unlikely to beinitiated from spliced transcripts or from functionally mono-cistronic transcripts, which may have arisen from the pro-moter in the 59UTR of hCATL A.
In order to unambiguously demonstrate the functionalityof this IRES, capped bicistronic mRNAs were generatedfrom pRDEF and pRDECF plasmids by in vitro transcription.Equal concentrations of these transcripts were then trans-fected into U87MG cells. This resulted in a significantlyhigher FLuc/RLuc ratio w3.2 fold (p-0.05)x as compared tothe control transcript (Figure 4D). Taken together, theseresults unequivocally demonstrated the presence of a func-tional IRES, which resulted in higher expression of thedownstream firefly cistron.
Discussion
CATL plays an important role in the development, progres-sion and metastasis of cancer and therefore its inhibitioncould prove useful in the management of this disease (Fradeet al., 2008; Fortenberry et al., 2010). The importance ofcombining CATL inhibition with chemotherapy as a morepromising therapeutic intervention in cancer has also beenhighlighted by Lankelma et al. (2010). Understanding themolecular regulatory mechanisms governing the expressionof this protease will thus have important therapeutic impli-cations. Our laboratory has previously demonstrated tran-
scriptional up regulation of CATL by vascular endothelialgrowth factor and wild type p53 in human glioblastoma cells(Keerthivasan et al., 2007; Katara et al., 2010). Our in vitroexperiments also revealed that multiple mRNA variantsencoding human CATL are differentially translated andhCATL AIII exhibited highest translational efficiency. Onthe contrary, hCATL A was translated with the lowest effi-ciency. Present study was performed to further elucidate thein vivo mechanism of human CATL regulation by thesevariants.
Here, we demonstrate that hCATL AIII exhibits compar-atively higher stability than hCATL A. Although it has beenobserved for other mRNAs, this is the first report describingdifferential stability of the cathepsin L splice variants(Hughes and Brady, 2005; Wang et al., 2005; Payton et al.,2007). Higher stability of hCATL AIII may be responsiblefor its elevated steady state levels in various human cell linesobserved in our previous study (Arora and Chauhan, 2002).Interestingly, the translational abilities of these two splicevariants in live cells were contrary to their in vitro potential.HCATL A exhibited a higher translation rate and its 59UTRwas sufficient to confer this advantage to a heterologousmRNA. We propose that stabilisation of this efficiently trans-lated but labile mRNA (hCATL A) may be a plausible mech-anism for CATL over expression. At the present state of ourknowledge, the reason for the instability of hCATL A is notclear but we speculate that a yet unidentified element in its59UTR may be responsible for its shorter half life.
In an attempt to elucidate the contradictory in vivo and invitro translational efficiency of hCATL A we analysed thenucleotide sequence of its 59UTR. As shown in Figure 3, itcould fold into a complex secondary stem loop structure (DGs -115.5 kcal/mol). Such stem loop structures in the 59UTRwith DGF-50 kcal/mol have been demonstrated to inhibitcap dependant ribosomal scanning (Kozak, 1986). Further
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

59UTR mediated regulation of hCATL expression 409
Article in press - uncorrected proof
Figure 4 Demonstration of IRES activity in hCATL A UTR.(A) In silico analysis of hCATL A UTR revealed that it contains an ‘IRES pattern’ in its 39 end. The full-length 59UTR was cloned in theintercistronic region of the bicistronic plasmid, pRDEF, in between DEMCV stem loop and firefly luciferase cistron, resulting in pRDECFconstruct. (B) U87MG or HepG2 cells transfected with 1 mg of the constructs pRDEF or pRDECF were lysed 48 h post transfectionfollowed by estimation of RLuc and FLuc activities. The FLuc and RLuc activities are shown separately as fold increase over control. Thevalues are presented as mean"SE of at least three independent experiments performed in triplicates. The FLuc activity is represented by h
and RLuc activity by j. (C) Five mg of total cellular RNA isolated from U87MG cells transiently transfected with pRDEF or pRDECFplasmids were reverse transcribed. This was then subjected to PCR using the primer pairs P1/P2 and P3/ P4. The positions of these primershave been indicated by arrows. Lane 1: RT-negative control; Lanes 3 and 5: PCR products using bicistronic DNA as templates; Lanes 4and 6: PCR products using RT as template and P1/P2 primers; Lanes 7 and 8: PCR products using RT as template and P3/P4 primers. Noamplification of any shorter transcript was seen in any of the lanes. For Northern blot analysis, 35 mg of total RNA from the above cellstransfected with pRDEF or pRDECF plasmids was resolved on 1% denaturing agarose gel and transferred to a nylon membrane. This blotwas then hybridised with a firefly cDNA radiolabelled probe. Single bicistronic transcript of expected size. UT-Untransfected cells. (D)Bicistronic capped mRNAs were generated by in vitro transcription using 1 mg of the linearised plasmids, pRDEF and pRDECF, as template.0.8 mg of capped and polyadenylated bicistronic mRNAs generated from pRDEF and pRDECF were transfected into U87MG cells usingLipofectamine. After 16 hours, cells were lysed and RLuc and FLuc activities were plotted similarly as in (B). Asterisk: significant withrespect to pRDEF.
