Embed Size (px)
Citation preview

Porcine reproductive and respiratory syndrome virus infectionactivates NOD2–RIP2 signal pathway in MARC-145 cells
Huiyuan Jing, Liurong Fang, Dang Wang, Zhen Ding, Rui Luo, Huanchun Chen,Shaobo Xiao n
State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China
a r t i c l e i n f o
Article history:Received 23 January 2014Returned to author for revisions12 February 2014Accepted 22 April 2014Available online 13 May 2014
Keywords:Porcine reproductive and respiratorysyndrome virus (PRRSV)NLRNOD2Inflammation
a b s t r a c t
Nucleotide-binding oligomerization domains (NOD)-like receptors (NLRs) evolve as a group of germline-encoded receptors that detect cytosolic pathogen-associated molecular patterns. Porcine reproductiveand respiratory syndrome virus (PRRSV) is an Arterivirus that has been devastating the swine industryworldwide. By examining the expression kinetics of ten selected NLRs, NOD2 and NLRP3 were found to becontinuously up-regulated in PRRSV-infected MARC-145 cells during 48 h of post-infection. Further studyrevealed that PRRSV infection enhanced the expression and phosphorylation of RIP2. Knockdown of NOD2and RIP2 by siRNA significantly decreased PRRSV-induced phosphorylation of NF-κB subunit p65, JNK, Erkand p38 MAPK, as well as the expression of IL-6, IL-8, TNF-α, and RANTES in MARC-145 cells. Moreover,increased expression of NOD2 and RIP2 mRNAwere observed in alveolar macrophages isolated from PRRSV-challenged piglets at 3, 7 and 10 day post-challenge. Collectively, our results revealed that PRRSV infectionactivates NOD2–RIP2 signaling pathway to induce pro-inflammatory response.
& 2014 Elsevier Inc. All rights reserved.
Introduction
Pattern-recognition receptors (PRRs) are groups of germline-encoded receptors that detect and respond rapidly to conservedbacterial- and viral-derived structures known as pathogen-associatedmolecular patterns (PAMPs) (Akira et al., 2006; Takeuchi and Akira,2010). To date, three main classes of PRRs have been described,namely Toll-like receptors (TLRs), retinoic acid inducible gene-I-likereceptors (RLRs), and nucleotide-binding oligomerization domain-like receptors (NLRs) (Bonardi et al., 2012). Compared with TLRs andRLRs, the functions of NLRs remain largely elusive (Lupfer andKanneganti, 2013b). The NLRs family proteins, characterized by atripartite architecture, own more members (23 in human and 34 inmouse) and participate in a diverse set of innate immune signalingpathways (Ting et al., 2008). The typical structure of NLRs reveal aC-terminal leucine-rich repeat (LRR) domain that is involved inpathogens recognition, a centrally located nucleotide-binding oligo-merization domain that mediates self-oligomerization and activation,and a N-terminal effector domainwhich is responsible for the proteininteraction and signal transduction (Shaw et al., 2010). Depend on thecomposition of their N-terminal effector domains, the NLRs family
can be divided into four subfamilies (Schroder and Tschopp, 2010).The NLRA and NLRB subfamily each consists of one member inhuman, CIITA and NAIP, respectively. The NLRP subfamily membersare featured by causing inflammasome assembly to activate caspase-1and lead to maturation of interleukin (IL)-1β and IL-18 (Schroder andTschopp, 2010). The NLRC subfamily proteins, such as NOD1 andNOD2, contain a caspase recruitment domain (CARD) and participatein initiation pro-inflammatory responses (Ting et al., 2010).
Despite the crucial role of NLRs in mounting immune responsesagainst bacterial infection have progressed remarkably, questionsconcerned with virus recognition function of most NLRs remains(Lupfer and Kanneganti, 2013a). To date, the NLRP3 inflammasomeis the best characterized member of the NLRP subfamily which isactivated by viruses including DNA viruses from the familiesPoxviridae, Herpesviridae, Adenoviridae and RNA viruses from thefamilies Orthomyxoviridae, Paramyxioviridae, Rhabdoviridae, Picor-naviridae, and Flaviviridae (Lupfer and Kanneganti, 2013a). Morerecently, NLRC subfamily member NOD2 has also been shown tofunction as a viral PRR and trigger multiple signaling in responseto virus invasion (Sabbah et al., 2009; Lupfer et al., 2013).
Porcine reproductive and respiratory syndrome (PRRS) is a viralinfectious disease characterized by severe reproductive failurein sows and respiratory distress in piglets and growing pigs(Albina, 1997; Meulenberg, 2000). The causative agent, PRRSvirus (PRRSV), is a single-stranded positive-sense RNA virusclassified within the family Arteriviridae (Rossow, 1998; Snijder
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/yviro
Virology
http://dx.doi.org/10.1016/j.virol.2014.04.0310042-6822/& 2014 Elsevier Inc. All rights reserved.
n Correspondence to: Laboratory of Infectious Diseases, College of VeterinaryMedicine, Huazhong Agricultural University, 1 Shi-zi-shan Street, Wuhan 430070,China. Tel.: þ86 27 8728 6884; fax: þ86 27 8728 2608.
E-mail address: [email protected] (S. Xiao).
Virology 458-459 (2014) 162–171
brought to you by COREView metadata, citation and similar papers at core.ac.uk
provided by Elsevier - Publisher Connector
and Meulenberg, 1998). Since its emergence in the late 1980s,PRRS has continuously been a threat to the global swine industry,causing high economic losses (Neumann et al., 2005). Unfortu-nately, both traditional control strategies and conventional vac-cines are insufficient to provide sustainable control of PRRS(Darwich et al., 2010; Meng, 2000; Murtaugh et al., 2010). A majorobstacle in the development of a successful PRRS vaccine is theunconventional immune response of pigs to the virus (Calzada-Nova et al., 2011; Kimman et al., 2009; Yoo et al., 2010), whichremains poorly characterized. A better understanding of the virus-host interactions in PRRSV infection will facilitate development ofmore effective control measures (Beura et al., 2010; Chen et al.,2010; Kim et al., 2010). Although previous studies have showedthat PRRSV infection regulated TLRs or RLRs-mediated immuneresponses (Beura et al., 2010; Liu et al., 2009; Luo et al., 2008; Sanget al., 2008a, 2008b; Zhang et al., 2013b), the role of NLRs in PRRSVimmunity remains largely unclear.
In this study, we investigated the expression dynamics of tenselected NLRs during PRRSV infection and demonstrated PRRSVinduced NOD2 and RIP2 expression in vitro and in vivo. We also
revealed that PRRSV activated NOD2–RIP2 pathway and utilizedthis pathway to induce inflammatory production.