analysis of this UTR revealed the presence of an IRES pat-tern. To test its functionality, the A UTR was cloned in theintercistronic region of a bicistronic vector, pRDEF, down-stream to a stem loop structure formed by an inactive EMCVIRES (DE), which was known to be inhibitory to ribosomalread-through (Johannes et al., 1999). Transfection of this
bicistronic plasmid resulted in a much stronger expression ofthe downstream FLuc cistron as compared to the empty vec-tor in U87MG (39-fold) and HepG2 cells (12-fold) (Figure4B). Others have also observed such tissue specific differ-ences in the readout from these plasmids (Borman et al.,1997; Ye et al., 1997; Creancier et al., 2000; Stoneley et al.,
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

410 S. Mittal et al.
Article in press - uncorrected proof
2000). Although DNA transfections with bicistronic vectorsare useful in determining whether a sequence contains anIRES, this method is considered inadequate because cellstransfected with such vector may give rise to transcripts gen-erated by aberrant splicing or cryptic promoters present inthe UTRs (Van Eden et al., 2004). The role of splicing wastherefore ruled out by performing RT-PCR using two sets ofprimers. We chose a primer pair (P1/P2), with the forwardprimer P1 from the region upstream to a chimeric intron, asthis intron is reported to contain splice donor sites (Holciket al., 2005). Another primer pair (P3/P4) was chosen fromRLuc and FLuc coding regions to rule out splicing in theintercistronic region. Northern blotting was also performedto look for shorter transcripts with lower expression levels.However, by both the techniques no shorter mRNA speciescould be detected (Figure 4C). Transfection of capped bicis-tronic mRNA in cells has been used to conclusively dem-onstrate the functionality of an IRES in the UTRs of severalmRNAs (Van Eden et al., 2004; Ray et al., 2006; Jiang etal., 2007). Using this strategy, we observed a 3.2-foldincrease (p-0.05) in FLuc/RLuc ratio from the UTR con-taining bicistronic mRNA (pRDECF) as compared to control,thereby confirming the functionality of IRES in the A UTR(Figure 4D). Such modest IRES activity with transfectionsof bicistronic capped mRNA has also been observed with c-myc and Smad59 UTRs (Stoneley et al., 2000; Shiroki et al.,2002). These authors speculated that some nuclear factorsmay be required for efficient IRES-mediated translation ini-tiation. Internal initiation of translation in cellular mRNAshas been shown to occur in response to a variety of condi-tions under which cap dependent translation is compromisedsuch as inflammation, angiogenesis and response to serum(Johannes and Sarnow, 1998; Johannes et al., 1999). CATLexpression is also induced by growth factors, tumour pro-moters, cAMP and various oncogenes (Troen et al., 1988;Kane and Gottesman, 1990; Collette et al., 2004). Furtherstudies to identify the conditions under which hCATL AIRES induces cathepsin L expression are currently inprogress.
The IRES dependent mechanism of translation is used bymany cellular mRNAs including those that encode for sometranscription, translation and growth factors (Iizuka et al.,1994; Teerink et al., 1995; Vagner et al., 1995; Gan andRhoads, 1996). Some IRES elements require at least a subsetof translation initiation factors and other RNA binding pro-teins to facilitate IRES mediated translation. Such IRES ele-ments do not support translation in cell free systems whichlack these non canonical RNA biding protein factors knownas IRES trans-activating factors (ITAFs). A number of cel-lular proteins such as polypyrimidine tract binding protein(PTB), poly (rC) binding protein, upstream of n-ras (unr),the lupus autoantigen (La), etc have been demonstrated tofunction as ITAFs (Fitzgerald and Semler, 2009). In this con-text it is noteworthy that Apaf-1 IRES was found be activein rabbit reticulocyte lysates only when the system was sup-plemented with PTB and unr (Mitchell et al., 2001). Simi-larly IRES activity of hepatitis C and polioviral mRNAs wasenhanced several folds after addition of La autoantigen (Ali
et al., 2000). Requirement of similar ITAF(s) may be one ofthe possible reasons for the absence of hCATL A IRES activ-ity in rabbit reticulocyte lyasate. This explains the compar-atively poor in vitro translational efficiency of this variantobserved in our earlier study (Arora and Chauhan, 2002).
In summary, our results demonstrate that the longest var-iant of human CATL, hCATL A, is efficiently translated invivo. This comparatively unstable variant contains a func-tional IRES, which contributes to increased translation fromthis mRNA in cancer cells. These results suggest that 59UTRmediated regulatory mechanisms are involved in modulatingmRNA stability and translation of hCATL A and therebyCATL expression. More importantly, it underscores a grow-ing appreciation that genes with critical cellular functions areregulated at multiple levels, to ensure the precise expressionof the encoded protein. Conceivably, targeting the nucleo-tides unique to the 59UTR region of only hCATL A by anti-sense/RNAi may prove useful in the management of clinicalconditions resulting from CATL over expression.
Materials and methods
Cell culture
U87MG, NIH3T3 and HepG2 cells were obtained from NCCS(National Center For Cell Science), Pune and grown in Dulbecco’smodified Eagle’s medium (Sigma, St. Louis, MO, USA) with glu-tamine, glucose and sodium pyruvate supplemented with 10% fetalcalf serum and 20 mg/ml ciprofloxacin, in a humidified atmosphereof 5% CO2 and 95% air at 378C.
Sequence analysis and secondary structure
predictions
The nucleotide sequence of hCATL A 59UTR was scanned for reg-ulatory motifs using a pattern matching software, ‘UTRScan’(Pesole et al., 2000). We also analysed this UTR for secondarystructures by the MFOLD web server using the default settings andthe temperature fixed at 378C (Zuker, 2003).
Construction of recombinant constructs
Various recombinant constructs used in this paper are illustrated inthe respective figures. The constructs pAutrL and pAIIIutrL havebeen described earlier (Arora and Chauhan, 2002). A common strat-egy was employed for cloning the A and AIII 59UTRs along withluciferase in pCDNA 3.1. The respective UTRs along with lucif-erase cDNA were removed from pAutrL and pAIIIutrL by Sac Idigestion (at the 39 end of luciferase). After blunt ending, these weredigested with HindIII and cloned into pCDNA 3.1 at EcoRV andHindIII sites. The resultant chimeric constructs were named aspAutrL/3.1 and pAIIIutrL/3.1.
For the generation of bicistronic construct, pRDECF, the fulllength 59UTR of hCATL A were cloned in the intercistronic regionof the pRDEF vector, downstream to the stem loop region, DE, atEcoRI and NcoI sites. The UTR of hCATL A was amplified usingprimers FCTSL27 and RCTSL311 containing EcoRI and NcoIrestriction sites. However, as NcoI was not unique in the pRDEFvector, we first subcloned the XhoI and XbaI digested fragmentfrom this vector into the pGL3C vector (Promega, Madison, WI,USA) and then used the resultant construct to ligate the PCR ampli-
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

59UTR mediated regulation of hCATL expression 411
Article in press - uncorrected proof
fied hCATL A UTR in EcoRI and NcoI sites. The resultant cloneswere again digested with XhoI and XbaI and ligated into pRDEFat the respective sites.