Results
PRRSV infection steadily up-regulates the expression of NOD2and NLRP3
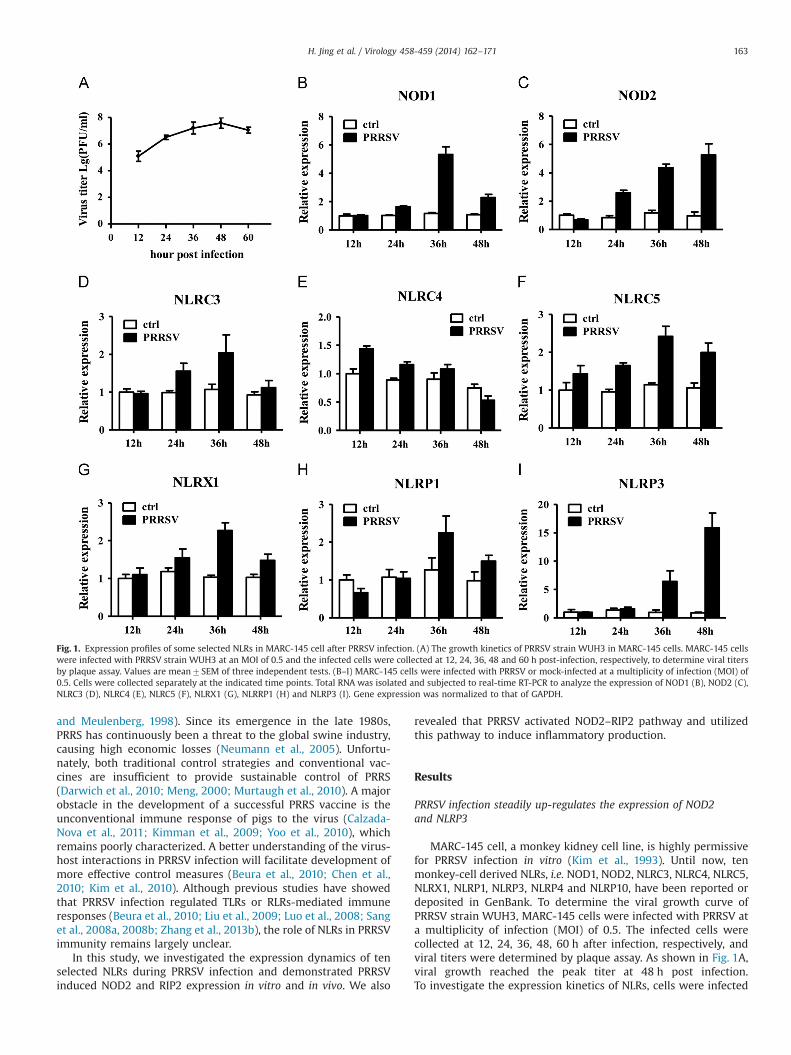
MARC-145 cell, a monkey kidney cell line, is highly permissivefor PRRSV infection in vitro (Kim et al., 1993). Until now, tenmonkey-cell derived NLRs, i.e. NOD1, NOD2, NLRC3, NLRC4, NLRC5,NLRX1, NLRP1, NLRP3, NLRP4 and NLRP10, have been reported ordeposited in GenBank. To determine the viral growth curve ofPRRSV strain WUH3, MARC-145 cells were infected with PRRSV ata multiplicity of infection (MOI) of 0.5. The infected cells werecollected at 12, 24, 36, 48, 60 h after infection, respectively, andviral titers were determined by plaque assay. As shown in Fig. 1A,viral growth reached the peak titer at 48 h post infection.To investigate the expression kinetics of NLRs, cells were infected
Fig. 1. Expression profiles of some selected NLRs in MARC-145 cell after PRRSV infection. (A) The growth kinetics of PRRSV strain WUH3 in MARC-145 cells. MARC-145 cellswere infected with PRRSV strain WUH3 at an MOI of 0.5 and the infected cells were collected at 12, 24, 36, 48 and 60 h post-infection, respectively, to determine viral titersby plaque assay. Values are mean7SEM of three independent tests. (B–I) MARC-145 cells were infected with PRRSV or mock-infected at a multiplicity of infection (MOI) of0.5. Cells were collected separately at the indicated time points. Total RNA was isolated and subjected to real-time RT-PCR to analyze the expression of NOD1 (B), NOD2 (C),NLRC3 (D), NLRC4 (E), NLRC5 (F), NLRX1 (G), NLRRP1 (H) and NLRP3 (I). Gene expression was normalized to that of GAPDH.
H. Jing et al. / Virology 458-459 (2014) 162–171 163
with PRRSV (MOI¼0.5) and collected at 12, 24, 36, and 48 h afterinfection, respectively, and subjected to real-time RT-PCR using theprimers listed in Supplemental Table 1. As shown in Fig. 1, PRRSVinfection robustly increased the mRNA expression of NOD1(Fig. 1B), NOD2 (Fig. 1C) and NLRP3 (Fig. 1I) to more than5-folds. In contrast, only marginal (less than 3-fold) changes ofother NLRs mRNA could be observed upon PRRSV infection(Fig. 1D–H). Of note, NOD2 was up-regulated at 24 h post-infec-tion, and increased at a steady-state level, with maximal produc-tion at 48 h post-infection; NLRP3 was up-regulated after 36 hpost-infection; the expression of NOD1 up-regulated nearly6-fold at 36 h post-infection but significantly decreased at 48 h
post-infection. We did not get stable results owning to lowexpression of NLRP4 and NLRP10 (data not shown). Consideringthat the possible role of NLRP3 to PRRSV-mediated IL-1β releasehas been reported recently (Zhang et al., 2013a), we focus oursubsequent investigations on defining the role of NOD2 duringPRRSV infection.
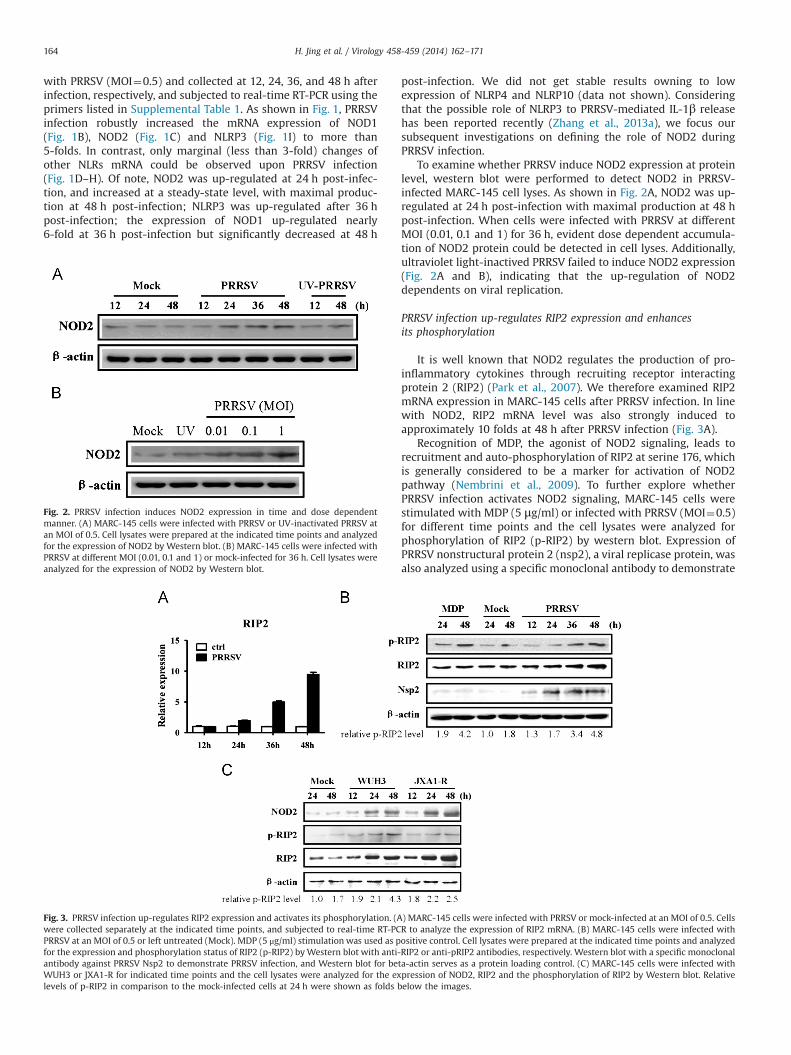
To examine whether PRRSV induce NOD2 expression at proteinlevel, western blot were performed to detect NOD2 in PRRSV-infected MARC-145 cell lyses. As shown in Fig. 2A, NOD2 was up-regulated at 24 h post-infection with maximal production at 48 hpost-infection. When cells were infected with PRRSV at differentMOI (0.01, 0.1 and 1) for 36 h, evident dose dependent accumula-tion of NOD2 protein could be detected in cell lyses. Additionally,ultraviolet light-inactived PRRSV failed to induce NOD2 expression(Fig. 2A and B), indicating that the up-regulation of NOD2dependents on viral replication.
PRRSV infection up-regulates RIP2 expression and enhancesits phosphorylation
It is well known that NOD2 regulates the production of pro-inflammatory cytokines through recruiting receptor interactingprotein 2 (RIP2) (Park et al., 2007). We therefore examined RIP2mRNA expression in MARC-145 cells after PRRSV infection. In linewith NOD2, RIP2 mRNA level was also strongly induced toapproximately 10 folds at 48 h after PRRSV infection (Fig. 3A).
Recognition of MDP, the agonist of NOD2 signaling, leads torecruitment and auto-phosphorylation of RIP2 at serine 176, whichis generally considered to be a marker for activation of NOD2pathway (Nembrini et al., 2009). To further explore whetherPRRSV infection activates NOD2 signaling, MARC-145 cells werestimulated with MDP (5 μg/ml) or infected with PRRSV (MOI¼0.5)for different time points and the cell lysates were analyzed forphosphorylation of RIP2 (p-RIP2) by western blot. Expression ofPRRSV nonstructural protein 2 (nsp2), a viral replicase protein, wasalso analyzed using a specific monoclonal antibody to demonstrate
Fig. 2. PRRSV infection induces NOD2 expression in time and dose dependentmanner. (A) MARC-145 cells were infected with PRRSV or UV-inactivated PRRSV atan MOI of 0.5. Cell lysates were prepared at the indicated time points and analyzedfor the expression of NOD2 by Western blot. (B) MARC-145 cells were infected withPRRSV at different MOI (0.01, 0.1 and 1) or mock-infected for 36 h. Cell lysates wereanalyzed for the expression of NOD2 by Western blot.