Actinomycin D treatment and Northern blotting
The NIH3T3 cells stably expressing hCATL A or hCATL AIII,maintained in G418 containing media, were plated a day prior totreatment. The next day, media was replaced with actinomycin D(Sigma) (5 mg/ml) containing media for 0, 2, 4 and 8 h followedby isolation of total cellular RNA using Tri Reagent (Sigma). Twen-ty micrograms of total RNA of each time point were resolved on1.2% denaturing agarose gel and transferred to a nylon membrane.The membrane was then hybridised with an wa-32Px labelled 473 bphCATL cDNA probe. After autoradiography, the CATL probe wasstripped and the same blot was hybridised with an wa-32Px-labelledmouse GAPDH probe for normalisation. Densitometric analysis ofthe resulting bands was performed for quantitation. A similar pro-cedure was applied for Northern analysis of U87MG cells transient-ly transfected with pRDEF or pRDECF plasmids. Forty-eight hourspost transfection, 35 mg of total RNA from these cells was used forblotting. An wa-32Px-labelled 450 bp firefly cDNA probe was gen-erated by PCR and used for hybridisation.
Cell labelling and immunoprecipitation
NIH3T3 cells stably expressing human cathepsin L splice variants,hCATL A or hCATL AIII were plated into six well plates at a celldensity of 2=105 cells/well. The next day cells were washed withPBS and starved in methionine free media. After an hour, cells werelabelled with methionine free media (FCS) containing 100 mCi/mlw35Sx methionine. After 2 h of labelling, cells were harvested in SDSlysis Buffer A. Equal numbers of counts (2=106 cpm) of radiola-belled proteins were incubated with 2 ml of rabbit antihumanproCATL polyclonal antibody at 4oC for 1 h. Then 30 ml of 20%protein A sepharose suspension was added and further incubated atRT with shaking for an additional hour. The immunoprecipitateswere washed and analysed on SDS PAGE and subjected to auto-radiography as described earlier (Chauhan et al., 1998).
Transient transfections
Cultured cells were plated into six-well plates one day prior to trans-fection. The next day, cells were transfected using Transfast Reagent(Promega) as per the manufacturer’s protocol. Forty-eight hoursafter transfection, the cells were harvested, lysed and assayed forluciferase activities as described earlier (Keerthivasan et al., 2007).Plasmid encoding renilla luciferase was transfected to normalise fortransfection efficiencies and beta galactosidase was used for nor-malisation in case of transfection of bicistronic constructs.
Reverse transcription and Real Time PCR analysis
Total RNA isolated from untransfected and transfected cells usingTri reagent was pretreated with DNAse I. A 1–5 mg sample of totalRNA were reverse transcribed using M-MuLV Reverse Transcrip-tase (MBI Fermentas, Vilnius, Lithuania) according to the manu-facturer’s instructions. Real-time PCR was performed in a Bio-Radi-Cycler (Bio-Rad, Hercules, CA, USA) using specific hCATL prim-ers from the coding region as described earlier (Keerthivasan et al.,2007). The relative mRNA levels of the hCATL transcripts werenormalised to reference transcript (18 srRNA) and relative foldchange was calculated using the 2-DDCT method.
RT-PCR was performed using two sets of primers: P1(sense) /P2(antisense), corresponding to the region upstream to the chimericintron in the bicistronic constructs and 59 end of FLuc, respectively,and P3 (sense)/P4 (antisense), corresponding to the 59 end of RLucand 39 end of FLuc, respectively. The respective sequences of theabove mentioned primers were: P1 (59-ACT TAA GCT GCA GAAGTT GGT C-39), P2 (59-CCA TCT TCC AGC GGA TAG AATGG-39), P3 (59-CGC AAG AAG ATG CAC CTG AT-39) and P4 (59-CGT CCA CAA ACA CAA CTC CTC-39). For construction ofpRDECF, the full length UTR of hCATL A was PCR amplifiedusing the sense primer FCTSL27, with EcoRI site (59-CGG AATTCG TGG AGT GCG CCT G-39) and the antisense primer,RCTSL311, with a NcoI site (59-AAC CAT GGG TGT AGG ATTCAT GT-39) for cloning into the respective sites of pREDF.
Transfection of bicistronic mRNAs
pRDEF and pRDECF plasmids were linearised with BamHI andsubsequently purified. One microgram of each of these plasmidswas then used for in vitro transcription to generate capped bicistro-nic mRNAs using AmpliCap-Max� T7 High Yield Message MakerKit (Epicentre Biotechnologies, Madison, WI, USA) according tothe manufacturer’s protocol. 0.8 mg of bicistronic mRNA was com-bined with 2 ml of lipofectamine (Invitrogen Corporation, Califor-nia, USA) and used for transfection into each well of a 24-wellplate as per the manufacturer’s recommendations. Sixteen hoursafter transfections, cells were harvested, lysed and assayed for lucif-erase activities.