Fig. 3. PRRSV infection up-regulates RIP2 expression and activates its phosphorylation. (A) MARC-145 cells were infected with PRRSV or mock-infected at an MOI of 0.5. Cellswere collected separately at the indicated time points, and subjected to real-time RT-PCR to analyze the expression of RIP2 mRNA. (B) MARC-145 cells were infected withPRRSV at an MOI of 0.5 or left untreated (Mock). MDP (5 μg/ml) stimulation was used as positive control. Cell lysates were prepared at the indicated time points and analyzedfor the expression and phosphorylation status of RIP2 (p-RIP2) byWestern blot with anti-RIP2 or anti-pRIP2 antibodies, respectively. Western blot with a specific monoclonalantibody against PRRSV Nsp2 to demonstrate PRRSV infection, and Western blot for beta-actin serves as a protein loading control. (C) MARC-145 cells were infected withWUH3 or JXA1-R for indicated time points and the cell lysates were analyzed for the expression of NOD2, RIP2 and the phosphorylation of RIP2 by Western blot. Relativelevels of p-RIP2 in comparison to the mock-infected cells at 24 h were shown as folds below the images.
H. Jing et al. / Virology 458-459 (2014) 162–171164
virus replication. As shown in Fig. 3B, the amount of phosphorylatedRIP2 increased markedly along with Nsp2 since 24 h after PRRSVinfection, suggested that PRRSV induced RIP2 phosphorylation was
virus replication dependent. Densitometry analysis showed that thelevel of p-RIP2 at 48 h post PRRSV infection was 4.8-fold higher thanthat of the mock-infected cells at 24 h. Compared with mock-infected
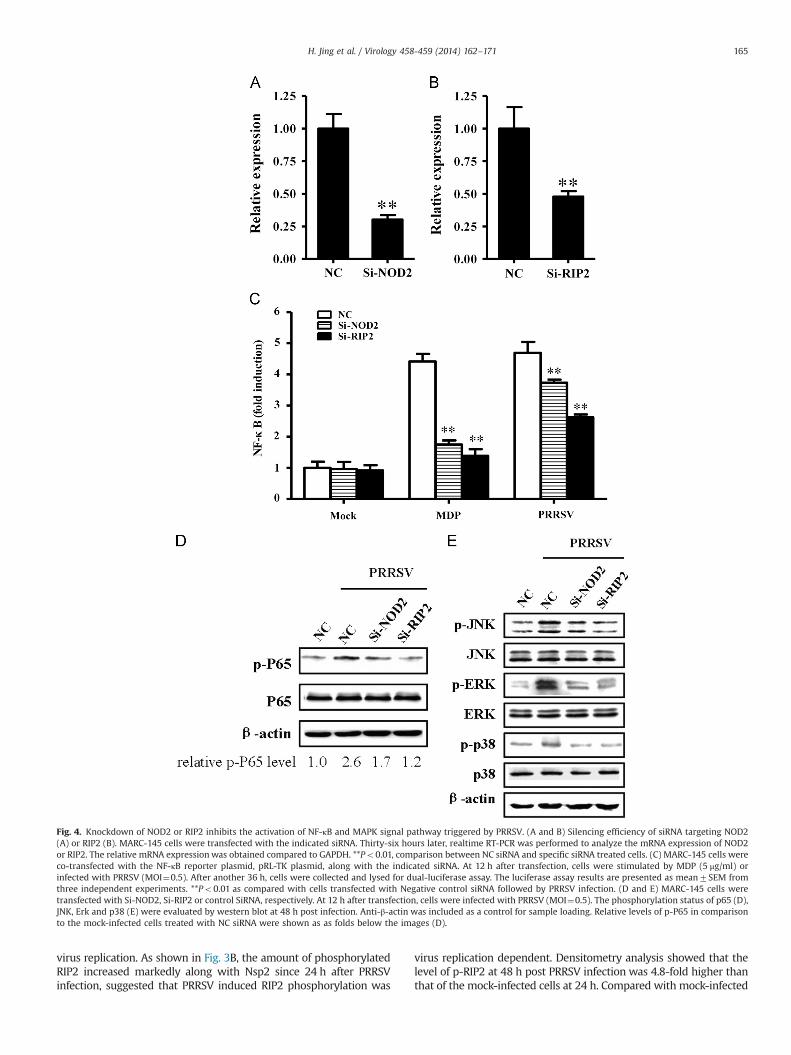
Fig. 4. Knockdown of NOD2 or RIP2 inhibits the activation of NF-κB and MAPK signal pathway triggered by PRRSV. (A and B) Silencing efficiency of siRNA targeting NOD2(A) or RIP2 (B). MARC-145 cells were transfected with the indicated siRNA. Thirty-six hours later, realtime RT-PCR was performed to analyze the mRNA expression of NOD2or RIP2. The relative mRNA expression was obtained compared to GAPDH. **Po0.01, comparison between NC siRNA and specific siRNA treated cells. (C) MARC-145 cells wereco-transfected with the NF-κB reporter plasmid, pRL-TK plasmid, along with the indicated siRNA. At 12 h after transfection, cells were stimulated by MDP (5 μg/ml) orinfected with PRRSV (MOI¼0.5). After another 36 h, cells were collected and lysed for dual-luciferase assay. The luciferase assay results are presented as mean7SEM fromthree independent experiments. **Po0.01 as compared with cells transfected with Negative control siRNA followed by PRRSV infection. (D and E) MARC-145 cells weretransfected with Si-NOD2, Si-RIP2 or control SiRNA, respectively. At 12 h after transfection, cells were infected with PRRSV (MOI¼0.5). The phosphorylation status of p65 (D),JNK, Erk and p38 (E) were evaluated by western blot at 48 h post infection. Anti-β-actin was included as a control for sample loading. Relative levels of p-P65 in comparisonto the mock-infected cells treated with NC siRNA were shown as as folds below the images (D).
H. Jing et al. / Virology 458-459 (2014) 162–171 165
cells at 24 h, the levels of p-RIP2 in PRRSV infected cells at 12, 24 and36 h were 1.3, 1.7 and 3.4-fold, respectively (Fig. 3B). MDP stimulationdid not alter RIP2 protein expression, nonetheless increased itsphosphorylation. Interestingly, although RIP2 protein level seemedto be rather constant within 24 h post infection, an obvious up-regulation was detected after 36 h which consistent with the mRNAdata shown in Fig. 3A.
Previous studies have demonstrated that virulence strain andattenuated strain of PRRSV had different ability to induce pro-inflammatory cytokines (Patel et al., 2010; Wang et al., 2013;Yu et al., 2013). To investigate whether there are strains differencein up-regulation and activation of NOD2 and RIP2, PRRSV JXA1-R(Han et al., 2009), an attenuated vaccine strain which wasextensively used in China, was tested. To this end, MARC-145 cellswere infected with WUH3 or JXA1-R for different time pointsand cell lysates were analyzed for expression of NOD2, RIP2 andphosphorylation of RIP2 by Western blot. As shown in Fig. 3C,JXA1-R infection also up-regulated the expression of NOD2 andRIP2, as well as the phosphorylation of RIP2, however, relativelower levels of RIP2 phosphorylation were detected in JXA1-R-infected cells than in WUH3-infected cells at 48 h post infection.
Knockdown of NOD2 and RIP2 inhibits the activation of NF-κBand MAPK signal pathway triggered by PRRSV
Following recruitment of RIP2 by NOD2, RIP2 undergoesautophosphorylation and ubiquitination and subsequent activatesNF-κB and MAPK pathway (Tigno-Aranjuez and Abbott, 2012).To investigate whether NOD2 and RIP2 were involved in PRRSV-induced NF-κB activities, siRNA-mediated interference was pre-formed. The knockdown efficiency of NOD2 and RIP2-specificsiRNA were over 50% (Fig. 4A and B). Next, MARC-145 cells wereco-transfected with the NF-κB-Luc, pRL-TK, along with siRNAtargeting NOD2, RIP2 or negative control siRNA, respectively.At 12 h after transfection, cells were stimulated by MDP or infectedwith PRRSV for 36 h. As expected, a 5 fold induction was observed
after MDP-treatment for 36 h. In addition, NOD2 and RIP2 specificsiRNA did not inhibit the basal NF-κB activity, whereas reducedPRRSV-induced NF-κB activities (Fig. 4C). We also detected thephosphorylation status of NF-κB subunit p65 (p-P65) in siRNA-transfected cells after PRRSV infection. As shown in Fig. 4D, thephosphorylation of endogenous p65 (Fig. 4D) induced by PRRSVwas inhibited to a similar extent by NOD2 and RIP2-specific siRNAin comparison to NC (negative control siRNA). These data indicatedthat NOD2 and RIP2 are involved in PRRSV-induced NF-κBactivities.
In parallel to the activation of NF-κB signaling pathway, theNOD2–RIP2 complex also triggers the phosphorylation of JNK, Erkand p38 MAPK to induce the transcription of pro-inflammatorygenes (Lee and Lee, 2012; Song et al., 2013; Ting et al., 2010;Yin et al., 2012; Yu et al., 2013). To better understand whetherinterference of NOD2 and RIP2 affects PRRSV-induced MAPKsignaling, we conducted siRNA-mediated reduction of NOD2 andRIP2 in MARC-145 cells and inoculated with PRRSV (MOI¼0.5) for48 h. As shown in Fig. 4E, while the total JNK, Erk and p38 MAPKwas not changed among Si-NOD2, Si-RIP2 and NC treatmentgroups, the phosphorylation status of JNK, Erk and p38 MAPKwere significantly inhibited. These results demonstrated thatNOD2 and RIP2 are in part responsible for PRRSV induced activa-tion of MAPK pathway in PRRSV infected MARC-145 cells.
NOD2 and RIP2 silencing impair PRRSV-induced expressionof NF-κB-dependent pro-inflammatory cytokines
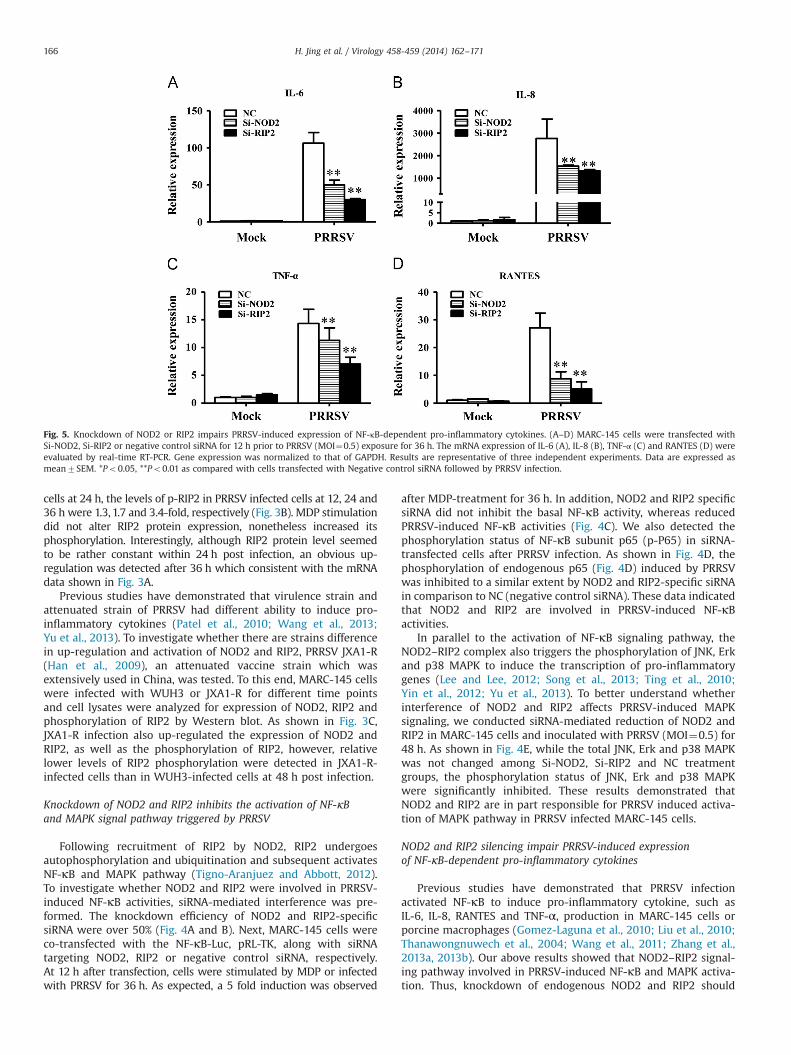
Previous studies have demonstrated that PRRSV infectionactivated NF-κB to induce pro-inflammatory cytokine, such asIL-6, IL-8, RANTES and TNF-α, production in MARC-145 cells orporcine macrophages (Gomez-Laguna et al., 2010; Liu et al., 2010;Thanawongnuwech et al., 2004; Wang et al., 2011; Zhang et al.,2013a, 2013b). Our above results showed that NOD2–RIP2 signal-ing pathway involved in PRRSV-induced NF-κB and MAPK activa-tion. Thus, knockdown of endogenous NOD2 and RIP2 should
Fig. 5. Knockdown of NOD2 or RIP2 impairs PRRSV-induced expression of NF-κB-dependent pro-inflammatory cytokines. (A–D) MARC-145 cells were transfected withSi-NOD2, Si-RIP2 or negative control siRNA for 12 h prior to PRRSV (MOI¼0.5) exposure for 36 h. The mRNA expression of IL-6 (A), IL-8 (B), TNF-α (C) and RANTES (D) wereevaluated by real-time RT-PCR. Gene expression was normalized to that of GAPDH. Results are representative of three independent experiments. Data are expressed asmean7SEM. *Po0.05, **Po0.01 as compared with cells transfected with Negative control siRNA followed by PRRSV infection.
H. Jing et al. / Virology 458-459 (2014) 162–171166
impair PRRSV-induced pro-inflammatory cytokine production. Totest this hypothesis, we assayed these cytokines mRNA expressionin MARC-145 cells treated with NOD2 or RIP2 specific siRNA priorto PRRSV infection. As shown in Fig. 5, a dramatic reducedexpression of IL-6 (Fig. 5A), IL-8 (Fig. 5B), RANTES (Fig. 5C) andTNF-α (Fig. 5D) could be observed in cells transfected with NOD2or RIP2-specific siRNA in comparison with cells transfected NCsiRNA. Taken collectively, these data confirmed that NOD2–RIP2signaling is involved in the efficient induction of pro-inflammatoryresponses by PRRSV.
PRRSV infection induces NOD2 and RIP2 expression in vivo
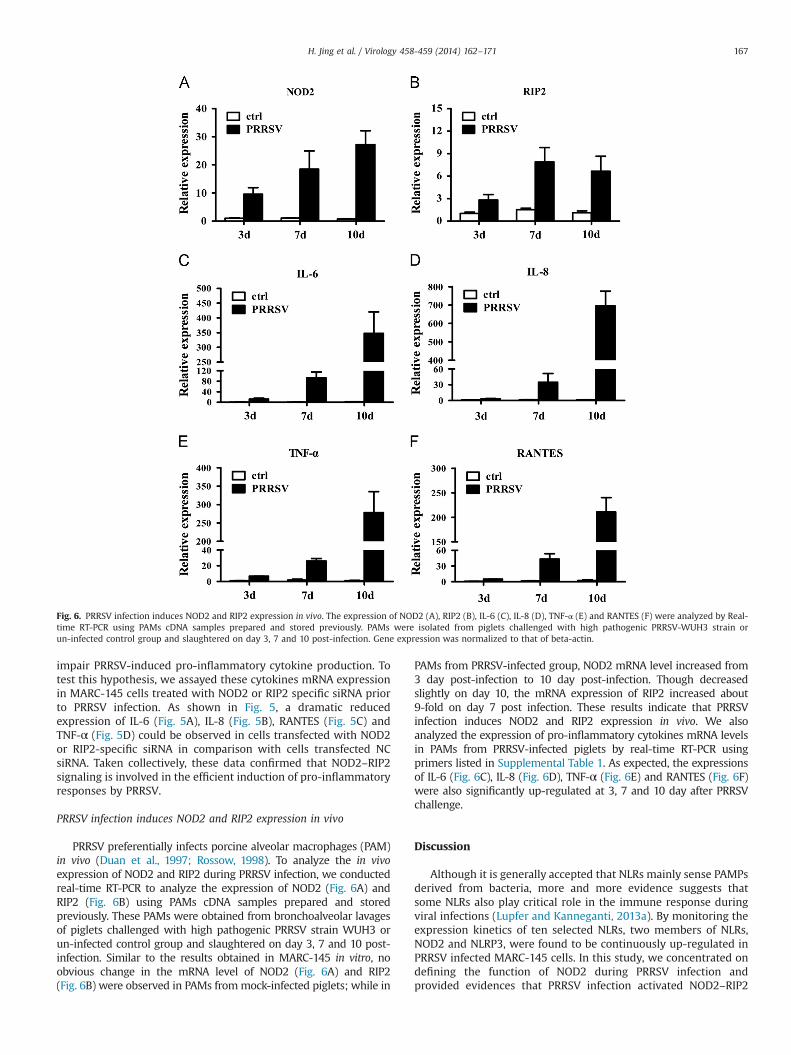
PRRSV preferentially infects porcine alveolar macrophages (PAM)in vivo (Duan et al., 1997; Rossow, 1998). To analyze the in vivoexpression of NOD2 and RIP2 during PRRSV infection, we conductedreal-time RT-PCR to analyze the expression of NOD2 (Fig. 6A) andRIP2 (Fig. 6B) using PAMs cDNA samples prepared and storedpreviously. These PAMs were obtained from bronchoalveolar lavagesof piglets challenged with high pathogenic PRRSV strain WUH3 orun-infected control group and slaughtered on day 3, 7 and 10 post-infection. Similar to the results obtained in MARC-145 in vitro, noobvious change in the mRNA level of NOD2 (Fig. 6A) and RIP2(Fig. 6B) were observed in PAMs frommock-infected piglets; while in
PAMs from PRRSV-infected group, NOD2 mRNA level increased from3 day post-infection to 10 day post-infection. Though decreasedslightly on day 10, the mRNA expression of RIP2 increased about9-fold on day 7 post infection. These results indicate that PRRSVinfection induces NOD2 and RIP2 expression in vivo. We alsoanalyzed the expression of pro-inflammatory cytokines mRNA levelsin PAMs from PRRSV-infected piglets by real-time RT-PCR usingprimers listed in Supplemental Table 1. As expected, the expressionsof IL-6 (Fig. 6C), IL-8 (Fig. 6D), TNF-α (Fig. 6E) and RANTES (Fig. 6F)were also significantly up-regulated at 3, 7 and 10 day after PRRSVchallenge.