Acknowledgments
We are thankful to Dr. S. Das, Department of Microbiology andCell Biology, Indian Institute of Science, Bangalore, India, for thekind gift of the bicistronic vector, pRDEF. This study was supportedby grants from Department of Science and Technology, Governmentof India, New Delhi. S. Mittal was a recipient of Research Asso-ciateship from the Indian Council of Medical Research, Governmentof India, New Delhi. Riyaz A. Mir is a recipient of Senior ResearchFellowship from the Indian Council of Medical Research, Govern-ment of India, New Delhi.
References
Ali, N., Pruijn, G.J., Kenan, D.J., Keene, J.D., and Siddiqui, A.(2000). Human La antigen is required for the hepatitis C virusinternal ribosome entry site-mediated translation. J. Biol. Chem.275, 27531–27540.
Arora, S. and Chauhan, S.S. (2002). Identification and characteri-zation of a novel human cathepsin L splice variant. Gene 293,123–131.
Bakhshi, R., Goel, A., Seth, P., Chhikara, P., and Chauhan, S.S.(2001). Cloning and characterization of human cathepsin L pro-moter. Gene 275, 93–101.
Borman, A.M., Le Mercier, P., Girard, M., and Kean, K.M. (1997).Comparison of picornaviral IRES-driven internal initiation oftranslation in cultured cells of different origins. Nucleic AcidsRes. 25, 925–932.
Chauhan, S.S., Goldstein, L.J., and Gottesman, M.M. (1991).Expression of cathepsin L in human tumors. Cancer Res. 51,1478–1481.
Chauhan, S.S., Popescu, N.C., Ray, D., Fleischmann, R., Gottesman,M.M., and Troen, B.R. (1993). Cloning, genomic organization,
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

412 S. Mittal et al.
Article in press - uncorrected proof
and chromosomal localization of human cathepsin L. J. Biol.Chem. 268, 1039–1045.
Chauhan, S.S., Ray, D., Kane, S.E., Willingham, M.C., and Gottes-man, M.M. (1998). Involvement of carboxy-terminal aminoacids in secretion of human lysosomal protease cathepsin L. Bio-chemistry 37, 8584–8594.
Collette, J., Ulku, A.S., Der, C.J., Jones, A., and Erickson, A.H.(2004). Enhanced cathepsin L expression is mediated by differ-ent Ras effector pathways in fibroblasts and epithelial cells. Int.J. Cancer 112, 190–199.
Creancier, L., Morello, D., Mercier, P., and Prats, A.C. (2000).Fibroblast growth factor 2 internal ribosome entry site (IRES)activity ex vivo and in transgenic mice reveals a stringent tissue-specific regulation. J. Cell. Biol. 150, 275–281.
Dhar, D., Roy, S., and Das, S. (2007). Translational control of theinterferon regulatory factor 2 mRNA by IRES element. NucleicAcids Res. 35, 5409–5421.
Fitzgerald, K.D. and Semler, B.L. (2009). Bridging IRES elementsin mRNAs to the eukaryotic translation apparatus. Biochim. Bio-phys. Acta 1789, 518–528.
Fortenberry, Y.M., Brandal, S., Bialas, R.C., and Church, F.C.(2010). Protein C inhibitor regulates both cathepsin L activityand cell-mediated tumour cell migration. Biochim. Biophys.Acta 1800, 580–590.
Frade, R., Rodrigues-Lima, F., Huang, S., Xie, K., Guillaume, N.,and Bar-Eli, M. (1998). Procathepsin-L, a proteinase that cleaveshuman C3 (the third component of complement), confers hightumorogenic and metastatic properties to human melanoma cells.Cancer Res. 58, 2733–2736.
Frade, R., Rousselet, N., and Jean, D. (2008). Intratumoral genedelivery of anti-cathepsin L single-chain variable fragment bylentiviral vector inhibits tumour progression induced by humanmelanoma cells. Cancer Gene Ther. 15, 591–604.
Gal, S. and Gottesman, M.M. (1988). Isolation and sequence of acDNA for human pro-(cathepsin L). Biochem. J. 253, 303–306.