Discussion
Although it is generally accepted that NLRs mainly sense PAMPsderived from bacteria, more and more evidence suggests thatsome NLRs also play critical role in the immune response duringviral infections (Lupfer and Kanneganti, 2013a). By monitoring theexpression kinetics of ten selected NLRs, two members of NLRs,NOD2 and NLRP3, were found to be continuously up-regulated inPRRSV infected MARC-145 cells. In this study, we concentrated ondefining the function of NOD2 during PRRSV infection andprovided evidences that PRRSV infection activated NOD2–RIP2
Fig. 6. PRRSV infection induces NOD2 and RIP2 expression in vivo. The expression of NOD2 (A), RIP2 (B), IL-6 (C), IL-8 (D), TNF-α (E) and RANTES (F) were analyzed by Real-time RT-PCR using PAMs cDNA samples prepared and stored previously. PAMs were isolated from piglets challenged with high pathogenic PRRSV-WUH3 strain orun-infected control group and slaughtered on day 3, 7 and 10 post-infection. Gene expression was normalized to that of beta-actin.
H. Jing et al. / Virology 458-459 (2014) 162–171 167

signal pathway. We also demonstrated that PRRSV utilized thispathway to induce pro-inflammatory cytokine production.
Among the various clinical signs of PRRSV-infection, interstitialpneumonia is considered as the one of the most remarkablefeatures (Rossow, 1998), suggesting that the inflammatoryresponse plays an important role in infection and pathogenesisinduced by PRRSV. A large number of studies have reported thatPRRSV infection induced inflammatory cytokines productionin vivo and in vitro, however, only a few of them, such as IL-15and RANTES, have been mechanically characterized (Fu et al.,2012; Gomez-Laguna et al., 2010; Liu et al., 2010; Wang et al.,2011). Interestingly, the induction of these cytokines mentionedabove during PRRSV infection all depend on NF-κB, the keytranscription factor for inflammation pathway (Fu et al., 2012;Wang et al., 2011). Indeed, previous studies have demonstratedthat PRRSV infection activated NF-κB pathway in MARC-145 cellsand PAMs at late phases of infection through mechanism(s) currently unclear (Lee and Kleiboeker, 2005). Our presentresults supported these observations and provided a new insightinto the mechanism by which PRRSV triggers NF-κB activities. Atthe same time, it should be noted that knockdown of NOD2 orRIP2 only partially impaired PRRSV-triggered NF-κB activities. Wespeculated that this was probably due to the activation of TLRssignaling. An obvious reason is that beside NLRs, other PRRs suchas TLRs and RLRs, were also involved in virus-induced inflamma-tory responses (Akira et al., 2006; Lupfer and Kanneganti, 2013a).Although previous studies have demonstrated that PRRSV antag-onized the RLRs pathway (Luo et al., 2008), the expressions ofcertain TLRs were up-regulated after PRRSV infection (Liu et al.,2009; Zhang et al., 2013b). But until now, in the course of PRRSVinfection, the specific TLR(s) associated with NF-κB activation haveyet unidentified and the cross-talks between NOD2 and TLRsmediated signaling has been documented with limited data inprevious studies. These are interesting issues and have beeninvestigating in our laboratory. On the other hand, it should benote that the interaction between PRRSV and NF-κB activities is adynamic process and PRRSV also encodes several nonstructuralproteins, such as nsp1α, nsp1β, nsp2, and nsp11, to negativelyregulate NF-κB activities and evade the host immune surveillancein certain conditions (Kim et al., 2010; Shi et al., 2011; Song et al.,2010; Sun et al., 2010; Yoo et al., 2010). PRRSV appears to acquirethe capability to redirect NF-κB activities for its own benefit. Theunderlying mechanism that could explain how PRRSV regulatesNF-κB activities to its own advantage awaits further clarification.
In this study, we found that PRRSV infection up-regulatedNOD2 expression in vitro and in vivo and activated NOD2–RIP2pathway. Some viruses have been reported to utilize NOD2–RIP2pathway to accelerate secondary bacterial invasion. For instance,murine norovirus-1 (MNV-1) infection induces NOD1 and NOD2expression, which contributes to lethality of secondary E. coliinfection (Kim et al., 2011). In addition, stimulation of humanprimary cells with Respiratory syncytial virus (RSV) and MDPinduces a synergy in proinflammatory cytokine production viaNOD2–RIP2 pathway, which might lead to increased inflammationand disease severity (Vissers et al., 2012). Most cases of PRRSVinfection in pigs are complicated by secondary opportunisticbacterial infections and Streptococcus suis, Mycoplasma hyopneu-moniaeare and Haemophilus parasuis are commonly isolated fromPRRSV infected pigs (Thanawongnuwech et al., 2000, 2004;Yu et al., 2012). While the exact mechanisms of interaction betweenPRRSV and secondary bacterial infection remain unclear, second-ary bacterial invasion might be in part responsible for systemicinflammation, lungs damage and even to animal death duringPRRSV infection (Zhang et al., 2013b). Interestingly, we found thatstimulation of PRRSV-primed MARC-145 cells with MDP resultedin a synergistic activation of NF-κB promoter, but not vice verse
(data not shown). Similar phenomenon was reported in RSV-infected human PBMCs (Vissers et al., 2012). Since NOD2 candetect distinct microbial PAMPs and trigger the activation ofinflammatory response, the exaggerated production of proinflam-matory cytokines via these pathways might contribute to theexacerbated clinical symptoms in combined infections of PRRSVand secondary pathogens (Qiao et al., 2011; Zhang et al., 2013b).
Apart from involvement of NF-κB activities and inflammatoryresponses, NOD2 has also been implicated in the regulation ofautophagy (Homer et al., 2012; Travassos et al., 2010). Autophagyis an evolutionarily conserved lysosome-dependent degradationpathway that acts in the maintenance of cellular homeostasis andplays important functions in viral replication and pathogenesis(Levine and Klionsky, 2004). The role of NOD2 in this process wasinitially proposed because NOD2 interacts and colocalized withATG16L1, a key regulator of the autophagy process (Travassoset al., 2010). More recently, Lupfer et al. further demonstrated arole for NOD2–RIP2 signaling in the regulation of influenza A virus-induced mitophagy, substantiating the link between NOD2 andautophagy (Lupfer et al., 2013). Previously, we and two other groupshave reported that PRRSV infection triggers the formation ofautophagosomes (Chen et al., 2012; Liu et al., 2012; Sun et al.,2012). These observations, together with the present study, not onlyimplied that there are some unidentified relationship betweenPRRSV-triggered autophagy, NOD2 up-regulation and the activatedthe NOD2–RIP2 signaling axis, but further reinforced the conceptthat NOD2 pathway plays multiple roles in viral immunity.
Although many studies have demonstrated a role for NOD2 inthe recognition of bacterial peptidoglycan, there is still no evi-dence for direct binding to their ligands (Moreira and Zamboni,2012). The role of NOD2 pathway in viral immunity was firstreported in 2009 by Sabbah et al. who demonstrated that NOD2can function as a cytoplasmic PRR to recognize synthetic or viralssRNA. Furthermore, treatment of cells with synthetic or viralssRNA resulted in higher expression of NOD2 (Sabbah et al., 2009).Although the activation mechanisms of NOD2 pathway by virusinfection are incompletely understood, virus-triggered IFN-β pro-duction has been demonstrated as a possible mechanism for virus-induced NOD2 up-regulation (Kim et al., 2011; Sabbah et al., 2009;Vissers et al., 2012). However, PRRSV is known as a poor inducerfor IFN (Calzada-Nova et al., 2011), it is therefore plausible tospeculate that PRRSV might encode one or more structural or non-structural to activate NOD2 pathway, even though no such reportas viral proteins are involved in NOD2 recognition thus far.