Gan, W. and Rhoads, R.E. (1996). Internal initiation of translationdirected by the 59untranslated region of the mRNA for eIF4G, afactor involved in the picornavirus-induced switch from cap-dependent to internal initiation. J. Biol. Chem. 271, 623–626.
Gocheva, V. and Joyce, J.A. (2007). Cysteine cathepsins and thecutting edge of cancer invasion. Cell Cycle 6, 60–64.
Goulet, B.S.L., Leduy, L., Bogyo, M., Weber, E., Chauhan, S.S.,and Nepveu, A. (2007). Increased expression and activity ofnuclear cathepsin L in cancer cells suggests a novel mechanismof cell transformation. Mol. Cancer Res. 5, 899–907.
Holcik, M., Graber, T., Lewis, S.M., Lefebvre, C.A., Lacasse, E.,and Baird, S. (2005). Spurious splicing within the XIAP 59 UTRoccurs in the Rluc/Fluc but not the betagal/CAT bicistronicreporter system. RNA 11, 1605–1609.
Hughes, T.A. and Brady, H.J. (2005). Expression of axin2 is regu-lated by the alternative 59-untranslated regions of its mRNA. J.Biol. Chem. 280, 8581–8588.
Iizuka, N., Najita, L., Franzusoff, A., and Sarnow, P. (1994). Cap-dependent and cap-independent translation by internal initiationof mRNAs in cell-free extracts prepared from Saccharomycescerevisiae. Mol. Cell. Biol. 14, 7322–7330.
Jiang, H., Coleman, J., Miskimins, R., Srinivasan, R., and Miski-mins, W.K. (2007). Cap-independent translation through the p2759-UTR. Nucleic Acids Res. 35, 4767–4778.
Johannes, G. and Sarnow, P. (1998). Cap-independent polysomalassociation of natural mRNAs encoding c-myc, BiP, and eIF4Gconferred by internal ribosome entry sites. RNA 4, 1500–1513.
Johannes, G., Carter, M.S., Eisen, M.B., Brown, P.O., and Sarnow,P. (1999). Identification of eukaryotic mRNAs that are translated
at reduced cap binding complex eIF4F concentrations using acDNA microarray. Proc. Natl. Acad. Sci. USA 96, 13118–13123.
Kane, S.E. and Gottesman, M.M. (1990). The role of cathepsin Lin malignant transformation. Semin. Cancer Biol. 1, 127–136.
Katara, R., Mir, R.A., Shukla, A.A., Tiwari, A., Singh, N., andChauhan, S.S. (2010). Wild type p53-dependent transcriptionalupregulation of cathepsin L expression is mediated by C/EBPa
in human glioblastoma cells. Biol. Chem. 391, 1031–1040.Keerthivasan, S., Keerthivasan, G., Mittal, S., and Chauhan, S.S.
(2007). Transcriptional upregulation of human cathepsin L byVEGF in glioblastoma cells. Gene 399, 129–136.
Kos, J. and Lah, T.T. (1998). Cysteine proteinases and their endog-enous inhibitors: target proteins for prognosis, diagnosis andtherapy in cancer. Oncol. Rep. 5, 1349–1361.
Kozak, M. (1986). Influences of mRNA secondary structure on ini-tiation by eukaryotic ribosomes. Proc. Natl. Acad. Sci. USA 83,2850–2854.
Lankelma, J.M., Voorend, D.M., Barwari, T., Koetsveld, J., Van derSpek, A.H., De Porto, A.P., Van Rooijen, G., and Van Noorden,C.J. (2010). Cathepsin L, target in cancer treatment? Life Sci.86, 225–233.
Mitchell, S.A., Brown, E.C., Coldwell, M.J., Jackson, R.J., and Wil-lis, A.E. (2001). Protein factor requirements of the Apaf-1 inter-nal ribosome entry segment: roles of polypyrimidine tractbinding protein and upstream of N-ras. Mol. Cell. Biol. 21,3364–3374.
Payton, S.G., Haska, C.L., Flatley, R.M., Ge, Y., and Matherly, L.H.(2007). Effects of 59 untranslated region diversity on the post-transcriptional regulation of the human reduced folate carrier.Biochim. Biophys. Acta 1769, 131–138.
Pesole, G., Liuni, S., Grillo, G., Licciulli, F., Larizza, A., Maka-lowski, W., and Saccone, C. (2000). UTRdb and UTRsite: spe-cialized databases of sequences and functional elements of 59
and 39 untranslated regions of eukaryotic mRNAs. Nucleic AcidsRes. 28, 193–196.