In summary, we report that NOD2 and RIP2 are inducibleduring PRRSV infection in vitro and in vivo. Our findings solidifythat NOD2–RIP2 signaling is involved in PRRSV induced pro-inflammatory response by influencing NF-кB and MAPK pathwayand subsequent production of inflammatory cytokines. In thecontext that many new functions and downstream events ofNOD2 pathway have been revealing, it is interesting that theactivated NOD2–RIP2 pathway during PRRSV infection might beassociated with the secondary bacterial invasion, autophagy andother processes. Dissection of these issues is important for betterunderstanding of numerous aspects of PRRSV pathogenesis includ-ing inflammation as well as cellular antiviral immunity.
Materials and methods
Cells, viruses, and regents
MARC-145 cells were cultured and maintained in Dulbecco'smodified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum(FBS), 100 U/mL penicillin, 10 μg/mLstreptomycin sulfate and then incubated at 37 1C in a humidified
H. Jing et al. / Virology 458-459 (2014) 162–171168

5% CO2 incubator. PRRSV strain WUH3 is a highly pathogenictype 2 (North American) PRRSV which was isolated from the brainof pigs suffering from the “high fever” syndrome in China at theend of 2006 (Li et al., 2009). JXA1-R is an attenuated vaccine strainderived from highly pathogenic PRRSV strain JXA1 (Han et al.,2009; Tian et al., 2007). PRRSV was amplified and titered in MARC-145 cells. UV inactive PRRSV were irradiated under short-wave(254 nm) ultraviolet light for 1 h. Loss of infectivity was confirmedby the inability of the UV light-exposed viruses to produce acytopathic effect on monolayers of MARC-145 cells. Polyclonalanti-bodies against p65, JNK, Erk, P38, RIP2 and their phosphory-lated forms were purchased from Cell Signaling Technology. NOD2polyclonal anti-body was purchased from Santa Cruz. Anti-β-actinwas purchased from Beyotime (China). PRRSV Nsp2 monoclonalantibody was produced from hybridoma cells derived from Sp2/0myeloma cells and spleen cells of BALB/c mice immunized withrecombinant Nsp2 protein of PRRSV strain WUH3. Muramyldipeptide (MDP) was purchased from Sigma-Aldrich.
Plaque assay for determination of PRRSV titers
MARC-145 cells were infected with PRRSV at a multiplicity ofinfection (MOI) of 0.5. Cells were collected at indicated time pointsand stored at �80 1C until analysis of viral replication by plaqueassay. Plaque assay was essentially performed as described pre-viously (Luo et al., 2011). Briefly, 95% confluent MARC-145 cellsgrown in six-well tissue culture plates were infected for 1 h with10-fold serial dilutions (1000 ml) of PRRSV-containing samples.After three washes with PBS (pH 7.4), cells were overlaid with 1.8%(w/v) Bacto agar mixed 1:1 with 2�DMEM containing 0.05 mg/mlneutral red. Plaques were counted 2 days post-infection. Theaverage plaque number and standard deviations were calculatedfrom three independent experiments. Virus titers were expressedas plaque forming units (PFU)/ml.
RNA exaction and quantitative real-time RT-PCR
MARC-145 cells grown in 24-well plates were infected withPRRSV or mock-infected at a multiplicity of infection (MOI) of 0.5.Total RNA were isolated at the indicated time points using TRIzolreagent (Invitrogen). Real-time RT-PCR was performed using SYBRGreen Real Time PCR Master Mix (Toyobo Biologics, Osaka, Japan)in the LightCycler 480 (Roche Molecular Biochemicals). Individualtranscripts in each sample were assayed three times. The PCRconditions were as follows: initial denaturation for 10 min at95 1C, followed by 40 cycles of 15 s at 95 1C, 15 s at 58 1C and40 s at 72 1C. The fold change in gene expression relative to normalwas calculated using the delta delta cycles to threshold (ΔΔCT)method. Primers (Supplemental Table 1) were designed using thePrimer Express software (version 3.0; Applied Biosystems,Carlsbad, CA).
Transfection and luciferase reporter assay
Transient transfection was performed using Lipofectamine2000 (Invitrogen). MARC-145 cells were seeded on 24-well platesat a density of 2–4�105 cells/ well and cultured until the cellsreached approximately 70–80% confluence, and were then trans-fected with the indicated plasmids or siRNA. For each transfection,0.2 μg of the NF-κB reporter plasmid (pNF-kB-Luc purchasedfrom Stratagene) along with 0.05 μg of pRL-TK and 80 nM ofsiRNA were used. Cell extracts were collected 36 h post PRRSVinfection. Firefly and Renilla luciferase activities were determinedusing the Dual-luciferase reporter assay system (Promega) accordingto the manufacturer's instructions. Values for each sample werenormalized using the Renilla luciferase values.
SiRNA-mediated interference
The siRNA targeting monkey NOD2, RIP2 or negative controlsiRNA were synthesized by GenePharma (China). Transient trans-fection of siRNA was performed using lipofectamine 2000 (Invi-trogen) according to the manufacturer's instructions. The dose ofsiRNA used for transfection was optimized in the primary experi-ments and no appreciable cellular toxicity was observed. SiRNAsequences used are as follows: Si-NOD2, sense 50-CAGCCCUGAU-GACAUUUCUTT-30, antisense 50-AGAAAUGUCAUCAGGGCUGTT-30;Si-RIP2, sense 50-AGAAUAUCCUAUUGGACAATT-30, antisense 50-UUGU-CCAAUAGGAUAUUCUTT-30; Negative control, sense 50-UUCUCCGAAC-GUGUCACGUTT-30, antisense 50-ACGUGACACGUUCGGAGAATT-30.
Western blot assay
Cell extracts were prepared by adding 200 μL 2� lysis bufferA (LBA) (65 mM Tris–HCl [pH 6.8], 4% sodium dodecyl sulfate,3% DL-dithiothreitol, and 40% glycerol). The extracts were boiled insodium dodecylsulphate (SDS) protein sample buffer (2% SDS,60 m M Tris–HCl [pH 6.8], 10% glycerol, 0.001% bromophenolblue,and 0.33% β-mercaptoethanol), then resolved by 10% acrylamidesodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were electroblotted onto a PVDFmembrane (Millipore, Billerica, MA). Membranes were blockedwith 10% (w/v) dried skim milk in tris-buffered saline containingTween 20 (TBST). Western blot was performed using anti-p65,p-p65, JNK, p-JNK, Erk, p-Erk, P38, p-P38, NOD2, RIP2 and p-RIP2Rabbit polyclonal antibodies. PRRSV Nsp2 monoclonal antibody(1:1000) used for the detection of Nsp2. β-Actin was detected withan anti-beta-actin monoclonal antibody (MAb) (Beyotime, China)to serve as loading control. Secondary antibodies, horseradishperoxidase-conjugated anti-mouse and anti-rabbit IgG antibody,were used at a dilution of 1:3000, and protein bands werevisualized using SuperSignal West Pio Luminol kit (Pierce).
Statistical analysis
All experiments were performed at least three times withreproducible results. Statistical analysis and generation of graphswere performed using GraphPad Prism software. Results arepresented as mean and standard error (SEM) from three indepen-dent experiments. The Student's t-test was used to determinestatistical significance. P-values less than 0.05 were consideredstatistically significant, and P-values less than 0.01 were consid-ered highly significant.
Acknowledgments
This work was supported by the National Basic ResearchProgram (973) of China (2014CB522700), the National NaturalSciences Foundation of China (31225027 and 31172326), the KeyGrant Project of Chinese Ministry of Education (313025), and theFundamental Research Funds for the Central Universities(2013PY043 and 2013PY055).
Appendix A. Supplementary material
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.virol.2014.04.031.
H. Jing et al. / Virology 458-459 (2014) 162–171 169

References
Akira, S., Uematsu, S., Takeuchi, O., 2006. Pathogen recognition and innateimmunity. Cell 124 (4), 783–801.
Albina, E., 1997. Epidemiology of porcine reproductive and respiratory syndrome(PRRS): an overview. Vet. Microbiol. 55 (1–4), 309–316.
Beura, L.K., Sarkar, S.N., Kwon, B., Subramaniam, S., Jones, C., Pattnaik, A.K., Osorio,F.A., 2010. Porcine reproductive and respiratory syndrome virus nonstructuralprotein 1beta modulates host innate immune response by antagonizing IRF3activation. J. Virol. 84 (3), 1574–1584.
Bonardi, V., Cherkis, K., Nishimura, M.T., Dangl, J.L., 2012. A new eye on NLRproteins: focused on clarity or diffused by complexity? Curr. Opin. Immunol. 24(1), 41–50.
Calzada-Nova, G., Schnitzlein, W.M., Husmann, R.J., Zuckermann, F.A., 2011. NorthAmerican porcine reproductive and respiratory syndrome viruses inhibit type Iinterferon production by plasmacytoid dendritic cells. J. Virol. 85 (6),2703–2713.
Chen, Q., Fang, L., Wang, D., Wang, S., Li, P., Li, M., Luo, R., Chen, H., Xiao, S., 2012.Induction of autophagy enhances porcine reproductive and respiratory syn-drome virus replication. Virus Res. 163 (2), 650–655.
Chen, Z., Zhou, X., Lunney, J.K., Lawson, S., Sun, Z., Brown, E., Christopher-Hennings, J.,Knudsen, D., Nelson, E., Fang, Y., 2010. Immunodominant epitopes in nsp2 ofporcine reproductive and respiratory syndrome virus are dispensable for replica-tion, but play an important role in modulation of the host immune response.J. Gen. Virol. 91 (Pt 4), 1047–1057.
Darwich, L., Diaz, I., Mateu, E., 2010. Certainties, doubts and hypotheses in porcinereproductive and respiratory syndrome virus immunobiology. Virus Res. 154(1–2), 123–132.
Duan, X., Nauwynck, H.J., Pensaert, M.B., 1997. Virus quantification and identifica-tion of cellular targets in the lungs and lymphoid tissues of pigs at differenttime intervals after inoculation with porcine reproductive and respiratorysyndrome virus (PRRSV). Vet. Microbiol. 56 (1–2), 9–19.
Fu, Y., Quan, R., Zhang, H., Hou, J., Tang, J., Feng, W.H., 2012. Porcine reproductiveand respiratory syndrome virus induces interleukin-15 through the NF-kappaBsignaling pathway. J. Virol. 86 (14), 7625–7636.
Gomez-Laguna, J., Salguero, F.J., Barranco, I., Pallares, F.J., Rodriguez-Gomez, I.M.,Bernabe, A., Carrasco, L., 2010. Cytokine expression by macrophages in the lungof pigs infected with the porcine reproductive and respiratory syndrome virus.J. Comp. Pathol. 142 (1), 51–60.
Han, W., Wu, J.J., Deng, X.Y., Cao, Z., Yu, X.L., Wang, C.B., Zhao, T.Z., Chen, N.H., Hu, H.H.,Bin, W., Hou, L.L., Wang, L.L., Tian, K.G., Zhang, Z.Q., 2009. Molecular mutationsassociated with the in vitro passage of virulent porcine reproductive andrespiratory syndrome virus. Virus Genes 38 (2), 276–284.
Homer, C.R., Kabi, A., Marina-Garcia, N., Sreekumar, A., Nesvizhskii, A.I., Nickerson,K.P., Chinnaiyan, A.M., Nunez, G., McDonald, C., 2012. A dual role for receptor-interacting protein kinase 2 (RIP2) kinase activity in nucleotide-bindingoligomerization domain 2 (NOD2)-dependent autophagy. J. Biol. Chem. 287(30), 25565–25576.
Kim, H.S., Kwang, J., Yoon, I.J., Joo, H.S., Frey, M.L., 1993. Enhanced replication ofporcine reproductive and respiratory syndrome (PRRS) virus in a homogeneoussubpopulation of MA-104 cell line. Arch. Virol. 133 (3–4), 477–483.
Kim, O., Sun, Y., Lai, F.W., Song, C., Yoo, D., 2010. Modulation of type I interferoninduction by porcine reproductive and respiratory syndrome virus and degra-dation of CREB-binding protein by non-structural protein 1 in MARC-145 andHeLa cells. Virology 402 (2), 315–326.
Kim, Y.G., Park, J.H., Reimer, T., Baker, D.P., Kawai, T., Kumar, H., Akira, S., Wobus, C.,Nunez, G., 2011. Viral infection augments Nod1/2 signaling to potentiatelethality associated with secondary bacterial infections. Cell Host Microbe 9(6), 496–507.
Kimman, T.G., Cornelissen, L.A., Moormann, R.J., Rebel, J.M., Stockhofe-Zurwieden, N.,2009. Challenges for porcine reproductive and respiratory syndrome virus (PRRSV)vaccinology. Vaccine 27 (28), 3704–3718.
Levine, B., Klionsky, D.J., 2004. Development by self-digestion: molecular mechan-isms and biological functions of autophagy. Dev. Cell 6 (4), 463–477.
Lee, S.M., Kleiboeker, S.B., 2005. Porcine arterivirus activates the NF-kappaBpathway through IkappaB degradation. Virology 342 (1), 47–59.
Lee, Y.J., Lee, C., 2012. Stress-activated protein kinases are involved in porcinereproductive and respiratory syndrome virus infection and modulate virus-induced cytokine production. Virology 427 (2), 80–89.
Li, B., Fang, L., Xu, Z., Liu, S., Gao, J., Jiang, Y., Chen, H., Xiao, S., 2009. Recombinationin vaccine and circulating strains of porcine reproductive and respiratorysyndrome viruses. Emerg. Infect. Dis. 15 (12), 2032–2035.
Liu, Y., Shi, W., Zhou, E., Wang, S., Hu, S., Cai, X., Rong, F., Wu, J., Xu, M., Li, L., 2010.Dynamic changes in inflammatory cytokines in pigs infected with highlypathogenic porcine reproductive and respiratory syndrome virus. Clin. VaccineImmunol. 17 (9), 1439–1445.
Liu, C.H., Chaung, H.C., Chang, H.L., Peng, Y.T., Chung, W.B., 2009. Expression of Toll-like receptor mRNA and cytokines in pigs infected with porcine reproductiveand respiratory syndrome virus. Vet. Microbiol. 136 (3–4), 266–276.
Liu, Q., Qin, Y., Zhou, L., Kou, Q., Guo, X., Ge, X., Yang, H., Hu, H., 2012. Autophagysustains the replication of porcine reproductive and respiratory virus in hostcells. Virology 429 (2), 136–147.
Luo, R., Fang, L., Jin, H., Jiang, Y., Wang, D., Chen, H., Xiao, S., 2011. Antiviral activityof type I and type III interferons against porcine reproductive and respiratorysyndrome virus (PRRSV). Antivir. Res. 91 (2), 99–101.
Luo, R., Xiao, S., Jiang, Y., Jin, H., Wang, D., Liu, M., Chen, H., Fang, L., 2008. Porcinereproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol. Immunol.45 (10), 2839–2846.
Lupfer, C., Kanneganti, T.D., 2013a. The expanding role of NLRs in antiviralimmunity. Immunol. Rev. 255 (1), 13–24.
Lupfer, C., Kanneganti, T.D., 2013b. Unsolved mysteries in NLR biology. Front.Immunol. 4, 285.
Lupfer, C., Thomas, P.G., Anand, P.K., Vogel, P., Milasta, S., Martinez, J., Huang, G.,Green, M., Kundu, M., Chi, H., Xavier, R.J., Green, D.R., Lamkanfi, M., Dinarello, C.A.,Doherty, P.C., Kanneganti, T.D., 2013. Receptor interacting protein kinase2-mediated mitophagy regulates inflammasome activation during virus infection.Nat. Immunol. 14 (5), 480–488.
Meng, X.J., 2000. Heterogeneity of porcine reproductive and respiratory syndromevirus: implications for current vaccine efficacy and future vaccine development.Vet. Microbiol. 74 (4), 309–329.
Meulenberg, J.J., 2000. PRRSV, the virus. Vet. Res. 31 (1), 11–21.Moreira, L.O., Zamboni, D.S., 2012. NOD1 and NOD2 signaling in infection and
inflammation. Front. Immunol. 3, 328.Murtaugh, M.P., Stadejek, T., Abrahante, J.E., Lam, T.T., Leung, F.C., 2010. The ever-
expanding diversity of porcine reproductive and respiratory syndrome virus.Virus Res. 154 (1–2), 18–30.
Nembrini, C., Kisielow, J., Shamshiev, A.T., Tortola, L., Coyle, A.J., Kopf, M., Marsland,B.J., 2009. The kinase activity of Rip2 determines its stability and consequentlyNod1- and Nod2-mediated immune responses. J. Biol. Chem. 284 (29),19183–19188.
Neumann, E.J., Kliebenstein, J.B., Johnson, C.D., Mabry, J.W., Bush, E.J., Seitzinger, A.H.,Green, A.L., Zimmerman, J.J., 2005. Assessment of the economic impact of porcinereproductive and respiratory syndrome on swine production in the United States.J. Am. Vet. Med. Assoc. 227 (3), 385–392.
Park, J.H., Kim, Y.G., McDonald, C., Kanneganti, T.D., Hasegawa, M., Body-Malapel,M., Inohara, N., Nunez, G., 2007. RICK/RIP2 mediates innate immune responsesinduced through Nod1 and Nod2 but not TLRs. J. Immunol. 178 (4), 2380–2386.
Patel, D., Nan, Y., Shen, M., Ritthipichai, K., Zhu, X., Zhang, Y.J., 2010. Porcinereproductive and respiratory syndrome virus inhibits type I interferon signalingby blocking STAT1/STAT2 nuclear translocation. J. Virol. 84 (21), 11045–11055.