Ray, P.S., Grover, R., and Das, S. (2006). Two internal ribosomeentry sites mediate the translation of p53 isoforms. EMBO Rep.7, 404–410.
Rescheleit, D.K., Rommerskirch, W.J., and Wiederanders, B.(1996). Sequence analysis and distribution of two new humancathepsin L splice variants. FEBS Lett. 394, 345–348.
Rousselet, N., Mills, L., Jean, D., Tellez, C., Bar-Eli, M., and Frade,R. (2004). Inhibition of tumorigenicity and metastasis of humanmelanoma cells by anti-cathepsin L single chain variable frag-ment. Cancer Res. 64, 146–151.
Seth, P., Mahajan, V.S., and Chauhan, S.S. (2003). Transcription ofhuman cathepsin L mRNA species hCATL B from a novel alter-native promoter in the first intron of its gene. Gene 321, 83–91.
Shiroki, K., Ohsawa, C., Sugi, N., Wakiyama, M., Miura, K., Wata-nabe, M., Suzuki, Y., and Sugano, S. (2002). Internal ribosomeentry site-mediated translation of Smad5 in vivo: requirementfor a nuclear event. Nucleic Acids Res. 30, 2851–2861.
Stoneley, M., Subkhankulova, T. Le, Quesne, J.P., Coldwell, M.J.,Jopling, C.L., Belsham, G.J., and Willis, A.E. (2000). Analysisof the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res.28, 687–694.
Teerink, H., Voorma, H.O., and Thomas, A.A.M. (1995). The humaninsulin like growth factor II leader 1 contains an internal ribo-somal entry site. Biochim. Biophys. Acta 1264, 403–408.
Troen, B.R., Ascherman, D., Atlas, D., and Gottesman, M.M.(1988). Cloning and expression of the gene for the major excret-ed protein of transformed mouse fibroblasts. A secreted lyso-
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM

59UTR mediated regulation of hCATL expression 413
Article in press - uncorrected proof
somal protease regulated by transformation. J. Biol. Chem. 263,254–261.
Urbich, C., Heeschen, C., Aicher, A., Sasaki, K., Bruhl, T., Farhadi,M.R., Vajkoczy, P., Hofmann, W.K., Peters, C., Pennacchio,L.A., et al. (2005). Cathepsin L is required for endothelial pro-genitor cell-induced neovascularization. Nat. Med. 11, 206–213.
Vagner, S., Gensac, M.-C., Maret, A., Bayard, F., Amalric, F., Prats,H., and Prats, A.-C. (1995). Alternative translation of humanfibroblast growth factor 2 mRNA occurs by internal entry ofribosomes. Mol. Cell. Biol. 15, 35–44.
Van Eden, M.E., Byrd, M.P., Sherrill, K.W., and Lloyd, R.E. (2004).Demonstrating internal ribosome entry sites in eukaryotic m-RNAs using stringent RNA test procedures. RNA 10, 720–730.
Wang, G., Guo, X., and Floros, J. (2005). Differences in the trans-lation efficiency and mRNA stability mediated by 59-UTR splicevariants of human SP-A1 and SP-A2 genes. Am. J. Physiol.Lung Cell. Mol. Physiol. 289, 497–508.
Ye, X., Fong, P., Iizuka, N., Choate, D., and Cavener, D.R. (1997).Ultrabithorax and Antennapedia 59 untranslated regions promotedevelopmentally regulated internal translation initiation. Mol.Cell. Biol. 17, 1714–1721.
Zuker, M. (2003). Mfold web server for nucleic acid folding andhybridization prediction. Nucleic Acids Res. 31, 3406–3415.
Received July 20, 2010; accepted December 13, 2010
Brought to you by | Columbia University Library The Burke Library New YorkAuthenticated | 128.59.62.83
Download Date | 9/9/12 3:32 AM
























![[VI]. Post-Transcriptional Processing and Post-Transcriptional Control of Gene Expression](https://img.pdfslide.us/doc/110x75/56815a87550346895dc7f921/vi-post-transcriptional-processing-and-post-transcriptional-control-of-gene.jpg)