Qiao, S., Feng, L., Bao, D., Guo, J., Wan, B., Xiao, Z., Yang, S., Zhang, G., 2011. Porcinereproductive and respiratory syndrome virus and bacterial endotoxin act insynergy to amplify the inflammatory response of infected macrophages. Vet.Microbiol. 149 (1–2), 213–220.
Rossow, K.D., 1998. Porcine reproductive and respiratory syndrome. Vet. Pathol. 35(1), 1–20.
Sabbah, A., Chang, T.H., Harnack, R., Frohlich, V., Tominaga, K., Dube, P.H., Xiang, Y.,Bose, S., 2009. Activation of innate immune antiviral responses by Nod2. Nat.Immunol. 10 (10), 1073–1080.
Sang, Y., Ross, C.R., Rowland, R.R., Blecha, F., 2008a. Toll-like receptor 3 activationdecreases porcine arterivirus infection. Viral. Immunol. 21 (3), 303–313.
Sang, Y., Yang, J., Ross, C.R., Rowland, R.R., Blecha, F., 2008b. Molecular identificationand functional expression of porcine Toll-like receptor (TLR) 3 and TLR7. Vet.Immunol. Immunopathol. 125 (1–2), 162–167.
Schroder, K., Tschopp, J., 2010. The inflammasomes. Cell 140 (6), 821–832.Shaw, P.J., Lamkanfi, M., Kanneganti, T.D., 2010. NOD-like receptor (NLR) signaling
beyond the inflammasome. Eur. J. Immunol. 40 (3), 624–627.Snijder, E.J., Meulenberg, J.J., 1998. The molecular biology of arteriviruses. J. Gen.
Virol. 79 (Pt 5), 961–979.Song, C., Krell, P., Yoo, D., 2010. Nonstructural protein 1alpha subunit-based
inhibition of NF-kappaB activation and suppression of interferon-beta produc-tion by porcine reproductive and respiratory syndrome virus. Virology 407 (2),268–280.
Song, S., Bi, J., Wang, D., Fang, L., Zhang, L., Li, F., Chen, H., Xiao, S., 2013. Porcinereproductive and respiratory syndrome virus infection activates IL-10 produc-tion through NF-kappaB and p38 MAPK pathways in porcine alveolar macro-phages. Dev. Comp. Immunol. 39 (3), 265–272.
Sun, Z., Chen, Z., Lawson, S.R., Fang, Y., 2010. The cysteine protease domain ofporcine reproductive and respiratory syndrome virus nonstructural protein2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 84(15), 7832–7846.
Sun, M.X., Huang, L., Wang, R., Yu, Y.L., Li, C., Li, P.P., Hu, X.C., Hao, H.P., Ishag, H.A.,Mao, X., 2012. Porcine reproductive and respiratory syndrome virus inducesautophagy to promote virus replication. Autophagy 8 (10), 1443–1447.
Takeuchi, O., Akira, S., 2010. Pattern recognition receptors and inflammation. Cell140 (6), 805–820.
Thanawongnuwech, R., Brown, G.B., Halbur, P.G., Roth, J.A., Royer, R.L., Thacker, B.J.,2000. Pathogenesis of porcine reproductive and respiratory syndrome virus-induced increase in susceptibility to Streptococcus suis infection. Vet. Pathol. 37(2), 143–152.
Thanawongnuwech, R., Thacker, B., Halbur, P., Thacker, E.L., 2004. Increasedproduction of proinflammatory cytokines following infection with porcinereproductive and respiratory syndrome virus and Mycoplasma hyopneumo-niae. Clin. Diagn. Lab. Immunol. 11 (5), 901–908.
Tian, K., Yu, X., Zhao, T., Feng, Y., Cao, Z., Wang, C., Hu, Y., Chen, X., Hu, D., Tian, X.,Liu, D., Zhang, S., Deng, X., Ding, Y., Yang, L., Zhang, Y., Xiao, H., Qiao, M.,Wang, B., Hou, L., Wang, X., Yang, X., Kang, L., Sun, M., Jin, P., Wang, S., Kitamura, Y.,Yan, J., Gao, G.F., 2007. Emergence of fatal PRRSV variants: unparalleled outbreaksof atypical PRRS in China and molecular dissection of the unique hallmark. PLoSOne 2 (6), e526.
H. Jing et al. / Virology 458-459 (2014) 162–171170

Tigno-Aranjuez, J.T., Abbott, D.W., 2012. Ubiquitination and phosphorylation in theregulation of NOD2 signaling and NOD2-mediated disease. Biochim. Biophys.Acta 1823 (11), 2022–2028.
Ting, J.P., Duncan, J.A., Lei, Y., 2010. How the noninflammasome NLRs function in theinnate immune system. Science 327 (5963), 286–290.
Ting, J.P., Lovering, R.C., Alnemri, E.S., Bertin, J., Boss, J.M., Davis, B.K., Flavell, R.A.,Girardin, S.E., Godzik, A., Harton, J.A., Hoffman, H.M., Hugot, J.P., Inohara, N.,Mackenzie, A., Maltais, L.J., Nunez, G., Ogura, Y., Otten, L.A., Philpott, D., Reed, J.C.,Reith, W., Schreiber, S., Steimle, V., Ward, P.A., 2008. The NLR gene family:a standard nomenclature. Immunity 28 (3), 285–287.
Travassos, L.H., Carneiro, L.A., Ramjeet, M., Hussey, S., Kim, Y.G., Magalhaes, J.G.,Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I.G., Allaoui, A., Jones, N.L.,Nunez, G., Girardin, S.E., Philpott, D.J., 2010. Nod1 and Nod2 direct autophagyby recruiting ATG16L1 to the plasma membrane at the site of bacterial entry.Nat. Immunol. 11 (1), 55–62.
Vissers, M., Remijn, T., Oosting, M., de Jong, D.J., Diavatopoulos, D.A., Hermans, P.W.,Ferwerda, G., 2012. Respiratory syncytial virus infection augments NOD2signaling in an IFN-beta-dependent manner in human primary cells. Eur. J.Immunol. 42 (10), 2727–2735.
Wang, R., Nan, Y., Yu, Y., Zhang, Y.J., 2013. Porcine reproductive and respiratorysyndrome virus Nsp1beta inhibits interferon-activated JAK/STAT signal trans-duction by inducing karyopherin-alpha1 degradation. J. Virol. 87 (9),5219–5228.
Wang, Y., Luo, R., Fang, L., Wang, D., Bi, J., Chen, H., Xiao, S., 2011. Porcinereproductive and respiratory syndrome virus (PRRSV) infection activateschemokine RANTES in MARC-145 cells. Mol. Immunol. 48 (4), 586–591.
Yin, S., Huo, Y., Dong, Y., Fan, L., Yang, H., Wang, L., Ning, Y., Hu, H., 2012. Activationof c-Jun NH(2)-terminal kinase is required for porcine reproductive andrespiratory syndrome virus-induced apoptosis but not for virus replication.Virus Res. 166 (1–2), 103–108.
Yoo, D., Song, C., Sun, Y., Du, Y., Kim, O., Liu, H.C., 2010. Modulation of host cellresponses and evasion strategies for porcine reproductive and respiratorysyndrome virus. Virus Res. 154 (1–2), 48–60.
Yu, J., Wu, J., Zhang, Y., Guo, L., Cong, X., Du, Y., Li, J., Sun, W., Shi, J., Peng, J., Yin, F.,Wang, D., Zhao, P., Wang, J., 2012. Concurrent highly pathogenic por-cine reproductive and respiratory syndrome virus infection accelerates Hae-mophilus parasuis infection in conventional pigs. Vet. Microbiol. 158 (3–4),316–321.
Yu, Y., Wang, R., Nan, Y., Zhang, L., Zhang, Y., 2013. Induction of STAT1 phosphor-ylation at serine 727 and expression of proinflammatory cytokines by porcinereproductive and respiratory syndrome virus. PLoS One 8 (4), e61967.
Zhang, K., Hou, Q., Zhong, Z., Li, X., Chen, H., Li, W., Wen, J., Wang, L., Liu, W., Zhong, F.,2013a. Porcine reproductive and respiratory syndrome virus activates inflamma-somes of porcine alveolar macrophages via its small envelope protein E. Virology442 (2), 156–162.
Zhang, L., Liu, J., Bai, J., Wang, X., Li, Y., Jiang, P., 2013b. Comparative expression ofToll-like receptors and inflammatory cytokines in pigs infected with differentvirulent porcine reproductive and respiratory syndrome virus isolates. Virol. J.10 (1), 135.
H. Jing et al. / Virology 458-459 (2014) 162–171 171





























